 Olivia R. Carroll
Olivia R. Carroll Amber L. Pillar
Amber L. Pillar Alexandra C. Brown
Alexandra C. Brown Min Feng
Min Feng Hui Chen
Hui Chen Chantal Donovan
Chantal Donovan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 16 March 2023
Sec. Respiratory Physiology and Pathophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1099719
This article is part of the Research Topic Women in Respiratory Physiology and Pathophysiology: 2022 View all 5 articles
Recent advances in mouse models of experimental asthma coupled with vast improvements in systems that assess respiratory physiology have considerably increased the accuracy and human relevance of the outputs from these studies. In fact, these models have become important pre-clinical testing platforms with proven value and their capacity to be rapidly adapted to interrogate emerging clinical concepts, including the recent discovery of different asthma phenotypes and endotypes, has accelerated the discovery of disease-causing mechanisms and increased our understanding of asthma pathogenesis and the associated effects on lung physiology. In this review, we discuss key distinctions in respiratory physiology between asthma and severe asthma, including the magnitude of airway hyperresponsiveness and recently discovered disease drivers that underpin this phenomenon such as structural changes, airway remodeling, airway smooth muscle hypertrophy, altered airway smooth muscle calcium signaling, and inflammation. We also explore state-of-the-art mouse lung function measurement techniques that accurately recapitulate the human scenario as well as recent advances in precision cut lung slices and cell culture systems. Furthermore, we consider how these techniques have been applied to recently developed mouse models of asthma, severe asthma, and asthma-chronic obstructive pulmonary disease overlap, to examine the effects of clinically relevant exposures (including ovalbumin, house dust mite antigen in the absence or presence of cigarette smoke, cockroach allergen, pollen, and respiratory microbes) and to increase our understanding of lung physiology in these diseases and identify new therapeutic targets. Lastly, we focus on recent studies that examine the effects of diet on asthma outcomes, including high fat diet and asthma, low iron diet during pregnancy and predisposition to asthma development in offspring, and environmental exposures on asthma outcomes. We conclude our review with a discussion of new clinical concepts in asthma and severe asthma that warrant investigation and how we could utilize mouse models and advanced lung physiology measurement systems to identify factors and mechanisms with potential for therapeutic targeting.
Asthma affects approximately 10% of the population and has higher prevalence in developed countries (To et al., 2012). The World Health Organization (WHO) reports that 455,000 deaths were attributed to asthma in 2019, and that asthma is the most common chronic disease affecting children (WHO, 2022). The global prevalence of asthma varies between sources, including the Global burden of disease, Centers for Disease Control and Prevention, the Global Initiative for Asthma, and European Respiratory Society/American Thoracic Society reports, at 224–309 million people, 7.8%, 1%–18%, and 5%–10%, respectively (Diseases and Injuries, 2020; Global Initiative for Asthma, 2021; Louis et al., 2022). Asthma in adulthood is both under- and over-diagnosed, which may be attributed to multiple definitions of the disease and suboptimal adherence to diagnostic guidelines (Aaron et al., 2018; Akinbami et al., 2020; Louis et al., 2022). Globally, around 3%–10% of asthma is classified as severe, which predominantly affects females (Busse et al., 2000; The ENFUMOSA cross, 2003; Larsson et al., 2018). A cross-sectional study reported that within a randomly selected cohort, 1.1% had severe asthma and of the asthmatic cohort, 9.5% had severe disease (Ronnebjerg et al., 2021).
The pathophysiology of asthma is underpinned by chronic airway inflammation. Genetic, epigenetic, environmental risk factors, and a combination of innate and adaptive immune cells (including macrophages, neutrophils, innate lymphoid cells, dendritic cells, helper T cells [TH]1, TH2, TH17 and TH22 and follicular helper T cells [TFH]) are linked with asthma pathogenesis and severity. Severe asthma is heterogeneous and can be broadly characterized by different mechanisms (endotypes; type-2 [T2] high, T2 low, or non-T2) and various clinical presentations (phenotypes) (Kuruvilla et al., 2019). These mechanisms and factors contribute to changes in histopathology, including increased airway collagen deposition, mucus cell hypersecretion metaplasia, airway smooth muscle (ASM) hypertrophy/hyperplasia, reduced lung function, and airway hyperresponsiveness (AHR), and ultimately cumulate into symptoms of chest tightness, wheezing and/or shortness of breath.
The T2 high endotype of asthma (commonly associated with the allergic phenotype) is comprised of two phases: the early (sensitization) and late (effector phase or challenge) phases. In the early phase, alarmins are released by epithelial cells in response to allergens and can induce cytokine production in type-2 innate lymphoid cells. Dendritic cells uptake allergens, migrate to lymph nodes, and present antigenic components to naïve T cells, which induces TH2 and TFH cell differentiation. Historically, TH2 cells have been recognized for their role in T2 cytokine production (interleukin [IL]-4, −5 and −13) in airway inflammation, airway eosinophil infiltration, immunoglobulin E class switching and AHR (Pope et al., 2001; Agrawal and Shao, 2010; Kudo et al., 2013; Doran et al., 2017). TFH cells are also implicated in mediating the early phase of allergic asthma. However, unlike TH2 cells, TFH cells predominantly produce IL-4 and IL-21 (Coquet et al., 2015; Kobayashi et al., 2017). TFH cells also produce IL-17 and have been shown to induce IgE class switching in severe asthma (Zaretsky et al., 2017). TH2 and TFH cells support IgE antibody production and B cell maturation. Deficiency in Bcl6, the master regulator in CD4+ T cells, reduces TFH numbers and IgE, but does not affect T2 cytokine responses or eosinophilic inflammation in the airways (Kobayashi et al., 2017), highlighting the function of these two T helper cell subsets in driving allergic asthma. This is reinforced by evidence of TH2 differentiation in response to allergens in the absence of IL-4 (Jankovic et al., 2000).
The late phase of allergic asthma is an established T2 response, characterized by eosinophil hematopoiesis, chemoattraction and activation, induced by granulocyte-macrophage colony-stimulating factor, IL-3 and -5. AHR presents at this stage, alongside features of epithelial damage, tissue remodeling, and chronic airway inflammation. Epithelial to mesenchymal transition presents after recurring exacerbations, which results in airways remodeling (Kudo et al., 2013). This, in conjunction with airway narrowing and tissue inflammation, contributes to symptomatic dyspnea, wheezing or whistling upon exhalation, and chest tightness (James and Wenzel, 2007; Wenzel, 2016; Quirt et al., 2018). Tissue resident mast cell numbers increase and express FcεRI, a high affinity IgE receptor. Basophil granulocytes are coated with IgE and IgE cross-linking occurs (He et al., 2013). Pro-inflammatory responses are then mediated by leukotrienes, prostaglandins, neuropeptides (substance P and neurokinin A), calcitonin gene related peptide, and histamine (Kim et al., 2018).
The T2 low endotype of asthma (TH1 and TH17 high) is associated with more severe disease, which may progress to become steroid-insensitive. Patients with severe asthma have lower atopy than patients with non-severe asthma, and IgE, blood eosinophil numbers and fractional exhaled nitric oxide (FeNO) do not differentiate severity (Moore et al., 2007; Guan et al., 2013). Notably, blood and airway neutrophil numbers have been shown to be increased in severe asthma (Crisford et al., 2021). Severe asthma presents with lower lung function and marked bronchodilator reversibility compared to mild-moderate disease. Symptoms typically present daily and disease duration is longer with sinusitis and pneumonia (Moore et al., 2007).
Respiratory infections increase susceptibility to severe asthma development, which can become steroid-insensitive (Wark et al., 2002; Hansbro et al., 2004; Cho et al., 2005). Airway epithelial cell bacterial colonization with Chlamydia pneumoniae, mycoplasma pneumoniae and staphylococcal endotoxins have been linked with asthma severity in both allergic and non-allergic endotypes. Furthermore, enterotoxins released by Staphylococcus aureus may act as sensitizing agents and superantigens that can activate T and B cells in human mucosa, that have been associated with severe asthma (Bachert and Zhang, 2012; Sintobin et al., 2019). Haemophilus influenzae is also implicated in the pathogenesis of severe disease (Simpson et al., 2007; Wood et al., 2010; Green et al., 2014). In addition to bacterial infections, respiratory viral infections such as respiratory syncytial virus (RSV), rhinovirus and influenza, have also been shown to contribute to asthma severity (Wark et al., 2002; Hansbro et al., 2008; Liao et al., 2016) and it is now well established that rhinovirus and RSV are risk factors for the development of atopic and non-atopic asthma, respectively (Mikhail and Grayson, 2019). Furthermore, rhinovirus, RSV, and influenza can exacerbate established asthma (Mikhail and Grayson, 2019), which can also become steroid-insensitive. Irrespective of the endotype or phenotype of asthma, the severity of asthma can be associated with increased potency of bronchoconstrictors but also an increase in the maximum contraction of ASM that can lead to complete airway closure in fatal asthma (Woolcock et al., 1998). Therefore, improving outcomes for patients requires an understanding of the mechanisms controlling airway reactivity and the influence of inflammatory mediators.
Changes in lung function, encompassing AHR and baseline airflow obstruction, in asthma and severe asthma are underpinned by many disease-driving factors. AHR is underpinned by hypersensitivity and hyperreactivity of smooth muscle to various stimuli [non-specific irritants, pharmacological agents (e.g., methacholine), and inflammatory mediators (e.g., histamine, leukotrienes from mast cells)], as well as impaired bronchodilator responses. AHR can occur on the background of inflammation and/or remodeling, however, this is not always the case and highlights that AHR can occur both dependently and independently of inflammation and remodeling in people with asthma and in mouse models of experimental disease, irrespective of disease severity.
Clinically, AHR is defined as a 20% decline in forced expiratory volume in 1 s (FEV1) after bronchoprovocation (Lotvall et al., 1998). The paradigm of AHR pathophysiology comprises tissue remodeling and structural changes to the airways and parenchyma. AHR can occur in non-asthmatics and thus may not always be indicative of asthma, however, AHR has been correlated with asthma severity (Deykin et al., 2007). ASM hypertrophy and hyperplasia, altered ASM calcium signaling, changes in epithelial cell composition, and nervous system activation, are also contributing factors to AHR. As both asthma and severe asthma are highly heterogenous, the contribution of one or more of these disease drivers to changes in lung function is patient-specific. However, persistent AHR can often coincides with airway remodeling, thus in severe asthma, and particularly in steroid-insensitive phenotypes, more severe AHR may arise from being unresponsive to current therapies.
Chronic inflammation drives smooth muscle tone, bronchospasm, edema, and mucus secretion. Airways mucus plugging also substantially contributes to asthma-related mortality rates (Worren et al., 2018). Other asthmatic phenotypes, such as obesity-associated asthma, can result in restrictive lung physiology as breathing at low lung tidal volumes increases AHR, where actin-myosin cross-linking in ASM may contribute to resistance from muscle stiffening (Fredberg et al., 1997). Neurological and psychological factors have also been linked to asthma presentation (Rosenblat et al., 2022).
It is also important to consider the contributions of different regions of the lung to AHR, including the small airways. In asthmatic patients, the small airways have greater inflammation (as measured by increased eosinophil numbers) than the larger airways (Hamid et al., 1997) and increased baseline resistance when compared to the small airways of non-asthmatics (Wagner et al., 1998). In vivo exposure of these small airways in asthmatic subjects to histamine resulted in greater increases in airway resistance and less reversibility with a non-selective β1 and β2-adrenoceptor agonist when compared to responses in non-asthmatic subjects (Wagner et al., 1998). Increased airway resistance can be associated with increased inflammation (Hamid et al., 1997) and airway wall remodeling (Burgel, 2011). In vitro studies have also shown that the small airways are more sensitive to contractile mediators (Mechiche et al., 2003), and less sensitive to β2-adrenoceptor agonists, than the large airways (Finney et al., 1985). However, whether there are differences in small airways AHR across the different endotypes and/or phenotypes of asthma, remains to be determined.
The lung structural changes of patients with asthma can include epithelial cell shedding, goblet cell hypertrophy, basement membrane thickening, increased ASM size (hypertrophy) and growth (hyperplasia), and increased vascularization (Hall and Agrawal, 2014; Szefler, 2015), that are further increased in severe asthma patients (Benayoun et al., 2003; Woodruff et al., 2004; Pepe et al., 2005). This relationship precipitated the dogma that AHR in asthma is caused by increased ASM hypertrophy and hyperplasia. However, some studies have shown that reducing the amount of ASM in the lungs of patients with asthma using bronchial thermoplasty does not improve overall lung function (Castro et al., 2010). Thus, more/larger ASM is not the sole factor contributing to AHR, which raises the question of what other factors contribute to increased AHR.
In asthma, ASM can be hyperproliferative, hypersecretory, and/or hypercontractile. ASM can be hypercontractile due to intrinsic changes in actin-myosin cross-linking, and can also release extracellular matrix factors (such as collagen types I and III, fibronectin, and elastin), cytokines that can promote inflammation (Belvisi et al., 1997; John et al., 1998; McKay et al., 2000; Hirst, 2003), and chemokines that can recruit immune cells (e.g., eotaxin). ASM heterogeneity may also be present between large and small airways, where ASM hypertrophy in large airways occurs in both non-fatal and fatal cases of asthma, and ASM hyperplasia occurs in both large and small airways in fatal asthma only (James et al., 2012). Importantly, both of these features are associated with increases in extracellular matrix deposition (James et al., 2012). However, whether these ASM changes are asthma endo/phenotype-specific is yet to be determined.
ASM tone and hypercontractility can be driven by calcium signaling pathways: the calcium oscillation pathway and the ‘calcium sensitivity’ pathway. These can be activated by contractile agonists such as methacholine, and these pathways are fundamentally similar in both human and mouse airways (Bai et al., 2007; Ressmeyer et al., 2010) (Figure 1). Calcium oscillations involve calcium release and reuptake between the sarcoplasmic reticulum (SR) and the cytosol. The release of calcium from the SR into the cytosol occurs through the activation of the inositol trisphosphate receptors (IP3R) and/or ryanodine receptors (RyR), whereas reuptake occurs via the sarco/endoplasmic reticulum Ca2+–ATPase (SERCA). Increases in cytosolic calcium result in the activation of calmodulin and myosin light chain kinase, resulting in myosin light chain phosphorylation and muscle contraction. In contrast, calcium sensitivity involves signaling through protein kinase C or rho-activated kinase, that can phosphorylate and inactivate myosin light chain phosphatase (MLCP). The resulting contractile apparatus becomes more sensitive to Ca2+ and contraction.
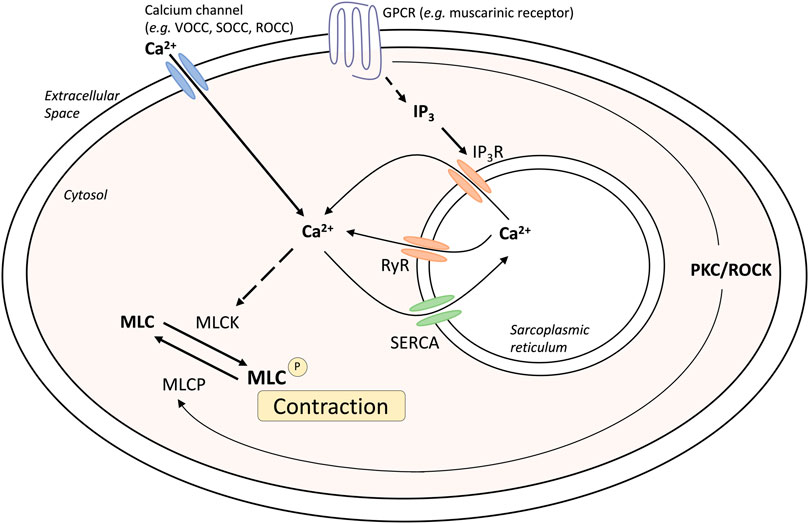
FIGURE 1. Mechanisms of airway smooth muscle contraction. Abbreviations: IP3, inositol triphosphate; GPCR, G-protein coupled receptor; MLC, myosin light chain; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; PKC, Protein kinase C; ROCC, receptor operated calcium channel; ROCK, Rho associated coiled-coil containing protein kinase; RyR, Ryanodine receptor; SERCA, Sarco/endoplasmic reticulum Ca(2+)-ATPase; SOCC, storage operated calcium channel; VOCC, voltage operated calcium channel.
Changes in intracellular Ca2+ levels can be detected using confocal or two-photon microscopy in ASM cells within precision cut lung slices (PCLS) loaded with fluorescent Ca2+ indicator dyes, such as Oregon green (Sanderson et al., 2008). This allows for assessment of contractile responses to agonists such as methacholine. Importantly, muscle contraction in response to methacholine occurs when calcium oscillations are abolished by treatment with caffeine and ryanodine to clamp intracellular calcium ([Ca2+]i) levels. Under these conditions, airway contraction is solely induced by increased Ca2+-sensitivity, whereby reduced MLCP activity maintains muscle tone (Bai and Sanderson, 2006).
Extracellular calcium can also contribute to ASM tone. Calcium can enter the cytosol through L-type voltage-operated Ca2+-channels in response to membrane K+ channel-dependent depolarization (Marthan et al., 1989), receptor-operated channels or ion channels, or store-operated calcium channels. Although calcium signaling has been found to be altered in disease states such as asthma and chronic obstructive pulmonary disease (COPD), this does not appear to be related to changes in receptor expression or levels, but rather increased sensitivity to the changes in calcium within the muscle.
It is now well established that inflammation and AHR can occur independently in people with asthma or severe asthma and in murine experimental asthma and severe asthma. However, in some cases, increased presence of inflammatory cells and inflammatory cytokines/chemokines, can contribute to increased AHR. Macrophages and neutrophils can increase AHR through their release of cytokines such as IL-13. Inflammatory cytokines and chemokines can also be released from structural cells, such as ASM, within the lungs. IL-13 can directly act on the IL-13R and IL-4R on ASM to increase inflammation by up-regulating pro-inflammatory cytokines, such as IL-1β and TNFα, and directly enhancing G-protein coupled receptor agonist-associated calcium signaling (Kibe et al., 2003; Tliba et al., 2003; Vargaftig and Singer, 2003). Furthermore, IL-13 can induce AHR through epithelial changes, including increased swelling, permeability, rigidity, and mucus production (Kirstein et al., 2010). IL-13-induced AHR has also been observed in response to non-calcium altering agonists such as potassium chloride, and it has been postulated that IL-13 can alter contractile machinery in ASM by enhancing sensitivity of myofilaments or rearranging structural cytoskeleton. IL-13 can also induce AHR without increasing inflammation (Yang et al., 2001; Venkayya et al., 2002), and this is thought to be via altering gene expression of other resident airway cells, including epithelial cells and ASM cells (Laporte et al., 2001; Lee et al., 2001; Kuperman et al., 2002; Venkayya et al., 2002).
Lung function measurements, including the forced oscillation technique (FOT), forced expiration (spirometry in humans, negative pressure-driven forced expiration in mice), and AHR to provocation, are commonly used as diagnostic tools in humans, and as tools to assess the severity of respiratory disease and response to treatments in pre-clinical models (predominantly in mice). FOT and forced expiration for the purposes of measuring baseline lung function has been addressed elsewhere (Devos et al., 2017; Bonnardel et al., 2019), thus, this review is focused on AHR.
In mice, AHR is commonly measured following provocation with a contractile stimulus (such as methacholine) and this is an invasive and terminal procedure. The most commonly used invasive plethysmographs to measure airway resistance to methacholine provocation in asthma/allergic airway disease mouse models are the Scireq flexiVent, eSpira EMMS Spirometry, and Buxco FinePointe Resistance and Compliance systems. Asthma/allergic airway disease models routinely use the Scireq flexiVent system to perform perturbations that assess Newtonian Resistance/central airways resistance (Rn), tissue elastance (H; measure of alveolar tissue stiffness) and damping (G; index of alveolar tissue restriction), and/or transpulmonary resistance (Rrs), elastance (Ers), and compliance (Crs), following provocation with nebulized methacholine (up to 100 mg/kg) (Kim et al., 2017a; Kim et al., 2017b; Piyadasa et al., 2018; Ambhore et al., 2019; Kalidhindi et al., 2019; Ali et al., 2020a; Pinkerton et al., 2022; Tu et al., 2022; Xiao et al., 2022). Increases in Rn, H, G, Rrs, and Ers, and decreased Crs, when compared to control (non-allergic) mice is commonly reported. Whilst the order and timing of perturbations is usually dependent on the stimulus used to induce asthma/allergic airways disease, the tidal volume is commonly set at between 8 and 10 mL/kg and the respiratory rate at 150 or 450 breaths/minute. The Buxco invasive plethysmograph also measures airway resistance following provocation with nebulized methacholine (up to 8 mg/kg) (Royce et al., 2011; Donovan et al., 2013; Royce et al., 2022), however, this system does not differentiate between central airways and transpulmonary measurements. Nevertheless, invasive plethysmography measures key features of AHR in response to provocation in experimental asthma models and provides sensitive detection of changes in lung function parameters that are highly representative of those that occur in patients with asthma and severe asthma.
AHR and its driving mechanisms can also be assessed using specialized ex vivo techniques. Airway tissues, such as bronchi and trachea, can be isolated from human lung resections or dissected from mice, and airway reactivity assessed to contractile stimuli in organ bath or myograph systems. Trachea isolated from mouse models of experimental asthma exhibit AHR to methacholine, which is a similar assessment to invasive plethysmography techniques (Donovan et al., 2013) and is a platform to assess airway reactivity ex vivo. Recent advances in human and mouse PCLS preparations have also allowed for assessment of AHR ex vivo, with PCLSs being able preserve the natural tissue architecture and interdependency of the ASM and the surrounding parenchyma. PCLS have also been used to assess changes in small airway reactivity and calcium signaling within ASM cells in response to contractile and bronchodilator stimuli (Donovan et al., 2013; Bourke et al., 2014; Donovan et al., 2015; Liu et al., 2017). Application of this technique in mouse models of experimental asthma has provided significant insights that complement and advance studies on isolated larger airways, where airway inflammation has been shown to alter airway reactivity (Sukkar et al., 2001; Tliba et al., 2003; Zhang et al., 2007). PCLSs from house dust mite antigen (HDM)-treated mice have increased contraction to methacholine (measured as a reduction in airway lumen area) compared to PBS control-treated mice (Liu et al., 2017). In contrast, PCLSs from ovalbumin (ova)-treated mice have reduced methacholine-induced contraction despite having increased AHR when measured in vivo and in isolated trachea (Donovan et al., 2013). Whilst this technique provides a unique platform to assess AHR and calcium signaling in ASM, it is important to consider that airway stiffness may impair airway contraction and confound assessment of AHR in models with robust airway remodeling.
It is possible to examine and measure some biologically relevant properties of ASM in isolated cell culture systems, such as changes in calcium signaling using calcium dyes (Bourke et al., 2011) and its hypercontractile properties using collagen gel preparations (as a surrogate for contraction) (Matsumoto et al., 2007; Bourke et al., 2011). The properties of ASM in vitro can be affected by different culture conditions, however it is now well accepted that isolated ASM cells from patients with asthma have altered contractile, proliferative and secretory function compared to those from subjects without asthma [reviewed in (Zuyderduyn et al., 2008)]. ASM from patients with asthma have increased contraction to histamine compared to ASM from subjects without asthma as measured by a decrease in collagen gel area (Matsumoto et al., 2007). However, whether these properties of ASM are also observed in cells isolated from mouse models of asthma remains to be determined and may provide a new platform to accelerate drug discovery. Lung-on-a-chip technology allows in vitro modeling and study of human musculature and may be a feasible approach for such studies. Engineered human airway musculature can recapitulate healthy and asthmatic bronchoconstriction and bronchodilation. Inhibition of Rho-mediated calcium sensitization and contraction in this model has been shown to decrease basal tone of bronchial smooth muscle and prevent hypercontraction (Nesmith et al., 2014).
Murine models of experimental asthma facilitate the investigation of mechanisms underpinning disease pathogenesis, the identification of novel therapeutic targets, and are platforms for evaluating the efficacy and safety of novel therapeutics before they enter clinical trials. However, mice do not develop asthma spontaneously and this necessitates the experimental induction of allergic responses in the airways to model this disease. Numerous models have been developed and utilize different allergens (ova, HDM, cockroach, pollen), routes of allergen administration (intraperitoneal, subcutaneous, intranasal, aerosol inhalation), and exposure protocols (single versus multiple consecutive exposures to allergen). Despite these differences, the principles of inducing experimental asthma are common and typically involve an initial allergen sensitization phase followed by a challenge phase where mice are re-exposed to the sensitizing agent to initiate allergic recall. These models have been successfully developed in multiple strains of mice, however, BALB/c strain mice are used most often owing to their TH2-prone immunological response, which allows these models to more accurately recapitulate hallmark features of allergic asthma including airway inflammation, airway remodeling, and AHR. Importantly, the presence and severity of these features are dependent on the type of allergen used to induce disease, the exposure protocol, and the measurement of AHR recorded (Table 1). In addition, the choice of equipment used to measure AHR can result in different outcomes, mainly in relation to airway resistance, in different regions of the lung (Rn, Rrs, airway resistance). Nevertheless, invasive techniques (e.g., Scireq flexiVent, Buxco systems and eSpira EMMS Spirometry) do provide more accurate recordings of resistance than non-invasive techniques (e.g., enhanced pause [Penh]) [comprehensively reviewed in (Bates and Irvin, 2003)]. Comparing AHR across experimental groups in different studies is challenging as the measurements of AHR are not directly interchangeable due to the equipment used and often mice have different baseline levels of resistance depending on the calibration of the machines. In some models presented in Table 1, the data are expressed as % change from baseline/saline which does allow for direct comparisons between experimental groups and different experiments in other publications. For transparency in future studies, presenting % change and raw data would allow for readers to directly compare the degree of AHR induced by different exposure protocols to determine the most appropriate model of experimental asthma to employ. This, along with strain, allergen and exposure protocol, is imperative to carefully consider the outcomes of interest against the endo/phenotype that each model can offer.

TABLE 1. Experimental asthma models.
The mostly widely used allergen that is used to induce experimental asthma is ova, derived from chicken egg protein. Ova is commonly administered systemically, via intraperitoneal or subcutaneous injection, in conjunction with the potent TH2-inducing adjuvant, aluminium hydroxide (Royce et al., 2011; Donovan et al., 2013; Essilfie et al., 2015; Kim et al., 2017b; Kim et al., 2019; Pinkerton et al., 2022) to drive allergic sensitization. This is followed at a later time by respiratory challenge with ova to initiate allergic recall responses and model allergen-induced exacerbations of established allergic disease. These challenges are often administered intranasally; however, a recent study highlighted that aerosol challenge, compared to intranasal challenge with ova, induces greater AHR in response to 50 mg/mL dose of methacholine suggesting that ova-induced experimental asthma responses are more potently induced by inhaled, rather than intranasal, ova challenge (Kim et al., 2019). Ova-induced models are renowned for their reproducibility and robust allergic inflammatory responses that recapitulate key disease features including airway inflammation, epithelial hypertrophy, goblet cell hyperplasia, and AHR (Essilfie et al., 2015; Kim et al., 2017b; Kim et al., 2019; Pinkerton et al., 2022). However, and despite this favorable set of outcomes for modeling asthma experimentally, there is contention about the clinical relevance of ova as an allergen, which is an important limitation of this model as it highlights a potential barrier for translation of findings into human disease. In addition, allergen sensitization via intraperitoneal injection in conjunction with an adjuvant does not reflect the natural course of allergen sensitization in asthma in humans. Ova models are also not associated with persistent airway remodeling, which is a key feature of human asthma, and the chosen allergen exposure regimen may induce disease features that are, or are not, sensitive to corticosteroid treatment, which further highlights potential limitations of ova-based asthma models. More recently, experimental asthma models have moved towards utilizing more clinically and physiologically relevant allergens and model protocols that are strongly linked with the human disease.
Sharing many similarities to human disease, mouse models of HDM-induced experimental asthma are characterized by eosinophil-dominant airway inflammation, airway remodeling, and AHR (Barry et al., 2013; Woo et al., 2018; Ali et al., 2020a; Tu et al., 2022). The immune response to HDM is also T2-driven, with increased IL-5, IL-13, IL-33, IL-25, and thymic stromal lymphopoietin contributing to eosinophils, type 2 innate lymphoid cells (ILC2s) and dendritic cell recruitment/activation in the lung [reviewed in (Jacquet, 2021)]. The hallmark features of disease are similar to ova-induced experimental asthma, however HDM-induced experimental asthma can be considered more clinically relevant as these models have allergic sensitization through the intranasal route, which can be achieved in the absence of an adjuvant and is a more realistic representation of what occurs in humans. Furthermore, HDM induces allergic airways disease through a combination of toll-like responses and enzymatic reactions [reviewed in (Abu Khweek et al., 2020)], which may be absent in ova models. The specific protocol of HDM antigen exposure is also an important consideration; with acute vs. chronic HDM exposure models leading to different disease outcomes (Woo et al., 2018) (Table 1). Furthermore, multiple preparations of HDM antigen can be used as an allergen (whole extract vs. components) and this represents an important limitation in published studies to date, where the type of HDM preparation that is used for experimentation is often not fully specified. In particular, the batch numbers of HDM allergens used, and whether the HDM protein concentration is determined by bicinchoninic acid assay (BCA) or Bradford assessment, is often omitted from manuscripts.
Another common environmental aeroallergen is fungus. Fungal asthma is poorly managed and causes frequent exacerbations and hospitalizations (Lehmann et al., 2017). Fungal asthma is considered predominately a T2 driven disease, however, it can be associated with T2 low endotypes of asthma (TH1 and TH17 high) (reviewed in [van Tilburg Bernardes et al., 2020)]. Alternaria alternata is an allergenic mold that is recognized as a potent aeroallergen and a common inducer of asthma exacerbations (Daines et al., 2021). When employed in murine models of fungal asthma, filtrates of A. alternata are used to sensitize and challenge mice. Recent studies have also shown that direct spore exposure, as opposed to traditional filtrate exposure, is more physiologically relevant and has the capacity to induce similar (and more potent) disease outcomes (Daines et al., 2021) (Table 1). Aspergillus fumigatus has also been used in combination with allergen/s to mimic disease outcomes. The double allergen model involves sensitization and challenge with dust mite (Dermatophagoides farinae) followed by intranasal exacerbation with A. fumigatus (Matsuse et al., 2013). This model has demonstrated key features of experimental asthma, including increased inflammation (macrophages, neutrophils, eosinophils and lymphocytes, IL-5, IL-13), and mucus secreting cells, and partial steroid-insensitivity to dexamethasone (decreased macrophages, eosinophils, and IL-5, but no effect on neutrophils, lymphocytes, IL-13, mucus-secreting cells) (Matsuse et al., 2013). Furthermore, triple allergen models of experimental asthma have been established to more accurately recapitulate the human scenario. These models involve sensitization (on day 0 and 5) with a triple allergen cocktail (dust mite [D. farinae], ragweed [Ambrosia artemisiifolia], and A. fumigatus) in the presence of an adjuvant, followed by intranasal administration of the triple allergen mixture on days 12, 13, and 14 (Chung et al., 2020). This model induces hallmark features of asthma including inflammation, remodeling (increased mucus secreting cells) and AHR (increased resistance to MCh; Scireq flexiVent).
The continual optimization of murine models of experimental asthma is an important and indispensable paradigm in the journey towards increasing our understanding of the underlying physiological mechanisms that drive asthma pathogenesis. Crucially, to address the heterogeneous nature of asthma, researchers have developed many different allergen-induced models that have allowed for recapitulation of different endotypes and phenotypes of asthma. Consequently, this has led to the development of experimental protocols that model severe, steroid insensitive or resistant phenotypes of asthma; allowing for the identification of mechanisms that may drive the onset of more severe disease.
Mouse models of experimental severe asthma that recapitulate features of human disease have provided invaluable insight into the possible mechanisms that are responsible for driving the increased disease severity and steroid insensitivity that are characteristic of severe asthma in humans (Table 2). Severe asthma is often characterized by an insensitivity to currently available therapeutics and the capacity to model this association experimentally is crucial. Improving our understanding of mechanisms underpinning steroid insensitivity (sometimes referred to as steroid resistance in publications) will identify novel targets for therapeutic development. Importantly, there is strong evidence that supports a link between specific respiratory infections and severe asthma exacerbations, whereby different infections can induce different phenotypes of severe disease (e.g., neutrophilic-dominant vs. eosinophil-dominant). Recent advances in murine models have incorporated these elements into the protocol design and have been optimized to model and investigate the effect of respiratory infections on disease outcomes (Essilfie et al., 2012).
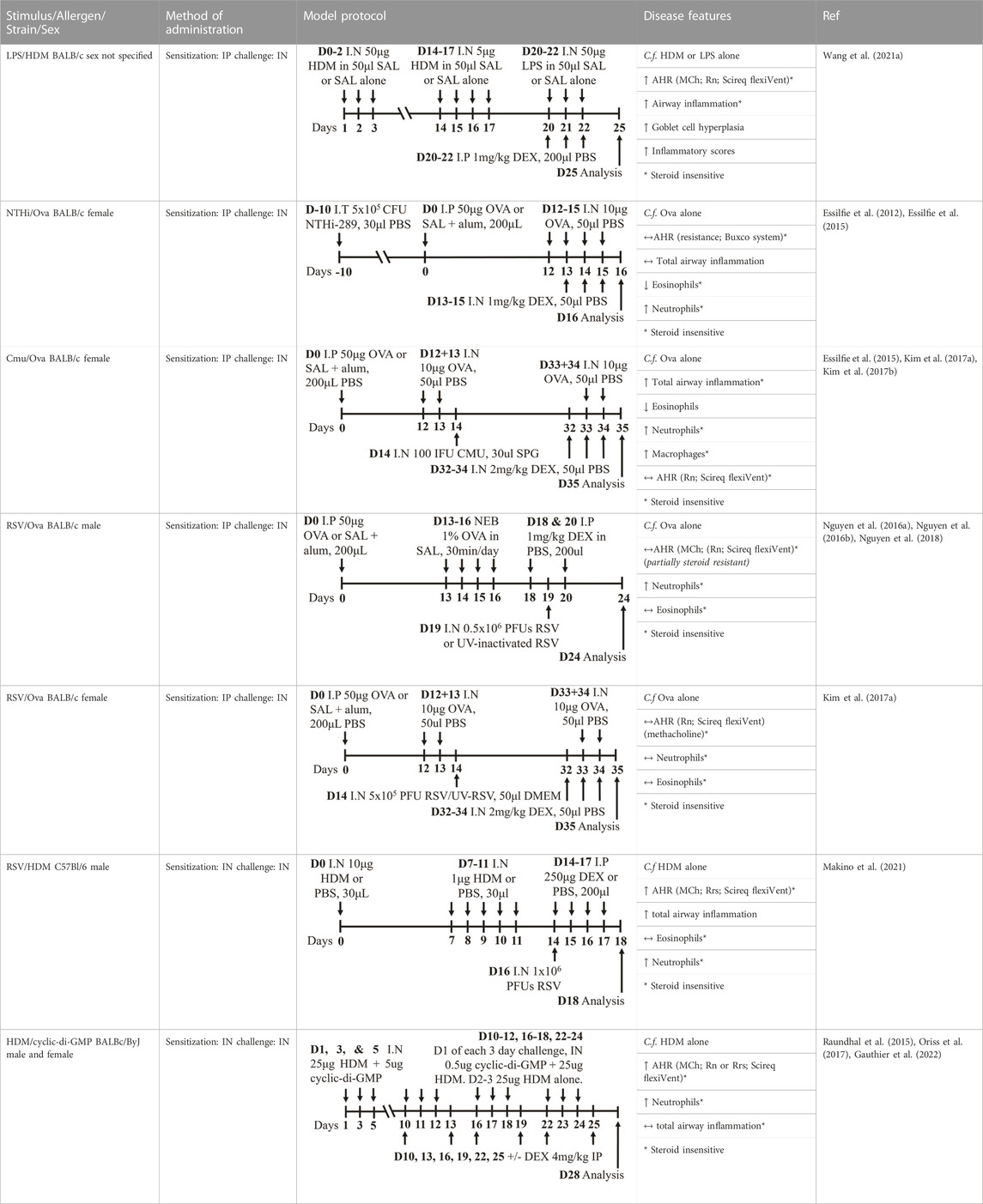
TABLE 2. Experimental mixed-exposure asthma models.
Infectious pathogens such as C. pneumoniae (Essilfie et al., 2015; Kim et al., 2017a; Kim et al., 2017b), H. influenzae (Essilfie et al., 2012; Essilfie et al., 2015; Kim et al., 2017a), RSV (Nguyen et al., 2016a; Nguyen et al., 2016b; Kim et al., 2017a; Nguyen et al., 2018), influenza A virus (Kim et al., 2017a), and rhinovirus (Bartlett et al., 2008) have been used to induce severe disease in mouse models of experimental asthma. These infections are often induced through natural respiratory routes after experimental asthma is established, which reflects the typical scenario of an infection-induced exacerbation of asthma. An important element of these infection-induced models is their capacity to model different phenotypes of severe disease, which provide a platform for the elucidation of broadly applicable and/or phenotype-specific mechanisms of pathogenesis. When infections with the natural mouse respiratory pathogens Chlamydia muridarum or non-typeable H. influenzae are induced in female BALB/c mice with ova-induced experimental asthma, this results in steroid-insensitive disease (AHR and total airway inflammation that are insensitive to dexamethasone treatment) with a switch from eosinophilic to neutrophil-dominant airway inflammation (Kim et al., 2017a). Importantly, these preclinical observations are validated by clinical observations that show an association between bacterial infection and neutrophil-dominant inflammation in the sputum of patients with asthma (Wood et al., 2010). Contrastingly, whilst viral infection (mouse-adapted influenza A virus or RSV), like with bacterial infection-induced models of severe asthma, also induce steroid-insensitive AHR, there is no switch towards neutrophil-dominant inflammation and total airway inflammation becomes partially steroid-insensitive (Kim et al., 2017a). Rhinoviruses are also important viral infections involved the increased risk of asthma and asthma exacerbations; however, mouse models of rhinovirus infections are challenging as the major group of rhinoviruses (that comprises 90% of all rhinovirus serotypes) requires human intercellular adhesion molecule-1 (ICAM-1) for replication, which is not present in mice. Nevertheless, minor group rhinoviruses, such as rhinovirus-1B, can replicate in wild-type BALB/c mice, and this has been shown to increase AHR in ova-induced experimental asthma associated with increased neutrophil responses (Bartlett et al., 2008). Whether this model of rhinovirus and ova-induced experimental asthma is steroid-insensitive is yet to be elucidated.
Viral infection models of severe asthma are characterized by steroid-insensitive eosinophilic airway inflammation and AHR (Kim et al., 2017a), and this agrees with previous studies showing that eosinophilic inflammation can be present despite moderate-to high-dose steroid treatment (Jayaram et al., 2005; Trevor and Deshane, 2014). Interestingly, in infection-induced models (bacterial and viral), aberrant microRNA-21 (miR-21) responses have been shown to drive steroid insensitivity in experimental severe asthma and therapeutically targeting miR-21 may offer a broadly effective treatment for infection-induced severe, steroid-insensitive asthma.
In addition, components of bacteria, such as lipopolysaccharide (LPS) or cyclic-di-GMP, have been utilized in HDM-induced models of experimental asthma to induce features of severe asthma (Raundhal et al., 2015; Gauthier et al., 2017; Oriss et al., 2017; Wang et al., 2021a; Gauthier et al., 2022). These models have provided significant mechanistic insight into the clinical observation that patients with severe asthma, particularly T2 low patients, are steroid-insensitive (Woodruff et al., 2009). Importantly, these studies together have identified key disease-causing and interconnected roles of dysregulated CXCR3 ligand (CXCL10)/TH1 cell recruitment/IFNγ/secretory leukocyte protease inhibitor (SLPI) responses in steroid-insensitive severe asthma (Raundhal et al., 2015; Gauthier et al., 2017; Oriss et al., 2017; Gauthier et al., 2022).
Collectively, infection- or bacterial component-induced models of severe asthma continue to provide important mechanistic insight into disease pathogenesis whilst also highlighting the complexity of severe disease. Considering the growing appreciation for the differences in immune responses between male and females, and the differences between sex hormones, genetics, and epigenetics in asthma [reviewed in (Chowdhury et al., 2021)], an important future direction will be to understand how sex hormones influence disease outcomes in these models. The role/s of sex hormones influencing asthma and AHR is yet to be fully elucidated however, there is observational data demonstrating that estrogen receptor β knockout male and female mice have increased AHR in a mixed allergen (ovalbumin, A. alternata, A. fumigatus, and D. farinae) model compared to wild type mixed allergen alone (Kalidhindi et al., 2019), however the specific roles of hormone receptors in asthma and the magnitude of AHR remains to be fully explored. It will also be important to determine how obesity influences immune responses to infection, both alone and in the context of asthma and AHR, and to understand whether the pathogenic mechanisms that have been identified in models of infection-induced disease (e.g., miR-21) are also relevant in obesity-associated disease.
Asthma can also overlap with COPD in patients, and these patients are currently referred to as having asthma-COPD overlap (ACO). Similar to severe asthma, patients with ACO have chronic airway inflammation that is linked with airway remodeling, decreased lung function, wheezing, and shortness of breath (Leung and Sin, 2017). The cellular composition of the airway inflammation in these patients is also heterogeneous, however, ACO has similar endotypes to severe asthma (T2 high, T2 low, or non-T2). Recent advances in mouse models of experimental ACO [reviewed in (Tu et al., 2021)] have provided important mechanistic insights into novel, disease-causing mechanisms in ACO and identified potential therapeutic targets that may also be applicable to other forms of asthma. Experimental ACO can be induced by chronic administration of HDM antigen and concurrent exposure to cigarette smoke after HDM-induced experimental asthma is established (Tu et al., 2022). Importantly, this model results in steroid insensitivity of AHR and other key disease features such as airway remodeling, which is reminiscent of ACO in humans (Dey et al., 2022). Comprehensive transcriptomic analysis of airway and parenchyma tissues in this model revealed robust increases in innate immune responses (increased SPI1, TFEC), complement responses, and disturbances in the expression of metabolism-associated factors (increased GLMP, PLSCR1, MTF1) (Tu et al., 2022). Significantly, therapeutic targeting of SPI1 suppressed some of the key features of experimental ACO, including steroid insensitive AHR (Tu et al., 2022). These data show that the combination of HDM antigen and cigarette smoke exposures drive changes in lung function that are not responsive to mainstay therapies and highlight the potential utility of this model for deciphering the specific mechanisms that cause this outcome.
Obesity has been linked with the development of asthma, however, the mechanisms underpinning this relationship remain largely unknown. The obese asthma phenotype represents a significant cohort of asthmatics who have more severe disease that is commonly refractory to mainstay therapies (Dixon et al., 2011). To improve our understanding of obese asthma and the mechanisms underpinning the severe, steroid insensitivity of this disease phenotype, multiple mouse models of obesity-associated asthma have been developed and optimized.
Obesity can be modeled in mice through genetic modifications (mono, polygenic mutations, transgenic mice), surgical or chemical induction, or, more commonly, by the administration of hypercaloric, specialized diets (Pinkerton et al., 2022). Diet-induced models of obesity lead to dietary imbalance, which is the most common cause for obesity in humans. Compared with genetically induced/modified models of obesity, the timeframe of diet-induced obesity is often protracted to model the natural and gradual accumulation of adiposity that is observed in obesity in humans. A range of different diets have been recognized to induce obesity in mice, including high-fat diets (HFD), high-sugar diets, and a combination of the two that represents a Western diet (high fat and high sugar). Whilst this array of diets allows for different facets of obesity and metabolic disorders to be modeled, it is important to consider the variations in the macro- and micronutrient profiles of these different diets. This variability highlights the lack of a standardized, well-defined diet for modeling obesity and may underpin the discrepancies in reported outcomes between studies. Therefore, to ensure reproducibility and consistency across investigations, it is important that the formulation of diets that are used to induce obesity are considered and specified.
These different models of obesity have facilitated investigations into the complex relationship between obesity and asthma (Kim et al., 2014; Pinkerton et al., 2022). HFD-induced obesity in mice (HFD; 60% energy derived from fat; Research Diets, D12492) is characterized by increased total body mass, inflammation (measured in terms of increased macrophages and ILC3s in the lung and increased macrophages in adipose tissues), and AHR in response to methacholine provocation compared to control chow (CC)-fed mice (Kim et al., 2014). Importantly, this obesity-induced AHR has been associated with NLRP3 and IL-17 pathway-dependent IL-17+ILC3 responses. In an independent and more recent study, HFD-induced obesity in mice (HFD; 60% energy derived from lipids, 15% energy from protein; Specialty Feeds, SF14-154), was also shown to be characterized by increased total body mass as well as adiposity where robust increases in the mass of major white adipose tissue pads were observed, including the parametrial, inguinal, and retroperitoneal fat pads compared to CC-fed mice (CC; 16% energy derived from lipids, 21% energy from protein; Specialty Feeds SF09-091; Table 3) (Pinkerton et al., 2022). Furthermore, when superimposed with ova-induced experimental asthma, HFD-induced obesity was shown to promote steroid-insensitive AHR but with no significant effect on allergic airway inflammation when compared to CC-fed, allergic mice. Significantly, this study also highlighted a novel association between increased T2 immune (concomitant IL-5 and IL-13 responses) and NLRP3 inflammasome responses in the airway in obesity-associated severe asthma, providing a potential mechanism and explanation as to why obese individuals experience more severe asthma that is refractory to current therapies (Pinkerton et al., 2022). Importantly, the pre-clinical findings from this study have been validated in clinical samples, demonstrating this model’s utility for increasing the understanding of the mechanistic interplay underpinning obesity-associated severe asthma in humans. However, whether therapeutic targeting of T2-induced, NLRP3-mediated responses in obesity-associated severe asthma is effective in humans, remains to be determined.
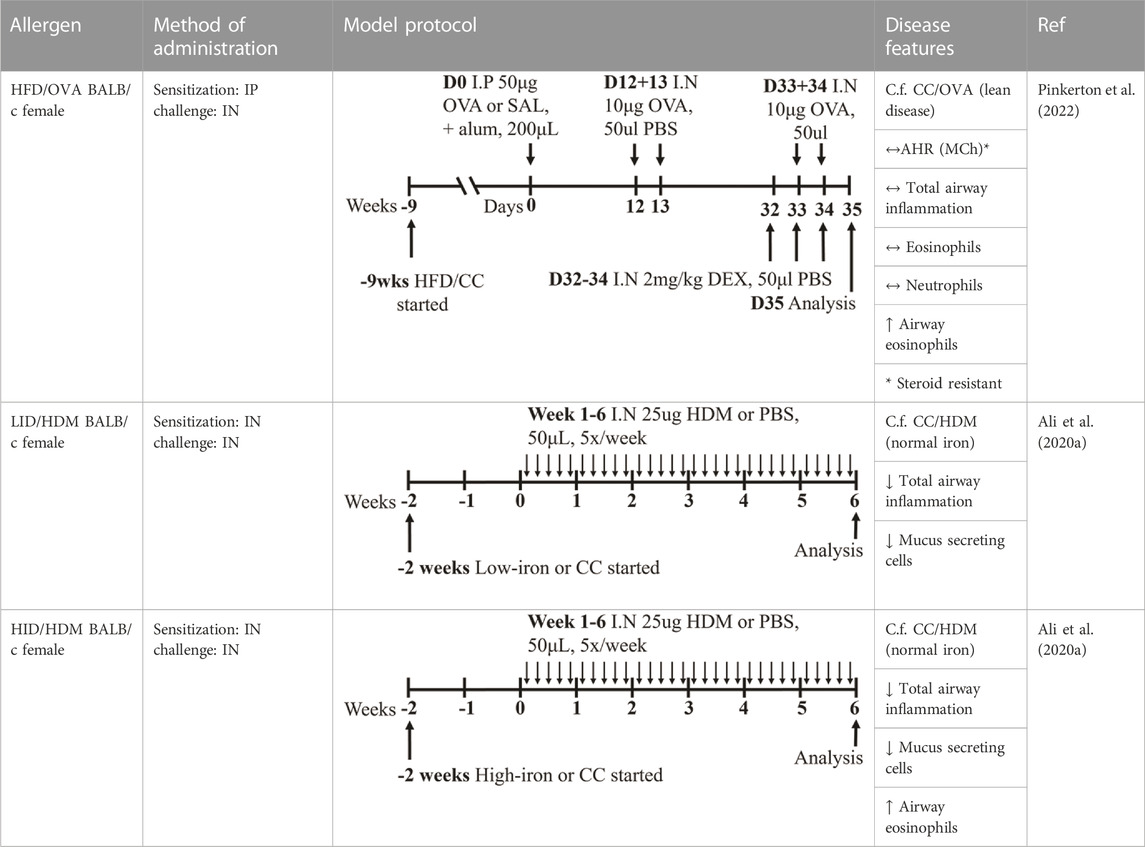
TABLE 3. Experimental asthma models (diet).
The paucity of knowledge relating to the impact of obesity on respiratory physiology, immune and inflammatory responses, and in the absence or presence of asthma, highlight essential avenues for future research and highlights the importance of refining and interrogating obesity-associated asthma models. Furthermore, it remains to be determined whether reducing adiposity can alter respiratory physiology and AHR, or whether specific dietary nutrients or metabolites can alter respiratory function, and these are important additional avenues for future study.
Increased lung iron status is linked to the pathogenesis and severity of asthma, cystic fibrosis, COPD, and idiopathic pulmonary fibrosis (Reid et al., 2007; Ghio et al., 2013; Cloonan et al., 2017; Ali et al., 2020a; Ali et al., 2020b). Interestingly, lower systemic iron status is also implicated in the development of asthma. Low systemic iron, as measured by exhaled breath condensate, is associated with asthma in both adults and children (Ramakrishnan and Borade, 2010; Vlasic et al., 2011; Brigham et al., 2015). Additionally, lower maternal iron status during pregnancy has been linked with poorer respiratory outcomes in children (Triche et al., 2011; Bedard et al., 2018). Maternal anemia in pregnancy is linked to early-life recurrent wheeze and poorer childhood lung function, as measured by FEV1, FVC, and physician-diagnosed asthma at age six (Triche et al., 2011; Bedard et al., 2018). Lower maternal hemoglobin concentration during pregnancy is also linked with elevated IgE and risk of allergic sensitization in children (Shaheen et al., 2017).
Intergenerational models of maternal systemic iron status have predominantly been studied in the context of brain development in offspring (Juul et al., 2019). A recent study modeling diet-induced reduction in systemic iron status during pregnancy found that lower maternal iron status resulted in offspring with impaired lung function at baseline, and increased AHR, airway inflammation, and small airway collagen deposition (Gomez et al., 2021). However, studies modeling altered iron status during pregnancy that examine lung function and structure in offspring are limited.
Genetically-induced changes to iron status provide invaluable enhancements to our understanding of physiological changes from altered iron status and are essential in modeling genetic disorders, such as hereditary hemochromatosis, which is an iron overload disorder that is caused by mutations in the Homeostatic Iron Regulator (HFE) gene. HFE−/− mouse models of iron overload have advanced our understanding of physiological processes involving iron isotype regulation and compositions of numerous organs and cell types (Albalat et al., 2021). A recent study used an iron overload model (Hamp knockout) to show that maternal hepcidin, rather than embryonic hepcidin, regulates embryonic iron levels (Sangkhae et al., 2020). Systemic iron deficiency has also been modeled using transthyretin-hepcidin transgenic mice that overexpress hepcidin (Nicolas et al., 2002), and ferroportin ablation in intestinal epithelial cells to eliminate intestinal iron absorption resulting in end-stage iron deficiency anemia (Schwartz et al., 2019).
Genetic disease models are valuable tools for defining mechanisms of pathogenesis via gain or loss of function experiments. However, protocols that involve diet-induced alterations to iron status are essential for comparing physiological processing of various forms of iron. This has been demonstrated by a study that compared ferrous and ferric iron absorption, whereby examination of the effects of diet-induced iron sufficient status (35 mg Fe/kg) and iron deficiency (2 and 20 mg Fe/kg) showed that ferric iron, compared to ferrous iron, may be a more effective iron supplement due to its slow release and reduced rate of absorption (Aslam et al., 2014).
Despite these advances, there remains limited understanding of the mechanisms underpinning the clinical associations between systemic iron deficiency during pregnancy and childhood asthma and allergy, which highlights the importance of utilizing intergenerational iron deficiency and asthma models to examine these associations. In this scenario, diet-induced models of iron deficiency are the most clinically relevant approach and would facilitate assessments of the effects of different iron supplementation formulae during pregnancy on respiratory outcomes in offspring.
The fetal origins of adult disease hypothesis proposes that the in utero environment is critical for determining an individual’s susceptibility to certain chronic diseases later in life (Barker, 1990). Notably, suboptimal nutritional status, such as undernutrition due to maternal smoking or maternal exposure to heavily polluted air, as well as toxins inhaled or ingested by pregnant mothers, play key roles in fetal underdevelopment and organ dysfunction after birth (Wang et al., 2020; Chen et al., 2021; Li et al., 2021). The latent and persistent effects of fetal programming may arise from epigenetic modifications that permanently change key regulators of a range of biological processes, including cellular metabolism (e.g., mitochondrial function) and immune responses (e.g., heightened pro-inflammatory mediator production) with or without additional external environmental stimuli after birth (Barker and Osmond, 1986; Petronis, 2010; Li et al., 2018; Wang et al., 2020; Chen et al., 2021).
In humans, lung morphogenesis begins at 3 to 4 weeks post-conception, followed by five stages of intense development (embryonic, pseudoglandular, canalicular, saccular, and alveolar) that occur up to 36 weeks post-conception, and completion of lung development during childhood and young adulthood. In utero environmental insults during any of the developmental stages can result in abnormal lung architecture (Li et al., 2021) and function (Harju et al., 2016; Lee et al., 2018), and increased susceptibility to asthma (Hazlehurst et al., 2021) and asthma exacerbations after birth (Gilliland et al., 2001). Tobacco cigarette smoke and particulate matter (PM2.5) are currently the most commonly occurring in utero toxins that impair lung function in offspring and increase their susceptibility to asthma (Li et al., 2018; Wang et al., 2021b; Chen et al., 2021).
Epidemiological studies suggest that maternal smoking and inhalation of heavily polluted air are linked to intrauterine underdevelopment, preterm birth, and small birth weight (Wang et al., 2020). As a result, lung development and function are inevitably affected (Hayatbakhsh et al., 2009; Joubert et al., 2016). Notably, low birth weight and impaired lung function are strongly linked with the increased prevalence of childhood wheezing and asthma (Turner et al., 2009; Xu et al., 2014). Both tobacco cigarette smoke and PM2.5 contain thousands of chemical substances, including free radicals, that possess strong oxidative properties. As a result, oxidative stress has been proposed as the common mechanism that translates maternal exposure to cigarette smoke and PM2.5 into inflammatory responses and pathology in the lung in offspring (Sukjamnong et al., 2017; Wang et al., 2021b). Indeed, maternal exposure to tobacco cigarette smoke and combustion-generated PM induced systemic oxidative stress in offspring and greater AHR (Wang et al., 2013; Zacharasiewicz, 2016; Wang et al., 2021b).
Increased pro-inflammatory cytokine responses, including that of IL-8, and TH2 hyperactivity reflected by increased IL-5, IL-9, and IL-13 levels, have been detected in the cord blood of newborns from smoking mothers (Noakes et al., 2003; Chahal et al., 2017). However, some infants born to smokers also show changes in toll-like receptor (TLR) 2 function and attenuation of TNF-α, IL-6, and IL-10 levels indicating decreased innate immune responses (Noakes et al., 2003). In animal models, T helper cells including TH1, TH2, TH17, and T regulatory cells were reduced in offspring born to mothers with in utero PM exposure (Wang et al., 2013). AHR is also present in the absence of additional ova challenge in adult mice suggesting that this is a prolonged effect of in utero exposure (Wang et al., 2021b). Alarmingly, these adverse effects in offspring still occur as a result of maternal inhalation of air pollutants that are present at levels considered safe, and these sequelae are not mitigated by migrating to a pollutant-free environment after conception (Wang et al., 2021b). Thus, the in utero effect on AHR most likely involves epigenetic changes and the maternally-derived mitochondrial DNA without modification of genomic profiles (Shrine et al., 2019). A meta-analysis of 13 cohort studies identified more than 3,000 CpG islands that are differentially methylated due to maternal smoking (Shrine et al., 2019). Some of these effects, such as hypermethylation of ESR1 and CpGs in IL-32, have been linked with the pathogenesis of asthma (Shrine et al., 2019). In this scenario, PM exposure can initially cause changes in maternal DNA methylation (Ferrari et al., 2019) that are then inherited by the offspring, which could explain why migrating to a pollutant-free area during pregnancy is not sufficient to mitigate the risk of asthma development in offspring (Wang et al., 2021b). Additionally, epigenetic modifications to the fetal genome during in utero PM exposure can further increase the risk of asthma, such as the antioxidant superoxide dismutase 2 (SOD2) promoter DNA methylation, and TMEM184A which is involved in the inflammatory response (Zhou et al., 2019; Parikh et al., 2022). In summary, maternal exposure to inhaled toxins, such as tobacco smoke and PMs, can increase the risk of asthma in offspring via multiple mechanisms, including impaired lung development and structure, oxidative stress, immune response, and epigenetic modifications.
Refinements and innovation in technologies to assess lung function in mouse models that recapitulate the key features of asthma and different endo/phenotypes of severe asthma have significantly advanced the understanding of disease-causing mechanisms. This has led to improvements in the clinical management of the disease, however, there remains many endo/phenotypes of severe asthma that have no effective treatment options. The use of highly representative mouse models (Tables 1–3) in combination with diet and environmental exposures, are key to increasing the understanding of mechanisms of pathogenesis, including drivers of AHR, and should include comprehensive immune profiling and remodeling assessments. These models coupled with technological advances in measuring respiratory physiology, will lead to the identification of new therapeutic targets that support the goal of achieving disease remission.
OC, AP, AB, MF, HC, and CD wrote the manuscript. All authors have read and approved the manuscript.
The authors would like to acknowledge the intellectual input and mentorship of Dr Richard Y. Kim and Distinguished Professor Brian G. G. Oliver (University of Technology Sydney), and Dr Henry M. Gomez and Professor Jay C. Horvat (Hunter Medical Research Institute and The University of Newcastle Australia).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aaron, S. D., Boulet, L. P., Reddel, H. K., and Gershon, A. S. (2018). Underdiagnosis and overdiagnosis of asthma. Am. J. Respir. Crit. Care Med. 198, 1012–1020. doi:10.1164/rccm.201804-0682CI
Abu Khweek, A., Kim, E., Joldrichsen, M. R., Amer, A. O., and Boyaka, P. N. (2020). Insights into mucosal innate immune responses in house dust mite-mediated allergic asthma. Front. Immunol. 11, 534501. doi:10.3389/fimmu.2020.534501
Agrawal, D. K., and Shao, Z. (2010). Pathogenesis of allergic airway inflammation. Curr. Allergy Asthma Rep. 10, 39–48. doi:10.1007/s11882-009-0081-7
Akinbami, L. J., Salo, P. M., Cloutier, M. M., Wilkerson, J. C., Elward, K. S., Mazurek, J. M., et al. (2020). Primary care clinician adherence with asthma guidelines: The national asthma survey of physicians. J. Asthma 57, 543–555. doi:10.1080/02770903.2019.1579831
Albalat, E., Cavey, T., Leroyer, P., Ropert, M., Balter, V., and Loreal, O. (2021). Hfe gene knock-out in a mouse model of hereditary hemochromatosis affects bodily iron isotope compositions. Front. Med. 8, 711822. doi:10.3389/fmed.2021.711822
Ali, M. K., Kim, R. Y., Brown, A. C., Donovan, C., Vanka, K. S., Mayall, J. R., et al. (2020). Critical role for iron accumulation in the pathogenesis of fibrotic lung disease. J. Pathol. 251, 49–62. doi:10.1002/path.5401
Ali, M. K., Kim, R. Y., Brown, A. C., Mayall, J. R., Karim, R., Pinkerton, J. W., et al. (2020). Crucial role for lung iron level and regulation in the pathogenesis and severity of asthma. Eur. Respir. J. 55, 1901340. doi:10.1183/13993003.01340-2019
Alqarni, S. A., Bineid, A., Ahmad, S. F., Al-Harbi, N. O., Alqahtani, F., Ibrahim, K. E., et al. (2022). Blockade of tyrosine kinase, LCK leads to reduction in airway inflammation through regulation of pulmonary Th2/treg balance and oxidative stress in cockroach extract-induced mouse model of allergic asthma. Metabolites 12, 793. doi:10.3390/metabo12090793
Ambhore, N. S., Kalidhindi, R. S. R., Loganathan, J., and Sathish, V. (2019). Role of differential estrogen receptor activation in airway hyperreactivity and remodeling in a murine model of asthma. Am. J. Respir. Cell Mol. Biol. 61, 469–480. doi:10.1165/rcmb.2018-0321OC
Aslam, M. F., Frazer, D. M., Faria, N., Bruggraber, S. F., Wilkins, S. J., Mirciov, C., et al. (2014). Ferroportin mediates the intestinal absorption of iron from a nanoparticulate ferritin core mimetic in mice. FASEB J. 28, 3671–3678. doi:10.1096/fj.14-251520
Bachert, C., and Zhang, N. (2012). Chronic rhinosinusitis and asthma: Novel understanding of the role of IgE 'above atopy. J. Intern Med. 272, 133–143. doi:10.1111/j.1365-2796.2012.02559.x
Bai, Y., and Sanderson, M. J. (2006). Modulation of the Ca2+ sensitivity of airway smooth muscle cells in murine lung slices. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L208–L221. doi:10.1152/ajplung.00494.2005
Bai, Y., Zhang, M., and Sanderson, M. J. (2007). Contractility and Ca2+ signaling of smooth muscle cells in different generations of mouse airways. Am. J. Respir. Cell Mol. Biol. 36, 122–130. doi:10.1165/rcmb.2006-0036OC
Barker, D. J. (1990). The fetal and infant origins of adult disease. BMJ 301, 1111. doi:10.1136/bmj.301.6761.1111
Barker, D. J. P., and Osmond, C. (1986). Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 327, 1077–1081. doi:10.1016/s0140-6736(86)91340-1
Barry, J., Loh, Z., Collison, A., Mazzone, S., Lalwani, A., Zhang, V., et al. (2013). Absence of toll–IL-1 receptor 8/single immunoglobulin IL-1 receptor–related molecule reduces house dust mite–induced allergic airway inflammation in mice. Am. J. Respir. Cell Mol. Biol. 49, 481–490. doi:10.1165/rcmb.2012-0425OC
Bartlett, N. W., Walton, R. P., Edwards, M. R., Aniscenko, J., Caramori, G., Zhu, J., et al. (2008). Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat. Med. 14, 199–204. doi:10.1038/nm1713
Bates, J. H., and Irvin, C. G. (2003). Measuring lung function in mice: The phenotyping uncertainty principle. J. Appl. Physiol. 94, 1297–1306. doi:10.1152/japplphysiol.00706.2002
Bedard, A., Lewis, S. J., Burgess, S., Henderson, A. J., and Shaheen, S. O. (2018). Maternal iron status during pregnancy and respiratory and atopic outcomes in the offspring: A mendelian randomisation study. BMJ Open Respir. Res. 5, e000275. doi:10.1136/bmjresp-2018-000275
Belvisi, M. G., Saunders, M. A., Haddad el, B., Hirst, S. J., Yacoub, M. H., Barnes, P. J., et al. (1997). Induction of cyclo-oxygenase-2 by cytokines in human cultured airway smooth muscle cells: Novel inflammatory role of this cell type. Br. J. Pharmacol. 120, 910–916. doi:10.1038/sj.bjp.0700963
Benayoun, L., Druilhe, A., Dombret, M. C., Aubier, M., and Pretolani, M. (2003). Airway structural alterations selectively associated with severe asthma. Am. J. Respir. Crit. Care Med. 167, 1360–1368. doi:10.1164/rccm.200209-1030OC
Bonnardel, E., Prevel, R., Campagnac, M., Dubreuil, M., Marthan, R., Berger, P., et al. (2019). Determination of reliable lung function parameters in intubated mice. Respir. Res. 20, 211. doi:10.1186/s12931-019-1177-9
Bourke, J. E., Bai, Y., Donovan, C., Esposito, J. G., Tan, X., and Sanderson, M. J. (2014). Novel small airway bronchodilator responses to rosiglitazone in mouse lung slices. Am. J. Respir. Cell Mol. Biol. 50, 748–756. doi:10.1165/rcmb.2013-0247OC
Bourke, J. E., Li, X., Foster, S. R., Wee, E., Dagher, H., Ziogas, J., et al. (2011). Collagen remodelling by airway smooth muscle is resistant to steroids and β₂-agonists. Eur. Respir. J. 37, 173–182. doi:10.1183/09031936.00008109
Brigham, E. P., McCormack, M. C., Takemoto, C. M., and Matsui, E. C. (2015). Iron status is associated with asthma and lung function in US women. PLoS One 10, e0117545. doi:10.1371/journal.pone.0117545
Burgel, P. R. (2011). The role of small airways in obstructive airway diseases. Eur. Respir. Rev. 20, 23–33. doi:10.1183/09059180.00010410
Busse, W. W., Banks-Schlegel, S., and Wenzel, S. E. (2000). Pathophysiology of severe asthma. J. Allergy Clin. Immunol. 106, 1033–1042. doi:10.1067/mai.2000.111307
Castro, M., Rubin, A. S., Laviolette, M., Fiterman, J., De Andrade Lima, M., Shah, P. L., et al. (2010). Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: A multicenter, randomized, double-blind, sham-controlled clinical trial. Am. J. Respir. Crit. Care Med. 181, 116–124. doi:10.1164/rccm.200903-0354OC
Chahal, N., McLain, A. C., Ghassabian, A., Michels, K. A., Bell, E. M., Lawrence, D. A., et al. (2017). Maternal smoking and newborn cytokine and immunoglobulin levels. Nicotine Tob. Res. 19, 789–796. doi:10.1093/ntr/ntw324
Chen, H., Oliver, B. G., Pant, A., Olivera, A., Poronnik, P., Pollock, C. A., et al. (2021). Particulate matter, an intrauterine toxin affecting foetal development and beyond. Antioxidants 10, 732. doi:10.3390/antiox10050732
Cho, Y. S., Kim, T. B., Lee, T. H., Moon, K. A., Lee, J., Kim, Y. K., et al. (2005). Chlamydia pneumoniae infection enhances cellular proliferation and reduces steroid responsiveness of human peripheral blood mononuclear cells via a tumor necrosis factor-alpha-dependent pathway. Clin. Exp. Allergy 35, 1625–1631. doi:10.1111/j.1365-2222.2005.02391.x
Chowdhury, N. U., Guntur, V. P., Newcomb, D. C., and Wechsler, M. E. (2021). Sex and gender in asthma. Eur. Respir. Rev. 30, 210067. doi:10.1183/16000617.0067-2021
Chung, S., Lee, Y. G., Karpurapu, M., Englert, J. A., Ballinger, M. N., Davis, I. C., et al. (2020). Depletion of microRNA-451 in response to allergen exposure accentuates asthmatic inflammation by regulating Sirtuin2. Am. J. Physiol. Lung Cell Mol. Physiol. 318, L921–L930. doi:10.1152/ajplung.00457.2019
Cloonan, S. M., Mumby, S., Adcock, I. M., Choi, A. M. K., Chung, K. F., and Quinlan, G. J. (2017). The "Iron"-y of iron overload and iron deficiency in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 196, 1103–1112. doi:10.1164/rccm.201702-0311PP
Coquet, J. M., Schuijs, M. J., Smyth, M. J., Deswarte, K., Beyaert, R., Braun, H., et al. (2015). Interleukin-21-Producing CD4(+) T cells promote type 2 immunity to house dust mites. Immunity 43, 318–330. doi:10.1016/j.immuni.2015.07.015
Crisford, H., Sapey, E., Rogers, G. B., Taylor, S., Nagakumar, P., Lokwani, R., et al. (2021). Neutrophils in asthma: The good, the bad and the bacteria. Thorax 76, 835–844. doi:10.1136/thoraxjnl-2020-215986
Daines, M., Pereira, R., Cunningham, A., Pryor, B., Besselsen, D. G., Liu, Y., et al. (2021). Novel mouse models of fungal asthma. Front. Cell Infect. Microbiol. 11, 683194. doi:10.3389/fcimb.2021.683194
Devos, F. C., Maaske, A., Robichaud, A., Pollaris, L., Seys, S., Lopez, C. A., et al. (2017). Forced expiration measurements in mouse models of obstructive and restrictive lung diseases. Respir. Res. 18, 123. doi:10.1186/s12931-017-0610-1
Dey, S., Lu, W., Weber, H. C., Young, S., Larby, J., Chia, C., et al. (2022). Differential airway remodeling changes were observed in patients with asthma COPD overlap compared to patients with asthma and COPD alone. Am. J. Physiol. Lung Cell Mol. Physiol. 323, L473–L483. doi:10.1152/ajplung.00137.2022
Deykin, A., Wechsler, M. E., Boushey, H. A., Chinchilli, V. M., Kunselman, S. J., Craig, T. J., et al. (2007). Combination therapy with a long-acting beta-agonist and a leukotriene antagonist in moderate asthma. Am. J. Respir. Crit. Care Med. 175, 228–234. doi:10.1164/rccm.200601-112OC
Diseases, G. B. D., and Injuries, C. (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the global burden of disease study 2019. Lancet 396, 1204–1222. doi:10.1016/S0140-6736(20)30925-9
Dixon, A. E., Pratley, R. E., Forgione, P. M., Kaminsky, D. A., Whittaker-Leclair, L. A., Griffes, L. A., et al. (2011). Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J. Allergy Clin. Immunol. 128, 508–515.e1-2. doi:10.1016/j.jaci.2011.06.009
Donovan, C., Royce, S. G., Esposito, J., Tran, J., Ibrahim, Z. A., Tang, M. L., et al. (2013). Differential effects of allergen challenge on large and small airway reactivity in mice. PLoS One 8, e74101. doi:10.1371/journal.pone.0074101
Donovan, C., Royce, S. G., Vlahos, R., and Bourke, J. E. (2015). Lipopolysaccharide does not alter small airway reactivity in mouse lung slices. PLoS One 10, e0122069. doi:10.1371/journal.pone.0122069
Doran, E., Cai, F., Holweg, C. T. J., Wong, K., Brumm, J., and Arron, J. R. (2017). Interleukin-13 in asthma and other eosinophilic disorders. Front. Med. 4, 139. doi:10.3389/fmed.2017.00139
The ENFUMOSA cross-sectional European multicentre study of the clinical phenotype of chronic severe asthma. European Network for Understanding Mechanisms of Severe Asthma. Eur. Respir. J. 2003, 22:470–477.
Essilfie, A-T., Simpson, J. L., Dunkley, M. L., Morgan, L. C., Oliver, B. G., Gibson, P. G., et al. (2012). Combined Haemophilus influenzae respiratory infection and allergic airways disease drives chronic infection and features of neutrophilic asthma. Thorax 67, 588–599. doi:10.1136/thoraxjnl-2011-200160
Essilfie, A. T., Horvat, J. C., Kim, R. Y., Mayall, J. R., Pinkerton, J. W., Beckett, E. L., et al. (2015). Macrolide therapy suppresses key features of experimental steroid-sensitive and steroid-insensitive asthma. Thorax 70, 458–467. doi:10.1136/thoraxjnl-2014-206067
Ferrari, L., Carugno, M., and Bollati, V. (2019). Particulate matter exposure shapes DNA methylation through the lifespan. Clin. Epigenetics 11, 129. doi:10.1186/s13148-019-0726-x
Finney, M. J., Karlsson, J. A., and Persson, C. G. (1985). Effects of bronchoconstrictors and bronchodilators on a novel human small airway preparation. Br. J. Pharmacol. 85, 29–36. doi:10.1111/j.1476-5381.1985.tb08827.x
Fredberg, J. J., Inouye, D., Miller, B., Nathan, M., Jafari, S., Raboudi, S. H., et al. (1997). Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am. J. Respir. Crit. Care Med. 156, 1752–1759. doi:10.1164/ajrccm.156.6.9611016
Gauthier, M., Chakraborty, K., Oriss, T. B., Raundhal, M., Das, S., Chen, J., et al. (2017). Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight 2, e94580. doi:10.1172/jci.insight.94580
Gauthier, M., Kale, S. L., Oriss, T. B., Scholl, K., Das, S., Yuan, H., et al. (2022). Dual role for CXCR3 and CCR5 in asthmatic type 1 inflammation. J. Allergy Clin. Immunol. 149, 113–124.e7. doi:10.1016/j.jaci.2021.05.044
Ghio, A. J., Roggli, V. L., Soukup, J. M., Richards, J. H., Randell, S. H., and Muhlebach, M. S. (2013). Iron accumulates in the lavage and explanted lungs of cystic fibrosis patients. J. Cyst. Fibros. 12, 390–398. doi:10.1016/j.jcf.2012.10.010
Gilliland, F. D., Li, Y. F., and Peters, J. M. (2001). Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am. J. Respir. Crit. Care Med. 163, 429–436. doi:10.1164/ajrccm.163.2.2006009
Gomez, H. M., Pillar, A. L., Brown, A. C., Kim, R. Y., Ali, M. K., Essilfie, A. T., et al. (2021). Investigating the links between lower iron status in pregnancy and respiratory disease in offspring using murine models. Nutrients 13, 4461. doi:10.3390/nu13124461
Green, B. J., Wiriyachaiporn, S., Grainge, C., Rogers, G. B., Kehagia, V., Lau, L., et al. (2014). Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS One 9, e100645. doi:10.1371/journal.pone.0100645
Guan, W. J., Zheng, J. P., Gao, Y., Jiang, C. Y., Xie, Y. Q., Shi, X., et al. (2013). Responsiveness to leukotriene D4 and methacholine for predicting efficacy of montelukast in asthma. J. Thorac. Dis. 5, 298–301. doi:10.3978/j.issn.2072-1439.2013.02.01
Hall, S., and Agrawal, D. K. (2014). Key mediators in the immunopathogenesis of allergic asthma. Int. Immunopharmacol. 23, 316–329. doi:10.1016/j.intimp.2014.05.034
Hamid, Q., Song, Y., Kotsimbos, T. C., Minshall, E., Bai, T. R., Hegele, R. G., et al. (1997). Inflammation of small airways in asthma. J. Allergy Clin. Immunol. 100, 44–51. doi:10.1016/s0091-6749(97)70193-3
Hansbro, N. G., Horvat, J. C., Wark, P. A., and Hansbro, P. M. (2008). Understanding the mechanisms of viral induced asthma: New therapeutic directions. Pharmacol. Ther. 117, 313–353. doi:10.1016/j.pharmthera.2007.11.002
Hansbro, P. M., Beagley, K. W., Horvat, J. C., and Gibson, P. G. (2004). Role of atypical bacterial infection of the lung in predisposition/protection of asthma. Pharmacol. Ther. 101, 193–210. doi:10.1016/j.pharmthera.2003.10.007
Harju, M., Keski-Nisula, L., Georgiadis, L., and Heinonen, S. (2016). Parental smoking and cessation during pregnancy and the risk of childhood asthma. BMC Public Health 16, 428. doi:10.1186/s12889-016-3029-6
Hayatbakhsh, M. R., Sadasivam, S., Mamun, A. A., Najman, J. M., Williams, G. M., and O'Callaghan, M. J. (2009). Maternal smoking during and after pregnancy and lung function in early adulthood: A prospective study. Thorax 64, 810–814. doi:10.1136/thx.2009.116301
Hazlehurst, M. F., Carroll, K. N., Loftus, C. T., Szpiro, A. A., Moore, P. E., Kaufman, J. D., et al. (2021). Maternal exposure to PM2.5 during pregnancy and asthma risk in early childhood: Consideration of phases of fetal lung development. Environ. Epidemiol. 5, e130. doi:10.1097/ee9.0000000000000130
He, S. H., Zhang, H. Y., Zeng, X. N., Chen, D., and Yang, P. C. (2013). Mast cells and basophils are essential for allergies: Mechanisms of allergic inflammation and a proposed procedure for diagnosis. Acta Pharmacol. Sin. 34, 1270–1283. doi:10.1038/aps.2013.88
Hirst, S. J. (2003). Regulation of airway smooth muscle cell immunomodulatory function: Role in asthma. Respir. Physiol. Neurobiol. 137, 309–326. doi:10.1016/s1569-9048(03)00155-1
Jacquet, A. (2021). Characterization of innate immune responses to house dust mite allergens: Pitfalls and limitations. Front. Allergy 2, 662378. doi:10.3389/falgy.2021.662378
James, A. L., Elliot, J. G., Jones, R. L., Carroll, M. L., Mauad, T., Bai, T. R., et al. (2012). Airway smooth muscle hypertrophy and hyperplasia in asthma. Am. J. Respir. Crit. Care Med. 185, 1058–1064. doi:10.1164/rccm.201110-1849OC
James, A. L., and Wenzel, S. (2007). Clinical relevance of airway remodelling in airway diseases. Eur. Respir. J. 30, 134–155. doi:10.1183/09031936.00146905
Jankovic, D., Kullberg, M. C., Noben-Trauth, N., Caspar, P., Paul, W. E., and Sher, A. (2000). Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4+ lymphocytes with a Th2 cytokine profile. J. Immunol. 164, 3047–3055. doi:10.4049/jimmunol.164.6.3047
Jayaram, L., Duong, M., Pizzichini, M. M., Pizzichini, E., Kamada, D., Efthimiadis, A., et al. (2005). Failure of montelukast to reduce sputum eosinophilia in high-dose corticosteroid-dependent asthma. Eur. Respir. J. 25, 41–46. doi:10.1183/09031936.04.00008104
John, M., Au, B. T., Jose, P. J., Lim, S., Saunders, M., Barnes, P. J., et al. (1998). Expression and release of interleukin-8 by human airway smooth muscle cells: Inhibition by Th-2 cytokines and corticosteroids. Am. J. Respir. Cell Mol. Biol. 18, 84–90. doi:10.1165/ajrcmb.18.1.2813
Joubert, B. R., Felix, J. F., Yousefi, P., Bakulski, K. M., Just, A. C., Breton, C., et al. (2016). DNA methylation in newborns and maternal smoking in pregnancy: Genome-wide consortium meta-analysis. Am. J. Hum. Genet. 98, 680–696. doi:10.1016/j.ajhg.2016.02.019
Juul, S. E., Derman, R. J., and Auerbach, M. (2019). Perinatal iron deficiency: Implications for mothers and infants. Neonatology 115, 269–274. doi:10.1159/000495978
Kalidhindi, R. S. R., Ambhore, N. S., Bhallamudi, S., Loganathan, J., and Sathish, V. (2019). Role of estrogen receptors α and β in a murine model of asthma: Exacerbated airway hyperresponsiveness and remodeling in ERβ knockout mice. Front. Pharmacol. 10, 1499. doi:10.3389/fphar.2019.01499
Kibe, A., Inoue, H., Fukuyama, S., Machida, K., Matsumoto, K., Koto, H., et al. (2003). Differential regulation by glucocorticoid of interleukin-13-induced eosinophilia, hyperresponsiveness, and goblet cell hyperplasia in mouse airways. Am. J. Respir. Crit. Care Med. 167, 50–56. doi:10.1164/rccm.2110084
Kim, D. I., Song, M-K., and Lee, K. Y. (2019). Comparison of asthma phenotypes in OVA-induced mice challenged via inhaled and intranasal routes. BMC Pulm. Med. 19, 241. doi:10.1186/s12890-019-1001-9
Kim, H. Y., Lee, H. J., Chang, Y. J., Pichavant, M., Shore, S. A., Fitzgerald, K. A., et al. (2014). Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med. 20, 54–61. doi:10.1038/nm.3423
Kim, M. J., Kim, Y. Y., Choi, Y. A., Baek, M. C., Lee, B., Park, P. H., et al. (2018). Elaeocarpusin inhibits mast cell-mediated allergic inflammation. Front. Pharmacol. 9, 591. doi:10.3389/fphar.2018.00591
Kim, R. Y., Horvat, J. C., Pinkerton, J. W., Starkey, M. R., Essilfie, A. T., Mayall, J. R., et al. (2017). MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase–mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 139, 519–532. doi:10.1016/j.jaci.2016.04.038
Kim, R. Y., Pinkerton, J. W., Essilfie, A. T., Robertson, A. A. B., Baines, K. J., Brown, A. C., et al. (2017). Role for NLRP3 inflammasome-mediated, IL-1β-dependent responses in severe, steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 196, 283–297. doi:10.1164/rccm.201609-1830OC
Kirstein, F., Horsnell, W. G., Kuperman, D. A., Huang, X., Erle, D. J., Lopata, A. L., et al. (2010). Expression of IL-4 receptor alpha on smooth muscle cells is not necessary for development of experimental allergic asthma. J. Allergy Clin. Immunol. 126, 347–354. doi:10.1016/j.jaci.2010.04.028
Kobayashi, T., Iijima, K., Dent, A. L., and Kita, H. (2017). Follicular helper T cells mediate IgE antibody response to airborne allergens. J. Allergy Clin. Immunol. 139, 300–313.e7. doi:10.1016/j.jaci.2016.04.021
Kudo, M., Ishigatsubo, Y., and Aoki, I. (2013). Pathology of asthma. Front. Microbiol. 4, 263. doi:10.3389/fmicb.2013.00263
Kuperman, D. A., Huang, X., Koth, L. L., Chang, G. H., Dolganov, G. M., Zhu, Z., et al. (2002). Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8, 885–889. doi:10.1038/nm734
Kuruvilla, M. E., Lee, F. E., and Lee, G. B. (2019). Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin. Rev. Allergy Immunol. 56, 219–233. doi:10.1007/s12016-018-8712-1
Laporte, J. C., Moore, P. E., Baraldo, S., Jouvin, M. H., Church, T. L., Schwartzman, I. N., et al. (2001). Direct effects of interleukin-13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am. J. Respir. Crit. Care Med. 164, 141–148. doi:10.1164/ajrccm.164.1.2008060
Larsson, K., Stallberg, B., Lisspers, K., Telg, G., Johansson, G., Thuresson, M., et al. (2018). Prevalence and management of severe asthma in primary care: An observational cohort study in Sweden (PACEHR). Respir. Res. 19, 12. doi:10.1186/s12931-018-0719-x
Lee, A., Leon Hsu, H. H., Mathilda Chiu, Y. H., Bose, S., Rosa, M. J., Kloog, I., et al. (2018). Prenatal fine particulate exposure and early childhood asthma: Effect of maternal stress and fetal sex. J. Allergy Clin. Immunol. 141, 1880–1886. doi:10.1016/j.jaci.2017.07.017
Lee, J. H., Kaminski, N., Dolganov, G., Grunig, G., Koth, L., Solomon, C., et al. (2001). Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am. J. Respir. Cell Mol. Biol. 25, 474–485. doi:10.1165/ajrcmb.25.4.4522
Lehmann, S., Sprünken, A., Wagner, N., Tenbrock, K., and Ott, H. (2017). Clinical relevance of IgE-mediated sensitization against the mould Alternaria alternata in children with asthma. Ther. Adv. Respir. Dis. 11, 30–39. doi:10.1177/1753465816680786
Leung, J. M., and Sin, D. D. (2017). Asthma-COPD overlap syndrome: Pathogenesis, clinical features, and therapeutic targets. BMJ 358, j3772. doi:10.1136/bmj.j3772
Li, G., Saad, S., Oliver, B. G., and Chen, H. (2018). Heat or burn? Impacts of intrauterine tobacco smoke and E-cigarette vapor exposure on the offspring's health outcome. Toxics 6, 43. doi:10.3390/toxics6030043
Li, R., Peng, X., Wu, Y., Lv, W., Xie, H., Ishii, Y., et al. (2021). Exposure to PM2.5 during pregnancy causes lung inflammation in the offspring: Mechanism of action of mogrosides. Ecotoxicol. Environ. Saf. 228, 112955. doi:10.1016/j.ecoenv.2021.112955
Liao, H., Yang, Z., Yang, C., Tang, Y., Liu, S., Guan, W., et al. (2016). Impact of viral infection on acute exacerbation of asthma in out-patient clinics: A prospective study. J. Thorac. Dis. 8, 505–512. doi:10.21037/jtd.2016.02.76
Liu, G., Cooley, M. A., Nair, P. M., Donovan, C., Hsu, A. C., Jarnicki, A. G., et al. (2017). Airway remodelling and inflammation in asthma are dependent on the extracellular matrix protein fibulin-1c. J. Pathol. 243, 510–523. doi:10.1002/path.4979
Lotvall, J., Inman, M., and O'Byrne, P. (1998). Measurement of airway hyperresponsiveness: New considerations. Thorax 53, 419–424. doi:10.1136/thx.53.5.419
Louis, R., Satia, I., Ojanguren, I., Schleich, F., Bonini, M., Tonia, T., et al. (2022). European respiratory society guidelines for the diagnosis of asthma in adults. Eur. Respir. J. 60, 2101585. doi:10.1183/13993003.01585-2021
Makino, A., Shibata, T., Nagayasu, M., Hosoya, I., Nishimura, T., Nakano, C., et al. (2021). RSV infection-elicited high MMP-12–producing macrophages exacerbate allergic airway inflammation with neutrophil infiltration. iScience 24, 103201. doi:10.1016/j.isci.2021.103201
Marthan, R., Martin, C., Amedee, T., and Mironneau, J. (1989). Calcium channel currents in isolated smooth muscle cells from human bronchus. J. Appl. Physiol. 66, 1706–1714. doi:10.1152/jappl.1989.66.4.1706
Matsumoto, H., Moir, L. M., Oliver, B. G., Burgess, J. K., Roth, M., Black, J. L., et al. (2007). Comparison of gel contraction mediated by airway smooth muscle cells from patients with and without asthma. Thorax 62, 848–854. doi:10.1136/thx.2006.070474
Matsuse, H., Fukushima, C., Fukahori, S., Tsuchida, T., Kawano, T., Nishino, T., et al. (2013). Differential effects of dexamethasone and itraconazole on Aspergillus fumigatus-exacerbated allergic airway inflammation in a murine model of mite-sensitized asthma. Respiration 85, 429–435. doi:10.1159/000345861
McKay, S., Hirst, S. J., Haas, M. B., de Jongste, J. C., Hoogsteden, H. C., Saxena, P. R., et al. (2000). Tumor necrosis factor-alpha enhances mRNA expression and secretion of interleukin-6 in cultured human airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 23, 103–111. doi:10.1165/ajrcmb.23.1.3765
Mechiche, H., Naline, E., Candenas, L., Pinto, F. M., Birembault, P., Advenier, C., et al. (2003). Effects of cysteinyl leukotrienes in small human bronchus and antagonist activity of montelukast and its metabolites. Clin. Exp. Allergy 33, 887–894. doi:10.1046/j.1365-2222.2003.01696.x
Mikhail, I., and Grayson, M. H. (2019). Asthma and viral infections: An intricate relationship. Ann. Allergy Asthma Immunol. 123, 352–358. doi:10.1016/j.anai.2019.06.020
Mookerjee, I., Solly, N. R., Royce, S. G., Tregear, G. W., Samuel, C. S., and Tang, M. L. (2006). Endogenous relaxin regulates collagen deposition in an animal model of allergic airway disease. Endocrinology 147, 754–761. doi:10.1210/en.2005-1006
Moore, W. C., Bleecker, E. R., Curran-Everett, D., Erzurum, S. C., Ameredes, B. T., Bacharier, L., et al. (2007). Characterization of the severe asthma phenotype by the national heart, lung, and blood institute's severe asthma research program. J. Allergy Clin. Immunol. 119, 405–413. doi:10.1016/j.jaci.2006.11.639
Nesmith, A. P., Agarwal, A., McCain, M. L., and Parker, K. K. (2014). Human airway musculature on a chip: An in vitro model of allergic asthmatic bronchoconstriction and bronchodilation. Lab. Chip 14, 3925–3936. doi:10.1039/c4lc00688g
Nguyen, T. H., Maltby, S., Eyers, F., Foster, P. S., and Yang, M. (2016). Bromodomain and extra terminal (BET) inhibitor suppresses macrophage-driven steroid-resistant exacerbations of airway hyper-responsiveness and inflammation. PLoS One 11, e0163392. doi:10.1371/journal.pone.0163392
Nguyen, T. H., Maltby, S., Simpson, J. L., Eyers, F., Baines, K. J., Gibson, P. G., et al. (2016). TNF-Α and macrophages are critical for respiratory syncytial virus–induced exacerbations in a mouse model of allergic airways disease. J. Immunol. 196, 3547–3558. doi:10.4049/jimmunol.1502339
Nguyen, T. H., Maltby, S., Tay, H. L., Eyers, F., Foster, P. S., and Yang, M. (2018). Identification of IFN-gamma and IL-27 as critical regulators of respiratory syncytial virus-induced exacerbation of allergic airways disease in a mouse model. J. Immunol. 200, 237–247. doi:10.4049/jimmunol.1601950
Nicolas, G., Bennoun, M., Porteu, A., Mativet, S., Beaumont, C., Grandchamp, B., et al. (2002). Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. 99, 4596–4601. doi:10.1073/pnas.072632499
Noakes, P. S., Holt, P. G., and Prescott, S. L. (2003). Maternal smoking in pregnancy alters neonatal cytokine responses. Allergy 58, 1053–1058. doi:10.1034/j.1398-9995.2003.00290.x
Oriss, T. B., Raundhal, M., Morse, C., Huff, R. E., Das, S., Hannum, R., et al. (2017). IRF5 distinguishes severe asthma in humans and drives Th1 phenotype and airway hyperreactivity in mice. JCI Insight 2, e91019. doi:10.1172/jci.insight.91019
Parikh, M. N., Brokamp, C., Rasnick, E., Ding, L., Mersha, T. B., Bowers, K., et al. (2022). Epigenome-wide association of neonatal methylation and trimester-specific prenatal PM2.5 exposure. Environ. Epidemiol. 6, e227. doi:10.1097/EE9.0000000000000227
Pepe, C., Foley, S., Shannon, J., Lemiere, C., Olivenstein, R., Ernst, P., et al. (2005). Differences in airway remodeling between subjects with severe and moderate asthma. J. Allergy Clin. Immunol. 116, 544–549. doi:10.1016/j.jaci.2005.06.011
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465, 721–727. doi:10.1038/nature09230
Pinkerton, J. W., Kim, R. Y., Brown, A. C., Rae, B. E., Donovan, C., Mayall, J. R., et al. (2022). Relationship between type 2 cytokine and inflammasome responses in obesity-associated asthma. J. Allergy Clin. Immunol. 149, 1270–1280. doi:10.1016/j.jaci.2021.10.003
Piyadasa, H., Hemshekhar, M., Altieri, A., Basu, S., van der Does, A. M., Halayko, A. J., et al. (2018). Immunomodulatory innate defence regulator (IDR) peptide alleviates airway inflammation and hyper-responsiveness. Thorax 73, 908–917. doi:10.1136/thoraxjnl-2017-210739
Pope, S. M., Brandt, E. B., Mishra, A., Hogan, S. P., Zimmermann, N., Matthaei, K. I., et al. (2001). IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J. Allergy Clin. Immunol. 108, 594–601. doi:10.1067/mai.2001.118600
Quirt, J., Hildebrand, K. J., Mazza, J., Noya, F., and Kim, H. (2018). AsthmaAllergy Asthma Clin. Immunol. 14, 50. doi:10.1186/s13223-018-0279-0
Ramakrishnan, K., and Borade, A. (2010). Anemia as a risk factor for childhood asthma. Lung India 27, 51–53. doi:10.4103/0970-2113.63605
Raundhal, M., Morse, C., Khare, A., Oriss, T. B., Milosevic, J., Trudeau, J., et al. (2015). High IFN-gamma and low SLPI mark severe asthma in mice and humans. J. Clin. Invest. 125, 3037–3050. doi:10.1172/JCI80911
Reid, D. W., Carroll, V., O'May, C., Champion, A., and Kirov, S. M. (2007). Increased airway iron as a potential factor in the persistence of Pseudomonas aeruginosa infection in cystic fibrosis. Eur. Respir. J. 30, 286–292. doi:10.1183/09031936.00154006
Ressmeyer, A. R., Bai, Y., Delmotte, P., Uy, K. F., Thistlethwaite, P., Fraire, A., et al. (2010). Human airway contraction and formoterol-induced relaxation is determined by Ca2+ oscillations and Ca2+ sensitivity. Am. J. Respir. Cell Mol. Biol. 43, 179–191. doi:10.1165/rcmb.2009-0222OC
Ronnebjerg, L., Axelsson, M., Kankaanranta, H., Backman, H., Radinger, M., Lundback, B., et al. (2021). Severe asthma in a general population study: Prevalence and clinical characteristics. J. Asthma Allergy 14, 1105–1115. doi:10.2147/JAA.S327659
Rosenblat, J. D., Husain, M. I., Lee, Y., McIntyre, R. S., Mansur, R. B., Castle, D., et al. (2022). The Canadian network for mood and anxiety treatments (CANMAT) task force report: Serotonergic psychedelic treatments for major depressive disorder. Can. J. Psychiatry 68, 5–21. doi:10.1177/07067437221111371
Royce, S. G., Dang, W., Yuan, G., Tran, J., El Osta, A., Karagiannis, T. C., et al. (2011). Resveratrol has protective effects against airway remodeling and airway hyperreactivity in a murine model of allergic airways disease. Pathobiol. Aging Age Relat. Dis. 1, 7134. doi:10.3402/pba.v1i0.7134
Royce, S. G., Licciardi, P. V., Beh, R. C., Bourke, J. E., Donovan, C., Hung, A., et al. (2022). Sulforaphane prevents and reverses allergic airways disease in mice via anti-inflammatory, antioxidant and epigenetic mechanisms. Cell Mol. Life Sci. 79, 579. doi:10.1007/s00018-022-04609-3
Sanderson, M. J., Delmotte, P., Bai, Y., and Perez-Zogbhi, J. F. (2008). Regulation of airway smooth muscle cell contractility by Ca2+ signaling and sensitivity. Proc. Am. Thorac. Soc. 5, 23–31. doi:10.1513/pats.200704-050VS
Sangkhae, V., Fisher, A. L., Chua, K. J., Ruchala, P., Ganz, T., and Nemeth, E. (2020). Maternal hepcidin determines embryo iron homeostasis in mice. Blood 136, 2206–2216. doi:10.1182/blood.2020005745
Schwartz, A. J., Converso-Baran, K., Michele, D. E., and Shah, Y. M. (2019). A genetic mouse model of severe iron deficiency anemia reveals tissue-specific transcriptional stress responses and cardiac remodeling. J. Biol. Chem. 294, 14991–15002. doi:10.1074/jbc.RA119.009578
Shaheen, S. O., Macdonald-Wallis, C., Lawlor, D. A., and Henderson, A. J. (2017). Haemoglobin concentrations in pregnancy and respiratory and allergic outcomes in childhood: Birth cohort study. Clin. Exp. Allergy 47, 1615–1624. doi:10.1111/cea.13034
Shrine, N., Portelli, M. A., John, C., Soler Artigas, M., Bennett, N., Hall, R., et al. (2019). Moderate-to-severe asthma in individuals of European ancestry: A genome-wide association study. Lancet Respir. Med. 7, 20–34. doi:10.1016/S2213-2600(18)30389-8
Simpson, J. L., Grissell, T. V., Douwes, J., Scott, R. J., Boyle, M. J., and Gibson, P. G. (2007). Innate immune activation in neutrophilic asthma and bronchiectasis. Thorax 62, 211–218. doi:10.1136/thx.2006.061358
Sintobin, I., Siroux, V., Holtappels, G., Pison, C., Nadif, R., Bousquet, J., et al. (2019). Sensitisation to staphylococcal enterotoxins and asthma severity: A longitudinal study in the EGEA cohort. Eur. Respir. J. 54, 1900198. doi:10.1183/13993003.00198-2019
Sukjamnong, S., Chan, Y. L., Zakarya, R., Saad, S., Sharma, P., Santiyanont, R., et al. (2017). Effect of long-term maternal smoking on the offspring's lung health. Am. J. Physiol. Lung Cell Mol. Physiol. 313, L416–l423. doi:10.1152/ajplung.00134.2017
Sukkar, M. B., Hughes, J. M., Armour, C. L., and Johnson, P. R. (2001). Tumour necrosis factor-alpha potentiates contraction of human bronchus in vitro. Respirol 6, 199–203. doi:10.1046/j.1440-1843.2001.00334.x
Szefler, S. J. (2015). Advances in pediatric asthma in 2014: Moving toward a population health perspective. J. Allergy Clin. Immunol. 135, 644–652. doi:10.1016/j.jaci.2014.12.1921
Tliba, O., Deshpande, D., Chen, H., Van Besien, C., Kannan, M., Panettieri, R. A., et al. (2003). IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br. J. Pharmacol. 140, 1159–1162. doi:10.1038/sj.bjp.0705558
To, T., Stanojevic, S., Moores, G., Gershon, A. S., Bateman, E. D., Cruz, A. A., et al. (2012). Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Public Health 12, 204. doi:10.1186/1471-2458-12-204
Trevor, J. L., and Deshane, J. S. (2014). Refractory asthma: Mechanisms, targets, and therapy. Allergy 69, 817–827. doi:10.1111/all.12412
Triche, E. W., Lundsberg, L. S., Wickner, P. G., Belanger, K., Leaderer, B. P., and Bracken, M. B. (2011). Association of maternal anemia with increased wheeze and asthma in children. Ann. Allergy Asthma Immunol. 106, 131–139.e1. doi:10.1016/j.anai.2010.11.007
Tu, X., Donovan, C., Kim, R. Y., Wark, P. A. B., Horvat, J. C., and Hansbro, P. M. (2021). Asthma-COPD overlap: Current understanding and the utility of experimental models. Eur. Respir. Rev. 30, 190185. doi:10.1183/16000617.0185-2019
Tu, X., Kim, R. Y., Brown, A. C., de Jong, E., Jones-Freeman, B., Ali, M. K., et al. (2022). Airway and parenchymal transcriptomics in a novel model of asthma and COPD overlap. J. Allergy Clin. Immunol. 150, 817–829.e6. doi:10.1016/j.jaci.2022.04.032
Turner, S. W., Young, S., Goldblatt, J., Landau, L. I., and Le Souëf, P. N. (2009). Childhood asthma and increased airway responsiveness: A relationship that begins in infancy. Am. J. Respir. Crit. Care Med. 179, 98–104. doi:10.1164/rccm.200805-804OC
Uwadiae, F. I., Pyle, C. J., Walker, S. A., Lloyd, C. M., and Harker, J. A. (2019). Targeting the ICOS/ICOS-L pathway in a mouse model of established allergic asthma disrupts T follicular helper cell responses and ameliorates disease. Allergy 74, 650–662. doi:10.1111/all.13602
van Tilburg Bernardes, E., Gutierrez, M. W., and Arrieta, M. C. (2020). The fungal microbiome and asthma. Front. Cell Infect. Microbiol. 10, 583418. doi:10.3389/fcimb.2020.583418
Vargaftig, B. B., and Singer, M. (2003). Leukotrienes mediate murine bronchopulmonary hyperreactivity, inflammation, and part of mucosal metaplasia and tissue injury induced by recombinant murine interleukin-13. Am. J. Respir. Cell Mol. Biol. 28, 410–419. doi:10.1165/rcmb.2002-0032OC
Venkayya, R., Lam, M., Willkom, M., Grunig, G., Corry, D. B., and Erle, D. J. (2002). The Th2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am. J. Respir. Cell Mol. Biol. 26, 202–208. doi:10.1165/ajrcmb.26.2.4600
Vlasic, V., Trifunovic, J., Cepelak, I., Nimac, P., Topic, R. Z., and Dodig, S. (2011). Urates in exhaled breath condensate of children with obstructive sleep apnea. Biochem. Med. 21, 139–144. doi:10.11613/bm.2011.022
Wagner, E. M., Bleecker, E. R., Permutt, S., and Liu, M. C. (1998). Direct assessment of small airways reactivity in human subjects. Am. J. Respir. Crit. Care Med. 157, 447–452. doi:10.1164/ajrccm.157.2.9611043
Wang, B., Chan, Y. L., Li, G., Ho, K. F., Anwer, A. G., Smith, B. J., et al. (2021). Maternal particulate matter exposure impairs lung health and is associated with mitochondrial damage. Antioxidants 10, 1029. doi:10.3390/antiox10071029
Wang, B., Chen, H., Chan, Y. L., Wang, G., and Oliver, B. G. (2020). Why do intrauterine exposure to air pollution and cigarette smoke increase the risk of asthma? Front. Cell Dev. Biol. 8, 38. doi:10.3389/fcell.2020.00038
Wang, L., Netto, K. G., Zhou, L., Liu, X., Wang, M., Zhang, G., et al. (2021). Single-cell transcriptomic analysis reveals the immune landscape of lung in steroid-resistant asthma exacerbation. Proc. Natl. Acad. Sci. 118, e2005590118. doi:10.1073/pnas.2005590118
Wang, P., You, D., Saravia, J., Shen, H., and Cormier, S. A. (2013). Maternal exposure to combustion generated PM inhibits pulmonary Th1 maturation and concomitantly enhances postnatal asthma development in offspring. Part Fibre Toxicol. 10, 29. doi:10.1186/1743-8977-10-29
Wark, P. A., Johnston, S. L., Moric, I., Simpson, J. L., Hensley, M. J., and Gibson, P. G. (2002). Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur. Respir. J. 19, 68–75. doi:10.1183/09031936.02.00226302
Wenzel, S. E. (2016). Emergence of biomolecular pathways to define novel asthma phenotypes. Type-2 immunity and beyond. Am. J. Respir. Cell Mol. Biol. 55, 1–4. doi:10.1165/rcmb.2016-0141PS
WHO (2022). The world health organisation - asthma. Available at: https://www.who.int/news-room/fact-sheets/detail/asthma.
Woo, L. N., Guo, W. Y., Wang, X., Young, A., Salehi, S., Hin, A., et al. (2018). A 4-week model of house dust mite (HDM) induced allergic airways inflammation with airway remodeling. Sci. Rep. 8, 6925. doi:10.1038/s41598-018-24574-x
Wood, L. G., Simpson, J. L., Hansbro, P. M., and Gibson, P. G. (2010). Potentially pathogenic bacteria cultured from the sputum of stable asthmatics are associated with increased 8-isoprostane and airway neutrophilia. Free Radic. Res. 44, 146–154. doi:10.3109/10715760903362576
Woodruff, P. G., Dolganov, G. M., Ferrando, R. E., Donnelly, S., Hays, S. R., Solberg, O. D., et al. (2004). Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am. J. Respir. Crit. Care Med. 169, 1001–1006. doi:10.1164/rccm.200311-1529OC
Woodruff, P. G., Modrek, B., Choy, D. F., Jia, G., Abbas, A. R., Ellwanger, A., et al. (2009). T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 180, 388–395. doi:10.1164/rccm.200903-0392OC
Woolcock, A. J., Dusser, D., and Fajac, I. (1998). Severity of chronic asthma. Thorax 53, 442–444. doi:10.1136/thx.53.6.442
Worren, M. K., Storesund, A. K., Sharma, R., Vintermyr, O. K., Gjerde, S., and Flaatten, H. K. (2018). Plugs of the air passages. Tidsskr. Nor. Laegeforen 138. doi:10.4045/tidsskr.18.0133
Xi, G. P., Zhang, Q., and Yin, J. (2021). Establishment and characterization of murine models of asthma and subcutaneous immunotherapy for Humulus pollen allergy. Immun. Inflamm. Dis. 9, 443–455. doi:10.1002/iid3.405
Xiao, S., Wang, Q., Gao, H., Zhao, X., Zhi, J., and Yang, D. (2022). Dexmedetomidine alleviates airway hyperresponsiveness and allergic airway inflammation through the TLR4/NF‑κB signaling pathway in mice. Mol. Med. Rep. 25, 74. doi:10.3892/mmr.2022.12590
Xie, Z., Sun, H., Li, X., Sun, W., and Yin, J. (2021). Alteration of lung tissues proteins in birch pollen induced asthma mice before and after SCIT. PLoS One 16, e0258051. doi:10.1371/journal.pone.0258051
Xu, X. F., Li, Y. J., Sheng, Y. J., Liu, J. L., Tang, L. F., and Chen, Z. M. (2014). Effect of low birth weight on childhood asthma: A meta-analysis. BMC Pediatr. 14, 275. doi:10.1186/1471-2431-14-275
Yang, M., Hogan, S. P., Henry, P. J., Matthaei, K. I., McKenzie, A. N., Young, I. G., et al. (2001). Interleukin-13 mediates airways hyperreactivity through the IL-4 receptor-alpha chain and STAT-6 independently of IL-5 and eotaxin. Am. J. Respir. Cell Mol. Biol. 25, 522–530. doi:10.1165/ajrcmb.25.4.4620
Zacharasiewicz, A. (2016). Maternal smoking in pregnancy and its influence on childhood asthma. ERJ Open Res. 2, 00042. doi:10.1183/23120541.00042-2016
Zaretsky, I., Atrakchi, O., Mazor, R. D., Stoler-Barak, L., Biram, A., Feigelson, S. W., et al. (2017). ICAMs support B cell interactions with T follicular helper cells and promote clonal selection. J. Exp. Med. 214, 3435–3448. doi:10.1084/jem.20171129
Zhang, Y., Cardell, L. O., and Adner, M. (2007). IL-1β induces murine airway 5-HT2A receptor hyperresponsiveness via a non-transcriptional MAPK-dependent mechanism. Respir. Res. 8, 29. doi:10.1186/1465-9921-8-29
Zhou, G., He, T., Huang, H., Feng, F., Liu, X., Li, Z., et al. (2019). Prenatal ambient air pollution exposure and SOD2 promoter methylation in maternal and cord blood. Ecotoxicol. Environ. Saf. 181, 428–434. doi:10.1016/j.ecoenv.2019.06.039
Keywords: asthma1, lung function2, airway hyperresponsiveness3, mouse models4, severe asthma5
Citation: Carroll OR, Pillar AL, Brown AC, Feng M, Chen H and Donovan C (2023) Advances in respiratory physiology in mouse models of experimental asthma. Front. Physiol. 14:1099719. doi: 10.3389/fphys.2023.1099719
Received: 16 November 2022; Accepted: 07 February 2023;
Published: 16 March 2023.
Reviewed by:
Jean-Francois Lauzon-Joset, Laval University, CanadaCopyright © 2023 Carroll, Pillar, Brown, Feng, Chen and Donovan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chantal Donovan, Y2hhbnRhbC5kb25vdmFuQHV0cy5lZHUuYXU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.