 Jan B. Parys
Jan B. Parys Fabien Van Coppenolle
Fabien Van Coppenolle- 1Laboratory for Molecular and Cellular Signaling, Department of Cellular and Molecular Medicine & Leuven Kanker Instituut, KU Leuven, Leuven, Belgium
- 2CarMeN Laboratory, INSERM, INRA, INSA Lyon, Université Claude Bernard Lyon 1, Lyon, France
- 3Groupement Hospitalier EST, Department of Cardiology, Hospices Civils de Lyon, Lyon, France
The heterotrimeric Sec61 protein complex forms the functional core of the so-called translocon that forms an aqueous channel in the endoplasmic reticulum (ER). The primary role of the Sec61 complex is to allow protein import in the ER during translation. Surprisingly, a completely different function in intracellular Ca2+ homeostasis has emerged for the Sec61 complex, and the latter is now accepted as one of the major Ca2+-leak pathways of the ER. In this review, we first discuss the structure of the Sec61 complex and focus on the pharmacology and regulation of the Sec61 complex as a Ca2+-leak channel. Subsequently, we will pay particular attention to pathologies that are linked to Sec61 mutations, such as plasma cell deficiency and congenital neutropenia. Finally, we will explore the relevance of the Sec61 complex as a Ca2+-leak channel in various pathophysiological (ER stress, apoptosis, ischemia-reperfusion) and pathological (type 2 diabetes, cancer) settings.
1 Introduction
Intracellular Ca2+ signaling forms the core of a ubiquitous signal transduction pathway controlling important cell biological processes, including fertilization, proliferation, development, learning and memory, muscle contraction, secretory behavior, metabolism, apoptosis, autophagy, etc. (Berridge et al., 2000; Bagur and Hajnóczky, 2017; Giorgi et al., 2018; Wacquier et al., 2019; Bootman and Bultynck, 2020). The activity of the various members of the so-called Ca2+-signaling toolkit and their mutual functional and/or structural interactions determine the spatio-temporal properties of the Ca2+ signals and thus their eventual physiological effect. This toolkit effectively consists of a plethora of Ca2+ pumps and exchangers, Ca2+ channels and Ca2+-binding proteins located in the cytosol, the plasma membrane or various organelles (Berridge et al., 2003).
The endoplasmic reticulum (ER) forms the largest intracellular Ca2+ store and the Ca2+ ions released from the ER play an important role in the occurrence of the above-mentioned complex spatio-temporal Ca2+ signals (Berridge, 2002; Lam and Galione, 2013). Members of two related families of Ca2+-release channels are expressed in the ER (and the SR, the sarcoplasmic reticulum), i.e. inositol 1,4,5-trisphosphate (IP3) receptors (IP3R) and ryanodine receptors (RyR). While the RyR is expressed in a predominant way in only a limited number of tissues, especially skeletal and cardiac muscle as well as the brain (Lanner, 2012), the IP3R is ubiquitously expressed (Foskett et al., 2007; Berridge, 2016; Prole and Taylor, 2019; Hamada and Mikoshiba, 2020). The latter are therefore considered the main ER Ca2+-release channels involved in intracellular Ca2+ signaling. To perform this function, they form large tetrameric structures (4 x ∼300 kDa) that are activated by IP3 and that additionally are exquisitely regulated by Ca2+ itself, by phosphorylation/dephosphorylation processes and by multiple protein-protein interactions (Foskett et al., 2007; Vanderheyden et al., 2009; Prole and Taylor, 2016; Prole and Taylor, 2019; Hamada and Mikoshiba, 2020).
Less well understood, though physiologically at least even important, are the so-called Ca2+-leak channels that, depending on the cell type and the intracellular milieu, modulate the basal permeability of the ER for Ca2+ (Camello et al., 2002; Sammels et al., 2010; Takeshima et al., 2015; Lemos et al., 2021). These Ca2+-leak channels can affect intracellular Ca2+ signaling in multiple ways (Lemos et al., 2021). First, even a very low level of leak activity will impact cell behavior in stress situations wherein the SERCA (sarco- and endoplasmic reticulum Ca2+ ATPase) pumps can no longer compensate for the Ca2+ leakage, thereby leading to decreased filling of the ER Ca2+ store. Second, if the latter occurs or if the endogenous leak activity is anyway exceeding the capacity of the SERCA pumps, the setpoint for the ER Ca2+ concentration ([Ca2+]ER) will lower and thus directly or indirectly lead to decreased IP3-dependent Ca2+ release. Finally, highly active Ca2+-leak channels may themselves produce small Ca2+ signals interfering in a positive or negative way with intracellular Ca2+ signaling.
Surprisingly, the Ca2+-leak channels form a large though very heterogenous group of proteins that share as their only common characteristic, their potency to increase the permeability of the ER membrane for Ca2+ (Lemos et al., 2021). Some of them are dysfunctional versions of proteins involved in physiological Ca2+ handling such as IP3R or RyR. Dysfunctions can originate from (excessive) post-translational modifications, protease-mediated cleavages or mutations. Ca2+ signaling events such as Ca2+ puffs and sparks can also be considered to result from Ca2+-leak activity, although the channels involved (IP3R and RyR, respectively) are not dysfunctional, and their activity strongly depend on the local environment, especially [Ca2+]cyt and [Ca2+]ER (Berridge, 2006; Cheng and Lederer, 2008; Konieczny et al., 2012). Even SERCA pumps can participate in Ca2+ leakage from the ER either by increased slippage of the pump (Inesi and de Meis, 1989) or when truncated (Chami et al., 2008). A second group of Ca2+-leak channels is formed by ion channels that are at least partially expressed in the ER, as some TRP, Orai and pannexin channels, members of the Bax-inhibitor 1 family or some less-studied proteins, such as mitsugumin 23. Finally, a third group consists of proteins that are well known for a physiological function that is absolutely unrelated to Ca2+ handling, but that have also been shown to function as Ca2+-leak channels.
The importance of some of the proteins of the latter group in Ca2+ handling has recently emerged (Lemos et al., 2021). Presenilins form a first example. Presenilins 1 and 2 are expressed in the ER and Golgi apparatus (Annaert et al., 1999). They represent the catalytic core of the γ-secretase complex, a protease involved in the cleavage of multiple proteins including the amyloid precursor protein (De Strooper et al., 1998). Presenilins have been shown to function as bona fide ER Ca2+-leak channels (Tu et al., 2006; Zatti et al., 2006; Bandara et al., 2013; Klec et al., 2019). However, other studies suggest that they regulate other Ca2+-handling proteins, e.g., IP3Rs (Shilling et al., 2012; Shilling et al., 2014). Of course, these two functions are not mutually exclusive. A second example, for which evidence of its role as a Ca2+-leak channel has accumulated over the years, is the Sec61 complex/translocon (hereafter called the ‘Sec61 complex’). Its primary function is of course related to protein import in the ER during translation (Lang et al., 2017; Gemmer and Förster, 2020), but its role in Ca2+ handling is increasingly evident and has recently also been linked to pathological situations. It will therefore be the subject of the present review.
2 Structure and role of the Sec61 complex in protein translation/translocation and in the ER-associated degradation process (ERAD)
2.1 Function of the Sec61 complex
In eukaryotic cells, some proteins must be translocated or inserted into the ER membrane. The precursors of these proteins are characterized by a signal peptide, a hydrophobic N-terminal sequence or a transmembrane helix of 20–30 amino acids (Simon and Blobel, 1991). The nascent polypeptide chain exits from the ribosome with its signal peptide. Its recognition by the signal recognition particle (SRP) causes elongation arrest (Grudnik et al., 2009). Subsequently, this ribosome-nascent chain complex is, in a GTP-dependent manner, targeted to the membrane via interactions between SRP and its receptor. After the ribosomes dock to the Sec61 complex, the signal peptide is released from the SRP, and the SRP dissociates from its receptor (for review, see Egea et al., 2005). Interestingly, protein translocation through the Sec61 complex is also intricately associated with the Sec62/Sec63 complex (Jung and Kim, 2021).
The transport of proteins in parallel with their translation, membrane insertion and processing via the Sec61 complex requires the coordinated action of different cofactors and enzymes. The functional core of the Sec61 complex in mammals is formed by three different subunits (α, β and γ), allowing the proteins to cross or insert into the ER membrane (for review, see Lang et al., 2017). This basic function is complemented by accessory components, which are physically associated with it (Grudnik et al., 2009; Dejgaard et al., 2010; Shen et al., 2012). These accessory components, including SPC (signal peptidase complex), OST (oligosaccharyltransferase), TRAM (translocating chain-associated membrane protein), Sec62/63 complex and TRAP (translocon-associated protein complex), assist the passage of proteins through the channel formed by the Sec61 complex, where they allow maturation of nascent chains by covalent modifications and their chaperone-like function (Haßdenteufel et al., 2018; Ichhaporia and Hendershot, 2021).
During translocation, nascent proteins are correctly folded by ER luminal chaperones. BiP (a Ca2+- and ATP-dependent HSP70 chaperone also named GRP78) is one of the major ER chaperones that assists nascent proteins in the folding process (Pobre et al., 2019). BiP is also involved in controlling Sec61 complex permeability during translocation (Lièvremont et al., 1997; Hamman et al., 1998; Schäuble et al., 2012; Hammadi et al., 2013). Other auxiliary proteins are also associated with the Sec61 complex, such as Sec63 (Müller et al., 2010; Lang et al., 2012; Conti et al., 2015) and ERj1 (Blau et al., 2005; Dudek et al., 2005). Calnexin, a lectin-type chaperone of the ER, allows the maturation and oligomerization of secretory glycoproteins, and may associate with the Sec61 complex core, at least transiently or in a substrate-specific manner (Chevet et al., 1999; Schnell and Hebert, 2003; Lakkaraju et al., 2012).
The tertiary structure of newly synthetized proteins is under the control of molecular chaperone proteins. Inappropriate folding and accumulation of misfolded proteins first trigger the unfolded protein response (UPR) leading to a decrease in protein synthesis and an increase in chaperone expression (Ma and Hendershot, 2001; Hwang and Qi, 2018; Zhang et al., 2019). In parallel, during ERAD, unfolded proteins are retrotranslocated from the ER to the cytoplasm, poly-ubiquitinated and degraded by the 26S proteasome (Needham et al., 2019). The main channel for retrograde transport of proteins during ERAD is formed by the multispanning ubiquitin ligase Hrd1 (Schoebel et al., 2017; Wu and Rapoport, 2018), although the Sec61 complex can also be involved (Römisch, 2017).
2.2 Architecture of the Sec61 complex
The transient nature of the many interactions at the Sec61 complex level made it difficult to determine its structure. Even in the case of the Sec61 complex core, the composition, architecture, and mechanism of operation remain difficult to determine. However, technical advances, particularly in the field of cryo-electron microscopy, have made it recently possible to increase the understanding of the heterotrimeric Sec61 complex.
Studies on human cells have shown that the main components of the Sec61 complex are Sec61 (α, β and γ), TRAP and OST (Pfeffer et al., 2014). A secondary structure can be defined at the subnanometer scale, including the protein-conducting channel (Pfeffer et al., 2015). It has not yet been clearly defined whether OST transiently associates with the Sec61 complex for a specific phase of translocation or whether different types of Sec61 complexes coexist in the cell. It has, however, been shown that initiation of translation strengthens the association between OST and Sec61 (Conti et al., 2015; Bai et al., 2018), which tends towards the first hypothesis. However, no such conclusive pattern could be revealed in vivo (Mahamid et al., 2016).
2.2.1 The pore
The main feature of the ER Sec61 complex is the existence of an aqueous pore across the membrane. Thanks to experiments using puromycin treatment, the Sec61 complex has been seen as a channel gated by the ribosome. Indeed, these ER transmembrane pores were detected by conductivity measurements, and the dependence of these channels on puromycin suggested that the nascent polypeptide chain must be released from the ribosome to allow the transmembrane passage of ions (Simon and Blobel, 1991). Since the channel is closed when the ribosomes are detached from the membrane, the passage of ions through the channel is dependent on both the nascent chain and the ribosome (Simon and Blobel, 1991; Van Coppenolle et al., 2004).
Since the lumen of the ER, and not the cytoplasm, forms a continuum with the Sec61 complex pore upon protein translocation, the ribosome itself must form the permeability barrier by strongly binding the cytoplasmic surface of the Sec61 complex and sealing the pore from there (Crowley et al., 1994).
The experiments conducted by Hamman et al. (Hamman et al., 1998) indicate that the ribosome remains in place on the Sec61 complex until the translation is complete. At this stage, the pore of the Sec61 complex linked to the ribosome had a diameter of 40–60 Å, potentially allowing Ca2+ leakage. In contrast, the pore of the Sec61 complex has an internal diameter of 9–15 Å when not bound to the ribosome (Hamman et al., 1997).
2.2.2 The components of the Sec61 complex
2.2.2.1 Sec61
Sec61 has a heterotrimeric structure forming a channel allowing the passage of proteins (Figure 1). The Sec61α subunit consists of 10 transmembrane helices, arranged in two pseudosymmetrical N- and C-terminal halves around a central pore, impermeable to passive ion flow. The Sec61β and Sec61γ subunits are located at the periphery of the Sec61 complex, and each has a transmembrane helix (Berg et al., 2004; Egea and Stroud, 2010; Li et al., 2016).
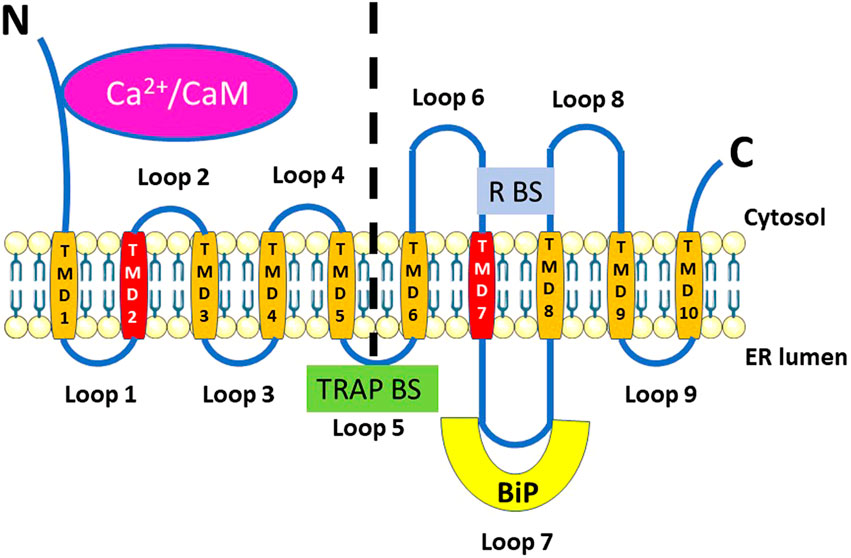
FIGURE 1. Structure of Sec61α, highlighting elements relevant for its function as ER Ca2+-leak channel. Sec61α is characterized by having 10 transmembrane domains (TMD1-10, in orange) arranged in two pseudo-symmetrical N- and C-terminal halves (delineated by the vertical dashed line) with both the N and C termini located in the cytosol. The two sterically adjacent TMD2 and TMD7 (in dark orange) form the lateral gate. The interaction of Ca2+/calmodulin (Ca2+/CaM) at the N-terminus and of BiP at loop 7 is shown. In addition, the luminal binding site for the translocon-associated protein complex (TRAP BS) on loop 5, at the interphase between the N- and C-terminal halves, and of the ribosome (R BS) on loops 6 and 8 are indicated.
During protein transport and translation, the ribosome binds to cytosolic loops 6 and 8 of Sec61α (Figure 1). A recent mechanistic model shows that Sec61 transiently opens to allow insertion of the transmembrane helix or nascent protein signal sequence into the ER membrane while remaining impermeable to low-molecular-weight proteins (Becker et al., 2009; Gogala et al., 2014; Park et al., 2014; Voorhees et al., 2014; Voorhees and Hegde, 2016).
The pore of the Sec61 complex formed by Sec61 completely encompasses the ER lipid bilayer through which the newly synthesized secretory proteins are translocated (Swanton and Bulleid, 2003). The heteromeric Sec61 complex forms the nucleus of this pore, which can be blocked by BiP on the luminal side. The ribosome binds to the cytosolic side, and the Sec61 complex can be considered an SRP-ribosome gated channel.
2.2.2.2 TRAP
TRAP is a heterotetrameric membrane protein complex, the role of which is to assist Sec61 with protein insertion and more specifically in the topogenesis of polytopic membrane proteins (Sommer et al., 2013). It is made up of three subunits (α, β, δ) forming a transmembrane helix and a luminal domain, while TRAPγ forms a set of four transmembrane helices with a small cytosolic domain (von Heijne and Gavel, 1988). The luminal part of TRAP binds Sec61α at the level of the two N- and C-terminal halves (Figure 1) and could thus influence the conformation of Sec61α and interact with the newly synthesized proteins that are translocated. Interestingly, in TRAPβ-depleted HeLa cells, thapsigargin induced via the Sec61 complex a smaller increase in [Ca2+]cyt than in control cells (Nguyen et al., 2018). It would therefore be interesting to compare in further experiments simultaneously [Ca2+]cyt and [Ca2+]ER to decipher whether the absence of TRAPβ decreases the Ca2+ leak and/or partly depletes the Ca2+ stores.
2.2.2.3 OST
The OST complex is made up of at least seven proteins. OST catalyzes the glycosylation of newly synthesized proteins (Kelleher and Gilmore, 2006). One of the subunits of OST is STT3. It occurs in two paralogous forms STT3A and STT3B, which are involved in glycosylation during and after translation respectively (Ruiz-Canada et al., 2009). They are associated with at least six accessory subunits of OST, the function or structure of which are not yet completely understood: ribophorin I, ribophorin II, OST48, DAD1, N33 (or IAP) and OST4. The N33 subunit of OST has oxidoreductase activity and is believed to increase glycosylation efficiency by slowing the conformational changes of glycoproteins (Mohorko et al., 2014). Recent findings using high-resolution cryoelectron microscopy revealed that OST binds to Sec61α and partially penetrates the lumen of the ER (Braunger et al., 2018; Ramírez et al., 2019). However, its interaction with the Sec61 complex has yet to be further determined.
2.2.2.4 Other important ER proteins
Calnexin, an intraluminal ER membrane protein acting as a chaperone during nascent protein conformation (Chen et al., 1995), may be linked to nascent polypeptide chains (Chen et al., 1995; Oliver et al., 1996; Tatu and Helenius, 1997). Calnexin therefore appears to be close to the Sec61 complex, but there is as yet no evidence of its participation in the complex.
Further ER proteins such as calreticulin, protein disulfide isomerase, BiP and ERp57 also transiently interact with the nascent protein chain (Nicchitta and Zheng, 1997; Oliver et al., 1997; Tatu and Helenius, 1997), but it was arbitrarily decided to consider only membrane proteins associated with the Sec61 complex.
2.2.3 The ribosome
Sec61α is located in the center of the Sec61 complex and binds the ribosome via its loops 6 and 8, close to the exit peptide of the ribosome (Raden et al., 2000; Song et al., 2000). While TRAP specifically binds a region of Sec61α, OST interacts with Sec61 via a larger interface in the ER membrane. On the cytosolic face of the Sec61 complex, the small cytosolic domains of TRAP and OST interact with the large ribosomal subunit via specific cytosolic contact sites (Silberstein and Gilmore, 1996; Wang and Dobberstein, 1999; Kriegler et al., 2020; Mohanty et al., 2020).
3 Sec61 complex, Ca2+ leakage and pharmacology
As some antibiotics inhibit translation by acting on the Sec61 complex, these compounds turned out to be very useful to modulate Sec61 complex opening, either inducing or inhibiting Ca2+ leakage. Nevertheless, it is important to note that acute pharmacological modulation of Sec61 complex opening with those antibiotics during short-term Ca2+-imaging experiments is too brief to significantly affect protein translation/translocation (Van Coppenolle et al., 2004). Indeed, these molecules act on the Ca2+ permeability of the Sec61 complex in the order of minutes, while they will take a longer time to act on protein translation in a detectable way. Similarly, during long-term treatment of cells with these compounds it is necessary to pay attention to use a concentration that is able to modify Sec61 complex permeability to Ca2+ without significantly affecting protein translation.
3.1 Three states for one complex
There are three known conformations of Sec61α: idle, intermediate and open. In its non-translating state, Sec61α binds to the ribosome with a plugged gate corresponding to the idle conformation (Voorhees et al., 2014). Ca2+ leakage via the Sec61 complex probably does not occur in this state (Wirth et al., 2003). After binding to the ribosome and just before signal peptide engagement, Sec61α moves toward the intermediate state (Bhadra et al., 2021). Here, the Sec61α complex “breathe” along the opened lateral gate (Li et al., 2016), which may trigger Ca2+ permeability. Once nascent protein translocation occurs through the channel, Ca2+ cannot cross the pore. After translocation, when the peptidic chain has been released, the Sec61 complex remains transiently in an open post-translocation state that allows Ca2+ leakage via the aqueous pore. Indeed, the time between the ribosome detaches from the Sec61 complex, and the reformation of a luminal plug corresponds to the time period during which Ca2+ leakage can occur through the wide-open pore with a 50 Å diameter. Subsequently, the Sec61 complex returns again to the idle state when the ribosomes come off (Bhadra et al., 2021).
3.2 Pharmacological aspects
Puromycin is a structural analog of phenylalanyl-tRNA, causing the formation of an abnormal peptidyl-puromycin. The newly synthetized polypeptide chains are therefore incomplete, which causes their detachment from the Sec61 complex and the inhibition of protein synthesis (Traut and Monro, 1964). At this level, the ribosome is still linked to the Sec61 complex (Pestova and Hellen, 2001; Van Coppenolle et al., 2004), and the pore is open, allowing Ca2+ leakage (Van Coppenolle et al., 2004; Wonderlin, 2009).
Concerning pactamycin, its mode of action is still debated. During translation, all tRNA substrates attach to three ribosome binding sites named A (aminoacyl), P (peptidyl) and E (exit). These three sites are located at the junction between the ribosomal subunit and the tRNA (Yusupov et al., 2001). Indeed, the first studies showed that pactamycin associates with the ribosome, thereby preventing tRNA from entering the P site. This would be due to a change in the conformation of the ribosome or by direct competition, thus inhibiting protein translation (Eida and Mahmud, 2019). Regardless, the peptidic chain is released in the presence of pactamycin during translation, allowing Ca2+ leakage (Al-Mawla et al., 2020).
Anisomycin is a translation inhibitor that acts by binding to a part of the 60S subunit (Hummel et al., 1987; Rodriguez-Fonseca et al., 1995) of the ribosome that has been suggested to be the peptidyl transferase center (Rodriguez-Fonseca et al., 1995; Iordanov et al., 1997). Anisomycin has been shown to inhibit puromycin reaction in an in vitro system (Ioannou et al., 1998). The inhibition of the peptidyl transferase activity by anisomycin prevents elongation of the peptide chain and any Ca2+ leakage through the Sec61 complex (Van Coppenolle et al., 2004; Hammadi et al., 2013).
Emetine interacts with the 40S ribosome subunit at the E site, inhibiting the progression of the ribosome on mRNA. This molecule is therefore an irreversible inhibitor of translation elongation, which stabilizes the ribosome/nascent chain complex, causing the pore of the Sec61 complex to close (Al-Mawla et al., 2020). It should be noted that emetine also seems to mobilize Ca2+ from the Golgi apparatus, although the Ca2+ channel involved was not identified (Gallegos-Gómez et al., 2018). The latter point should be taken into consideration when using the compound.
Cycloheximide is commonly used for control purposes. This antibiotic acts as an inhibitor of elongation, binding the E site of the ribosome without any modulation of Sec61 complex permeability to Ca2+ (Van Coppenolle et al., 2004).
It is now also possible to modulate the permeability of the Sec61 complex with molecules other than antibiotics. Indeed, recently, the Mycobacterium ulcerans exotoxin mycolactone has been shown to enhance ER Ca2+ leakage via the Sec61 complex (Bhadra et al., 2021). The mechanisms of action of mycolactone on the Sec61 complex are detailed in Section 3.5.
Another molecule of interest is eeyarestatin, a chemical inhibitor of protein degradation via the ERAD system (Wang et al., 2008; Wang et al., 2009) and of translocation of nascent polypeptides into the ER via the Sec61 complex (Cross et al., 2009). Recently, eeyarestatin analogs have also been found to mediate ER Ca2+ leakage via the Sec61 complex (Gamayun et al., 2019). In this interesting study, eeyarestatin was proposed to bind Sec61α in the open state, where it would prevent the closure of the lateral gate, keeping Sec61α in a Ca2+-permeable state.
The list of drugs acting on the Ca2+ permeability of the Sec61 complex is gradually increasing, thereby widening the scope of studies in this field.
3.3 The Sec61 complex: A new functional, ER Ca2+-leak channel
As mentioned above (see Section 2.2.1), the pore of the Sec61 complex is the largest in the ER, with a diameter of 40–60 Å in the ribosome-bound state and a smaller diameter of 9–15 Å in the ribosome-free state (Hamman et al., 1997; Hamman et al., 1998). Subsequently, the role of the Sec61 complex as an ER Ca2+-leak channel emerged thanks to the work of Wonderlin’s group showing that small polarized molecules could cross the ER membrane through the Sec61 complex (Heritage and Wonderlin, 2001; Roy and Wonderlin, 2003). We therefore hypothesized that Ca2+ ions would also move across the ER membrane via the Sec61 complex. To verify this hypothesis, the main experimental problem was how to best open the Sec61 complex pore to directly measure ER Ca2+ leakage. Similar to Wonderlin and collaborators, we started by using puromycin to, for the first time, directly detect Ca2+ leakage through the Sec61 complex in mouse pancreatic acinar cells (Lomax et al., 2002).
In subsequent experiments, we continued the investigation of the cell physiological role of the Sec61 complex as an ER Ca2+ leak channel. Using human cancerous prostate cells (the LNCaP cell line), we determined that puromycin induced Ca2+ leakage through the Sec61 complex independently of IP3Rs or RyRs (Van Coppenolle et al., 2004). Moreover, for this study, we systematically used anisomycin to counteract puromycin’s action to control the specificity of action of these compounds on the Sec61 complex. In a similar way, other studies used pactamycin instead of puromycin and emetine instead of anisomycin (Ong et al., 2007; Al-Mawla et al., 2020). The fundamental cell physiological role of the Sec61 complex was elucidated in a second study using LNCaP cells (Flourakis et al., 2006). The [Ca2+]ER at rest is the result of the equilibrium between Ca2+ uptake mediated by the SERCA pumps and passive Ca2+ leakage through Ca2+-leak channels. Inhibition of SERCA pumps by the specific inhibitor thapsigargin therefore fully reveals the passive Ca2+ release through ER Ca2+-leak channels, as it is not compensated by reuptake. The measured Ca2+ leak is, in those conditions, the result of all the ER Ca2+-leak channels open at this stage. This leak appeared to be mostly mediated by the Sec61 complex. Moreover, similar results were obtained using EGTA-AM (a cytosolic Ca2+ chelator) to enhance Ca2+ release. Altogether, these data highlight the role of the Sec61 complex as a major Ca2+-leak channel in the LNCaP cell model. In addition, it is well known that ER Ca2+ depletion can activate SOCs (store-operated channels). Interestingly, we have also demonstrated that Ca2+ release that occurs via the Sec61 complex activates SOCs. This was the first characterization of store-operated Ca2+ entry triggered by passive Ca2+-leak channels (Flourakis et al., 2006).
Giunti et al. elegantly studied the efflux of Ca2+ from ER-derived rat liver microsomal vesicles (Giunti et al., 2007). They detected two basal passive pathways for Ca2+ efflux. One of them is carried by the Sec61 complex since the leakage was stimulated by puromycin perfusion. The second efflux pathway is more mysterious and requires counterion influx. It does not involve inactive SERCA pumps, Bcl-2 proteins, or known Ca2+ channels such as IP3Rs or RyRs. Interestingly, the authors also noticed that the efflux was largest in the rough microsomal subfractions, which are enriched in the Sec61 complex. Thanks to this approach, this work provides strong evidence for the role of the Sec61 complex as a functional ER Ca2+-leak channel at the organellar level.
In addition to these studies, we should mention the interesting work of Layhadi et al. (Layhadi and Fountain, 2017). They highlighted in THP-1 macrophages the role of the Sec61 complex in the control of the [Ca2+]cyt at rest. In the absence of extracellular Ca2+, inhibition of the Sec61 complex with anisomycin reduced the resting [Ca2+]cyt. In addition, the authors demonstrated that inhibition of the Sec61 complex by anisomycin enhances the response to ADP (via P2Y receptors) in human primary macrophages. Altogether, this work clearly indicates that the Sec61 complex is not only an ER Ca2+-leak channel but also acts, at least in macrophages, as a regulator of ER Ca2+ content and of the [Ca2+]cyt at resting state.
Taken together, various studies have demonstrated that the Sec61 complex is a functional ER Ca2+-leak channel in several cell types. Nevertheless, the Ca2+-leakage rate depends on the dynamics of plug binding/dissociating the pore. An important question, therefore, is which factors will control Ca2+ leakage?
3.4 Regulation of Sec61 complex Ca2+ leakage from the ER by Ca2+-binding proteins
A permanently open ER Ca2+-leak channel would in the long run form a hazard for the cell as both ER Ca2+ levels and cytosolic Ca2+ levels control many aspects of cell function, including the occurrence of ER stress and/or the induction of cell death (Orrenius et al., 2003; Pinton et al., 2008; Krebs et al., 2015; La Rovere et al., 2016; Danese et al., 2021). It can therefore be expected that intra-ER and/or cytosolic proteins sensing the local [Ca2+] control the Ca2+ flux through the Sec61 complex.
3.4.1 The regulating role of BiP
As stated above (see Section 2.1), BiP is an important ER intraluminal heat shock protein. It has a chaperone function towards newly synthesized proteins, controls the protein flux through the Sec61 complex, and activates the UPR in ER stress conditions (Pobre et al., 2019). Moreover, it participates in the regulation of intracellular Ca2+ homeostasis by acting as an intraluminal Ca2+-binding protein (Lièvremont et al., 1997), by stabilizing the type 1 IP3R (Higo et al., 2010) and by controlling ER Ca2+ leakage through the Sec61 complex (Schäuble et al., 2012; Hammadi et al., 2013). During ER stress, BiP preferentially targets accumulating unfolded or misfolded proteins, thereby releasing the three canonical ER-stress sensors, inositol-requiring enzyme 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which consequently become free to activate the UPR (Ron and Walter, 2007). In addition, during ER stress, BiP will also dissociate from both the IP3R and the Sec61 complex, leading to complex changes in ER Ca2+ handling. This includes first a decreased Ca2+ release from the ER and subsequently an increased Ca2+ release that can lead to cell apoptosis (Kiviluoto et al., 2013).
In relation to the Sec61 complex, BiP is an allosteric effector of the Sec61 channel, supporting the translocation of proteins through it (Lang et al., 2017). A BiP-binding site on a minihelix located in loop 7 of Sec61α (Figure 1) appears to be involved in the gating of the Sec61 complex and in the regulation of ER Ca2+ leakage through the Sec61 complex (Schäuble et al., 2012; Schorr et al., 2015). The expression in human HeLa cells of the Sec61α-Y344H mutant deficient in BiP binding (see Section 4.1) increased ER Ca2+ leakage (Schäuble et al., 2012). In the same study, an increased Ca2+ leak was also observed in conditions in which BiP was made unavailable, either via siRNA-mediated gene silencing or via induction of protein misfolding by short-term treatment with dithiothreitol or tunicamycin. This effect could not be mimicked by other chaperones, such as protein disulfide isomerase, calreticulin or GRP94, indicating the specificity of BiP compared to other chaperones. However, in this role, BiP is assisted by two of its intraluminal co-chaperones of the Hsp40 family, ERj3 and ERj6. siRNA-induced depletion of HeLa cells of either one of these two proteins led to increased Sec61-mediated Ca2+ leakage out of the ER (Schorr et al., 2015). This regulation was specific for ERj3 and ERj6 as depleting the Hsp40 proteins ERj1, Sec63 (also known as ERj2), ERj5 and ERj7 had no such effect.
The control of ER Ca2+ leakage by BiP has interesting functional consequences for the cell. First, as the presence of unfolded or misfolded proteins results in the release of BiP from the Sec61 complex, cells with a particularly high protein synthesis rate (e.g., β cells of the pancreas or hepatocytes) are particularly prone to experience increased Ca2+ leakage out of the ER, potentially leading to apoptosis (Hammadi et al., 2013; Kiviluoto et al., 2013; Schorr et al., 2015; Lang et al., 2017; Lemos et al., 2021) (Figure 2). In addition, since BiP is dependent on ATP for its function, it was proposed (Lang et al., 2019) that low [ATP]ER will lead to decreased BiP activity and thus increased Ca2+ leakage out of the ER. The subsequent decrease in [Ca2+]ER and increase in [Ca2+]cyt will then activate the ADP/ATP exchanger of the ER (Klein et al., 2018) to replenish ER ATP content.
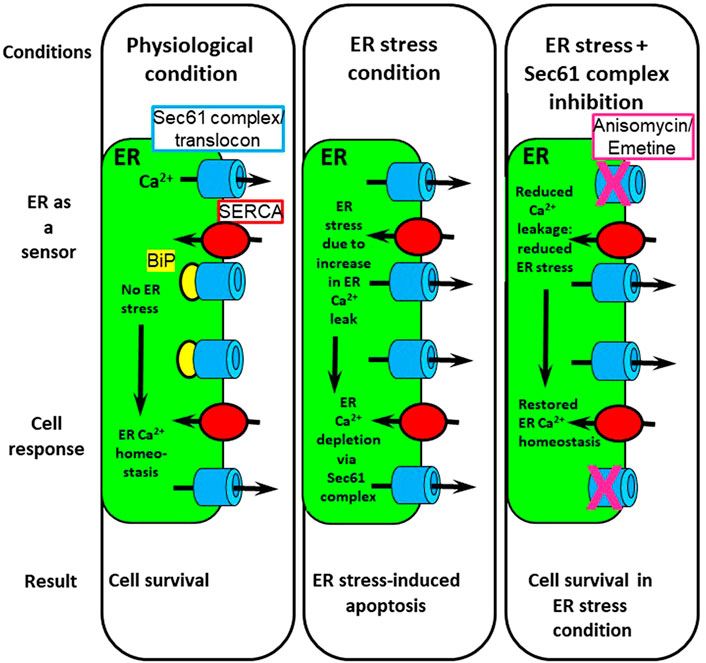
FIGURE 2. Involvement of the Sec61 complex/translocon in cell survival. In physiological condition, ER Ca2+ homeostasis is due to a balance between Ca2+ leak and Ca2+ re-uptake by SERCA pumps. ER stress triggers Ca2+ depletion of the ER Ca2+ stores via an increase in Ca2+ leakage through the Sec61 complex. Pharmacological inhibition of the Sec61 complex with anisomycin or emetine during ER stress restores ER Ca2+ homeostasis and protects cells from apoptosis. Adapted from Cassel et al., Plos One, 2016 (Cassel et al., 2016) licensed under a Creative Commons Attribution 4.0 International License.
In conclusion, BiP, the luminal plug of the Sec61 complex, might attach and detach from the pore with particular dynamics determining the Ca2+-leakage rate depending on physiological cell conditions (such as ER stress, BiP expression or rate of translation).
3.4.2 The regulatory role of calmodulin (CaM)
CaM is a ubiquitously expressed cytosolic Ca2+-binding protein. It contains four EF-hand motifs with an affinity for Ca2+ in the physiological range (Kd’s between 0.5 and 5 µM), making CaM ideally suited as an intracellular Ca2+ sensor (Chin and Means, 2000). CaM therefore modulates a plethora of proteins, including various Ca2+ pumps and channels.
It is thus particularly interesting that Ca2+ leakage through the Sec61 complex is also modulated by CaM (Erdmann et al., 2011). Sec61α contains in its N-terminal domain a high-affinity binding site (a.a. 19–32) that specifically interacts with Ca2+/CaM but does not interact with apoCaM (Figure 1). Electrophysiological experiments determined that CaM induced Ca2+-dependent closure of the Sec61 channel. The reverse also holds, as treatment with the CaM antagonists ophiobolin A or trifluoperazine enhanced Ca2+ leakage. Moreover, molecular modeling suggested that Ca2+/CaM binds in the gap between the ribosome and the Sec61 complex.
3.5 Modulation of Sec61-mediated Ca2+ leakage by bacterial toxins
Mycolactone is an exotoxin produced by Mycobacterium ulcerans that causes Buruli ulcer, a chronic skin necrosis (Demangel et al., 2009; Sarfo et al., 2016; Demangel and High, 2018). Pre-ulcerative and ulcerative lesions are painless, often delaying curative actions (En et al., 2008). Mycobacterium ulcerans infections lead to the suppression of the immune system allowing the multiplication of bacteria in the skin (Yotsu et al., 2018).
Many molecular targets of mycolactone have been identified, including the Sec61 complex (Baron et al., 2016; Morel et al., 2018; Gérard et al., 2020; Demangel, 2021; O’Keefe et al., 2022). Mycolactone inhibits Sec61-dependent protein translation/translocation into the ER (Hall et al., 2014). This has been linked to the mechanism of immunosuppression induced by mycolactone via inhibition of T cell activation and antigen presentation (Baron et al., 2016; Grotzke et al., 2017). Since the Sec61 complex is a target of mycolactone, the question arose as to the potential effects of this toxin on ER Ca2+ permeability. This aspect has been studied by Bhadra et al. (Bhadra et al., 2021). Using the HEK293 and HCT116 cell lines, they demonstrated that mycolactone enhances ER Ca2+ leakage via the Sec61 complex. The absence of Ca2+ depletion in cells expressing Sec61α mutants, resistant to mycolactone binding, corroborates the direct action of the toxin on the complex. Interestingly, they postulate that among the three known conformations of Sec61α, idle, intermediate and open (see Section 3.1), mycolactone stabilizes the intermediate conformation, locking the Sec61 complex in a Ca2+-permeable conformation.
3.6 The regulatory role of Sec62
Evidence exists that for post-translational protein translocation, the Sec61 complex associates with the Sec62/Sec63 complex. Sec62 is a 399 a. a. protein characterized by having both its N- and C-terminal regions in the cytosol, flanking the two transmembrane domains (TMD) connected by a short intraluminal loop (Daimon et al., 1997; Tyedmers et al., 2000).
Analysis of various prostate cancer cell lines demonstrated that reduced Sec62 protein levels lead to larger Ca2+ leakage out of the ER and reduced cell viability while increased Sec62 expression correlated with protection against thapsigargin-induced apoptosis (Greiner et al., 2011). These results strongly suggest that Sec62 is an inhibitor of ER Ca2+ leakage, most likely via a direct or indirect action on the Sec61 complex. Subsequent work indicated that Ca2+ affected the binding of the C terminus of Sec62 to the N terminus of Sec61α (Linxweiler et al., 2013). Moreover, mutation of a putative Ca2+-binding motif (a.a. 308–319) in the C terminus of Sec62, increased thapsigargin-induced Ca2+ leakage out of the ER. Taking into account that Sec62 silencing and treatment with CaM antagonists lead to the same phenotype, the authors propose a model in which Sec62 binding to Sec61 is relieved by Ca2+, allowing Ca2+/CaM to occupy its binding site and thus to inhibit ER Ca2+ leakage through the Sec61 complex. Obviously, another possible mechanism would involve the recruitment of BiP at the luminal side, subsequent to Sec62/Sec63 binding.
Taken together, the regulation of Sec61 complex-mediated Ca2+ leakage by multiple Ca2+-dependent proteins may indicate the importance of keeping Ca2+ leakage under tight control, or may indicate that depending on the situation, e.g., the presence or absence of ER stress, the cell relies on a different mechanism for controlling Ca2+ leakage out of the ER.
4 Involvement of Sec61 complex-mediated ER Ca2+ leakage in pathological conditions
4.1 SEC61 mutations and related pathologies
Several pathogenic mutations have been detected, as well in the genes coding for the proteins forming the Sec61 complex as in those coding for proteins associated with the complex (Lang et al., 2017; Sicking et al., 2021). These mutations result in various clinical phenotypes, including type 2 diabetes mellitus (T2D), immunodeficiency, tubulo-interstitial kidney disease and neutropenia (for mutations in SEC61A1). The occurrence of polycystic liver disease and colorectal cancer has been linked to SEC61B and that of glioblastoma multiforme, hepatocellular and renal cell carcinoma to SEC61C expression levels. Interestingly, mutations in proteins associated with the SEC61 complex recapitulate some of the above-mentioned diseases (T2D, polycystic liver disease) but can also lead to other diseases, such as the so-called congenital disorder of glycosylation (Lang et al., 2017; Sicking et al., 2021). Obviously, these mutations can affect each of the various aspects of Sec61 complex function and do not necessarily imply dysfunctional cellular Ca2+ handling. Interestingly, however, a number of those mutations were determined to impact ER Ca2+ leakage in mice (Schäuble et al., 2012) and humans (Schubert et al., 2018; Van Nieuwenhove et al., 2020) (Table 1).
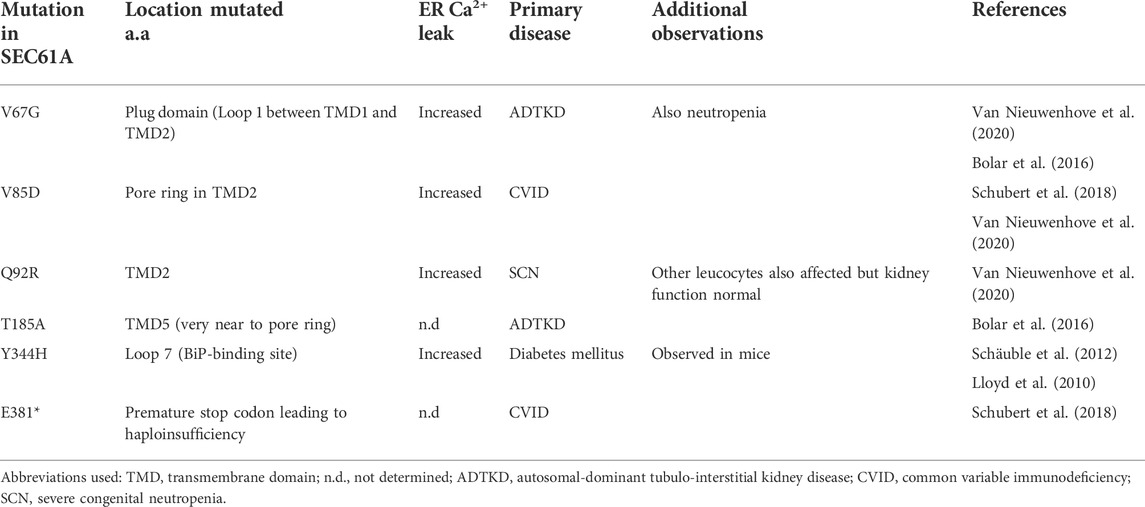
TABLE 1. Identified SEC61A1 disease mutations and their effects on the ER Ca2+ leak. Description of SEC61A mutations leading to various diseases affecting kidney function, immunological behavior and/or metabolism, with special attention to the structural and functional consequences for the ER Ca2+ leak. TMD2 forms part of the lateral gate, while the pore ring is the name of the central constriction of Sec61α, which in its closed conformation is occupied by Loop 1, called the plug domain.
Common variable immunodeficiency (CVID) is a group of diseases of various origins that are generally characterized by impaired B-cell differentiation/function, resulting in low levels of antibody production and, consequently, recurrent infections. Two mutations in SEC61A1, a heterozygous missense mutation (V85D) and a nonsense mutation (E381*), have been linked to CVID (Schubert et al., 2018). SEC61A1-V85D, when expressed in HeLa cells, not only impaired protein translocation to the ER, but also resulted in a severe depletion of the ER Ca2+ store due to increased Ca2+ leakage out of the ER. SEC61A1-V85D expression selectively impaired the survival of plasma cells. This was due to the induction of unresolvable ER stress, leading to terminal UPR.
Autosomal dominant severe congenital neutropenia (SCN) forms another genetically heterogeneous group characterized by differentiation arrest in the formation of granulocytes. Mutations in over 20 genes are already involved in this pathology, while for many patients the responsible mutation has not yet been characterized. In a recent study, the point mutation Q92R in SEC61A1 was identified in a patient with severe congenital neutropenia (Van Nieuwenhove et al., 2020). Expression of SEC61A1-Q92R in HL-60 cells induced a 30% depletion of the ER Ca2+ store compared to wild-type SEC61A1. This was similar to the depletion observed when expressing in the same cells SEC61A1-V67G or SEC61A1-T185A, two heterozygous missense mutations already shown to result in autosomal-dominant tubulo-interstitial kidney disease (ADTKD) (Bolar et al., 2016). Additionally, SEC61A1-Q92R patient cells demonstrated an increased UPR and were more prone to apoptosis.
Interestingly, the ADTKD patients expressing the V67G mutation also displayed some neutropenia (Bolar et al., 2016). Similarly, the patient with the Q92R mutation was not only characterized by SCN, but also displayed B-cell maturation defects, reminiscent of the patients with SEC61A mutations leading to CVID, though harbored normal kidney function (Van Nieuwenhove et al., 2020).
Finally, investigation in a mouse model for T2D indicated that the missense mutation Y344H in loop 7 of murine Sec61α was linked to the occurrence in β cells of continuing ER stress leading to apoptosis and the subsequent development of T2D (Lloyd et al., 2010). The same mice also displayed hypercholesterolemia, hypertriglyceridemia, hepatomegaly, steatosis and, in older animals, hepatic cirrhosis. These observations are also relevant for humans, since the motif is fully conserved between mice and humans. Subsequent expression of Sec61α-Y344H in human HeLa cells led to increased ER Ca2+ leakage (Schäuble et al., 2012). Moreover, this mutation impairs binding of BiP to Sec61α, consistent with the observation that BiP limits ER Ca2+ leakage (see Section 3.4.1). Furthermore, these results strongly suggest that the gating of the Sec61 complex by BiP occurs via its binding to loop 7 of Sec61α. Interestingly, deletion of ERj6, one of the proteins assisting BiP in its regulation of Sec61 complex-mediated Ca2+ leakage, results in both mice (Ladiges et al., 2005) and humans (Synofzik et al., 2014) in pancreatic β cell failure and T2D, suggesting that increased Ca2+ leakage might form part of the mechanism involved.
It is presently not understood how the various SEC61A1 mutations form the origin of different clinical phenotypes, but this may be due to a combination of various functional effects, including but not limited to dysfunctional Ca2+ handling, and destabilization of the protein leading to lower expression levels. It is, however, clear that, with the exception of Y344H, which affects BiP binding, the other mutations linked to dysfunctional Ca2+ handling (V67G, V85D, Q92R, Y344H) are thus far all located in or near TMD2 (Table 1). V67G is located in the loop between TMD1 and TMD2 that forms a plug that seals and stabilizes the pore during the closed state (Linxweiler et al., 2013). V85D and Q92R, on the other hand, introduce a charge in the hydrophobic TMD2, which will likely affect the pore conformation and modify Ca2+-channel properties. Moreover, TMD2 and TMD7 form the so-called lateral gate that can allow/prevent lateral access to the central pore (Sicking et al., 2021).
In view of the structure of SEC61A, it may be expected that other mutations will be discovered that also affect its Ca2+ handling, which will help our understanding of the Ca2+-leak pathway.
4.2 Involvement of Sec61-mediated Ca2+ leakage in ER stress and UPR in disease
As detailed earlier, ER stress is due to either accumulation of unfolded or misfolded proteins in the ER lumen or due to Ca2+-store depletion. The UPR is an adaptive phenomenon aimed at reducing the unfolded protein burden (Hammadi et al., 2013; Hetz et al., 2020). A prolonged UPR is associated with ERAD. Unfolded proteins are retrotranslocated from the ER lumen to the cytoplasm, where they are degraded by the 26S proteasome (Wu and Rapoport, 2018). Prolonged ER stress will lead to apoptosis. Among all the signaling transduction pathways of ER stress and UPR, it is interesting here to focus on BiP overexpression. In the absence of ER stress, BiP maintains IRE1, PERK and ATF6 (Behnke et al., 2015; Pobre et al., 2019) in an inactive state. BiP also plugs the pore of the luminal side of the Sec61 complex (Hamman et al., 1997; Haigh and Johnson, 2002; Alder et al., 2005) and stabilizes the type 1 IP3R (Higo et al., 2010). During ER stress and UPR, a reorientation of BiP occurs: the chaperone binds to unfolded proteins and thus unhooks from the Sec61 complex, type 1 IP3R, IRE1, PERK and ATF6, concomitantly triggering Ca2+ leakage from the ER via the Sec61 complex (Figure 2). Very recently, using GCamP6, a genetically encoded Ca2+ indicator tethered to the ER membrane, evidence was presented that during the early phase of the UPR, the Sec61 complex evoked Ca2+ signals (Feliziani et al., 2022). Interestingly, these Ca2+ signals had physiological significance, as they contributed to PERK activation.
Ca2+ dysregulations as well as UPR perturbations have been associated with many diseases such as T2D (Özcan et al., 2006; Hotamisligil, 2010), cardiac pathologies (Minamino and Kitakaze, 2010; Hammadi et al., 2013; Luo et al., 2015) and cancer (Tsai and Weissman, 2010; Hammadi et al., 2013). Further questions arise now. Are ER stress and Ca2+ homeostasis independent mechanisms? What could be the link between these two mechanisms, and how could they be involved in these pathologies?
The involvement of the Sec61 complex in pathological contexts is beginning to be investigated. In the following sections, we will focus on cancer, T2D, cardiac ischemia-reperfusion and stunned myocardium with the Sec61 complex as a potential therapeutic target.
4.2.1 Sec61 complex and cancer
Resistance to apoptosis is one of the major hallmarks of cancerous cells. New therapeutic strategies must focus on the induction of cell death. There is a direct link between ER Ca2+ homeostasis and cell survival. Any disruption of Ca2+ exchange between the ER and cytoplasm will have important consequences on the initiation of apoptosis. Cell death due to ER stress and a prolonged UPR is linked to ER Ca2+-store depletion (Zhang and Kaufman, 2008; Peters and Raghavan, 2011). In this context, the main route through which Ca2+ is released from the ER lumen during ER stress to switch the cell survival/apoptosis balance in favor of apoptosis is unknown. The Sec61 complex appears to be a good candidate to explain the mechanism of Ca2+-store depletion associated with ER stress and UPR.
The role of the Sec61 complex in cell death and ER stress has been investigated in the human cancerous cell line LNCaP (Hammadi et al., 2013). In this study, its involvement in ER stress as an ER Ca2+-leak channel was assessed using multiple ER stress/UPR inducers: brefeldin A, dithiothreitol and tunicamycin to trigger UPR as well as thapsigargin and puromycin to deplete ER Ca2+ stores in the absence of misfolded protein accumulation. In each condition, anisomycin both reduced ER stress and prevented depletion of the ER Ca2+ store, demonstrating that the Sec61 complex acts as a major ER Ca2+-leak channel during ER stress/UPR. Remarkably, inhibition of Sec61 complex-mediated Ca2+ leakage by anisomycin also inhibits apoptosis triggered by Ca2+ store depletion induced by thapsigargin and puromycin. As anisomycin can also activate a number of protein kinases, a puromycin/anisomycin pair (or equivalent) should always be used in these types of studies. Since the effect of one molecule blocks that of the other, their use allows us to rule out any non-specific action that would impede the correct interpretation of the results. The study by Hammadi et al. (Hammadi et al., 2013) therefore showed for the first time that the Sec61 complex controls ER Ca2+ content in stressed conditions. This finding indicates that pharmacological modulation of the Sec61 complex could promote cell survival.
Returning to the apoptosis resistance of cancer cells, BiP has been correlated with cancer malignancy (Virrey et al., 2008; Ni et al., 2011). Indeed, most cancerous cells overexpress BiP compared to normal cells. A study by Reddy et al. (Reddy et al., 2003) highlighted the link between BiP overexpression and drug resistance in cancer cells. Other studies clearly demonstrate that BiP is involved in the resistance of breast cancer cells to etoposide (an inhibitor of topoisomerase 2, used in chemotherapy) (Mandic et al., 2003) and to apoptosis (Fu et al., 2007). Moreover, BiP is overexpressed in ER stress, during UPR and in cancer cells (Ni et al., 2011; Farshbaf et al., 2020). On the one hand, BiP seals the luminal pore of the Sec61 complex (Hamman et al., 1998) and thus inhibits ER Ca2+ leakage (Schäuble et al., 2012; Hammadi et al., 2013). On the other hand, UPR and ER stress trigger BiP overexpression (Hammadi et al., 2013). Therefore, apoptosis resistance of cancerous cells could be partly due to a reduced ER Ca2+ leak at resting state involving the tandem Sec61 complex/BiP. Pharmacological modulation of the Ca2+ leak though the Sec61 complex could thus form a potential approach to force apoptosis-resistant cancer cells to undergo cell death and to enhance chemotherapy efficiency.
4.2.2 Protection of human pancreatic islets from lipotoxicity by modulation of the Sec61 complex
T2D is associated with pancreatic β cell dysfunction and insulin resistance. This pathology is a major concern in health care worldwide. Its prevalence and incidence are rising, especially in developed countries such as western Europe (Khan et al., 2020). One of the major causes of T2D is a high-fat, high-sucrose diet. Indeed, excessive consumption of free fatty acids (FFAs) is correlated with a high risk for developing T2D (Risérus et al., 2009). In this context, palmitate is one of the main FFA in blood (Dey et al., 2007; Hommelberg et al., 2011; Leamy et al., 2014) that triggers deleterious effects called “lipotoxicity” (Bensellam et al., 2012). Palmitate-induced β cell lipotoxicity is associated with apoptosis involving numerous mechanisms, such as reactive oxygen species (ROS) production (Cnop, 2008; Fonseca et al., 2011), inflammation (Eguchi et al., 2012) and autophagy (Martino et al., 2012). Chronic exposure to FFA enhances ER stress/UPR in β cells (Özcan et al., 2006). In T2D, high insulin protein synthesis also triggers ER stress/UPR (Song et al., 2008; Åkerfeldt et al., 2008), as evidenced by ER stress marker overexpression (Back and Kaufman, 2012; Papa, 2012). Similarly, chemical chaperones used in a mouse model of T2D reduce ER stress and restore glucose homeostasis (Özcan et al., 2006). On the other hand, dysregulation of Ca2+ homeostasis is also associated with defective insulin release (Gwiazda et al., 2009). Interestingly, Cnop et al. (Cnop et al., 2014) analyzed the transcriptome of human islets after palmitate treatment. They demonstrated an enhanced level of SEC61α and BIP transcripts. Taken together, these data support the involvement of the Sec61 complex and Ca2+ leakage from the ER in palmitate-induced lipotoxicity in pancreatic β cells.
This hypothesis has been checked in the MIN6B1 cell line, obtained from a mouse insulinoma (Miyazaki et al., 1990; Ishihara et al., 1993). In these studies, the authors established and characterized a pancreatic β cell line that exhibits morphological and physiological characteristics (glucose metabolism and glucose-stimulated insulin secretion (GSIS)) similar to normal β cells. Physiologically more relevant, pharmacological modulation of the Sec61 complex was also performed in human pancreatic islets obtained from non-diabetic donors (Cassel et al., 2016). In this study, inhibition of the Sec61 complex with anisomycin prevented palmitate-induced ER Ca2+ depletion and reduced ER stress. It is well known that under physiological conditions, glucose stimulates insulin secretion by β cells. The GSIS assay is a method to investigate the physiological functionality of islets. One of the deleterious effects of palmitate-induced lipotoxicity is the decrease in insulin secretion in response to glucose. Using the GSIS method, it has been shown that the inhibition of the Sec61 complex by anisomycin restores glucose-induced insulin secretion in human islets treated with palmitate (Cassel et al., 2016).
In conclusion, Ca2+ leakage through the Sec61 complex appears to be a key element involved in T2D. One of the mechanisms by which palmitate causes lipotoxicity in pancreatic β cells occurs via ER stress associated with an increase in ER Ca2+-store depletion through the Sec61 complex. Therefore, pharmacological modulation of Sec61 opening could be a promising strategy for the treatment of ER stress-linked pathologies such as T2D (Figure 2).
4.2.3 Cardioprotective role of the modulation of Sec61-mediated Ca2+ leakage during heart ischemia-reperfusion and cardiac infarcts
Cardiac infarction is one of the leading causes of mortality. Obstruction of a coronary artery induces ischemia in an area at risk, which is responsible for lesions due to apoptosis of cardiomyocytes. Reflow triggers further reperfusion injuries (Hausenloy et al., 2016). The role of ER Ca2+ handling in the regulation of heart rate, myocardial contraction, blood pressure and blood flow is the subject of considerable investigation. Ca2+ signaling is central for heart function, through its physiological role in excitation-contraction coupling as well as by its detrimental impact during heart failure and myocardial ischemia-reperfusion. During this latter condition, it is well accepted that the cytosolic accumulation of Ca2+ due to ER Ca2+ depletion via Ca2+-leak channels results in mitochondrial Ca2+ overload, which can trigger the opening of the mitochondrial permeability transition pore (mPTP), leading to cell death (Ovize et al., 2010; Al-Mawla et al., 2020; Ramachandra et al., 2020). During ischemia, the decrease in O2 uptake induces cellular acidosis, stimulating Na+/H+ exchange (for review, see Allen and Xiao, 2003). As a consequence, Na+ influx will be counterbalanced by the activity of the Na+/Ca2+ exchanger, further contributing to the [Ca2+]cyt increase during ischemia. The initial Ca2+ release via ER Ca2+-leak channels is a crucial step in the cascade of events responsible for Ca2+ dysregulation and cell death. The dynamics and the amplitude of Ca2+ exchanged between subcellular compartments and especially between the ER and mitochondria are capital determinants of cell fate. Nevertheless, the overall signaling pathways are still not fully understood.
The role of the Sec61 complex in myocardial infarction has been investigated in mice (Al-Mawla et al., 2020). Mice were subjected to ischemia/reperfusion (I/R) protocols. In a complementary way, in vitro experiments were conducted on primary mouse cardiomyocytes in hypoxia/reperfusion (H/R) protocols, mimicking I/R at the cellular level. In these cells, the Sec61 complex was first shown to function as a Ca2+-leak channel. In cardiomyocytes, it is well known that during excitation-contraction coupling, type 2 RyRs are involved in Ca2+ release via a Ca2+-induced Ca2+ release mechanism. Interestingly, Sec61 complex activation mobilizes a RyR-independent Ca2+ pool that affects neither contraction nor RyR-dependent Ca2+ stores (Al-Mawla et al., 2020). These data are compatible with a compartmentalization of the Ca2+ stores: a puromycin-sensitive Ca2+ pool where Sec61 complexes are located (probably the ER) and a caffeine-sensitive Ca2+ pool containing RyR channels, located in the SR. Pharmacological pre-activation of the Sec61 complex with puromycin induces a preventive ER Ca2+ release from RyR-independent stores that consequently decreases the rate of Ca2+ increase in the cytosol as well as mitochondrial Ca2+ overload and mPTP opening during hypoxia. These data explain how puromycin, applied before H/R (pre-conditioning), significantly reduces cell death. In vivo cardioprotective experiments show that pharmacological modulation of the Sec61 complex (pre-conditioning) protects the mouse heart from I/R injury and reduces infarct size after I/R (Al-Mawla et al., 2020).
In conclusion, the Sec61 complex and its Ca2+ leak form a new paradigm in cardioprotection and in I/R injuries by functionally uncoupling Ca2+-dependent contraction from Ca2+-dependent cell fate.
4.2.4 Pharmacological inhibition of the Sec61 complex improves contractile recovery in stunned myocardium
In the heart, ER stress and cytosolic Ca2+ overload also occur in myocardial stunning (Bolli and Marbán, 1999; Mariángelo et al., 2020). This contractile dysfunction occurs after brief episodes of ischemia with negative consequences on myocardium contraction despite the absence of cell death. Recently, an interesting article highlighted the beneficial effects of Sec61 complex inhibition (with emetine) in Ca2+ dysregulation and post-ischemic contractile dysfunction in stunned myocardium (Mariángelo et al., 2022). First, the authors show that inhibition of the Sec61 complex prior to I/R, prevents stunning-induced ER stress in rat hearts and improves post-ischemic mechanical recovery in stunned myocardium. In addition, Sec61 complex blockage reduced the I/R-induced increase in diastolic Ca2+ in mouse hearts.
Altogether, these data point out the capital role of Ca2+ leakage via the Sec61 complex in stunned myocardium. This work reinforces the potential therapeutic value of pharmacological modulation of Ca2+ leakage via the Sec61 complex to cope with the deleterious consequences of cardiac I/R.
5 Future perspectives and concluding remarks
From the above it thus appears that the Sec61 complex plays an important role in intracellular Ca2+ homeostasis beyond its central role in protein translocation through the ER membrane. This role in Ca2+ homeostasis is due to its functioning as a major ER Ca2+-leak channel. An uncontrolled Ca2+ leak from the ER to cytosol or mitochondria would be highly disruptive for normal cell behavior, and the Sec61 complex Ca2+-leak function is consequently controlled by various mechanisms. Channel closure is physiologically achieved by BiP from the luminal side (see Section 3.4.1) and by Ca2+/CaM from the cytosolic side (see Section 3.4.2). These regulatory mechanisms allow the Sec61 complex to modulate Ca2+ signaling, both in physiological and in patho (physio)logical settings.
As mentioned above, the ER Ca2+ content results from the dynamic equilibrium existing between Ca2+ release from the ER and its reuptake by SERCA pumps. The activity of both the Ca2+-release channels and SERCAs is finely regulated and depends to a large degree on [Ca2+]cyt and [Ca2+]ER (Berridge et al., 2003; Berridge, 2016), so that the ER Ca2+-store content is regulated in a dynamic way. Inhibition of the SERCA pumps by specific inhibitors (e.g., thapsigargin) incapacitates Ca2+ reuptake, uncovering the (normally compensated) activity of the ER Ca2+-leak channels. How does Ca2+ leakage via the Sec61 complex now fit in ER Ca2+ homeostasis? To answer this question, we can consider two scenarios.
In the first scenario, we assume in the cell a single, non-compartmentalized Ca2+ pool. This is, for example, the case in the human prostate cancer cell line LNCaP. In this cellular model, it has been shown that the acute activation of the Sec61 complex by puromycin induces partial emptying of a non-compartmentalized Ca2+ store. The rate of Ca2+ release is slower than that of IP3R- or RyR-mediated Ca2+ release (Van Coppenolle et al., 2004). After the puromycin response, the [Ca2+]ER stabilizes to a lower value than at resting state. This value results from a new balance between Ca2+ leakage (via an undefined number of Sec61 complexes kept open by puromycin as well as by other types of ER Ca2+-leak channels that are presumably present) and SERCA reuptake activity. One element to consider is that during the acute action of puromycin (or emetine), only the Sec61 complexes involved in translation are sensitive to puromycin. Complexes that do not participate in translation remain impermeable to Ca2+. During chronic puromycin or emetine perfusion, their concentration should therefore be chosen so that it does modulate the ER Ca2+ levels without any effect on translation (Hammadi et al., 2013). Beyond that, the cytotoxic effects of these antibiotics will take over. Thus, contrary to what we might initially consider given the pore diameter of the Sec61 complex, puromycin, at the concentration used, whether acute or chronic, does not cause a total emptying of the ER Ca2+ stores. This does not exclude the possibility that other modulatory mechanisms can also act on the Sec61 complex during long-term perfusion. This point requires further investigation. Subsequently, the residual ER Ca2+-store content can be mobilized following IP3R or RyR activation, thereby generating a new Ca2+ steady-state at an even lower value (Van Coppenolle et al., 2004). At this point, it is important to note that the activation of the Ca2+-leak channels, the IP3Rs, the RyRs or the inhibition of SERCAs do not manage to completely drain the ER. Complete release of ER Ca2+ will only be possible with Ca2+ ionophores, e.g., ionomycin.
A second scenario takes into account the compartmentalization of the Ca2+ store in an ER and an SR Ca2+ pool, which can function in an either partially or totally independent way. This case was discussed in primary mouse cardiomyocytes (Al-Mawla et al., 2020) in Section 4.2.3. The modes of action of puromycin and emetine will be similar to those described in the previous paragraph. However, in such a cellular model, there is a functional dichotomy between ER and SR. Thus, the action of emetine, acting on the Sec61 complex of the ER, would have no significant impact on the caffeine-sensitive pool of the SR, expressing RyRs and thus controlling excitation-contraction coupling.
ER stress has a complex relationship with intracellular Ca2+ handling (Kiviluoto et al., 2013; Carreras-Sureda et al., 2018; Groenendyk et al., 2021). On the one hand, one of the consequences of unalleviated ER stress is that it leads to increased Ca2+ release from the ER, though on the other hand, ER Ca2+-store depletion is also a trigger for ER stress. Independent of its origin, in ER stress conditions, BiP will detach from its various physiological binding partners to help alleviate ER stress by binding to unfolded or misfolded proteins as well as to allow the UPR to start. Consequently, the regulation of ER Ca2+ leakage through the Sec61 complex is abrogated as BiP dissociates, and Ca2+ leakage increases. This increase in Ca2+ leakage is a factor that can lead to cell death. As we have highlighted above (see Section 4.2), inhibition of Sec61 Ca2+ leakage can antagonize these effects and lead to cell survival (Figure 2).
As Sec61 complex-mediated Ca2+ leakage appears dysregulated in various pathological situations, we pose that Sec61 forms a valid therapeutic target and that its modulation, activation in cancer (see section 4.2.1) and inhibition in T2D (see section 4.2.2) can lead to beneficial effects. Although more complex, Sec61 complex modulation can also have a role in cardioprotection (see section 4.2.3 and section 4.2.4).
From all these studies, what therapeutic conclusions, based on pharmacological modulation of Ca2+ leakage via the Sec61 complex, can be inferred? First, it is necessary to determine, on a case-by-case basis, the concentration and time of action of molecules of interest that do not significantly alter protein translation while acting on Ca2+ permeability. As ER Ca2+ leakage modulates the cellular death/survival balance, we must therefore consider the intended objective of the treatments. In cancer, it will be to promote cell death, but in other cases (e.g., during ischemic stress (heart, brain, kidney) or gluco-lipotoxicity (T2D)), it will be to promote cell survival. In the case of cancer, it would therefore likely be necessary to use Ca2+-leakage inducers, which could be used in combination with existing therapies to act preferentially on cancer cells rather than on healthy cells. We assume that a small dose of these molecules could already tip the scales towards cell death. In the case of T2D or stunned myocardium, however, it is likely that longer-term treatments would be more effective. Conversely, in the framework of cardiac I/R in a mouse model, a puromycin bolus has already shown its efficacy, both in vivo and in vitro.
Moreover, we anticipate that future research will uncover the role of Sec61 complex-mediated Ca2+ leakage in additional pathological situations. Since the basis of neurodegenerative diseases lies in the accumulation of unfolded proteins and Ca2+ dysregulation, neuroprotective strategies involving the regulation of Ca2+-leak channels in general and the Sec61 complex in particular will probably emerge. Obvious possible candidates include neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and Creutzfeld-Jacob disease. The role of intracellular Ca2+ in general and of ER Ca2+ in particular (Ureshino et al., 2019; da Costa et al., 2020; Lim et al., 2021; Callens et al., 2021; Kovacs et al., 2021; Guan et al., 2021; Huang et al., 2022; Ge et al., 2022; Kim et al., 2022) as well as of ER stress and accumulation of misfolded proteins (da Costa et al., 2020; Guan et al., 2021; Kim et al., 2022; Ren et al., 2021; Yasmeen et al., 2022; Shacham et al., 2019; Chakraborty et al., 2005) have indeed already been demonstrated in several neurodegenerative diseases.
Expanding from T2D, we can also expect a role for Sec61 complex-mediated Ca2+ leakage in other diseases in which a relation with ER stress and Ca2+ signaling was demonstrated, such as non-alcoholic fatty liver disease (Lebeaupin et al., 2018; Chen et al., 2021).
A separate avenue for future research constitutes lysosomal diseases. Indeed, the ER and lysosomes are in close connection and at least small Ca2+ signals originating from IP3R can feed into lysosomes (Atakpa et al., 2018). It is therefore possible that Ca2+ ions leaking from the ER by the Sec61 complex can similarly impact lysosomal Ca2+. This understanding could be relevant for various lysosomal storage diseases, including Niemann-Pick type C disease and Gaucher disease, in which ER and/or lysosomal Ca2+ appears dysregulated (Lloyd-Evans and Platt, 2011; Liu and Lieberman, 2019).
Finally, until now, only a limited number of mutations in Sec61 genes and in other components of the complex have been described (see section 4.1). We therefore anticipate that future work will uncover additional (human) mutations, which will shed new light on the mechanism of action, regulation and physiological and pathological roles of the Sec61 complex in Ca2+ homeostasis.
Taken together, these findings strengthen the evidence that Sec61 complex-mediated Ca2+ leakage plays an important role in intracellular Ca2+ homeostasis, is involved in major pathologies when performing in an unregulated way, and thus forms a promising therapeutic target. Further research on the topic will therefore be of great importance to fully elucidate its physiological as well as its patho (physio)logical role.
Author contributions
JBP and FVC contributed equally at the conceptualization, writing, correction and finalization of the manuscript and both authors approved the final version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Åkerfeldt M. C., Howes J., Chan J. Y., Stevens V. A., Boubenna N., McGuire H. M., et al. (2008). Cytokine-induced β-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 57 (11), 3034–3044. doi:10.2337/db07-1802
Al-Mawla R., Ducrozet M., Tessier N., Païta L., Pillot B., Gouriou Y., et al. (2020). Acute induction of translocon-mediated Ca2+ leak protects cardiomyocytes against ischemia/reperfusion injury. Cells 9 (5), 1319. doi:10.3390/cells9051319
Alder N. N., Shen Y., Brodsky J. L., Hendershot L. M., Johnson A. E. (2005). The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 168 (3), 389–399. doi:10.1083/jcb.200409174
Allen D. G., Xiao X. H. (2003). Role of the cardiac Na+/H+ exchanger during ischemia and reperfusion. Cardiovasc. Res. 57 (4), 934–941. doi:10.1016/s0008-6363(02)00836-2
Annaert W. G., Levesque L., Craessaerts K., Dierinck I., Snellings G., Westaway D., et al. (1999). Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J. Cell Biol. 147 (2), 277–294. doi:10.1083/jcb.147.2.277
Atakpa P., Thillaiappan N. B., Mataragka S., Prole D. L., Taylor C. W. (2018). IP3 receptors preferentially associate with ER-lysosome contact sites and selectively deliver Ca2+ to lysosomes. Cell Rep. 25 (11), 3180–3193. e7. doi:10.1016/j.celrep.2018.11.064
Back S. H., Kaufman R. J. (2012). Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793. doi:10.1146/annurev-biochem-072909-095555
Bagur R., Hajnóczky G. (2017). Intracellular Ca2+ sensing: Its role in calcium homeostasis and signaling. Mol. Cell 66 (6), 780–788. doi:10.1016/j.molcel.2017.05.028
Bai L., Wang T., Zhao G., Kovach A., Li H. (2018). The atomic structure of a eukaryotic oligosaccharyl transferase complex. Nature 555 (7696), 328–333. doi:10.1038/nature25755
Bandara S., Malmersjö S., Meyer T. (2013). Regulators of calcium homeostasis identified by inference of kinetic model parameters from live single cells perturbed by siRNA. Sci. Signal. 6 (283), ra56. doi:10.1126/scisignal.2003649
Baron L., Paatero A. O., Morel J. D., Impens F., Guenin-Macé L., Saint-Auret S., et al. (2016). Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 213 (13), 2885–2896. doi:10.1084/jem.20160662
Becker T., Bhushan S., Jarasch A., Armache J. P., Funes S., Jossinet F., et al. (2009). Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 326 (5958), 1369–1373. doi:10.1126/science.1178535
Behnke J., Feige M. J., Hendershot L. M. (2015). BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 427 (7), 1589–1608. doi:10.1016/j.jmb.2015.02.011
Bensellam M., Laybutt D. R., Jonas J. C. (2012). The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 364 (1), 1–27. doi:10.1016/j.mce.2012.08.003
Berg B., Clemons W. M., Collinson I., Modis Y., Hartmann E., Harrison S. C., et al. (2004). X-ray structure of a protein-conducting channel. Nature 427 (6969), 36–44. doi:10.1038/nature02218
Berridge M. J., Bootman M. D., Roderick H. L. (2003). Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4 (7), 517–529. doi:10.1038/nrm1155
Berridge M. J. (2006). Calcium microdomains: Organization and function. Cell Calcium 40 (5-6), 405–412. doi:10.1016/j.ceca.2006.09.002
Berridge M. J., Lipp P., Bootman M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1 (1), 11–21. doi:10.1038/35036035
Berridge M. J. (2002). The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 32 (5), 235–249. doi:10.1016/s0143416002001823
Berridge M. J. (2016). The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96 (4), 1261–1296. doi:10.1152/physrev.00006.2016
Bhadra P., Dos Santos S., Gamayun I., Pick T., Neumann C., Ogbechi J., et al. (2021). Mycolactone enhances the Ca2+ leak from endoplasmic reticulum by trapping Sec61 translocons in a Ca2+ permeable state. Biochem. J. 478 (22), 4005–4024. doi:10.1042/BCJ20210345
Blau M., Mullapudi S., Becker T., Dudek J., Zimmermann R., Penczek P. A., et al. (2005). ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat. Struct. Mol. Biol. 12 (11), 1015–1016. doi:10.1038/nsmb998
Bolar N. A., Golzio C., Živná M., Hayot G., Van Hemelrijk C., Schepers D., et al. (2016). Heterozygous loss-of-function SEC61A1 mutations cause autosomal-dominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am. J. Hum. Genet. 99 (1), 174–187. doi:10.1016/j.ajhg.2016.05.028
Bolli R., Marbán E. (1999). Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 79 (2), 609–634. doi:10.1152/physrev.1999.79.2.609
Bootman M. D., Bultynck G. (2020). Fundamentals of cellular calcium signaling: A primer. Cold Spring Harb. Perspect. Biol. 12 (1), a038802. doi:10.1101/cshperspect.a038802
Braunger K., Pfeffer S., Shrimal S., Gilmore R., Berninghausen O., Mandon E. C., et al. (2018). Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360 (6385), 215–219. doi:10.1126/science.aar7899
Callens M., Kraskovskaya N., Derevtsova K., Annaert W., Bultynck G., Bezprozvanny I., et al. (2021). The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer’s disease. Biochim. Biophys. Acta. Mol. Cell Res. 1868 (6), 118997. doi:10.1016/j.bbamcr.2021.118997
Camello C., Lomax R., Petersen O. H., Tepikin A. V. (2002). Calcium leak from intracellular stores—The enigma of calcium signalling. Cell Calcium 32 (5), 355–361. doi:10.1016/s0143416002001926
Carreras-Sureda A., Pihán P., Hetz C. (2018). Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 70, 24–31. doi:10.1016/j.ceca.2017.08.004
Cassel R., Ducreux S., Alam M. R., Dingreville F., Berlé C., Burda-Jacob K., et al. (2016). Protection of human pancreatic islets from lipotoxicity by modulation of the translocon. PLoS One 11 (2), e0148686. doi:10.1371/journal.pone.0148686
Chakraborty C., Nandi S., Jana S. (2005). Prion disease: A deadly disease for protein misfolding. Curr. Pharm. Biotechnol. 6 (2), 167–177. doi:10.2174/1389201053642321
Chami M., Oulès B., Szabadkai G., Tacine R., Rizzuto R., Paterlini-Bréchot P. (2008). Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 32 (5), 641–651. doi:10.1016/j.molcel.2008.11.014
Chen C. C., Hsu L. W., Chen K. D., Chiu K. W., Chen C. L., Huang K. T. (2021). Emerging roles of calcium signaling in the development of non-alcoholic fatty liver disease. Int. J. Mol. Sci. 23 (1), 256. doi:10.3390/ijms23010256
Chen W., Helenius J., Braakman I., Helenius A. (1995). Cotranslational folding and calnexin binding during glycoprotein synthesis. Proc. Natl. Acad. Sci. U. S. A. 92 (14), 6229–6233. doi:10.1073/pnas.92.14.6229
Cheng H., Lederer W. J. (2008). Calcium sparks. Physiol. Rev. 88 (4), 1491–1545. doi:10.1152/physrev.00030.2007
Chevet E., Wong H. N., Gerber D., Cochet C., Fazel A., Cameron P. H., et al. (1999). Phosphorylation by CK2 and MAPK enhances calnexin association with ribosomes. EMBO J. 18 (13), 3655–3666. doi:10.1093/emboj/18.13.3655
Chin D., Means A. R. (2000). Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 10 (8), 322–328. doi:10.1016/s0962-8924(00)01800-6
Cnop M., Abdulkarim B., Bottu G., Cunha D. A., Igoillo-Esteve M., Masini M., et al. (2014). RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63 (6), 1978–1993. doi:10.2337/db13-1383
Cnop M. (2008). Fatty acids and glucolipotoxicity in the pathogenesis of Type 2 diabetes. Biochem. Soc. Trans. 36 (3), 348–352. doi:10.1042/BST0360348
Conti B. J., Devaraneni P. K., Yang Z., David L. L., Skach W. R. (2015). Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 58 (2), 269–283. doi:10.1016/j.molcel.2015.02.018
Cross B. C. S., McKibbin C., Callan A. C., Roboti P., Piacenti M., Rabu C., et al. (2009). Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 122 (23), 4393–4400. doi:10.1242/jcs.054494
Crowley K. S., Liao S., Worrell V. E., Reinhart G. D., Johnson A. E. (1994). Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell 78 (3), 461–471. doi:10.1016/0092-8674(94)90424-3
da Costa C. A., Manaa W. E., Duplan E., Checler F. (2020). The endoplasmic reticulum stress/unfolded protein response and their contributions to Parkinson’s disease physiopathology. Cells 9 (11), 2495. doi:10.3390/cells9112495
Daimon M., Susa S., Suzuki K., Kato T., Yamatani K., Sasaki H. (1997). Identification of a human cDNA homologue to the Drosophila translocation protein 1 (Dtrp1). Biochem. Biophys. Res. Commun. 230 (1), 100–104. doi:10.1006/bbrc.1996.5892
Danese A., Leo S., Rimessi A., Wieckowski M. R., Fiorica F., Giorgi C., et al. (2021). Cell death as a result of calcium signaling modulation: A cancer-centric prospective. Biochim. Biophys. Acta. Mol. Cell Res. 1868 (8), 119061. doi:10.1016/j.bbamcr.2021.119061
De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., et al. (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391 (6665), 387–390. doi:10.1038/34910
Dejgaard K., Theberge J. F., Heath-Engel H., Chevet E., Tremblay M. L., Thomas D. Y. (2010). Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J. Proteome Res. 9 (4), 1763–1771. doi:10.1021/pr900900x
Demangel C., High S. (2018). Sec61 blockade by mycolactone: A central mechanism in Buruli ulcer disease. Biol. Cell 110 (11), 237–248. doi:10.1111/boc.201800030
Demangel C. (2021). Immunity against Mycobacterium ulcerans - the subversive role of mycolactone. Immunol. Rev. 301 (1), 209–221. doi:10.1111/imr.12956
Demangel C., Stinear T. P., Cole S. T. (2009). Buruli ulcer: Reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat. Rev. Microbiol. 7 (1), 50–60. doi:10.1038/nrmicro2077
Dey D., Pal B. C., Biswas T., Roy S. S., Bandyopadhyay A., Mandal S. K., et al. (2007). A lupinoside prevented fatty acid induced inhibition of insulin sensitivity in 3T3 L1 adipocytes. Mol. Cell. Biochem. 300 (1), 149–157. doi:10.1007/s11010-006-9378-1
Dudek J., Greiner M., Müller A., Hendershot L. M., Kopsch K., Nastainczyk W., et al. (2005). ERj1p has a basic role in protein biogenesis at the endoplasmic reticulum. Nat. Struct. Mol. Biol. 12 (11), 1008–1014. doi:10.1038/nsmb1007
Egea P. F., Stroud R. M. (2010). Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc. Natl. Acad. Sci. U. S. A. 107 (40), 17182–17187. doi:10.1073/pnas.1012556107
Egea P. F., Stroud R. M., Walter P. (2005). Targeting proteins to membranes: Structure of the signal recognition particle. Curr. Opin. Struct. Biol. 15 (2), 213–220. doi:10.1016/j.sbi.2005.03.007
Eguchi K., Manabe I., Oishi-Tanaka Y., Ohsugi M., Kono N., Ogata F., et al. (2012). Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 15 (4), 518–533. doi:10.1016/j.cmet.2012.01.023
Eida A. A., Mahmud T. (2019). The secondary metabolite pactamycin with potential for pharmaceutical applications: Biosynthesis and regulation. Appl. Microbiol. Biotechnol. 103 (11), 4337–4345. doi:10.1007/s00253-019-09831-x
En J., Goto M., Nakanaga K., Higashi M., Ishii N., Saito H., et al. (2008). Mycolactone is responsible for the painlessness of Mycobacterium ulcerans infection (Buruli ulcer) in a murine Study. Infect. Immun. 76 (5), 2002–2007. doi:10.1128/IAI.01588-07
Erdmann F., Schäuble N., Lang S., Jung M., Honigmann A., Ahmad M., et al. (2011). Interaction of calmodulin with Sec61α limits Ca2+ leakage from the endoplasmic reticulum. EMBO J. 30 (1), 17–31. doi:10.1038/emboj.2010.284
Farshbaf M., Khosroushahi A. Y., Mojarad-Jabali S., Zarebkohan A., Valizadeh H., Walker P. R. (2020). Cell surface GRP78: An emerging imaging marker and therapeutic target for cancer. J. Control. Release 328, 932–941. doi:10.1016/j.jconrel.2020.10.055
Feliziani C., Fernandez M., Quassollo G., Holstein D., Bairo S. M., Paton J. C., et al. (2022). Ca2+ signalling system initiated by endoplasmic reticulum stress stimulates PERK activation. Cell Calcium 106, 102622. doi:10.1016/j.ceca.2022.102622
Flourakis M., Van Coppenolle F., Lehen’kyi V., Beck B., Skryma R., Prevarskaya N. (2006). Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store operated channels activation. FASEB J. 20 (8), 1215–1217. doi:10.1096/fj.05-5254fje
Fonseca S. G., Gromada J., Urano F. (2011). Endoplasmic reticulum stress and pancreatic β cell death. Trends Endocrinol. Metab. 22 (7), 266–274. doi:10.1016/j.tem.2011.02.008
Foskett J. K., White C., Cheung K. H., Mak D. O. D. (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87 (2), 593–658. doi:10.1152/physrev.00035.2006
Fu Y., Li J., Lee A. S. (2007). GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation–induced apoptosis. Cancer Res. 67 (8), 3734–3740. doi:10.1158/0008-5472.CAN-06-4594
Gallegos-Gómez M. L., Greotti E., López-Méndez M. C., Sánchez-Vázquez V. H., Arias J. M., Guerrero-Hernández A. (2018). The trans Golgi region is a labile intracellular Ca2+ store sensitive to emetine. Sci. Rep. 8, 17143. doi:10.1038/s41598-018-35280-z
Gamayun I., O’Keefe S., Pick T., Klein M. C., Nguyen D., McKibbin C., et al. (2019). Eeyarestatin compounds selectively enhance Sec61-mediated Ca2+ leakage from the endoplasmic reticulum. Cell Chem. Biol. 26 (4), 571–583. e6. doi:10.1016/j.chembiol.2019.01.010
Ge M., Zhang J., Chen S., Huang Y., Chen W., He L., et al. (2022). Role of calcium homeostasis in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 18, 487–498. doi:10.2147/NDT.S350939
Gemmer M., Förster F. (2020). A clearer picture of the ER translocon complex. J. Cell Sci. 133 (3), jcs231340. doi:10.1242/jcs.231340
Gérard S. F., Hall B. S., Zaki A. M., Corfield K. A., Mayerhofer P. U., Costa C., et al. (2020). Structure of the inhibited state of the Sec translocon. Mol. Cell 79 (3), 406–415. e7. doi:10.1016/j.molcel.2020.06.013
Giorgi C., Danese A., Missiroli S., Patergnani S., Pinton P. (2018). Calcium dynamics as a machine for decoding signals. Trends Cell Biol. 28 (4), 258–273. doi:10.1016/j.tcb.2018.01.002
Giunti R., Gamberucci A., Fulceri R., Bánhegyi G., Benedetti A. (2007). Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: A mechanistic study on rat liver microsomes. Arch. Biochem. Biophys. 462 (1), 115–121. doi:10.1016/j.abb.2007.03.039
Gogala M., Becker T., Beatrix B., Armache J. P., Barrio-Garcia C., Berninghausen O., et al. (2014). Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506 (7486), 107–110. doi:10.1038/nature12950
Greiner M., Kreutzer B., Lang S., Jung V., Cavalié A., Unteregger G., et al. (2011). Sec62 protein level is crucial for the ER stress tolerance of prostate cancer. Prostate 71 (10), 1074–1083. doi:10.1002/pros.21324
Groenendyk J., Agellon L. B., Michalak M. (2021). Calcium signaling and endoplasmic reticulum stress. Int. Rev. Cell Mol. Biol. 363, 1–20. doi:10.1016/bs.ircmb.2021.03.003
Grotzke J. E., Kozik P., Morel J. D., Impens F., Pietrosemoli N., Cresswell P., et al. (2017). Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc. Natl. Acad. Sci. U. S. A. 114 (29), E5910–E5919. doi:10.1073/pnas.1705242114
Grudnik P., Bange G., Sinning I. (2009). Protein targeting by the signal recognition particle. Biol. Chem. 390 (8), 775–782. doi:10.1515/BC.2009.102
Guan P. P., Cao L. L., Yang Y., Wang P. (2021). Calcium ions aggravate Alzheimer’s disease through the aberrant activation of neuronal networks, leading to synaptic and cognitive deficits. Front. Mol. Neurosci. 14, 757515. doi:10.3389/fnmol.2021.757515
Gwiazda K. S., Yang T. L. B., Lin Y., Johnson J. D. (2009). Effects of palmitate on ER and cytosolic Ca2+ homeostasis in β-cells. Am. J. Physiol. Endocrinol. Metab. 296 (4), E690–E701. doi:10.1152/ajpendo.90525.2008
Haigh N. G., Johnson A. E. (2002). A new role for BiP: Closing the aqueous translocon pore during protein integration into the ER membrane. J. Cell Biol. 156 (2), 261–270. doi:10.1083/jcb.200110074
Hall B. S., Hill K., McKenna M., Ogbechi J., High S., Willis A. E., et al. (2014). The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog. 10 (4), e1004061. doi:10.1371/journal.ppat.1004061
Hamada K., Mikoshiba K. (2020). IP3 receptor plasticity underlying diverse functions. Annu. Rev. Physiol. 82, 151–176. doi:10.1146/annurev-physiol-021119-034433
Hammadi M., Oulidi A., Gackière F., Katsogiannou M., Slomianny C., Roudbaraki M., et al. (2013). Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 27 (4), 1600–1609. doi:10.1096/fj.12-218875
Hamman B. D., Chen J. C., Johnson E. E., Johnson A. E. (1997). The aqueous pore through the translocon has a diameter of 40–60 Å during cotranslational protein translocation at the ER membrane. Cell 89 (4), 535–544. doi:10.1016/s0092-8674(00)80235-4
Hamman B. D., Hendershot L. M., Johnson A. E. (1998). BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 92 (6), 747–758. doi:10.1016/s0092-8674(00)81403-8
Hausenloy D. J., Barrabes J. A., Bøtker H. E., Davidson S. M., Di Lisa F., Downey J., et al. (2016). Ischaemic conditioning and targeting reperfusion injury: A 30 year voyage of discovery. Basic Res. Cardiol. 111 (6), 70. doi:10.1007/s00395-016-0588-8
Haßdenteufel S., Johnson N., Paton A. W., Paton J. C., High S., Zimmermann R. (2018). Chaperone-mediated Sec61 channel gating during ER import of small precursor proteins overcomes Sec61 inhibitor-reinforced energy barrier. Cell Rep. 23 (5), 1373–1386. doi:10.1016/j.celrep.2018.03.122
Heritage D., Wonderlin W. F. (2001). Translocon pores in the endoplasmic reticulum are permeable to a neutral, polar molecule. J. Biol. Chem. 276 (25), 22655–22662. doi:10.1074/jbc.M102409200
Hetz C., Zhang K., Kaufman R. J. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21 (8), 421–438. doi:10.1038/s41580-020-0250-z
Higo T., Hamada K., Hisatsune C., Nukina N., Hashikawa T., Hattori M., et al. (2010). Mechanism of ER stress-induced brain damage by IP3 Receptor. Neuron 68 (5), 865–878. doi:10.1016/j.neuron.2010.11.010
Hommelberg P. P. H., Plat J., Sparks L. M., Schols A. M. W. J., van Essen A. L. M., Kelders M. C. J. M., et al. (2011). Palmitate-induced skeletal muscle insulin resistance does not require NF-κB activation. Cell. Mol. Life Sci. 68 (7), 1215–1225. doi:10.1007/s00018-010-0515-3
Hotamisligil G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140 (6), 900–917. doi:10.1016/j.cell.2010.02.034
Huang D. X., Yu X., Yu W. J., Zhang X. M., Liu C., Liu H. P., et al. (2022). Calcium signaling regulated by cellular membrane systems and calcium homeostasis perturbed in Alzheimer’s disease. Front. Cell Dev. Biol. 10, 834962. doi:10.3389/fcell.2022.834962
Hummel H., Böck A., Bock A. (1987). 23S ribosomal RNA mutations in halobacteria conferring resistance to the anti-80S ribosome targeted antibiotic anisomycin. Nucleic Acids Res. 15 (6), 2431–2443. doi:10.1093/nar/15.6.2431
Hwang J., Qi L. (2018). Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends biochem. Sci. 43 (8), 593–605. doi:10.1016/j.tibs.2018.06.005
Ichhaporia V. P., Hendershot L. M. (2021). Role of the HSP70 co-chaperone SIL1 in health and disease. Int. J. Mol. Sci. 22 (4), 1564. doi:10.3390/ijms22041564
Inesi G., de Meis L. (1989). Regulation of steady state filling in sarcoplasmic reticulum: Roles of back-inhibition, leakage, and slippage of the calcium pump. J. Biol. Chem. 264 (10), 5929–5936. doi:10.1016/s0021-9258(18)83639-0
Ioannou M., Coutsogeorgopoulos C., Synetos D. (1998). Kinetics of inhibition of rabbit reticulocyte peptidyltransferase by anisomycin and sparsomycin. Mol. Pharmacol. 53 (6), 1089–1096.
Iordanov M. S., Pribnow D., Magun J. L., Dinh T. H., Pearson J. A., Chen S. L., et al. (1997). Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the α-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 17 (6), 3373–3381. doi:10.1128/mcb.17.6.3373
Ishihara H., Asano T., Tsukuda K., Katagiri H., Inukai K., Anai M., et al. (1993). Pancreatic β cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia 36 (11), 1139–1145. doi:10.1007/BF00401058
Jung S. J., Kim H. (2021). Emerging view on the molecular functions of Sec62 and Sec63 in protein translocation. Int. J. Mol. Sci. 22 (23), 12757. doi:10.3390/ijms222312757
Kelleher D. J., Gilmore R. (2006). An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16 (4), 47R–62R. doi:10.1093/glycob/cwj066
Khan M. A. B., Hashim M. J., King J. K., Govender R. D., Mustafa H., Al Kaabi J. (2020). Epidemiology of type 2 diabetes – global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 10 (1), 107–111. doi:10.2991/jegh.k.191028.001
Kim S., Kim D. K., Jeong S., Lee J. (2022). The common cellular events in the neurodegenerative diseases and the associated role of endoplasmic reticulum stress. Int. J. Mol. Sci. 23 (11), 5894. doi:10.3390/ijms23115894
Kiviluoto S., Vervliet T., Ivanova H., Decuypere J. P., De Smedt H., Missiaen L., et al. (2013). Regulation of inositol 1, 4, 5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 1833 (7), 1612–1624. doi:10.1016/j.bbamcr.2013.01.026
Klec C., Madreiter-Sokolowski C. T., Stryeck S., Sachdev V., Duta-Mare M., Gottschalk B., et al. (2019). Glycogen synthase kinase 3β controls presenilin-1-mediated endoplasmic reticulum Ca2+ leak directed to mitochondria in pancreatic islets and β-cells. Cell. Physiol. biochem. 52 (1), 57–75. doi:10.33594/000000005
Klein M. C., Zimmermann K., Schorr S., Landini M., Klemens P. A. W., Altensell J., et al. (2018). AXER is an ATP/ADP exchanger in the membrane of the endoplasmic reticulum. Nat. Commun. 9 (1), 3489. doi:10.1038/s41467-018-06003-9
Konieczny V., Keebler M. V., Taylor C. W. (2012). Spatial organization of intracellular Ca2+ signals. Semin. Cell Dev. Biol. 23 (2), 172–180. doi:10.1016/j.semcdb.2011.09.006
Kovacs G., Reimer L., Jensen P. H. (2021). Endoplasmic reticulum-based calcium dysfunctions in synucleinopathies. Front. Neurol. 12, 742625. doi:10.3389/fneur.2021.742625
Krebs J., Agellon L. B., Michalak M. (2015). Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 460 (1), 114–121. doi:10.1016/j.bbrc.2015.02.004
Kriegler T., Kiburg G., Hessa T. (2020). Translocon-associated protein complex (TRAP) is crucial for co-translational translocation of pre-proinsulin. J. Mol. Biol. 432 (24), 166694. doi:10.1016/j.jmb.2020.10.028
La Rovere R. M. L., Roest G., Bultynck G., Parys J. B. (2016). Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 60 (2), 74–87. doi:10.1016/j.ceca.2016.04.005
Ladiges W. C., Knoblaugh S. E., Morton J. F., Korth M. J., Sopher B. L., Baskin C. R., et al. (2005). Pancreatic β-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54 (4), 1074–1081. doi:10.2337/diabetes.54.4.1074
Lakkaraju A. K., Abrami L., Lemmin T., Blaskovic S., Kunz B., Kihara A., et al. (2012). Palmitoylated calnexin is a key component of the ribosome–translocon complex. EMBO J. 31 (7), 1823–1835. doi:10.1038/emboj.2012.15
Lam A. K. M., Galione A. (2013). The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys. Acta 1833 (11), 2542–2559. doi:10.1016/j.bbamcr.2013.06.004
Lang S., Benedix J., Fedeles S. V., Schorr S., Schirra C., Schäuble N., et al. (2012). Different effects of Sec61α, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 125 (8), 1958–1969. doi:10.1242/jcs.096727
Lang S., Nguyen D., Pfeffer S., Förster F., Helms V., Zimmermann R. (2019). Functions and mechanisms of the human ribosome-translocon complex. Subcell. Biochem. 93, 83–141. doi:10.1007/978-3-030-28151-9_4
Lang S., Pfeffer S., Lee P. H., Cavalié A., Helms V., Förster F., et al. (2017). An update on Sec61 channel functions, mechanisms, and related diseases. Front. Physiol. 8, 887. doi:10.3389/fphys.2017.00887
Lanner J. T. (2012). Ryanodine receptor physiology and its role in disease. Adv. Exp. Med. Biol. 740, 217–234. doi:10.1007/978-94-007-2888-2_9
Layhadi J. A., Fountain S. J. (2017). Influence of ER leak on resting cytoplasmic Ca2+ and receptor-mediated Ca2+ signalling in human macrophage. Biochem. Biophys. Res. Commun. 487 (3), 633–639. doi:10.1016/j.bbrc.2017.04.106
Leamy A. K., Egnatchik R. A., Shiota M., Ivanova P. T., Myers D. S., Brown H. A., et al. (2014). Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J. Lipid Res. 55 (7), 1478–1488. doi:10.1194/jlr.M050237
Lebeaupin C., Vallée D., Hazari Y., Hetz C., Chevet E., Bailly-Maitre B. (2018). Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 69 (4), 927–947. doi:10.1016/j.jhep.2018.06.008
Lemos F. O., Bultynck G., Parys J. B. (2021). A comprehensive overview of the complex world of the endo- and sarcoplasmic reticulum Ca2+-leak channels. Biochim. Biophys. Acta. Mol. Cell Res. 1868 (7), 119020. doi:10.1016/j.bbamcr.2021.119020
Li L., Park E., Ling J., Ingram J., Ploegh H., Rapoport T. A. (2016). Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature 531 (7594), 395–399. doi:10.1038/nature17163
Lièvremont J. P., Rizzuto R., Hendershot L., Meldolesi J. (1997). BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J. Biol. Chem. 272 (49), 30873–30879. doi:10.1074/jbc.272.49.30873
Lim D., Dematteis G., Tapella L., Genazzani A. A., Calì T., Brini M., et al. (2021). Ca2+ handling at the mitochondria-ER contact sites in neurodegeneration. Cell Calcium 98, 102453. doi:10.1016/j.ceca.2021.102453
Linxweiler M., Schorr S., Schäuble N., Jung M., Linxweiler J., Langer F., et al. (2013). Targeting cell migration and the endoplasmic reticulum stress response with calmodulin antagonists: A clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer 13, 574. doi:10.1186/1471-2407-13-574
Liu E. A., Lieberman A. P. (2019). The intersection of lysosomal and endoplasmic reticulum calcium with autophagy defects in lysosomal diseases. Neurosci. Lett. 697, 10–16. doi:10.1016/j.neulet.2018.04.049
Lloyd D. J., Wheeler M. C., Gekakis N. (2010). A point mutation in Sec61alpha1 leads to diabetes and hepatosteatosis in mice. Diabetes 59 (2), 460–470. doi:10.2337/db08-1362
Lloyd-Evans E., Platt F. M. (2011). Lysosomal Ca2+ homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 50 (2), 200–205. doi:10.1016/j.ceca.2011.03.010
Lomax R. B., Camello C., Van Coppenolle F., Petersen O. H., Tepikin A. V. (2002). Basal and physiological Ca2+ leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J. Biol. Chem. 277 (29), 26479–26485. doi:10.1074/jbc.M201845200
Luo T., Kim J. K., Chen B., Abdel-Latif A., Kitakaze M., Yan L. (2015). Attenuation of ER stress prevents post-infarction-induced cardiac rupture and remodeling by modulating both cardiac apoptosis and fibrosis. Chem. Biol. Interact. 225, 90–98. doi:10.1016/j.cbi.2014.10.032
Ma Y., Hendershot L. M. (2001). The unfolding tale of the unfolded protein response. Cell 107 (7), 827–830. doi:10.1016/s0092-8674(01)00623-7
Mahamid J., Pfeffer S., Schaffer M., Villa E., Danev R., Kuhn Cuellar L., et al. (2016). Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 351 (6276), 969–972. doi:10.1126/science.aad8857
Mandic A., Hansson J., Linder S., Shoshan M. C. (2003). Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 278 (11), 9100–9106. doi:10.1074/jbc.M210284200
Mariángelo J. I. E., Román B., Silvestri M. A., Salas M., Vittone L., Said M., et al. (2020). Chemical chaperones improve the functional recovery of stunned myocardium by attenuating the endoplasmic reticulum stress. Acta Physiol. 228 (2), e13358. doi:10.1111/apha.13358
Mariángelo J. I. E., Valverde C. A., Vittone L., Said M., Mundiña-Weilenmann C. (2022). Pharmacological inhibition of translocon is sufficient to alleviate endoplasmic reticulum stress and improve Ca2+ handling and contractile recovery of stunned myocardium. Eur. J. Pharmacol. 914, 174665. doi:10.1016/j.ejphar.2021.174665
Martino L., Masini M., Novelli M., Beffy P., Bugliani M., Marselli L., et al. (2012). Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human Pancreatic Islets. PLoS One 7 (5), e36188. doi:10.1371/journal.pone.0036188
Minamino T., Kitakaze M. (2010). ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 48 (6), 1105–1110. doi:10.1016/j.yjmcc.2009.10.026
Miyazaki J. I., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., et al. (1990). Establishment of a pancreatic β cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology 127 (1), 126–132. doi:10.1210/endo-127-1-126
Mohanty S., P Chaudhary B., Zoetewey D. (2020). Structural Insight into the mechanism of N-linked glycosylation by oligosaccharyltransferase. Biomolecules 10 (4), 624. doi:10.3390/biom10040624
Mohorko E., Owen R. L., Malojčić G., Brozzo M. S., Aebi M., Glockshuber R. (2014). Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure 22 (4), 590–601. doi:10.1016/j.str.2014.02.013
Morel J. D., Paatero A. O., Wei J., Yewdell J. W., Guenin-Macé L., Van Haver D., et al. (2018). Proteomics reveals scope of mycolactone-mediated Sec61 blockade and distinctive stress signature. Mol. Cell. Proteomics 17 (9), 1750–1765. doi:10.1074/mcp.RA118.000824
Müller L., de Escauriaza M. D., Lajoie P., Theis M., Jung M., Müller A., et al. (2010). Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 21 (5), 691–703. doi:10.1091/mbc.e09-08-0730
Needham P. G., Guerriero C. J., Brodsky J. L. (2019). Chaperoning endoplasmic reticulum–associated degradation (ERAD) and protein conformational diseases. Cold Spring Harb. Perspect. Biol. 11 (8), a033928. doi:10.1101/cshperspect.a033928
Nguyen D., Stutz R., Schorr S., Lang S., Pfeffer S., Freeze H. H., et al. (2018). Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat. Commun. 9, 3765. doi:10.1038/s41467-018-06188-z
Ni M., Zhang Y., Lee A. S. (2011). Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 434 (2), 181–188. doi:10.1042/BJ20101569
Nicchitta C. V., Zheng T. (1997). Regulation of the ribosome–membrane junction at early stages of presecretory protein translocation in the mammalian endoplasmic reticulum. J. Cell Biol. 139 (7), 1697–1708. doi:10.1083/jcb.139.7.1697
O’Keefe S., High S., Demangel C. (2022). Biochemical and biological assays of mycolactone-mediated inhibition of Sec61. Methods Mol. Biol. 2387, 163–181. doi:10.1007/978-1-0716-1779-3_16
Oliver J. D., Hresko R. C., Mueckler M., High S. (1996). The Glut 1 glucose transporter interacts with calnexin and calreticulin. J. Biol. Chem. 271 (23), 13691–13696. doi:10.1074/jbc.271.23.13691
Oliver J. D., van der Wal F. J., Bulleid N. J., High S. (1997). Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 275 (5296), 86–88. doi:10.1126/science.275.5296.86
Ong H. L., Liu X., Sharma A., Hegde R. S., Ambudkar I. S. (2007). Intracellular Ca2+ release via the ER translocon activates store-operated calcium entry. Pflugers Arch. 453 (6), 797–808. doi:10.1007/s00424-006-0163-5
Orrenius S., Zhivotovsky B., Nicotera P. (2003). Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 4 (7), 552–565. doi:10.1038/nrm1150
Ovize M., Baxter G. F., Di Lisa F., Ferdinandy P., Garcia-Dorado D., Hausenloy D. J., et al. (2010). Postconditioning and protection from reperfusion injury: Where do we stand? Position Paper from the working group of cellular biology of the heart of the European Society of Cardiology. Cardiovasc. Res. 87 (3), 406–423. doi:10.1093/cvr/cvq129
Özcan U., Yilmaz E., Özcan L., Furuhashi M., Vaillancourt E., Smith R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313 (5790), 1137–1140. doi:10.1126/science.1128294
Papa F. R. (2012). Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harb. Perspect. Med. 2 (9), a007666. doi:10.1101/cshperspect.a007666
Park E., Ménétret J. F., Gumbart J. C., Ludtke S. J., Li W., Whynot A., et al. (2014). Structure of the SecY channel during initiation of protein translocation. Nature 506 (7486), 102–106. doi:10.1038/nature12720
Pestova T. V., Hellen C. U. (2001). Functions of eukaryotic factors in initiation of translation. Cold Spring Harb. Symp. Quant. Biol. 66, 389–396. doi:10.1101/sqb.2001.66.389
Peters L. R., Raghavan M. (2011). Endoplasmic reticulum calcium depletion impacts chaperone secretion, innate immunity, and phagocytic uptake of cells. J. Immunol. 187 (2), 919–931. doi:10.4049/jimmunol.1100690
Pfeffer S., Burbaum L., Unverdorben P., Pech M., Chen Y., Zimmermann R., et al. (2015). Structure of the native Sec61 protein-conducting channel. Nat. Commun. 6, 8403. doi:10.1038/ncomms9403
Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., et al. (2014). Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5 (1), 3072. doi:10.1038/ncomms4072
Pinton P., Giorgi C., Siviero R., Zecchini E., Rizzuto R. (2008). Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27 (50), 6407–6418. doi:10.1038/onc.2008.308
Pobre K. F. R., Poet G. J., Hendershot L. M. (2019). The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 294 (6), 2098–2108. doi:10.1074/jbc.REV118.002804
Prole D. L., Taylor C. W. (2016). Inositol 1, 4, 5‐trisphosphate receptors and their protein partners as signalling hubs. J. Physiol. 594 (11), 2849–2866. doi:10.1113/JP271139
Prole D. L., Taylor C. W. (2019). Structure and function of IP3 receptors. Cold Spring Harb. Perspect. Biol. 11 (4), a035063. doi:10.1101/cshperspect.a035063
Raden D., Song W., Gilmore R. (2000). Role of the cytoplasmic segments of Sec61alpha in the ribosome-binding and translocation-promoting activities of the Sec61 complex. J. Cell Biol. 150 (1), 53–64. doi:10.1083/jcb.150.1.53
Ramachandra C. J. A., Hernandez-Resendiz S., Crespo-Avilan G. E., Lin Y. H., Hausenloy D. J. (2020). Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 57, 102884. doi:10.1016/j.ebiom.2020.102884
Ramírez A. S., Kowal J., Locher K. P. (2019). Cryo–electron microscopy structures of human oligosaccharyltransferase complexes OST-A and OST-B. Science 366 (6471), 1372–1375. doi:10.1126/science.aaz3505
Reddy R. K., Mao C., Baumeister P., Austin R. C., Kaufman R. J., Lee A. S. (2003). Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 278 (23), 20915–20924. doi:10.1074/jbc.M212328200
Ren H., Zhai W., Lu X., Wang G. (2021). The cross-Links of endoplasmic reticulum stress, autophagy, and neurodegeneration in Parkinson's disease. Front. Aging Neurosci. 13, 691881. doi:10.3389/fnagi.2021.691881
Risérus U., Willett W. C., Hu F. B. (2009). Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 48 (1), 44–51. doi:10.1016/j.plipres.2008.10.002
Rodriguez-Fonseca C., Amils R., Garrett R. A. (1995). Fine structure of the peptidyl transferase centre on 23 S-like rRNAs deduced from chemical probing of antibiotic-ribosome complexes. J. Mol. Biol. 247 (2), 224–235. doi:10.1006/jmbi.1994.0135
Römisch K. (2017). A case for Sec61 channel involvement in ERAD. Trends biochem. Sci. 42 (3), 171–179. doi:10.1016/j.tibs.2016.10.005
Ron D., Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8 (7), 519–529. doi:10.1038/nrm2199
Roy A., Wonderlin W. F. (2003). The permeability of the endoplasmic reticulum is dynamically coupled to protein synthesis. J. Biol. Chem. 278 (7), 4397–4403. doi:10.1074/jbc.M207295200
Ruiz-Canada C., Kelleher D. J., Gilmore R. (2009). Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136 (2), 272–283. doi:10.1016/j.cell.2008.11.047
Sammels E., Parys J. B., Missiaen L., De Smedt H., Bultynck G. (2010). Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell Calcium 47 (4), 297–314. doi:10.1016/j.ceca.2010.02.001
Sarfo F. S., Phillips R., Wansbrough-Jones M., Simmonds R. E. (2016). Recent advances: Role of mycolactone in the pathogenesis and monitoring of Mycobacterium ulcerans infection/Buruli ulcer disease. Cell. Microbiol. 18 (1), 17–29. doi:10.1111/cmi.12547
Schäuble N., Lang S., Jung M., Cappel S., Schorr S., Ulucan Ö., et al. (2012). BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J. 31 (15), 3282–3296. doi:10.1038/emboj.2012.189
Schnell D. J., Hebert D. N. (2003). Protein translocons: Multifunctional mediators of protein translocation across membranes. Cell 112 (4), 491–505. doi:10.1016/s0092-8674(03)00110-7
Schoebel S., Mi W., Stein A., Ovchinnikov S., Pavlovicz R., DiMaio F., et al. (2017). Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548 (7667), 352–355. doi:10.1038/nature23314
Schorr S., Klein M. C., Gamayun I., Melnyk A., Jung M., Schäuble N., et al. (2015). Co-chaperone specificity in gating of the polypeptide conducting channel in the membrane of the human endoplasmic reticulum. J. Biol. Chem. 290 (30), 18621–18635. doi:10.1074/jbc.M115.636639
Schubert D., Klein M. C., Hassdenteufel S., Caballero-Oteyza A., Yang L., Proietti M., et al. (2018). Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 subunit (SEC61A1). J. Allergy Clin. Immunol. 141 (4), 1427–1438. doi:10.1016/j.jaci.2017.06.042
Shacham T., Sharma N., Lederkremer G. Z. (2019). Protein misfolding and ER stress in Huntington’s disease. Front. Mol. Biosci. 6, 20. doi:10.3389/fmolb.2019.00020
Shen K., Arslan S., Akopian D., Ha T., Shan S. O. (2012). Activated GTPase movement on an RNA scaffold drives cotranslational protein targeting. Nature 492 (7428), 271–275. doi:10.1038/nature11726
Shilling D., Mak D. O. D., Kang D. E., Foskett J. K. (2012). Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J. Biol. Chem. 287 (14), 10933–10944. doi:10.1074/jbc.M111.300491
Shilling D., Müller M., Takano H., Mak D. O. D., Abel T., Coulter D. A., et al. (2014). Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 34 (20), 6910–6923. doi:10.1523/JNEUROSCI.5441-13.2014
Sicking M., Lang S., Bochen F., Roos A., Drenth J. P. H., Zakaria M., et al. (2021). Complexity and specificity of Sec61-channelopathies: Human diseases affecting gating of the Sec61 complex. Cells 10 (5), 1036. doi:10.3390/cells10051036
Silberstein S., Gilmore R. (1996). Biochemistry, molecular biology, and genetics of the oligosaccharyltransferase. FASEB J. 10 (8), 849–858. doi:10.1096/fasebj.10.8.8666161
Simon S. M., Blobel G. (1991). A protein-conducting channel in the endoplasmic reticulum. Cell 65 (3), 371–380. doi:10.1016/0092-8674(91)90455-8
Sommer N., Junne T., Kalies K. U., Spiess M., Hartmann E. (2013). TRAP assists membrane protein topogenesis at the mammalian ER membrane. Biochim. Biophys. Acta 1833 (12), 3104–3111. doi:10.1016/j.bbamcr.2013.08.018
Song B., Scheuner D., Ron D., Pennathur S., Kaufman R. J. (2008). CHOP deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 118 (10), 3378–3389. doi:10.1172/JCI34587
Song W., Raden D., Mandon E. C., Gilmore R. (2000). Role of Sec61alpha in the regulated transfer of the ribosome-nascent chain complex from the signal recognition particle to the translocation channel. Cell 100 (3), 333–343. doi:10.1016/s0092-8674(00)80669-8
Swanton E., Bulleid N. J. (2003). Protein folding and translocation across the endoplasmic reticulum membrane. Mol. Membr. Biol. 20 (2), 99–104. doi:10.1080/0968768031000069241
Synofzik M., Haack T. B., Kopajtich R., Gorza M., Rapaport D., Greiner M., et al. (2014). Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Genet. 95 (6), 689–697. doi:10.1016/j.ajhg.2014.10.013
Takeshima H., Venturi E., Sitsapesan R. (2015). New and notable ion-channels in the sarcoplasmic/endoplasmic reticulum: Do they support the process of intracellular Ca2+ release? J. Physiol. 593 (15), 3241–3251. doi:10.1113/jphysiol.2014.281881
Tatu U., Helenius A. (1997). Interactions between newly synthesized glycoproteins, calnexin and a network of resident chaperones in the endoplasmic reticulum. J. Cell Biol. 136 (3), 555–565. doi:10.1083/jcb.136.3.555
Traut R. R., Monro R. E. (1964). The puromycin reaction and its relation to protein synthesis. J. Mol. Biol. 10 (1), 63–72. doi:10.1016/s0022-2836(64)80028-0
Tsai Y. C., Weissman A. M. (2010). The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer 1 (7), 764–778. doi:10.1177/1947601910383011
Tu H., Nelson O., Bezprozvanny A., Wang Z., Lee S. F., Hao Y. H., et al. (2006). Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126 (5), 981–993. doi:10.1016/j.cell.2006.06.059
Tyedmers J., Lerner M., Bies C., Dudek J., Skowronek M. H., Haas I. G., et al. (2000). Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc. Natl. Acad. Sci. U. S. A. 97 (13), 7214–7219. doi:10.1073/pnas.97.13.7214
Ureshino R. P., Erustes A. G., Bassani T. B., Wachilewski P., Guarache G. C., Nascimento A. C., et al. (2019). The interplay between Ca2+ signaling pathways and neurodegeneration. Int. J. Mol. Sci. 20 (23), 6004. doi:10.3390/ijms20236004
Van Coppenolle F., Vanden Abeele F., Slomianny C., Flourakis M., Hesketh J., Dewailly E., et al. (2004). Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 117 (18), 4135–4142. doi:10.1242/jcs.01274
Van Nieuwenhove E., Barber J. S., Neumann J., Smeets E., Willemsen M., Pasciuto E., et al. (2020). Defective Sec61α1 underlies a novel cause of autosomal dominant severe congenital neutropenia. J. Allergy Clin. Immunol. 146 (5), 1180–1193. doi:10.1016/j.jaci.2020.03.034
Vanderheyden V., Devogelaere B., Missiaen L., De Smedt H., Bultynck G., Parys J. B. (2009). Regulation of inositol 1, 4, 5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim. Biophys. Acta 1793 (6), 959–970. doi:10.1016/j.bbamcr.2008.12.003
Virrey J. J., Dong D., Stiles C., Patterson J. B., Pen L., Ni M., et al. (2008). Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 6 (8), 1268–1275. doi:10.1158/1541-7786.MCR-08-0060
von Heijne G., Gavel Y. (1988). Topogenic signals in integral membrane proteins. Eur. J. Biochem. 174 (4), 671–678. doi:10.1111/j.1432-1033.1988.tb14150.x
Voorhees R. M., Fernández I. S., Scheres S. H. W., Hegde R. S. (2014). Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 157 (7), 1632–1643. doi:10.1016/j.cell.2014.05.024
Voorhees R. M., Hegde R. S. (2016). Structure of the Sec61 channel opened by a signal sequence. Science 351 (6268), 88–91. doi:10.1126/science.aad4992
Wacquier B., Voorsluijs V., Combettes L., Dupont G. (2019). Coding and decoding of oscillatory Ca2+ signals. Semin. Cell Dev. Biol. 94, 11–19. doi:10.1016/j.semcdb.2019.01.008
Wang L., Dobberstein B. (1999). Oligomeric complexes involved in translocation of proteins across the membrane of the endoplasmic reticulum. FEBS Lett. 457 (3), 316–322. doi:10.1016/s0014-5793(99)01075-3
Wang Q., Li L., Ye Y. (2008). Inhibition of p97-dependent protein degradation by eeyarestatin I. J. Biol. Chem. 283 (12), 7445–7454. doi:10.1074/jbc.M708347200
Wang Q., Mora-Jensen H., Weniger M. A., Perez-Galan P., Wolford C., Hai T., et al. (2009). ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. U. S. A. 106 (7), 2200–2205. doi:10.1073/pnas.0807611106
Wirth A., Jung M., Bies C., Frien M., Tyedmers J., Zimmermann R., et al. (2003). The Sec61p complex is a dynamic precursor activated channel. Mol. Cell 12 (1), 261–268. doi:10.1016/s1097-2765(03)00283-1
Wonderlin W. F. (2009). Constitutive, translation-independent opening of the protein-conducting channel in the endoplasmic reticulum. Pflugers Arch. 457 (4), 917–930. doi:10.1007/s00424-008-0545-y
Wu X., Rapoport T. A. (2018). Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 53, 22–28. doi:10.1016/j.ceb.2018.04.004
Yasmeen N., Datta M., Kumar V., Alshehri F. S., Almalki A. H., Haque S. (2022). Deciphering the link between ERUPR signaling and microRNA in pathogenesis of Alzheimer’s disease. Front. Aging Neurosci. 14, 880167. doi:10.3389/fnagi.2022.880167
Yotsu R. R., Suzuki K., Simmonds R. E., Bedimo R., Ablordey A., Yeboah-Manu D., et al. (2018). Buruli ulcer: A review of the current knowledge. Curr. Trop. Med. Rep. 5 (4), 247–256. doi:10.1007/s40475-018-0166-2
Yusupov M. M., Yusupova G. Z., Baucom A., Lieberman K., Earnest T. N., Cate J. H., et al. (2001). Crystal structure of the ribosome at 5.5 Å resolution. Science 292 (5518), 883–896. doi:10.1126/science.1060089
Zatti G., Burgo A., Giacomello M., Barbiero L., Ghidoni R., Sinigaglia G., et al. (2006). Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 39 (6), 539–550. doi:10.1016/j.ceca.2006.03.002
Zhang I. X., Raghavan M., Satin L. S. (2019). The endoplasmic reticulum and calcium homeostasis in pancreatic β cells. Endocrinology 161 (2), bqz028. doi:10.1210/endocr/bqz028
Keywords: Sec61 complex/translocon, reticular Ca2+-leak channel, ER stress, cell survival, apoptosis, intracellular Ca2+ homeostasis
Citation: Parys JB and Van Coppenolle F (2022) Sec61 complex/translocon: The role of an atypical ER Ca2+-leak channel in health and disease. Front. Physiol. 13:991149. doi: 10.3389/fphys.2022.991149
Received: 11 July 2022; Accepted: 20 September 2022;
Published: 06 October 2022.
Edited by:
Richard Zimmermann, Saarland University, GermanyReviewed by:
Agustín Guerrero-Hernández, Instituto Politécnico Nacional de México (CINVESTAV), MexicoMartin Jung, Saarland University, Germany
Copyright © 2022 Parys and Van Coppenolle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan B. Parys, amFuLnBhcnlzQGt1bGV1dmVuLmJl; Fabien Van Coppenolle, ZmFiaWVuLnZhbi1jb3BwZW5vbGxlQHVuaXYtbHlvbjEuZnI=
†These authors have contributed equally to this work