 Y. H. Tompkins
Y. H. Tompkins G. Liu
G. Liu W. K. Kim
W. K. Kim- Department of Poultry Science, University of GA, Athens, GA, United States
The effects of hydrogen peroxide (H2O2) on the osteogenic differentiation of primary chicken mesenchymal stem cells (MSCs) were investigated. MSCs were subjected to an osteogenic program and exposed to various concentrations of H2O2 for 14 days. Results showed that high concentrations of H2O2 (200 and 400 nM) significantly increased pro-apoptotic marker CASP8 expression and impaired osteogenic differentiation, as indicated by decreased mRNA expression levels of osteogenesis-related genes and reduced in vitro mineralization. In contrast, long-term H2O2 exposure promoted basal expression of adipogenic markers at the expense of osteogenesis in MSCs during osteogenic differentiation, and increased intracellular reactive oxygen species (ROS) production, as well as altered antioxidant enzyme gene expression. These findings suggest that long-term H2O2-induced ROS production impairs osteogenic differentiation in chicken MSCs under an osteogenic program.
Introduction
Modern commercial poultry production is strictly operated based on balanced nutrition and optimized environmental conditions. However, oxidative stress is ubiquitous in broiler production systems. For example, unbalanced nutrition, pathogen infection, and poor environmental conditions including heat stress, ammonia exposure, and flock density, can induce oxidative stress in broilers (Mishra and Jha, 2019; Ali Hassan and Li, 2021; Chauhan et al., 2021). Oxidative stress has been reported as a negative factor in broiler performance, healthy growth, and production quality (Mishra and Jha, 2019; Surai et al., 2019), representing an unbalanced condition between the production of reactive oxygen species (ROS) and the antioxidant defense systems (Sies et al., 2017). In broiler production, in addition to management-associated physiological oxidative stress, infectious agents are key factors that can cause severe oxidative stress in broilers (Zorov et al., 2014; Forrester et al., 2018).
Bone growth depends on the activity of bone-related cells, where osteoblasts are important cells involved in bone formation (Hambli, 2014). Osteoblast originates from mesenchymal stem cells (Zomorodian and Baghaban Eslaminejad, 2012). There is an ongoing interest in using mesenchymal stem cells (MSCs) as a study model to understand cell physiology, and etiology of bone disease (Svoradova et al., 2021), and the effect of oxidative stress on mammal MSCs has been well noted (Denu and Hematti, 2016). Hydrogen peroxide (H2O2), a non-radical ROS, has been extensively studied for its effects (Sharma et al., 2012). It is one of the most common endogenous byproducts of mitochondrial respiration that is present in the avian system (Ojano-Dirain et al., 2007). At a cellular level, ROS level is tightly regulated by the antioxidant defense system and is critical for MSCs multipotency (Atashi et al., 2015). A low basal level of ROS is a critical mediator in pathophysiological responses (Maraldi et al., 2015), while differentiated MSCs presented a higher intracellular ROS production which is essential for cell survival and early differentiation (Hu et al., 2018). However, the uncontrollable high levels of ROS not only impair the cell membrane fluidity and permeability but are also responsible for oxidation damage to DNA, RNA, protein, and lipid damage in mitochondria, leading to cellular senescence including cell dysfunction, cellular injury and cell apoptosis (Denu and Hematti, 2016). For cellular homeostasis, endogenous scavengers such as enzymatic proteins, including superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalases (CAT), and non-enzymatic antioxidants, such as vitamins and trace minerals (Hu et al., 2018), are all work together for controlling intracellular ROS-related stress by removing and converting excessive ROS (Denu and Hematti, 2016).
Relatively high level of ROS can directly interact with critical signaling molecules in essential osteogenic pathways, negatively impacts bone homeostasis (Atashi et al., 2015). Oxidative stress induced by ROS over-production has been considered a pathogenic factor involved in human skeletal disorders (Sharma et al., 2015). Moreover, in response to oxidative stress, ROS can activate extracellular signal-regulated kinases that modulate nuclear factor kappa light chain enhancer of activated B-cell (NF-κB) and NF-E2 p45-related factor 2 (NRF2) signaling pathways, which are critical for regulating inflammation, cellular redox status, and bone homeostasis by regulating osteoblast or osteoclast differentiation and activity (Sun et al., 2015; Yuan et al., 2017). For example, in the mouse cell line, ROS induced MSCs commit to adipogenesis rather than osteogenesis at the transcriptional level (Lin et al., 2018). However, the response of chicken MSCs to high ROS levels and the effect of ROS production on avian osteoblastic differentiation are not well understood. Understanding the impact of ROS on MSC terminal fate and differentiation capacity is important for developing novel strategies for prevent, anticipate, or revert oxidative stress-induced leg problems in chicken production. Therefore, in the current study, H2O2 was used as a stimulator of oxidative stress in MSCs culture. This study aimed to investigate the effects of H2O2 on the osteogenic differentiation of chicken MSCs isolated from broiler compact bones.
Materials and methods
Animal use and ethics statement
The study was carried out in compliance with the ARRIVE guidelines. All experiment protocols and animal use were approved by the Institutional Animal Care and Use Committee at the University of Georgia, Athens, GA.
Isolation of broiler MSCs
MSCs were isolated using previously described methods (Adhikari et al., 2018). Briefly, legs from one-day old chicks were obtained after cervical dislocation. The leg tissue was soaked in alcohol for a minute and then dried with Kimwipes (Kimberly Clark, Irving, TX, United States). Muscle was removed, and long bones were harvested. The long bones were kept in high glucose Dulbecco’s Modified Eagle’s medium (DMEM; contains 4.5 g/L glucose, 25 mM HEPES, sodium pyruvate, and without L-glutamine; 15–018-cv, Corning, Corning, NY, United States) until muscle and cartilage tissues were completely removed using a scalpel and scissors in a bio-safety cabinet (NuAire, Plymouth, MN, United States). The bones were placed in washing buffer to cut off the metaphysis. Only tibia diaphysis and femur diaphysis were kept for cell isolation. Washing buffer contained 2% fetal bovine serum (FBS) (Hyclone Laboratories Inc., Logan, UT) in Dulbecco’s phosphate-buffer saline (PBS) (Corning). The bones were cracked with a scalpel, bone marrow was flushed out with washing buffer, and bone marrow was discarded. The bones were chopped into small fragments and suspended in a 50 mL tube containing a 10 mL digestion medium consisting of 100 IU/mL penicillin, 100 ug/mL streptomycin, 0.25% collagenase (Sigma-Aldrich, St. Louis, MO, USA), 20% FBS, and high glucose DMEM. The tubes were placed in a 37°C incubated with an orbital shaker set at 180 rpm for 60 min (VWR, Radnor, PA, United States). The digested bone solution was filtered with a 40 μm cell strainer (Thermo Fisher Scientific, Waltham, MA, United States) set over a 50 ml tube to remove the bone fragments, and then the filtered medium was centrifuged at 1,200 rpm for 10 min. The supernatant was discarded and the cell pellet was resuspended in 20 ml growth medium consisting of DMEM with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.292 mg/mL L-glutamine (Thermo Fisher Scientific), and 10 ml resuspend cells were plated in a 100 mm cell culture dishes (Corning). Cells were incubated at 37°C in a humidified incubator (NuAire) containing 5% CO2. Half of the medium was replaced with fresh growth medium after 24 h of culture, and the culture medium was changed every two days thereafter. For cell passing, when the cells reached 80% confluency, they were washed twice with 5 mL pre-warmed PBS, dissociated with 1.5 mL 0.1% Trypsin-EDTA (Corning) for 2 min at 37°C, and subcultured with cell density of 25,000 cells/cm2 in 100 mm cell culture dishes.
Viability of cultured chicken MSCs with H2O2 exposure
The viability of cells was determined using cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) kits (Cayman Chemical, Ann Arbor, MI, USA). Cells were seeded at a concentration of 5 × 104 cells/100 μL in differentiation medium per well with 96-well black wall culture plates. The treatment of MSCs with various concentrations of H2O2 (50, 100, 200, 400, and 800 nM; H2O2, 30% (w/w) solution, Sigma-Aldrich) during culture was also examined. The H2O2 stock was diluted by PBS, and the same volume of diluted H2O2 solution was added to the culture medium. MSCs were cultured without H2O2 but PBS (osteogenic differentiation medium with 0 nM H2O2) as a control. Cells were incubated with different levels of H2O2 treatments for 6, 24, and 48 h in the dark. The MTT viability assay was not performed for longer periods of time because high cell density led to high absorbance readings that impaired the accuracy of detection. DMEM with 10% MTT was added and incubated for 4 h, after wich the culture medium was completely discarded. The formed formazan was dissolved with 100 µl dimethyl sulfoxide (DMSO; Sigma-Aldrich) to produce a purple color, and the plates were then placed on an orbital shaker (VWR) set at low speed for 5 min. The absorbance was measured at 570 nm using a microplate reader (BioTek, Winooski, VT, United states).
Intracellular reactive oxygen species detection
The intracellular ROS levels were examined using the DCFDA/H2DCFDA cellular ROS assay kits (Abcam Cambridge, MA, United states)) according to the manufacturer’s instruction. Briefly, as per manufacturer’s instructions, 3 × 104 cells were seeded in a black clear-flat-bottom 96-well microplate and allowed to adhere overnight. The cells were treated with or without H2O2 treatment for 2 h, 6 h, 24 h, and 48 h, but not for longer periods of time as the ROS detection required relatively low cell density for accurate results. After the corresponding treatments, the medium was removed, 100 µL/well of 1× washing buffer was added to remove any residual material, and then 100 µL/well of the diluted DCFDA solution was added to stain for 45 min at 37°C in the culture incubator. After removing the DCFDA solution, the cells were rinsed once with a washing buffer, 100 µL/well of washing buffer was then added for microplate measurement. The fluorescence density was measured with a microplate reader (Spectramax M5, Molecular Devices, San Jose, CA) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. Images were obtained using a fluorescence microscope (Keyence bz-X8000, Keyence Corp., Osaka, Japan) at ×10 magnification.
Osteogenic differentiation
MSC cells were expanded to passage 4 for osteogenic differentiation study. After selecting the proper concentration for H2O2 treatment based on the MTT assay, the cells were seeded at a density of 8 × 104 cells per well in 0.2% gelatin-coated (Alfa Aesar, Ward Hill, MA USA) 24-well cell culture plates (Corning), and cultured in growth medium at 37°C in the cell culture incubator (NuAire) until 100% confluency. The cells were then treated with osteogenic differentiation medium containing high glucose DMEM with 10–7 M dexamethasone (Sigma-Aldrich), 10 mM β-glycerophosphate (Sigma-Aldrich), 50 μg/mL ascorbate (Sigma-Aldrich), 5% FBS, and 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.292 mg/mL L-glutamine (Thermo Fisher Scientific) to induce osteogenic differentiation. Cells cultured in growth medium served as the negative control. Culture medium was replaced with fresh pre-warmed differentiation medium daily. The cells underwent differentiation for 6 h, 24 h, 48 h, 72 h, 96 h, 5 days, 6 days, 10 days and 14 days.
Alizarin red S staining and mineral deposit quantification
The degree of mineralization of chicken MSCs was determined using Alizarin red S staining (Adhikari et al., 2018). Briefly, the cells were seeded at a density of 8 × 104 cells per well in 0.2% gelatin-coated (Alfa Aesar) 24-well cell culture plates (Corning), and cultured in a growth medium at 37°C in the cell culture incubator (NuAire) until 100% confluency. The cells were then exposed to H2O2 in osteogenic differentiation medium for 6, 10, and 14 days. On each day of staining, the cells were fixed with 10% neutral buffered formalin for 1 h and then stained with 0.2% Alizarin red S (Sigma-Aldrich, St. Louis, MO) in distilled water for 45 min at room temperature. After rinsing with distilled water, images of cell culture plates were captured in ×2 magnification using a microscope with a camera (Keyence bz-×8000, Keyence). Mineralized nodules were labeled as dark red spots. To quantify the mineral deposition, the stained cells were solubilized with 200 µl of 10% acetic acid per well and incubated for 30 min with low-speed shaking on an orbital shaker (VWR). After the monolayer was loosely attached, the cells were gently scraped from the plate and transferred to a 1.5 mL microcentrifuge tube. The microcentrifuge tubes containing the cells were then vortexed vigorously for 40 s and heated to 85 °C for 10 min. The tubes were transferred on ice to cool down for 5 min and then centrifuged at 20,000 g for 15 min. After that, 150 µl of the supernatant was aliquoted to a new 1.5 ml microcentrifuge tube and the pH was neutralized with 60 µl 10% ammonium hydroxide. After supernatant neutralization, 50 µl of each sample was loaded into an opaque-walled transparent bottom 96-well plate and read at OD 405 nm using a microplate reader (BioTek) for Alizarin red S staining quantification (Serguienko et al., 2018).
Von kossa staining and quantification
The degree of mineralization of chicken MSCs was determined using the von Kossa staining (Adhikari et al., 2018). Cells were seeded at a density of 8 × 104 cells per well in 0.2% gelatin-coated (Alfa Aesar) 24-well cell culture plates (Corning), and cultured in growth medium at 37°C until 100% confluency. Then the cells were exposed to H2O2 in osteogenic differentiation medium for 6, 10, and 14 days. At different time points, the cell culture plates were washed three times with PBS and then fixed with 0.1% glutaraldehyde (G5882, Sigma-Aldrich) in PBS (pH 7.0) for 15 min at room temperature. After discarding the fixation buffer, the cells were washed three times with distilled water and then incubated in 5% silver nitrate (Sigma-Aldrich) for 30 min. The silver nitrate solution was discarded, and the cells were washed with distilled water at least three times, air-dried, and exposed to bright light until black color developed in areas of calcification. Images of cell culture plates were captured at ×2 magnification using a microscope with a camera (Keyence bz-×8000, Keyence). Mineralized nodules were observed as dark brown to black spots. The stained plates were quantified using the area fractions method with the ImageJ program (National Institutes of Health, Bethesda, MD, USA). Three images from each well were analyzed, and the mean area fraction from each well was used for statistical analysis.
RNA isolation, cDNA synthesis, and real-time polymerase chain reaction (qRT-PCR) analysis
The cell culture process for RNA isolation was the same as the osteogenic differentiation. Briefly, MSC cells were expanded to passage 4 and seeded at a density of 8 × 104 cells per well in 0.2% gelatin-coated (Alfa Aesar) 24-well cell culture plates (Corning), and cultured in growth medium until they reached100% confluency. The cells were differentiated for 6 h, 24 h, 48 h, 72 h, 96 h, 5 days, 6 days, 10 days and 14 days, with or without H2O2 treatment. At each time point, total RNA was extracted from the cells using QIAzol lysis reagents (Qiagen 79,306, Germantown, MD, USA) according to the manufacturer’s instructions. The Nano-Drop 1000 Spectrophotometer (ThermoFisher Scientific) was used to determine the quantity of extracted RNA. cDNA was synthesized from 2000 ng of total RNA using high-capacity cDNA reverse transcription kits (Thermo Fisher Scientific). Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) was used to measure mRNA expression. Primers were designed using the Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The specificity of primers was validated by PCR product sequencing; the details of primer sequences used for the experiment are presented in Table 1. Primer quality was verified through melting curve analysis and gel electrophoresis in this study. The qRT-PCR was performed on an Applied Biosystems StepOnePlus™ (Thermo Fisher Scientific) with iTaq™ universal SYBR Green Supermix (BioRad, Hercules, CA, United states) using the following conditions for all genes: 95°C for 10 min followed 40 cycles at 95°C for 15 s, annealing temperature (Table 1) for 20 s, and extending at 72°C for 1 min.
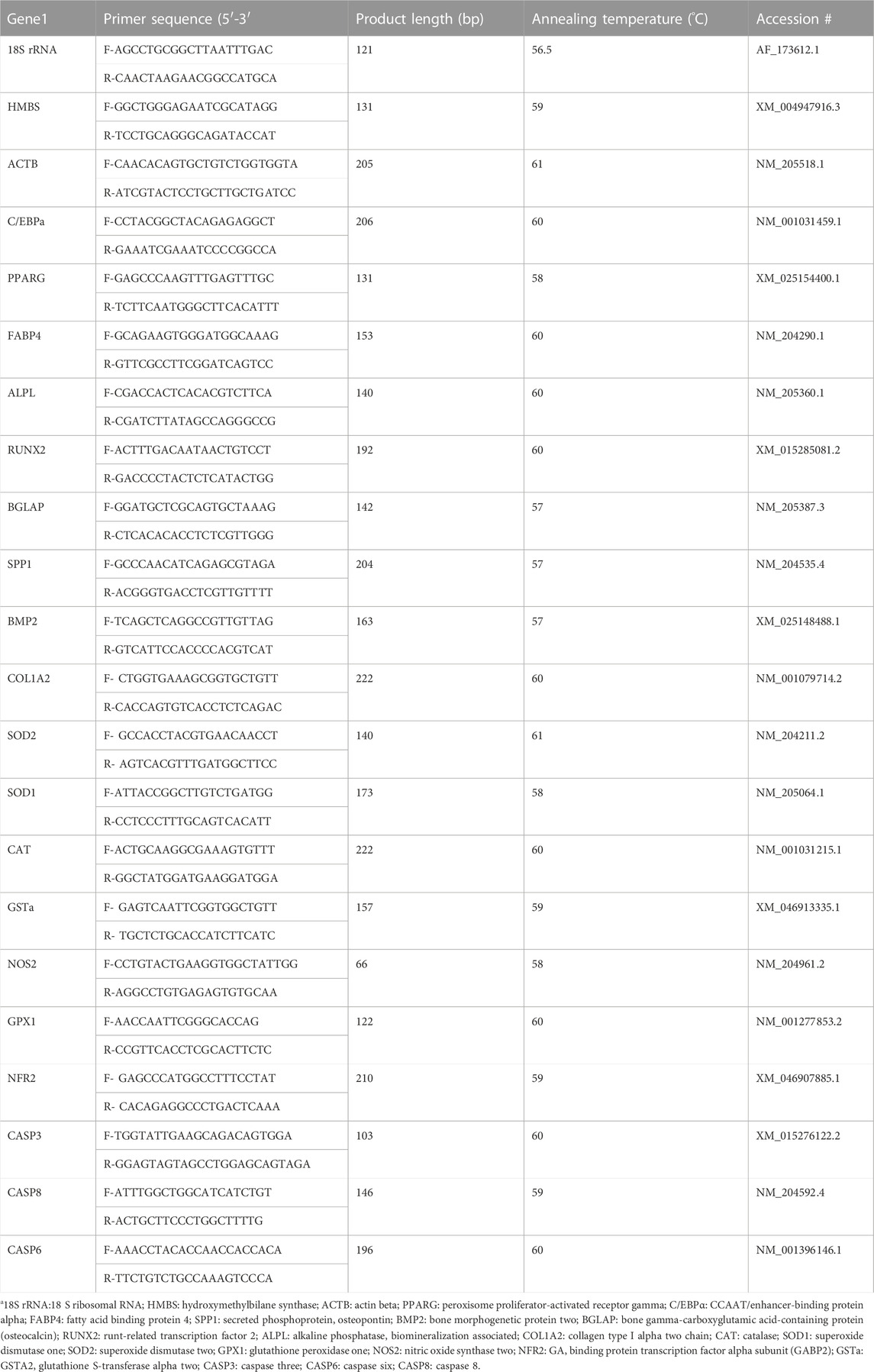
TABLE 1. Nucleotide sequences of the primers used for quantitative real-time RT-PCR.
The geometric means of the cycle threshold (Ct) values of three housekeeping genes, including hydroxymethylbilane synthase (HMBS), 18 S ribosomal RNA (18S rRNA), and actin beta (ACTB) were used for normalization. The stability of the housekeeping genes was confirmed by their consistent Ct values among the treatments (p > 0.1) and also assessed by statistical algorithms by software program NormFinder (Version 0.953; https://moma.dk/normfinder-software) (Wan et al., 2011). Peroxisome proliferator-activated receptor gamma (PPARG), adipose tissue fatty acid binding protein 4 (FABP4), and CCAAT enhancer binding protein alpha (CEBPA) were used as early markers of adipogenic differentiation, and alkaline phosphatase-biomineralization associated (ALPL), bone gamma-carboxyglutamate protein (BGLAP), runt-related transcription factor 2 (RUNX2), secreted phosphoprotein 1 (SPP1), collagen type I alpha two chain (COL1A2), bone–specific alkaline phosphatase (ALP), and bone morphogenetic protein 2 (BMP2) were used as osteogenic marker genes in the bone marrow. Nuclear factor kappa B subunit 1 (NFKB1) and antioxidant enzyme protein coding genes including catalase (CAT), superoxide dismutase type 1 (SOD1), superoxide dismutase type 2 (SOD2), glutathione peroxidase 1 (GPX1), glutathione S-transferase alpha 2 (GSTA2), and nitric oxide synthase 2 (NOS2) were used to determine the antioxidant enzyme activity and oxidative stress status. Pro-apoptotic marker genes, such as Caspase 3 (CASP3), Caspase 8 (CASP8), and Caspase 6 (CASP6), were used to assess the cell apoptosis. Samples were run in triplicate, and relative gene expression data were analyzed using the 2−ΔΔCt formula (Livak and Schmittgen, 2001). The expression levels of the other treatment groups were presented as fold changes relative to the average ΔCT value for each gene in the control group.
Statistical analysis
All experimental data were expressed as means with standard errors of the mean (SEM). The data were tested for homogeneity of variances and normality of studentized residuals. The differences between the treatment groups were analyzed using one-way ANOVA, and the means were statistically analyzed using Tukey’s test using JMP Pro14 (SAS Institute, Cary, NC, USA). Statistical significance was set at p ≤ 0.05, and values of 0.05 ≤ p ≤ 0.1 were also presented to show a trend towards statistical significance (Serdar et al., 2021).
Results
Cell viability, cell apoptosis, and intracellular ROS production with exogenous H2O2 exposure
Cell viability was measured by MTT assay after H2O2 exposure (Figure 1). The viability of chicken MSCs exposed to H2O2 showed a different rate of decline among doses. Chicken MSCs were cultured with different concentrations (50–800 nM) of H2O2 in osteogenic differentiation medium for 6 h showed a non-cytotoxic effect (p > 0.05; Figure 1A). At 24 h of treatment, 800 nM of H2O2 significantly reduced cell viability (p < 0.05; Figure 1B) by approximately 70% compared to the untreated control. After 48 h, 800 nM of H2O2 reduced cell viability by approximately 90% compared to the untreated control (p < 0.05; Figure 1C). No statistically significant change in cell viability was observed with treatment concentrations lower than 800 nM. Treatment doses above 400 nM significantly reduced the number of cells to an extent that was not adequate to conduct the experiment. Therefore, doses below 400 nM were selected for further studies. The final treatment concentrations of 100, 200, and 400 nM of H2O2 were selected as the treatment doses for the following experiments.
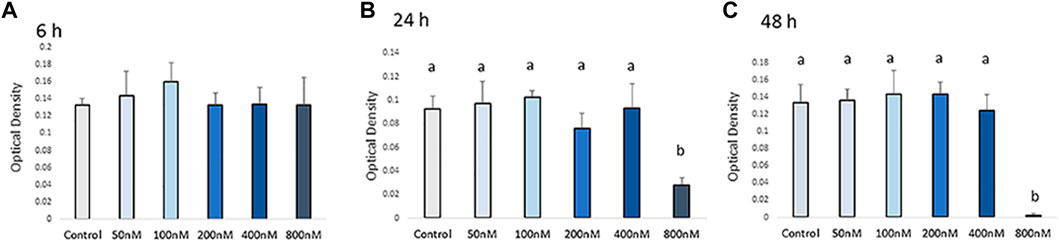
FIGURE 1. Effects of H2O2 on the cell viability. Cells were treated with the indicated concentrations of H2O2 for 6 h (A), 24 h (B) and 48 h (C). The graphs show changes in cellular growth as assessed by MTT assays. The MTT assay showed that exposure to concentrations higher than 400 nM of H2O2 can reduce cell viability. Therefore, the appropriate H2O2 concentration was screened out and final treatment concentrations of 100 nM, 200 nM and 400 nM of H2O2 were selected as the treatments dose for the following experiments. a, b Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test, p < 0.05; data are shown as mean ± SEM of four independent replicates (n = 4).
Intracellular ROS production in chicken MSCs after H2O2 exposure was monitored using a cellular DCFDA assay (Figures 2A,B). A significant increase in intracellular ROS production was detected in 100, 200, and 400 nM H2O2-treated chicken MSCs after 6 h compared to the control (p < 0.05; Figure 2B), with the highest upregulated ROS response observed at 100 nM of H2O2. After 12 h of H2O2 exposure, there was a significant difference in ROS production between 100 nM and 400 nM groups (p < 0.05; Figure 2B). However, intracellular ROS signal were not significantly affected by H2O2 treatment after 24 and 48 h of H2O2 exposure. Based on these data, the effective treatment duration is 12 h following exogenous H2O2 exposure. To minimized damage to the cell membrane and reduce variations in results due to cell death and reduced cell number caused by prolonged severe stress, the frequency of medium change was set to be every 24 h.
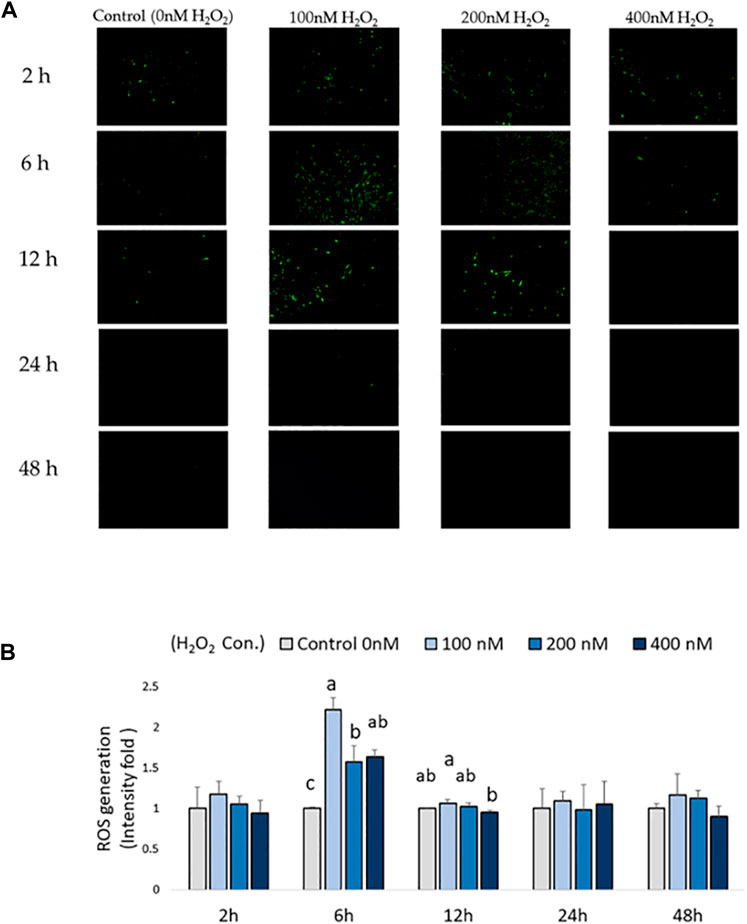
FIGURE 2. Effects of H2O2-induced reactive oxygen species (ROS) production in chicken MSCs. 3 × 104 cells were seeded in a black clear-flat-bottom 96-well microplate and allowed to adhere overnight. Cells were then treated with the indicated concentrations of H2O2 for 2 h, 6 h, 12 h, 24 h and 48 h. ROS levels in MSCs were measured using a DCFDA/H2DCFDA cellular ROS assay kit. MSCs cultured without H2O2 but PBS (osteogenic differentiation medium with 0 nM H2O2) were used as the control. (A) Figures were selected as representative images from the DCFDA/H2DCFDA cellular ROS assay at different time points. MSCs cultured without any treatments or DCFDA/H2DCFDA was used to set the background adjustment. (B) Quantitative analysis was performed by measuring fluorescence intensity. Each value represents the mean ± SEM of three independent replicates (n = 3). MSCs cultured in osteogenic differentiation medium without H2O2 treatment (0 nM H2O2) were used as the control, and data were present as fold-change normalized to the fluorescence intensity level of the control. a, ab, b, c Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test, p < 0.05.
At low concentrations of H2O2 treatment, mRNA expression of pro-apoptotic markers, including CASP-3, CASP-6 and CASP-8 remained unchanged in chicken MSCs during the early differentiation stage from 6 h to day 5 (p > 0.05; Figure 3). This suggests that the effect of H2O2 on the osteogenic differentiation of MSCs was not due to a cytotoxic effect causing cell apoptosis at the early differentiation stage. However, 200 nM H2O2 significantly increased mRNA expression of CASP-8 compared to the control on day 6 (p < 0.05; Figure 3), and 400 nM H2O2 numerically increased expression of CASP-8 (p < 0.05; Figure 3). Neither of the higher treatment doses changed the expression of CASP-3 or CASP-6 on day 6. On day 10 and day 14, high cycle threshold (Ct > 38) value indicating the expression of CASP-6 and CASP-8 were not detected.
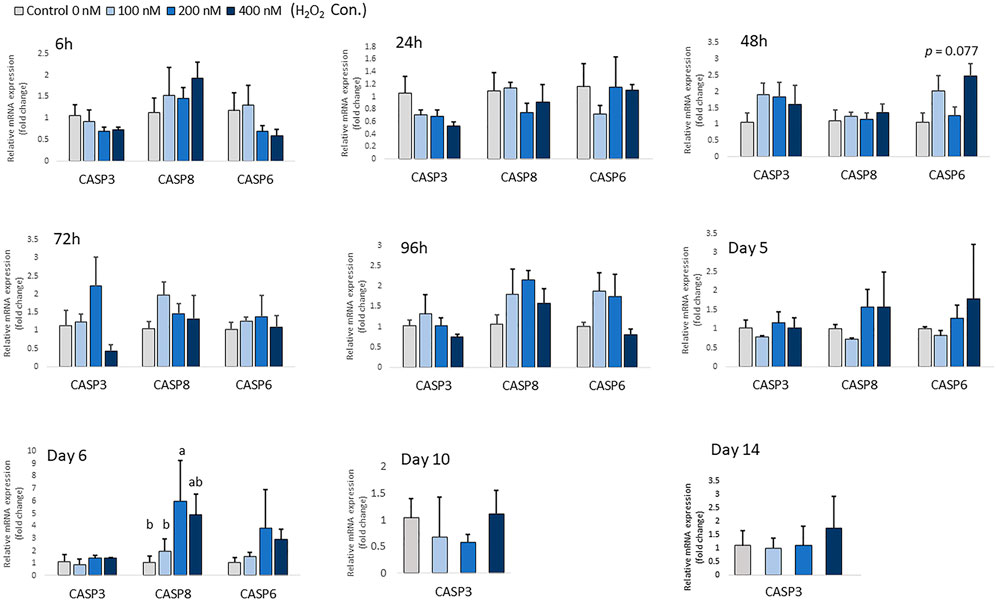
FIGURE 3. Effects of H2O2 on mRNA expression of apoptosis markers in chicken MSCs. Differentiation cells were treated with the indicated concentrations of H2O2 for 6 h, 24 h, 48 h, 72 h, 96 h, 5 days, 6 days, 10 days and 14 days. Each value represents the mean ± SEM of three independent experiments (n = 3). CASP3: caspase three; CASP6: caspase six; CASP8: caspase eight; a, ab, b, c Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test, p < 0.05.
Altered gene expression of antioxidant enzyme in response to extracellular H2O2 exposure
There was no significant change in the expression of antioxidant enzyme mRNA after 6 h of differentiation, except for expression of NOS2, which showed a trend of decreasing with higher H2O2 treatment doses (p = 0.087; Figure 4). At 24 h of treatment, the mRNA expression of SOD1 was decreased by the highest concentration of H2O2 (400 nM) (p < 0.05; Figure 4). At 48 h of treatment, 100 nM H2O2 augmented the expression of CAT compared to the control. After 5 days of treatment and differentiation, the mRNA expression of GPX1 was upregulated by 400 nM H2O2 compared to the control (p < 0.05). 200 nM H2O2 significantly increased mRNA expression of CAT compared to the 400 nM H2O2 treatment group (p < 0.05). After 6 days of H2O2 treatment, 400 nM H2O2 significantly upregulated mRNA expression of SOD2 compared to the 100 nM H2O2 group (p < 0.05). There was a trend of increasing expression of NRF2 with 200 and 400 nM H2O2 treatments (p = 0.077). After 14 days of differentiation, a trend of increasing mRNA level of CAT (p = 0.068) was observed with 400 nM H2O2 treatment. However, expression of SOD2, GSTa and NRF2 was not detected on day 10 and day 14 of differentiation. In conclusion, depending on the different treatment concentration of H2O2, the expression of antioxidant enzyme gene was altered at the later stage of differentiation.
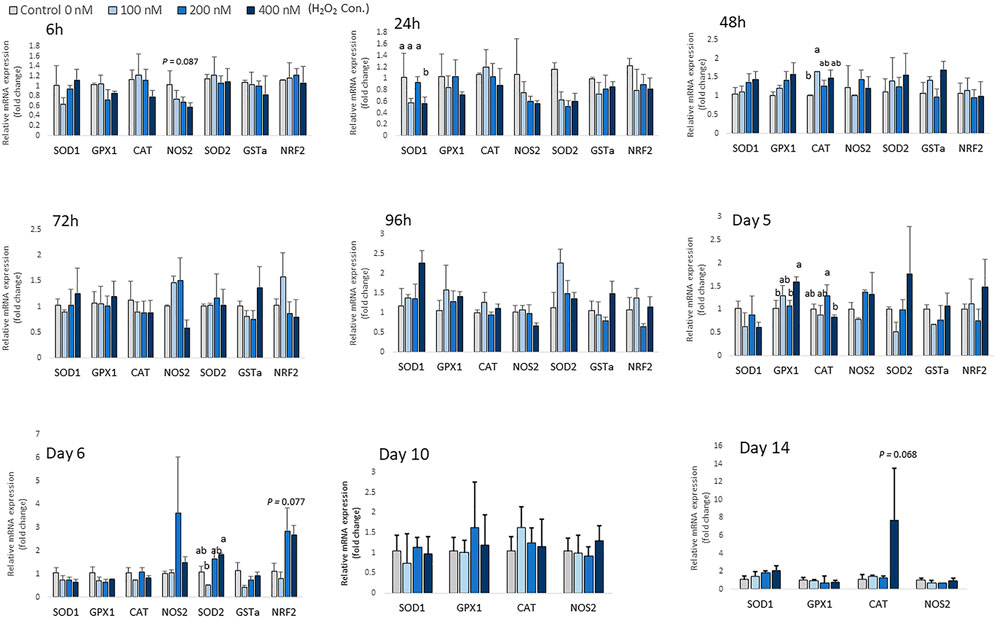
FIGURE 4. Effects of H2O2 on mRNA expression of antioxidant enzymes in chicken MSCs. Cells were treated with the indicated concentrations of H2O2 for 6 h, 24 h, 48 h, 72 h, 96 h, 5 days, 6 days, 10 days and 14 days. The expression of SOD2, GSTa and NRF2 was not detected on day 10 and day 14 of differentiation. Each value represents the mean ± SEM of three independent experiments (n = 3). CAT: catalase; SOD1: superoxide dismutase one; SOD2: superoxide dismutase two; GPX1: glutathione peroxidase one; NOS2: nitric oxide synthase two; NFR2: GA binding protein transcription factor alpha subunit (GABP2); GSTa: glutathione S-transferase alpha two; a, ab, b Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test, p < 0.05.
Effect of H2O2 on osteogenesis in chicken MSCs
Chicken MSCs were treated with various concentrations of H2O2 in osteogenic differentiation medium for 14 days, the effects on osteogenic differentiation varied at different time points (Figure 5). At the beginning of differentiation, there was a significant decrease in SPP1 (p < 0.05) and a numerically decreased mRNA expression of BMP2 (p = 0.053) after 6 h of H2O2 treatment. In the following days, the mRNA expression of osteogenic marker genes was unchanged. After 96 h of treatment, cells exposure to 400 nM H2O2 showed significantly higher mRNA expression of ALP (p < 0.05), BGLAP (p < 0.05) and SPP1 (p < 0.05), and 400 nM H2O2 tended to increase the expression of BMP2 (p = 0.092) and Col1A2 (p = 0.060). After 5 days of H2O2 exposure, mRNA expression of BGLAP (p < 0.05), ALP (p < 0.05) and Col1A2 (p < 0.05) was suppressed by 200 nM H2O2 compared to the control. In contrast, mRNA expression of BGLAP was upregulated by 400 nM H2O2 compared to the other treatment groups (p < 0.05) and the expression of ALP was increased by 400 nM H2O2 (p < 0.05) compared to the low treatment doses of 100 and 200 nM H2O2. After 6 days of daily treatment, mRNA expression of ALP (p < 0.05), BGLAP (p < 0.05) and Col1A2 (p < 0.05) was reduced by H2O2 treatments, and there was a trend of decreasing in SPP1 (p = 0.066) with H2O2 treatments. On day 10, the expression of BMP2 was significantly reduced by H2O2 treatments compared to the control (p < 0.05), and there was a trend of decreasing in RUNX2 (p = 0.079) with H2O2 treatments. On day 14, 100 nM H2O2 significantly reduced the expression of BMP2 compared to the control (p < 0.05), and 400 nM H2O2 significantly reduced expression of Col1A2 compared to the control (p < 0.05). There was also a trend of decreasing in SPP1 (p = 0.092) with H2O2 treatments on day 14. However, ALP expression was not detected on day 10 and day 14 of differentiation.
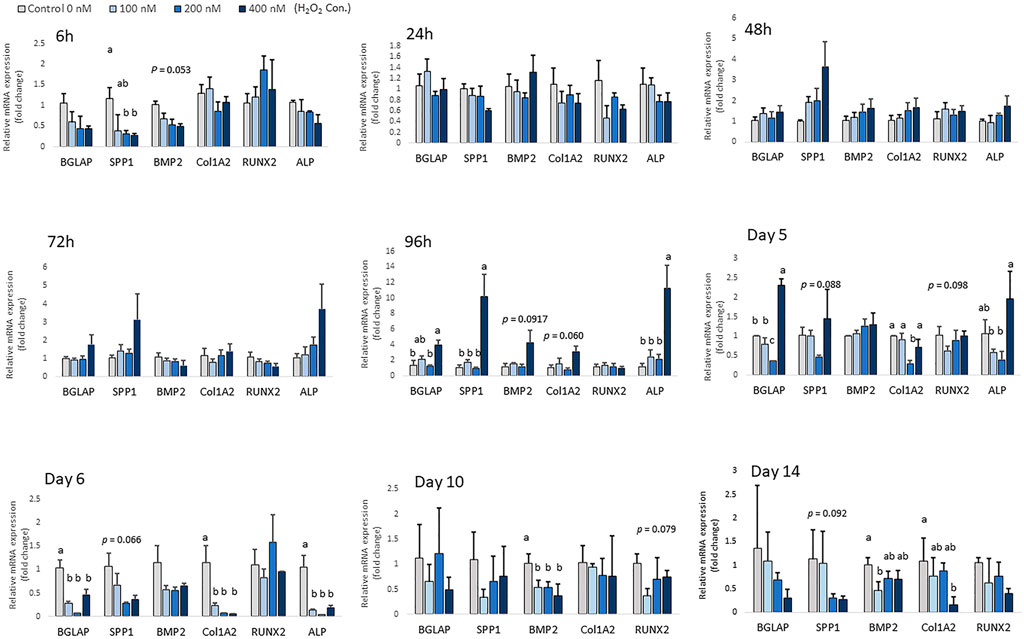
FIGURE 5. Effects of H2O2 on mRNA expression of osteogenic differentiation markers in chicken MSCs. Cells were treated with the indicated concentrations of H2O2 in differentiation medium for 6 h, 24 h, 48 h, 72 h, 96 h, 5 days, 6 days, 10 days and 14 days. Each value represents the mean ± SEM of three independent experiments (n = 3). SPP1: secreted phosphoprotein, osteopontin; BMP2: bone morphogenetic protein two; BGLAP: bone gamma-carboxyglutamic acid-containing protein (osteocalcin); RUNX2: runt-related transcription factor 2; ALP: alkaline phosphatase, biomineralization associated; Col1A2: collagen type I alpha two chain. a, ab, b, c Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test, p < 0.05.
In parallel with the mRNA expression mentioned above, the inhibition of osteogenic differentiation was characterized by a reduction in mineral accumulation after 6 days, 10 days and 14 days of differentiation. The effects of H2O2 on the mineralization were visualized using Alizarin red staining (Figure 6A). The optical density (O.D.) value result showed that 400 nM of H2O2 significantly reduced the O.D. value by 30% compared to the control after 6 days of H2O2 exposure (p < 0.05; Figure 6B). There were no statistically significant changes after 10 and 14 days of differentiation, but smaller mineralized crystals were observed with a higher dose of H2O2 treatment, and 400 nM H2O2 led a decrease in mineralization by 20% and 40%. The extracellular calcium (black crystals) content was quantified by von Kossa staining (Figure 7). After 6 days of differentiation, there were smaller and fewer extensive crystals and less mineralized matrix with higher doses of H2O2 treatment. The colorimetric analysis showed a significant decrease in O.D. with 400 nM H2O2 treatment (p < 0.05; Figure 7B). After 10 days of H2O2 treatment, a lower number of mineralized nodules and smaller size of crystals were observed with higher concentrations of H2O2 treatment; the O.D. showed that 200 and 400 nM H2O2 led to a significant decrease in mineralization, with the least mineral deposition observed in the 400 nM H2O2 group (p < 0.05; Figure 7B). After 14 days of differentiation, 400 nM H2O2 significantly suppressed mineralization compared to the other groups (p < 0.05; Figure 7B). In conclusion, the different doses of H2O2 treatment resulted in varying effects on osteogenic gene expression at different time points, while mineralization was significantly reduced by H2O2 treatment, with the greatest reduction observed in the high treatment concentration groups after 10 and 14 days of differentiation.
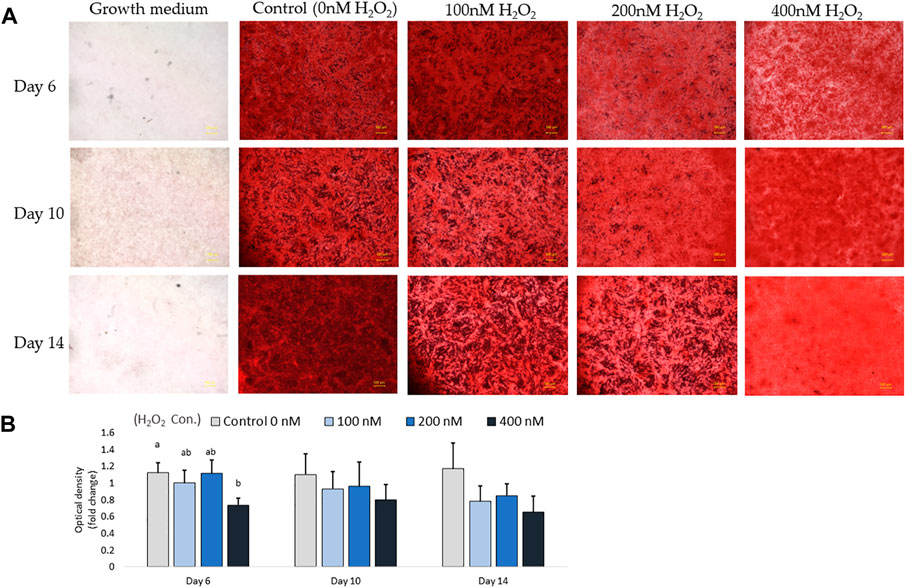
FIGURE 6. Alizarin red S staining for mineralization on day 6, day 10 and day 14. Images were randomly acquired at ×2 magnification. The calcified nodules appeared bright red in color. Mineral deposit quantification was conducted, with each value representing the mean ± SEM of three independent experiments (n = 3). a, b Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test at each time points, respectively, p < 0.05.
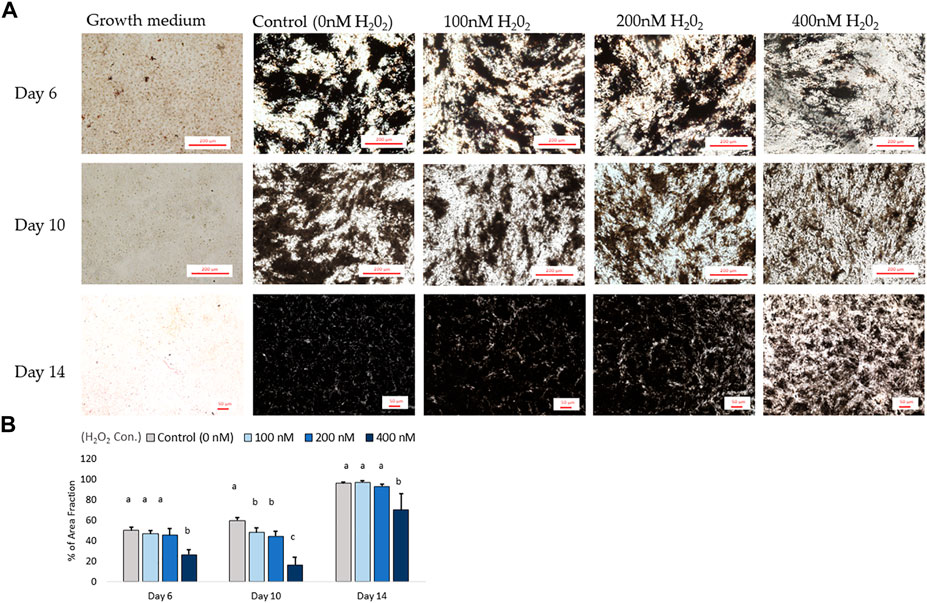
FIGURE 7. The von Kossa staining results for mineralization on day 6, day 10 and day 14. (A) Images were randomly acquired in ×4 magnification for day 6 and 10 images. On day 14, the figure was acquired at ×2 magnification due to mineralization interrupting autofocus using higher magnification lenses. Four images per well were analyzed. Black objects indicates phosphate and calcium deposition. ImageJ analysis quantified the percent area fraction for each treatment based on three independently sampled experiments of each species, with each value representing the mean ± SEM of three independent experiments (n = 3). a, b Treatments with different letters indicate a significantly difference between treatments using Tukey’s HSD test at each time points, respectively; p < 0.05.
Meanwhile, under the condition of osteogenic induction differentiation, the expression of adipogenic-related genes was altered with H2O2 treatment. Decreases in PPARG expression with 200 nM and 400 nM H2O2 treatment doses were observed at 6 h (p < 0.05; Supplementary Figure). Afterward, H2O2 reduced the expression of PPARG (p = 0.053; Supplementary Figure), CEBPA (p < 0.05) and FABP4 (p < 0.05) after 24 h of osteogenic differentiation. In contrast, with prolonged treatment periods, 400 nM H2O2 significantly elevated the expression of PPARG (p < 0.05) and FABP4 (p < 0.05) after 96 h of treatment. A similar expression pattern was observed after 5 days of H2O2 treatment under osteogenic differentiation condition, where 400 nM H2O2 significantly increased the expression of PPARG (p < 0.05), CEBPA (p < 0.05) compared to the control; the expression level of FABP4 was significantly elevated by 200nM and 400 nM H2O2 treatment (p < 0.05) compared to the control. After 6 days of H2O2 treatment, 200 and 400 nM H2O2 treatments increased the expression of PPARG (p < 0.05; Supplementary Figure) compared to the control; 400 nM H2O2 treatment drastically increased the expression of FABP4 (p < 0.05; Supplementary Figure) compared to other groups. However, 100 nM H2O2 reduced expression of CEBPA (p < 0.05; Supplementary Figure) compared to the control. Adipogenic expression was not significantly changed by H2O2 treatment on day 10 and day 14.
Discussion
Cell fate with the presence of oxidative stress can vary depending on the cell types, treatment intensity, duration, dosage, and the cell differentiation status (Denu and Hematti, 2016). Moreover, several studies have pointed out that undifferentiated stem cells have superior antioxidant defense than differentiated cells; for example, undifferentiated MSCs are known to have relatively low levels of intracellular ROS and high levels of glutathione in the human cell line (Valle-Prieto and Conget, 2010). Studies have shown that mouse embryonic stem cells exhibit high antioxidant activity and stress-resistance, but several antioxidant and cellular resistance genes are downregulated during differentiation (Saretzki et al., 2004). Under normal circumstances during mineralization, MSCs differentiate and their expression of mineralization factors increases (Blair et al., 2017). The size of mineral crystals increases during bone mineralization, and the collagen fibers become more organized and condensed (Blair et al., 2017). Studies on other cell types have indicated that H2O2 solutions have a significant effect on collagen production (Nashchekina et al., 2021), as previous studies have reported the increased activities of oxidative stress was linked to decreased collagen synthesis in fibroblasts (Siwik et al., 2001), human cartilage (Altindag et al., 2007), and chick embryo tissue culture (Ramp et al., 1987). Moreover, the chicken embryo tissue culture has also indicated that multiple exposures to H2O2 markedly inhibit collagen synthesis (Ramp et al., 1987). In this report, exogenous H2O2 (100–400 nM) suppressed the osteoblastic mineralization of chicken compact bone-derived MSCs, manifested by reduced osteogenic differentiation gene markers and less mineral deposition. The decreased expression of Col1A2 after 6 days of H2O2 treatment supported the hypothesis that H2O2-induced oxidative stress can directly interrupt type 1 collagen production. Moreover, the concentration of H2O2 in the present study is much lower than the H2O2 doses used in human and mouse stem cell studies (Nouri et al., 2019), demonstrating that chicken compact bone-derived MSCs are relatively sensitive to oxidative stress compared to other cell types in chicken (chicken cardiomyocytes: 0.2 mM H2O2 (Jiang et al., 2005; Wan et al., 2016); chicken cardiac cells, 0.2–2.0 mM H2O2; and chicken epithelial cells, 300 μM H2O2 (Lin et al., 2016)). Interestingly, a previous study indicated that ROS production did not influence the aging process of avian fibroblast cells (Strecker et al., 2010). Comparing the blood redox state markers of 78 free-living avian species revealed that relatively long-lived bird species had high levels of antioxidants status (especially total antioxidant status and total glutathione) and low levels of ROS (Xia and Møller, 2018). With the rapid growth and relatively short lifespan of broilers, it is likely that high levels of ROS due to oxidative stress may occur in a great extent in broiler production. Chicken MSCs-differentiated osteoblasts are particularly susceptible to oxidative stress, making the negative impact of ROS production on bone homeostasis a potential factor in the development of skeletal abnormalities.
MSCs have the ability to differentiate into various cell phenotype types, which are controlled by transcription factors such as PPARG, RUNX2 and SOX9, which regulate adipogenesis, osteogenesis and chondrogenesis, respectively (Robert et al., 2020). In particular, adipogenesis and osteogenesis have a reciprocal relationship (Takada et al., 2007; Robert et al., 2020). For example, in human and mouse primary MSCs, PPARG2 insufficiency resulted in increased osteogenesis of osteoblast (Takada et al., 2007), while depletion of RUNX2 promoted adipogenesis (Enomoto et al., 2004). ROS level in MSCs also plays a crucial role in determining their differentiation potential (Ho et al., 2013; Denu and Hematti, 2016). Previous studies have shown that mRNA expression of antioxidant enzymes such as SOD, CAT, and GPX is upregulated during adipogenesis in human MSCs (Ho et al., 2013). In this study, prolonged exposure to H2O2 increased cellular oxidative stress and increased the basal expression of adipogenic differentiation markers at the later stages of differentiation, which was accompanied by decreased mineralization. This is consistent with previous studies that have shown that H2O2 exposure altered the differentiation potential in human and mouse MSCs or cell lines (Ho et al., 2013; Lin et al., 2018). In addition, by analyzing genome-wide gene expression profiling, Menssen et al. (Menssen et al., 2011) reported an upregulated CASP8 level during adipogenic differentiation in human bone marrow-derived MSCs. Therefore, the increased expression of CASP8 on day 6 might have been due to adipogenic differentiation of chicken MSCs, rather than apoptosis directly caused by H2O2-induced oxidative stress. Moreover, the effect of H2O2 treatment on regulating cell differentiation has been observed in different types of cells, where sublethal doses of oxidative stress induce morphological alterations (Shadel and Horvath, 2015; Diebold and Chandel, 2016; Infante and Rodriguez, 2018). For example, prolonged H2O2 treatment activates NF-κB transcriptional activity while stimulating brown adipogenesis during myogenic differentiation in mice satellite cells (Morozzi et al., 2017). Therefore, at least in part, oxidative stress is a factor for the dysfunction of bone tissue, not only by causing cell death, but also by interrupting MSCs differentiation capacity and decreasing the osteogenic ability directly.
In the current study, several osteogenic differentiation markers were significantly upregulated with the highest H2O2 treatment dose after 4 and 5 days of treatment, and then drastically dropped after 6 days of treatment. We made several hypotheses to explain the upregulated and downregulated expression patterns. Firstly, studies pointed out that the cellular effects of ROS may differ depending on the cell differentiation stage due to the difference between progenitor cells and mature cells (Khalid et al., 2020). For example, during the initial differentiation process, MSCs commit to pre-osteoblasts while actively proliferating (Infante and Rodriguez, 2018). Over the later stage of differentiation, the pre-osteoblasts can further mature into non-proliferating osteoblasts that start matrix secretion, maturation, and mineralization (Infante and Rodriguez, 2018). Studies showed that H2O2 treatment significantly enhance bone marrow MSCs proliferation and migration ability (Pendergrass et al., 2013). Human and mouse studies revealed a low level of intracellular ROS and high levels of antioxidants in undifferentiated MSCs (Hu et al., 2018). In contrast, differentiated MSCs show a higher level of ROS and lower activity of antioxidative enzymes (Hu et al., 2018). Therefore, it is important to distinguish the multiple roles of ROS in pre-osteoblast differentiation and osteoblast maturation. Secondly, we hypothesize that ROS over-production mediated the cell cycle and caused cell prematurity. ROS is a fundamental signal in many signaling pathways metabolisms (Shadel and Horvath, 2015). The low level of ROS allowed reversible oxidative modifications until the ROS production overwhelm its antioxidant capacity, which leads to severe cellular damage (Diebold and Chandel, 2016). Redox status plays a vital role in the cell cycle, and accumulated intracellular ROS can force MSCs to undergo cellular senescence, substantially interrupt stem cells differentiation (Brandl et al., 2011). The expression of GPX, CAT and SOD2 changed in current study, indicating an altered oxidation-reduction status. SOD2, which plays a vital role in regulating mitochondrial stress and osteoblastogenesis, was upregulated (Gao et al., 2018). This helped reduce mRNA over-expression of PPARG and FABP-4 in diabetic mouse models (Sen et al., 2015). In the current study, the increased expression of SOD2 suggested that cells were actively suppressing adipogenic differentiation under oxidative stress. However, we speculated that long-term exposure cells to higher levels of exogenous H2O2 stimulated intracellular ROS production and promoted pre-osteoblast commitment at the early differentiation stage, but also led to the accumulation of ROS, which resulted in cell prematurity and apoptosis, decreasing mineral accrual at the later stage. The molecular mechanisms by which ROS affects the avian cell cycle, however, are largely unexplored and require further investigation.
Another hypothesis is that exogenous H2O2 stimulated a regeneration response. H2O2 is well-known ROS signaling intermediate in response to tissue injury (Sanchez-de-Diego et al., 2019). ROS activates signaling that influences vital pathways, such as Wnt or TGF/BMP pathways, which are response to fracture healing and tissue repairment mechanism by regulating osteogenic differentiation of stem cells (Wang et al., 2017; Sheppard et al., 2022). MSCs can migrate to the sites of injury in response to various stimuli, including cytokines or growth factors, and differentiate into tissue-specific cell types to repair the damaged region (Merimi et al., 2021). Additionally, ROS can regulate the activation of BMPs and RUNX2 pathways in MSCs during the repair process by mediating the activity of NF-κB signaling (Mandal et al., 2011). Therefore, in the present study, the increased expression of bone formation marks ALP, SPP1 and BGLAP after 5 days of differentiation may suggested that the high ROS level stimulated a cell repair-response, which recruited MSCs to differentiate into osteoblasts to maintain cell population and homeostasis. However, continuous oxidative stress cause MSCs to commit apoptosis, ultimately reducing mineralization.
Oxidative stress has been linked to many bone-related diseases in humans and mammals (Reis and Ramos, 2021). In broilers, at least 30% of birds showed poor locomotion during the fast growth period (Wideman, 2016; Zhang et al., 2020), which interfered chicken’ accessibility to feed and water, predominantly reducing the growth and causing an economic loss in production. Generally, cultured cells have higher throughput and shorter turnaround times than in vivo study models (Dawson et al., 2014), and the response of avian stem cells to different stress stimuli has been widely studied in the context of growth and physiology (Ali Hassan and Li, 2021; Xu et al., 2022). Therefore, understanding oxidative stress in a cell model is essential for a better understanding of bone pathogenic process in chickens. Tibial dyschondroplasia (TD) and bacterial osteomyelitis (BCO) are two common skeletal abnormalities in the broiler production that cause bones deformation and lameness (Hartcher and Lum, 2019). Previous studies have indicated that BCO is initiated by mechanical micro-fracturing, followed by bacterial colonization and bone degradation, leading to necrosis (Wideman, 2016). Mitochondrial dysfunction and apoptosis are also involved in BCO in broilers (Ferver et al., 2021). Although there is no direct evidence linking oxidative stress and BCO pathogenesis, higher levels of oxidative stress in response to local infection have been reported in human patients with chronic osteomyelitis (Massaccesi et al., 2022). The pathology of osteomyelitis is characterized by localized inflammation, bone mineral loss, and structural damage, which share similarities with broiler BCO (Grbic et al., 2014). Furthermore, a recent study on femoral necrosis pathogenicity also reported an abnormal increased in lipid metabolism and decreases in bone formation in a chicken femoral head necrosis disease model (Fan et al., 2021). Therefore, by gathering all the evidence above, we proposed that oxidative stress could potentially be a co-factor involved in chicken bone necrosis. Moreover, tibial dyschondroplasia (TD) is another common bone abnormality in fast-growing broilers, characterized by tibial bone deformities with non-vascular, non-mineralized growth plates (Mehmood et al., 2017; Zhang et al., 2019). It is a chondrogenesis-related growth plate development disease that is highly associated with premature and apoptosis of cells (Mehmood, 2018). There is an known relationship between TD and oxidative stress induced by thiram, which reduces liver antioxidation capability and damages liver function (Li et al., 2007). Altered systemic antioxidant activity has also been reported in broilers with TD (Zhang et al., 2018; Huang et al., 2021). Although the function of osteoblast has not been fully illustrated in broiler TD model, osteoblasts are responsible for forming the type I collagen matrix surrounding vasculature buds, and osteoblast and osteocyte have direct association with chondrocyte maturation and hypotrophy (Zhang et al., 2020). Based on this information, we hypothesize that TD may be partially associated with systemic oxidative stress. Studies have shown that mycoplasma (M.) can produce H2O2 and superoxide radicals, which induce oxidative stress in the respiratory epithelium and directly affect bone metabolism (Shimizu, 2016). Clinical signs of M. synoviae infection include joint lesions in avian species (Osorio et al., 2007). These results provide evidence of the pathogenicity of mycoplasmas on bone integrity and support our current results showing that high level and long-term effects of H2O2 negatively regulate osteoblast cell activity.
Although many questions remain, ever-growing numbers of observations regarding chicken bone disorders and avian bone health rapidly shape our understanding of various topics, such as metabolic regulation and the pathogenesis of bone disorders in broilers (Porto et al., 2015). The knowledge of ROS generation and antioxidant defense systems has generated a great deal of interest due to its potential applications in animal production, but it remains to be profoundly explored in chicken stem cell models. In conclusion, the concentration of ROS is in a dynamic equilibrium and is modulated by cellular processes that produce and eliminate ROS. Cellular effects of ROS may vary depending on the differentiation stage of the cells. Treatment with H2O2 altered the expression of cellular antioxidant enzyme gene, and long-term treatment with H2O2 inhibited osteogenic biomineralization and decreased the expression of osteogenic differentiation markers in chicken MSCs. The impaired osteogenic differentiation potential was associated with an increased potential for adipogenesis in chicken MSCs under oxidative stress, highlighting that cellular oxidative stress caused by exogenous H2O2 accumulation modulates stem cell differentiation capacity.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at the University of Georgia, Athens, GA.
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. YT and WK conceived and designed this study. YT and GL contributed to sample collection. YT contributed to data analyses. The paper was written through contribution and critical review of the manuscript by all authors (YT, GL, and WK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1124355/full#supplementary-material
References
Adhikari R., Chen C., Waters E., West F. D., Kim W. K. (2018). Isolation and differentiation of mesenchymal stem cells from broiler chicken compact bones. Front. Physiol. 9, 1892. doi:10.3389/fphys.2018.01892
Ali Hassan N., Li Z. (2021). Oxidative stress in broiler chicken and its consequences on meat quality. Int. J. Life Sci. Res. Archive 1 (1), 045–054. doi:10.53771/ijlsra.2021.1.1.0054
Altindag O., Erel O., Aksoy N., Selek S., Celik H., Karaoglanoglu M. (2007). Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol. Int. 27 (4), 339–344. doi:10.1007/s00296-006-0247-8
Atashi F., Modarressi A., Pepper M. S. (2015). The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 24 (10), 1150–1163. doi:10.1089/scd.2014.0484
Blair H. C., Larrouture Q. C., Li Y., Lin H., Beer-Stoltz D., Liu L., et al. (2017). Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Eng. Part B Rev. 23 (3), 268–280. doi:10.1089/ten.TEB.2016.0454
Brandl A., Meyer M., Bechmann V., Nerlich M., Angele P. (2011). Oxidative stress induces senescence in human mesenchymal stem cells. Exp. Cell Res. 317 (11), 1541–1547. doi:10.1016/j.yexcr.2011.02.015
Chauhan S. S., Rashamol V. P., Bagath M., Sejian V., Dunshea F. R. (2021). Impacts of heat stress on immune responses and oxidative stress in farm animals and nutritional strategies for amelioration. Int. J. Biometeorol. 65 (7), 1231–1244. doi:10.1007/s00484-021-02083-3
Dawson J. I., Kanczler J., Tare R., Kassem M., Oreffo R. O. (2014). Concise review: Bridging the gap: Bone regeneration using skeletal stem cell-based strategies - where are we now? Stem Cells 32 (1), 35–44. doi:10.1002/stem.1559
Denu R. A., Hematti P. (2016). Effects of oxidative stress on mesenchymal stem cell biology. Oxid. Med. Cell Longev. 2016, 2989076. doi:10.1155/2016/2989076
Diebold L., Chandel N. S. (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 100, 86–93. doi:10.1016/j.freeradbiomed.2016.04.198
Enomoto H., Furuichi T., Zanma A., Yamana K., Yoshida C., Sumitani S., et al. (2004). Runx2 deficiency in chondrocytes causes adipogenic changes in vitro. J. Cell Sci. 117 (3), 417–425. doi:10.1242/jcs.00866
Fan R., Liu K., Zhou Z. (2021). Abnormal lipid profile in fast-growing broilers with spontaneous femoral head necrosis. Front. Physiol. 12, 685968. doi:10.3389/fphys.2021.685968
Ferver A., Greene E., Wideman R., Dridi S. (2021). Evidence of mitochondrial dysfunction in bacterial chondronecrosis with osteomyelitis-affected broilers. Front. Vet. Sci. 8, 640901. doi:10.3389/fvets.2021.640901
Forrester S. J., Kikuchi D. S., Hernandes M. S., Xu Q., Griendling K. K. (2018). Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 122 (6), 877–902. doi:10.1161/CIRCRESAHA.117.311401
Gao J., Feng Z., Wang X., Zeng M., Liu J., Han S., et al. (2018). SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 25 (2), 229–240. doi:10.1038/cdd.2017.144
Grbic R., Miric D. J., Kisic B., Popovic L., Nestorovic V., Vasic A. (2014). Sequential analysis of oxidative stress markers and vitamin C status in acute bacterial osteomyelitis. Mediat. Inflamm. 2014, 975061. doi:10.1155/2014/975061
Hambli R. (2014). Connecting mechanics and bone cell activities in the bone remodeling process: An integrated finite element modeling. Front. Bioeng. Biotechnol. 2, 6. doi:10.3389/fbioe.2014.00006
Hartcher K. M., Lum H. K. (2019). Genetic selection of broilers and welfare consequences: A review. World's Poult. Sci. J. 76 (1), 154–167. doi:10.1080/00439339.2019.1680025
Ho P. J., Yen M. L., Tang B. C., Chen C. T., Yen B. L. (2013). H2O2 accumulation mediates differentiation capacity alteration, but not proliferative decline, in senescent human fetal mesenchymal stem cells. Antioxid. Redox Signal 18 (15), 1895–1905. doi:10.1089/ars.2012.4692
Hu C., Zhao L., Peng C., Li L. (2018). Regulation of the mitochondrial reactive oxygen species: Strategies to control mesenchymal stem cell fates ex vivo and in vivo. J. Cell Mol. Med. 22 (11), 5196–5207. doi:10.1111/jcmm.13835
Huang S. C., Cao Q. Q., Cao Y. B., Yang Y. R., Xu T. T., Yue K., et al. (2021). Morinda officinalis polysaccharides improve meat quality by reducing oxidative damage in chickens suffering from tibial dyschondroplasia. Food Chem. 344, 128688. doi:10.1016/j.foodchem.2020.128688
Infante A., Rodriguez C. I. (2018). Osteogenesis and aging: Lessons from mesenchymal stem cells. Stem Cell Res. Ther. 9 (1), 244. doi:10.1186/s13287-018-0995-x
Jiang B., Xiao W., Shi Y., Liu M., Xiao X. (2005). Heat shock pretreatment inhibited the release of Smac/DIABLO from mitochondria and apoptosis induced by hydrogen peroxide in cardiomyocytes and C2C12 myogenic cells. Cell Stress Chaperones 10 (3), 252–262. doi:10.1379/csc-124r.1
Khalid S., Yamazaki H., Socorro M., Monier D., Beniash E., Napierala D. (2020). Reactive oxygen species (ROS) generation as an underlying mechanism of inorganic phosphate (Pi)-induced mineralization of osteogenic cells. Free Radic. Biol. Med. 153, 103–111. doi:10.1016/j.freeradbiomed.2020.04.008
Li J., Bi D., Pan S., Zhang Y. (2007). Effect of diet with thiram on liver antioxidant capacity and tibial dyschondroplasia in broilers. Br. Poult. Sci. 48 (6), 724–728. doi:10.1080/00071660701665858
Lin C. H., Li N. T., Cheng H. S., Yen M. L. (2018). Oxidative stress induces imbalance of adipogenic/osteoblastic lineage commitment in mesenchymal stem cells through decreasing SIRT1 functions. J. Cell Mol. Med. 22 (2), 786–796. doi:10.1111/jcmm.13356
Lin X., Jiang S., Jiang Z., Zheng C., Gou Z. (2016). Effects of equol on H2O2-induced oxidative stress in primary chicken intestinal epithelial cells. Poult. Sci. 95 (6), 1380–1386. doi:10.3382/ps/pew034
Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Mandal C. C., Ganapathy S., Gorin Y., Mahadev K., Block K., Abboud H. E., et al. (2011). Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 433 (2), 393–402. doi:10.1042/BJ20100357
Maraldi T., Angeloni C., Giannoni E., Sell C. (2015). Reactive oxygen species in stem cells. Oxid. Med. Cell Longev. 2015, 159080. doi:10.1155/2015/159080
Massaccesi L., Galliera E., Pellegrini A., Banfi G., Corsi Romanelli M. M. (2022). Osteomyelitis, oxidative stress and related biomarkers. Antioxidants (Basel) 11 (6), 1061. doi:10.3390/antiox11061061
Mehmood K. (2018). Tetramethylpyrazine mitigates toxicity and liver oxidative stress in tibial dyschondroplasia chickens. Pak. Veterinary J. 38 (01), 76–80. doi:10.29261/pakvetj/2018.015
Mehmood K., Zhang H., Iqbal M. K., Rehman M. U., Shahzad M., Li K., et al. (2017). In vitro effect of apigenin and danshen in tibial dyschondroplasia through inhibition of heat-shock protein 90 and vascular endothelial growth factor expressions in avian growth plate cells. Avian Dis. 61 (3), 372–377. doi:10.1637/11641-032817-RegR
Menssen A., Haupl T., Sittinger M., Delorme B., Charbord P., Ringe J. (2011). Differential gene expression profiling of human bone marrow-derived mesenchymal stem cells during adipogenic development. BMC Genomics 12, 461. doi:10.1186/1471-2164-12-461
Merimi M., El-Majzoub R., Lagneaux L., Moussa Agha D., Bouhtit F., Meuleman N., et al. (2021). The therapeutic potential of mesenchymal stromal cells for regenerative medicine: Current knowledge and future understandings. Front. Cell Dev. Biol. 9, 661532. doi:10.3389/fcell.2021.661532
Mishra B., Jha R. (2019). Oxidative stress in the poultry gut: Potential challenges and interventions. Front. Vet. Sci. 6, 60. doi:10.3389/fvets.2019.00060
Morozzi G., Beccafico S., Bianchi R., Riuzzi F., Bellezza I., Giambanco I., et al. (2017). Oxidative stress-induced S100B accumulation converts myoblasts into Brown adipocytes via an NF-κB/YY1/miR-133 axis and NF-κB/YY1/BMP-7 axis. Cell Death Differ. 24 (12), 2077–2088. doi:10.1038/cdd.2017.132
Nashchekina Y., Nikonov P., Mikhailova N., Nashchekin A. (2021). Collagen scaffolds treated by hydrogen peroxide for cell cultivation. Polym. (Basel) 13 (23), 4134. doi:10.3390/polym13234134
Nouri F., Nematollahi-Mahani S. N., Shari fi A. M. (2019). Preconditioning of mesenchymal stem cells with non-toxic concentration of hydrogen peroxide against oxidative stress induced cell death: The role of hypoxia-inducible factor-1. Adv. Pharm. Bull. 9 (1), 76–83. doi:10.15171/apb.2019.010
Ojano-Dirain C., Tinsley N. B., Wing T., Cooper M., Bottje W. G. (2007). Membrane potential and H2O2 production in duodenal mitochondria from broiler chickens (Gallus gallus domesticus) with low and high feed efficiency. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 147 (4), 934–941. doi:10.1016/j.cbpa.2007.02.029
Osorio C., Fletcher O. J., Abdul-Aziz T., Gonder E., Tilley B., Ley D. H. (2007). Pneumonia of Turkey breeder hens associated with mycoplasma synoviae. Avian Dis. 51 (3), 791–796. doi:10.1637/0005-2086(2007)51[791:POTBHA]2.0.CO;2
Pendergrass K. D., Boopathy A. V., Seshadri G., Maiellaro-Rafferty K., Che P. L., Brown M. E., et al. (2013). Acute preconditioning of cardiac progenitor cells with hydrogen peroxide enhances angiogenic pathways following ischemia-reperfusion injury. Stem Cells Dev. 22 (17), 2414–2424. doi:10.1089/scd.2012.0673
Porto M. L., Rodrigues B. P., Menezes T. N., Ceschim S. L., Casarini D. E., Gava A. L., et al. (2015). Reactive oxygen species contribute to dysfunction of bone marrow hematopoietic stem cells in aged C57BL/6 J mice. J. Biomed. Sci. 22, 97. doi:10.1186/s12929-015-0201-8
Ramp W. K., Arnold R. R., Russell J. E., Yancey J. M. (1987). Hydrogen peroxide inhibits glucose metabolism and collagen synthesis in bone. J. Periodontol. 58 (5), 340–344. doi:10.1902/jop.1987.58.5.340
Reis J., Ramos A. (2021). In sickness and in health: The oxygen reactive species and the bone. Front. Bioeng. Biotechnol. 9, 745911. doi:10.3389/fbioe.2021.745911
Robert A. W., Marcon B. H., Dallagiovanna B., Shigunov P. (2020). Adipogenesis, osteogenesis, and chondrogenesis of human mesenchymal stem/stromal cells: A comparative transcriptome approach. Front. Cell Dev. Biol. 8, 561. doi:10.3389/fcell.2020.00561
Sanchez-de-Diego C., Valer J. A., Pimenta-Lopes C., Rosa J. L., Ventura F. (2019). Interplay between BMPs and reactive oxygen species in cell signaling and pathology. Biomolecules 9 (10), 534. doi:10.3390/biom9100534
Saretzki G., Armstrong L., Leake A., Lako M., von Zglinicki T. (2004). Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 22 (6), 962–971. doi:10.1634/stemcells.22-6-962
Sen S., Domingues C. C., Rouphael C., Chou C., Kim C., Yadava N. (2015). Genetic modification of human mesenchymal stem cells helps to reduce adiposity and improve glucose tolerance in an obese diabetic mouse model. Stem Cell Res. Ther. 6, 242. doi:10.1186/s13287-015-0224-9
Serdar C. C., Cihan M., Yucel D., Serdar M. A. (2021). Sample size, power and effect size revisited: Simplified and practical approaches in pre-clinical, clinical and laboratory studies. Biochem. Med. Zagreb. 31 (1), 010502. doi:10.11613/BM.2021.010502
Serguienko A., Wang M. Y., Myklebost O. (2018). Real-time vital mineralization detection and quantification during in vitro osteoblast differentiation. Biol. Proced. Online 20, 14. doi:10.1186/s12575-018-0079-4
Shadel G. S., Horvath T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163 (3), 560–569. doi:10.1016/j.cell.2015.10.001
Sharma P., Jha A. B., Dubey R. S., Pessarakli M. (2012). Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 1–26. doi:10.1155/2012/217037
Sharma T., Islam N., Ahmad J., Akhtar N., Beg M. (2015). Correlation between bone mineral density and oxidative stress in postmenopausal women. Indian J. Endocrinol. Metab. 19 (4), 491–497. doi:10.4103/2230-8210.159053
Sheppard A. J., Barfield A. M., Barton S., Dong Y. (2022). Understanding reactive oxygen species in bone regeneration: A glance at potential therapeutics and bioengineering applications. Front. Bioeng. Biotechnol. 10, 836764. doi:10.3389/fbioe.2022.836764
Shimizu T. (2016). Inflammation-inducing factors of mycoplasma pneumoniae. Front. Microbiol. 7, 414. doi:10.3389/fmicb.2016.00414
Sies H., Berndt C., Jones D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi:10.1146/annurev-biochem-061516-045037
Siwik D. A., Pagano P. J., Colucci W. S. (2001). Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 280 (1), C53–C60. doi:10.1152/ajpcell.2001.280.1.C53
Strecker V., Mai S., Muster B., Beneke S., Burkle A., Bereiter-Hahn J., et al. (2010). Aging of different avian cultured cells: Lack of ROS-induced damage and quality control mechanisms. Mech. Ageing Dev. 131 (1), 48–59. doi:10.1016/j.mad.2009.11.005
Sun Y. X., Xu A. H., Yang Y., Li J. (2015). Role of Nrf2 in bone metabolism. J. Biomed. Sci. 22, 101. doi:10.1186/s12929-015-0212-5
Surai P. F., Kochish , Fisinin V. I., Kidd M. T. (2019). Antioxidant defence systems and oxidative stress in poultry biology: An update. Antioxidants (Basel) 8 (7), 235. doi:10.3390/antiox8070235
Svoradova A., Zmrhal V., Venusova E., Slama P. (2021). Chicken mesenchymal stem cells and their applications: A mini review. Anim. (Basel) 11 (7), 1883. doi:10.3390/ani11071883
Takada I., Suzawa M., Matsumoto K., Kato S. (2007). Suppression of PPAR transactivation switches cell fate of bone marrow stem cells from adipocytes into osteoblasts. Ann. N. Y. Acad. Sci. 1116, 182–195. doi:10.1196/annals.1402.034
Valle-Prieto A., Conget P. A. (2010). Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. 19 (12), 1885–1893. doi:10.1089/scd.2010.0093
Wan C., Xiang J., Li Y., Guo D. (2016). Differential gene expression patterns in chicken cardiomyocytes during hydrogen peroxide-induced apoptosis. PLoS One 11 (1), e0147950. doi:10.1371/journal.pone.0147950
Wan H., Yuan W., Ruan M., Ye Q., Wang R., Li Z., et al. (2011). Identification of reference genes for reverse transcription quantitative real-time PCR normalization in pepper (Capsicum annuum L.). Biochem. Biophys. Res. Commun. 416 (1-2), 24–30. doi:10.1016/j.bbrc.2011.10.105
Wang T., Zhang X., Bikle D. D. (2017). Osteogenic differentiation of periosteal cells during fracture healing. J. Cell Physiol. 232 (5), 913–921. doi:10.1002/jcp.25641
Wideman R. F. (2016). Bacterial chondronecrosis with osteomyelitis and lameness in broilers: A review. Poult. Sci. 95 (2), 325–344. doi:10.3382/ps/pev320
Xia C., Møller A. P. (2018). Long-lived birds suffer less from oxidative stress. Avian Res. 9 (1), 41. doi:10.1186/s40657-018-0133-6
Xu J., Strasburg G. M., Reed K. M., Velleman S. G. (2022). Thermal stress and selection for growth affect myogenic satellite cell lipid accumulation and adipogenic gene expression through mechanistic target of rapamycin pathway. J. Anim. Sci. 100 (8), skac001. doi:10.1093/jas/skac001
Yuan Z., Zhang J., Huang Y., Zhang Y., Liu W., Wang G., et al. (2017). NRF2 overexpression in mesenchymal stem cells induces stem-cell marker expression and enhances osteoblastic differentiation. Biochem. Biophys. Res. Commun. 491 (1), 228–235. doi:10.1016/j.bbrc.2017.07.083
Zhang H., Mehmood K., Jiang X., Li Z., Yao W., Zhang J., et al. (2019). Identification of differentially expressed MiRNAs profile in a thiram-induced tibial dyschondroplasia. Ecotoxicol. Environ. Saf. 175, 83–89. doi:10.1016/j.ecoenv.2019.03.043
Zhang H., Mehmood K., Li K., Rehman M. U., Jiang X., Huang S., et al. (2018). Icariin ameliorate thiram-induced tibial dyschondroplasia via regulation of WNT4 and VEGF expression in broiler chickens. Front. Pharmacol. 9, 123. doi:10.3389/fphar.2018.00123
Zhang H., Wang Y., Mehmood K., Chang Y. F., Tang Z., Li Y. (2020). Treatment of tibial dyschondroplasia with traditional Chinese medicines: "Lesson and future directions. Poult. Sci. 99 (12), 6422–6433. doi:10.1016/j.psj.2020.08.055
Zomorodian E., Baghaban Eslaminejad M. (2012). Mesenchymal stem cells as a potent cell source for bone regeneration. Stem Cells Int. 2012, 980353. doi:10.1155/2012/980353
Keywords: bone health, oxidative stress, chicken MSCs, cellular ROS, cell differentiation
Citation: Tompkins YH, Liu G and Kim WK (2023) Impact of exogenous hydrogen peroxide on osteogenic differentiation of broiler chicken compact bones derived mesenchymal stem cells. Front. Physiol. 14:1124355. doi: 10.3389/fphys.2023.1124355
Received: 15 December 2022; Accepted: 19 January 2023;
Published: 26 January 2023.
Edited by:
Anthony Pokoo-Aikins, Toxicology and Mycotoxin Research Unit, U.S. National Poultry Research Center, Agricultural Research Service (USDA), United StatesReviewed by:
Frank Idan, Kwame Nkrumah University of Science and Technology, GhanaXiao Lin, Northwestern Polytechnical University, China
Copyright © 2023 Tompkins, Liu and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. K. Kim, d2traW1AdWdhLmVkdQ==