 Alejandro Moreno-Domínguez
Alejandro Moreno-Domínguez Olaia Colinas1,2,3
Olaia Colinas1,2,3 Tarik Smani
Tarik Smani José López-Barneo
José López-Barneo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 03 March 2023
Sec. Vascular Physiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1142354
This article is part of the Research TopicVascular Pathophysiology in HypoxiaView all 7 articles
An adequate supply of oxygen (O2) is essential for most life forms on earth, making the delivery of appropriate levels of O2 to tissues a fundamental physiological challenge. When O2 levels in the alveoli and/or blood are low, compensatory adaptive reflexes are produced that increase the uptake of O2 and its distribution to tissues within a few seconds. This paper analyzes the most important acute vasomotor responses to lack of O2 (hypoxia): hypoxic pulmonary vasoconstriction (HPV) and hypoxic vasodilation (HVD). HPV affects distal pulmonary (resistance) arteries, with its homeostatic role being to divert blood to well ventilated alveoli to thereby optimize the ventilation/perfusion ratio. HVD is produced in most systemic arteries, in particular in the skeletal muscle, coronary, and cerebral circulations, to increase blood supply to poorly oxygenated tissues. Although vasomotor responses to hypoxia are modulated by endothelial factors and autonomic innervation, it is well established that arterial smooth muscle cells contain an acute O2 sensing system capable of detecting changes in O2 tension and to signal membrane ion channels, which in turn regulate cytosolic Ca2+ levels and myocyte contraction. Here, we summarize current knowledge on the nature of O2 sensing and signaling systems underlying acute vasomotor responses to hypoxia. We also discuss similarities and differences existing in O2 sensors and effectors in the various arterial territories.
Blood oxygen tension (PO2) is an important local factor in the regulation of circulation (Wadsworth, 1994; López-Barneo et al., 1999; Weir et al., 2005; Jackson, 2016). Arteries and arterioles are able to sense changes in PO2 in the blood or surrounding tissues and acutely respond with a vasomotor response (constriction or dilation) that depends on the vascular territory. Resistance pulmonary arteries constrict in response to a decrease in alveolar PO2 (alveolar hypoxia), thereby diverting blood flow from poorly ventilated lung regions towards areas with a higher PO2 to optimize ventilation/perfusion ratio. In conditions of persistent hypoxemia (low blood PO2), hypoxic pulmonary vasoconstriction (HPV) contributes to the pathogenesis of pulmonary hypertension (Harder et al., 1985; Weir et al., 2005; Waypa et al., 2006; Sylvester et al., 2012; Sommer et al., 2016). Unlike resistance pulmonary arteries, conduit pulmonary vessels and most systemic arteries (in particular in the skeletal muscle as well as in coronary and cerebral territories) dilate in response to hypoxia. Hypoxic vasodilation (HVD) also has a relevant physiologic role as it favors the perfusion of O2-deprived tissues (Detar, 1980; Marriott and Marshall, 1990; Wadsworth, 1994; Weir et al., 1997; Jackson, 2016).
Despite their considerable physiological relevance, the mechanisms underlying HPV and HVD have remained elusive and are still under debate. While substances released from the vascular endothelium (such as nitric oxide) can modulate vessel diameter, numerous studies have nevertheless demonstrated the existence of endothelium-independent hypoxic vasomotor responses and suggested the existence of an acute O2 sensor in vascular smooth muscle (VSM) (Hales, 1985; Madden et al., 1992; Pearce, 1995; Jackson, 2016). Indeed, a growing body of experimental work indicates that, in the case of HPV, the intrinsic O2 sensing mechanism in VSM involves the mitochondria, which appear to function as O2 sensors by initiating a redox-signaling pathway that leads to the activation of downstream effectors regulating vascular tone (Weir et al., 2005; Waypa et al., 2006). In this review, we briefly examine the most important mechanisms by which different VSM cells detect and respond to acute changes in O2 tension, focusing on the similarities and differences between the sensing and signaling pathways underlaying HPV and HVD. Given the limitations of scope and space, many relevant aspects of the extensive literature on the role of VSM in the pathophysiology of pulmonary and systemic vessels are considered sparingly, with readers referred to cited papers for a more in-depth explanation.
HPV is a fundamental homeostatic mechanism that evolved to generate acute adaptive compensatory responses to regional changes in ventilation of the lung. HPV is better observed in the most penetrating and smaller caliber resistance arteries close to the alveolar region, although other segments of the pulmonary circulation (including the pulmonary veins) also constrict in response to hypoxia. In contrast, hypoxia induces vasodilation in the proximal conducting vessels (main trunk and first branches of the pulmonary artery), as it does in the systemic vasculature. HPV is rapidly and reversibly triggered within a few seconds in response to hypoxia (Madden et al., 1992; Archer and Michelakis, 2002). The vasomotor response exhibits two phases when alveolar hypoxia is maintained: an initial acute phase involving rapid vascular contraction and an increase in vascular resistance, with subsequent partial relaxation lasting about 30 min. This phase is followed by a second phased involving a sustained increase in vascular tone. Both phases of HPV have been described in terms of changes in pulmonary arterial pressure at constant blood flow in isolated rat lungs or pulmonary lobes, as well as via the measurement of the isometric force exerted by isolated pulmonary arteries (Bennie et al., 1991; Leach et al., 1994; Weissmann et al., 1995; Weissmann et al., 2004; Robertson et al., 2001) (for an extended review see Sylvester et al., 2012). Although some studies have suggested that endothelium-derived vasoactive substances mediate HPV (Holden and McCall, 1984), the role attributed to the pulmonary vascular endothelium in HPV is largely regulatory (for an extended review see Grimmer and Kuebler, 2017). This notion is based on the observations that isolated myocytes (Madden et al., 1992; Zhang et al., 1997; Sham et al., 2000) and endothelium-denuded pulmonary arterial rings (Yuan et al., 1990; Bennie et al., 1991; Archer et al., 1998) are capable of contracting when exposed to hypoxia, indicating that HPV is a mechanism intrinsic to pulmonary artery smooth muscle cells (PASMCs). However, the magnitude of the pressor response can be modulated by different circulating mediators from the vascular endothelium and the autonomic nervous system (Archer et al., 1989; Frostell et al., 1991; Brimioulle et al., 1997; Weissmann et al., 2001).
The HPV reflex implies the presence of a functional acute O2-sensitive system in PASMCs. This sensor is able to detect decreases in alveolar O2 and to signal effector elements responsible for triggering membrane depolarization and extracellular Ca2+ entry (and eventually Ca2+ release from intracellular stores), resulting in a subsequent increase in vascular tone. In what follows, we briefly summarize the sensing/signaling systems reported so far which, although subject to debated, seem to have garnered experimental support.
The levels of alveolar hypoxia capable of triggering HPV do not seem to inhibit mitochondrial cellular respiration to a degree sufficient to compromise ATP production (Waypa and Schumacker, 2002; Sommer et al., 2016). Nonetheless, it has been suggested that small changes in the cytosolic AMP/ATP ratio could act as a signaling mechanism, inducing the activation of AMP kinase with a constant intracellular ATP concentration. This, in turn, would contribute to the release of Ca2+ from intracellular stores via cyclic ADP-ribose (cADPR, cyclic adenosine diphosphate ribose) through its interaction with ryanodine receptors (RyR) in the sarcoplasmic reticulum. Although it is not yet established whether AMP kinase directly elicits cADPR-dependent Ca2+ release, it has been proposed that this could result from phosphorylation by AMP kinase of ADP-ribosyl cyclase, which catalyzes the cyclization of NAD+ to produce cADPR, and a subsequent increase in cADPR accumulation. AMP kinase could also increase the sensitivity of the Ca2+ release process to cADPR due to the phosphorylation of RyRs or an intermediate cADPR-binding protein (Leach et al., 2000; Evans et al., 2005). As the production of cADPR is also correlated with NADH levels, accumulation of the two nucleotides in response to hypoxia would represent an AMP kinase-mediated O2-sensing and signaling system (Wilson et al., 2001). In contrast to this proposal, it has also been shown that selective inhibition of cADPR-induced Ca2+ release has no effect on the initial acute phase of HPV, suggesting that AMP kinase activation contributes mainly to the sustained phase of HPV (Dipp and Evans, 2001; Moudgil et al., 2005).
NADPH oxidase (NOX) is capable of producing reactive oxygen species (ROS) in proportion to PO2 levels by catalyzing the transfer of electrons from NADPH to O2, and has therefore been suggested to work as an O2 sensor (Mohazzab et al., 1995; Marshall et al., 1996). However, although the loss of functional NOX dramatically lowers ROS production in NOX2-deficient mice, HPV is still preserved in these animals (Archer et al., 1999). Protein kinase C (PKC) has also been suggested to regulate HPV via NOX activation (Weissmann et al., 1999). In this regard, several reports in the literature described a possible direct interaction between mitochondria and NOX, where PKC-dependent ROS release from mitochondria could activate NOX under hypoxic conditions, leading to further increases in ROS levels, as well as ROS-dependent increases in intracellular Ca2+, and in PASMC contraction (Rathore et al., 2008). However, the direct effects of mitochondrial ROS on PKC activity have not yet been investigated. In contrast, it was proposed that glucose-6-phosphate dehydrogenase (G6PD), an enzyme that participates in the pentose phosphate pathway catalyzing the production of NADPH, is responsible for NOX activation in hypoxia, as G6PD-deficient mice exhibit a reduced HPV response (Gupte et al., 2010). In all of these cases, NOXs are considered to play an important role in HPV either as O2 sensors per se or as part of the hypoxic signaling cascade.
Rho kinase is a major mediator of Ca2+ sensitization, capable of inducing PASMC contraction by inhibiting myosin light-chain phosphatase in a Ca2+-independent manner (Somlyo and Somlyo, 2003). Hypoxia, acting through Rho kinase, is capable of accentuating HPV by increasing phosphorylation of the light chain and augmenting contraction (Wang et al., 2001). Evidence that Rho kinase is involved in sustained HPV comes from studies in isolated arteries and in perfused rat lungs, where Y-27632, a Rho kinase inhibitor, preferentially inhibited the sustained contraction phase of HPV, but had no impact on the acute hypoxic contraction (Robertson et al., 2000; Wang et al., 2001; Fagan et al., 2004). The hypoxic activation of Rho kinase was suggested to be dependent on ROS, as superoxide could trigger Rho kinase-mediated Ca2+ sensitization and vasoconstriction in rat pulmonary arteries (Knock et al., 2009). However, the role of this kinase in eliciting a specific HPV response is unclear, as Rho kinase activity is also present in systemic vascular beds (Numaguchi et al., 1999; Mukai et al., 2001), which dilate in response to hypoxia. Therefore, Rho kinase seems to contribute to a modulatory system that determines the magnitude of HPV but is not part of the acute O2 sensing pathway.
There is general consensus that mitochondria in PASMCs play a major role as a sensor and signaling system of alveolar hypoxia. Given their high O2 consumption and ability to generate ROS, as well as their content in numerous metabolic intermediaries and proximity to the plasma membrane and the sarcoplasmic reticulum, mitochondria are ideal candidates for having an essential O2 sensing and signaling function (Weir et al., 2005; Waypa et al., 2010; Sommer et al., 2016). Mitochondria can work as early detection systems for the conjugation of O2 supply and demand, optimizing the ventilation/perfusion ratio through of HPV response in the case of pulmonary resistance arteries.
Mitochondria involvement in pulmonary vascular sensitivity to O2 was initially postulated after it was verified that certain proximal inhibitors of the electron transport chain (ETC) triggered an acute, hypoxia-like, pressor response in PASMCs (Rounds and McMurtry, 1981). On the other hand, changes in the equilibrium of redox pairs able to interact with sulfhydryl groups of proteins were proposed to contribute to the O2-dependent physiological control of pulmonary vascular smooth muscle tone (Weir et al., 1985). Subsequently, a mitochondrial-based redox O2 sensor in PASMCs was suggested by several investigators (Archer et al., 1993; Waypa et al., 2001; Waypa et al., 2010; Weissmann et al., 2003; Wang et al., 2007; Song et al., 2017). O2-regulated K+ channels (Post et al., 1992; Yuan et al., 1993), potentially modulated by redox changes, were also described in PASMCs (Archer and Michelakis, 2002; Cogolludo et al., 2006; Mittal et al., 2012; Olschewski and Weir, 2015). Although it is generally agreed that mitochondria-dependent redox changes mediate the effects of hypoxia on membrane ion channels, a long-standing debate has ensued on whether hypoxia increases or decreases mitochondrial ROS production as well as on what might be the source of Ca2+ required for myocyte contraction (either extracellular influx or the release from sarcoplasmic reticulum) (Michelakis et al., 2002; Waypa et al., 2006; Wang and Zheng, 2010; Veit et al., 2015). Using PASMCs transfected with genetically encoded fluorescent probes, it was shown that hypoxia increases ROS in the cytosol and the mitochondrial intermembrane space (IMS) but decreases ROS within the matrix (Waypa et al., 2010). In our view, discrepancies regarding the type of ROS signal generated in PASMCs during hypoxia are probably a consequence of the different methodologies used to measure ROS production by the cells and the various organ/cellular preparations studied. The various methods of ROS detection used can also be particularly sensitive to different radical species (e.g., O2−, H2O2 or OH−), which could interact in different ways with ion channels (Sahoo et al., 2014). In addition, experimental observations across numerous studies were based on the extracellular application of redox reagents which may not properly mimic the changes in intracellular ROS signals generated during hypoxia (Reeve et al., 1995; Michelakis et al., 2004; Sommer et al., 2010; Waypa et al., 2010).
In the context of the current discussion, it is relevant to mention that an integrated mitochondrial-to-membrane signaling (MMS) model was proposed to explain acute O2 sensing by chemoreceptor glomus cells in the carotid body (CB), the prototypical acute O2 sensing organ in mammals. CB glomus cells are innervated by sensory fibers connected to brainstem centers that trigger a reflex hyperventilatory response and increased cardiac output in response to hypoxemia. In the MMS model, hypoxia produces a decrease in the activity of cytochrome c oxidase (CCO), thereby leading to a backlog of electrons along the ETC, with an increase in the reduced quinone (QH2)/quinone (Q) ratio and a slowdown of mitochondrial complex (MC) I. These changes result in the accumulation of NADH and production of a rapid and reversible increase in ROS at the mitochondrial intermembrane space (possibly H2O2 generated in MCI and MCIII). After equilibration with the cytosol, these signals modulate plasmalemmal ion channels to induce cell depolarization. Monitoring mitochondrial ROS production with the genetic probes described above for PASMC (Waypa et al., 2010) showed that, in parallel with the signaling increase in ROS at the IMS and cytosol, hypoxia produces a fast and highly reversible decrease in ROS at the mitochondrial matrix, which is interpreted as a non-selective signal derived from the activity of matrix dehydrogenases (Fernández-Agüera et al., 2015; Arias-Mayenco et al., 2018; Moreno Domínguez et al., 2020).
The MMS model of acute O2 sensing was initially suggested given that mice deficient in NDUFS2, a core subunit essential for MCI assembly and NADH dehydrogenase activity (Kashani-Poor et al., 2001; Baradaran et al., 2013), showed selective abolition of CB glomus cell sensitivity to hypoxia and the HVR (Fernández-Agüera et al., 2015; Arias-Mayenco et al., 2018). Interestingly, it was also shown that silencing Ndufs2 expression with interfering RNA (siRNA) selectively blocked the contractile response of PASMCs to hypoxia without affecting responses to other vasoconstrictor agents (Dunham-Snary et al., 2019). Moreover, mice lacking the gene coding the Rieske iron-sulfur protein (RISP) in cultured PASMC show a disruption of MCIII with inhibition of hypoxic ROS production and attenuation of the acute increase in right ventricular systolic pressure during hypoxia (Waypa et al., 2013). Recent studies on CB cells using the same mouse model showed a loss of both the hypoxic NADH and IMS ROS signals along with glomus cell responsiveness to hypoxia. However, in RISP-deficient mice as well as in MCI-deficient mice, modulation of the matrix ROS by hypoxia is maintained, suggesting that this signal is not directly involved in acute O2 sensing (Arias-Mayenco et al., 2018; Cabello-Rivera et al., 2022). It seems, therefore, that the mitochondrial-based O2 sensing process in PASMCs share many properties with the MMS model described for CB glomus cells (Figure 1). In agreement with this, mice without the gene coding for COX4I2, an atypical MCIV subunit isoform highly expressed in the lung and in glomus cells which may modulate the apparent affinity of CCO for O2, show a reduction of HPV (Sommer et al., 2017) as well as the hypoxic activation of CB glomus cells (Moreno-Domínguez et al., 2020). In addition to COX4I2, other MCIV subunits may contribute to modulating the sensitivity of mitochondria for O2 in PASMCs (Figure 1).
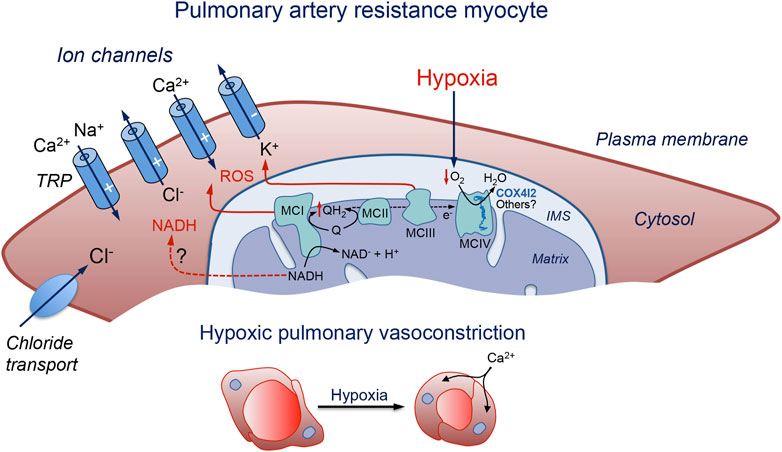
FIGURE 1. Schematic representation of a pulmonary artery resistance myocyte illustrating the molecular pathways proposed to be involved in acute pulmonary vasoconstriction. Hypoxia slows down the mitochondrial electron transport chain and causes the generation of signaling molecules (ROS and possibly NADH) which modulate membrane ion channels to produce cell depolarization, Ca2+ influx, and contraction. IMS: intermembrane space; MC: mitochondrial complex; NADH: reduced form of the nicotinamide adenine dinucleotide; Q: ubiquinone; QH2: reduced ubiquinone; ROS: reactive oxygen species; TRP: transient receptor potential channel. The symbols (−) and (+) indicate inhibition or activation of the channels during hypoxia, respectively. See text for further details.
It is well established that HPV, at least the initial phase, requires PASMC membrane depolarization due to the modulation of several types of ion channels. Hypoxia-induced membrane depolarization leads to the opening of L-type Ca2+ channels, Ca2+ influx, and myocyte contraction. Numerous ion channel types expressed in the PASMC plasma membrane are susceptible to modification by the cellular redox state and have been identified as potential targets of hypoxic vasoconstriction downstream of the hypoxia-induced mitochondrial signals. Although ROS seem to be the main molecule signaling hypoxia in PASMCs, NADH accumulation (another mitochondrial signal generated during hypoxia in CB chemoreceptor cells (Moreno-Domínguez et al., 2020)), may also have a relevant signaling role. The most significant ion channel classes which are the target of the hypoxia-signaling pathway mediating HPV are described below.
Several groups have shown that acute hypoxia can inhibit voltage-gated (Kv) channel activity in dispersed PASMCs and reported a contribution of these channels to HPV (Post et al., 1992; Yuan et al., 1993; Hulme et al., 1999; Archer et al., 2004). Some studies have shown that members of the two-pore domain potassium channel family (e.g., TASK-1) are also inhibited by moderate hypoxia in human PASMCs (Olschewski et al., 2006), although these channels do not seem to be absolutely required for HPV of murine intra-pulmonary arteries (Murtaza et al., 2017). However, TASK-1 channels appear to play a prominent role in regulating membrane potential in human PASMCs since mutations in the KCNK3 gene (coding TASK-1 channels) have been reported in patients with sporadic and familial pulmonary hypertension (see Lambert et al., 2018).
The O2-sensitive voltage-dependent K+ current in PASMCs has been proposed to be slowly inactivating, sensitive to 4-AP, and resistant to charybdotoxin. This profile would exclude several types of K+ channels (such as BKCa, Kv1.2, Kv1.6, Kv1.4 or Kv4.3) as HPV mediators (Archer et al., 2004). The existence of several subtypes of PASMCs according to the density of Kv (delayed rectifier) and maxi K+ channel expression has been reported. Myocytes containing predominantly Kv channels are mostly found in resistance arteries, whereas conduit pulmonary arteries contain myocytes predominantly expressing maxi K+ channels or a mixture of Kv and maxi-K+ channels (Archer et al., 1996, 2004; Smani et al., 2001). Inhibition of Kv channels was shown to initiate membrane depolarization, activation of voltage-dependent Ca2+ channels, and vasoconstriction (Nelson and Quayle, 1995) (Figure 1). On the other hand, downregulation of Kv channels (e.g., Kv1.5) was associated with a depolarized PASMC phenotype and pulmonary hypertension (Yuan et al., 1998; Remillard et al., 2007; for review see Lambert et al., 2018). However, the mechanism of Kv inhibition under hypoxic conditions has not been fully elucidated. In line with the hypothesis suggesting that an increase in ROS concentration serves as an activating mechanism of HPV, some studies have verified that the application of H2O2 mimics the hypoxia-induced depolarization in mouse PASMCs, inhibiting the current mediated by Kv channels (Sommer et al., 2017). However, it must be kept in mind that H2O2 can interact with many different redox-susceptible sites in ion channels (Sahoo et al., 2014; Mori et al., 2016). As mentioned above, the extracellular application of H2O2 does not necessarily mimic the effect of this reagent when it is generated by intracellular ROS-producing organelles (see López-Barneo and Ortega-Sáenz, 2022). Intracellular NADH, which binds to Kv channel ß subunits with relatively high affinity (Kd 1 μM) (Liu et al., 2001), is known to inhibit the activity of several subclasses of Kv channels (Tipparaju et al., 2005; Kilfoil et al., 2019), that are modulated by hypoxia in pulmonary resistance myocytes (Archer et al., 1998; Moral-Sanz et al., 2016). However, whether NADH accumulation has a relevant role as a hypoxic signaling molecule in PASMCs remains to be determined.
Ca2+-permeant cationic TRP channels, in particular TRPC1, TRPC6 and TRPV4, are highly expressed in resistance pulmonary myocytes where they contribute to Ca2+ entry and contraction (Remillard and Yuan, 2006; Reyes et al., 2018). TRPC channels can form complexes with Stim1/2 to mediate store-operated Ca2+ entry. The first, rapid phase of HPV, is abolished in TRPC6 knockout mice, while knockdown of Stim1 inhibits the second phase (Weissmann et al., 2006; Lu et al., 2009). Pharmacological blockade of TRPC6 channels also inhibits the acute phase of HPV (Urban et al., 2012). The relevance of TRPC6 channels for HPV is further supported by the fact that siRNA-mediated downregulation of TRPC6 expression inhibits the proliferation of PASMCs (Yu et al., 2003), while single nucleotide mutations of the Trpc6 gene are associated with idiopathic pulmonary hypertension (Yu et al., 2009). A significant decrease in HPV is also observed in mice deficient of TRPV4 channels, which can form heteromeric assemblies with TRPC6 channels (Goldenberg et al., 2015). Direct activation of TRP channels secondary to the increase in ROS levels could constitute a complementary (or in some myocytes alternative) mechanism to K+ channel-mediated depolarization and cytosolic Ca2+ influx during hypoxia. TRP channel-mediated cationic currents may also produce myocyte depolarization and activate L-type Ca2+ channels (Sommer et al., 2016) (Figure 1). Acute O2 sensing by CB glomus cells is unaltered in mice lacking TRPC6 channels (Torres-Torrelo et al., 2021), which suggests that although pulmonary myocytes and CB chemoreceptor cells may share similar mitochondrial-based O2 sensing and signaling mechanisms, the effectors of the hypoxic response may differ in these cell types.
Voltage-dependent L-type Ca2+ channels play a leading role in HPV given that they mediate the rise in cytosolic [Ca2+] during hypoxia and in this manner couple PASMC excitation to contraction. Indeed, the L-type Ca2+ channel density in rabbit pulmonary resistance myocytes is almost twice that in conduit myocytes (Franco-Obregón and López-Barneo, 1996). However, it remains unclear whether Ca2+ channels are regulated by O2 tension or their activation during HPV is secondary to cell depolarization produced by hypoxia-induced modulation of other channel types (e.g., inhibition of K+ channels and/or activation of TRP channels) (Figure 1). The presence of two different types of myocytes in the pulmonary arterial tree whose voltage-gated Ca2+ channels are modulated by PO2 was previously reported. In this way, Ca2+ currents in conduit myocytes, which predominate in the main arterial trunk and large branches, are inhibited by low PO2, while in resistance myocytes, concentrated in the smaller arterial branches, hypoxia increases the amplitude of the Ca2+ current (Franco-Obregón and López-Barneo, 1996).
The differential modulation by hypoxia of L-type Ca2+ channels in myocytes along the pulmonary arterial tree is strongly supported by the altered effects of low PO2 on cytosolic Ca2+ homeostasis in intact non-dialyzed VSM cells from the corresponding pulmonary arterial segments. As observed in other cells types, arterial myocytes (pulmonary and systemic) in vitro exhibit spontaneous oscillations of intracellular [Ca2+] (Ca2+ spikes), which largely represent the rapid release of Ca2+ from intracellular stores (Berridge, 1993). These Ca2+ spikes are modulated by transmembrane Ca2+ influx through L-type Ca2+ channels (see Blatter and Wier, 1992; Berridge, 1993; Franco-Obregón et al., 1995; Ureña et al., 1996). The removal of extracellular Ca2+ produces a rapid decrease in basal cytosolic [Ca2+] accompanied by either a decrease in the frequency of spikes or even their complete abolition (Figure 2A). In contrast, exposure of the cells to high external K+, presumably causing membrane depolarization and Ca2+ entry, produces an elevation in basal cytosolic [Ca2+], which is accompanied by a slight augmentation in Ca2+ spike frequency and a decrease in amplitude, or even suppression, of the spikes (Figure 2B). In most pulmonary conduit myocytes, as well as in myocytes of systemic arteries (see Section 3.3.2), hypoxia has a similar effect to that of removal of external Ca2+ (Figure 2C). The action of hypoxia on conduit myocytes is also opposite to that of high K+ (Figure 2D). In resistance myocytes, hypoxia raises basal cytosolic Ca2+ with a decrease in spike amplitude (Figure 2E); these effects are countered by the application of nifedipine (Figure 2F). The mechanism by which lowering PO2 differentially regulates L-type Ca2+ channels expressed in PASMCs is not known. It has been reported that the pore-forming subunit of L-type channels (α1C or Cav 1.2) expressed in the heart has specific cysteine residues whose oxidation could increase the probability of channel opening (Hool, 2015; Muralidharan et al., 2016). This fact would explain the increase in voltage-dependent Ca2+current in pulmonary resistance myocytes. It is possible that in conduction myocytes, or in myocytes of systemic arteries (see Section 3.3.2) Ca2+ channels are formed by α-subunit variants or auxiliary subunits that modify their sensitivity to mitochondrial signals (ROS and NADH).
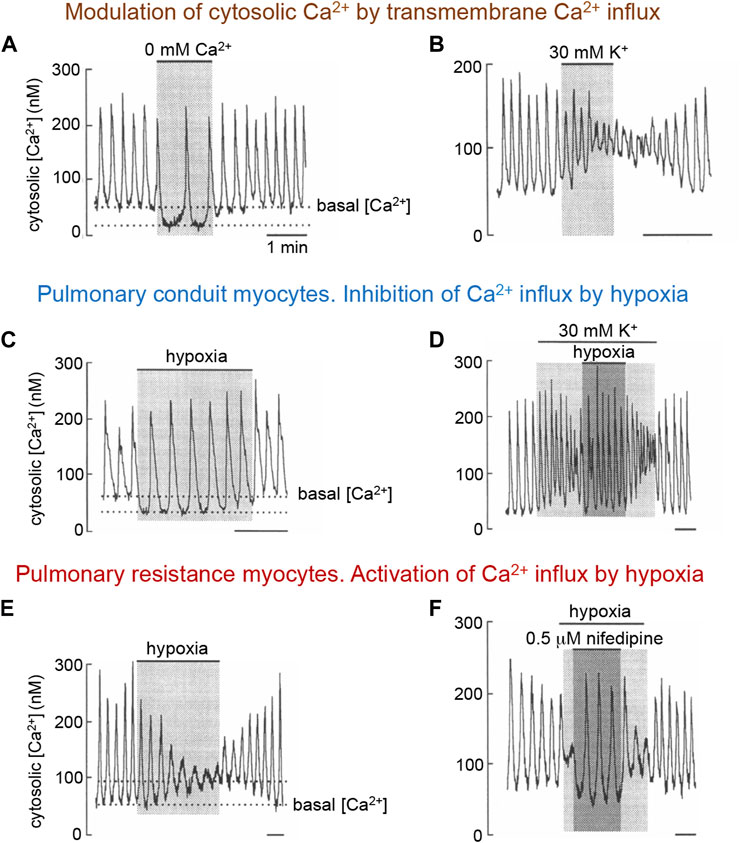
FIGURE 2. Modulation of spontaneous cytosolic Ca2+ spikes in rabbit pulmonary artery myocytes by transmembrane Ca2+ influx and hypoxia. (A, B). Changes in basal Ca2+ and spike frequency and amplitude in a pulmonary resistance myocyte upon removal of extracellular Ca2+ or depolarization with high extracellular K+ (≥30 mM). Similar effects of Ca2+ removal or K+-induced depolarization are observed in pulmonary conduit myocytes. (C, D), Decrease in basal cytosolic Ca2+ and changes in spikes characteristics in resting and depolarized conduit myocyte during exposure to hypoxia. (E, F). Increase in basal cytosolic Ca2+ and changes in spike characteristics in a resistance pulmonary myocyte. Blockade of Ca2+ channels counteract the effect of hypoxia. In all figures application of hypoxia was done by switching from a bathing solution with PO2∼150 mmm Hg to another with PO2 ∼15 mm Hg. Time calibration bars in all panels is 1 min. Modified from Ureña et al., 1996.
The intracellular Cl− concentration in VSM cells is unusually high (∼50 mM or even higher) due to active Cl− transport, with the Cl− equilibrium potential (near 0 mV) in these cells being more positive than the resting potential (∼−50 to −60 mV) (Yuan, 1997; Smani et al., 2001; Hübner et al., 2015; Leblanc et al., 2015). Therefore, activation of Cl− channels normally leads to Cl− efflux and cell depolarization, causing increased intracellular Ca2+ and contraction. Over the last few years, Cl− channels have gained prominence as contributors to HPV and pulmonary hypertension. Ca2+-activated Cl− channels are highly expressed in resistance PASMCs, where spontaneous cytosolic Ca2+ spikes or the pharmacologically-induced release of Ca2+ from internal stores generate inward (depolarizing) Cl− currents. In contrast, increases in cytosolic [Ca2+] most frequently produce outward (hyperpolarizing) K+ currents in conduit myocytes (Smani et al., 2001) (Figure 3). These data suggest that hypoxic vasoconstriction in distal PASMCs due to Ca2+ influx or Ca2+ release from internal stores is potentiated by Cl− channel activation. In favor of the prominent role played by Cl− channels in HPV and the development of pulmonary hypertension are the upregulation by sustained hypoxia of Ca2+-activated Cl− channels in PASMCs (Sun et al., 2012) along with the induction of Cl− transporters and increased intracellular [Cl−] in chronically hypoxic rats (Sun et al., 2021). Moreover, the expression of TMEM16A, a member of the transmembrane protein 16 family which mediates most of the Ca2+-activated Cl− current in PASMCs (Manoury et al., 2010), is upregulated in pulmonary hypertension and contributes to the enhanced pulmonary vasoconstriction associated with this pathological condition (Forrest et al., 2012; Leblanc et al., 2015; Maurer et al., 2020; Sun et al., 2021).
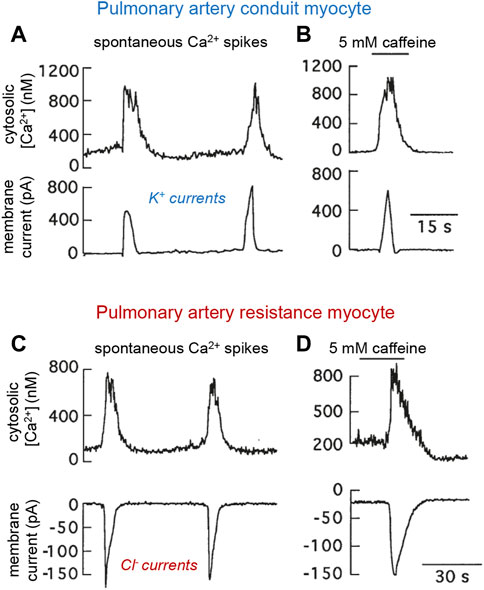
FIGURE 3. Spontaneous and caffeine-induced spikes of cytosolic Ca2+ and parallel modifications of transmembrane ionic currents in patch clamped rabbit pulmonary arterial myocytes. (A, B). Conduit myocyte that generated outward K+ currents. (C, D). Resistance myocyte in which the Ca2+ oscillations and caffeine induced inward Cl− currents. Experiments done using the standard external solution and an internal solution containing Fura-2 and 22 mM ClNa. In all recordings membrane potential was −58.9 mV. Caffeine was added to the external solution. Modified from Smani et al., 2001.
Taken together, the available experimental evidence suggests that the effector pathways mediating HPV are complex, as they do not depend on the modulation of a specific ion channel type. However, the level of expression of the main ion channel classes mediating HPV is higher in resistance myocytes than in conduit myocytes. It seems that, as occurs in CB chemoreceptor cells (see López-Barneo and Ortega-Sáenz, 2022), signaling molecules produced during hypoxia (ROS and possibly also NADH among others) interact promiscuously with different types of ion channels. Interestingly, the primary effects elicited by hypoxia on some ion channels in resistance myocytes (e.g., cell depolarization or Ca2+ release from stores) are potentiated by the activation of other ion channel classes to jointly produce a robust increase in cytosolic [Ca2+] and contraction.
There is consensus that O2 is a potent vasoactive agent and a major regulator of systemic vascular tone in vivo (Jackson, 2016). In contrast with resistance pulmonary arteries, a decrease in PO2 elicits relaxation and vasodilatation in systemic arteries, which increases O2 supply to tissues (Detar, 1980; Marriott and Marshall, 1990; Wadsworth, 1994). HVD is a generalized response, although it is particularly potent in brain, heart and skeletal muscle arterial territories. Together with other mechanisms (Gordon et al., 2007) increased blood flow during hypoxia probably contributes to neurovascular coupling - a dilation of small blood vessels whereby O2 availability is increased in the most active brain regions. Similarly, relaxation is the most common effect of low PO2 on the coronary arteries, which transport oxygenated blood to the myocardium. This response is of critical physiologic importance because it contributes to adjusting the amount of O2 supplied to the working heart to meet its metabolic needs, as any imbalance between O2 delivery and demand can lead to myocardial damage. For O2 to participate in the local regulation of blood flow in these tissues, small arteries and arterioles in the microcirculation must be able to sense acute changes in PO2 and trigger HVD. The cellular location of the O2 sensor and the mechanism underlying the physiological response to low PO2 nevertheless remains largely unknown (Jackson, 2016).
Different cellular sites, including the arteriolar muscular wall, the endothelial layer, erythrocytes, or extravascular cells, such as parenchymal and nerve cells, have been postulated to sense reductions in the PO2 levels and signal systemic arterial myocytes to elicit vasorelaxation (Jackson, 2016). It is well known that the vascular endothelium contributes to vasodilation by releasing potent relaxing factors that regulate smooth muscle contractility (Fredericks et al., 1994; Liu and Flavahan, 1997; Lynch et al., 2006). However, numerous studies have proposed VSM cells as a primary site for sensing decreased PO2 independently of metabolic factors released from the endothelium or surrounding tissues (Dart and Standen, 1995; Jackson, 2000; Smani et al., 2002; Gauthier, 2006). Indeed, it is well established that hypoxia can reversibly induce relaxation of endothelial-denuded arterial rings precontracted by exposure to high extracellular K+ (Smani et al., 2002) (Figures 4A,B).
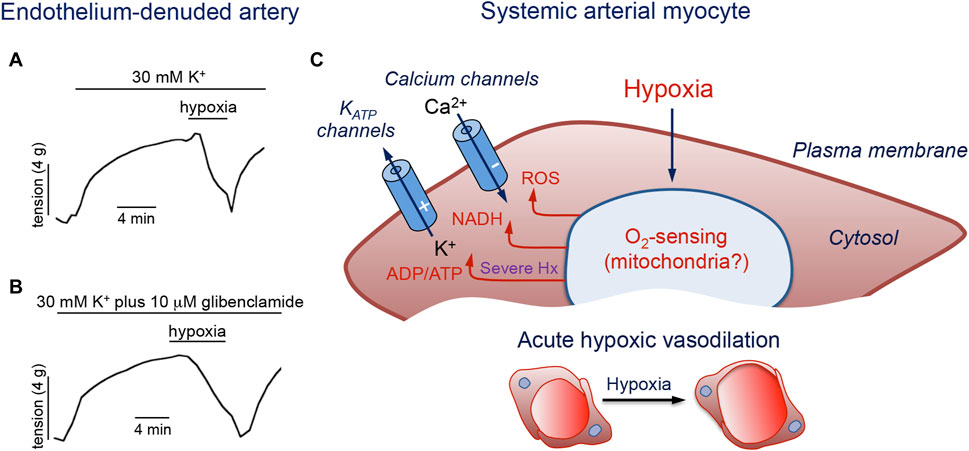
FIGURE 4. Molecular pathways involved in hypoxic vasodilation. (A, B). Reduction of tension by hypoxia in endothelium-denuded porcine coronary arterial rings precontracted with 30 mM K+ or with 30 mM K+ plus 10 μM glibenclamide to block KATP channels. Modified from Smani et al., 2002. (C). Schematic representation of a systemic artery myocyte. Hypoxia (Hx) acting on an O2 sensor generates intracellular signals (ROS, NADH and increased ADP/ATP ratio) that modulate membrane ion channels to produce cell hyperpolarization and inhibition of Ca2+ influx. The symbols (−) and (+) indicate inhibition or activation of the channels during hypoxia, respectively. See text for further details.
As mentioned above (see Section 2.2.4), a growing body of work implicates the mitochondria as the major organelles responsible for acute O2 sensing and signaling in PASMCs, and similar mechanisms may also operate in systemic arterial VSM (Figure 4C). In this regard, it was reported that ROS generation increases during hypoxia in renal arteries (Michelakis et al., 2002). Experiments using genetically encoded roGPF sensors showed hypoxia-induced cytosolic and mitochondrial IMS ROS signals in systemic myocytes similar to those recorded in PASMC (Waypa et al., 2010). An oxidized stated with decreases in the [NADPH]/[NADP+] ratio was observed in hypoxic bovine coronary arteries, while HVD was attenuated by the thiol-reducing agent dithiothreitol (Gupte and Wolin, 2006). Therefore, although the mechanism of acute O2 sensing in systemic arterial myocytes is not known, it is likely that, as in the pulmonary circulation, mitochondria also play an important role. Since hypoxia has antagonistic functional effects on pulmonary and systemic VSM cells, the notion that both cell types share similar mitochondrial-based O2 sensing and signaling mechanisms would imply that they have differences in the downstream pathways and effector elements (Waypa et al., 2010). In this respect, it is relevant to note differences in ion channel types and densities between resistance and conduit pulmonary myocytes (see Section 2.3), given that acute hypoxic vasodilation is observed in conduit pulmonary arteries as well as in systemic arteries. Indeed, as shown in Figures 5A,B for single human and pig coronary arterial myocytes, hypoxia inhibits Ca2+ influx in a manner similar to that observed in conduit PASMCs (see Figures 2A–F). In coronary myocytes with spontaneous cytosolic Ca2+ oscillations, the effect of hypoxia is mimicked by the removal of extracellular Ca2+ (Figure 5A, left) or blockade of L-type Ca2+ channels with nifedipine (Figure 5A, right). Remarkably, hypoxia has no effect on cytosolic Ca2+ levels in the presence of nifedipine. In quiescent myocytes, hypoxia reversibly inhibits depolarization-induced Ca2+ influx (Figure 5B), thus further suggesting a direct action on the opening of Ca2+ channels (Figure 4C) (see below).
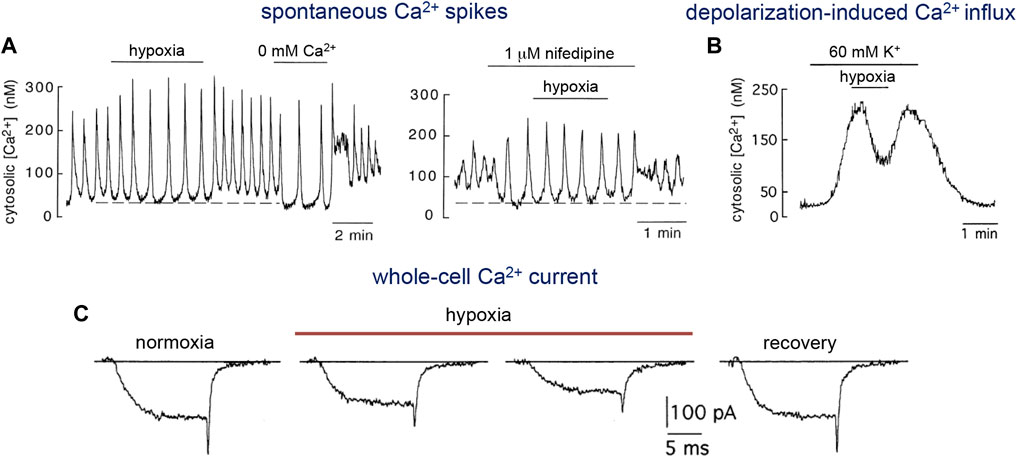
FIGURE 5. Inhibition of L-type Ca2+ channel activity by hypoxia. (A). Response to hypoxia of a non-dialyzed human coronary artery myocyte which generated spontaneously intracellular Ca2+ oscillations. Left. Reduction of basal cytosolic Ca2+ (dotted line) and increase in the amplitude of the oscillations by hypoxia and removal of extracellular Ca2+ (0 mM Ca2+ and 1 mM EGTA added). Right. Blockade of L-type Ca2+ channels with nifedipine produced the same effect of hypoxia or the removal of external Ca2+. Note that hypoxia did not have any effect in the presence of nifedipine. Modified from Smani et al., 2002. (B). Reversible reduction of cytosolic Ca2+ in a Fura-2 loaded quiescent pig coronary myocyte in which membrane voltage-dependent Ca2+ channels and Ca2+ influx were activated by depolarization with high extracellular K+. From López-Barneo et al., 1999. (C). Ca2+ currents recorded from a rabbit celiac arterial myocyte during 15 ms step depolarizations to +10 mV from a holding potential of −80 mV. After exposure to hypoxia (PO2 ∼15 mmHg) there is a fast and progressive inhibition of current amplitude. Reversibility is illustrated by the recovery trace. Modified from Franco-Obregon et al., 1995.
Over the last decades two major effectors have been associated with hypoxia-induced VSM relaxation: ATP-sensitive K+ (KATP) channels and voltage-sensitive L-type Ca2+ channels. Membrane hyperpolarization caused by the activation of KATP channels or the inhibition of voltage-dependent Ca2+ channel opening can both lead to a decrease in Ca2+ influx and to VSM relaxation. However, these mechanisms may not be mutually exclusive, and interactions between them are highly probable (Pearce, 1995). In addition, other intracellular processes, including decreases in intracellular pH, elevations of inorganic phosphate, or activation of Kv7 channels (Taggart and Wray, 1998; Hedegaard et al., 2014), have also been suggested to contribute to hypoxic vasorelaxation.
Numerous studies have suggested that KATP channels are involved in HVD in the skeletal and coronary circulations (Daut et al., 1990; Jackson, 1993; Marshall et al., 1993; Dart and Standen, 1995). However, other reports have shown that KATP currents remain stable during hypoxia because ATP concentrations do not drop to levels required for the channel activation (Jackson, 2000; Quayle et al., 2006). In agreement with these observations, it was also demonstrated that the hypoxia-induced decrease in resting tension of coronary or femoral arterial rings is unaffected by the inhibition of KATP channels with glibenclamide (specific inhibitor of KATP channels) (Smani et al., 2002; Quayle et al., 2006). In rat gracilis pressurized arteries, KATP channel blockers reduce membrane potential hyperpolarization and vasodilation in response to moderate and severe, but not mild, hypoxia (Frisbee et al., 2002). Currently, the most accepted view is that in most vascular territories intracellular ATP levels and the activity of KATP channels are not directly modified by physiological levels of hypoxia. However, KATP channel modulation by other paracrine and endocrine factors can contribute to the regulation of VSM cell excitability and contraction.
Several studies on systemic arterial smooth muscle have provided compelling evidence that HVD is triggered through mechanisms that decrease voltage-gated Ca2+ influx independently of KATP channel activation. As shown above (see Figures 4A,B), hypoxia induces acute relaxation of precontracted coronary arterial rings by mechanisms not involving the endothelium or KATP channel activation (Smani et al., 2002). In addition, inhibition of Ca2+ influx by hypoxia has been observed in intact, non-dialyzed myocytes from human, porcine and rabbit systemic arteries as well as in conduit PASMCs (see Figures 2A–F; Figures5A,B). The inhibition of L-type Ca2+ channels by hypoxia was initially demonstrated in patch-clamped freshly dispersed VSM cells from celiac arteries (Franco-Obregón et al., 1995). The effect of hypoxia on L-type Ca2+ currents is highly reversible and occurs within a physiological range of PO2 values without affecting currents mediated by T-type Ca2+ channels (Franco-Obregón et al., 1995; López-Barneo et al., 1999; Saitoh et al., 2006) (Figure 5C). To our knowledge, inhibition of L-type Ca2+ currents by hypoxia has been observed in VSM cells from several territories (celiac, femoral, coronary, cerebral, cheek pouch, and main pulmonary arteries) and species (human, pig, rabbit, rat and hamster), where hypoxemia normally produces vasodilatation (Marriot and Marshall, 1990; Pearce et al., 1992; Franco-Obregon et al., 1995; Herrera and Walker, 1998; Welsh et al., 1998; Smani et al., 2002; Quayle et al., 2006). Additionally, hypoxia decreases the amplitude of currents mediated by recombinant human L-type cardiovascular Ca2+ channel α1C subunit expressed in human embryonic kidney (HEK) cells in a manner indistinguishable from that observed in native smooth muscle L-type Ca2+ channels (Fearon et al., 1997). Moreover, inhibition of Ca2+ currents by hypoxia with characteristics similar to those reported in VSM cells has also been reported for rat and guinea pig ventricular myocytes (Hool, 2000; Rosa et al., 2012).
The mechanism whereby hypoxia modulates L-type Ca2+ channels is unknown although it seems that redox signals play a relevant role. Several groups have shown that whole-cell currents generated by recombinant Ca2+ channels (rabbit and human α1C subunit) expressed in mammalian cell lines are reduced in amplitude by lipophilic oxidizers of sulfhydryl groups, with these effects reverted by agents that reduce disulfide bonds (Chiamvimonvat et al., 1995; Fearon et al., 1999). Single channel recordings indicated that redox reagents alter channel gating without affecting permeation (Chiamvimonvat et al., 1995). These studies also reported that pre-treatment of the cells with oxidizing agents prevents inhibition of the Ca2+ current by hypoxia (Fearon et al., 1999). In agreement with these data, macroscopic Ca2+ currents and Ca2+ channel gating currents in guinea pig ventricular myocytes are inhibited by oxidants in a manner that is prevented by dithiothreitol (Lacampagne et al., 1995). These observations suggest that the pore-forming α-subunit of the cardiovascular L-type Ca2+ channel contains functionally important sulfhydryl groups that modulate gating. Studies performed using mutated α1C subunits with replacement of specific cysteine residues have shown that the L-type Ca2+ channels are indeed redox sensors although the effects of ROS on these channels are a matter of controversy (Hool, 2015; Muralidharan et al., 2016). Direct modification by oxidants of thiol groups in purified L-type Ca2+ channels reconstituted in proteoliposomes increases channel open probability, while reducing agents have the opposite effect (Hool, 2015). Therefore, it seems that redox reagents have different effects on Ca2+ channels reconstituted in bilayers compared with the same recombinant channel subunit expressed in mammalian cells (Chiamvimonvat et al., 1995; Fearon et al., 1999) or native Ca2+ channels (Lacampagne et al., 1995). It is possible that redox agents used in the different experimental conditions interact with distinct extra and intracellular cysteine residues existing in the α1C subunit which results in opposite effects on channel gating. It is also conceivable that ROS actions in cellular models are also mediated by intrinsic molecules associated with the α1C subunit.
Regarding the effect of hypoxia, it has been shown that L-type Ca2+ channels reconstituted in liposomes are insensitive to hypoxia, which suggests that, as discussed above, an independent O2 sensor (possibly mitochondria) is required for the hypoxic modulation of channel function. It is likely that besides ROS, hypoxia generates other signals (e.g., NADH) that also contribute to the physiological regulation of L-type Ca2+ channel gating. In this respect, it is relevant to note that hemeoxygenase (HO), a broadly distributed enzyme that degrades heme and produces CO, was been suggested to directly modulate cardiac L-type Ca2+ channels by binding directly to the C-terminal end of the α1C subunit (Rosa et al., 2012). Pharmacological CO production has also been shown to inhibit recombinant (α1C-subunit) Ca2+ channels expressed in mammalian cells, although the effect was proposed to be mediated by the ability of CO to promote production of ROS in mitochondria (Scragg et al., 2008). To our knowledge, vasomotor responses to hypoxia in HO knockout mice have not been studied; however, we have shown that HO-2 is not required for CB responsiveness to hypoxia (Ortega-Sáenz et al., 2006).
Acute O2 sensing by VSM cells plays a fundamental role in the control of blood vessel diameter in response to the level of O2 tension in the nearby environment, this being a chemosensory function necessary to trigger compensatory adaptive responses. In this review we have focused on the study of two classic vasomotor responses to hypoxia in which our group has carried out considerable experimental work: HPV and HVD. HPV depends on the vasoconstriction of small caliber pulmonary arteries (resistance vessels) which contain a specific and varied set of plasmalemmal ion channels that trigger extracellular Ca2+ influx and contraction during hypoxia. Hypoxia can also directly or indirectly induce Ca2+ release from intracellular stores. Whilst much progress has been made during the last decades in defining the molecular processes underlying HPV, uncertainty remains about the O2-sensing mechanism and the signals that modulate ion channel activity. While it appears that mitochondria act as O2 sensors in pulmonary resistance myocytes, it remains to be unequivocally demonstrated, however, that ROS and other mitochondrial signals are directly responsible for modulating membrane ion channels. Moreover, although important advances have been made recently, the molecular determinants of mitochondrial specialization in lung arterial myocytes, in particular those responsible for the apparent low O2 affinity of cytochrome c oxidase, remain to be fully clarified. In addition to its physiological value, HPV is of pathophysiological interest since, if maintained, it can lead to pulmonary hypertension. Given the medical importance of this pathology, it is important to continue to make progress in understanding how the initial (acute) activation of myocytes by hypoxia leads to the cellular proliferation and vascular remodeling phenomena characteristic of pulmonary hypertension.
In contrast to resistance myocytes, myocytes in conduction pulmonary arteries relax with hypoxia; a response that is similar to the HVD seen in most systemic arteries. Although HVD has a fundamental physiological role, especially in the coronary and cerebral circulations, its molecular determinants remain poorly understood. The nature of the acute O2 sensing apparatus in systemic myocytes and how the sensor signals to membrane ion channels are still to be defined. Existing data suggest that inhibition of L-type Ca2+ channels by hypoxia plays a key role in HVD, but further experimental data are needed to definitively clarify this signaling pathway. The molecular description of HVD may provide new pharmacological targets of interest in clinical conditions such as coronary and cerebral ischemia or arterial hypertension. This pharmacological development may acquire even greater medical relevance if it is taken into account that inhibition by hypoxia of Ca2+ channels in cardiac myocytes, which share the pore-forming α1C subunit with arterial myocytes, may have a cardioprotective role in the event of cardiac ischemia. The fields of HPV and HVD face relevant and highly attractive challenges that are important in fundamental research, but which also have clear translational implications in cardiocirculatory pathophysiology and pharmacology, which must be addressed in future experimental work.
AM-D and JL-B prepared the first draft of the manuscript and figures. AM-D, JL-B, OC, TS, and JU contributed to the writing of the final version of the paper. All authors contributed to the article and approved the submitted version.
This research was supported by the Andalusian Government (FEDER Andalucía 2014–2020, 2018 Call, US-1255654), the Spanish Ministries of Science and Innovation and Health (Grants SAF 2016-74990-R and PID 2019-106410RB-I00 funded by MCIN/AEI/10.13039/501100011033), and the European Research Council (ERC Advanced Grant PRJ201502629).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Archer, S. L., Huang, J., Henry, T., Peterson, D., and Weir, E. K. (1993). A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 73 (6), 1100–1112. doi:10.1161/01.res.73.6.1100
Archer, S. L., Huang, J. M. C., Reeve, H. L., Hampl, V., Tolarova, S., Michelakis, E. D., et al. (1996). Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ. Res. 78 (3), 431–442. doi:10.1161/01.res.78.3.431
Archer, S. L., and Michelakis, E. D. (2002). The mechanism(s) of hypoxic pulmonary vasoconstriction: Potassium channels, redox O2 sensors, and controversies. News Physiol. Sci. 17, 131–137. doi:10.1152/nips.01388.2002
Archer, S. L., Reeve, H. L., Michelakis, E. D., Puttagunta, L., Waite, R., Nelson, D. P., et al. (1999). O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc. Natl. Acad. Sci. U. S. A. 96 (14), 7944–7949. doi:10.1073/pnas.96.14.7944
Archer, S. L., Souil, E., Dinh-Xuan, A. T., Schremmer, B., Mercier, J. C., El Yaagoubi, A., et al. (1998). Molecular identification of the role of voltage gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin. Invest. 101 (11), 2319–2330. doi:10.1172/JCI333
Archer, S. L., Tolins, J. P., Raij, L., and Weir, E. K. (1989). Hypoxic pulmonary vasoconstriction is enhanced by inhibition of the synthesis of an endothelium derived relaxing factor. Biochem. Biophys. Res. Commun. 164 (3), 1198–1205. doi:10.1016/0006-291x(89)91796-8
Archer, S. L., Wu, X. C., Thébaud, B., Nsair, A., Bonnet, S., Tyrrell, B., et al. (2004). Preferential expression and function of voltage-gated, O2-sensitive kchannels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: Ionic diversity in smooth muscle cells. Circ. Res. 95 (3), 308–318. doi:10.1161/01.RES.0000137173.42723.fb
Arias-Mayenco, I., Gonzalez-Rodriguez, P., Torres-Torrelo, H., Gao, L., Fernandez-Aguera, M. C., et al. (2018). Acute O2 sensing: Role of coenzyme QH2/Q ratio and mitochondrial ROS compartmentalization. Cell. Metab. 28 (1), 145–158. doi:10.1016/j.cmet.2018.05.009
Baradaran, R., Berrisford, J. M., Minhas, G. S., and Sazanov, L. A. (2013). Crystal structure of the entire respiratory complex I. Nature 494 (7438), 443–448. doi:10.1038/nature11871
Bennie, R. E., Packer, C. S., Powell, D. R., Jin, N., and Rhoades, R. A. (1991). Biphasic contractile response of pulmonary artery to hypoxia. Am. J. Physiol. 261, L156–L163. doi:10.1152/ajplung.1991.261.2.L156
Berridge, M. J. (1993). Inositol trisphosphate and calcium signalling. Nature 361 (6410), 315–325. doi:10.1038/361315a0
Blatter, L. A., and Wier, W. G. (1992). Agonist-induced [Ca2+]i waves and Ca(2+)-induced Ca2+ release in mammalian vascular smooth muscle cells. Am. J. Physiol. 263, H576–H586. doi:10.1152/ajpheart.1992.263.2.H576
Brimioulle, S., Vachiéry, J. L., Brichant, J. F., Delcroix, M., Lejeune, P., and Naeije, R. (1997). Sympathetic modulation of hypoxic pulmonary vasoconstriction in intact dogs. Cardiovasc Res. 34 (2), 384–392. doi:10.1016/s0008-6363(97)00028-x
Cabello-Rivera, D., Ortega-Saenz, P., Gao, L., Muñoz-Cabello, A. M., Bonilla-Henao, V., Schumacker, P. T., et al. (2022). Oxygen regulation of breathing is abolished in mitochondrial complex III-deficient arterial chemoreceptors. Proc. Natl. Acad. Sci. U. S. A. 119 (39), e2202178119. doi:10.1073/pnas.2202178119
Chiamvimonvat, N., O'Rourke, B., Kamp, T. J., Kallen, R. G., Hofmann, F., Flockerzi, V., et al. (1995). Functional consequences of sulfhydryl modification in the pore-forming subunits of cardiovascular Ca2+ and Na+ channels. Circ. Res. 76 (3), 325–334. doi:10.1161/01.res.76.3.325
Cogolludo, A., Frazziano, G., Cobeno, L., Moreno, L., Lodi, F., Villamor, E., et al. (2006). Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann. N. Y. Acad. Sci. 1091, 41–51. doi:10.1196/annals.1378.053
Dart, C., and Standen, N. B. (1995). Activation of ATP-dependent K+ channels by hypoxia in smooth muscle cells isolated from the pig coronary artery. J. Physiol. 483, 29–39. doi:10.1113/jphysiol.1995.sp020565
Daut, J., Maier-Rudolph, W., von Beckerath, N., Mehrke, G., Günther, K., and Goedel-Meinen, L. (1990). Hypoxic dilation of coronary arteries is mediated by ATP-sensitive potassium channels. Science 247 (4948), 1341–1344. doi:10.1126/science.2107575
Detar, R. (1980). Mechanism of physiological hypoxia-induced depression of vascular smooth muscle contraction. Am. J. Physiol. 238 (6), H761–H769. doi:10.1152/ajpheart.1980.238.6.H761
Dipp, M., and Evans, A. M. (2001). Cyclic ADP-ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circ. Res. 89 (1), 77–83. doi:10.1161/hh1301.093616
Dunham-Snary, K. J., Wu, D., Potus, F., Sykes, E. A., Mewburn, J. D., Charles, R. L., et al. (2019). Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 124 (12), 1727–1746. doi:10.1161/CIRCRESAHA.118.314284
Evans, A. M., Mustard, K. J., Wyatt, C. N., Peers, C., Dipp, M., Kumar, P., et al. (2005). Does AMP-activated protein kinase couple inhibition of mitocondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? J. Biol. Chem. 280 (50), 41504–41511. doi:10.1074/jbc.M510040200
Fagan, K. A., Oka, M., Bauer, N. R., Gebb, S. A., Ivy, D. D., Morris, K. G., et al. (2004). Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hyper-tension in mice by inhibition of Rho-kinase. Am. J. Physiol. Lung Cell. Mol. Physiol. 287 (4), L656–L664. doi:10.1152/ajplung.00090.2003
Fearon, I. M., Palmer, A. C., Balmforth, A. J., Ball, S. G., Mikala, G., Schwartz, A., et al. (1997). Hypoxia inhibits the recombinant α1C subunit of the human cardiac L-type Ca2+ channel. J. Physiol. 500, 551–556. doi:10.1113/jphysiol.1997.sp022041
Fearon, I. M., Palmer, A. C., Balmforth, A. J., Ball, S. G., Varadi, G., and Peers, C. (1999). Modulation of recombinant human cardiac L-type Ca2+ channels α1C subunits by redox reagents and hypoxia. J. Physiol. 514, 629–637. doi:10.1111/j.1469-7793.1999.629ad.x
Fernández-Agüera, M. C., Gao, L., González-Rodriguez, P., Pintado, C. O., Arias-Mayenco, I., García-Flores, P., et al. (2015). Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab. 22 (5), 825–837. doi:10.1016/j.cmet.2015.09.004
Forrest, A. S., Joyce, T. C., Huebner, M. L., Ayon, R. J., Wiwchar, M., Joyce, J., et al. (2012). Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am. J. Physiol. Cell. Physiol. 303 (12), C1229–C1243. doi:10.1152/ajpcell.00044.2012
Franco-Obregon, A., and Lopez-Barneo, J. (1996). Differential oxygen sensitivity of calcium channels in rabbit smooth muscle cells of conduit and resistance pulmonary arteries. J. Physiol. 491, 511–518.
Franco-Obregon, A., Ureña, J., and Lopez-Barneo, J. (1995). Oxygen-sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proc. Natl. Acad. Sci. U. S. A. 92 (10), 4715–4719. doi:10.1073/pnas.92.10.4715
Fredericks, K. T., Liu, Y., and Lombard, J. H. (1994). Response of extraparenchymal resistance arteries of rat skeletal muscle to reduced PO2. Am. J. Physiol. 267, H706–H715. doi:10.1152/ajpheart.1994.267.2.H706
Frisbee, J. C., Maier, K. G., Falck, J. R., Roman, R. J., and Lombard, J. H. (2002). Integration of hypoxic dilation signaling pathways for skeletal muscle resistance arteries. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283 (2), R309–R319. doi:10.1152/ajpregu.00741.2001
Frostell, C., Fratacci, M. D., Wain, J. C., Jones, R., and Zapol, W. M. (1991). Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation 83 (6), 2038–2047. doi:10.1161/01.cir.83.6.2038
Gauthier, K. M. (2006). Hypoxia-induced vascular smooth muscle relaxation: Increased ATP-sensitive K+ efflux or decreased voltage-sensitive Ca2+ influx? Am. J. Physiol. Heart Circ. Physiol. 291 (1), H24–H25. doi:10.1152/ajpheart.00260.2006
Goldenberg, N. M., Wang, L., Ranke, H., Liedtke, W., Tabuchi, A., and Kuebler, W. M. (2015). TRPV4 is required for hypoxic pulmonary vasoconstriction. Anesthesiology 122 (6), 1338–1348. doi:10.1097/ALN.0000000000000647
Gordon, G. R. J., Mulligan, S. J., and MacVicar, B. A. (2007). Astrocyte control of the cerebrovasculature. Glia 55 (12), 1214–1221. doi:10.1002/glia.20543
Grimmer, B., and Kuebler, W. M. (2017). The endothelium in hypoxic pulmonary vasoconstriction. J. Appl. Physiol. (1985) 123 (6), 1635–1646. doi:10.1152/japplphysiol.00120.2017
Gupte, R. S., Rawat, D. K., Chettimada, S., Cioffi, D. L., Wolin, M. S., Gerthoffer, W. T., et al. (2010). Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J. Biol. Chem. 285 (25), 19561–19571. doi:10.1074/jbc.M109.092916
Gupte, S. A., and Wolin, M. S. (2006). Hypoxia promotes relaxation of bovine coronary arteries through lowering cytosolic NADPH. Am. J. Physiol. Heart Circ. Physiol. 290 (6), H2228–H2238. doi:10.1152/ajpheart.00615.2005
Hales, C. A. (1985). The site and mechanism of oxygen sensing for the pulmonary vessels. Chest 88 (4), 235S–240S. doi:10.1378/chest.88.4_supplement.235s
Harder, D. R., Madden, J. A., and Dawson, C. (1985). A membrane electrical mechanism for hypoxic vasoconstriction of small pulmonary arteries from cat. Chest 88 (4), 233S–235S. doi:10.1378/chest.88.4_supplement.233s
Hedegaard, E. R., Nielsen, B. D., Kun, A., Hughes, A. D., Krøigaard, C., Mogensen, S., et al. (2014). Kv7 channels are involved in hypoxia-induced vasodilatation of porcine coronary arteries. Br. J. Pharmacol. 171 (1), 69–82. doi:10.1111/bph.12424
Herrera, G. M., and Walker, B. R. (1998). Involvement of L-type calcium channels in hypoxic relaxation of vascular smooth muscle. J. Vasc. Res. 35 (4), 265–273. doi:10.1159/000025593
Holden, W. E., and McCall, E. (1984). Hypoxia-induced contractions of porcine pulmonary artery strips depend on intact endothelium. Exp. Lung Res. 7 (2), 101–112. doi:10.3109/01902148409069671
Hool, L. C. (2015). How does the heart sense changes in oxygen tension: A role for ion channels? Antioxid. Redox Signal 22 (6), 522–536. doi:10.1089/ars.2014.5880
Hool, L. C. (2000). Hypoxia increases the sensitivity of the L-type Ca(2+) current to beta-adrenergic receptor stimulation via a C2 region-containing protein kinase C isoform. Circ. Res. 87 (12), 1164–1171. doi:10.1161/01.res.87.12.1164
Hübner, C. A., Schroeder, B. C., and Ehmke, H. (2015). Regulation of vascular tone and arterial blood pressure: Role of chloride transport in vascular smooth muscle. Pflugers Arch. 467 (3), 605–614. doi:10.1007/s00424-014-1684-y
Hulme, J. T., Coppock, E. A., Felipe, A., Martens, J. R., and Tamkun, M. M. (1999). Oxygen sensitivity of cloned voltage-gated K(+) channels expressed in the pulmonary vasculature. Circ. Res. 85 (6), 489–497. doi:10.1161/01.res.85.6.489
Jackson, W. F. (2016). Arteriolar oxygen reactivity: Where is the sensor and what is the mechanism of action? J. Physiol. 594 (18), 5055–5077. doi:10.1113/JP270192
Jackson, W. F. (1993). Arteriolar tone is determined by activity of ATP-sensitive potassium channels. Am. J. Physiol. 265, H1797–H1803. doi:10.1152/ajpheart.1993.265.5.H1797
Jackson, W. F. (2000). Hypoxia does not activate ATP-sensitive K+ channels in arteriolar muscle cells. Microcirculation 7 (2), 137–145. doi:10.1038/sj.mn.7300102
Kashani-Poor, N., Zwicker, K., Kerscher, S., and Brandt, U. (2001). A central functional role for the 49-kDa subunit within the catalytic core of mitochondrial complex I. J. Biol. Chem. 276 (26), 24082–24087. doi:10.1074/jbc.M102296200
Kilfoil, P. J., Chapalamadugu, K. C., Hu, X., Zhang, D., Raucci, F. J., Tur, J., et al. (2019). Metabolic regulation of Kv channels and cardiac repolarization by Kvβ2 subunits. J. Mol. Cell. Cardiol. 137, 93–106. doi:10.1016/j.yjmcc.2019.09.013
Knock, G. A., Snetkov, V. A., Shaifta, Y., Connolly, M., Drndarski, S., Noah, A., et al. (2009). Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic. Biol. Med. 46 (5), 633–642. doi:10.1016/j.freeradbiomed.2008.11.015
Lacampagne, A., Duittoz, A., Bolanos, P., Peineau, N., and Argibay, J. A. (1995). Effect of sulfhydryl oxidation on ionic and gating currents associated with L-type Ca2+ channels in isolated Guinea-pig ventricular myocytes. Cardiovasc Res. 30 (5), 799–806. doi:10.1016/0008-6363(95)00128-x
Lambert, M., Capuano, V., Olschewski, A., Sabourin, J., Nagaraj, C., Girerd, B., et al. (2018). Ion channels in pulmonary hypertension: A therapeutic interest? Int. J. Mol. Sci. 19 (10), 3162. doi:10.3390/ijms19103162
Leach, R. M., Robertson, T. P., Twort, C. H., and Ward, J. P. (1994). Hypoxic vasoconstriction in rat pulmonary and mesenteric arteries. Am. J. Physiol. 266, L223–L231. doi:10.1152/ajplung.1994.266.3.L223
Leach, R. M., Sheehan, D. W., Chacko, V. P., and Sylvester, J. T. (2000). Energy state, pH, and vasomotor tone during hypoxia in precontracted pulmonary and femoral arteries. Am. J. Physiol. Lung Cell. Mol. Physiol. 278 (2), L294–L304. doi:10.1152/ajplung.2000.278.2.L294
Leblanc, N., Forrest, A. S., Ayon, R. J., Wiwchar, M., Angermann, J. E., Pritchard, H. A., et al. (2015). Molecular and functional significance of Ca(2+)-activated Cl(-) channels in pulmonary arterial smooth muscle. Pulm. Circ. 5 (2), 244–268. doi:10.1086/680189
Liu, Q., and Flavahan, N. A. (1997). Hypoxic dilatation of porcine small coronary arteries: Role of endothelium and KATP-channels. Br. J. Pharmacol. 120 (4), 728–734. doi:10.1038/sj.bjp.0700939
Liu, S. Q., Jin, H., Zacarias, A., Srivastava, S., and Bhatnagar, A. (2001). Binding of pyridine nucleotide coenzymes to the beta-subunit of the voltage-sensitive K+channel. J. Biol. Chem. 276 (15), 11812–11820. doi:10.1074/jbc.M008259200
Lopez-Barneo, J., and Ortega-Saenz, P. (2022). Mitochondrial acute oxygen sensing and signaling. Crit. Rev. Biochem. Mol. Biol. 57 (2), 205–225. doi:10.1080/10409238.2021.2004575
Lopez-Barneo, J., Pardal, R., Montoro, R. J., Smani, T., Garcia-Hirschfeld, J., and Ureña, J. (1999). K+ and Ca2+ channel activity and cytosolic [Ca2+] in oxygen-sensing tissues. Respir. Physiol. 115 (2), 215–227. doi:10.1016/s0034-5687(99)00016-x
Lu, W., Wang, J., Peng, G., Shimoda, L. A., and Sylvester, J. T. (2009). Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 297 (1), L17–L25. doi:10.1152/ajplung.00063.2009
Lynch, F. M., Austin, C., Heagerty, A. M., and Izzard, A. S. (2006). Adenosine and hypoxic dilation of rat coronary small arteries: Roles of the ATP-sensitive potassium channel, endothelium, and nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 290 (3), H1145–H1150. doi:10.1152/ajpheart.00314.2005
Madden, J. A., Vadula, M. S., and Kurup, V. P. (1992). Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Am. J. Physiol. 263, L384–L393. doi:10.1152/ajplung.1992.263.3.L384
Manoury, B., Tamuleviciute, A., and Tammaro, P. (2010). TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J. Physiol. 588, 2305–2314. doi:10.1113/jphysiol.2010.189506
Marriott, J. F., and Marshall, J. M. (1990). Differential effects of hypoxia upon contractions evoked by potassium and noradrenaline in rabbit arteries in vitro. J. Physiol. 422, 1–13. doi:10.1113/jphysiol.1990.sp017968
Marshall, C., Mamary, A. J., Verhoeven, A. J., and Marshall, B. E. (1996). Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am. J. Respir. Cell. Mol. Biol. 15 (5), 633–644. doi:10.1165/ajrcmb.15.5.8918370
Marshall, J. M., Thomas, T., and Turner, L. (1993). A link between adenosine, ATP-sensitive K+ channels, potassium and muscle vasodilatation in the rat in systemic hypoxia. J. Physiol. 472, 1–9. doi:10.1113/jphysiol.1993.sp019931
Maurer, D. S., Zabini, D., Nagaraj, C., Sharma, N., Lengyel, M., Nagy, B. M., et al. (2020). Endothelial dysfunction following enhanced TMEM16A activity in human pulmonary arteries. Cells 9 (9), 1984. doi:10.3390/cells9091984
Michelakis, E. D., Hampl, V., Nsair, A., Wu, X., Harry, G., Haromy, A., et al. (2002). Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ. Res. 90 (12), 1307–1315. doi:10.1161/01.res.0000024689.07590.c2
Michelakis, E. D., Thebaud, B., Weir, E. K., and Archer, S. L. (2004). Hypoxic pulmonary vasoconstriction: Redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J. Mol. Cell. Cardiol. 37 (6), 1119–1136. doi:10.1016/j.yjmcc.2004.09.007
Mittal, M., Gu, X. Q., Pak, O., Pamenter, M. E., Haag, D., Fuchs, D. B., et al. (2012). Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 52 (6), 1033–1042. doi:10.1016/j.freeradbiomed.2011.12.004
Mohazzab, K. M., Fayngersh, R. P., Kaminski, P. M., and Wolin, M. S. (1995). Potential role of NADH oxidoreductase-derived reactive O2 species in calf pulmonary arterial PO2-elicited responses. Am. J. Physiol. 269, L637–L644. doi:10.1152/ajplung.1995.269.5.L637
Moral-Sanz, J., Mahmoud, A. D., Ross, F. A., Eldstrom, J., Fedida, D., Hardie, D. G., et al. (2016). AMP-activated protein kinase inhibits Kv 1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J. Physiol. 594 (17), 4901–4915. doi:10.1113/JP272032
Moreno-Dominguez, A., Ortega-Saenz, P., Gao, L., Colinas, O., Garcia-Flores, P., Bonilla-Henao, V., et al. (2020). Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal. 13 (615), eaay9452. doi:10.1126/scisignal.aay9452
Mori, Y., Takahashi, N., Polat, O. K., Kurokawa, T., Takeda, N., and Inoue, M. (2016). Redox-sensitive transient receptor potential channels in oxygen sensing and adaptation. Pflugers Arch. 468 (1), 85–97. doi:10.1007/s00424-015-1716-2
Moudgil, R., Michelakis, E. D., and Archer, S. L. (2005). Hypoxic pulmonary vasoconstriction. (1985) 98 (1), 390–403. doi:10.1152/japplphysiol.00733.2004
Mukai, Y., Shimokawa, H., Matoba, T., Kandabashi, T., Satoh, S., Hiroki, J., et al. (2001). Involvement of rho-kinase in hypertensive vascular disease: A novel therapeutic target in hypertension. FASEB J. 15 (6), 1062–1064. doi:10.1096/fj.00-0735fje
Muralidharan, P., Cserne Szappanos, H., Ingley, E., and Hool, L. (2016). Evidence for redox sensing by a human cardiac calcium channel. Sci. Rep. 6, 19067. doi:10.1038/srep19067
Murtaza, G., Mermer, P., Goldenberg, A., Pfeil, U., Paddenberg, R., Weissmann, N., et al. (2017). TASK-1 potassium channel is not critically involved in mediating hypoxic pulmonary vasoconstriction of murine intra-pulmonary arteries. PLoS One. 12 (3), e0174071. doi:10.1371/journal.pone.0174071[
Nelson, M. T., and Quayle, J. M. (1995). Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 268 (41), C799–C822. doi:10.1152/ajpcell.1995.268.4.C799
Numaguchi, K., Eguchi, S., Yamakawa, T., Motley, E. D., and Inagami, T. (1999). Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circ. Res. 85 (1), 5–11. doi:10.1161/01.res.85.1.5
Olschewski, A., Li, Y., Tang, B., Hanze, J., Eul, B., Bohle, R. M., et al. (2006). Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ. Res. 98 (8), 1072–1080. doi:10.1161/01.RES.0000219677.12988.e9
Olschewski, A., and Weir, E. K. (2015). Redox regulation of ion channels in the pulmonary circulation. Antioxid. Redox Signal 22 (6), 465–485. doi:10.1089/ars.2014.5899
Ortega-Saenz, P., Pascual, A., Gomez-Diaz, R., and Lopez-Barneo, J. (2006). Acute oxygen sensing in heme oxygenase-2 null mice. J. Gen. Physiol. 128 (4), 405–411. doi:10.1085/jgp.200609591
Pearce, W. J., Ashwal, S., Long, D. M., and Cuevas, J. (1992). Hypoxia inhibits calcium influx in rabbit basilar and carotid arteries. Am. J. Physiol. 262, H106–H113. doi:10.1152/ajpheart.1992.262.1.H106
Pearce, W. J. (1995). Mechanisms of hypoxic cerebral vasodilatation. Pharmacol. Ther. 65 (1), 75–91. doi:10.1016/0163-7258(94)00058-b
Post, J. M., Hume, J. R., Archer, S. L., and Weir, E. K. (1992). Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am. J. Physiol. 262, C882–C890. doi:10.1152/ajpcell.1992.262.4.C882
Quayle, J. M., Turner, M. R., Burrell, H. E., and Kamishima, T. (2006). Effects of hypoxia, anoxia, and metabolic inhibitors on KATP channels in rat femoral artery myocytes. Am. J. Physiol. Heart Circ. Physiol. 291 (1), H71–H80. doi:10.1152/ajpheart.01107.2005
Rathore, R., Zheng, Y. M., Niu, C. F., Liu, Q. H., Korde, A., Ho, Y. S., et al. (2008). Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 45 (9), 1223–1231. doi:10.1016/j.freeradbiomed.2008.06.012
Reeve, H. L., Weir, E. K., Nelson, D. P., Peterson, D. A., and Archer, S. L. (1995). Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue. Exp. Physiol. 80 (5), 825–834. doi:10.1113/expphysiol.1995.sp003890
Remillard, C. V., Tigno, D. D., Platoshyn, O., Burg, E. D., Brevnova, E. E., Conger, D., et al. (2007). Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell. Physiol. 292 (5), C1837–C1853. doi:10.1152/ajpcell.00405.2006
Remillard, C. V., and Yuan, J. X. (2006). TRP channels, CCE, and the pulmonary vascular smooth muscle. Microcirculation 13 (8), 671–692. doi:10.1080/10739680600930313
Reyes, R. V., Castillo-Galan, S., Hernandez, I., Herrera, E. A., Ebensperger, G., and Llanos, A. J. (2018). Revisiting the role of TRP, orai, and ASIC channels in the pulmonary arterial response to hypoxia. Front. Physiol. 9, 486. doi:10.3389/fphys.2018.00486
Robertson, T. P., Dipp, M., Ward, J. P., Aaronson, P. I., and Evans, A. M. (2000). Inhibition of sustained hypoxic vasoconstriction by Y-27632 in isolated intrapulmonary arteries and perfused lung of the rat. Br. J. Pharmacol. 131 (1), 5–9. doi:10.1038/sj.bjp.0703537
Robertson, T. P., Ward, J. P., and Aaronson, P. I. (2001). Hypoxia induces the release of a pulmonary selective, Ca2+-sensitising, vasoconstrictor from the perfused rat lung. Cardiovasc Res. 50 (1), 145–150. doi:10.1016/s0008-6363(01)00192-4
Rosa, A. O., Movafagh, S., Cleemann, L., and Morad, M. (2012). Hypoxic regulation of cardiac Ca2+ channel: Possible role of haem oxygenase. J. Physiol. 590 (17), 4223–4237. doi:10.1113/jphysiol.2012.236570
Rounds, S., and McMurtry, I. F. (1981). Inhibitors of oxidative ATP production cause transient vasoconstriction and block subsequent pressor responses in rat lungs. Circ. Res. 48 (3), 393–400. doi:10.1161/01.res.48.3.393
Sahoo, N., Hoshi, T., and Heinemann, S. H. (2014). Oxidative modulation of voltage-gated potassium channels. Antioxid. Redox Signal 21 (6), 933–952. doi:10.1089/ars.2013.5614
Saitoh, S., Zhang, C., Tune, J. D., Potter, B., Kiyooka, T., Rogers, P. A., et al. (2006). Hydrogen peroxide: A feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler. Thromb. Vasc. Biol. 26 (12), 2614–2621. doi:10.1161/01.ATV.0000249408.55796.da
Scragg, J. L., Dallas, M. L., Wilkinson, J. A., Varadi, G., and Peers, C. (2008). Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J. Biol. Chem. 283 (36), 24412–24419. doi:10.1074/jbc.M803037200
Sham, J. S., Crenshaw, B. R., Deng, L. H., Shimoda, L. A., and Sylvester, J. T. (2000). Effects of hypoxia in porcine pulmonary arterial myocytes: Roles of Kv channel and endothelin-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 279 (2), L262–L272. doi:10.1152/ajplung.2000.279.2.L262
Smani, T., Hernandez, A., Ureña, J., Castellano, A. G., Franco-Obregon, A., Ordoñez, A., et al. (2002). Reduction of Ca2+ channel activity by hypoxia in human and porcine coronary myocytes. Cardiovasc Res. 53 (1), 97–104. doi:10.1016/s0008-6363(01)00422-9
Smani, T., Iwabuchi, S., Lopez-Barneo, J., and Ureña, J. (2001). Differential segmental activation of Ca2+-dependent Cl- and K+ channels in pulmonary arterial myocytes. Cell. Calcium 29 (6), 369–377. doi:10.1054/ceca.2001.0199
Somlyo, A. P., and Somlyo, A. V. (2003). Ca2+ sensitivity of smooth muscle and non-muscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83 (4), 1325–1358. doi:10.1152/physrev.00023.2003
Sommer, N., Huttemann, M., Hüttemann, M., Pak, O., Scheibe, S., Knoepp, F., et al. (2017). Mitochondrial complex IV subunit 4 isoform 2 is essential for acute pulmonary oxygen sensing. Circ. Res. 121 (4), 424–438. doi:10.1161/CIRCRESAHA.116.310482
Sommer, N., Pak, O., Schorner, S., Derfuss, T., Krug, A., Gnaiger, E., et al. (2010). Mitochondrial cytochrome redox states and respiration in acute pulmonary oxygen sensing. Eur. Respir. J. 36 (5), 1056–1066. doi:10.1183/09031936.00013809
Sommer, N., Strielkov, I., Pak, O., and Weissmann, N. (2016). Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 47 (1), 288–303. doi:10.1183/13993003.00945-2015
Song, T., Zheng, Y. M., and Wang, Y. X. (2017). Cross talk between mitochondrial reactive oxygen species and sarcoplasmic reticulum calcium in pulmonary arterial smooth muscle cells. Adv. Exp. Med. Biol. 967, 289–298. doi:10.1007/978-3-319-63245-2_17
Sun, H., Paudel, O., and Sham, J. S. (2021). Increased intracellular Cl- concentration in pulmonary arterial myocytes is associated with chronic hypoxic pulmonary hypertension. Am. J. Physiol. Cell. Physiol. 321 (2), C297–C307. doi:10.1152/ajpcell.00172.2021
Sun, H., Xia, Y., Paudel, O., Yang, X. R., and Sham, J. S. (2012). Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: A mechanism contributing to enhanced vasoreactivity. J. Physiol. 590, 3507–3521. doi:10.1113/jphysiol.2012.232520
Sylvester, J. T., Shimoda, L. A., Aaronson, P. I., and Ward, J. P. (2012). Hypoxic pulmonary vasoconstriction. Physiol. Rev. 92 (1), 367–520. doi:10.1152/physrev.00041.2010
Taggart, M. J., and Wray, S. (1998). Hypoxia and smooth muscle function: Key regulatory events during metabolic stress. J. Physiol. 509, 315–325. doi:10.1111/j.1469-7793.1998.315bn.x
Tipparaju, S. M., Saxena, N., Liu, S. Q., Kumar, R., and Bhatnagar, A. (2005). Differential regulation of voltage-gated K+ channels by oxidized and reduced pyridine nucleotide coenzymes. Am. J. Physiol. Cell. Physiol. 288 (2), C366–C376. doi:10.1152/ajpcell.00354.2004
Torres-Torrelo, H., Ortega-Saenz, P., Gao, L., and Lopez-Barneo, J. (2021). Lactate sensing mechanisms in arterial chemoreceptor cells. Nat. Commun. 12 (1), 4166. doi:10.1038/s41467-021-24444-7
Urban, N., Hill, K., Wang, L., Kuebler, W. M., and Schaefer, M. (2012). Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell. Calcium 51 (2), 194–206. doi:10.1016/j.ceca.2012.01.001
Ureña, J., Franco-Obregon, A., and Lopez-Barneo, J. (1996). Contrasting effects of hypoxia on cytosolic Ca2+ spikes in conduit and resistance myocytes of the rabbit pulmonary artery. J. Physiol. 496, 103–109. doi:10.1113/jphysiol.1996.sp021668
Veit, F., Pak, O., Brandes, R. P., and Weissmann, N. (2015). Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: Focus on ion channels. Antioxid. Redox Signal 22 (6), 537–552. doi:10.1089/ars.2014.6234
Wadsworth, R. M. (1994). Vasoconstrictor and vasodilator effects of hypoxia. Trends Pharmacol. Sci.15 15, 47–53. doi:10.1016/0165-6147(94)90109-0
Wang, Q. S., Zheng, Y. M., Dong, L., Ho, Y. S., Guo, Z., and Wang, Y. X. (2007). Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic. Biol. Med. 42 (5), 642–653. doi:10.1016/j.freeradbiomed.2006.12.008
Wang, Y. X., and Zheng, Y. M. (2010). Role of ROS signaling in differential hypoxic Ca2+ and contractile responses in pulmonary and systemic vascular smooth muscle cells. Respir. Physiol. Neurobiol. 174 (3), 192–200. doi:10.1016/j.resp.2010.08.008
Wang, Z., Jin, N., Ganguli, S., Swartz, D. R., Li, L., and Rhoades, R. A. (2001). Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am. J. Respir. Cell. Mol. Biol. 25 (5), 628–635. doi:10.1165/ajrcmb.25.5.4461
Waypa, G. B., Chandel, N. S., and Schumacker, P. T. (2001). Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 88 (12), 1259–1266. doi:10.1161/hh1201.091960
Waypa, G. B., Guzy, R., Mungai, P. T., Mack, M. M., Marks, J. D., Roe, M. W., et al. (2006). Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 99 (9), 970–978. doi:10.1161/01.RES.0000247068.75808.3f
Waypa, G. B., Marks, J. D., Guzy, R., Mungai, P. T., Schriewer, J., Dokic, D., et al. (2010). Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 106 (3), 526–535. doi:10.1161/CIRCRESAHA.109.206334
Waypa, G. B., Marks, J. D., Guzy, R. D., Mungai, P. T., Schriewer, J. M., Dokic, D., et al. (2013). Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 187 (4), 424–432. doi:10.1164/rccm.201207-1294OC
Waypa, G. B., and Schumacker, P. T. (2002). O2 sensing in hypoxic pulmonary vasoconstriction: The mitochondrial door re-opens. Respir. Physiol. Neurobiol. 132 (1), 81–91. doi:10.1016/s1569-9048(02)00051-4
Weir, E. K., Eaton, J. W., and Chesler, E. (1985). Redox status and pulmonary vascular reactivity. Chest 88 (4), 249S–252S. doi:10.1378/chest.88.4_supplement.249s
Weir, E. K., Lopez-Barneo, J., Buckler, K. J., and Archer, S. L. (2005). Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353 (19), 2042–2055. doi:10.1056/NEJMra050002
Weir, E. K., Reeve, H. L., Cornfield, D. N., Tristani-Firouzi, M., Peterson, D. A., and Archer, S. L. (1997). Diversity of response in vascular smooth muscle cells to changes in oxygen tension. Kidney Int. 51 (2), 462–466. doi:10.1038/ki.1997.62
Weissmann, N., Akkayagil, E., Quanz, K., Schermuly, R. T., Ghofrani, H. A., Fink, L., et al. (2004). Basic features of hypoxic pulmonary vasoconstriction in mice. Respir. Physiol. Neurobiol. 139 (2), 191–202. doi:10.1016/j.resp.2003.10.003
Weissmann, N., Dietrich, A., Fuchs, B., Kalwa, H., Ay, M., Dumitrascu, R., et al. (2006). Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. U. S. A. 103 (50), 19093–19098. doi:10.1073/pnas.0606728103
Weissmann, N., Ebert, N., Ahrens, M., Ghofrani, H. A., Schermuly, R. T., Hänze, J., et al. (2003). Effects of mitochondrial inhibitors and uncouplers on hypoxic vasoconstriction in rabbit lungs. Am. J. Respir. Cell. Mol. Biol. 29 (6), 721–732. doi:10.1165/rcmb.2002-0217OC
Weissmann, N., Grimminger, F., Walmrath, D., and Seeger, W. (1995). Hypoxic vasoconstriction in buffer-perfused rabbit lungs. Respir. Physiol. 100 (2), 159–169. doi:10.1016/0034-5687(94)00133-k
Weissmann, N., Voswinckel, R., Hardebusch, T., Rosseau, S., Ghofrani, H. A., Schermuly, R., et al. (1999). Evidence for a role of protein kinase C in hypoxic pulmonary vasoconstriction. Am. J. Physiol. 276 (1), L90–L95. doi:10.1152/ajplung.1999.276.1.L90
Weissmann, N., Winterhalder, S., Nollen, M., Voswinckel, R., Quanz, K., Ghofrani, H. A., et al. (2001). NO and reactive oxygen species are involved in biphasic hypoxic vasoconstriction of isolated rabbit lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 280 (4), L638–L645. doi:10.1152/ajplung.2001.280.4.L638
Welsh, D. G., Jackson, W. F., and Segal, S. S. (1998). Oxygen induces electromechanical coupling in arteriolar smooth muscle cells: A role for L-type Ca2+ channels. Am. J. Physiol. 274 (6), H2018–H2024. doi:10.1152/ajpheart.1998.274.6.H2018
Wilson, H. L., Dipp, M., Thomas, J. M., Lad, C., Galione, A., and Evans, A. M. (2001). ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase act as a redox sensor. A primary role for cyclic ADP-ribose in hypoxic pulmonary vasoconstriction. J. Biol. Chem. 276 (14), 11180–11188. doi:10.1074/jbc.M004849200
Yu, Y., Keller, S. H., Remillard, C. V., Safrina, O., Nicholson, A., Zhang, S. L., et al. (2009). A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119 (17), 2313–2322. doi:10.1161/CIRCULATIONAHA.108.782458
Yu, Y., Sweeney, M., Zhang, S., Platoshyn, O., Landsberg, J., Rothman, A., et al. (2003). PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am. J. Physiol. Cell. Physiol. 284 (2), C316–C330. doi:10.1152/ajpcell.00125.2002
Yuan, J. X., Aldinger, A. M., Juhaszova, M., Wang, J., Conte, J. V., Gaine, S. P., et al. (1998). Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98 (14), 1400–1406. doi:10.1161/01.cir.98.14.1400
Yuan, X. J., Goldman, W. F., Tod, M. L., Rubin, L. J., and Blaustein, M. P. (1993). Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am. J. Physiol. 264, L116–L123. doi:10.1152/ajplung.1993.264.2.L116
Yuan, X. J. (1997). Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am. J. Physiol. 272, L959–L968. doi:10.1152/ajplung.1997.272.5.L959
Yuan, X. J., Tod, M. L., Rubin, L. J., and Blaustein, M. P. (1990). Contrasting effects of hypoxia on tension in rat pulmonary and mesenteric arteries. Am. J. Physiol. 259, H281–H289. doi:10.1152/ajpheart.1990.259.2.H281
Keywords: acute O2 sensing, hypoxic pulmonary vasoconstriction, hypoxic arterial vasodilation, vascular smooth muscle, mitochondria, ion channels
Citation: Moreno-Domínguez A, Colinas O, Smani T, Ureña J and López-Barneo J (2023) Acute oxygen sensing by vascular smooth muscle cells. Front. Physiol. 14:1142354. doi: 10.3389/fphys.2023.1142354
Received: 11 January 2023; Accepted: 21 February 2023;
Published: 03 March 2023.
Edited by:
Dewan S. Majid, Tulane University, United StatesReviewed by:
Christelle guibert, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2023 Moreno-Domínguez, Colinas, Smani, Ureña and López-Barneo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José López-Barneo, bGJhcm5lb0B1cy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.