
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 02 March 2023
Sec. Membrane Physiology and Membrane Biophysics
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1122444
This article is part of the Research TopicChloride Homeostasis in Animal Cell PhysiologyView all 6 articles
 Zhiyu Wang
Zhiyu Wang Kaylee Choi*
Kaylee Choi*Chloride homeostasis is critical in the physiological functions of the central nervous system (CNS). Its concentration is precisely regulated by multiple ion-transporting proteins such as chloride channels and transporters that are widely distributed in the brain cells, including neurons and glia. Unlike ion transporters, chloride channels provide rapid responses to efficiently regulate ion flux. Some of chloride channels are also permeable to selected organic anions such as glutamate and γ-aminobutyric acid, suggesting neuroexcitatory and neuroinhibitory functions while gating. Dysregulated chloride channels are implicated in neurological disorders, e.g., ischemia and neuroinflammation. Modulation of chloride homeostasis through chloride channels has been suggested as a potential therapeutic approach for neurological disorders. The drug design for CNS diseases is challenging because it requires the therapeutics to traverse the blood-brain-barrier. Small molecules are a well-established modality with better cell permeability due to their lower molecular weight and flexibility for structure optimization compared to biologics. In this article, we describe the important roles of chloride homeostasis in each type of brain cells and introduce selected chloride channels identified in the CNS. We then discuss the contribution of their dysregulations towards the pathogenesis of neurological disorders, emphasizing the potential of targeting chloride channels as a therapeutic strategy for CNS disease treatment. Along with this literature survey, we summarize the small molecules that modulate chloride channels and propose the potential strategy of optimizing existing drugs to brain-penetrants to support future CNS drug discovery.
As the most abundant anion, chloride performs multiple physiological functions in the cells, where it maintains cellular homeostasis (Verkman and Galietta, 2021). In the central nervous system (CNS), chloride participates in multiple events to support neuronal functions. For instance, chloride regulates postsynaptic inhibition involved in neural coding through GABAA receptors (GABAARs; GABA, γ-aminobutyric acid) (Doyon et al., 2016). Its concentration gradient directly impacts neuronal excitation and inhibition (Mahadevan et al., 2014). In addition, chloride mediates the physiological properties of the CNS-supporting cells, glia. Glial cells comprise astrocytes, microglia, and oligodendrocytes. Astrocytes are the most abundant glial cells in the brain, where they surround neurons to provide physical structures, maintain ion balances, regulate a blood flow, participate in neural repairs, and release and uptake neurotransmitters. These functions are regulated by chloride in multiple dimensions. For instance, the strength of Cl− current is associated with the activity of glutamate transporters in astrocytes (Wilson and Mongin, 2019). Under transient ischemic stress, chloride is maintained at a dynamic balance through multiple chloride-transporting mechanisms to prevent astrocytic swelling (Engels et al., 2021). Microglia, ‘brain macrophages’, keep sensing the microenvironment with a ramified morphology at the resting stage. Upon the recognition of foreign invaders such as pathogens or inflammatory molecules, they are activated to initiate immune responses with an alternation into amoeboid morphology initiating neuroinflammation. This activation with a consequent morphology change is tightly regulated by chloride. Chloride, to be specific, participates in the membrane stretch during ramification of microglia and the associated tyrosine-phosphorylation signaling pathway (Eder et al., 1998). Its influx also provokes lamellipodium formation, suggesting the critical role in microglia migration towards foreign species (Zierler et al., 2008). Upon activation, chloride mediates phagocytosis and the release of proinflammatory cytokines from microglia, suggesting the therapeutic potential of chloride channel modulators for microglia-involved neurodegenerative diseases (Schlichter et al., 1996; Eder et al., 1998; Novarino et al., 2004; Zierler et al., 2008). Oligodendrocytes are the myelinating glia in the CNS. They assemble myelin sheath along nerve cell axons, reducing internodal membrane capacitance and facilitating rapid conduction of electrical impulses (Stassart et al., 2018). Its proliferation, development, and maturation require chloride homeostasis (Magalhães and Rivera, 2016).
Chloride homeostasis is regulated by multiple chloride-transporting proteins including ion channels and transporters. Due to genetic disorders, acute injuries, or inflammation, however, these functional proteins are dysregulated, contributing towards the pathophysiology of numerous neurological disorders such as epilepsy, autism, ataxia, hyperekplexia, and neuropathic pain (Funk et al., 2008; Kahle et al., 2008; Tyzio et al., 2014; Wu et al., 2016; Wu et al., 2022). Modulation of chloride homeostasis in the CNS has been suggested as a promising therapeutic approach to resolve chloride disturbance and associated pathological disorders.
The drug design for CNS diseases is challenging because it requires the therapeutics to traverse the blood-brain-barrier (BBB). Compared to biologics, small molecules exhibit better permeability to BBB and cellular membranes and offer flexibility for hit discovery and lead optimization. In the past years, tremendous efforts have been made to modulate chloride homeostasis through chloride transporting proteins. Unlike the transporters, chloride channels provide fast responses to efficiently regulate ion flux driven by electrochemical gradient. In addition, various chloride channels are permeable to larger anions such as GABA and glutamate, exhibiting neuroinhibitory and neuroexcitatory effects while gating (Park et al., 2009; Lee et al., 2010; Woo et al., 2012). These features suggest a promising strategy to regulate chloride-involved neurological disorders through chloride channels.
Since chloride channels show distinct properties between the CNS and peripheral systems (Zhang et al., 2004), herein, we describe the important roles of chloride homeostasis in each type of brain cells and introduce selected chloride channels in the CNS, emphasizing the contributions of their dysregulations towards the pathogenesis of CNS disorders. We also discuss targeting chloride channels as a therapeutic strategy for CNS disease treatments and review the small molecules that modulate chloride homeostasis and associated neurological disorders. From medicinal chemistry perspective, we calculate the physicochemical properties of these molecules and propose potential strategies to optimize specific physicochemical parameters through structural modification, supporting future CNS drug discovery.
ClC channels are expressed on plasma membranes, intracellular organelles, and vesicles, where they regulate chloride gradients for various cellular functions. For instance, ClC-2 regulates intracellular chloride concentration of hippocampal pyramidal neurons through chloride extrusion based on its inward rectifying property (Rinke et al., 2010; Földy et al., 2010). The gating mechanisms of ClCs have been reported with their protein structures (Accardi, 2015; Poroca et al., 2017). Briefly, the subunit of dimeric ClC channels harbors an ion pore that is modulated by the protonation-deprotonation cycle of a glutamate gate. This cycle is voltage-dependent and can be initiated by repulsion or protonation when voltage navigates a permeant anion or a proton in. ClC family comprises nine members that can be divided into chloride channels (ClC-1, -2, -Ka, and -Kb) and Cl−/H+ exchangers (ClC-3 through -7). The contributions of ClCs in physiology and disease progressions have been extensively reviewed by Jentsch and colleagues (Jentsch and Pusch, 2018). In this section, we focus on the roles of ClC-1 and -2 in neurodegenerative diseases.
In the CNS, ClC-1 is distributed in the hippocampus, brain stem nuclei, thalamic nuclei, and frontal neocortex, participating in physiological processes (Chen et al., 2013). ClC-1 in the CNS exhibits features distinct from ClC-1 in muscle tissue. For example, ClC-1 in astrocytes exhibits less dependence on voltage and extracellular Cl− than that in skeletal muscle (Zhang et al., 2004). In the CNS, ClC-1 contributes to neuronal network maturation and neuronal excitability, suggesting its important role in preventing neurological disorders such as epilepsy (Rahmati et al., 2018). Furthermore, a parallel exome sequencing of 237 ion channel genes verifies that ClC-1 is involved in the pathogenesis of epilepsy (Chen et al., 2013).
Several drugs targeting ClC-1 have been developed to treat neuromuscular diseases. Acetazolamide influences the voltage-dependent gating of ClC-1, elevating open probability and chloride conductance (Eguchi et al., 2006). Acetazolamide, however, contains a primary sulfonamide group (see Table 1), which might impact the permeability across BBB (Wang et al., 2021). NMD Pharma, a clinical-stage biotech company, developed NMD670, a small-molecule inhibitor against ClC-1, recently granted Orphan Drug Designation by the Food and Drug Administration (FDA) for the treatment of myasthenia gravis. Myasthenia gravis is caused by autoimmunity against nicotinic acetylcholine receptors in skeletal muscle endplates in most cases (Koneczny and HerbstGravis, 2019). Therefore, the expected effect of NMD670 would be due to increased muscle excitability by blocking muscle ClC-1.

TABLE 1. Summary of chloride channels involved in neurological disorders and their small-molecule modulators with physiochemical properties.
In comparison to ClC-1, ClC-2 is abundantly expressed in the CNS, where it is triggered by negative membrane voltage, cellular volume change, increased intracellular Cl−, or extracellular acidification (Grunder et al., 1992; Jordt and Jentsch, 1997), modulating chloride efflux, neuroexcitation, myelination, and signaling transduction (Sik et al., 2000; Niemeyer et al., 2004). In hippocampal neurons, ClC-2 mediates chloride currents, a substantial part of the background conductance. The loss of ClC-2 in interneurons induces a dramatic increase of excitability, causing inhibition of principal neurons, thereby reducing overall network excitability (Rinke et al., 2010). In glia, ClC-2 has been identified as a positive modulator of oligodendrocyte maturation from precursor cells and subsequent myelin formation, repairing myeline-associated neurological disorders (Hou et al., 2018). Its function has been further demonstrated in aging study that identified the neuroprotective role of ClC-2 in the hippocampus (Cortez et al., 2010). In addition, ClC-2 is expressed in the end feet of astrocytes surrounding blood vessels, where it regulates chloride ion and blood flows (Sı́k et al., 2000).
Due to the wide distribution in the CNS, dysregulated ClC-2 may lead to multiple neurological disorders. ClC-2 mutation, for instance, has been suggested to be a cause of epilepsy (D'Agostino et al., 2004) although the mechanism needs further elucidation. In addition, aged ClC-2 KO mice exhibit perturbed neurotransmission patterns and increased excitation associated with astrocyte activation and neuronal degeneration (Cortez et al., 2010). Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a disease that causes seizures and developmental delay in early life, followed by a deterioration of motor functions and intellectual abilities. Pathology study identifies vacuolations in the myelin and astrocytes of MLC patients, suggesting that the disturbed ion homeostasis might be the reason for MLC development (van der Knaap et al., 1996; Duarri et al., 2011). Mutation on GLIALCAM is one explanation for MLC pathogenesis (López-Hernández et al., 2011). GlialCAM is a molecule that targets ClC-2 to cell junctions and increases ClC-2-mediated current, altering its functional properties (Jeworutzki et al., 2012). Aberrant GlialCAM, however, fails to target ClC-2 to cell junctions, leading to MLC disease. This observation is consistent with the animal study that shows ClC-2 KO mice develop widespread vacuolation in the white matter of the brain and spinal cord, which might be related to defective oligodendrocytes (Blanz et al., 2007).
AK-42 is a small molecule that inhibits ClC-2 with nanomolar potency (IC50 = 17 ± 1 nM) and rapidly and reversibly blocks ClC-2 currents. It displays unprecedented selectivity over ClC-1 and exhibits no off-target engagement against a panel of other common channels, receptors, and transporters expressed in brain tissue (Koster et al., 2020). This development provides a precise tool for future investigation on chloride-involved neurophysiology and the discovery of ClC-2-related therapeutics. In addition, peptide inhibitors have been developed as a pharmacological tool to probe ClC-2 structure/function (Thompson et al., 2009). To deliver the peptide therapeutics across BBB, brain-penetrating molecular transport vectors, such as BBB shuttle peptides, have been developed (Oller-Salvia et al., 2016). These brain-permeable peptides conjugated with therapeutics can traverse BBB through diverse mechanisms.
Ca2+-activated Cl− channels (CaCCs) are activated by intracellular Ca2+, exhibiting an outwardly rectifying current-voltage relationship at relatively low Ca2+ concentration while displaying a linear current-voltage relationship at higher Ca2+ concentration (Huang et al., 2012a).
Among the multiple family members in anoctamin (ANO, also known as TMEM16) channels, ANO1 and ANO2 are considered as CaCCs with defined physiological functions (Oh and Jung, 2016). In the CNS, they have been identified in the cerebellar cortex, hippocampus, and olfactory bulb, where they modulate synaptic transmissions and olfaction (Rasche et al., 2010; Huang et al., 2012b; Zhang et al., 2015). For instance, ANO1 plays a role in the network of inhibitory interneurons in the cerebellar cortex, while ANO2 may modulate the inhibitory input to Purkinje cells (Zhang et al., 2015). During CNS development, ANO1 also involves in the maturation of radial glial cells contributing to cortex development (Hong et al., 2019).
ANOs also participate in neurological diseases. For instance, ANO1 is overexpressed in various cancer cells including glioblastoma. Under this pathological condition, activation of tyrosine kinases and G protein-coupled receptors increases intracellular Ca2+ concentration in glioblastoma cells, triggering the gating of ANO1 (Kang et al., 2010). ANO1 also promotes cancer progression by stimulating the signaling pathway of cell proliferation (Britschgi et al., 2013; Liu et al., 2014). Suppression of ANO1 activity inhibits migration and invasion of these glioblastoma cell lines, indicating its therapeutic value (Lee et al., 2016). In addition, ANO1 elevates the excitability of dorsal-root ganglion neurons under inflammatory or neuropathic conditions, suggesting that ANO1 inhibitors can be developed as novel analgesics (Lee et al., 2014; Pineda-Farias et al., 2015). More recently, ANO2 has been identified as an autoimmune target in multiple sclerosis (Ayoglu et al., 2016).
Multiple ANO1 inhibitors have been developed and their therapeutic values have been investigated in neurological disorders. CaCCinh-A01 inhibits ANO1 with IC50 at 7.40 µM (Table 1) (Liu et al., 2015). This inhibitor also blocks another CaCC, Best1 with similar IC50. In comparison, T16Ainh-A01 partially inhibits ANO1 but has no activity on Best1 (Liu et al., 2015). Inhibition of ANO1 activity by CaCCinh-A01 and T16Ainh-A01 has been demonstrated as a tool to generate analgesia in nerve injury pain (Pineda-Farias et al., 2015). In addition, CaCCinh-A01 also preserves BBB integrity, attenuates brain infract size and neurological deficits after ischemic stroke, indicating that ANO1 may become a potential target for ischemic stroke (Liu et al., 2019).
Best1 is distributed in the olfactory bulb, hippocampus, and cerebellum, expressed in both neurons and astrocytes (Park et al., 2009). Best1 is activated by an increase of Ca2+ and induces Cl− flux across cell membrane. This action leads to membrane depolarization or hyperpolarization, depending on the equilibrium potential. Best1 also plays distinct roles in the brain, where it exhibits permeabilities for several other monovalent anions, including Br−, I−, SCN−, HCO3−, and NO3− (Qu and Hartzell, 2008; O'Driscoll et al., 2009). In addition, Best1 in astrocytes has been reported to regulate larger anions including GABA (Lee et al., 2010), one major inhibitory neurotransmitter, and glutamate (Park et al., 2009; Woo et al., 2012), one excitatory neurotransmitter mediated and recycled by astrocytes.
Interestingly, the expression and functions of Best1 exhibit altered patterns in astrocytes under pathological conditions. Resting astrocytes do not synthesize GABA but express Best1 at microdomains (astrocytic membrane protrusions enwrapping synaptic terminals) to regulate glutamate release targeting NMDA receptors (Oh and Lee, 2017). In Alzheimer’s disease (AD), however, astrocytes that surround Aβ plaque are activated to maintain brain homeostasis. This action triggers the synthesis of GABA in astrocytes and the redistribution of Best1 from perisynaptic microdomains to soma, from which GABA is released by astrocytes through Best1 (Oh and Lee, 2017). The GABA further diminishes the spike probability and synaptic plasticity, impacting learning and memory function (Jo et al., 2014). This evidence highlights the role of Best1 in neuron-glia crosstalk through GABA as a gliotransmitter in AD. Upregulated Best1 with associated increase of chloride currents has been observed in dorsal root ganglia after peripheral nerve axotomy and spinal nerve ligation (Boudes et al., 2009; Pineda-Farias et al., 2015). Also, Best1 KO mice exhibit decreased neurite outgrowth velocity in cultured injured sensory neurons, suggesting a positive role in regeneration (Oh and Lee, 2017).
Sodium phenylbutyrate (4-PBA) appears to act as a chaperone to improve Best1 protein folding and rescue the function of Best1 mutants, thereby improving the chloride conductance (Liu et al., 2020).
CFTR, a cAMP-dependent ion channel, transports chloride and bicarbonate in the epithelial cells of airways, gastrointestinal and reproductive organs (Verkman and Galietta, 2021). Aberrant CFTR results in cystic fibrosis (CF) and subsequent impaired fluid and pH homeostasis, contributing to the pathology in the lungs, pancreas, livers, intestine, and testis (Verkman and Galietta, 2021). Interestingly, CNS complications occur more frequently in CF patients than other lung transplant recipients (Goldstein et al., 2000), suggesting CFTR may exist in the CNS and participate in neuronal functions. Ex-vivo study has identified the expression of CFTR in hypothalamus, thalamus, amygdala, and limbic system (Mulberg et al., 1995; Mulberg et al., 1998; Weyler et al., 1999), the areas regulating metabolism, food intake, sex differentiation, and energy expenditure. This distribution seems to explain the symptoms of growth failure and malnutrition in CF patients.
At the cellular level, CFTR expression has been observed in both neurons and glia (Liu et al., 2006a; Guo et al., 2009) and its expression shows different patterns depending on brain development stage (Marcorelles et al., 2014). Patients with CF show axonal dystrophy and detectable amyloid precursor protein (Goldstein et al., 2000), implying that CFTR not only performs fundamental functions in cell maturation during brain development but also contributes to neurological disorders. Decreased expression of CFTR, for instance, has been observed in the astrocytes differentiated from patients with frontotemporal dementia type 3 (Chandrasekaran et al., 2021). In AD, CFTR gene expression is downregulated in the hypothalamus, suggesting a potential role in the regulation of metabolic function during neurodegeneration (Lahousse et al., 2003). Mutation in this gene leads to exaggerated proinflammatory responses in AD (Lahousse et al., 2003). In-vitro study demonstrates that CFTR suppresses apoptosis of glioma cells; inhibition of CFTR function or expression suppresses the glioma cell viability, whereas overexpression of CFTR shows an opposite impact (Zhao et al., 2020). This observation is consistent with the immunohistochemistry study on the samples collected from glioblastoma patients, from which the expression level of CFTR is significantly increased (Zhao et al., 2020).
More than 2000 CFTR mutants that impact protein synthesis and stability have been identified (Veit et al., 2016). To restore the function of CFTR, two types of modulators have been developed. “Correctors” are the small molecules directly interacting with mutant CFTR to repair protein folding and improve stability. In contrast, “potentiators” correct the dysregulation by improving channel gating. For instance, ivacaftor, a CFTR potentiator, improves the chloride transport by directly binding to CFTR to mediate gating, thereby restoring protein functions (Eckford et al., 2012). Lumacaftor and tezacaftor, CFTR correctors, act as chaperones during protein folding and increase protein trafficking to the cell membrane, thereby improving protein stability (Eckford et al., 2012; Ridley and Condren, 2020). The combinations of lumacaftor/ivacaftor and tezacaftor/ivacaftor have been approved by FDA for CF treatments.
Cell volume is maintained at a dynamic equilibrium through dedicated mechanisms during transmembrane fluxes of ions and nutrients, and synthesis/degradation of macromolecules. Among multiple volume-regulatory proteins, volume-regulated anion channel (VRAC) is activated by cell swelling and transports anions including Cl− along electrochemical gradients. This action leads to the efflux of water to counteract with cell swelling. In the CNS, VRAC gating triggers the release of organic osmolytes such as glutamate, impacting neuronal excitability (Kasuya and Nureki, 2022).
VRAC is a heteromeric protein and activated by cell swelling. Five isoforms named LRRC8A-E have been identified. Functional VRAC is formed by multiple LRRC8 proteins including the essential LRRC8A and at least one other family member among LRRC8B-E (Hyzinski-García et al., 2014; Voss et al., 2014). The expression ratio and different combinations of LRRC8 isoforms in this complex result in diverse properties, playing a unique role in the release of different neurotransmitters such as glutamate, aspartate, GABA, and taurine. In astrocytes, for example, the LRRC8A/D complex appears to regulate the release of uncharged osmolytes, while LRRC8A/C/D/E complex is responsible for charged molecules (Lutter et al., 2017; Schober et al., 2017).
Under pathological conditions such as traumatic brain injury, stroke, hyponatremia, and epilepsy, astrocytes swell, invading extracellular space and impacting neuronal functions (Barron et al., 1988; Manley et al., 2000; Fabene et al., 2006). These events result in the buildup of glutamate and aspartate in the extracellular space, persistently depolarizing neurons. VRACs appear to play an important role during these neurological disorders. For instance, VRACs are activated by astrocytic swelling during stroke and mediate the release of excitatory amino acids (Kimelberg, 2005) (Basarsky et al., 1999). Administration of DCPIB, a specific VRAC inhibitor, reduces infarct size in reversible middle cerebral artery occlusion and the release of glutamate in the ischemic cortical penumbra, suggesting neuroprotective effects in brain ischemia. As a fully charged anion at physiological pH, however, DCPIB is not able to traverse BBB (Zhang et al., 2008). In comparison, another VRAC inhibitor, tamoxifen, has been reported to penetrate BBB and reduce brain infarction in the stroke mouse model (Kimelberg et al., 2003). Some natural products, such as phloretin, also exhibit inhibition of VRAC and associated astrocytic amino acid release (Abdullaev et al., 2006).
The activation of GABAARs triggers an alternation of electrochemical potential, exerting inhibitory functions to regulate neuronal excitability in the CNS. GABAARs are pentameric receptor proteins composed of at least three different proteins collected from 19 subunits, α1-6, β1-3, γ1-3, δ, ε, θ, π, ρ1-3 (Brickley and Mody, 2012). The different combination of these subunits results in varying isoforms with diverse functions (Mortensen et al., 2011; Phulera et al., 2018). The predominant synaptic GABAARs are composed of two α1-subunits, two β2-subunits, and one γ2-subunit (Zhu et al., 2018), in which γ2-subunit is a major component and drives receptor clustering at synapse (Essrich et al., 1998). In contrast, the subunit composition of extrasynaptic GABAARs has the high occurrence of α4, α5, α6, and δ subunits (Brickley and Mody, 2012). Synaptic GABAARs interact with GABA with a low affinity to generate phase conductance that inhibits postsynaptic currents in a transient and rapid manner while extrasynaptic GABAARs mediate tonic conductance in the presence of ambient GABA with a high affinity, leading to a persistent inhibition (Lee and Maguire, 2014).
Disturbances in synaptic and extrasynaptic GABAARs result in multiple neurological disorders. For instance, patients with early Parkinson’s disease (PD) develop non-motor symptoms such as sleep disturbance, olfactory loss, and gastrointestinal abnormalities, which are related to the deficits of GABAergic system (Błaszczyk, 2016). There is also a correlation between genetic alteration of GABAARs and neurodevelopmental disorders such as fragile X syndrome, Rett syndrome, and Dravet syndrome (Braat and Kooy, 2015). Mutation in extrasynaptic δ-GABAARs leads to diminished tonic inhibition and epileptic seizures (Chuang and Reddy, 2018). Recent study has also identified that antipsychotic-free patients with schizophrenia have lower extrasynaptic α5-GABAARs in the hippocampus, which is not seen in antipsychotic-treated schizophrenia patients, highlighting the potential of GABAergic modulators as therapeutic targets for schizophrenia (Marques et al., 2021).
The diverse GABAAR subunits create more opportunities for the development of selective modulators. As GABAARs regulate neurotransmitters, they are targets of widely-used sedative and hypnotic drugs including barbiturates and benzodiazepines, which interact with the interface between α and γ subunits of GABAARs (May et al., 2013). Ligand binding locks the GABAARs into a conformation with a better exposure to GABA to potentiate inhibitory signals (Phulera et al., 2018). The same site is targeted by inverse agonists such as β-carbolines, which have an effect opposite to that of anxiolytic benzodiazepines.
In addition, inverse agonists selective to the α5 subunit of the GABAAR have been reported to enhance cognition without anxiogenic and convulsant effects, highlighting the therapeutic potentials to treat memory impairment associated with AD and related dementias (Sternfeld et al., 2004). Gaboxadol is a selective agonist for GABAARs that contain δ subunits, which are mainly localized in thalamic neurons. Gaboxadol improves sleeping conditions in the Phase III clinical trial to treat primary insomnia (Wafford and Ebert, 2006). However, gaboxadol is no longer in clinical development due to limited or variable efficacy and psychiatric side effects (Roth et al., 2010).
Maxi anion channels (MACs) are highly effective electrogenic chloride-transporting systems, involved in multiple physiological events. MACs are widely expressed throughout the body and triggered by osmotic cell swelling, apoptosis, ischemia, and hypoxia (Sabirov and Okada, 2009). Comparing to other chloride channels, MACs exhibit large single-channel conductance, functioning as a highly efficient anion-transporting system, and permeability to large organic anions including pyruvate, glutamate, and ATP due to their wide pore (Jalonen, 1993; Dutta et al., 2004; Liu et al., 2008a). MACs play multiple roles in the CNS. In astrocytes, MACs regulate cell volume against swelling (Jalonen, 1993). Under ischemic or osmotic stress, MACs serve as a major ATP- and glutamate-releasing pathway in astrocytes (Liu et al., 2006b; Liu et al., 2008b), modulating glutamatergic synaptic transmission and microglia activation (Xiang et al., 2006).
Multiple studies have described small molecules that modulate MAC functions. L-644-711 blocks MAC in cultured astrocytes and regulates cell volume (Jalonen, 1993). This activity alleviates brain edema resulting from traumatic injury and hypoosmotic hyponatremia (Barron et al., 1988; Trachtman and Cragoe, 1989). Deltamethrin, a type II pyrethroid pesticide, inhibits MAC activity by decreasing open probability (Forshaw et al., 1993). In addition, classical anion-channel blockers such as NPPB, SITS, DIDS, and DPC inhibit MAC activity (Sabirov and Okada, 2009). In contrast, tamoxifen, a VRAC blocker used to treat breast cancer, activates MAC (Sabirov and Okada, 2005). Ivermectin and pentobarbitone significantly activate MAC, providing a rationale for effective therapy against pyrethroid-induced neurotoxicity (Forshaw et al., 2000).
Chloride channels in the CNS not only directly modulate neuronal excitability, but also indirectly impact neuronal functions through the gliotransmitters released from astrocytes via gating organic anions such as glutamate and GABA, as depicted in Figure 1. In neurological disorders, dysregulated chloride channels release excessive neurotransmitters, causing neuronal impairment. Our literature survey highlights astrocytic swelling that impacts brain function. This section further discusses the impacts of dysregulated chloride channels during astrocytic swelling and elucidates their contributions to the pathogenesis of neurological disorders. Along with a summary of the small molecules that modulate chloride channels, we also propose the potential strategy of optimizing exiting drugs to brain-penetrants, supporting future CNS drug discovery.
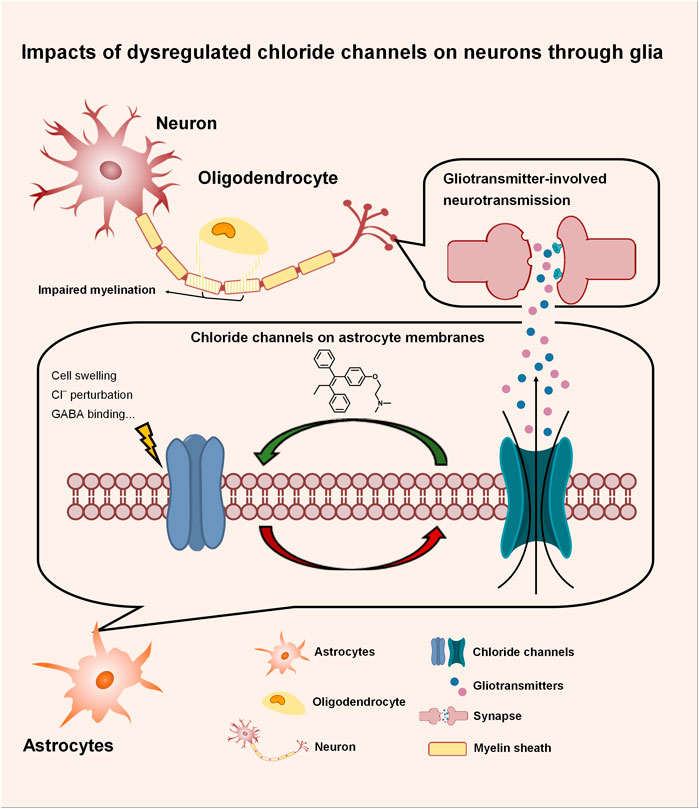
FIGURE 1. Impacts of dysregulated chloride channels on neurons through glia. Chloride channels in the CNS not only directly modulate neuronal excitability, but also indirectly impact neuronal functions through the gliotransmitters released from astrocytes via gating organic anions such as glutamate and GABA. In neurological disorders, dysregulated chloride channels release excessive neurotransmitters, causing neuronal impairment.
Astrocytes participate in fundamental roles in the CNS, where they maintain ion homeostasis, provide essential nutrients, and mediate neuronal excitability through gliotransmitters such as glutamate, GABA, and ATP. In physiological conditions, astrocytes release gliotransmitters to the network with neurons upon receptor activation (Gordon et al., 2005), osmotic perturbation (Darby et al., 2003), and deprivation of extracellular Ca2+ (Suadicani et al., 2006). During neurological disorders such as ischemia, however, ion-transporting systems of astrocytes and BBB endothelial cells are dysregulated, contributing to astrocytic swelling and vasogenic edema. Astrocytes are the major cell type that swells in gray matter (Kimelberg, 2000). As the most abundant cells in the CNS, astrocyte swelling significantly invades extracellular space, elevating intracranial pressure, reducing blood flow, and subsequently leading to tissue damage. As the CNS is encased within a rigid skull unlike other tissues, edema in the brain is life-threatening (Lafrenaye and Simard, 2019). In addition to these direct impacts, astrocytic swelling also dysregulates its own cellular function and induces secondary neurotoxicity. Astrocytes initiate cellular machinery against swelling to re-establish their pre-swelling volume by losing intracellular ions and excitatory amino acids such as glutamate, which induces excitotoxicity and subsequent neuronal injury (Kimelberg, 2000). As discussed in the last section, multiple chloride channels such as VRAC and Best1 have been suggested to modulate the release of excitatory amino acids, highlighting their roles in neuronal function impairments during neurological disorders and the therapeutic value of chloride-channel modulators.
In addition to the regulations of membrane potential and gating anions, chloride channels also participate in cell apoptosis through endoplasmic reticulum (ER) stress. Ischemia reperfusion injury and aging generate reactive oxygen species (Octavia et al., 2012; Liochev, 2013), which in turn activate VRACs, further inducing ER stress and downstream apoptosis (Shen et al., 2014). The role of ER stress in the pathogenesis of neurological disorders including AD, PD, and amyotrophic lateral sclerosis, is well documented (Lindholm et al., 2006). Treatment of chloride-channel blockers appears to prevent apoptosis through ER-stress pathway, reinforcing their therapeutic values against neurodegenerative diseases (Shen et al., 2014).
Designing molecules to traverse the BBB is a challenging hurdle in CNS drug discovery. The BBB is a layer that prevents hydrophilic substances, charged molecules, and proteins from entering into the extracellular fluid of the CNS from the circulating blood to protect brain tissues from pathogens and other neurotoxins. The BBB exchanges brain-necessary substances such as glucose, amino acids, and ions through selective and active transporting systems. Certain small molecules may also diffuse passively through the BBB and enter the brain. However, the BBB has a dedicated efflux system composed of breast cancer resistance protein (BCRP) and multiple drug resistance 1 (MDR1), restricting their substrates from the CNS.
In past decades, tremendous efforts have been made to regulate chloride homeostasis. Although most drugs were developed to target the peripheral systems, they provide a valuable reference for the drug discovery/repurpose towards CNS diseases. To support structure optimization for future endeavors, we summarized the small molecules that modulate chloride channels with appealing activity and specificity in Table 1 and calculated their physicochemical properties to identify the property to be optimized. cLogP stands for calculated logarithm of partition coefficient P. It is the ratio of compound concentration in a mixture of two immiscible solvents (such as water and n-octanol) at equilibrium, evaluating how hydrophobic a compound is. Hydrogen bond donors (HBDs) are the electronegative atoms, such as O or N, covalently bonded to hydrogens that can be donated. HBDs provide opportunities to molecular recognition, structural stability, drug partition, and permeability (Coimbra JTSFeghali et al., 2021). However, these polar moieties decrease the affinity toward the hydrophobic membrane and increase the energy penalty required to desolvate the molecule from water (Alex et al., 2011). Consequently, HBD is considered as one important parameter in medicinal chemistry. Polar surface area (PSA) is the surface area of all polar atoms in one molecule. CNS drugs usually have a relatively low PSA value than non-CNS drugs. These parameters have been integrated in multiple algorithms to predict compound permeability to BBB (Wager et al., 2010; Gupta et al., 2019). The preferred ranges of these parameters are as followed: 2 < cLogP < 4, HBD < 3, PSA < 90, and molecular weight (MW) < 450 (Wager et al., 2010).
By analyzing the structures shown in Table 1, we noticed that some molecules are built with the moieties that are not favorable to CNS penetration, such as carboxylate and primary sulfonamide. Several strategies have been proposed for structural modification of small molecules to improve BBB penetration: increasing lipophilicity, reducing hydrogen bond donor capacity, reducing PSA, enhancing rigidity, and reducing pKa (Xiong et al., 2021). Herein, we discuss the possible structure optimization of selected chloride channel mediators from Table 1 as case study and evaluate these modifications using physicochemical parameters.
Eszopiclone has one methyl group on the piperazine ring as highlighted in Scheme 1A, which is not directly involved in binding interactions with its target based on previous docking studies (Hanson et al., 2008). Substitution of a methyl group with other aliphatic chains such as an ethyl or propyl group improves cLogP value from 1.25 to 1.78 or 2.31, respectively. In contrast, elexacaftor in Scheme 1B shows high cLogP, PSA, and MW. The binding study has demonstrated that the sulfonamide and amide groups of elexacaftor form hydrogen bonds with CFTR. Elexacaftor also interacts with transmembrane helix through electrostatic and van der Waals interactions (Fiedorczuk and Chen, 2022), suggesting that all the moieties of elexacaftor orientate into one docking position and contribute to binding interactions. In this case, we propose a design to replace the pyrazole ring in the middle with a thiazole ring, in which the heteroatoms may serve as hydrogen bond accepters. In addition, the aromatic system of thiazole may also retain this moiety into a planer shape and provide hydrophobicity contributing to binding interaction with CFTR. To determine whether the molecule retains the comparable pose to fit in the binding pocket, we compare the 3D structures of elexacaftor before and after the modification. As shown in Scheme 1C, the molecules superimpose from the pyridine moiety to the pyrazole ring, suggesting the modified compound can fit in the same binding pocket with a similar binding pattern as elexacaftor. In addition to direct modifications of molecule structure, bio-isosteric replacement also can create a new molecule with similar biological properties to the parent compound but with optimized bioavailability, which has been commonly adopted in medicinal chemistry. For instance, carboxylate imparts significant polarity, thereby strongly impacting pharmacokinetics and drug distribution across BBB. Carboxylic acid may also undergo glucuronidation during phase II metabolism facilitating renal clearance, possibly causing another issue for CNS-drug design. We thereby devise an optimization of the carboxylic acid moiety in DCPIB to its bio-isostere oxadiazolone in Scheme 1D. The cLogP value is then optimized from 7.14 to 4.60.
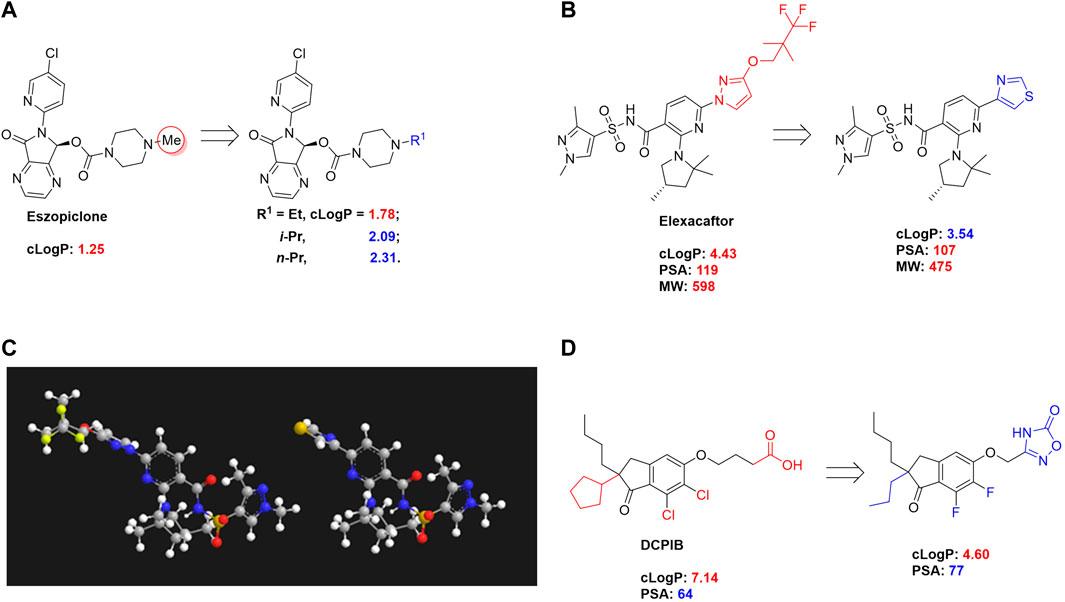
SCHEME 1. Examples of structure optimization to improve physicochemical properties. (A) Eszopiclone: methyl group (highlighted in red cycle) is replaced with a different group represented by R1. cLogP value is improved with substitution by ethyl or propyl groups. (B) Elexacaftor: this design replaces the pyrazole in the middle with a thiazole ring to retain the molecule orientation to the target. (C) Molecule orientation before (left) and after (right) structure modification for elexacaftor. (D) DCPIB: bio-isosteric replacement of carboxylic acid with oxadiazolone.
The rationale of proposed structure optimizations in the case studies above is solely based on the calculated physicochemical properties and previous binding studies. Minor changes on compound structure could change molecule’s orientation and thereby significantly impact compound activity. Thus, in vitro as well as in vivo assessments would be required to validate compound potency after structure modifications.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
All authors are employed by Amgen Inc., and their research and authorship of this article were completed within the scope of their employment with Amgen Inc.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
4-PBA, phenylbutyrate; Aβ, beta-amyloid; AD, Alzheimer’s disease; ANO, anoctamin; BBB, blood-brain-barrier; BCRP, breast cancer resistance protein; Best1, bestrophin 1; CaCC, Ca2+-gated chloride channel; CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; cLogP, calculated logarithm of partition coefficient P; CNS, central nervous system; ClC, voltage-gated chloride channel; ER, endoplasmic reticulum; FDA, food and drug administration; GABA, γ-aminobutyric acid; HBA, hydrogen bond acceptor; HBD, hydrogen bond Donor; HD, Huntington disease; MAC, maxi anion channel; MDR1, multiple drug resistance 1; MLC, megalencephalic leukoencephalopathy with subcortical cysts; MW, molecular weight; PD, Parkinson’s disease; THIP, 4,5,6,7-tetrahydroisoxazolopyridin-3-ol; TPSA, topological polar surface area; VRAC, volume regulated anion channel.
Abdullaev, I. F., Rudkouskaya, A., Schools, G. P., Kimelberg, H. K., and Mongin, A. A. (2006). Pharmacological comparison of swelling-activated excitatory amino acid release and Cl-currents in cultured rat astrocytes. J. Physiol. 572 (3), 677–689. doi:10.1113/jphysiol.2005.103820
Accardi, A. (2015). Structure and gating of CLC channels and exchangers. J. Physiol. 593 (18), 4129–4138. doi:10.1113/JP270575
Alex, A., Millan, D. S., Perez, M., Wakenhut, F., and Whitlock, G. A. (2011). Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. MedChemComm 2 (7), 669–674. doi:10.1039/c1md00093d
Ayoglu, B., Mitsios, N., Kockum, I., Khademi, M., Zandian, A., Sjöberg, R., et al. (2016). Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 113 (8), 2188–2193. doi:10.1073/pnas.1518553113
Barron, K. D., Dentinger, M. P., Kimelberg, H. K., Nelson, L. R., Bourke, R. S., Keegan, S., et al. (1988). Ultrastructural features of a brain injury model in cat. I. Vascular and neuroglial changes and the prevention of astroglial swelling by a fluorenyl (aryloxy) alkanoic acid derivative (L-644,711). Acta neuropathol. 75 (3), 295–307. doi:10.1007/BF00690538
Basarsky, T. A., Feighan, D., and MacVicar, B. A. (1999). Glutamate release through volume-activated channels during spreading depression. J. Neurosci. 19 (15), 6439–6445. doi:10.1523/JNEUROSCI.19-15-06439.1999
Blanz, J., Schweizer, M., Auberson, M., Maier, H., Muenscher, A., Hübner, C. A., et al. (2007). Leukoencephalopathy upon disruption of the chloride channelchannel ClC-2. J. Neurosci. 27 (24), 6581–6589. doi:10.1523/JNEUROSCI.0338-07.2007
Błaszczyk, J. W. (2016). Parkinson's disease and neurodegeneration: GABA-collapse hypothesis. Front. Neurosci. 10, 269. doi:10.3389/fnins.2016.00269
Boudes, M., Sar, C., Menigoz, A., Hilaire, C., Péquignot, M. O., Kozlenkov, A., et al. (2009). Best1 is a gene regulated by nerve injury and required for Ca2+-activated Cl-current expression in axotomized sensory neurons. J. Neurosci. 29 (32), 10063–10071. doi:10.1523/JNEUROSCI.1312-09.2009
Braat, S., and Kooy, R. F. (2015). The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86 (5), 1119–1130. doi:10.1016/j.neuron.2015.03.042
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABA(A) receptors: Their function in the CNS and implications for disease. Neuron 73 (1), 23–34. doi:10.1016/j.neuron.2011.12.012
Britschgi, A., Bill, A., Brinkhaus, H., Rothwell, C., Clay, I., Duss, S., et al. (2013). Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. U. S. A. 110 (11), E1026–E1034. doi:10.1073/pnas.1217072110
Chandrasekaran, A., Dittlau, K. S., Corsi, G. I., Haukedal, H., Doncheva, N. T., Ramakrishna, S., et al. (2021). Astrocytic reactivity triggered by defective autophagy and metabolic failure causes neurotoxicity in frontotemporal dementia type 3. Stem Cell Rep. 16 (11), 2736–2751. doi:10.1016/j.stemcr.2021.09.013
Chen, T. T., Klassen, T. L., Goldman, A. M., Marini, C., Guerrini, R., and Noebels, J. L. (2013). Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology 80 (12), 1078–1085. doi:10.1212/WNL.0b013e31828868e7
Chuang, S. H., and Reddy, D. S. (2018). Genetic and molecular regulation of extrasynaptic GABA-A receptors in the brain: Therapeutic insights for epilepsy. J. Pharmacol. Exp. Ther. 364 (2), 180–197. doi:10.1124/jpet.117.244673
Coimbra Jts, , Feghali, R., Ribeiro, R. P., Ramos, M. J., and Fernandes, P. A. (2021). The importance of intramolecular hydrogen bonds on the translocation of the small drug piracetam through a lipid bilayer. RSC Adv. 11 (2), 899–908. doi:10.1039/d0ra09995c
Cortez, M. A., Li, C., Whitehead, S. N., Dhani, S. U., D'Antonio, C., Huan, L. J., et al. (2010). Disruption of ClC-2 expression is associated with progressive neurodegeneration in aging mice. Neuroscience 167 (1), 154–162. doi:10.1016/j.neuroscience.2010.01.042
D'Agostino, D., Bertelli, M., Gallo, S., Cecchin, S., Albiero, E., Garofalo, P. G., et al. (2004). Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology 63 (8), 1500–1502. doi:10.1212/01.wnl.0000142093.94998.1a
Darby, M., Kuzmiski, J. B., Panenka, W., Feighan, D., and MacVicar, B. A. (2003). ATP released from astrocytes during swelling activates chloride channels. J. neurophysiology 89 (4), 1870–1877. doi:10.1152/jn.00510.2002
Doyon, N., Vinay, L., Prescott, S. A., and De Koninck, Y. (2016). Chloride regulation: A dynamic equilibrium crucial for synaptic inhibition. Neuron 89 (6), 1157–1172. doi:10.1016/j.neuron.2016.02.030
Duarri, A., Lopez de Heredia, M., Capdevila-Nortes, X., Ridder, M. C., Montolio, M., López-Hernández, T., et al. (2011). Knockdown of MLC1 in primary astrocytes causes cell vacuolation: A MLC disease cell model. Neurobiol. Dis. 43 (1), 228–238. doi:10.1016/j.nbd.2011.03.015
Dutta, A. K., Sabirov, R. Z., Uramoto, H., and Okada, Y. (2004). Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J. Physiol. 559 (3), 799–812. doi:10.1113/jphysiol.2004.069245
Eckford, P. D., Li, C., Ramjeesingh, M., and Bear, C. E. (2012). Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 287 (44), 36639–36649. doi:10.1074/jbc.M112.393637
Eder, C., Klee, R., and Heinemann, U. (1998). Involvement of stretch-activated Cl-channels in ramification of murine microglia. J. Neurosci. 18 (18), 7127–7137. doi:10.1523/JNEUROSCI.18-18-07127.1998
Eguchi, H., Tsujino, A., Kaibara, M., Hayashi, H., Shirabe, S., Taniyama, K., et al. (2006). Acetazolamide acts directly on the human skeletal muscle chloride channel. Muscle Nerve 34 (3), 292–297. doi:10.1002/mus.20585
Engels, M., Kalia, M., Rahmati, S., Petersilie, L., Kovermann, P., van Putten, M., et al. (2021). Glial chloride homeostasis under transient ischemic stress. Front. Cell Neurosci. 15, 735300. doi:10.3389/fncel.2021.735300
Essrich, C., Lorez, M., Benson, J. A., Fritschy, J. M., and Lüscher, B. (1998). Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1 (7), 563–571. doi:10.1038/2798
Fabene, P. F., Weiczner, R., Marzola, P., Nicolato, E., Calderan, L., Andrioli, A., et al. (2006). Structural and functional MRI following 4-aminopyridine-induced seizures: A comparative imaging and anatomical study. Neurobiol. Dis. 21 (1), 80–89. doi:10.1016/j.nbd.2005.06.013
Falk-Petersen, C. B., Tsonkov, T. M., Nielsen, M. S., Harpsøe, K., Bundgaard, C., Frølund, B., et al. (2020). Discovery of a new class of orthosteric antagonists with nanomolar potency at extrasynaptic GABAA receptors. Sci. Rep. 10 (1), 10078. doi:10.1038/s41598-020-66821-0
Fiedorczuk, K., and Chen, J. (2022). Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 378 (6617), 284–290. doi:10.1126/science.ade2216
Földy, C., Lee, S-H., Morgan, R. J., and Soltesz, I. (2010). Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat. Neurosci. 13 (9), 1047–1049. doi:10.1038/nn.2609
Forshaw, P. J., Lister, T., and Ray, D. E. (1993). Inhibition of a neuronal voltage-dependent chloride channel by the type II pyrethroid, deltamethrin. Neuropharmacology 32 (2), 105–111. doi:10.1016/0028-3908(93)90089-l
Forshaw, P. J., Lister, T., and Ray, D. E. (2000). The role of voltage-gated chloride channels in type II pyrethroid insecticide poisoning. Toxicol. Appl. Pharmacol. 163 (1), 1–8. doi:10.1006/taap.1999.8848
Funk, K., Woitecki, A., Franjic-Würtz, C., Gensch, T., Möhrlen, F., and Frings, S. (2008). Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol. pain 4, 32. doi:10.1186/1744-8069-4-32
Goldstein, A. B., Goldstein, L. S., Perl, M. K., Haug, M. T., Arroliga, A. C., and Stillwell, P. C. (2000). Cystic fibrosis patients with and without central nervous system complications following lung transplantation. Pediatr. Pulmonol. 30 (3), 203–206. doi:10.1002/1099-0496(200009)30:3<203:aid-ppul4>3.0.co;2-5
Gordon, G. R., Baimoukhametova, D. V., Hewitt, S. A., Rajapaksha, W. R., Fisher, T. E., and Bains, J. S. (2005). Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci. 8 (8), 1078–1086. doi:10.1038/nn1498
Grunder, S., Thiemann, A., Pusch, M., and Jentsch, T. J. (1992). Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 360 (6406), 759–762. doi:10.1038/360759a0
Guo, Y., Su, M., McNutt, M. A., and Gu, J. (2009). Expression and distribution of cystic fibrosis transmembrane conductance regulator in neurons of the human brain. J. Histochem. Cytochem. official J. Histochem. Soc. 57 (12), 1113–1120. doi:10.1369/jhc.2009.953455
Gupta, M., Lee, H. J., Barden, C. J., and Weaver, D. F. (2019). The blood–brain barrier (BBB) score. J. Med. Chem. 62 (21), 9824–9836. doi:10.1021/acs.jmedchem.9b01220
Hanson, S. M., Morlock, E. V., Satyshur, K. A., and Czajkowski, C. (2008). Structural requirements for eszopiclone and zolpidem binding to the gamma-aminobutyric acid type-A (GABAA) receptor are different. J. Med. Chem. 51 (22), 7243–7252. doi:10.1021/jm800889m
Hoestgaard-Jensen, K., Dalby, N. O., Krall, J., Hammer, H., Krogsgaard-Larsen, P., Frølund, B., et al. (2014). Probing α4βδ GABAA receptor heterogeneity: Differential regional effects of a functionally selective α4β1δ/α4β3δ receptor agonist on tonic and phasic inhibition in rat brain. J. Neurosci. 34 (49), 16256–16272. doi:10.1523/JNEUROSCI.1495-14.2014
Hong, G. S., Lee, S. H., Lee, B., Choi, J. H., Oh, S. J., Jang, Y., et al. (2019). ANO1/TMEM16A regulates process maturation in radial glial cells in the developing brain. Proc. Natl. Acad. Sci. U. S. A. 116 (25), 12494–12499. doi:10.1073/pnas.1901067116
Hou, X., Zhang, R., Wang, J., Li, Y., Li, F., Zhang, Y., et al. (2018). CLC-2 is a positive modulator of oligodendrocyte precursor cell differentiation and myelination. Mol. Med. Rep. 17 (3), 4515–4523. doi:10.3892/mmr.2018.8439
Huang, F., Wong, X., and Jan, L. Y. (2012). International union of basic and clinical pharmacology. LXXXV: Calcium-activated chloride channels. Pharmacol. Rev. 64 (1), 1–15. doi:10.1124/pr.111.005009
Huang, W. C., Xiao, S., Huang, F., Harfe, B. D., Jan, Y. N., and Jan, L. Y. (2012). Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron 74 (1), 179–192. doi:10.1016/j.neuron.2012.01.033
Hyzinski-García, M. C., Rudkouskaya, A., and Mongin, A. A. (2014). LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J. Physiol. 592 (22), 4855–4862. doi:10.1113/jphysiol.2014.278887
Jalonen, T. (1993). Single-channel characteristics of the large-conductance anion channel in rat cortical astrocytes in primary culture. Glia 9 (3), 227–237. doi:10.1002/glia.440090308
Jentsch, T. J., and Pusch, M. (2018). CLC chloride channels and transporters: Structure, function, physiology, and disease. Physiol. Rev. 98 (3), 1493–1590. doi:10.1152/physrev.00047.2017
Jeon, D., Ryu, K., Jo, S., Kim, I., and Namkung, W. (2022). VI-116, A novel potent inhibitor of VRAC with minimal effect on ANO1. Int. J. Mol. Sci. 23 (9), 5168. doi:10.3390/ijms23095168
Jeworutzki, E., López-Hernández, T., Capdevila-Nortes, X., Sirisi, S., Bengtsson, L., Montolio, M., et al. (2012). GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron 73 (5), 951–961. doi:10.1016/j.neuron.2011.12.039
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat. Med. 20 (8), 886–896. doi:10.1038/nm.3639
Jordt, S. E., and Jentsch, T. J. (1997). Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 16 (7), 1582–1592. doi:10.1093/emboj/16.7.1582
Kahle, K. T., Staley, K. J., Nahed, B. V., Gamba, G., Hebert, S. C., Lifton, R. P., et al. (2008). Roles of the cation–chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 4 (9), 490–503. doi:10.1038/ncpneuro0883
Kang, S. S., Han, K. S., Ku, B. M., Lee, Y. K., Hong, J., Shin, H. Y., et al. (2010). Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 70 (3), 1173–1183. doi:10.1158/0008-5472.CAN-09-2886
Kasuya, G., and Nureki, O. (2022). Recent advances in the structural biology of the volume-regulated anion channel LRRC8. Front. Pharmacol. 13, 896532. doi:10.3389/fphar.2022.896532
Kimelberg, H. K. (2005). Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50 (4), 389–397. doi:10.1002/glia.20174
Kimelberg, H. K., Jin, Y., Charniga, C., and Feustel, P. J. (2003). Neuroprotective activity of tamoxifen in permanent focal ischemia. J. Neurosurg. 99 (1), 138–142. doi:10.3171/jns.2003.99.1.0138
Kimelberg, H. K. (2000). Review: Cell volume in the CNS: Regulation and implications for nervous system function and pathology. Neurosci. 6 (1), 14–25. doi:10.1177/107385840000600110
Koneczny, I., and HerbstGravis, R. M. (2019). Myasthenia gravis: Pathogenic effects of autoantibodies on neuromuscular architecture. Cells 8 (7), 671. doi:10.3390/cells8070671
Koster, A. K., Reese, A. L., Kuryshev, Y., Wen, X., McKiernan, K. A., Gray, E. E., et al. (2020). Development and validation of a potent and specific inhibitor for the CLC-2 chloride channel. Proc. Natl. Acad. Sci. U. S. A. 117 (51), 32711–32721. doi:10.1073/pnas.2009977117
Lafrenaye, A. D., and Simard, J. M. (2019). Bursting at the seams: Molecular mechanisms mediating astrocyte swelling. Int. J. Mol. Sci. 20 (2), 330. doi:10.3390/ijms20020330
Lahousse, S. A., Stopa, E. G., Mulberg, A. E., and de la Monte, S. M. (2003). Reduced expression of the cystic fibrosis transmembrane conductance regulator gene in the hypothalamus of patients with Alzheimer's disease. J. Alzheimer's Dis JAD. 5 (6), 455–462. doi:10.3233/jad-2003-5605
Lee, B., Cho, H., Jung, J., Yang, Y. D., Yang, D. J., and Oh, U. (2014). Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol. pain 10, 5. doi:10.1186/1744-8069-10-5
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., et al. (2010). Channel-mediated tonic GABA release from glia. Science 330 (6005), 790–796. doi:10.1126/science.1184334
Lee, V., and Maguire, J. (2014). The impact of tonic GABAA receptor-mediated inhibition on neuronal excitability varies across brain region and cell type. Front. Neural Circuits 8, 3. doi:10.3389/fncir.2014.00003
Lee, Y-S., Lee, J. K., Bae, Y., Lee, B-S., Kim, E., Cho, C-H., et al. (2016). Suppression of 14-3-3γ-mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci. Rep. 6 (1), 26413. doi:10.1038/srep26413
Lindholm, D., Wootz, H., and Korhonen, L. (2006). ER stress and neurodegenerative diseases. Cell Death Differ. 13 (3), 385–392. doi:10.1038/sj.cdd.4401778
Liochev, S. I. (2013). Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 60, 1–4. doi:10.1016/j.freeradbiomed.2013.02.011
Liu, G. J., Kalous, A., Werry, E. L., and Bennett, M. R. (2006). Purine release from spinal cord microglia after elevation of calcium by glutamate. Mol. Pharmacol. 70 (3), 851–859. doi:10.1124/mol.105.021436
Liu, H-T., Tashmukhamedov, B. A., Inoue, H., Okada, Y., and Sabirov, R. Z. (2006). Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54 (5), 343–357. doi:10.1002/glia.20400
Liu, H-T., Toychiev, A. H., Takahashi, N., Sabirov, R. Z., and Okada, Y. (2008). Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res. 18 (5), 558–565. doi:10.1038/cr.2008.49
Liu, H. T., Sabirov, R. Z., and Okada, Y. (2008). Oxygen-glucose deprivation induces ATP release via maxi-anion channels in astrocytes. Purinergic Signal. 4 (2), 147–154. doi:10.1007/s11302-007-9077-8
Liu, J., Liu, Y., Ren, Y., Kang, L., and Zhang, L. (2014). Transmembrane protein with unknown function 16A overexpression promotes glioma formation through the nuclear factor-κB signaling pathway. Mol. Med. Rep. 9 (3), 1068–1074. doi:10.3892/mmr.2014.1888
Liu, J., Taylor, R. L., Baines, R. A., Swanton, L., Freeman, S., Corneo, B., et al. (2020). Small molecules restore bestrophin 1 expression and function of both dominant and recessive bestrophinopathies in patient-derived retinal pigment epithelium. Investigative Ophthalmol. Vis. Sci. 61 (5), 28. doi:10.1167/iovs.61.5.28
Liu, P. Y., Zhang, Z., Liu, Y., Tang, X. L., Shu, S., Bao, X. Y., et al. (2019). TMEM16A inhibition preserves blood-brain barrier integrity after ischemic stroke. Front. Cell Neurosci. 13, 360. doi:10.3389/fncel.2019.00360
Liu, Y., Liu, Z., and Wang, K. (2021). The Ca2+-activated chloride channel ANO1/TMEM16A: An emerging therapeutic target for epithelium-originated diseases? Acta Pharm. Sin. B 11 (6), 1412–1433. doi:10.1016/j.apsb.2020.12.003
Liu, Y., Zhang, H., Huang, D., Qi, J., Xu, J., Gao, H., et al. (2015). Characterization of the effects of Cl⁻ channel modulators on TMEM16A and bestrophin-1 Ca²⁺ activated Cl⁻ channels. Pflügers Archiv - Eur. J. Physiology 467 (7), 1417–1430. doi:10.1007/s00424-014-1572-5
López-Hernández, T., Ridder, M. C., Montolio, M., Capdevila-Nortes, X., Polder, E., Sirisi, S., et al. (2011). Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 88 (4), 422–432. doi:10.1016/j.ajhg.2011.02.009
Lutter, D., Ullrich, F., Lueck, J. C., Kempa, S., and Jentsch, T. J. (2017). Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J. Cell Sci. 130 (6), 1122–1133. doi:10.1242/jcs.196253
Maertens, C., Droogmans, G., Verbesselt, R., and Nilius, B. (2002). Block of volume-regulated anion channels by selective serotonin reuptake inhibitors. Naunyn-Schmiedeberg's Archives Pharmacol. 366 (2), 158–165. doi:10.1007/s00210-002-0567-5
Maertens, C., Wei, L., Voets, T., Droogmans, G., and Nilius, B. (1999). Block by fluoxetine of volume-regulated anion channels. Br. J. Pharmacol. 126 (2), 508–514. doi:10.1038/sj.bjp.0702314
Magalhães, A. C., and Rivera, C. (2016). NKCC1-Deficiency results in abnormal proliferation of neural progenitor cells of the lateral ganglionic eminence. Front. Cell. Neurosci. 10, 200. doi:10.3389/fncel.2016.00200
Mahadevan, V., Pressey, J. C., Acton, B. A., Uvarov, P., Huang, M. Y., Chevrier, J., et al. (2014). Kainate receptors coexist in a functional complex with KCC2 and regulate chloride homeostasis in hippocampal neurons. Cell Rep. 7 (6), 1762–1770. doi:10.1016/j.celrep.2014.05.022
Manley, G. T., Fujimura, M., Ma, T., Noshita, N., Filiz, F., Bollen, A. W., et al. (2000). Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 6 (2), 159–163. doi:10.1038/72256
Marcorelles, P., Friocourt, G., Uguen, A., Ledé, F., Férec, C., and Laquerrière, A. (2014). Cystic fibrosis transmembrane conductance regulator protein (CFTR) expression in the developing human brain: Comparative immunohistochemical study between patients with normal and mutated CFTR. J. Histochem. Cytochem. official J. Histochem. Soc. 62 (11), 791–801. doi:10.1369/0022155414546190
Marques, T. R., Ashok, A. H., Angelescu, I., Borgan, F., Myers, J., Lingford-Hughes, A., et al. (2021). GABA-A receptor differences in schizophrenia: A positron emission tomography study using [(11)C]Ro154513. Mol. psychiatry 26 (6), 2616–2625. doi:10.1038/s41380-020-0711-y
May, A. C., Fleischer, W., Kletke, O., Haas, H. L., and Sergeeva, O. A. (2013). Benzodiazepine-site pharmacology on GABAA receptors in histaminergic neurons. Br. J. Pharmacol. 170 (1), 222–232. doi:10.1111/bph.12280
Mortensen, M., Patel, B., and Smart, T. G. (2011). GABA potency at GABA(A) receptors found in synaptic and extrasynaptic zones. Front. Cell Neurosci. 6, 1. doi:10.3389/fncel.2012.00001
Mulberg, A. E., Resta, L. P., Wiedner, E. B., Altschuler, S. M., Jefferson, D. M., and Broussard, D. L. (1995). Expression and localization of the cystic fibrosis transmembrane conductance regulator mRNA and its protein in rat brain. J. Clin. Invest. 96 (1), 646–652. doi:10.1172/JCI118080
Mulberg, A. E., Weyler, R. T., Altschuler, S. M., and Hyde, T. M. (1998). Cystic fibrosis transmembrane conductance regulator expression in human hypothalamus. Neuroreport 9 (1), 141–144. doi:10.1097/00001756-199801050-00028
Niemeyer, M. I., Yusef, Y. R., Cornejo, I., Flores, C. A., Sepulveda, F. V., and Cid, L. P. (2004). Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol. Genomics 19 (1), 74–83. doi:10.1152/physiolgenomics.00070.2004
Novarino, G., Fabrizi, C., Tonini, R., Denti, M. A., Malchiodi-Albedi, F., Lauro, G. M., et al. (2004). Involvement of the intracellular ion channel CLIC1 in microglia-mediated beta-amyloid-induced neurotoxicity. J. Neurosci. 24 (23), 5322–5330. doi:10.1523/JNEUROSCI.1170-04.2004
O'Driscoll, K. E., Leblanc, N., Hatton, W. J., and Britton, F. C. (2009). Functional properties of murine bestrophin 1 channel. Biochem. biophysical Res. Commun. 384 (4), 476–481. doi:10.1016/j.bbrc.2009.05.008
Octavia, Y., Brunner-La Rocca, H. P., and Moens, A. L. (2012). NADPH oxidase-dependent oxidative stress in the failing heart: From pathogenic roles to therapeutic approach. Free Radic. Biol. Med. 52 (2), 291–297. doi:10.1016/j.freeradbiomed.2011.10.482
Oh, S. J., and Lee, C. J. (2017). Distribution and function of the bestrophin-1 (Best1) channel in the brain. Exp. Neurobiol. 26 (3), 113–121. doi:10.5607/en.2017.26.3.113
Oh, U., and Jung, J. (2016). Cellular functions of TMEM16/anoctamin. Pflugers Archiv Eur. J. physiology 468 (3), 443–453. doi:10.1007/s00424-016-1790-0
Oller-Salvia, B., Sánchez-Navarro, M., Giralt, E., and Teixidó, M. (2016). Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 45 (17), 4690–4707. doi:10.1039/c6cs00076b
Park, H., Oh, S. J., Han, K. S., Woo, D. H., Park, H., Mannaioni, G., et al. (2009). Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J. Neurosci. 29 (41), 13063–13073. doi:10.1523/JNEUROSCI.3193-09.2009
Pedemonte, N., Bertozzi, F., Caci, E., Sorana, F., Di Fruscia, P., Tomati, V., et al. (2020). Discovery of a picomolar potency pharmacological corrector of the mutant CFTR chloride channel. Sci. Adv. 6 (8), eaay9669. doi:10.1126/sciadv.aay9669
Phulera, S., Zhu, H., Yu, J., Claxton, D. P., Yoder, N., Yoshioka, C., et al. (2018). Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABA(A) receptor in complex with GABA. eLife 7, e39383. doi:10.7554/eLife.39383
Pineda-Farias, J. B., Barragán-Iglesias, P., Loeza-Alcocer, E., Torres-López, J. E., Rocha-González, H. I., Pérez-Severiano, F., et al. (2015). Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Mol. pain 11, 41. doi:10.1186/s12990-015-0042-1
Poroca, D. R., Pelis, R. M., and Chappe, V. M. (2017). ClC channels and transporters: Structure, physiological functions, and implications in human chloride channelopathies. Front. Pharmacol. 8, 151. doi:10.3389/fphar.2017.00151
Puia, G., Ducic, I., Vicini, S., and Costa, E. (1992). Molecular mechanisms of the partial allosteric modulatory effects of bretazenil at gamma-aminobutyric acid type A receptor. Proc. Natl. Acad. Sci. U. S. A. 89 (8), 3620–3624. doi:10.1073/pnas.89.8.3620
Qu, Z., and Hartzell, H. C. (2008). Bestrophin Cl-channels are highly permeable to HCO3. Am. J. physiology Cell physiology 294 (6), C1371–C1377. doi:10.1152/ajpcell.00398.2007
Rahmati, N., Hoebeek, F. E., Peter, S., and De Zeeuw, C. I. (2018). Chloride homeostasis in neurons with special emphasis on the olivocerebellar system: Differential roles for transporters and channels. Front. Cell Neurosci. 12, 101. doi:10.3389/fncel.2018.00101
Rasche, S., Toetter, B., Adler, J., Tschapek, A., Doerner, J. F., Kurtenbach, S., et al. (2010). Tmem16b is specifically expressed in the cilia of olfactory sensory neurons. Chem. senses 35 (3), 239–245. doi:10.1093/chemse/bjq007
Richter, G., Liao, V. W. Y., Ahring, P. K., and Chebib, M. (2020). The Z-drugs zolpidem, zaleplon, and eszopiclone have varying actions on human GABAA receptors containing γ1, γ2, and γ3 subunits. Front. Neurosci. 14, 599812. doi:10.3389/fnins.2020.599812
Ridley, K., and Condren, M. (2020). Elexacaftor-Tezacaftor-Ivacaftor: The first triple-combination cystic fibrosis transmembrane conductance regulator modulating therapy. J. Pediatr. Pharmacol. Ther. JPPT official J. PPAG. 25 (3), 192–197. doi:10.5863/1551-6776-25.3.192
Rinke, I., Artmann, J., and Stein, V. (2010). ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J. Neurosci. 30 (13), 4776–4786. doi:10.1523/JNEUROSCI.6299-09.2010
Roth, T., Lines, C., Vandormael, K., Ceesay, P., Anderson, D., and Snavely, D. (2010). Effect of gaboxadol on patient-reported measures of sleep and waking function in patients with primary insomnia: Results from two randomized, controlled, 3-month studies. J. Clin. sleep Med. JCSM official Publ. Am. Acad. Sleep Med. 6 (1), 30–39. doi:10.5664/jcsm.27707
Sabirov, R. Z., and Okada, Y. (2005). ATP release via anion channels. Purinergic Signal. 1 (4), 311–328. doi:10.1007/s11302-005-1557-0
Sabirov, R. Z., and Okada, Y. (2009). The maxi-anion channel: A classical channel playing novel roles through an unidentified molecular entity. J. Physiological Sci. 59 (1), 3–21. doi:10.1007/s12576-008-0008-4
Schlichter, L. C., Sakellaropoulos, G., Ballyk, B., Pennefather, P. S., and Phipps, D. J. (1996). Properties of K+ and Cl-channels and their involvement in proliferation of rat microglial cells. Glia 17 (3), 225–236. doi:10.1002/(SICI)1098-1136(199607)17:3<225:AID-GLIA5>3.0.CO;2-#
Schober, A. L., Wilson, C. S., and Mongin, A. A. (2017). Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J. Physiol. 595 (22), 6939–6951. doi:10.1113/JP275053
Shen, M., Wang, L., Wang, B., Wang, T., Yang, G., Shen, L., et al. (2014). Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: Involvement of CHOP through wnt. Cell Death Dis. 5 (11), e1528–e. doi:10.1038/cddis.2014.479
Shen, M. R., Droogmans, G., Eggermont, J., Voets, T., Ellory, J. C., and Nilius, B. (2000). Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol. 529 (2), 385–394. doi:10.1111/j.1469-7793.2000.00385.x
Sik, A., Smith, R. L., and Freund, T. F. (2000). Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience 101 (1), 51–65. doi:10.1016/s0306-4522(00)00360-2
Sı́k, A., Smith, R. L., and Freund, T. F. (2000). Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience 101 (1), 51–65. doi:10.1016/s0306-4522(00)00360-2
Stassart, R. M., Möbius, W., Nave, K. A., and Edgar, J. M. (2018). The axon-myelin unit in development and degenerative disease. Front. Neurosci. 12, 467. doi:10.3389/fnins.2018.00467
Sternfeld, F., Carling, R. W., Jelley, R. A., Ladduwahetty, T., Merchant, K. J., Moore, K. W., et al. (2004). Selective, orally active gamma-aminobutyric acidA alpha5 receptor inverse agonists as cognition enhancers. J. Med. Chem. 47 (9), 2176–2179. doi:10.1021/jm031076j
Suadicani, S. O., Brosnan, C. F., and Scemes, E. (2006). P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 26 (5), 1378–1385. doi:10.1523/JNEUROSCI.3902-05.2006
Thompson, C. H., Olivetti, P. R., Fuller, M. D., Freeman, C. S., McMaster, D., French, R. J., et al. (2009). Isolation and characterization of a high affinity peptide inhibitor of ClC-2 chloride channels. J. Biol. Chem. 284 (38), 26051–26062. doi:10.1074/jbc.M109.031724
Trachtman, H., and Cragoe, E. J. (1989). Hyponatremia-induced brain edema in Guinea pigs is reduced by treatment with the novel anion transport inhibitor L-644,711. Life Sci. 45 (22), 2141–2147. doi:10.1016/0024-3205(89)90080-5
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343 (6171), 675–679. doi:10.1126/science.1247190
van der Knaap, M. S., Barth, P. G., Vrensen, G. F., and Valk, J. (1996). Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta neuropathol. 92 (2), 206–212. doi:10.1007/s004010050510
Van der Plas, S. E., Kelgtermans, H., De Munck, T., Martina, S. L. X., Dropsit, S., Quinton, E., et al. (2018). Discovery of N-(3-Carbamoyl-5,5,7,7-tetramethyl-5,7-dihydro-4H-thieno[2,3-c]pyran-2-yl)-lH-pyrazole-5-carboxamide (GLPG1837), a novel potentiator which can open class III mutant cystic fibrosis transmembrane conductance regulator (CFTR) channels to a high extent. J. Med. Chem. 61 (4), 1425–1435. doi:10.1021/acs.jmedchem.7b01288
Van Goor, F., Hadida, S., Grootenhuis, P. D., Burton, B., Cao, D., Neuberger, T., et al. (2009). Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U. S. A. 106 (44), 18825–18830. doi:10.1073/pnas.0904709106
Van Goor, F., Hadida, S., Grootenhuis, P. D., Burton, B., Stack, J. H., Straley, K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U. S. A. 108 (46), 18843–18848. doi:10.1073/pnas.1105787108
Veit, G., Avramescu, R. G., Chiang, A. N., Houck, S. A., Cai, Z., Peters, K. W., et al. (2016). From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 27 (3), 424–433. doi:10.1091/mbc.E14-04-0935
Veit, G., Vaccarin, C., and Lukacs, G. L. (2021). Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 20 (5), 895–898. doi:10.1016/j.jcf.2021.03.011
Verkman, A. S., and Galietta, L. J. V. (2021). Chloride transport modulators as drug candidates. Am. J. Physiol. Cell Physiol. 321 (6), C932–C946. doi:10.1152/ajpcell.00334.2021
Voss, F. K., Ullrich, F., Münch, J., Lazarow, K., Lutter, D., Mah, N., et al. (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344 (6184), 634–638. doi:10.1126/science.1252826
Wafford, K. A., and Ebert, B. (2006). Gaboxadol-a new awakening in sleep. Curr. Opin. Pharmacol. 6 (1), 30–36. doi:10.1016/j.coph.2005.10.004
Wager, T. T., Hou, X., Verhoest, P. R., and Villalobos, A. (2010). Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 1 (6), 435–449. doi:10.1021/cn100008c
Wang, X., Liu, B., Searle, X., Yeung, C., Bogdan, A., Greszler, S., et al. (2018). Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic acid (ABBV/GLPG-2222), a potent cystic fibrosis transmembrane conductance regulator (CFTR) corrector for the treatment of cystic fibrosis. J. Med. Chem. 61 (4), 1436–1449. doi:10.1021/acs.jmedchem.7b01339
Wang, Z., Wang, Y., Pasangulapati, J. P., Stover, K. R., Liu, X., Schier, S., et al. (2021). Design, synthesis, and biological evaluation of furosemide analogs as therapeutics for the proteopathy and immunopathy of Alzheimer's disease. Eur. J. Med. Chem. 222, 113565. doi:10.1016/j.ejmech.2021.113565
Weyler, R. T., Yurko-Mauro, K. A., Rubenstein, R., Kollen, W. J., Reenstra, W., Altschuler, S. M., et al. (1999). CFTR is functionally active in GnRH-expressing GT1-7 hypothalamic neurons. Am. J. physiology 277 (3), C563–C571. doi:10.1152/ajpcell.1999.277.3.C563
Wilson, C. S., and Mongin, A. A. (2019). The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 689, 33–44. doi:10.1016/j.neulet.2018.01.012
Woo, D. H., Han, K. S., Shim, J. W., Yoon, B. E., Kim, E., Bae, J. Y., et al. (2012). TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151 (1), 25–40. doi:10.1016/j.cell.2012.09.005
Wu, H., Che, X., Tang, J., Ma, F., Pan, K., Zhao, M., et al. (2016). The K(+)-Cl(-) cotransporter KCC2 and chloride homeostasis: Potential therapeutic target in acute central nervous system injury. Mol. Neurobiol. 53 (4), 2141–2151. doi:10.1007/s12035-015-9162-x
Wu, Q., Akhter, A., Pant, S., Cho, E., Zhu, J. X., Garner, A., et al. (2022). Ataxia-linked SLC1A3 mutations alter EAAT1 chloride channel activity and glial regulation of CNS function. J. Clin. Investigation 132 (7), e154891. doi:10.1172/JCI154891
Xiang, Z., Chen, M., Ping, J., Dunn, P., Lv, J., Jiao, B., et al. (2006). Microglial morphology and its transformation after challenge by extracellular ATP in vitro. J. Neurosci. Res. 83 (1), 91–101. doi:10.1002/jnr.20709
Xiong, B., Wang, Y., Chen, Y., Xing, S., Liao, Q., Chen, Y., et al. (2021). Strategies for structural modification of small molecules to improve blood–brain barrier penetration: A recent perspective. J. Med. Chem. 64 (18), 13152–13173. doi:10.1021/acs.jmedchem.1c00910
Zhang, W., Schmelzeisen, S., Parthier, D., Frings, S., and Möhrlen, F. (2015). Anoctamin calcium-activated chloride channels may modulate inhibitory transmission in the cerebellar cortex. PloS one 10 (11), e0142160. doi:10.1371/journal.pone.0142160
Zhang, X. D., Morishima, S., Ando-Akatsuka, Y., Takahashi, N., Nabekura, T., Inoue, H., et al. (2004). Expression of novel isoforms of the CIC-1 chloride channel in astrocytic glial cells in vitro. Glia 47 (1), 46–57. doi:10.1002/glia.20024
Zhang, Y., Zhang, H., Feustel, P. J., and Kimelberg, H. K. (2008). DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp. Neurol. 210 (2), 514–520. doi:10.1016/j.expneurol.2007.11.027
Zhao, M., Zhang, J., Huang, W., Dong, J., Guo, J., U, K. P., et al. (2020). CFTR promotes malignant glioma development via up-regulation of Akt/Bcl2-mediated anti-apoptosis pathway. J. Cell. Mol. Med. 24 (13), 7301–7312. doi:10.1111/jcmm.15300
Zhi, Y., Liu, J., Kuang, P., Zhang, X., Xu, Z., Chen, Y., et al. (2022). Novel DCPIB analogs as dual inhibitors of VRAC/TREK1 channels reduced cGAS-STING mediated interferon responses. Biochem. Pharmacol. 199, 114988. doi:10.1016/j.bcp.2022.114988
Zhu, S., Noviello, C. M., Teng, J., Walsh, R. M., Kim, J. J., and Hibbs, R. E. (2018). Structure of a human synaptic GABAA receptor. Nature 559 (7712), 67–72. doi:10.1038/s41586-018-0255-3
Keywords: chloride homeostasis, chloride channels, central nervous system, small molecules, neurological disorders
Citation: Wang Z and Choi K (2023) Pharmacological modulation of chloride channels as a therapeutic strategy for neurological disorders. Front. Physiol. 14:1122444. doi: 10.3389/fphys.2023.1122444
Received: 12 December 2022; Accepted: 09 February 2023;
Published: 02 March 2023.
Edited by:
Jinwei Zhang, University of Exeter, United KingdomReviewed by:
Tenpei Akita, Hamamatsu University School of Medicine, JapanCopyright © 2023 Wang and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaylee Choi, a2F5bGVlLmNob2lAYW1nZW4uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.