
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Physiol. , 08 February 2023
Sec. Mitochondrial Research
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1106662
This article is part of the Research Topic Mitochondrial Disorders and Cardiovascular Diseases View all 7 articles
 Pablo Sánchez-Aguilera1,2
Pablo Sánchez-Aguilera1,2 Camila López-Crisosto1
Camila López-Crisosto1 Ignacio Norambuena-Soto1
Ignacio Norambuena-Soto1 Christian Penannen1Jumo Zhu2
Christian Penannen1Jumo Zhu2 Nils Bomer2
Nils Bomer2 Matijn F. Hoes3,4,5Peter Van Der Meer2
Matijn F. Hoes3,4,5Peter Van Der Meer2 Mario Chiong1
Mario Chiong1 B. Daan Westenbrink2*†
B. Daan Westenbrink2*† Sergio Lavandero1,6*†
Sergio Lavandero1,6*†A physiological increase in cardiac workload results in adaptive cardiac remodeling, characterized by increased oxidative metabolism and improvements in cardiac performance. Insulin-like growth factor-1 (IGF-1) has been identified as a critical regulator of physiological cardiac growth, but its precise role in cardiometabolic adaptations to physiological stress remains unresolved. Mitochondrial calcium (Ca2+) handling has been proposed to be required for sustaining key mitochondrial dehydrogenase activity and energy production during increased workload conditions, thus ensuring the adaptive cardiac response. We hypothesized that IGF-1 enhances mitochondrial energy production through a Ca2+-dependent mechanism to ensure adaptive cardiomyocyte growth. We found that stimulation with IGF-1 resulted in increased mitochondrial Ca2+ uptake in neonatal rat ventricular myocytes and human embryonic stem cell-derived cardiomyocytes, estimated by fluorescence microscopy and indirectly by a reduction in the pyruvate dehydrogenase phosphorylation. We showed that IGF-1 modulated the expression of mitochondrial Ca2+ uniporter (MCU) complex subunits and increased the mitochondrial membrane potential; consistent with higher MCU-mediated Ca2+ transport. Finally, we showed that IGF-1 improved mitochondrial respiration through a mechanism dependent on MCU-mediated Ca2+ transport. In conclusion, IGF-1-induced mitochondrial Ca2+ uptake is required to boost oxidative metabolism during cardiomyocyte adaptive growth.
Cardiac hypertrophy is a type of cardiac remodeling characterized by the enlargement of the heart in response to a repetitive or sustained increase in cardiac workload, classified as either physiological or pathological (Hill and Olson, 2008). Physiological cardiac hypertrophy develops during exercise or pregnancy and is characterized by mild reversible cardiac growth with conserved or increased contractility (Nakamura and Sadoshima, 2018). In contrast, pathological cardiac hypertrophy is an excessive cardiac growth observed in most cardiovascular diseases due to an unremitting increase in cardiac workload, which results in progressive and irreversible contractile dysfunction, cell death, and fibrosis (Nakamura and Sadoshima, 2018). There is little agreement on the mechanisms that determine both hypertrophic phenotypes, which could explain the lack of efficient treatments for pathological remodeling that often leads to heart failure (Hill and Olson, 2008).
Both hypertrophies are related to different signaling pathways and have a wholly opposed metabolic signature and energetic balance at the cardiomyocyte level (Brown et al., 2017). There is a consensus that physiological cardiac hypertrophy requires the activation of IGF-1 receptor signaling and mitochondrial function to be adaptive (Troncoso et al., 2014; Nijholt et al., 2022). In contrast, mechanical stress and catecholamines/angiotensin II overstimulation have been associated with Ca2+ mishandling and mitochondrial dysfunction, which are common features of pathological cardiac hypertrophy (Crilley et al., 2003; Nakamura and Sadoshima, 2018). Mitochondrial function and the energetic status of cardiomyocytes appear to be critical factors determining the developing hypertrophic phenotype.
Ca2+ exerts a delicate control of the energetic balance in cardiomyocytes. It promotes ATP consumption during contraction and increases mitochondrial ATP production to meet cardiac energy demands (Glancy and Balaban, 2012). This process occurs during each action potential when the sarcoplasmic reticulum releases Ca2+ that is partially absorbed by the mitochondria, enhancing the pyruvate metabolism and the tricarboxylic acid (TCA) cycle activity, thus promoting ATP synthesis (Denton, 2009). To reach the mitochondrial matrix, Ca2+ crosses the inner mitochondrial membrane through the mitochondrial Ca2+ uniporter (MCU) complex, strategically positioned in the vicinity of the junctional sarcoplasmic reticulum-mitochondrial associations (De La Fuente et al., 2016). The MCU complex (MCUC) is a Ca2+ transporter formed by the pore-forming subunit MCU, its paralog MCUB and the essential MCU regulator (EMRE) (Fan et al., 2020). In addition, MCU-mediated Ca2+-transport is regulated by mitochondrial Ca2+ uptake 1 and 2 (MICU1/2) and the MCU regulator 1 (MCUR1), which respectively control the channel gating and the conductance of MCU (Tomar et al., 2016; Wang et al., 2020).
Recently, the MCUC was linked to physiological cardiac hypertrophy. MCU protein levels increased in parallel with cardiac growth in two models of adaptive cardiac hypertrophy (Zaglia et al., 2017). Moreover, cardiac-specific overexpression of MCU in mice with pathological hypertrophy increased mitochondrial respiration and recovered cardiac performance, thereby resembling physiological cardiac growth (Suarez et al., 2018; Liu et al., 2021). There is plenty of evidence that mitochondrial dysfunction and Ca2+ mishandling contribute to the pathological cardiac hypertrophy (Gorski et al., 2015; Ljubojevic-Holzer et al., 2020; Liu et al., 2021). However, little attention has been paid to mitochondrial Ca2+ handling and its influence on oxidative metabolism in models of physiological cardiac hypertrophy. To address this question, we studied the effect of the physiological hypertrophic inductor IGF-1 on mitochondrial Ca2+ handling and oxidative metabolism in cultured cardiomyocytes.
NRVMs were obtained from 2 to 3 days old Sprague-Dawley rats. The isolation and culture were developed according to Galvez et al. (Galvez et al., 2001). Briefly, neonatal rats were decapitated, hearts extracted, and atria removed. Ventricles were washed in sterile Hank’s medium (Sigma-Aldrich, San Luis, MI, United States) at 37°C and submitted to enzymatic digestion. Cardiomyocytes were purified by a pre-pleated step for 2 h at 37°C to remove fibroblasts and afterward cultured in maintenance medium (DME:M199, 4:1; Sigma-Aldrich) supplemented with 15% fetal bovine serum and 0.1 mM bromodeoxyuridine (Sigma-Aldrich).
HUES9 human embryonic stem cells from Harvard Stem cells institute were differentiated into cardiomyocytes using a small molecule-derived approach (Hoes et al., 2018; Bomer et al., 2020). Briefly, HUES9 cells were maintained in Essential 8 medium (A1517001; Thermo Fisher Scientific, Waltham, MA, United States) before differentiation to cardiomyocytes was initiated, which was achieved by culturing HUES9 cells in RPMI1640 medium (21875-034, Life Technologies, Carlsbad, CA, United States) supplemented with 1x B27 minus insulin (Life Technologies, Carlsbad, CA, United States) and 6 μmol/L CHIR99021 (13122, Cayman Chemical, Ann Arbor, MI, United States). After 2 days, medium was refreshed with RPMI1640 supplemented with 1x B27 minus insulin and 2 μmol/L Wnt-C59 (5148, Tocris Bioscience, Bristol, United Kingdom). After 2 days, medium was changed to CDM3 medium (RPMI1640 plus 213 μg/mL ascorbic acid 2-phosphate plus 500 μg/mL recombinant human albumin). On day 8 after induction of differentiation, spontaneously contracting cardiomyocytes were observed, which were subsequently purified by replacing the medium to glucose-free CDM3 medium supplemented with 5 mmol/L sodium DL-lactate (CDM3L; Sigma-Aldrich). Ultimately, this resulted in >99% pure spontaneously beating cardiomyocyte cultures. The hES-CMs were suitable for experimentation ∼30 days after the differentiation protocol started.
Cardiomyocytes were stimulated with 10 nM recombinant human IGF-1 + 0.1% BSA (Thermo Fisher Scientific, Waltham, MA, United States) or 0.1% BSA in CDM3 or maintenance medium (hES-CMs or NRVMs respectively) between 15 min and 48 h depending on the experimental setup.
HEK-293T cells were cultured at 37°C, and 5% CO2 in Dulbecco modified Eagle medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal calf serum (Sigma-Aldrich). At 70% confluence, the cells were transfected with Fugene HD (Promega, Madison, WI, United States) together with pCMV∆8.91-transfer plasmid, VSV-G-packaging plasmid, and pLKO.1-plasmid expressing the genetically encoded mitochondrial Ca2+ indicator Mitycam provided by Dr. Adam Cohen (pMOS028, Addgene, plasmid #163046). The medium was replaced with CDM3 after 24 h and the supernatant containing the viral particles was harvested and filtered with a 0.45 nm filter after 48 h hES-CMs were incubated with CDM3 supplemented with 40% clean viral supernatant for 24 h. The next day, the medium was replaced by CMD3. After 48 h, cells were available for experiments.
Transfected hES-CMs with the Ca2+ indicator Mitycam were incubated in Tyrode medium (130 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.2) and mounted at 37°C in the Olympus IX-71 inverted microscope DeltaVision Elite (Olympus, Tokyo, Japan). Mitycam fluorescence (excitation/emission spectra: 498/515) was recorded at baseline (5 min) and during electric stimulation (2 Hz, 6 min) in the presence of 10 nM isoproterenol (Sigma-Aldrich).
Mitochondrial Ca2+ uptake was determined using the Ca2+ indicator Rhod2-AM (excitation/emission spectra: 575/675. Invitrogen, Waltham, MA, United States) (Maxwell et al., 2018). Briefly, cells were incubated with Rhod2-AM in Tyrode medium for 30 min at room temperature, washed, and de-esterified for another 30 min. Subsequently, NRVMs were permeabilized with 0.005% saponin for 30 s followed by the replacement for an internal medium without Ca2+. Cells were mounted in the Zeiss LSM 700 laser scanning confocal microscope (Carl Zeiss AG, Oberkochen, Germany), and the baseline Rhod2 fluorescence was recorded for 20 s. The medium was replaced by an internal medium with 2 µM free Ca2+, and the signal was recorded for 100 s.
For both cell types (hES-CMs and NRVMs), the images were processed and analyzed in the open-source software Fiji (Schindelin et al., 2012). For imaging analysis, the background signal was subtracted. Mitycam fluorescence signal was expressed as 1—(F/F0) and Rhod2 fluorescence signal as F/F0.
Cardiomyocytes were lysed in RIPA buffer supplemented with protease inhibitors (Roche, Basile, Switzerland), phosphatase inhibitors (Sigma-Aldrich), sodium orthovanadate (Sigma-Aldrich), and phenylmethylsulphonyl fluoride (Roche). Subsequently, cells were centrifuged at 12,000 g for 10 min at 4°C. The supernatant was recovered, and the isolated protein was quantified using the BCA protein assay (Thermo Fisher Scientific). The protein extract was cooked at 95°C for 5 min in SDS-PAGE loading buffer (Thermo Fisher Scientific) and stored at −20°C.
Protein electrophoresis was carried out by standard methods. Briefly, SDS PAGE was loaded with 10 µg of protein extract. Proteins were separated by electrophoresis at 80 mV in running buffer and electro-transferred to a PVDF membrane at 0.45 A for 90 min in transference buffer. Primary antibodies were diluted in blocking buffer (TBS, 0.1% Tween, and 5% bovine serum albumin) in a 1:1,000 ratio and incubated overnight at 4°C. Membranes were washed in TBS-0.1% tween and incubated in blocking buffer with the secondary antibody (anti-mouse or anti-rabbit, Sigma-Aldrich) for 1 h in a 1:5,000 ratio. REVERT™ Total protein staining was used as loading control (LI-COR Lincoln, NE, United States). Western blot detection was carried out in a chemiluminescence detector by standard methods.
Cells were lysed with TRIzol® (Invitrogen, Carlsbad, CA, United States) following the manufacturer’s instructions. The mRNA was quantified using a NanoDrop 2000 machine (Thermo Fisher Scientific). The cDNA was synthesized with the Quantitect RT kit (Qiagen N.V., Hilden, Germany) following the manufacturer’s instructions. All primers were designed in Primer-Blast software (NCBI, Bethesda, MD, United States) and internally validated. qPCR was carried out using the SYBR® Green Master Mix (Bio-Rad, Hercules, CA, United States) in the thermocycler CFX384 Touch Real-Time PCR Detection System (Bio-Rad).
Cardiomyocytes were incubated with 20 nM tetramethylrhodamine ethyl ester (TMRE+, Invitrogen; excitation/emission spectra: 553/577) in Tyrode medium at 37°C for 30 min. Cardiomyocytes were mounted in the Olympus IX-71 inverted microscope DeltaVision Elite. Baseline fluorescence was recorded for ∼200 s and subsequently, exposed to 10 mM Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) for ∼1,000 s. The noise signal was subtracted. TMRE+ fluorescence was expressed as F—Fmin (relative fluorescence unit, RFU).
hES-CMs were seeded in Seahorse assay plates at a density of 0.25 x 105 cells/well. 24 h before the experiment, cardiomyocytes were stimulated with IGF-1 or IGF-1 + 10 µM ru360 (Merck, Darmstadt, Germany). Oxygen consumption rate was determined by the Seahorse XF Cell Mito Stress Test programmed in the extracellular metabolic flux analyzer Agilent Seahorse XF96 (Agilent Technologies, Santa Clara, CA, United States). 20 min before the assay, CDM3 medium was replaced by Seahorse assay medium XF (Agilent Technologies) supplemented with 10 mM glucose and 1 mM sodium pyruvate at 37°C without CO2 control. The standard stress protocol was performed (Hoes et al., 2018), and the oxygen consumption rate was normalized by protein content determined by BCA protein assay.
Cell viability was determined using the Real-Time-Glo™ MT Cell viability assay Kit (Promega, Madison, WI, United States) following the manufacturer’s instructions. Briefly, the NanoLuc® enzyme and the viability substrate were 1x diluted and equilibrated at 37°C in CDM3 medium or CDM3 medium +10 µM ru360 (experimental media). hES-CMs were seeded in a clear bottom 96 well plate. CDM3 medium was replaced by the corresponding experimental medium and the luminescence was recorded from 0 to 48 h in the Synergy H1 plate reader (BioTek, Winooski, VT, United States). The signal was corrected by the protein content in each well.
NRVMs were seeded in gelatin-coated coverslips in a maintenance medium and stimulated with IGF-1 for 48 h. Subsequently, NRVMs were fixed in PBS + 4% paraformaldehyde for 15 min. Afterward, cells were incubated with rhodamine-phalloidin (1:500, Thermo Fisher Scientific) and Hoescht (1:1,000, Thermo Fisher Scientific) for 1 h at room temperature. Coverslips were mounted in slides with DAKO mounting medium (Agilent Technologies). The images were acquired using the Zeiss LSM 700 laser scanning confocal microscope. The relative cell area was quantified using the software Fiji. 50 to 100 cells were analyzed per condition.
All the results were expressed as mean ± SE from at least three independent assays. For values with normal distribution and equal variances was used t-test or one-way ANOVA was followed by the Tukey post-hoc test for multiple comparisons. For comparing groups without normal distribution was used U Mann-Whitney test or the Kruskal–Wallis test followed by the Dunn post-hoc test for multiple comparisons. A p-value < 0.05 was considered for statistical differences. The software GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, United States) was used for data analysis and visualization.
To study mitochondrial Ca2+ handling under conditions of physiological hypertrophy, we studied Ca2+ uptake kinetics of NRVMs in response to IGF-1 stimulation. We first corroborated the activation of IGF-1 receptor-dependent signaling in these cells and observed a time-dependent increase in AKT phosphorylation (Supplementary Figure S1). IGF-1 also promoted a clear hypertrophic response in NRVMs (Supplementary Figure S2). To assess the effect of IGF-1 on mitochondrial Ca2+ uptake we made use of the Ca2+ indicator Rhod2, restricting the fluorescence signal into the mitochondria by a permeabilization step with saponin (Figure 1A). IGF-1-treated NRVMs significantly improved their Ca2+ uptake capacity after an extracellular pulse of 2 µM free Ca2+ (Figures 1B, C), observed as higher maximal fluorescence and area under the curve than the control condition (Figure 1D). As a control, we blocked mitochondrial Ca2+ uptake with the MCU inhibitor ruthenium red (RuRed), confirming the role of the MCUC on the observed effect (Figures 1C, D). The rise in mitochondrial Ca2+ concentration activates the pyruvate dehydrogenase (PDH) phosphatase, which dephosphorylates and activates the PDH complex (Denton, 2009). We found that IGF-1 stimulation significantly reduced the PDH phosphorylation by 0.55 ± 0.04-fold over the control condition (Figure 1E).
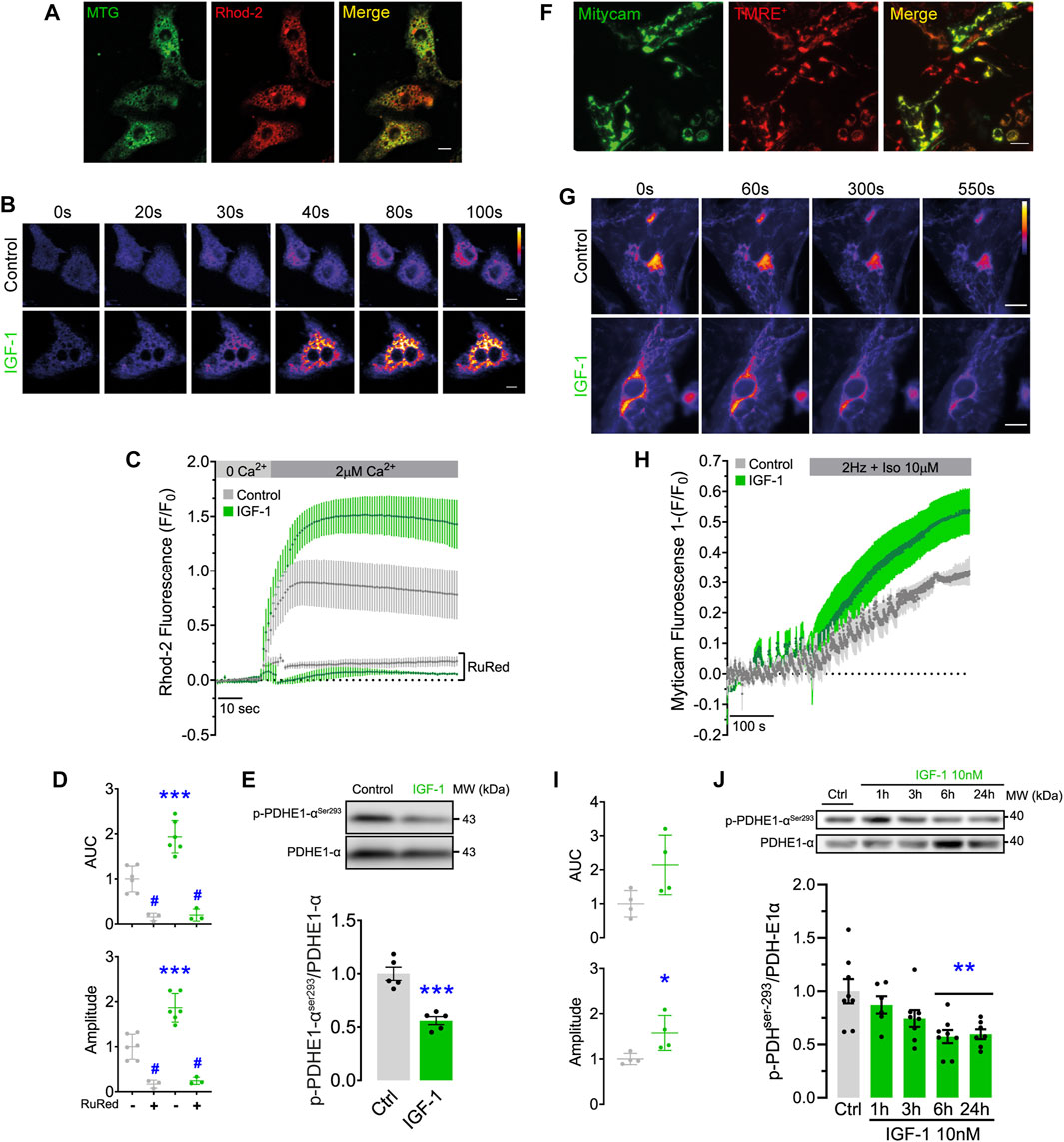
FIGURE 1. IGF-1 increases mitochondrial Ca2+ uptake and reduces the PDH phosphorylation in NRVMs and hES-CMs. (A) Rhod2 and Mitotracker Green fluorescence pattern. Scale bar: 20 µm. (B) Representative confocal images of Rhod2 fluorescence were recorded for 100 s in permeabilized NRVMs treated with IGF-1 for 24 h and exposed to a pulse of 2 μM free Ca2+ (n = 3–6). Scale bar: 20 µm. (C) Rhod2 fluorescence kinetic obtained from B, including ruthenium red (RuRed, 5 μM) treated conditions. (D) Area under the curve and maximal fluorescence (amplitude) registered in C for each condition. (E) Representative Western blot and quantification of phosphorylated PDH in serine 293 in NRVMs treated with IGF-1 for 24 h (n = 6). *p < 0.05 vs. control **p < 0.01 vs. control. ***p < 0.001 vs. control; #p < 0.05 vs. non-RuRed treated condition. (F) TMRE+ and Mitycam fluorescence pattern. Scale bar: 10 µm. (G) Representative images of Mitycam fluorescence were recorded for 550 s in hES-CMs treated with IGF-1 for 24 h and exposed to an electric field stimulation (2 Hz) and isoproterenol (Iso, 10 µM). (n = 3—6). Scale bar: 10 µm. (H) Mitycam fluorescence kinetic. (I) Area under the curve and maximum fluorescence (amplitude) determined from H for each condition (n = 4). (J) Representative Western blot and quantification of phosphorylated PDH in serine 293 between 1 and 24 h of IGF-1 stimulation (n = 7—8).
To generalize the results found in NRVMs, we replicated our findings in hES-CMs. Similar to NRVMs, IGF-1 induced AKT phosphorylation in these cells (Supplementary Figure S1). To measure the mitochondrial Ca2+ uptake, hES-CMs were infected with a lentiviral vector containing the genetically encoded Ca2+ indicator Mitycam, whose expression is restricted to mitochondria. As shown in Figure 1F, the Mitycam signal has an almost identical fluorescence pattern to TMRE+, a potentiometric dye that accumulates within the mitochondrial matrix. We promoted Ca2+ release from the sarcoplasmic reticulum by using electric field stimulation in presence of β-adrenergic agonist isoproterenol (Iso). We observed a drop in Mitycam fluorescence during electric stimulation, which was more pronounced at the end of the protocol in the cells treated with IGF-1 (Figures 1G–I), suggesting a greater capacity for Ca2+ uptake. As well as in NRVMs, IGF-1 stimulation significantly reduced the PDH phosphorylation (∼43% y ∼41% at 6 and 24 h of incubation, respectively) compared to non-treated cells (Figure 1J), indicative of a higher PDH activity.
Overall, these data suggest that IGF-1 enhances mitochondrial Ca2+ handling and pyruvate metabolism in both cellular models, indicating a conserved response in both species.
The limiting pathway of mitochondrial Ca2+ influx is the MCUC. Its function is mainly regulated by changes in the expression of the MCUC subunits affecting its Ca2+ sensitivity, gating, or the stability of the complex (Figure 2A) (Feno et al., 2021). In hES-CMs, IGF-1 did not change either MCU mRNA (Figure 2B) or protein levels (Figure 2C) at any of the evaluated time points. However, compatible with a higher MCU conductance, we observed a significant reduction of MICU1 mRNA levels at 24 and 48 h (0.59 ± 0.05 and 0.61 ± 0.04-fold over control conditions, respectively) and increased mRNA levels of MCUR1 at 6 h (1.41 ± 0.1-fold over control condition) (Figure 2D).
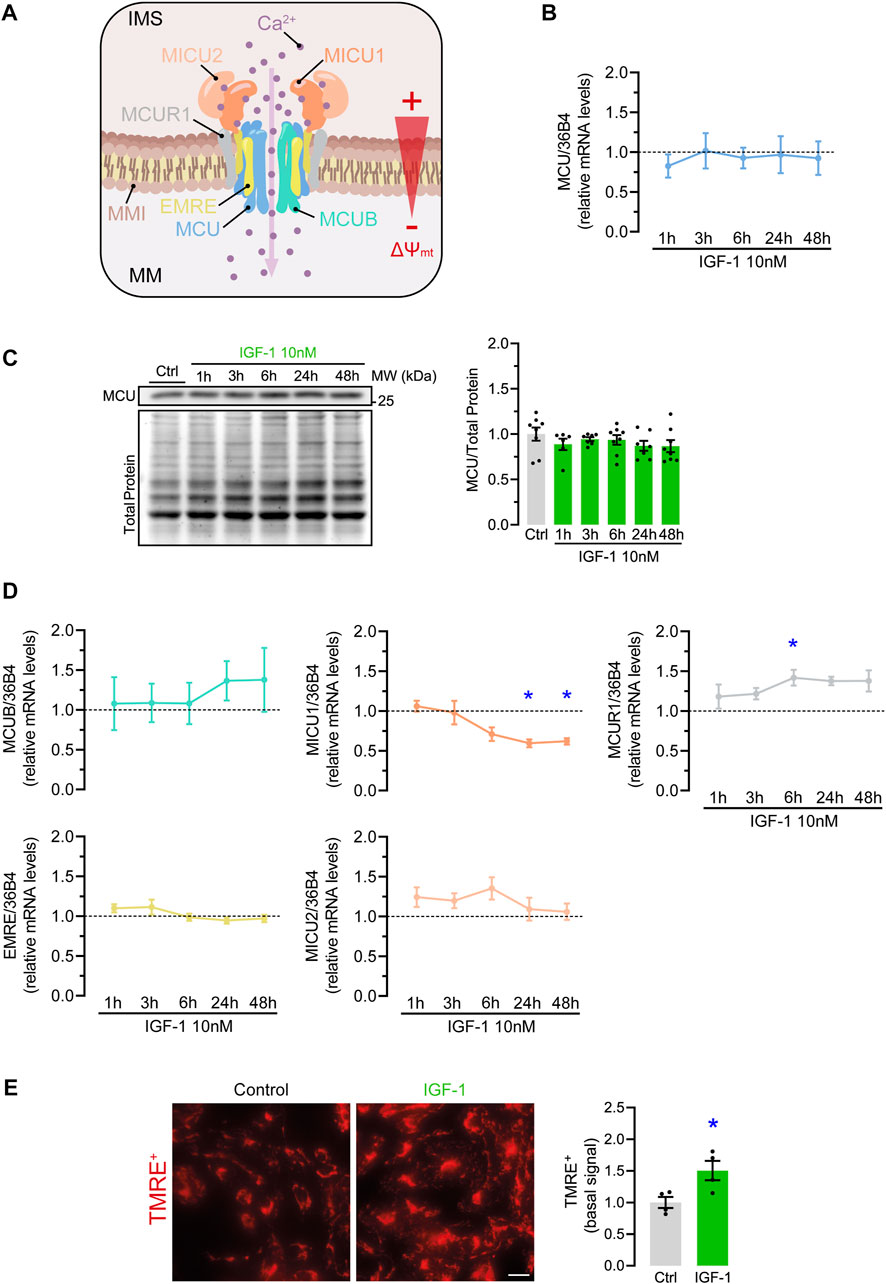
FIGURE 2. Effect of IGF-1 on MCU complex elements expression and ∆Ψmt in hES-CMs. (A) Scheme of MCU complex components (MCU, MCUB, SMDT1, MICU1, MICU2, MCUR1) inserted in the inner mitochondrial membrane (IMM) and a representation of the ∆Ψmt between the intermembrane space (IMS) and the mitochondrial matrix (MM); (B) Relative mRNA and (C) protein levels of MCU in hES-CMs stimulated with IGF-1 for 1–48 h (n = 6—8); (D) Relative mRNA levels of MCU complex regulatory subunits (MCUB, SMDT1, MICU1, MICU2, MCUR1) in hES-CMs stimulated with IGF-1 for 1–48 h (n = 6—8). The dotted line represents the mRNA levels of the untreated (control) condition; (E) Relative levels of basal TMRE+ in cells treated with IGF-1 for 24 h *p < 0.05 vs. control. Scale bar: 10 µm.
Next to MCUC composition, Ca2+ transport also depends on an electric gradient between the inter-membrane space and the mitochondrial matrix, known as mitochondrial membrane potential (∆Ψmt). This is the main driving force for Ca2+ influx through the MCUC (Figure 2A) (Nicholls and Crompton, 1980). Therefore, an increment of ∆Ψmt will promote a higher MCU-mediated Ca2+ transport. To test this hypothesis, we determined the basal ∆Ψmt in TMRE+-loaded cardiomyocytes and found that, indeed, IGF-1 increased the basal ∆Ψmt by 1.5 ± 0.09-fold over the control condition (Figure 2E). Altogether, these results indicate that IGF-1 enhances mitochondrial Ca2+ handling by a mechanism that combines changes in the MCUC and the driving force for mitochondrial Ca2+ influx.
The elevation of mitochondrial Ca2+ concentrations promotes the activity of PDH and the TCA cycle, indirectly increasing oxygen consumption rate (OCR) and ATP synthesis (Williams et al., 2015). Based on this premise, we investigated the effect of IGF-1 on the OCR of hES-CMs, using an extracellular metabolic flux analyzer. As expected, IGF-1 increased basal (1.37 ± 0.10-fold), ATP-linked (1.45 ± 0.14-fold), and maximal respiration (1.29 ± 0.09-fold), including the proton leak (1.71 ± 0.12-fold) (Figures 3A, B). To confirm whether the mitochondrial Ca2+ could play a role in the IGF-1-mediated mitochondrial respiration, we evaluated the OCR in hES-CMs co-incubated with the specific MCU inhibitor ru360. Remarkably, ru360 completely prevented the changes in OCR induced by IGF-1, showing similar values as the control condition (Figures 3A–C), except for the proton leak, which remained high (1.48 ± 0.15-fold over the control condition). It is important to note that in all evaluated time points, ru360 did not reduce cell viability (Supplementary Figure S3). Altogether, these results suggest that IGF-1 increases mitochondrial respiration by a mechanism dependent on MCU-mediated Ca2+ uptake.
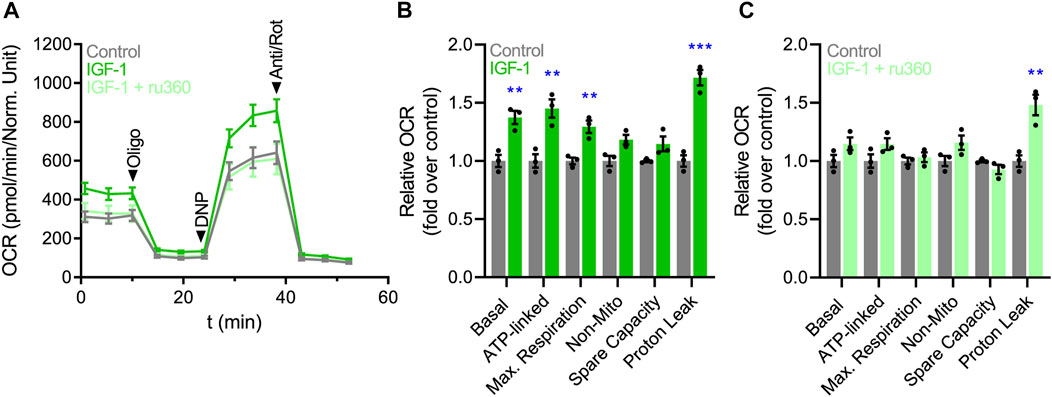
FIGURE 3. IGF-1 increases mitochondrial respiration by a mechanism dependent on mitochondrial Ca2+ uptake. (A) Representative plot of OCR over time. (B) Quantification of the relative changes in OCR (basal respiration, ATP-linked respiration, maximal respiration, non-mitochondrial respiration, spare capacity, and proton leak) in hES-CMs stimulated with IGF-1 (10 nM for 24 h) or vehicle. (C) Quantification of the relative changes in OCR in hES-CMs stimulated with ru360 (10 µM) or vehicle respiration (n = 3).
Our study showed that IGF-1 boosts mitochondrial Ca2+ uptake and stimulates Ca2+-sensitive mitochondrial dehydrogenases, which enhances oxidative metabolism during cardiomyocyte growth in both NRVMs and hES-CMs. Moreover, we propose that IGF-1 regulates mitochondrial Ca2+ uptake through modifications in MCUC composition and increasing ∆Ψmt. This study suggest that physiological cardiomyocyte growth is strictly linked to mitochondrial function and energy homeostasis and highlight Ca2+ as the main signal to allow the energetic adaptation.
Ca2+ allows the real-time adaptation of mitochondrial function to meet energy demands during increased cardiomyocyte workload (Wescott et al., 2019). This occurs even under pathological cardiac remodeling where MCU upregulation sustains mitochondrial and cardiac function and its downregulation exacerbates the pathological phenotype (Wang et al., 2022). This is possible because part of the released Ca2+ during contraction is absorbed by mitochondria, directly stimulating the activity of the isocitrate and α-ketoglutarate dehydrogenases, and indirectly, the PDH activity, which together will increase ATP synthesis (Glancy and Balaban, 2012). We postulated that IGF-1 promotes mitochondrial Ca2+ uptake to sustain the energetic requirements of a growing cardiomyocyte. In line with this hypothesis, we observed that the activation of the IGF-1 signaling pathway increased mitochondrial Ca2+ uptake in both cell types (Figures 1C, H). Moreover, IGF-1 reduced phospho-PDH, which indicates higher enzymatic activity and pyruvate oxidation and, indirectly, higher mitochondrial Ca2+ concentration (Figures 1E,J).
Our findings add new evidence on the subacute effect of IGF-1 on mitochondrial Ca2+ handling, a controversial area of research. For instance, Gutiérrez et al. concluded that the stimulation with 100 nM IGF-1 do not affect mitochondrial Ca2+ uptake in NRVMs (Gutiérrez et al., 2014). However, this could be explained because of the usage of supraphysiological IGF-1 concentrations or the utilization of a Ca2+ indicator with a substantial noise signal. By contrast, Troncoso et al. showed that 10 nM IGF-1 enhanced mitochondrial function in a Ca2+-dependent manner during nutritional starvation in NRVMs, which is in line with our findings (Troncoso et al., 2012).
Concerning PDH, its phosphorylation status has often been used as an accepted readout of mitochondrial Ca2+ content, being employed in several studies to check the effect of the genetic manipulation of MCU components on mitochondrial Ca2+ content (Pan et al., 2013; Luongo et al., 2015; Kwong et al., 2018). Here, we show that IGF-1 regulates PDH activity, possibly by a Ca2+-dependent mechanism. However, this data should be taken with caution because PDH is also regulated by the pyruvate dehydrogenase kinase that is insensitive to Ca2+ (Matsuhashi et al., 2015).
Mitochondrial Ca2+ uptake is a highly regulated process that is governed by factors related to the MCUC and the electrochemical gradient for Ca2+ transport (Williams et al., 2013). A change in any of the MCUC components could affect the conductance or stability of the complex (Vais et al., 2015; Payne et al., 2017; Lambert et al., 2019; Van Keuren et al., 2020). In addition, the ∆Ψmt mainly generated by the electron transport chain represents the main driving force for MCU-mediated Ca2+ transport (Nicholls and Crompton, 1980). Interestingly, we found that IGF-1 modified both factors, the MCUC components expression and ∆Ψmt (Figure 2).
There are several combinations of the MCUC composition that are compatible with enhanced MCU activity, which could explain the higher mitochondrial Ca2+ uptake. As illustrated in other studies, MICU1 loss-of-function increased the basal mitochondrial Ca2+ content (Rao et al., 2020; Kohlschmidt et al., 2021; Singh et al., 2022) and PDH activity (Rao et al., 2020). This could be explained by the fact that MICU1 acts as the gatekeeper of the MCU channel, limiting Ca2+ uptake at low cytosolic Ca2+ concentrations (<∼1, 3 μM) (Payne et al., 2017). Therefore, a reduction in MICU1 could lead to higher resting mitochondrial Ca2+ concentrations, increasing PDH activity. Regarding MICUR1 function, several reports indicated that the inclusion of MCUR1 in the MCUC enhances the Ca2+ transport, although there is no consensus on the mechanism (Mallilankaraman et al., 2012; Tomar et al., 2016). Paupe et al. proposed that MCUR1 acted as a scaffold factor for cytochrome C oxidase, which indirectly promotes mitochondrial Ca2+ transport by increasing the ∆Ψmt (Paupe et al., 2015). Interestingly, this theory agrees with the higher ∆Ψmt found in our experiments.
Wescott et al. showed that mitochondrial Ca2+ increments are necessary and sufficient for maintaining the ∆Ψmt and ATP levels of adult cardiomyocytes during situations of high energetic demand (Wescott et al., 2019). Based on this mechanism, we proposed that mitochondrial Ca2+ influx promoted by IGF-1 will potentiate metabolism and respiration to sustain the energy demands imposed by the hypertrophic stimulus. As hypothesized, we found that IGF-1 significantly enhanced ATP-linked respiration in a Ca2+-dependent manner (Figure 3). Other studies also found that IGF-1 increases OCR in NRVMs (Troncoso et al., 2012; del Campo et al., 2014), especially when mitochondria are stimulated with pyruvate/malate (Tigchelaar et al., 2016). Interestingly, the hormone triiodothyronine, another well-described physiological hypertrophic inductor, also increases mitochondrial Ca2+ uptake, OCR, and ATP synthesis (Tawfik et al., 2022). In addition, we also found that IGF-1 promoted a significant increase in the proton leak relative to control cells, which did not respond to ru360 incubation. During conditions of high energy demand, the elevation of proton leak is a protective response to reduce the production of excessive reactive oxygen species (Cadenas, 2018), which could be the case of cells under hypertrophic stimulation. However, we should take these values with caution because absolute proton leak values were low, and they did not represent a significant percentage of the maximal respiration for each condition (control: 14.7 ± 3.74%; IGF-1: 13.6 ± 3.44%; IGF-1 + ru360: 16.3 ± 4.83%).
In summary, this study shows that IGF-1 boosts mitochondrial Ca2+ handling and improves the oxidative metabolism of cardiomyocytes during physiological growth. In addition, we identified changes in the MCUC components and ∆Ψmt as possible regulatory mechanisms of the IGF-1-mediated mitochondrial Ca2+ uptake. These findings shed new light on the regulatory factors of cardiomyocyte remodeling, highlighting mitochondrial Ca2+ handling and MCUC as potential therapeutic targets for the prevention and treatment of pathologies related to energetic imbalance, such as pathological cardiac hypertrophy and heart failure.
The major limitation of this study is the absence of a causal relationship between the changes observed in MCUC stoichiometry and the increments in mitochondrial Ca2+ uptake promoted by IGF-1. Further studies should focus on determining the role of MICU1 and MCUR1 on IGF-1-mediated mitochondrial Ca2+ uptake, using genetic approaches of gain and loss of function of these proteins. An additional limitation is the absence of direct estimations of mitochondrial ATP production, which could demonstrate that IGF-1 is responsible to maintain the energy balance during cardiomyocyte hypertrophy. Despite these limitations, our study provides deeper insight into the regulation of hypertrophy in two different cardiomyocyte models and highlights the role of mitochondrial function in the process.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Universidad de Chile, Facultad Ciencias Quimicas y Farmaceuticas, Bioethics Committee.
Conceptualization, PS-A, SL, MC, and BW; supervision, SL, BW, and MC; performed the experiments, PS-A, CL-C, IN-S, JZ, and NB; formal analysis, PS-A, CL-C, and IN-S; contributed protocols/reagents/materials/analysis tools, SL, BW, NB, MH, and PvdM; writing-original draft preparation, PS-A; writing-review and editing, PS-A, SL, CP, MC, BW, PvdM, MH, and NB; visualization, PS-A; funding acquisition, SL and BW.
BW is supported by The Netherlands Organization for Scientific Research (NWO VENI, grant 016.176.147), the Netherlands Heart Foundation Senior Clinical Scientist Grant (2019T064), and CVON DOUBLE DOSE (grant 2020-8005). SL is supported by FONDAP 15130011 and FONDECYT 1200490.
The authors thank Adam Cohen for sharing and realizing the genetically encoded Ca2+ indicator Mitycam. Also, thanks to Susanne Feringa, Renate Jagersma, Christiane Baierl, Fidel Albornoz, and Gindra Latorre for their help with hES-CMs and NRVMs culture.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SM declared a past co-authorship with the authors PSA, NB, PVDM and DW to the handling editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1106662/full#supplementary-material
Bomer N., Grote Beverborg N., Hoes M. F., Streng K. W., Vermeer M., Dokter M. M., et al. (2020). Selenium and outcome in heart failure. Eur. J. Heart Fail 22, 1415–1423. doi:10.1002/ejhf.1644
Brown D. A., Perry J. B., Allen M. E., Sabbah H. N., Stauffer B. L., Shaikh S. R., et al. (2017). Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 14, 238–250. doi:10.1038/nrcardio.2016.203
Cadenas S. (2018). Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 1859, 940–950. doi:10.1016/j.bbabio.2018.05.019
Crilley J. G., Boehm E. A., Blair E., Rajagopalan B., Blamire A. M., Styles P., et al. (2003). Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 41, 1776–1782. doi:10.1016/s0735-1097(02)03009-7
De La Fuente S., Fernandez-Sanz C., Vail C., Agra E. J., Holmstrom K., Sun J., et al. (2016). Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. J. Biol. Chem. 291, 23343–23362. doi:10.1152/ajpendo.00146.2013
del Campo A., Parra V., Vásquez-Trincado C., Gutiérrez T., Morales P. E., López-Crisosto C., et al. (2014). Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am. J. Physiology Endocrinol. Metabolism 306, E1–E13. doi:10.1152/ajpendo.00146
Denton R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochimica Biophysica Acta (BBA) - Bioenergetics 1787, 1309–1316. doi:10.1016/j.bbabio.2009.01.005
Fan M., Zhang J., Tsai C. W., Orlando B. J., Rodriguez M., Xu Y., et al. (2020). Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 582, 129–133. doi:10.1038/s41586-020-2309-6
Feno S., Rizzuto R., Raffaello A., Vecellio Reane D. (2021). The molecular complexity of the Mitochondrial Calcium Uniporter. Cell Calcium 93, 102322. doi:10.1016/j.ceca.2020.102322
Galvez A., Morales M. P., Eltit J. M., Ocaranza P., Carrasco L., Campos X., et al. (2001). A rapid and strong apoptotic process is triggered by hyperosmotic stress in cultured rat cardiac myocytes. Cell Tissue Res. 304, 279–285. doi:10.1007/s004410100358
Glancy B., Balaban R. S. (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. doi:10.1021/bi2018909
Gorski P. A., Ceholski D. K., Hajjar R. J. (2015). Altered myocardial calcium cycling and energetics in heart failure--a rational approach for disease treatment. Cell Metab. 21, 183–194. doi:10.1016/j.cmet.2015.01.005
Gutiérrez T., Parra V., Troncoso R., Pennanen C., Contreras-Ferrat A., Vasquez-Trincado C., et al. (2014). Alteration in mitochondrial Ca(2+) uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun. Signal. CCS 12, 68. doi:10.1186/s12964-014-0068-4
Hill J. A., Olson E. N. (2008). Cardiac plasticity. N. Engl. J. Med. 358, 1370–1380. doi:10.1056/NEJMra072139
Hoes M. F., Grote Beverborg N., Kijlstra J. D., Kuipers J., Swinkels D. W., Giepmans B. N. G., et al. (2018). Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 20, 910–919. doi:10.1002/ejhf.1154
Kohlschmidt N., Elbracht M., Czech A., Hausler M., Phan V., Topf A., et al. (2021). Molecular pathophysiology of human MICU1 deficiency. Neuropathol. Appl. Neurobiol. 47, 840–855. doi:10.1111/nan.12694
Kwong J. Q., Huo J., Bround M. J., Boyer J. G., Schwanekamp J. A., Ghazal N., et al. (2018). The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI insight 3, e121689. doi:10.1172/jci.insight.121689
Lambert J. P., Luongo T. S., Tomar D., Jadiya P., Gao E., Zhang X., et al. (2019). MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 140, 1720–1733. doi:10.1161/CIRCULATIONAHA.118.037968
Liu T., Yang N., Sidor A., O'Rourke B. (2021). MCU overexpression rescues inotropy and reverses heart failure by reducing SR Ca2+ leak. Circulation Res. 128, 1191–1204. doi:10.1161/CIRCRESAHA.120.318562
Ljubojevic-Holzer S., Herren A. W., Djalinac N., Voglhuber J., Morotti S., Holzer M., et al. (2020). CaMKIIδC drives early adaptive Ca2+ change and late eccentric cardiac hypertrophy. Circ. Res. 127, 1159–1178. doi:10.1161/CIRCRESAHA.120.316947
Luongo T. S., Lambert J. P., Yuan A., Zhang X., Gross P., Song J., et al. (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12, 23–34. doi:10.1016/j.celrep.2015.06.017
Mallilankaraman K., Cárdenas C., Doonan P. J., Chandramoorthy H. C., Irrinki K. M., Golenár T., et al. (2012). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343. doi:10.1038/ncb2622
Matsuhashi T., Hishiki T., Zhou H., Ono T., Kaneda R., Iso T., et al. (2015). Activation of pyruvate dehydrogenase by dichloroacetate has the potential to induce epigenetic remodeling in the heart. J. Mol. Cell. Cardiol. 82, 116–124. doi:10.1016/j.yjmcc.2015.02.021
Maxwell J. T., Tsai C.-H., Mohiuddin T. A., Kwong J. Q. (2018). Analyses of mitochondrial calcium influx in isolated mitochondria and cultured cells. J. Vis. Exp. JoVE, 57225. doi:10.3791/57225
Nakamura M., Sadoshima J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407. doi:10.1038/s41569-018-0007-y
Nicholls D. G., Crompton M. (1980). Mitochondrial calcium transport. FEBS Lett. 111, 261–268. doi:10.1016/0014-5793(80)80806-4
Nijholt K. T., Sanchez-Aguilera P. I., Voorrips S. N., de Boer R. A., Westenbrink B. D. (2022). Exercise: A molecular tool to boost muscle growth and mitochondrial performance in heart failure? Eur. J. Heart Fail 24, 287–298. doi:10.1002/ejhf.2407
Pan X., Liu J., Nguyen T., Liu C., Sun J., Teng Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. doi:10.1038/ncb2868
Paupe V., Prudent J., Dassa E. P., Rendon O. Z., Shoubridge E. A. (2015). CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 21, 109–116. doi:10.1016/j.cmet.2014.12.004
Payne R., Hoff H., Roskowski A., Foskett J. K. (2017). MICU2 restricts spatial crosstalk between InsP3R and MCU channels by regulating threshold and gain of MICU1-mediated inhibition and activation of MCU. Cell Rep. 21, 3141–3154. doi:10.1016/j.celrep.2017.11.064
Rao G., Dwivedi S. K. D., Zhang Y., Dey A., Shameer K., Karthik R., et al. (2020). MicroRNA-195 controls MICU1 expression and tumor growth in ovarian cancer. EMBO Rep. 21, e48483. doi:10.15252/embr.201948483
Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi:10.1038/nmeth.2019
Singh R., Bartok A., Paillard M., Tyburski A., Elliott M., Hajnoczky G. (2022). Uncontrolled mitochondrial calcium uptake underlies the pathogenesis of neurodegeneration in MICU1-deficient mice and patients. Sci. Adv. 8, eabj4716. doi:10.1126/sciadv.abj4716
Suarez J., Cividini F., Scott B. T., Lehmann K., Diaz-Juarez J., Diemer T., et al. (2018). Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 293, 8182–8195. doi:10.1074/jbc.RA118.002066
Tawfik I., Gottschalk B., Jarc A., Bresilla D., Rost R., Obermayer-Pietsch B., et al. (2022). T3-induced enhancement of mitochondrial Ca(2+) uptake as a boost for mitochondrial metabolism. Free Radic. Biol. Med. 181, 197–208. doi:10.1016/j.freeradbiomed.2022.01.024
Tigchelaar W., de Jong A. M., Bloks V. W., van Gilst W. H., de Boer R. A., Silljé H. H. W. (2016). Hypertrophy induced KIF5B controls mitochondrial localization and function in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 97, 70–81. doi:10.1016/j.yjmcc.2016.04.005
Tomar D., Dong Z., Shanmughapriya S., Koch D. A., Thomas T., Hoffman N. E., et al. (2016). MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 15, 1673–1685. doi:10.1016/j.celrep.2016.04.050
Troncoso R., Ibarra C., Vicencio J. M., Jaimovich E., Lavandero S. (2014). New insights into IGF-1 signaling in the heart. Trends Endocrinol. metabolism TEM 25, 128–137. doi:10.1016/j.tem.2013.12.002
Troncoso R., Vicencio J. M., Parra V., Nemchenko A., Kawashima Y., Del Campo A., et al. (2012). Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 93, 320–329. doi:10.1093/cvr/cvr321
Vais H., Tanis J. E., Müller M., Payne R., Mallilankaraman K., Foskett J. K. (2015). MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metab. 22, 533–535. doi:10.1016/j.cmet.2015.09.015
Van Keuren A. M., Tsai C.-W., Balderas E., Rodriguez M. X., Chaudhuri D., Tsai M.-F. (2020). Mechanisms of EMRE-dependent MCU opening in the mitochondrial calcium uniporter complex. Cell Rep. 33, 108486. doi:10.1016/j.celrep.2020.108486
Wang C., Jacewicz A., Delgado B. D., Baradaran R., Long S. B. (2020). Structures reveal gatekeeping of the mitochondrial Ca(2+) uniporter by MICU1-MICU2. Elife 9, e59991. doi:10.7554/eLife.59991
Wang P., Shangcheng X., Jiqian X., Yanguo X., Yan L., Huiliang Z., et al. (2022). Elevated MCU expression by CaMKIIδB limits pathological cardiac remodeling. Circulation 145, 1067–1083. doi:10.1161/CIRCULATIONAHA.121.055841
Wescott A. P., Kao J. P. Y., Lederer W. J., Boyman L. (2019). Voltage-energized calcium-sensitive ATP production by mitochondria. Nat. Metab. 1, 975–984. doi:10.1038/s42255-019-0126-8
Williams G. S. B., Boyman L., Chikando A. C., Khairallah R. J., Lederer W. J. (2013). Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U. S. A. 110, 10479–10486. doi:10.1073/pnas.1300410110
Williams G. S. B., Boyman L., Lederer W. J. (2015). Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 78, 35–45. doi:10.1016/j.yjmcc.2014.10.019
Zaglia T., Ceriotti P., Campo A., Borile G., Armani A., Carullo P., Mongillo M. (2017). Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. U. S. A. 114, E9006–E9015. doi:10.1073/pnas.1708772114
Keywords: insulin-like growth factor 1 (IGF-1), MCU complex, mitochondrial calcium handling, physiological cardiac hypertrophy, neonatal rat ventricular myocytes (NRVMs), human embryonic stem cell derived-cardiomyocytes (hES-CMs)
Citation: Sánchez-Aguilera P, López-Crisosto C, Norambuena-Soto I, Penannen C, Zhu J, Bomer N, Hoes MF, Van Der Meer P, Chiong M, Westenbrink BD and Lavandero S (2023) IGF-1 boosts mitochondrial function by a Ca2+ uptake-dependent mechanism in cultured human and rat cardiomyocytes. Front. Physiol. 14:1106662. doi: 10.3389/fphys.2023.1106662
Received: 24 November 2022; Accepted: 23 January 2023;
Published: 08 February 2023.
Edited by:
Wenjuan Xing, Air Force Medical University, ChinaReviewed by:
Youyou Li, China Agricultural University, ChinaCopyright © 2023 Sánchez-Aguilera, López-Crisosto, Norambuena-Soto, Penannen, Zhu, Bomer, Hoes, Van Der Meer, Chiong, Westenbrink and Lavandero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. Daan Westenbrink, Yi5kLndlc3RlbmJyaW5rQHVtY2cubmw=; Sergio Lavandero, c2xhdmFuZGVyQHVjaGlsZS5jbA==
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.