 Rebecca Berggren-Nylund1Martin Ryde2
Rebecca Berggren-Nylund1Martin Ryde2 Anna Löfdahl1
Anna Löfdahl1 Arturo Ibáñez-Fonseca1Monica Kåredal3
Arturo Ibáñez-Fonseca1Monica Kåredal3 Gunilla Westergren-Thorsson1
Gunilla Westergren-Thorsson1 Ellen Tufvesson2
Ellen Tufvesson2 Anna-Karin Larsson-Callerfelt1*
Anna-Karin Larsson-Callerfelt1*- 1Lung Biology, Department of Experimental Medical Science, Lund University, Lund, Sweden
- 2Respiratory Medicine, Allergology and Palliative Medicine, Department of Clinical Sciences Lund, Lund University, Lund, Sweden
- 3Division of Occupational and Environmental Medicine, Lund University, Lund, Sweden
Introduction: Chronic lung disorders involve pathological alterations in the lung tissue with hypoxia as a consequence. Hypoxia may influence the release of inflammatory mediators and growth factors including vascular endothelial growth factor (VEGF) and prostaglandin (PG)E2. The aim of this work was to investigate how hypoxia affects human lung epithelial cells in combination with profibrotic stimuli and its correlation to pathogenesis.
Methods: Human bronchial (BEAS-2B) and alveolar (hAELVi) epithelial cells were exposed to either hypoxia (1% O2) or normoxia (21% O2) during 24 h, with or without transforming growth factor (TGF)-β1. mRNA expression of genes and proteins related to disease pathology were analysed with qPCR, ELISA or immunocytochemistry. Alterations in cell viability and metabolic activity were determined.
Results: In BEAS-2B and hAELVi, hypoxia significantly dowregulated genes related to fibrosis, mitochondrial stress, oxidative stress, apoptosis and inflammation whereas VEGF receptor 2 increased. Hypoxia increased the expression of Tenascin-C, whereas both hypoxia and TGF-β1 stimuli increased the release of VEGF, IL-6, IL-8 and MCP-1 in BEAS-2B. In hAELVi, hypoxia reduced the release of fibroblast growth factor, epidermal growth factor, PGE2, IL-6 and IL-8, whereas TGF-β1 stimulus significantly increased the release of PGE2 and IL-6. TGF-β1 stimulated BEAS-2B cells showed a decreased release of VEGF-A and IL-8, while TGF-β1 stimulated hAELVi cells showed a decreased release of PGE2 and IL-8 during hypoxia compared to normoxia. Metabolic activity was significantly increased by hypoxia in both epithelial cell types.
Discussion: In conclusion, our data indicate that bronchial and alveolar epithelial cells respond differently to hypoxia and profibrotic stimuli. The bronchial epithelium appears more responsive to changes in oxygen levels and remodelling processes compared to the alveoli, suggesting that hypoxia may be a driver of pathogenesis in chronic lung disorders.
1 Introduction
Chronic lung diseases, such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), are severe lung conditions with no cures or effective treatments available. COPD is one of the leading causes of death worldwide (Quaderi and Hurst, 2018). Although the two diseases are very different in nature, a common feature is the lack of sufficient oxygen supply resulting in hypoxic milieus in the lung. In COPD, airflow obstructions and remodelling of small airways and emphysema, with destruction of alveolar capillaries and alveolar epithelial cells, result in decreased oxygen transport and alveolar hypoxia (Siafakas et al., 2007). In IPF, there is ongoing remodelling processes with excessive extracellular matrix (ECM) synthesis and deposition resulting in dense fibrotic tissue with alveolar epithelial dysfunction and hypoxia (Barratt and Millar, 2014; Senavirathna et al., 2018). Epithelial cells cover all inner surfaces and serve as a line of defence and barrier and are in direct contact with altered oxygen levels (Shukla et al., 2020; Selo et al., 2021a). The concentration of inhaled oxygen decreases from 21% throughout the respiratory tree to about 13.6% in the alveoli due to the higher water vapour and carbon dioxide concentrations in the alveoli. The bronchoepithelium is supplied with blood from the systemic circulation whereas the alveolar epithelium has an important role in respiration and is supplied by the pulmonary circulation. Hypoxic pulmonary vasoconstriction occurs in the pulmonary circulation in response to low regional partial pressure of oxygen to redirect the gas exchange to better ventilated areas thereby maintaining regional perfusion/ventilation matching in the lung. The reduced oxygen levels can occur due to bronchoconstriction or remodelling of the airways and thereby cause hypoxic conditions also in the bronchial epithelium as well as hypoxia in the alveoli. In studies with primary cells obtained from both COPD and IPF patients, epithelial cells have been seen to undergo epithelial to mesenchymal transition and induced fibrosis through a cross talk between epithelial cells and fibroblasts which have been associated with persistent airway remodelling (Sun et al., 2014; Nishioka et al., 2015; Yao et al., 2021; Mallikarjuna et al., 2022). The involvement of these cells in hypoxic condition and remodelling processes needs further attention.
A known marker of hypoxia is hypoxia inducible factor (HIF), which is a transcription factor regulating oxygen homeostasis. It regulates the expression of hypoxia-targeted genes associated with apoptosis, migration, fibrosis, inflammation and angiogenesis (Xu and Dong, 2016). Angiogenesis is promoted by hypoxia through upregulated synthesis of growth factors, such as vascular endothelial growth factor (VEGF) (Harkness et al., 2014). Hypoxia has previously mainly been studied in different cancer diseases and less in detail in ongoing remodelling processes in chronic lung diseases, highlighting the need for more research into this area. In COPD, pulmonary vascular remodelling is common and comorbidities in cardiovascular disease have negative impact on COPD prognosis (Faner et al., 2014). Lung tissue remodelling as peribronchial fibrosis, increased bronchiolar vascularisation and airway vessel thickening is frequently observed in COPD patients (Harkness et al., 2014; Mercado et al., 2015). Similar events are observed in lung tissue from patients with IPF with remodelling of pulmonary arteries, altered microvasculature and correlated to reduced lung function (Barratt and Millar, 2014; Gaikwad et al., 2022). Transforming growth factor (TGF)-β1 is a key molecule of regulating ECM production and linked to fibrosis. More recently it has been seen to regulate the expression of VEGF, inducing bronchiolar angiogenesis (Willems-Widyastuti et al., 2011). However, the effect of TGF-β on bronchial and alveolar epithelial cells during hypoxia is less known. Hypoxia has previously been shown to be involved in pathways driven by inflammation relevant for disease pathology (Bartels et al., 2013).
Hence, the aim of this study was to investigate how hypoxia and TGF-β stimuli affects the expression of cellular stress, inflammatory and remodelling markers on mRNA and protein levels in human bronchial and alveolar epithelial cells. Our findings indicate that hypoxia significantly decreased genes related to fibrosis, mitochondrial and oxidative stress, apoptosis and inflammation whereas VEGFR2 was increased in the epithelial cells. Epithelial cells responded differently to hypoxia and profibrotic stimuli with an altered release of growth factors and inflammatory mediators, where the bronchial epithelium appeared more responsive to changes in oxygen levels and remodelling processes compared to the alveoli.
2 Materials and methods
2.1 Epithelial cell cultures
The bronchial cell line BEAS-2B was cultured in RPMI 1640 Medium (Thermo Fisher Scientific, Waltham, MA, United States) supplemented with 10% Foetal Clone III serum (FCIII, HyClone Laboratories, Marlborough, MA, United States), 1% sodium pyruvate (Stock 100 mM, Sigma-Aldrich, Darmstadt, Germany) and 1% Antibiotic/Antimycotic (Thermo Fisher Scientific) at 20% O2, 5% CO2. BEAS-2B cells were used in passage 27-31. The alveolar type I cell line Cl-hAELVi was cultured according to manufacturer’s instructions (InSCREENeX, Braunschweig, Germany), where culture flasks were precoated with 2.5 mL coating solution (InSCREENeX) overnight at 37°C before seeding. Basal huAEC medium with 6% supplements (InSCREENeX) was used for culturing hAELVi at 37°C, 20% O2, 5% CO2. The hAELVi cells were used in passage 14-16.
2.2 Hypoxic and normoxic exposures of epithelial cells
6-well cell culture plates (Nunclon Delta Surface, Thermo Scientific) were cultured with 200,000 cells in 2 mL medium per well until 70%–80% confluence. Medium was replaced with new medium with or without addition of TGF-
Post exposure, the plates were immediately put on ice and cell culture medium was collected. Selected wells for RNA extraction were washed with Phosphate Buffer Saline (PBS) and lysed with RLT buffer (Qiagen, Hilden, Germany) supplemented with 1%
For immunocytochemistry, BEAS-2B and hAELVi were cultured with 15,000 cells/well on 4-well glass Millicell EZ slides (Merck, Sigma Aldrich) at 37°C, 20% O2, 5% CO2 for 24 h. Medium was exchanged, and the slides were exposed to either normoxia or hypoxia. After 24 h, the medium was immediately aspirated, and cells were fixed with 4% formaldehyde for 15 min at room temperature (RT). Slides were washed and stored in PBS at 4°C.
2.3 Quantification of mRNA
The collected cell lysates for RNA were pooled within each individual experiment. The RNA purification was conducted using the Qiagen RNeasy Mini Kit (Ref 74104, Qiagen) according to the manufacturer’s instructions. RNA concentration was determined using Nanodrop 2000 Spectrophotometer (Thermo Fisher Scientific). RNA was converted into cDNA according to instructions of iScript cDNA Synthesis Kit (Bio-Rad, Cat# 1708891). cDNA samples mixed with primers in iTaq universal SYBR® Green Supermix (Bio-Rad, Cat# 1725124) were added to a MicroAmp Fast 96-Well Reaction Plate (Thermo Fisher Scientific). The housekeeping genes used were Glyceraldehyde 3-phosphate dehydrogenase (GAPDH),
2.4 Quantification of total protein amount
Protein concentration in cell lysates was determined using a Pierce bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The absorption was measured at 562 nm with a microplate reader (Multiskan GO, Thermo Fisher Scientific) and total protein concentrations were calculated.
2.5 Enzyme linked immunosorbent assay
The release of VEGF-A and VEGF-C after 24 h exposure of normoxic or hypoxic condition was analysed in the cell culture medium using Human VEGF Quantikine ELISA kit DVE00 and Human VEGF-C Quantikine ELISA kit DVC00 (both from R&D Systems, Minneapolis, MN, United States), according to manufacturer’s instructions. The absorption was measured at 450 and 570 nm (as reference) with a microplate reader (Multiskan GO, Thermo Fisher Scientific). The lower detection limit of quantification was 15.6 pg/mL for VEGF-A and 55 pg/mL for VEGF-C.
The release of prostaglandin E2 (PGE2) was analysed with PGE2 ELISA kit from Cayman Chemical, Ann Arbor, Michigan, United States, according to manufacturer’s instructions. The absorption was measured at 450 nm with a microplate reader (Multiskan GO, Thermo Fisher Scientific). The assay detection limit for PGE2 was 7.8 pg/mL. The total amount of released growth factor was normalized to total protein concentration amount for each individual sample.
2.6 Multiplex analysis
The release of several cytokines and growth factors were analysed in cell culture medium from cells exposed to 24 h normoxic or hypoxic conditions using multiplexed Luminex discovery assays from Bio-Techne (Minneapolis, MN, United States) on a Luminex platform (Bio-Plex 200, Bio-Rad Life Science, Hercules, CA) according to the instructions given by the manufacturer. The following cytokines were analysed: monocyte chemotactic protein-1 (MCP-1), interleukin (IL)-1b, IL-6, IL-8, fibroblast growth factor-basic (FGF-basic, also known as FGF-2), hepatocyte growth factor (HGF), vascular endothelial growth factor receptor 2 (VEGFR2), VEGFR3, epidermal growth factor (EGF) and Tenascin C. The calibration curves were fitted using a five-point regression model and the results were evaluated in the Bio-Plex Manager Software 6.0 (Bio-Rad). The lower limit of detection was 35 (MCP-1), 1.5 (FGF basic), 3.6 (IL-6), 140 (VEGFR2), 9.0 (EGF), 14 (IL-1β), 3.1 (IL-8), 49 (Tenascin C), 60 (VEGFR3) pg/mL, respectively. The total amount of released mediator was normalised to total protein amount for each individual sample.
2.7 Immunocytochemistry staining
Immunocytochemistry staining was performed on BEAS-2B and hAELVi cells for the protein expression of HIF2
2.8 Cell viability using Lactate dehydrogenase assay
BEAS-2B and hAELVi were cultured with 7,000 cells/well in a 96-well Standard Flat base Plates (Sarstedt, Nümbrecht, Germany) and exposed to hypoxic and normoxic conditions for 24 h. A Lactate dehydrogenase (LDH) quantification was conducted using a Cytotoxicity Detection Kit (Roche, Sigma Aldrich) in accordance with the manufacturer’s instructions. 1% Triton X-100 (Sigma Aldrich) was added to some of the wells (high controls) as well as controls with only medium (low controls). The following formula was used to determine cytotoxicity:
2.9 Metabolic activity—Water soluble tetrazolium salt test
The cells culture plates with cells prepared for the LDH assay were further analysed for metabolic activity using a Water-soluble tetrazolium salt 1 (WST-1) test (Sigma-Aldrich). Remnants of cell medium were removed and 200
2.10 Quantification of cell amount
Cell amount was determined as previously described (Westergren-Thorsson et al., 2018). BEAS2B and hAELVi were plated 7,000 cells/well in 96-well Standard Flat base Plates (Sarstedt) overnight and then exposed to either hypoxic or normoxic condition for 24 h. Control of the plates were fixed at the start of the exposure as a comparison for cell growth and the exposed plates were fixed after 24 h. Cells were fixed in 1% glutaraldehyde (Sigma-Aldrich), stained with 0.1% crystal violet (Sigma-Aldrich) and stained with 0.1% crystal violet (Sigma-Aldrich) for 30 min. Excess staining solution was washed away and cells were permeabilised overnight with 1% Triton X100 at 4°C (Merck, Darmstadt, Germany). Changes in cell amount were quantified with a microplate reader (Multiskan GO, Thermo Fisher Scientific), measuring absorbance at 595 nm in collected cell suspension.
2.11 Data presentation and statistical analysis
All statistical analyses were performed using the software GraphPad Prism 9.3.1 (San Diego, United States). Four independent in vitro experiments were performed with two biological replicates in each experiment (n = 8 for each exposure), unless otherwise stated. Samples for qPCR were pooled generating 4 individual samples. The data is presented as mean with standard deviation (SD) for normoxia and hypoxia for each cell type. Student’s t-test was used to compare statistical significance between two groups for the LDH and WST-1 data. One-way ANOVA followed by post-hoc analysis with Fisher’s LSD test were used for the analysis of VEGF, PGE2 and multiplex data. qPCR data was analysed with One sample t-test where normoxia was set to 1. Statistical significance was determined at p < 0.05 and indicated in the figures as *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
3 Results
3.1 Hypoxia induced altered gene expression in epithelial cells
The oxygen concentration in the air of the hypoxic chamber was confirmed as hypoxic condition verifying the oxygen settings. No large variations were observed following hypoxic or normoxic exposures in expression of the housekeeping genes in either BEAS-2B or hAELVi. Epithelial cells exposed to hypoxia for 24 h showed altered gene expression levels in comparison to normoxic exposure. Markers of hypoxia (HIF-1
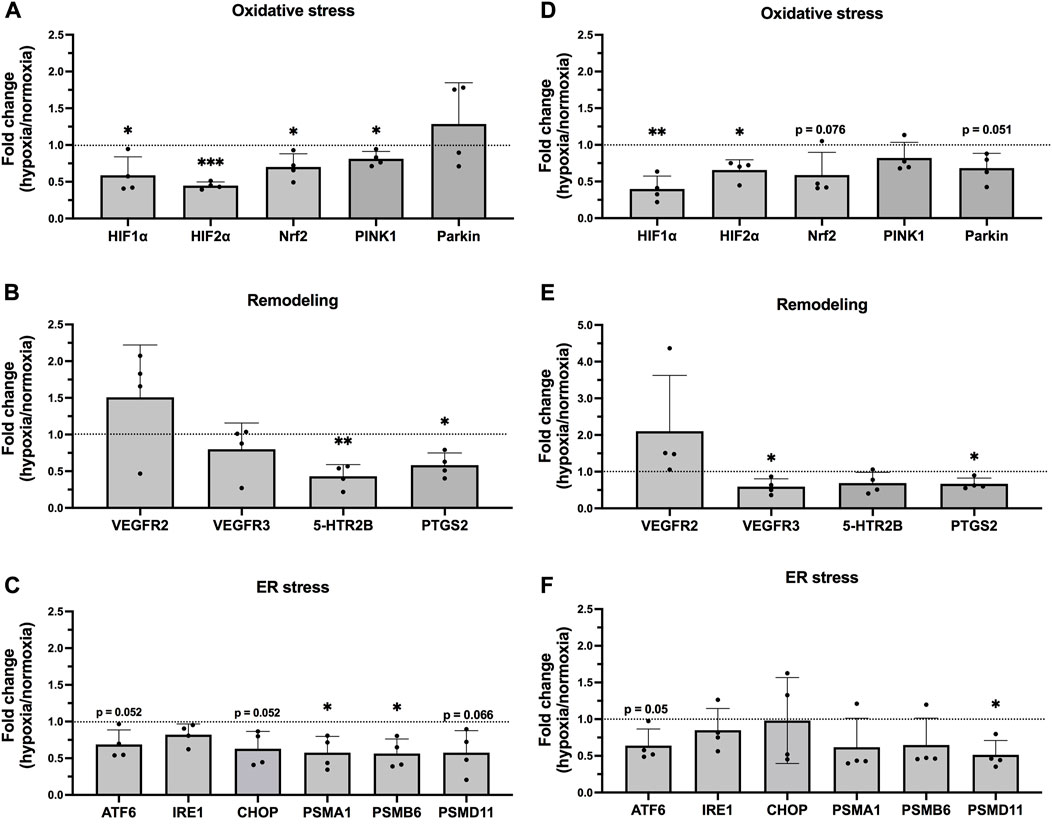
FIGURE 1. mRNA levels in epithelial cells exposed to hypoxia. Hypoxia-target genes in BEAS-2B (A–C) and hAELVi (D–F). Data is based on four individual experiments (n = 4). The data is presented as the ratio of hypoxia and normoxia with mean
In BEAS-2B cells, HIF1
3.2 Expression of HIF2
To further explore the change of HIF2
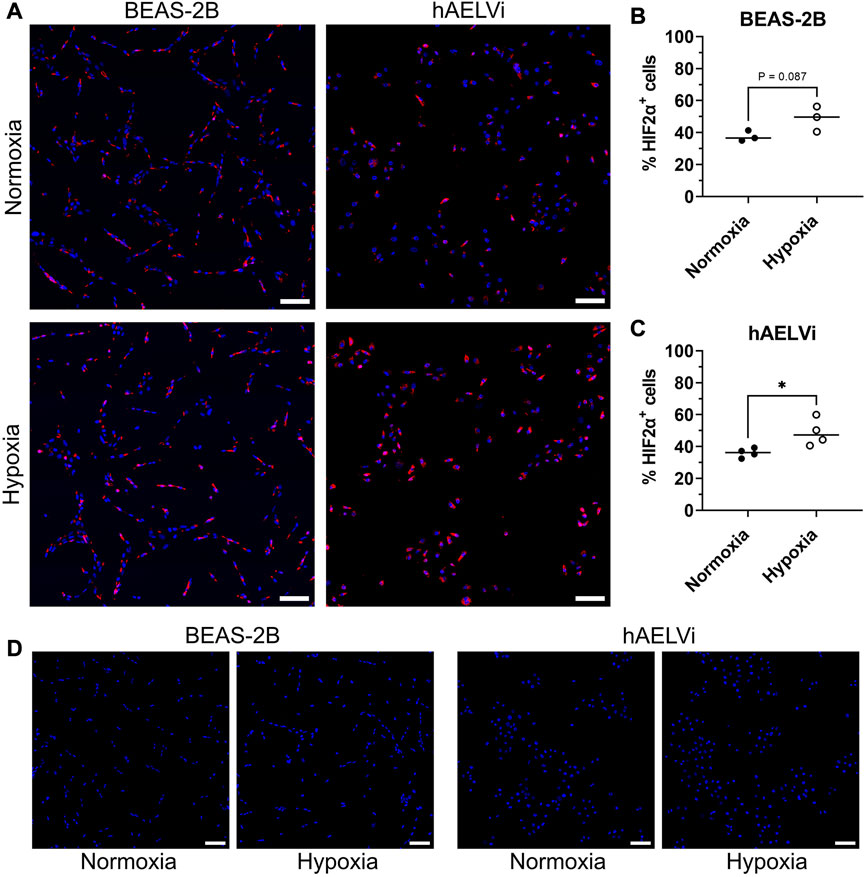
FIGURE 2. Immunofluorescence staining of HIF2
3.3 Effect of hypoxia and profibrotic stimuli on release of growth factors
The release of VEGF-A and VEGF-C was differentially affected following treatment with hypoxia and/or TGF-
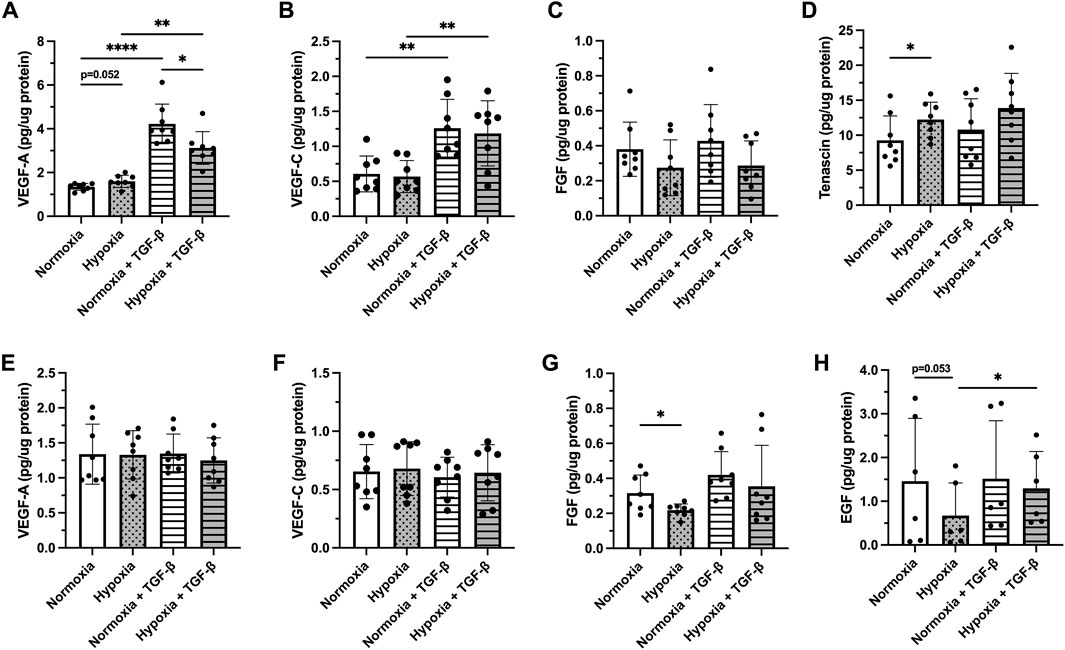
FIGURE 3. Release of following growth factors: Vascular endothelial growth factor (VEGF)-A (A,E) and VEGF-C (B,F), fibroblast growth factor (FGF) basic (C,G), Tenascin C (D) and epidermal growth factor [EGF, (H)] were investigated in BEAS-2B (A–D) and hAELVi (E–H) cells exposed 24 h to normoxia or hypoxia with or without TGF-β1 (10 ng/ml) stimuli. The total amount of released growth factor was normalized to total protein amount for each individual sample. Data is presented from four individual experiments, including two biological replicates from each experiment (n = 8 in total). The data is presented as mean
3.4 Expression of VEGFR3 in epithelial cells
To evaluate if VEGFR expression was altered at transcriptomic level after hypoxia exposure, immunocytochemistry for VEGFR2 and VEGFR3 was performed in BEAS-2B and hAELVi cells after 24 h of exposure. Comparisons were made between cells exposed to normoxia or hypoxia. Expression of VEGFR2 was not detectable in the epithelial cells (data not shown). VEGFR3 was detected in both normoxic and hypoxic culture conditions for both cell lines, with expression seen in both the cytoplasm and in the nucleus (Figures 4A–D). Quantification of number of VEGFR3 positive cells did not show a significant alteration in the expression of VEGFR3 after hypoxia exposure in BEAS-2B (Figure 4B) and hAELVi cells (Figure 4C).
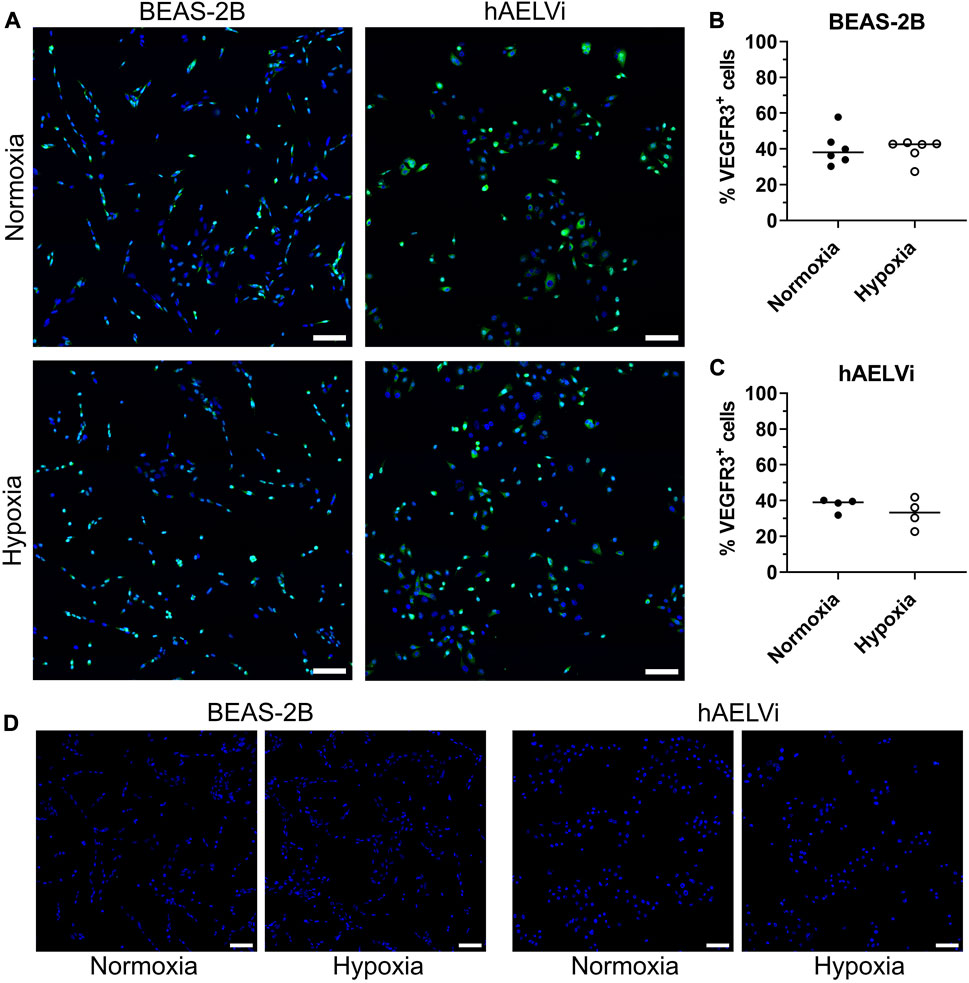
FIGURE 4. Immunofluorescence staining of VEGFR3 in BEAS-2B and hAELVi cells exposed to normoxia or hypoxia exposure (A). DAPI (blue) and VEGFR3 (green). Scale bar = 100 µm. Analysis of VEGFR3+cells in BEAS-2B (B) and hAELVi (C). Control staining without the anti-VEGFR3 primary antibody (D). DAPI (blue) and VEGFR3 (green). Scale bar = 100 µm.
3.5 Effect of hypoxia and profibrotic stimuli on the release of inflammatory mediators
The levels of inflammatory mediators secreted from the epithelial cells were higher in BEAS-2B for IL-6, IL-8, and MCP-1 at normoxia, compared to hAELVi. MCP-1 was not detectable in hAELVi. In contrast, hAELVi produced significantly higher amounts of PGE2 in normoxia, compared to BEAS-2B (Figures 5A–G). Hypoxia increased the release of IL-8 (p < 0.05, Figure 5B) and MCP-1 (p < 0.01; Figure 5D), while TGF-β1 significantly increased the release of PGE2 (p < 0.05, Figure 5A), IL-6 (p < 0.001; Figure 5B), IL-8 (p < 0.01; Figure 5C) and MCP-1 (p < 0.01, Figure 5D) in BEAS-2B cells. TGF-b stimulated cells though showed a decreased release of IL-8 (p < 0.05; Figure 5C) during hypoxia compared to normoxia.
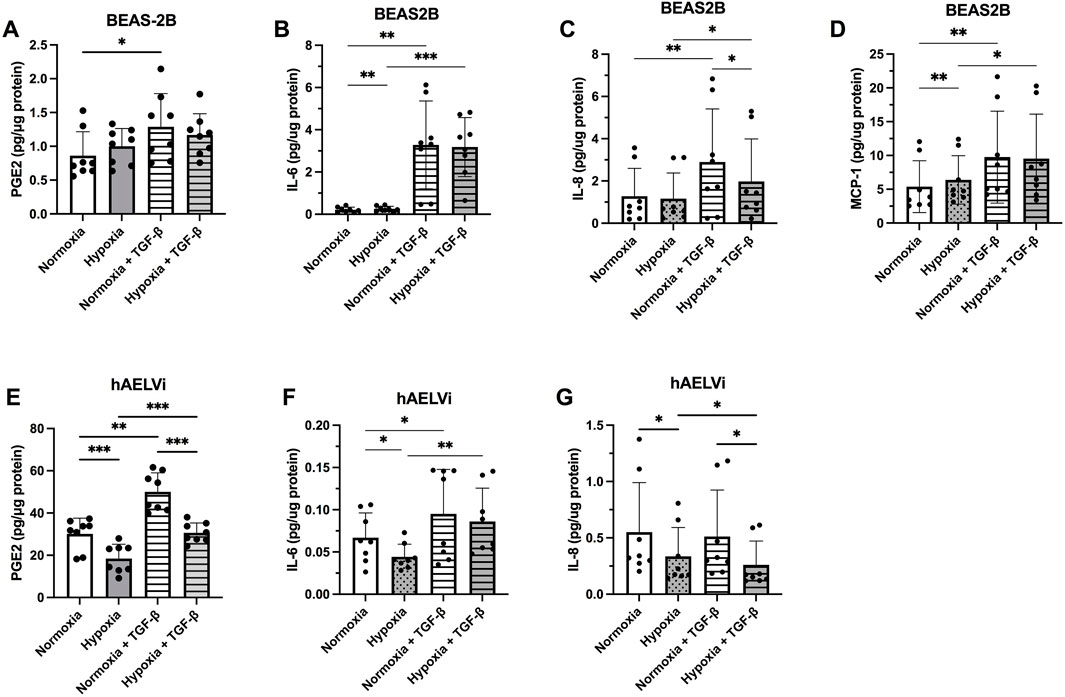
FIGURE 5. Release of the inflammatory mediators prostaglandin E2 (PGE2) (A,E), Interleukin (IL)-6 (B,F), IL-8 (C,G) and Monocyte chemoattractant protein (MCP)-1 (D) from BEAS-2B (A–D) and hAELVi (E–G) cells 24 h post hypoxia exposure, with or without 10 ng/mL of TGF-
In hAELVi, hypoxia significantly reduced the release of PGE2 (p < 0.001; Figure 5E), IL-6 (p < 0.05, Figure 5F), and IL-8 (p < 0.05; Figure 5G), whereas TGF-β1 stimulus significantly increased the release of PGE2 (p < 0.001; Figure 5E) and IL-6 (p < 0.05, Figure 5F). TGF-β1 stimulated cells though showed a decreased release of PGE2 (p < 0.001; Figure 5E) and IL-8 (p < 0.05; Figure 5G) during hypoxia compared to normoxia. MCP-1 was not detected in the hAELVi cells and the release of HGF was not detected in either BEAS-2B or hAELVi in the multiplex analysis.
3.6 Effect of hypoxia on cell viability, metabolic activity and cell amount
Cell cytotoxicity was examined in epithelial cells exposed to 24 h normoxia or hypoxia, through LDH assay to examine the effect of cultured conditions (Figure 6). There were no significant differences in LDH release between hypoxic and normoxic conditions in either BEAS-2B (Figure 6A) or hAELVi cells (Figure 6D). hAELVi showed a higher release of LDH compared to BEAS-2B independently of hypoxia exposure. Metabolic activity was higher in BEAS-2B compared to hAELVi (p < 0.0001) during both normoxic and hypoxic conditions (Figures 6B, E). Metabolic activity was significantly increased in both BEAS-2B (p < 0.0001; Figure 6C) and hAELVi (p < 0.0001; Figure 6F) after hypoxia exposure. There were no significant changes in cell amount between normoxic and hypoxic conditions in either BEAS-2B (Figure 6E) or hAELVi (Figure 6F).
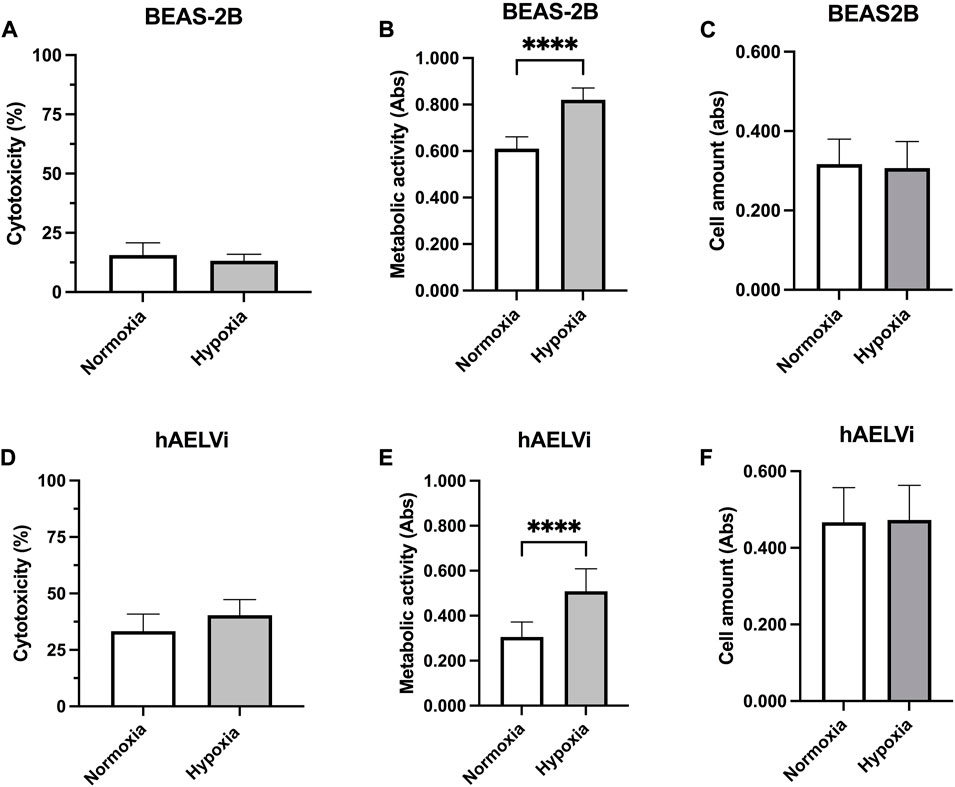
FIGURE 6. Cell viability after hypoxia exposure. Cytotoxicity measurements of BEAS-2B (A) and hAELVi (D) after 24 h exposure to either normoxia or hypoxia. Samples were related to cells treated with 1% Triton X-100, set to 100% cytotoxicity. Measured metabolic activity in BEAS-2B (B) and hAELVi (E) cells exposed to normoxia or hypoxia for 24 h. Cell amount in BEAS-2B (C) and hAELVi (F) after 24 h exposure to either normoxia or hypoxia. Data is based on 2-3 individual experiments with biological replicates (n = 6–8) for BEAS-2B and (n = 11–13) for hAELVi. Data is presented as mean
4 Discussion
The current study investigated how hypoxia (1% O2) affects human bronchial and alveolar epithelial cells and the expression of markers for angiogenesis, cellular stress, inflammation and remodelling processes. These selected markers are of importance in mechanisms involved in the pathogenesis of pulmonary diseases and may give a greater insight on the regulation of these factors during changes in oxygen levels in the lung. We found that hypoxia influenced gene expression markers associated with oxidative and cellular stress, inflammation and ECM remodelling in both BEAS-2B and hAELVi cells. Stimulation with profibrotic TGF-
Hypoxia is known to induce the transcription factors HIF-1α and HIF-2α by regulating the expression of hypoxia-target genes. In the current study, we observed a significant downregulation of HIF-1α and HIF-2
As elevated levels of HIF are associated with angiogenesis and pulmonary vascular remodelling (Harkness et al., 2014; Xu and Dong, 2016; Dai et al., 2018), we investigated if hypoxia induced expression of VEGFR2 relevant for angiogenesis and VEGF3R relevant for lymphangiogenesis. The epithelial cells expressed low amounts of VEGFR2 and VEGFR3. Expression of VEGFR2 at mRNA level was increased in both BEAS-2B and hAELVi in the current study, whereas VEGFR3 was significantly decreased in hAELVi at both mRNA and protein level after hypoxia exposure. Both BEAS-2B and hAELVi released the associated receptor ligands VEGF-A and VEGF-C, which has previously not been shown for hAELVi. VEGF-C stimulates lymphangiogenesis by binding to VEGFR3 (Schwager and Detmar, 2019) and HIF-1
In addition to angiogenesis, other markers related to remodelling and inflammatory processes were investigated in the context of hypoxia and profibrotic stimuli. TGF-β1 and hypoxia may act in synergistic ways by inducing each other in events involved in fibrosis and inflammation (McMahon et al., 2006; Mallikarjuna et al., 2022). Hypoxia has in general an important role in normal repair processes. Injury to the epithelium that cause low oxygen supply may induce the epithelial cells to secrete mediators to recruit inflammatory cells to the repair process and re-epithelialisation. These inflammatory cells have high metabolic demands for oxygen in the repair process. HIF-1 can influence the cellular inflammatory response by switching metabolism to glycolysis (Papandreou et al., 2006) and thereby reduce oxygen consumption. HIF-1 activation and downstream signalling induce an inflammatory response, proliferation, survival, growth factor release, and matrix synthesis in the repair process. However, if the hypoxia becomes chronic HIF-1 may accumulate resulting in pathological events as fibrosis by increased myofibroblast differentiation and excessive matrix production (Hong et al., 2014). As a synergistic effect, TGF-β1 may also induce HIF-1α via protein stabilisation (McMahon et al., 2006) through an increase in HIF-1α protein translation via the PI3K pathway and mitogen-activated protein kinase pathway (Maynard and Ohh, 2007). In the current study, secretion of tenascin C was upregulated in BEAS-2B cells following hypoxia. Tenascin C is suggested as a prognostic marker for fibrosis (Bhattacharyya et al., 2016; Bhattacharyya et al., 2022). Synovial fibroblasts showed an increased expression of tenascin C both at mRNA and protein level during hypoxic conditions (Tojyo et al., 2008). In alveolar hAELVi cells, hypoxia decreased the release of FGF and EGF in the current study. EGF is known to promote epithelial to mesenchymal transition and FGF is linked to development of fibrosis. In contrast to the bronchial epithelial cells our obtained data on release of growth factors imply that hypoxia does not promote a fibrotic event by directly targeting the alveolar epithelial cells. The inflammatory markers IL-6, IL-8 and MCP-1 are recognised as drivers in pulmonary fibrosis but also in COPD (Burgoyne et al., 2021). In the present study, TGF-β1 increased the release of IL-6, IL-8 and MCP-1 in bronchial epithelial cells and IL-6 in alveolar epithelial cells. Hypoxia increased the release of IL-6 and MCP-1 in bronchial epithelial cells whereas the release of IL-6 and IL-8 were decreased in the alveolar cells. Previous studies in endothelial cells have seen an increased release of IL-6 and IL-8 in response to hypoxia (Tamm et al., 1998). A major difference in inflammatory profile between bronchial and alveolar cells was the release of PGE2. hAELVi cells produced excessive amounts of PGE2, which was significantly increased by TGF-β1 stimulus and profoundly reduced by hypoxia exposure. In line with the reduced PGE2 levels, the COX-2 enzyme responsible for PGE2 synthesis was downregulated by hypoxia at mRNA level in both cell types in the current study. PGE2 has been shown to have anti-inflammatory effects in the lung (Dackor et al., 2011; Barnthaler et al., 2017) and to be reduced by hypoxia in human alveolar macrophages (Hempel et al., 1996). The high amount of PGE2 produced by the hAELVi cells may counterbalance other pro-inflammatory events induced by hypoxia or profibrotic stimuli, as shown by Barnthaler et al. (2017) in alveolar A549 epithelial cells and by Birrell et al. (2015) in different in vitro and in vivo models.
Patients with COPD have shown deviating expressions of genes related to ER stress, lysosomes, oxidative stress, inflammation and apoptosis when compared to healthy non-smokers (Mercado et al., 2015; Eapen et al., 2018; Weidner et al., 2018; Beghe et al., 2019; Sun et al., 2019). In the current study, hypoxia reduced expression of genes related to oxidative stress (nrf2), mitochondrial function (PINK and Parkin) and ER stress (PSMA1, PSMB6 and PSMD) in the epithelial cells, with a more pronounced effect on oxidative stress and mitochondrial dysfunction in BEAS-2B compared to hAELVi. Oxidative stress is an important mechanism in the pathogenesis of lung diseases as oxidative stress activates different signalling pathways to maintain homeostasis due to elevated levels of reactive oxygen species (ROS). Inducement of ROS causes cellular imbalances inducing ER-stress. However, disturbed mitochondrial function has been associated with unfolded protein responses in which expression of CHOP, PINK1 and Parkin usually are elevated. PINK1 is implicated in the inhibition of ROS production by recruiting Parkin to the dysfunctional mitochondria inducing mitophagy (Park et al., 2018; Kaspar et al., 2021). In this study, both cell lines had significantly higher metabolic activity in hypoxia compared to normoxia, which may be due to altered mitochondrial function. The increase in mitochondrial activity is consistent with the decrease of markers correlated to mitochondrial dysfunction (PINK1 and Parkin) and ER-stress observed at mRNA level. This data implies that hypoxia induced changes on mitochondrial activity, which could also be seen as a sign of viability, a result consistent with the unaffected apoptosis with decreased blc2 mRNA level observed for hypoxia exposed BEAS-2B and hAEVLi cells. Increased metabolic activity may indicate an increase in proliferative capacity, although no significant alterations in cell amounts were detected after 24 h in the present study, suggesting that the epithelial cells did not proliferate more in hypoxia. Increased proliferation rate has previously been observed in human lung fibroblasts and vascular smooth muscle cells after exposure to 3% O2 (Tamm et al., 1998) and in vivo exposure to hypoxia (10% O2) in a mice model induced proliferation of bronchial club epithelial cells (Torres-Capelli et al., 2016). Hypoxia via HIF-1 activation may also induce autophagy as a cell survival response to avoid cell death (Mazure and Pouyssegur, 2010). Li et al. (2022), showed in different cell lines that autophagy-dependent clearance of mitochondria decreased consumption of oxygen to preserve cell viability. Altogether, these findings imply that hypoxia is associated with altered mitochondrial function in lung epithelial cells.
A strength, but also a weakness, of these in vitro studies is the usage of non-carcinogenic bronchiolar and alveolar cell lines (Selo et al., 2021b). BEAS-2B shows both epithelial and mesenchymal characteristic morphology with a gene expression profile predominant of mesenchymal stem cell surface markers compared to epithelial cells (Han et al., 2020). The more recently obtained cell line hAELVi mimics the morphology and cell constitution of the alveolar lung tissue with a profile towards type 1 pneumocytes due to the tight intracellular junctions with high transepithelial electrical resistance (Kletting et al., 2018). In the current study we analysed both gene expression and protein levels after 24 h. An increase or decrease in gene expression could have appeared earlier that we were not able to detect due to unstable or degraded mRNA or the opposite that the cells have to be exposed during a longer time period. However, in vitro studies with primary epithelial cells at different time points of exposure followed up with transcriptomic and proteomic profiling analyses are necessary for verification of the results and inclusion of histological analysis of lung material from patients with chronic lung disorders to further understand how hypoxia and TGF-β1 affect pathological events.
5 Conclusion
The results from this study show that hypoxia alters the expression of several crucial mechanisms involved in pulmonary diseases by downregulating markers involved in oxidative stress and mitochondrial dysfunction, altered inflammatory response and tissue remodelling in both alveolar and bronchial cells. The response to profibrotic stimuli was altered in the presence of hypoxia with more pronounced effects in the bronchial epithelium, indicating that the bronchial epithelium is more responsive to changes in oxygen levels and remodelling processes compared to the alveoli. One reason of this differences in response to hypoxia could be the high amount of protective PGE2 that is produced by the alveolar epithelium, another that the bronchial epithelium is responding faster to alterations in O2 to maintain homeostasis whereas the alveoli is more adaptive. By mimicking the hypoxic response in a controlled in vitro setting pathological mechanisms can be investigated and linked to disease onset and progression.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
RB-N: Methodology, formal analysis, investigation, writing—original draft. MR: Methodology, investigation, formal analysis, writing—review and editing. AL: Methodology, formal analysis, writing—review and editing. MK: Methodology, investigation, formal analysis, writing—review and editing. AI-F: Methodology, formal analysis, writing—review and editing. GW-T: Resources, writing—review and editing, funding acquisition. ET: Resources, methodology, writing—review and editing, funding acquisition, formal analysis, supervision. A-KL-C: Resources, Methodology, investigation, formal analysis, writing—original draft, writing—review and editing, funding acquisition, supervision.
Funding
This work was supported by the Swedish Heart-Lung foundation (2020/0847, 2021/0382, and 2022/0847); Lund University Medical Faculty, Lund University library to cover the APC, Birgit and Sven Håkan Ohlsson Foundation, the Crafoord foundation (grant number 20200989 and 20221034) and Alfred Österlund foundation. Part of this work was also funded by the EMPIR 18HLT02 AeroTox project. The EMPIR program is co-financed by the Participating States and from the European Union’s Horizon 2020 research and innovation program. We are also grateful to the Fundación Ramón Areces (Spain; grant number BEVP33S12276) for the funding for AI-F postdoctoral position.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1094245/full#supplementary-material
References
Bankhead, P., Loughrey, M. B., Fernandez, J. A., Dombrowski, Y., McArt, D. G., Dunne, P. D., et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7 (1), 16878. doi:10.1038/s41598-017-17204-5
Barnthaler, T., Maric, J., Platzer, W., Konya, V., Theiler, A., Hasenohrl, C., et al. (2017). The role of PGE2 in alveolar epithelial and lung microvascular endothelial crosstalk. Sci. Rep. 7 (1), 7923. doi:10.1038/s41598-017-08228-y
Barratt, S., and Millar, A. (2014). Vascular remodelling in the pathogenesis of idiopathic pulmonary fibrosis. QJM 107 (7), 515–519. doi:10.1093/qjmed/hcu012
Bartels, K., Grenz, A., and Eltzschig, H. K. (2013). Hypoxia and inflammation are two sides of the same coin. Proc. Natl. Acad. Sci. U S A 110 (46), 18351–18352. doi:10.1073/pnas.1318345110
Beghe, B., Fabbri, L. M., Garofalo, M., Schito, M., Verduri, A., Bortolotti, M., et al. (2019). Three-year hospitalization and mortality in elderly smokers with chronic obstructive pulmonary disease or chronic Heart failure. Respiration 97 (3), 223–233. doi:10.1159/000492286
Bhattacharyya, S., Midwood, K. S., and Varga, J. (2022). Tenascin-C in fibrosis in multiple organs: Translational implications. Semin. Cell Dev. Biol. 128, 130–136. doi:10.1016/j.semcdb.2022.03.019
Bhattacharyya, S., Wang, W., Morales-Nebreda, L., Feng, G., Wu, M., Zhou, X., et al. (2016). Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 7, 11703. doi:10.1038/ncomms11703
Birrell, M. A., Maher, S. A., Dekkak, B., Jones, V., Wong, S., Brook, P., et al. (2015). Anti-inflammatory effects of PGE2 in the lung: Role of the EP4 receptor subtype. Thorax 70 (8), 740–747. doi:10.1136/thoraxjnl-2014-206592
Burgoyne, R. A., Fisher, A. J., and Borthwick, L. A. (2021). The role of epithelial damage in the pulmonary immune response. Cells 10 (10), 2763. doi:10.3390/cells10102763
Clerici, C., and Planes, C. (2009). Gene regulation in the adaptive process to hypoxia in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 296 (3), L267–L274. doi:10.1152/ajplung.90528.2008
Dackor, R. T., Cheng, J., Voltz, J. W., Card, J. W., Ferguson, C. D., Garrett, R. C., et al. (2011). Prostaglandin E₂ protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 301 (5), L645–L655. doi:10.1152/ajplung.00176.2011
Dai, Z., Zhu, M. M., Peng, Y., Machireddy, N., Evans, C. E., Machado, R., et al. (2018). Therapeutic targeting of vascular remodeling and right Heart failure in pulmonary arterial hypertension with a HIF-2α inhibitor. Am. J. Respir. Crit. care Med. 198 (11), 1423–1434. doi:10.1164/rccm.201710-2079OC
Eapen, M. S., Hansbro, P. M., Larsson-Callerfelt, A-K., Jolly, M. K., Myers, S., Sharma, P., et al. (2018). Chronic obstructive pulmonary disease and lung cancer: Underlying Pathophysiology and new therapeutic modalities. Drugs 78 (16), 1717–1740. doi:10.1007/s40265-018-1001-8
Faner, R., Cruz, T., Lopez-Giraldo, A., and Agusti, A. (2014). Network medicine, multimorbidity and the lung in the elderly. Eur. Respir. J. 44 (3), 775–788. doi:10.1183/09031936.00078714
Fu, X., and Zhang, F. (2018). Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp. Ther. Med. 16 (6), 4553–4561. doi:10.3892/etm.2018.6785
Gaikwad, A. V., Lu, W., Dey, S., Bhattarai, P., Chia, C., Larby, J., et al. (2022). Vascular remodelling in idiopathic pulmonary fibrosis patients and its detrimental effect on lung physiology: Potential role of endothelial-to-mesenchymal transition. ERJ Open Res. 8 (1), 00571–02021. doi:10.1183/23120541.00571-2021
Han, X., Na, T., Wu, T., and Yuan, B. Z. (2020). Human lung epithelial BEAS-2B cells exhibit characteristics of mesenchymal stem cells. PLoS One 15 (1), e0227174. doi:10.1371/journal.pone.0227174
Harkness, L. M., Kanabar, V., Sharma, H. S., Westergren-Thorsson, G., and Larsson-Callerfelt, A. K. (2014). Pulmonary vascular changes in asthma and COPD. Pulm. Pharmacol. Ther. 29 (2), 144–155. doi:10.1016/j.pupt.2014.09.003
Hempel, S. L., Monick, M. M., and Hunninghake, G. W. (1996). Effect of hypoxia on release of IL-1 and TNF by human alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 14 (2), 170–176. doi:10.1165/ajrcmb.14.2.8630267
Hogg, J. C. (2004). Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 364 (9435), 709–721. doi:10.1016/S0140-6736(04)16900-6
Hong, W. X., Hu, M. S., Esquivel, M., Liang, G. Y., Rennert, R. C., McArdle, A., et al. (2014). The role of hypoxia-inducible factor in wound healing. Adv. Wound Care (New Rochelle) 3 (5), 390–399. doi:10.1089/wound.2013.0520
Kaspar, S., Oertlin, C., Szczepanowska, K., Kukat, A., Senft, K., Lucas, C., et al. (2021). Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Isr. Sci. Adv. 7 (22), eabf0971. doi:10.1126/sciadv.abf0971
Kletting, S., Barthold, S., Repnik, U., Griffiths, G., Loretz, B., Schneider-Daum, N., et al. (2018). Co-culture of human alveolar epithelial (hAELVi) and macrophage (THP-1) cell lines. Altex 35 (2), 211–222. doi:10.14573/altex.1607191
Lee, S. H., Lee, S. H., Kim, C. H., Yang, K. S., Lee, E. J., Min, K. H., et al. (2014). Increased expression of vascular endothelial growth factor and hypoxia inducible factor-1α in lung tissue of patients with chronic bronchitis. Clin. Biochem 47 (7-8), 552–559. doi:10.1016/j.clinbiochem.2014.01.012
Li, J., Zhang, T., Ren, T., Liao, X., Hao, Y., Lim, J. S., et al. (2022). Oxygen-sensitive methylation of ULK1 is required for hypoxia-induced autophagy. Nat. Commun. 13 (1), 1172. doi:10.1038/s41467-022-28831-6
Mallikarjuna, P., Zhou, Y., and Landstrom, M. (2022). The synergistic cooperation between TGF-beta and hypoxia in cancer and fibrosis. Biomolecules 12 (5), 635. doi:10.3390/biom12050635
Maynard, M. A., and Ohh, M. (2007). The role of hypoxia-inducible factors in cancer. Cell Mol. Life Sci. 64 (16), 2170–2180. doi:10.1007/s00018-007-7082-2
Mazure, N. M., and Pouyssegur, J. (2010). Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 22 (2), 177–180. doi:10.1016/j.ceb.2009.11.015
McMahon, S., Charbonneau, M., Grandmont, S., Richard, D. E., and Dubois, C. M. (2006). Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 281 (34), 24171–24181. doi:10.1074/jbc.M604507200
Mercado, N., Ito, K., and Barnes, P. J. (2015). Accelerated ageing of the lung in COPD: New concepts. Thorax 70 (5), 482–489. doi:10.1136/thoraxjnl-2014-206084
Morfoisse, F., Kuchnio, A., Frainay, C., Gomez-Brouchet, A., Delisle, M. B., Marzi, S., et al. (2014). Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1α-independent translation-mediated mechanism. Cell Rep. 6 (1), 155–167. doi:10.1016/j.celrep.2013.12.011
Nishioka, M., Venkatesan, N., Dessalle, K., Mogas, A., Kyoh, S., Lin, T. Y., et al. (2015). Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir. Res. 16 (1), 72. doi:10.1186/s12931-015-0232-4
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L., and Denko, N. C. (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3 (3), 187–197. doi:10.1016/j.cmet.2006.01.012
Park, Y. S., Choi, S. E., and Koh, H. C. (2018). PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol. Lett. 284, 120–128. doi:10.1016/j.toxlet.2017.12.004
Pasupneti, S., Tian, W., Tu, A. B., Dahms, P., Granucci, E., Gandjeva, A., et al. (2020). Endothelial HIF-2α as a key endogenous mediator preventing emphysema. Am. J. Respir. Crit. Care Med. 202 (7), 983–995. doi:10.1164/rccm.202001-0078OC
Quaderi, S. A., and Hurst, J. R. (2018). The unmet global burden of COPD. Glob. Health Epidemiol. Genom 3, e4. doi:10.1017/gheg.2018.1
Schwager, S., and Detmar, M. (2019). Inflammation and lymphatic function. Front. Immunol. 10, 308. doi:10.3389/fimmu.2019.00308
Selo, M. A., Sake, J. A., Kim, K-J., and Ehrhardt, C. (2021). In vitro and ex vivo models in inhalation biopharmaceutical research — Advances, challenges and future perspectives. Adv. Drug Deliv. Rev. 177, 113862. doi:10.1016/j.addr.2021.113862
Selo, M. A., Sake, J. A., Kim, K. J., and Ehrhardt, C. (2021). In vitro and ex vivo models in inhalation biopharmaceutical research - advances, challenges and future perspectives. Adv. Drug Deliv. Rev. 177, 113862. doi:10.1016/j.addr.2021.113862
Senavirathna, L. K., Huang, C., Yang, X., Munteanu, M. C., Sathiaseelan, R., Xu, D., et al. (2018). Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci. Rep. 8 (1), 2709. doi:10.1038/s41598-018-21073-x
Shukla, S. D., Walters, E. H., Simpson, J. L., Keely, S., Wark, P. A. B., O'Toole, R. F., et al. (2020). Hypoxia-inducible factor and bacterial infections in chronic obstructive pulmonary disease. Respirology 25 (1), 53–63. doi:10.1111/resp.13722
Siafakas, N. M., Antoniou, K. M., and Tzortzaki, E. G. (2007). Role of angiogenesis and vascular remodeling in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct Pulmon Dis. 2 (4), 453–462.
Sun, C., Zhu, M., Yang, Z., Pan, X., Zhang, Y., Wang, Q., et al. (2014). LL-37 secreted by epithelium promotes fibroblast collagen production: A potential mechanism of small airway remodeling in chronic obstructive pulmonary disease. Lab. Investig. 94 (9), 991–1002. doi:10.1038/labinvest.2014.86
Sun, X., Feng, X., Zheng, D., Li, A., Li, C., Li, S., et al. (2019). Ergosterol attenuates cigarette smoke extract-induced COPD by modulating inflammation, oxidative stress and apoptosis in vitro and in vivo. Clin. Sci. (Lond). 133 (13), 1523–1536. doi:10.1042/CS20190331
Tamm, M., Bihl, M., Eickelberg, O., Stulz, P., Perruchoud, A. P., and Roth, M. (1998). Hypoxia-induced interleukin-6 and interleukin-8 production is mediated by platelet-activating factor and platelet-derived growth factor in primary human lung cells. Am. J. Respir. Cell Mol. Biol. 19 (4), 653–661. doi:10.1165/ajrcmb.19.4.3058
Tojyo, I., Yamaguchi, A., Nitta, T., Yoshida, H., Fujita, S., and Yoshida, T. (2008). Effect of hypoxia and interleukin-1beta on expression of tenascin-C in temporomandibular joint. Oral Dis. 14 (1), 45–50. doi:10.1111/j.1601-0825.2006.01344.x
Torres-Capelli, M., Marsboom, G., Li, Q. O., Tello, D., Rodriguez, F. M., Alonso, T., et al. (2016). Role of Hif2α oxygen sensing pathway in bronchial epithelial club cell proliferation. Sci. Rep. 6, 25357. doi:10.1038/srep25357
Uchida, T., Rossignol, F., Matthay, M. A., Mounier, R., Couette, S., Clottes, E., et al. (2004). Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 279 (15), 14871–14878. doi:10.1074/jbc.M400461200
Weidner, J., Jarenbäck, L., Åberg, I., Westergren-Thorsson, G., Ankerst, J., Bjermer, L., et al. (2018). Endoplasmic reticulum, Golgi, and lysosomes are disorganized in lung fibroblasts from chronic obstructive pulmonary disease patients. Physiol. Rep. 6 (5), e13584. doi:10.14814/phy2.13584
Westergren-Thorsson, G., Bagher, M., Andersson-Sjoland, A., Thiman, L., Lofdahl, C. G., Hallgren, O., et al. (2018). VEGF synthesis is induced by prostacyclin and TGF-beta in distal lung fibroblasts from COPD patients and control subjects: Implications for pulmonary vascular remodelling. Respirology 23 (1), 68–75. doi:10.1111/resp.13142
Willems-Widyastuti, A., Alagappan, V. K., Arulmani, U., Vanaudenaerde, B. M., de Boer, W. I., Mooi, W. J., et al. (2011). Transforming growth factor-beta 1 induces angiogenesis in vitro via VEGF production in human airway smooth muscle cells. Indian J. Biochem Biophys. 48 (4), 262–269.
Xu, C., and Dong, W. (2016). Role of hypoxia-inducible factor-1α in pathogenesis and disease evaluation of ulcerative colitis. Exp. Ther. Med. 11 (4), 1330–1334. doi:10.3892/etm.2016.3030
Yao, L., Zhou, Y., Li, J., Wickens, L., Conforti, F., Rattu, A., et al. (2021). Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 297 (3), 101096. doi:10.1016/j.jbc.2021.101096
Yasuo, M., Mizuno, S., Kraskauskas, D., Bogaard, H. J., Natarajan, R., Cool, C. D., et al. (2011). Hypoxia inducible factor-1α in human emphysema lung tissue. Eur. Respir. J. 37 (4), 775–783. doi:10.1183/09031936.00022910
Keywords: BEAS-2B, hAELVi, fibrosis, growth factors, hypoxia, inflammation, lung epithelium
Citation: Berggren-Nylund R, Ryde M, Löfdahl A, Ibáñez-Fonseca A, Kåredal M, Westergren-Thorsson G, Tufvesson E and Larsson-Callerfelt A-K (2023) Effects of hypoxia on bronchial and alveolar epithelial cells linked to pathogenesis in chronic lung disorders. Front. Physiol. 14:1094245. doi: 10.3389/fphys.2023.1094245
Received: 09 November 2022; Accepted: 02 March 2023;
Published: 13 March 2023.
Edited by:
Sukhwinder Singh Sohal, University of Tasmania, AustraliaReviewed by:
Bokara Kiran Kumar, Centre for Cellular and Molecular Biology (CCMB), IndiaDeepak A. Deshpande, Thomas Jefferson University, United States
Jill Johnson, Aston University, United Kingdom
Copyright © 2023 Berggren-Nylund, Ryde, Löfdahl, Ibáñez-Fonseca, Kåredal, Westergren-Thorsson, Tufvesson and Larsson-Callerfelt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna-Karin Larsson-Callerfelt, YW5uYS1rYXJpbi5sYXJzc29uX2NhbGxlcmZlbHRAbWVkLmx1LnNl