 Lily Den Hartog
Lily Den Hartog Atsushi Asakura
Atsushi Asakura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 27 September 2022
Sec. Striated Muscle Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.984373
This article is part of the Research TopicMechanisms in Duchenne Muscular Dystrophy Pathophysiology and TreatmentView all 10 articles
This review focuses upon the implications of the Notch signaling pathway in muscular dystrophies, particularly Duchenne muscular dystrophy (DMD): a pervasive and catastrophic condition concerned with skeletal muscle degeneration. Prior work has defined the pathogenesis of DMD, and several therapeutic approaches have been undertaken in order to regenerate skeletal muscle tissue and ameliorate the phenotype. There is presently no cure for DMD, but a promising avenue for novel therapies is inducing muscle regeneration via satellite cells (muscle stem cells). One specific target using this approach is the Notch signaling pathway. The canonical Notch signaling pathway has been well-characterized and it ultimately governs cell fate decision, cell proliferation, and induction of differentiation. Additionally, inhibition of the Notch signaling pathway has been directly implicated in the deficits seen with muscular dystrophies. Here, we explore the connection between the Notch signaling pathway and DMD, as well as how Notch signaling may be targeted to improve the muscle degeneration seen in muscular dystrophies.
Muscular dystrophies are a group of inherited disorders that involve progressive muscle weakness and degeneration of skeletal muscle (Lovering et al., 2005). Duchenne muscular dystrophy (DMD) is one of the most severe forms of inherited muscular dystrophy, proving to be lethal in 100% of cases, and is also the most prevalent, with an incidence of one per 5,136 male births (Crisafulli et al., 2020; Yao et al., 2021). This qualifies it as one of the most widespread recessive disorders among the human population (Mah, 2016). Becker muscular dystrophy (BMD) is also prevalent, but somewhat less common and less severe (Duan et al., 2021). Since there is currently no cure for DMD, it is crucial that further research be done in order to identify and optimize potential treatments and therapies (Guiraud et al., 2015).
DMD is caused by mutations of the dystrophin/DMD gene, which is located at the Xp21 locus on the X chromosome (Venugopal and Pavlakis, 2022). Because DMD is an X-linked disorder, it almost exclusively affects males, with females acting as asymptomatic carriers (Crisafulli et al., 2020). The dystrophin gene is the largest known human gene, containing 79 exons and spanning >2,200 kb (Gao and McNally, 2015). Secondary to its large size is its complex mutational spectrum; it has a high spontaneous mutation rate and there are >7,000 known mutations of the gene, with 1/3 of DMD cases resulting from sporadic mutations (Mah, 2016; Venugopal and Pavlakis, 2022). In current research, mutation rate is the probability that a unit length of DNA will mutate over time. Thus, the dystrophin gene has a high mutation rate, in part due to its huge gene size (Balin and Cascalho, 2010).
Deletion mutations account for 60%–70% of DMD cases, and point mutations and exonic duplications are also common (Gao and McNally, 2015; Duan et al., 2021). Dystrophin codes for the protein dystrophin, whose production can be limited and even eliminated by dystrophin mutation (Duan et al., 2021; Yao et al., 2021). Out-of-frame dystrophin mutations or premature stop-codon mutations typically lead to complete loss of the dystrophin protein and are more severe, while in-frame mutations that lead to the synthesis of a partially functional truncated protein produce milder forms of muscular dystrophy, such as BMD (Mah, 2016; Duan et al., 2021).
Dystrophin is located at the sarcolemma (cell membrane) of skeletal muscle cells and cardiomyocytes, interacting with a group of peripheral membrane and transmembrane proteins to form the dystrophin-associated glycoprotein complex (DGC) (Jiang et al., 2014). In healthy muscle, the DGC provides structural stability to skeletal and heart muscle, participates in transmembrane signaling, and plays a role in the vasomotor response to physical activity (Mah, 2016). In dystrophic muscle, however, loss of dystrophin diminishes the DGC, leading to weakness of the cytoskeleton, sarcolemmal lesions, and increased membrane fluidity (Wilschut et al., 2012; Venugopal and Pavlakis, 2022). This causes abnormal calcium influx and inflammation, which has catastrophic ramifications including the altered composition of structural glycoproteins in the extracellular matrix, activation of proteases and proinflammatory cytokines, ischemic injury, oxidative and nitrosative stress, and mitochondrial dysfunction (Duan et al., 2021). As a result of this loss in myofiber integrity, resident muscle stem cells undergo fibrogenesis rather than myogenesis, causing aberrant collagen deposition and subsequent necrosis (Wilschut et al., 2012; Kornegay, 2017). Through repeated cycles of necrosis and regeneration, muscle is gradually replaced with fibrous connective tissue and fat, producing the phenotypic characteristics of DMD (Venugopal and Pavlakis, 2022).
Skeletal muscle regeneration is carried out by the proliferation of quiescent muscle satellite cells (muscle stem cells) and the differentiation of myofibers. Activation of the Notch pathway is required to maintain the quiescent state of muscle satellite cells, and disruption of the Notch pathway leads to disruption of muscle satellite cell maintenance, thereby impairing muscle regeneration. Hence, disruption of the Notch pathway is one of the mechanisms of skeletal muscle disease. Currently, three muscle disease genes that interact with the Notch pathway have recently been identified—Jag2, MEGF10, and POGLUT1—whose mutation has been noted in muscular dystrophies. This review introduces the pathogenesis and treatment strategies for DMD, and discusses the biology of the Notch pathway and genetics in muscular dystrophies.
DMD symptoms occur as early as 2–3 years of age and the disorder causes death prior to the third or fourth decade of life, usually around ages 19–25 (Mah, 2016; Duan et al., 2021; Iftikhar et al., 2021). DMD initially presents with symptoms of difficulties climbing stairs, a waddling gait, toe walking, Gowers’ sign, and frequent falls (Birnkrant et al., 2018b; Duan et al., 2021). The physiological presentation of DMD goes on to primarily include progressive muscle degeneration, proximal weakness, and joint contractures (Mah, 2016; Coppens et al., 2021). Due to this, DMD patients are typically wheelchair-dependent by 10–12 years of age (Duan et al., 2021). DMD eventually brings about numerous secondary complications, including scoliosis, respiratory insufficiency, and cardiac issues (Mah, 2016). Dilated cardiomyopathy, myocardial necrosis, conduction defects, and arrhythmias are very common as well, with cardiorespiratory failure being the leading cause of mortality in DMD patients (Mah, 2016; Yao et al., 2021). This can be attributed to the absence of dystrophin in cardiomyocytes, which causes a loss of contractile function (Yucel et al., 2018). Additionally, though most DMD patients do not fit the criteria for intellectual disability, most do have some degree of cognitive impairment (Yucel et al., 2018). Many DMD patients do have a below-average IQ, as studies have reported that 20% of DMD patients have an IQ lower than 70 and 44% of DMD patients have learning disabilities (Yucel et al., 2018; Mohamadian et al., 2022). DMD patients also have been shown to be at increased risk for neurobehavioral comorbidities, such as attention-deficit/hyperactivity disorder, autism spectrum disorder, epilepsy, and anxiety (Mohamadian et al., 2022). There has been a suggestion of genotype-phenotype association with regards to dystrophin mutation and neurodevelopmental manifestations, as Ricotti et al. found that patients with distal mutations in the dystrophin gene were more likely to have neurodevelopmental problems, intellectual disability, memory deficits, and decreased grey matter volume. However, this study also demonstrated that emotional and behavioral problems were equally distributed among patients with proximal versus distal dystrophin mutations, calling into question this genotype-phenotype association (Ricotti et al., 2016). Cognitive impairments seen in DMD patients have typically been thought of as non-progressive, though recent studies have suggested otherwise (Bagdatlioglu et al., 2020). Studies in a mouse model of DMD have demonstrated that long-term memory and anxiety behaviors do worsen with age, which indicates that dystrophin deficiency causes progressive cognitive impairment greater than that naturally seen with aging, and furthermore that the reduced life expectancy of human DMD patients may mask their potential for progressive cognitive impairment (Bagdatlioglu et al., 2020).
DMD is medically evaluated in several manners, including via laboratory work, muscle biopsy, gene analysis, electromyography, and cardiac testing (Venugopal and Pavlakis, 2022). The possibility of newborn screening has been extensively discussed, and a recent survey revealed that most DMD physicians would see a benefit in newborn screening and feel that the DMD care community is ready for this (Armstrong et al., 2022). This may be of greatly advantageous, as the majority of DMD physicians also indicated that they would recommend initiating therapies much earlier than the typical age at which DMD is currently diagnosed (Armstrong et al., 2022). However, this has not yet become standard (Birnkrant et al., 2018b). The diagnosis of DMD is usually confirmed after symptoms are present via a laboratory test for serum creatine kinase (CK), a muscle enzyme that, when elevated, reflects ongoing muscle damage (Duan et al., 2021). Serum CK is often elevated in DMD patients before the development of clinical symptoms, as levels peak by age two and its level is typically 10–20x greater than the upper limit of normal (Venugopal and Pavlakis, 2022). Other muscle enzymes, including aldolase, alanine transaminase (ALT), and aspartate aminotransferase (AST), are often elevated as well, though not used as a diagnostic measure (Duan et al., 2021). Though serial muscle biopsies from DMD patients are not usually necessary for diagnosis, they do reveal multiple dystrophic changes, which include disorganization, scattered degeneration, connective tissue proliferation, inflammation, muscle fiber necrosis, and extensive deposits of adipose tissue in place of muscle (Guiraud et al., 2015; Coppens et al., 2021; Venugopal and Pavlakis, 2022). Gene analysis of DMD patients shows a complete absence of the dystrophin gene, and dystrophin immunoblotting may be utilized in order to predict the severity of the disease (Venugopal and Pavlakis, 2022). In examining muscles, electromyography can detect nonspecific myopathic features associated with muscular dystrophy (Venugopal and Pavlakis, 2022). Finally, cardiac testing is used to evaluate for complications frequently seen with DMD; electrocardiogram reveals characteristic changes, telemetry identifies conduction abnormalities, and echocardiogram shows evidence of dilated cardiomyopathy, which is present in nearly all DMD patients by the time they reach their twenties (Venugopal and Pavlakis, 2022).
Despite its prevalence and severity, there is currently no cure for DMD and most available therapies simply act to manage symptoms and prolong ambulation and lifespan (Birnkrant et al., 2018b; Yao et al., 2021). As laid out clearly in a multi-part review by Birnkrant et al., a multidisciplinary approach to treatment is critical, typically led by a neuromuscular specialist. Corticosteroids and supportive measures have remained the standard of care over the past 30 years, since the molecular basis of DMD was defined (Motohashi et al., 2019). Corticosteroids, usually prednisone or deflazacort, are typically initiated around 4–5 years of age and provide numerous benefits for DMD patients, including deceleration of myofiber necrosis, improved pulmonary function, delayed development of scoliosis, prolonged independent ambulation, reduced progression of cardiomyopathy, improved mortality, and stabilization of muscle strength and function (Mah, 2016; Birnkrant et al., 2018b; Iftikhar et al., 2021; Venugopal and Pavlakis, 2022). That said, there is a slew of adverse side effects associated with corticosteroid use, including short stature, obesity, and cataracts, deeming it a suboptimal treatment (Mah, 2016). Corticosteroids also greatly increase the probability that DMD patients develop osteoporosis and skeletal fractures, including vertebral compression fractures, for which they are already at an elevated risk (Birnkrant et al., 2018a). Corticosteroid treatment is typically supplemented by supportive interventions that act to prolong survival and lessen symptom severity. To account for cardiac troubles, DMD patients are typically treated with angiotensin-converting enzyme (ACE) inhibitors and beta-blockers in order to slow the progression of cardiomyopathy and attempt to prevent heart failure (Venugopal and Pavlakis, 2022). It is recommended that spirometry be initiated when the patient is 5–6 years old, and noninvasive positive pressure ventilation and effective airway clearance are used to manage respiratory issues (Mah, 2016; Birnkrant et al., 2018a). Orthopedic management and physiotherapy are employed with the primary goals of minimizing joint contractures, maintaining a straight spine, and promoting bone health (Birnkrant et al., 2018a). Finally, endocrine and gastrointestinal management are also crucial to the appropriate treatment of DMD patients (Birnkrant et al., 2018b).
Recent scientific advances have garnered the potential for novel therapies to fight numerous neuromuscular diseases, including DMD. Currently, many therapeutic approaches aim to rescue dystrophin expression in skeletal muscle with hopes of improving function in dystrophic muscle, promoting muscle hypertrophy, and reducing muscle wasting (Cossu and Sampaolesi, 2007; Vieira et al., 2015). The goal of these therapies is not to cure DMD, but rather to improve severe DMD phenotypes to mimic more mild phenotypes, similar to BMD (Tominari and Aoki, 2022). A common limitation in the quest for effective DMD therapies is that there is no specific set of recommended or required outcome measures, making goals ambiguous (Shimizu-Motohashi et al., 2019). With regard to rescuing dystrophin expression, the level of dystrophin required for clinical efficacy has remained unclear; while restoring 15%–20% of normal dystrophin levels in mouse models has shown improvement, 33%–40% has been reported as necessary for improvement in dog models (Sun et al., 2020). In humans, restoring 30% of normal dystrophin levels has been suggested as the standard for notable improvement (Sun et al., 2020). However, the relevance of this is still unknown, as some patients with low levels of dystrophin have been shown to maintain relatively normal muscle function, and studies in DMD patients that have extremely low but still detectable levels of dystrophin have suggested that even a small amount of dystrophin protein can mitigate skeletal muscle deficits (Nakamura et al., 2016; Waldrop et al., 2018).
Leading approaches in novel DMD therapies thus far have been gene-based, including gene replacement, endogenous gene repair, exon skipping, and read-through therapies (Yao et al., 2021). Multiple issues with these techniques have arisen, including immune response to the vector, difficulty delivering genes to post-mitotic muscle fibers, and the fact that many of these techniques only have potential for treating DMD patients with particular genetic mutations, of which there are several (Cossu and Sampaolesi, 2007; Iftikhar et al., 2021). Other methods being explored include upregulation of utrophin, inhibition of histone deacetylase, antagonization of the myostatin pathway, and increased angiogenesis (Cossu and Sampaolesi, 2007; Verma et al., 2018; Sun et al., 2020). Agents have also been explored in order to target downstream pathological changes seen in DMD, such as fibrosis, inflammation, ischemia, oxidative stress, loss of calcium homeostasis, and muscle atrophy (Yao et al., 2021). Though exon-skipping and read-through therapies in particular have shown promise, they have their drawbacks and other approaches have shown limited success in preclinical and clinical trials thus far; therefore, there is a distinct need for further research and development.
Of note, several model organisms have been used to study DMD and potential therapies, the most common of which is the mdx mouse. The mdx mouse has a mutation in the dystrophin gene itself, similar to human DMD patients (Yucel et al., 2018). However, mdx mice do not exhibit a shortened lifespan, severe muscle degeneration, or other key features of DMD, such as cardiomyopathy (Yucel et al., 2018; Gao et al., 2019). Their milder phenotype is likely attributable to the high regenerative capacity of mouse muscle, as well as the fact that utrophin is still active, whose expression may compensate for the lack of dystrophin (Plantié et al., 2015; Gao et al., 2019). The utrophin protein has a remarkably similar structure and function to dystrophin in linking the sarcolemma to the cytoskeleton, but is primarily expressed in skeletal muscle during fetal development and, in healthy muscle, is downregulated prior to birth (Yucel et al., 2018). Utrophin is usually upregulated in skeletal muscle when dystrophin is absent and has even been proposed as an alternative target protein to dystrophin, due to their similarities (Yao et al., 2021). However, due to key functional differences in the protein structure, utrophin cannot fully compensate for the loss of dystrophin in human DMD patients (Yucel et al., 2018). In order to account for this in murine models, double knockout (dKO) mice models have been utilized, which, in addition to knocking out dystrophin, also knock out one of a variety of genes that play important roles in myogenesis and muscle function (Yucel et al., 2018). Namely, dystrophin/utrophin dKO mice have been employed in studies, as they more closely resemble the clinical manifestations of DMD and have been deemed more suitable for gene therapy testing (Gao et al., 2019). Furthermore, a golden retriever muscular dystrophy (GRMD) dog model has come into use, as it better aligns with the progressive course of human DMD than most mouse models (Kornegay, 2017). GRMD studies have not always substantiated findings in mdx mouse studies, and have also illuminated various side effects of potential treatments that are not seen in mdx mice (Kornegay, 2017). GRMD has also been crucial in investigating the speed of disease progression, as RNA sequencing has been employed to identify biomarkers for this (Brinkmeyer-Langford et al., 2018). Despite their utility, practical considerations do limit the use of GRMD models, especially dog availability and expense (Kornegay, 2017). Finally, drosophila and zebrafish models of DMD have been studied, as these organisms have modeled many human diseases over the last several decades and comparative genomic studies have demonstrated sequence conservation of dystrophin in both organisms (Plantié et al., 2015). In particular, zebrafish sapje mutants have been useful, as they contain a mutation at dystrophin and do not express compensatory utrophin, so they exhibit features more similar to human DMD pathology (Plantié et al., 2015).
Because muscular dystrophies, including DMD, are characterized by progressive degeneration of skeletal muscle, the structure and function of skeletal muscle must be understood in order to develop effective therapeutic strategies. In humans, skeletal muscle comprises approximately 40% of total body weight (Frontera and Ochala, 2015). It is responsible for converting chemical energy into mechanical energy, hence generating force and power to produce movement and perform voluntary functions (Frontera and Ochala, 2015). Skeletal muscle is necessary for the maintenance of homeostasis and other bodily functions as well, including respiration and metabolic function (Joanisse et al., 2017; Sousa-Victor et al., 2022).
Skeletal muscle is heterogeneous tissue, composed of different types of muscle fibers (Joanisse et al., 2017). Its architecture involves a specific arrangement of these muscle fibers, along with surrounding connective tissue (Frontera and Ochala, 2015). Skeletal muscle fibers are formed through the fusion of single cells and contain several hundred post-mitotic nuclei in their mature form (Tedesco et al., 2010). This multinucleation presents a hurdle to the treatment of deficient skeletal muscle, as therapies face the task of restoring proper gene expression in hundreds of millions of post-mitotic nuclei (Tedesco et al., 2010).
Skeletal muscle tissue has the ability to regenerate new muscle fibers upon indication by homeostatic demand, hypertrophy, or need for repair secondary to injury, exercise, or disease (Tedesco et al., 2010; Sato et al., 2012). The regenerative capacity of skeletal muscle declines with aging (Sousa-Victor et al., 2022). The stem cells of postnatal muscle, known as satellite cells, are responsible for >99% of the regenerative potential of adult skeletal muscle (Conboy et al., 2003; Bröhl et al., 2012; Joanisse et al., 2017). Other progenitors, including pericytes, endothelial cells, and interstitial cells, have a limited amount of regenerative potential as well (Tedesco et al., 2010; Verma et al., 2018).
Satellite cells are crucial to the regenerative capacity of skeletal muscle tissue; when they dysfunction, the skeletal muscle loses its regenerative ability, leading to the degeneration reminiscent of DMD (Asakura et al., 2002; Bröhl et al., 2012; Sato et al., 2012; Fuchs and Blau, 2020). During prenatal development, some muscle progenitor cells migrate into position as mononuclear cells between the basal lamina of the muscle fiber and its sarcolemma; these cells ultimately constitute the satellite cell pool and are defined by their unique molecular location (Tedesco et al., 2010; Wilschut et al., 2012). Satellite cells are mononuclear and comprise 2.5%–6% of all nuclei for any given skeletal muscle fiber (Tedesco et al., 2010). They play important roles both in establishing and growing muscle during development and in maintaining muscle in adults via regeneration (Bröhl et al., 2012). Satellite cells are distinct from other types of progenitors in that they are limited to myogenesis, rather than possessing a broader multilineage differentiation potential (Asakura et al., 2001; 2002; Wilschut et al., 2012).
Satellite cells can adopt several different states, including quiescence, activation, proliferation, and differentiation (Li et al., 2021). Initially, during the perinatal and postnatal periods, satellite cells are proliferative (Bröhl et al., 2012). Then, in healthy adult muscle, satellite cells maintain the essential feature of quiescence under homeostatic conditions (Bröhl et al., 2012). Quiescence is common to stem cells and is defined by reversible mitotic arrest: quiescent cells do not divide, but rather are still able to re-enter the cell cycle and proliferate upon stimulation (Bjornson et al., 2011). Quiescence permits self-renewal, allowing for the maintenance of stem cell populations throughout the lifetime of an animal (Bjornson et al., 2011). Furthermore, quiescence involves reduced metabolic activity, so that satellite cells are protected against endogenous stress from DNA replication and cellular respiration (Bjornson et al., 2011). On a genetic level, quiescent cells highly express the paired box 7 (Pax7) gene, which has been shown to be the most reliable marker for satellite cells (Mourikis et al., 2012; Fuchs and Blau, 2020; Starosta and Konieczny, 2021; Sousa-Victor et al., 2022). Mice null for Pax7 are notably deficient in satellite cells, and misregulation of Pax7 has been implicated in DMD (Gayraud-Morel et al., 2012; Coppens et al., 2021). Aside from satellite cells, Pax7 is not expressed by any other cell type in muscle tissue (Fuchs and Blau, 2020).
Upon injury or due to homeostatic demand, satellite cells leave their quiescent state, re-entering the cell cycle and downregulating Pax7 (Dumont et al., 2015; Kann et al., 2021). At this point, they instead express myogenic regulatory factors (MRFs), including Myf5 and MyoD, which promote cell activation and the cells become myoblasts, poised to regenerate muscle tissue (Mourikis et al., 2012; Wilschut et al., 2012; Sousa-Victor et al., 2022). MRFs are specifically expressed in skeletal muscle lineage, and when transfected into certain other cell types, MRFs can induce them to adopt a myogenic fate (Biressi et al., 2013). Cell ablation studies have demonstrated that Myf5 and MyoD can independently initiate myogenic differentiation, thus deeming them myogenic determination genes (Biressi et al., 2013). Myf5 is the first MRF to be expressed during mammalian development (Biressi et al., 2013). Though MyoD is only expressed in a small percentage of quiescent satellite cells, all progenitors of satellite cells transcribe MyoD prenatally and express it prior to the first cell division (Kanisicak et al., 2009; Gayraud-Morel et al., 2012). Other MRFs include Mrf4, which only acts as a determination gene during embryonic development, and myogenin, which is never co-expressed with Pax7 and acts as a differentiation factor downstream of MyoD and Myf5 (Kottlors and Kirschner, 2010; Gayraud-Morel et al., 2012; Biressi et al., 2013).
Importantly, the satellite cell population is heterogenous, and subpopulations serve distinct functions (Dumont et al., 2015). Some primarily perform asymmetric divisions, which are more prevalent during homeostasis in order to generate myogenic progenitors while maintaining the satellite cell pool (Dumont et al., 2015; Sousa-Victor et al., 2022). Others predominantly perform symmetric divisions, which are more common after indication by injury or breakdown, as they act to expand the satellite cell pool (Dumont et al., 2015; Sousa-Victor et al., 2022). Satellite cells maintain a dynamic balance between symmetric and asymmetric division in accordance with the present needs of the muscle, and which type of division that satellite cells undergo is primarily governed by two factors: polarity and orientation (Dumont et al., 2015). Asymmetric division is driven by unequal distribution of polarity proteins, specifically members of the Partitioning-defective protein (PAR) family, which establish cell polarity in several different cell types (Dumont et al., 2015). With regard to orientation, symmetric divisions typically occur in a planar orientation, while asymmetric divisions usually occur in an apical-basal orientation (Kuang et al., 2007). Furthermore, different subpopulations of satellite cells express genes associated with quiescence and myogenesis to varying degrees (Dumont et al., 2015). Though MyoD is consistently expressed in all activated satellite cells, there are populations of Myf5-positive and Myf5-negative satellite cells present in adult muscle; studies using fluorescent protein tagging have revealed that approximately 10% of satellite cells constitute a subpopulation that never expresses Myf5 during development (Kuang et al., 2007; Dumont et al., 2015; Sousa-Victor et al., 2022). Interestingly, these Myf5-negative cells have shown a greater ability to self-renew and are more stem-like as compared to their Myf5-positive counterparts (Kuang et al., 2007; Sousa-Victor et al., 2022). Also, satellite cells demonstrate Pax7 at different levels, and subpopulations of satellite cells that express higher levels of Pax7 are less prone to differentiation (Rocheteau et al., 2012). This is not surprising since Pax7 is so intimately associated with quiescence and proliferation.
The intrinsic regulatory mechanisms that govern quiescence, cell cycle progression, and cell fate determination of satellite cells are influenced by extrinsic cues (Mourikis et al., 2012; Dumont et al., 2015; Verma et al., 2018). These cues involve proximity to endothelial cells, as well as the specialized local niche between the myofiber sarcolemma and basal lamina in which satellite cells reside (Dumont et al., 2015; Verma et al., 2018). During quiescence, the basal lamina physically separates satellite cells from other local niche cells (Kann et al., 2021). Muscular injury induces changes in this niche, namely disruption of the basal lamina, which exposes satellite cells to the growth factors and signaling molecules that they are normally sequestered from (Kann et al., 2021). This external cue subsequently promotes satellite cell activation and proliferation.
Though it has been established that satellite cells drive muscle regeneration and that impaired regeneration drives the phenotype of DMD, the specific role of satellite cells in this impaired regeneration has been disputed (Dumont and Rudnicki, 2016).
Some studies have reported that satellite cell number remains the same or is even elevated in dystrophic muscle as compared with age-matched controls (Kottlors and Kirschner, 2010; Jiang et al., 2014). Further, some studies have suggested that impaired regeneration occurs secondary to the surrounding pathogenic environment rather than intrinsic issues with the regenerative capacity of satellite cells (Boldrin et al., 2015). This would indicate that lack of satellite cells is not the culprit for impaired regeneration. Similarly, other studies have suggested that insufficient regeneration may be caused by failure of differentiation rather than a lack of self-renewal and proliferation (Kottlors and Kirschner, 2010). This presents a direct link between dystrophin deficiency and impaired regeneration, as dystrophin is crucial for defining cell polarity, which strongly influences whether satellite cell division is asymmetric or symmetric and subsequently whether differentiation and myogenesis occur (Chang et al., 2016; Dumont and Rudnicki, 2016). Dystrophin influences polarity by interacting with MARK2, a cell polarity-regulating kinase (Chang et al., 2016). When dystrophin is deficient in DMD muscle, there is reduced expression of MARK2, which leads to loss of PARD3 polarization, a member of the PAR family (Chang et al., 2016). Consequently, asymmetric satellite cell divisions wither, so fewer myogenic progenitors are produced and muscle regeneration is impaired (Chang et al., 2016; Dumont and Rudnicki, 2016). This link between dystrophin and polarity demonstrates a direct role for dystrophin in the loss of regenerative capacity seen in DMD, and supports the idea that exhaustion of the satellite pool may not be the cause for impaired regeneration.
On the contrary, several studies have indicated that satellite cell pool exhaustion precipitates loss of regenerative capacity, and satellite cells do remain a key target for therapies combatting DMD (Wilschut et al., 2012). Due to their constant activation in an attempt to compensate for the unrelenting muscle degeneration of DMD, the satellite cell pool becomes depleted in dystrophic tissue, leading to failure of muscle repair and accelerated disease progression (Morgan and Zammit, 2010; Sacco et al., 2010; Wilschut et al., 2012; Jiang et al., 2014; Mu et al., 2015). Hence, rescuing satellite cell functionality has been a key target for DMD therapies (Liu et al., 2021).
The year 1990 saw the first successful satellite cell transplant into a boy with DMD, which achieved dystrophin production (Law et al., 1990). Though clinical trials throughout the 1990s demonstrated safety, no significant functional benefit was identified (Cossu and Sampaolesi, 2007). Approaches to satellite cell transplantation are still being explored in order to account for their drawbacks. For example, the heterogeneity of cultured satellite cells makes them somewhat unreliable in transfer therapies, so they have been cultured in various systems or on different mediums in order to select satellite cells that can best survive, engraft, and repopulate (Sun et al., 2020). Due to the lack of adequate success with satellite cell transfer thus far, the quest for optimization of this approach persists.
Though satellite cells have been looked to as a superior option, as they have self-renewal capacity and differentiation potential, myoblast-transfer therapies have been explored as well, as they have a high ability to generate muscle fibers (Shimizu-Motohashi et al., 2019). Pioneer myoblast-transfer studies demonstrated that intramuscular injection of healthy myoblasts into mdx mice resulted in fusion with host fibers and extensive dystrophin production (Tedesco et al., 2010). These early suggestions of success led to several clinical trials in the early 1990s–2000s that unfortunately failed for various reasons, including poor survival, limited migration of donor cells after transplantation, and immune responses that caused the rejection of the donor cells (Tedesco et al., 2010; Shimizu-Motohashi et al., 2019). In the interim, numerous approaches with different transplantation techniques have been attempted to sidestep these concerns, including employing conditional proliferation-dependent suicide agents in order to combat the oncogenic potential of immortalized cells (Sun et al., 2020). However, ample success has not yet been achieved with myoblasts (Sakai et al., 2017).
In addition to satellite cells and myoblasts, several different myogenic progenitors have been investigated as potential candidates in transfer therapies as well, including mesenchymal stem cells, CD133+ cells, mesangioblasts, embryonic stem cells, and induced pluripotent stem cells (Sakai et al., 2017; Shimizu-Motohashi et al., 2019; Tominari and Aoki, 2022). Despite the established efficacy of exon-skipping and read-through therapies, cell transplantation therapies are applicable to more patients, as they are not specific to patients with certain types of mutations (Tominari and Aoki, 2022). Cell transfer therapies have both explored using genetically unmodified cells from healthy donors, which are mutation-free but carry a greater risk of immune reaction, and using autologous genetically-corrected cells, which require gene manipulation prior to transplantation but carry a much lower risk of immune reaction (Shimizu-Motohashi et al., 2019; Duan et al., 2021). Clinical trials in human patients thus far have primarily focused on cells from healthy donors, while preclinical trials in animals have employed the autologous genetically-corrected cell approach (Shimizu-Motohashi et al., 2019). As discussed with regards to satellite cell- and myoblast-transfer therapies, promise for these approaches has been demonstrated but they have not been overwhelmingly successful thus far.
Satellite cells are an important target to consider in novel therapies since their depletion is a strong potential cause for DMD progression. Therefore, it is essential that we deepen our understanding of the various pathways that regulate their function. One of these regulators is the Notch signaling system, whose dysfunction has been shown to specifically deplete satellite cells, as is seen in DMD (Jiang et al., 2014). The Notch signaling pathway is highly conserved in both vertebrates and invertebrates and regulates cell proliferation, cell fate decisions, and induction of differentiation (Bröhl et al., 2012; Sato et al., 2012; Starosta and Konieczny, 2021). The Notch pathway serves important roles in both embryonic development and in adults (Luo et al., 2005). During development, Notch signaling is crucial for formation of healthy muscle, as elimination of the pathway has been shown to lead to premature differentiation of myogenic progenitors and ultimately to the development of small and weak muscle groups (Bröhl et al., 2012; Mourikis et al., 2012). Furthermore, embryonic knockouts of various components of the Notch pathway genes have been shown to be lethal in mice, deeming it essential for embryonic development (McIntyre et al., 2020). In adults, Notch signaling is crucial for tissue renewal and maintenance in multiple organ systems, with one of its primary roles being regulation of myogenesis and the regeneration of skeletal muscle tissue (Conboy and Rando, 2002; Sato et al., 2012; Coppens et al., 2021). With old age, Notch signaling decreases in skeletal muscle, leading to an age-related decline in the proliferative ability of satellite cells and subsequently to lower regenerative potential (Conboy et al., 2003). The pathway is context-dependent, altering based on factors such as cell type, timing, and mode of signaling, so that it can respond to the current needs of the organism (Kuroda et al., 1999).
In order to promote the regeneration of skeletal muscle in the adult, Notch signaling regulates satellite cells by suppressing myogenic differentiation (Mourikis et al., 2012). When Notch is inactivated, MyoD activates satellite cells causing their differentiation. However, activated Notch promotes the expression of Hes and Hey family genes, particularly Hes1, Hey1, and HeyL (Sakai et al., 2017; D’Souza et al., 2008). These genes suppress MyoD, and therefore suppress differentiation while promoting satellite cell proliferation and self-renewal in order to maintain the satellite cell pool, as depicted in Figure 1 (Gioftsidi et al., 2022). Maintenance of the satellite cell pool then augments the regenerative ability of the skeletal muscle. Unsurprisingly, active Notch signaling is associated with the maintenance of quiescence; in quiescent satellite cells, Notch signaling is high and Notch target genes are more highly expressed to promote self-renewal and proliferation, whilst MyoD and differentiation are suppressed (Fukada et al., 2007; Bjornson et al., 2011; Baghdadi et al., 2018; Kann et al., 2021; Sousa-Victor et al., 2022). Several studies in both mdx mice and in satellite cell-derived myoblasts have also demonstrated that constitutive Notch activation upregulates Pax7, which is associated with satellite cell quiescence and self-renewal (Wen et al., 2012; Jiang et al., 2014).
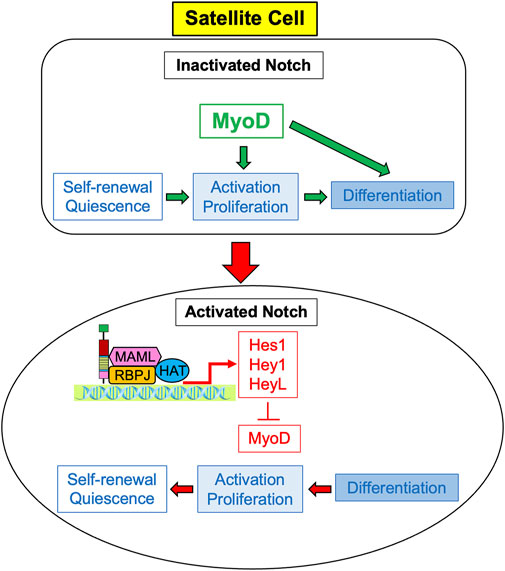
FIGURE 1. Notch signaling induces proliferation of satellite cells. Expression of MyoD causes activation, cell the proliferation and differentiation of satellite cells, so they ultimately cease cell division and differentiate into myocytes. Activated Notch signaling results in the expression of Notch target genes Hes1, Hey 1, and HeyL, which inhibit the transcription of MyoD. Without MyoD, satellite cells predominately proliferate rather than differentiate, and ultimately self-renew to return to quiescent cells.
It should be noted that Notch signaling plays a role in muscle cells beyond just satellite cells, as well. Importantly, Notch1 may be activated in myotubes, which serves to upregulate the expression of Notch ligands and enhance the regenerative capacity of adjacent satellite cells (Bi et al., 2016). This aspect enhances the ability of myotubes as a stem cell niche. Furthermore, it has been shown that in disuse and diabetes, multinucleated myofibers express Notch2 via activation by the Notch ligand Dll4 from the microvascular endothelium (Fujimaki et al., 2022). This ultimately leads to the progression of muscle atrophy seen in these conditions, making it a therapeutic target (Fujimaki et al., 2022).
A molecular scheme for the Notch signaling pathway has been established, lending insight into ways in which it can be targeted in therapies. Notch receptors are large type I transmembrane proteins comprised of an extracellular domain, a single transmembrane domain, and an intracellular region (Figure 2; Sato et al., 2012). There are four different Notch receptors in humans, Notch1-4, which are structurally distinct. Their extracellular domain includes a signal peptide, 29–36 epidermal growth factor (EGF) repeats, three conserved cysteine-rich Lin12-Notch repeats (LNR), and a heterodimerization domain (HD); together, the LNR and HD constitute the negative regulatory region (NRR), which keeps the receptor “off” when there are no ligands present (Sato et al., 2012). The Notch intracellular domain (NICD) consists of a recombining binding protein - J (RBPJ) association module domain (RAM), nuclear localization signals (NLS), seven ankyrin repeats (ANK), a transcriptional activation domain (TAD), and a C-terminal proline-glutamic acid/serine threonine-rich motif (PEST) (Sato et al., 2012). Importantly, Notch3 and Notch4 lack a TAD (Fujimaki et al., 2017).
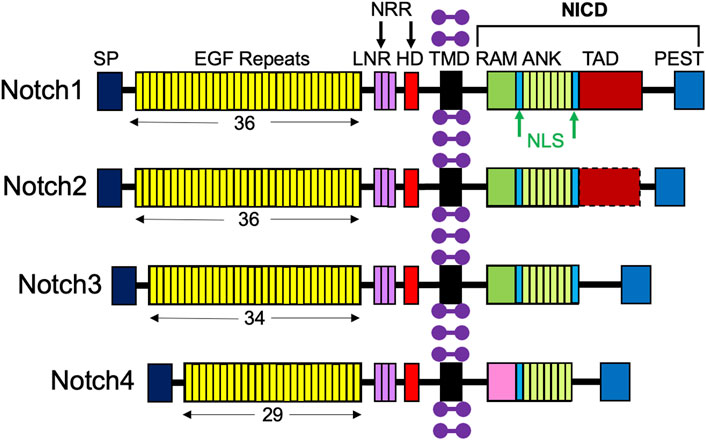
FIGURE 2. Structures of the four Notch receptors. The four Notch receptors identified in humans are depicted with their structural components: SP, signal peptide; EGF, repeats indicated by the number below; LNR, LIN12-Notch repeats; HD, heterodimerization domain; NRR, negative regulatory region; TMD, transmembrane domain; RAM, RBPJ-associated module; NLS, nuclear localization signal; ANK, ankyrin repeats; TAD, transcriptional activation domain; PEST (proline/glutamic acid/serine threonine-rich motif), and NICD, Notch intracellular domain. Purple dumbbells represent surrounding membrane phospholipids.
Like Notch receptors, Notch ligands are also type 1 cell surface proteins with multiple tandem EGF repeats in their extracellular domains (Figure 3; D’Souza et al., 2008). There are five Notch ligands in humans, Jagged1, 2 (Jag1, 2) and Delta-like 1, 3, and 4 (Dll1, 3, 4), which each have unique structures. Notch ligands are presented on neighboring niche cells and activate Notch receptors through a juxtracrine pathway (Coppens et al., 2021; Kann et al., 2021). Of note, there has been evidence to demonstrate cis-inhibitory interactions between Notch ligands and receptors located on the same cell (Coppens et al., 2021).
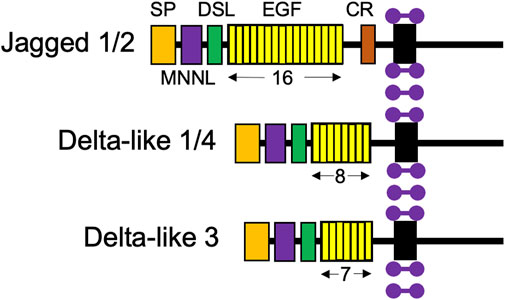
FIGURE 3. Structures of the five Notch ligand proteins. The five Notch ligands identified in humans are depicted with their structural components: SP, signal peptide; MNNL, modular N-terminal Notch ligand; DSL, Delta, Serrate, and LAG-2 domains; EGF, repeats indicated by number below; and CR, cysteine-rich domain. Purple dumbbells represent surrounding membrane phospholipids.
After the ligand binds to the extracellular domain of the Notch receptor, a signal transduction cascade is initiated, known as the canonical Notch signaling pathway, depicted in Figure 4 (McIntyre et al., 2020). Initially, the receptor undergoes a series of proteolytic cleavages, first by an ADAM-family metalloproteinase in the extracellular domain and then by γ-secretase in the transmembrane region, in order to ultimately liberate the Notch intracellular domain (NICD) (McIntyre et al., 2020). This step presents an opportunity for regulation of the Notch pathway; if cells are treated with DAPT, γ-secretase is inhibited, NICD will therefore not be liberated, and transcription of target genes will not be activated (Dong et al., 2021). Once liberated under normal conditions, NICD translocates to the nucleus and associates with a DNA-binding protein that includes RBPJ (Nandagopal et al., 2018; Kann et al., 2021). Of note, RBPJ is also known as the CSL (CBF1/Suppressor of Hairless/LAG-1) (McIntyre et al., 2020). Without NICD, RBPJ binds transcriptional corepressors to inhibit the transcription of target genes (Bjornson et al., 2011). When NICD is present, it binds RBPJ, displaces the corepressors, and recruits coactivators, including Mastermind (Mastermind-like in mammals) and histone acetyltransferases to assemble a transcriptional complex and activate transcription of Notch target genes, including Hes1, Hey1, and HeyL (Figure 1, 2; McIntyre et al., 2020; D’Souza et al., 2008; Bröhl et al., 2012; Sato et al., 2012; Baghdadi et al., 2018). Intracellular Notch activity is regulated by protein turnover that occurs at the PEST at the C-terminal domain of the NICD, as the NICD rapidly degrades when targeted by ubiquitylation (Andersson et al., 2011).
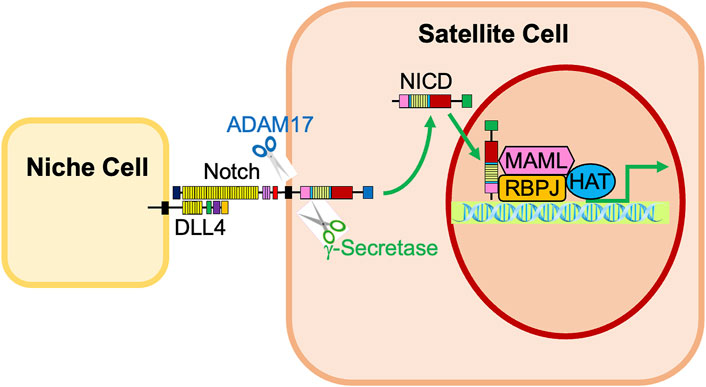
FIGURE 4. Activation of Notch signaling in satellite cells by neighboring niche cells. Notch receptors expressed in satellite cells are activated by interaction with Notch ligands, such as Dll4, on neighboring niche cells. The Notch receptor is cleaved by ADAM17 and γ-secretase, liberating the Notch Intracellular Domain (NICD). The NICD translocates into the nucleus, associates with RBPJ, and recruits transcriptional coactivators, including Mastermind (MAML) and histone acetyltransferases (HAT). This complex promotes the activation of Notch target genes Hes1, Hey1, and HeyL.
Multiple studies have demonstrated that the Notch receptors and ligands are not functionally equivalent, having varied downstream effects on self-renewal, proliferation, and differentiation of satellite cells (Preuße et al., 2015; Nandagopal et al., 2018). These differences are important to understand, especially when considering how to optimally target Notch in novel therapies.
With regards to receptors, prior studies have demonstrated that Notch1 and Notch3 have different and even contradictory roles, with Notch3 acting as a repressor for Notch1 (Low et al., 2018). Whereas activity of the canonical Notch signaling pathway typically leads to an increase in proliferation of satellite cells, experiments have shown that cells only expressing Notch3 proliferate less than controls and, similarly, that deficiency of Notch3 even leads to increased muscle regeneration and higher numbers of satellite cells (Kitamoto and Hanaoka, 2010). Additionally, Fujimaki et al. demonstrated that knockout of either Notch1 or Notch2 in satellite cells in mice prevents proliferation and self-renewal, indicating that they are necessary for the maintenance of the satellite cell pool and adult muscle regeneration (Fujimaki et al., 2017). On the contrary, mice deficient in Notch3 were viable and fertile, and even exhibited elevated numbers of quiescent satellite cells and greater proliferative ability, again suggesting that Notch3 acts to negatively regulate satellite cell proliferation (Fujimaki et al., 2017).
Numerous studies have specifically evaluated the ability of ligands Dll1, Dll4, Jag1, and Jag2 to induce Notch signaling, with results proving to be somewhat contradictory (Sakai et al., 2017). In human cells, in vitro studies have shown that Dll4 and Jag1 more strongly induce Notch signaling than does Dll1 (Sakai et al., 2017). Moreover, differences between Dll1 and Dll4 have been identified with respect to both the manner in which they activate Notch1 and their downstream effects, and it has been established that they are unable to replace the function of one another in many tissues (Preuße et al., 2015). First, Dll4 is presented to satellite cells by adjacent myofibers during quiescence, whereas Dll1 is expressed by differentiating cells that provide a self-renewing signal during regeneration (Yartseva et al., 2020; Kann et al., 2021). Second, Dll1 and Dll4 activate Notch1 in distinct manners: Dll1 activates Notch1 in discrete, frequency-modulated pulses, while Dll4 activates Notch1 in a sustained, amplitude-modulated manner (Nandagopal et al., 2018). Satellite cells are able to discriminate between these two types of signals using dynamics, with the Dll1 signal primarily upregulating Hes1 and the Dll4 signal primarily upregulating Hey1 and HeyL (Nandagopal et al., 2018; Zhang et al., 2021). Different ligands also affect satellite cell activity differently in distinct environments as a stem cell niche. Recent work has demonstrated that proximity to blood vessels is associated with satellite cell self-renewal as a vascular niche, specifically when Dll4 induces quiescence, therefore creating a vascular niche for satellite cell maintenance (Verma et al., 2018).
The absence of Notch signaling precipitates impaired muscle regeneration and phenotypic characteristics reminiscent of DMD (Siebel and Lendahl, 2017). Lin et al. showed that conditional knockout mice whose endogenous Notch signaling was blocked in the satellite cell compartment acquired several features of muscular dystrophy, namely impaired muscle regeneration (Lin et al., 2013). Furthermore, inhibition of Notch signaling in satellite cells led to reduced self-renewal capacity and proliferation as well as increased differentiation of myoblasts, which together exhaust the satellite cell pool (Lin et al., 2013; Verma et al., 2018; Fiore et al., 2020). Together, these results suggest that Notch signaling promotes processes necessary for muscle tissue regeneration, and it is thus likely that impaired Notch signaling contributes to the pathogenic mechanisms of DMD (Tables 1, 2). On that note, insufficient Notch signaling has been directly implicated in DMD, as recent works demonstrated that DMD muscle tissue contains decreased Notch signaling (Table 1; Church et al., 2014; Starosta and Konieczny, 2021), although an earlier work showed upregulation of Notch signaling genes in mdx mice (Table 1; Turk et al., 2005). Jiang et al. showed that satellite cells in the skeletal muscle of mdx mice exhibit reduced expression of multiple Notch receptors, ligands, and target genes such as Hes1, Hey1 and HeyL (Table 1; Jiang et al., 2014). Matrix metalloproteinase-9 (MMP9) is an extracellular protease involved in tissue remodeling, inflammation, and development of interstitials in skeletal muscle (Hindi et al., 2013). Gene knockout of Mmp9 increases the expression of Notch ligands and receptors, and Notch target genes in skeletal muscle of mdx mice, and dramatically improves myopathy and augments myofiber regeneration in mdx mice (Table 1; Hindi et al., 2013).

TABLE 1. Aberrant expression of Notch signaling genes in muscular dystropjy.

TABLE 2. Phenotypical changes of muscular dystrophy via modulation of Notch signaling.
Fiore et al. demonstrated that loss of or pharmacological inhibition of protein kinase C-θ (PKCθ), which modulates various signaling pathways in muscle, led to increased Notch signaling in mdx mice, and consequently improved muscle regeneration and reduced muscle wasting. This study also noted that inhibition of PKCθ acted to upregulate the expression level of Pax7 and Notch1; together, these results demonstrate an avenue for increasing Notch pathway activity to ameliorate the DMD phenotype (Table 1; Fiore et al., 2020).
Specific components of the Notch pathway, namely ligands Jag1 and Jag2, have been implicated in the dysfunction seen in muscular dystrophy. One large study identified the over-representation of pathogenic Jag2 variants in patients with genetically-unsolved muscular dystrophy (Table 2; Coppens et al., 2021). Also, the downregulation of Jag2 in murine myoblasts has been connected with the downregulation of other components of the Notch pathway, suggesting its importance (Coppens et al., 2021). Jag1 has been closely tied to DMD, especially in a GRMD DMD model dog (Table 2; Vieira et al., 2015). Whole genome studies have uncovered an SNP in the promoter region of GRMD Jag1 that creates a novel myogenin binding site (Vieira et al., 2015; Gioftsidi et al., 2022). This allows for greater expression of Jag1 via myogenin, a transcription factor in the MyoD family, which leads to greater proliferative potential (Vieira et al., 2015; Gioftsidi et al., 2022). Not only has greater proliferative capacity been observed with overexpression of Jag1, but this intervention can even ameliorate the DMD phenotype in GRMD, marking it as a potential therapeutic target and indicating that Jag1 may act as a genetic modifier of DMD (Vieira et al., 2015; Gioftsidi et al., 2022). Similarly, overexpression of Jag1 in the sapje, a zebrafish model of DMD, produced a fiber organization resembling the wild-type phenotype (Vieira et al., 2015; Gioftsidi et al., 2022).
Despite promising evidence for restoration of Notch signaling or upregulation of components of the Notch pathway to benefit DMD, other reports have been inconsistent. In one study, genes for Notch signaling were downregulated in mdx satellite cells and Notch1 activation was sufficient to rescue the self-renewal deficiencies of mdx satellite cells, but failed to improve muscle pathology (Tables 1, 2; Jiang et al., 2014). Another study concluded that though Notch inhibition does produce slight functional defects in dystrophic muscle, Notch activation does not significantly improve muscle regeneration in mouse models (Table 2; Church et al., 2014). So, despite some promising experiments, further research is needed to clearly elucidate the link between muscular dystrophy and Notch signaling and set the stage for effective therapies.
To add to these contradictory findings, some studies have found overexpression of Notch signaling to actually be associated with the DMD phenotype, whereas deficiency of Notch signaling is usually thought to precede the satellite cell pool depletion implicated in DMD (Mu et al., 2015). Studies in dystrophin/utrophin dKO mice have exhibited sustained inflammation, impaired muscle regeneration, and rapid depletion and senescence of satellite cells associated with overactivation of Notch signaling genes (Table 1; Mu et al., 2015). The reasoning for this is that Notch signaling may repress myogenesis, which causes it to adversely affect muscle regeneration (Mu et al., 2015). Subsequent experiments showed that intramuscular injection of DAPT, a γ-secretase inhibitor, acted to inhibit Notch signaling and consequently upregulated expression of Pax7 and MyoD, and also improved the histopathology of dystrophic muscle (Table 2; Mu et al., 2015). This suggests that activated Notch signaling may participate in the pathology of DMD, and that downregulation of Notch may be an effective therapeutic approach (Mu et al., 2015). Importantly, these experiments utilized dKO mice, rather than the mdx mouse model that has been more frequently employed. It has been suggested that findings in dKO mice may more accurately reflect phenomena in human DMD, as these models more accurately mimic DMD pathology (Gao et al., 2019).
Notably, overexpression of Notch exhibits context-dependent effects, depending upon whether it is induced in satellite cells or in differentiated cells (Table 2; Bi et al., 2016). For example, Bi et al. found that overexpression of Notch signaling in satellite cells causes dedifferentiation into quiescent satellite cells, which causes defects in muscle growth and regeneration. However, myotube-specific constitutive Notch activation actually improves regeneration in aged and dystrophic muscles (Table 2; Bi et al., 2016).
With regards to DMD treatment development, Notch signaling has been targeted in prior attempts at transplant-based therapeutic approaches. For instance, for engraftment to be successful, cells must be expanded ex vivo since a single donor muscle biopsy does not provide enough cells to meaningfully affect the muscle mass of a DMD patient (Parker et al., 2012). The way in which satellite cells are cultured meaningfully affects their effectiveness after transplant for the repair of dystrophic muscle (Parker et al., 2012; Parker and Tapscott, 2013). Studies have demonstrated that ex vivo expansion on tissue culture plates coated with Dll1-IgG fusion protein inhibits differentiation and increases levels of genes normally expressed in satellite cells, which leads to more effective engraftment and regeneration (Parker et al., 2012; Parker and Tapscott, 2013). Therefore, approaches incorporating components of the Notch pathway may be beneficial in transplant-based therapies.
Furthermore, therapeutic applications of the Notch pathway have been explored with regard to other topics in developmental and cancer biology (Zohorsky and Mequanint, 2021). Some studies have tried delivering soluble ligands in an attempt to activate Notch signaling, though these attempts have been largely unsuccessful (Zohorsky and Mequanint, 2021). Hence, the focus is being directed to the embedded and immobilized delivery of Notch ligands in order to facilitate activation of the endogenous pathway (Zohorsky and Mequanint, 2021). Thus far, Notch ligands have been immobilized to two-dimensional surfaces to examine the behavior of various cell types, including satellite cells (Parker et al., 2012; Safaee et al., 2017; Zohorsky and Mequanint, 2021). This idea has been employed in practice to respond to several maladies. For instance, one study found that the delivery of Jag1 to wounds was able to enhance wound healing (Chigurupati et al., 2007). Another found that delivery of Jag1-containing hydrogels inhibited myofibroblast differentiation in order to counteract cardiac fibrosis and expedite cardiac repair (Boopathy et al., 2015). Utilizing a different approach, another group found that implementing soluble Jag1 via stents could inhibit Notch signaling and subsequently prevent restenosis in vein grafts (Zhou et al., 2015). Hence, research thus far has demonstrated success in targeting the Notch pathway via multiple approaches in order to achieve varied goals, suggesting promise for targeting the Notch pathway in DMD treatments. It should be noted that the Notch pathway has been successfully targeted in cancer therapies as well, given the wealth of Notch research done through the lens of cancer biology and the implication of abnormal activation and expression of the Notch pathway in cancers, especially breast cancer and liver cancer (Zhang et al., 2019; Jia et al., 2021). Over the past 10 years, several new classes of drugs have emerged to therapeutically target Notch in cancer, acting to limit Notch signaling to reduce the pathway’s pro-oncogenic effects (Moore et al., 2020). Delivery of γ-secretase inhibitors such as DAPT has been effective, as well as receptor/ligand antibodies and Notch transcription complex inhibitors (Moore et al., 2020; Jia et al., 2021). A pro-inflammatory cytokine, IL-1β, is known to promote cell cycle progression of non-dystrophic myogenic cells but not DMD myogenic cells. Jag1, which is induced by IL-1β in DMD but not by normal cells, reduces the proliferation and differentiation of myogenic cells. Therefore, up-regulation of Jag1 by IL-1β plays a crucial role in the loss of muscle regeneration capacity of DMD muscles (Table 2; Nagata et al., 2017). Finally, inhibition of Notch and TGF-β promotes myogenic differentiation of human DMD-derived induced Pluripotent Stem Cells (iPSCs) (Table 2; Choi et al., 2012). With respect to muscular dystrophy treatment, the opposite approach would likely be taken, attempting to increase Notch activity rather than decrease it as is done in cancer treatments.
Finally, mutations in several components of the Notch pathway have been implicated in other various hereditary disorders, in addition to muscular dystrophies (Table 2). Missense mutations in Notch ligands Jag1, Dll1, and Dll4 have been linked to Alagille syndrome I (ALGS), nonspecific neurodevelopmental disorders, and Adams-Oliver Syndrome 6 (AOS6), respectively (Coppens et al., 2021). CGG repeat expansions in the noncoding region of the NOTCH2NLC gene, a Notch inhibitor (Fiddes et al., 2018), can enhance Notch signaling. These expansions have been associated with several diseases, including multiple system atrophy (MSA), leukoencephalopathy, and forms of dementia including Alzheimer’s disease and frontotemporal dementia (Fiddes et al., 2018; Ogasawara et al., 2020). CGG repeats of NOTCH2NLC have also been implicated as a causative factor in neuronal intranuclear inclusion disease (NIID) and oculopharyngodistal myopathy (OPDM), both neurodegenerative diseases that involve progressive muscle weakness (Table 2; Ogasawara et al., 2020). Moreover, mutations at splice sites in different Notch receptors and ligands have also been shown to influence Notch signaling and have been implied in various pathologic phenotypes (Vargas-Franco et al., 2022). The splicing mechanism is somewhat unclear, though one study linked hnRNPL, a splicing regulator, to the Notch signaling pathway in several ways; overexpression of a partner of hnRNPL in zebrafish has been shown to destabilize the NICD and inhibit Notch signaling, studies in mice demonstrated that hnRNPL level is increased when Dll3 (a Notch inhibitory ligand) is lost, the fly homolog of hnRNPL (smooth) genetically modifies Notch, and hnRNPL downstream RNA targets include multiple components of the Notch pathway, including receptors Notch2 and Notch3 (Vargas-Franco et al., 2022). Though the specific mechanism remains unknown, possible splicing defects in Notch pathway components have been associated with many specific diseases. ALGS is normally caused by mutations in the Jag1, and heterozygous mutation at a splicing site just before exon 11 has been identified as a culprit (Zhu et al., 2021). Though much less frequently, ALGS is also caused by mutations in the Notch2, and a splice site mutation within the ankyrin repeats leads to decreased Notch signaling, playing a role in the pathology (Kamath et al., 2012). A variant in Notch4 has been associated with schizophrenia, and recent analyses suggested that polymorphisms affecting the alternative splicing of Notch4 may increase schizophrenia susceptibility (Shayevitz et al., 2012). Splice site mutations in Notch3 have been linked to CASADIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) as a potential source of pathology (Lundkvist et al., 2005). A splice site variant of Dll4, which includes an in-frame insertion of 12 bp, has been associated with a variable neurodevelopmental phenotype (Fischer-Zirnsak et al., 2019). Lastly, studies in zebrafish have demonstrated that different splice variants of deltaC, a Notch ligand, cannot replace the function of one another during midline formation and somitogenesis (Mara et al., 2008).
It should be noted that there are several interactions between the Notch pathway and other components in satellite cells, which work together to govern satellite cell proliferation. Multiple EGF-like domains 10 (MEGF10) is a transmembrane receptor expressed in both developing muscle satellite cells and myoblasts, which has exhibited marked similarity to Notch (Saha et al., 2017; Draper et al., 2019; Li et al., 2021). MEGF10 regulates myogenesis in conjunction with the Notch pathway, and MEGF10 deficiency displays several characteristics similar to Notch deficiency (Draper et al., 2019; Li et al., 2021). Biallelic loss of function of MEGF10 causes MEGF10 myopathy or Early-onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD), which involves areflexia, respiratory distress, and dysphagia (Table 2; Logan et al., 2011; Saha et al., 2017). A suggested cellular mechanism for disease in MEGF10 myopathy is slower proliferation and migration of satellite cells (Li et al., 2021). This leads to reduced MyoD expression and subsequent defects in myogenesis, which contributes to impaired skeletal muscle regeneration after injury seen in MEGF10 myopathy (Li et al., 2021). Several similarities and relationships have been identified between MEGF10 and the Notch pathway. First, sequence alignment of mammalian MEGF10 and its drosophila homolog, Drpr, have highlighted the conservation of domains that are characteristic of the Notch ligands (Draper et al., 2019). Drpr deficiency has been shown to lead to muscle abnormalities in flies. In addition, flies that overexpress mouse MEGF10 or fly Drpr display developmental arrest (Draper et al., 2019). In mice, there has been a suggestion for interaction between MEGF10 and the Notch pathway in regulating myogenesis (Table 2; Saha et al., 2017). Further, it has been shown that knockdown of Megf10 results in downregulation of Notch1, Notch2, Notch3, and Hes1, hence suggesting that the extracellular domain of MEGF10 may act as a ligand to activate Notch signaling (Holterman et al., 2007). Also, MEGF10 myopathy involves impaired tyrosine phosphorylation, which causes impaired interaction between MEGF10 and the Notch signaling pathway (Li et al., 2021). Similarly to Notch, deficits seen with MEGF10 myopathy have suggested MEGF10 as a target for potential therapies for muscle diseases; rescuing MEGF10 may have therapeutic implications with regard to ameliorating impaired muscle regeneration (Li et al., 2021).
Protein glycosylation is one of the major regulatory mechanisms of the signaling pathway (Pandey et al., 2021). The extracellular domain of the Notch receptor is modified with O-fucose and O-glucose glycans, and this glycosylation is crucial for the activity of the pathway (Takeuchi et al., 2018). POGLUT1 is involved in the post-translational modification and function of Notch receptors and ligands by reducing O-glucosyltransferase activity on Notch, receptors and ligands and missense mutations in POGLUT1 were identified in a family with autosomal recessive limb-girdle muscular dystrophy (LGMD) (Table 2; Servián-Morilla et al., 2016). Primary myoblasts from patients with POGLUT1 mutations demonstrate slow proliferation, a decreased pool of quiescent satellite cells and a decreased Notch signaling in their muscle tissues (Table 1; Servián-Morilla et al., 2016). Studies have also identified potential interactions between POGLUT1 and Jag1, though the biological relevance is still unclear (Pandey et al., 2021).
α-Dystroglycan is a glycosylated protein and a key component of the dystrophin-glycoprotein complex (DCG), as it is essential for normal basement membrane development and muscle maintenance (Servián-Morilla et al., 2016). Its extracellular subunit is modified with O-linked glycans, which is necessary for the proliferation of satellite cells. One study focused on ribitol-phosphate modification, which is necessary for functional maturation of α-dystroglycan (Tokuoka et al., 2022). Cytidine-diphosphate (CDP-ribitol) is a donor substrate for ribitol-phosphate modification , and proof-of-concept work indicated that supplementation therapy with CDP-ribitol could accelerate development of therapeutic agents for diseases involving glycosylation defects, including DMD (Tokuoka et al., 2022). These results suggest that modulations of Notch protein and pathway may be a promising therapeutic target for muscular dystrophies.
Due to their prevalence and severity, it is imperative that we continue to search for effective therapies for muscular dystrophies, including DMD. The pathogenesis of DMD and other muscular dystrophies has been well-characterized, and there has been a convincing indication that depletion of the satellite cell pool is implicated in the degeneration that defines the muscular dystrophy phenotype. By targeting the Notch signaling pathway, therapies have the potential to selectively increase the proliferation of satellite cells, thereby hopefully ameliorating the muscular dystrophy phenotype and dramatically improving the quality of life for those living with the condition. Prior work has demonstrated some success with satellite cell-transfer therapies, as well as with targeted upregulation of certain components of the Notch signaling pathway, including the ligand Jag1. Further research is necessary for the optimization of these therapies and the exploration of different manners in which the power of the Notch signaling pathway can be harnessed in order to combat muscular dystrophies.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by NIHR01AR062142, NIHR21AR070319, and Regenerative Medicine Minnesota (RMM) Grant to AA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andersson E. R., Sandberg R., Lendahl U. (2011). Notch signaling: Simplicity in design, versatility in function. Development 138, 3593–3612. doi:10.1242/dev.063610
Armstrong N., Schrader R., Fischer R., Crossnohere N. (2022). Duchenne expert physician perspectives on Duchenne newborn screening and early Duchenne care. Am. J. Med. Genet. Part C Semin. Med. Genet. n/a. doi:10.1002/ajmg.c.31993
Asakura A., Komaki M., Rudnicki M. A. (200). Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 68, 245–253. doi:10.1046/j.1432-0436.2001.680412.x
Asakura A., Seale P., Girgis-Gabardo A., Rudnicki M. A. (2002) Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 159, 123–134. doi:10.1083/jcb.200202092
Bagdatlioglu E., Porcari P., Greally E., Blamire A. M., Straub V. W. (2020). Cognitive impairment appears progressive in the mdx mouse. Neuromuscul. Disord. 30, 368–388. doi:10.1016/j.nmd.2020.02.018
Baghdadi M. B., Castel D., Machado L., Fukada S.-I., Birk D. E., Relaix F., et al. (2018). Reciprocal signalling by Notch-Collagen V-CALCR retains muscle stem cells in their niche. Nature 557, 714–718. doi:10.1038/s41586-018-0144-9
Balin S. J., Cascalho M. (2010). The rate of mutation of a single gene. Nucleic Acids Res. 38, 1575–1582. doi:10.1093/nar/gkp1119
Bi P., Yue F., Sato Y., Wirbisky S., Liu W., Shan T., et al. (2016). Stage-specific effects of Notch activation during skeletal myogenesis. Elife 5, e17355, doi:10.7554/eLife.17355
Biressi S., Bjornson C. R. R., Carlig P. M. M., Nishijo K., Keller C., Rando T. A. (2013). Myf5 expression during fetal myogenesis defines the developmental progenitors of adult satellite cells. Dev. Biol. 379, 195–207. doi:10.1016/j.ydbio.2013.04.021
Birnkrant D. J., Bushby K., Bann C. M., Alman B. A., Apkon S. D., Blackwell A., et al. (2018a). Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet. Neurol. 17, 347–361. doi:10.1016/S1474-4422(18)30025-5
Birnkrant D. J., Bushby K., Bann C. M., Apkon S. D., Blackwell A., Brumbaugh D., et al. (2018b). Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet. Neurol. 17, 251–267. doi:10.1016/S1474-4422(18)30024-3
Bjornson C. R. R., Cheung T. H., Liu L., Tripathi P. V., Steeper K. M., Rando T. A. (2011). Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242. doi:10.1002/stem.773
Boldrin L., Zammit P. S., Morgan J. E. (2015). Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 14, 20–29. doi:10.1016/j.scr.2014.10.007
Boopathy A. V., Martinez M. D., Smith A. W., Brown M. E., García A. J., Davis M. E. (2015). Intramyocardial delivery of notch ligand-containing hydrogels improves cardiac function and angiogenesis following infarction. Tissue Eng. Part A 21, 2315–2322. doi:10.1089/ten.TEA.2014.0622
Brinkmeyer-Langford C., Chu C., Balog-Alvarez C., Yu X., Cai J. J., Nabity M., et al. (2018). Expression profiling of disease progression in canine model of Duchenne muscular dystrophy. PLoS One 13, e0194485, doi:10.1371/journal.pone.0194485
Bröhl D., Vasyutina E., Czajkowski M. T., Griger J., Rassek C., Rahn H.-P., et al. (2012). Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on notch signals. Dev. Cell 23, 469–481. doi:10.1016/j.devcel.2012.07.014
Chang N. C., Chevalier F. P., Rudnicki M. A. (2016). Satellite cells in muscular dystrophy - lost in polarity. Trends Mol. Med. 22, 479–496. doi:10.1016/j.molmed.2016.04.002
Chigurupati S., Arumugam T. V., Son T. G., Lathia J. D., Jameel S., Mughal M. R., et al. (2007). Involvement of notch signaling in wound healing. PLoS One 2, e1167, doi:10.1371/journal.pone.0001167
Choi I. Y., Lim H. T., Che Y. H., Lee G., Kim Y. J. (2012). Inhibition of the combinatorial signaling of transforming growth factor-beta and NOTCH promotes myotube formation of human pluripotent stem cell-derived skeletal muscle progenitor cells. Cells. 10, 1649, doi:10.3390/cells10071649
Church J. E., Trieu J., Chee A., Naim T., Gehrig S. M., Lamon S., et al. (2014). Alterations in Notch signalling in skeletal muscles from mdx and dko dystrophic mice and patients with Duchenne muscular dystrophy. Exp. Physiol. 99, 675–687. doi:10.1113/expphysiol.2013.077255
Conboy I. M., Conboy M. J., Smythe G. M., Rando T. A. (2003). Notch-mediated restoration of regenerative potential to aged muscle. Science 302, 1575–1577. doi:10.1126/science.1087573
Conboy I. M., Rando T. A. (2002). The regulation of notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 3, 397–409. doi:10.1016/s1534-5807(02)00254-x
Coppens S., Barnard A. M., Puusepp S., Pajusalu S., Õunap K., Vargas-Franco D., et al. (2021). A form of muscular dystrophy associated with pathogenic variants in JAG2. Am. J. Hum. Genet. 108, 1164–1856. doi:10.1016/j.ajhg.2021.04.018
Cossu G., Sampaolesi M. (2007). New therapies for Duchenne muscular dystrophy: Challenges, prospects and clinical trials. Trends Mol. Med. 13, 520–526. doi:10.1016/j.molmed.2007.10.003
Crisafulli S., Sultana J., Fontana A., Salvo F., Messina S., Trifirò G. (2020). Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 15, 141, doi:10.1186/s13023-020-01430-8
Dong Z., Huo J., Liang A., Chen J., Chen G., Liu D. (2021). Gamma-Secretase Inhibitor (DAPT), a potential therapeutic target drug, caused neurotoxicity in planarian regeneration by inhibiting Notch signaling pathway. Sci. Total Environ. 781, 146735, doi:10.1016/j.scitotenv.2021.146735
Draper I., Saha M., Stonebreaker H., Salomon R. N., Matin B., Kang P. B. (2019). The impact of Megf10/Drpr gain-of-function on muscle development in Drosophila. FEBS Lett. 593, 680–696. doi:10.1002/1873-3468.13348
D’Souza B., Miyamoto A., Weinmaster G. (2008). The many facets of Notch ligands. Oncogene 27, 5148–5167. doi:10.1038/onc.2008.229
Duan D., Goemans N., Takeda S., Mercuri E., Aartsma-Rus A. (2021). Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 7, 13, doi:10.1038/s41572-021-00248-3
Dumont N. A., Rudnicki M. A. (2016). Targeting muscle stem cell intrinsic defects to treat Duchenne muscular dystrophy. Npj Regen. Med. 1, 16006, doi:10.1038/npjregenmed.2016.6
Dumont N. A., Wang Y. X., Rudnicki M. A. (2015). Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 142, 1572–1581. doi:10.1242/dev.114223
Fiddes I. T., Lodewijk G. A., Mooring M., Bosworth C. M., Ewing A. D., Mantalas G. L., et al. (2018). Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173, 1356–1369. doi:10.1016/j.cell.2018.03.051e22
Fiore P. F., Benedetti A., Sandonà M., Madaro L., De Bardi M., Saccone V., et al. (2020). Lack of PKCθ promotes regenerative ability of muscle stem cells in chronic muscle injury. Int. J. Mol. Sci. 21, E932, doi:10.3390/ijms21030932
Fischer-Zirnsak B., Segebrecht L., Schubach M., Charles P., Alderman E., Brown K., et al. (2019). Haploinsufficiency of the notch ligand DLL1 causes variable neurodevelopmental disorders. Am. J. Hum. Genet. 105, 631–639. doi:10.1016/j.ajhg.2019.07.002
Frontera W. R., Ochala J. (2015). Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 96, 183–195. doi:10.1007/s00223-014-9915-y
Fuchs E., Blau H. M. (2020). Tissue stem cells: Architects of their niches. Cell Stem Cell 27, 532–556. doi:10.1016/j.stem.2020.09.011
Fujimaki S., Matsumoto T., Muramatsu M., Nagahisa H., Horii N., Seko D., et al. (2022). The endothelial Dll4–muscular Notch2 axis regulates skeletal muscle mass. Nat. Metab. 4, 180–189. doi:10.1038/s42255-022-00533-9
Fujimaki S., Seko D., Kitajima Y., Yoshioka K., Tsuchiya Y., Masuda S., et al. (2017). Notch1 and Notch2 coordinately regulate stem cell function in the quiescent and activated states of muscle satellite cells. Stem Cells 36, 278–285. doi:10.1002/stem.2743
Fukada S., Uezumi A., Ikemoto M., Masuda S., Segawa M., Tanimura N., et al. (2007). Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 25, 2448–2459. doi:10.1634/stemcells.2007-0019
Gao Q. Q., McNally E. M. (2015). The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 5, 1223–1239. doi:10.1002/cphy.c140048
Gao X., Tang Y., Amra S., Sun X., Cui Y., Cheng H., et al. (2019). Systemic investigation of bone and muscle abnormalities in dystrophin/utrophin double knockout mice during postnatal development and the mechanisms. Hum. Mol. Genet. 28, 1738–1751. doi:10.1093/hmg/ddz012
Gayraud-Morel B., Chrétien F., Jory A., Sambasivan R., Negroni E., Flamant P., et al. (2012). Myf5 haploinsufficiency reveals distinct cell fate potentials for adult skeletal muscle stem cells. J. Cell Sci. 125, 1738–1749. doi:10.1242/jcs.097006
Gioftsidi S., Relaix F., Mourikis P. (2022). The Notch signaling network in muscle stem cells during development, homeostasis, and disease. Skelet. Muscle 12, 9, doi:10.1186/s13395-022-00293-w
Guiraud S., Aartsma-Rus A., Vieira N. M., Davies K. E., van Ommen G.-J. B., Kunkel L. M. (2015). The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genomics Hum. Genet. 16, 281–308. doi:10.1146/annurev-genom-090314-025003
Hindi S. M., Shin J., Ogura Y., Li H., Kumar A. (2013). Matrix metalloproteinase-9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS One. 8, e72121, doi:10.1371/journal.pone.0072121
Holterman C. E., Le Grand F., Kuang S., Seale P., Rudnicki M. A. (2007). Megf10 regulates the progression of the satellite cell myogenic program. J. Cell Biol. 179, 911–922. doi:10.1083/jcb.200709083
Iftikhar M., Frey J., Shohan M. J., Malek S., Mousa S. A. (2021). Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol. Ther. 220, 107719, doi:10.1016/j.pharmthera.2020.107719
Jia H., Wang Z., Zhang J., Feng F. (2021). γ-Secretase inhibitors for breast cancer and hepatocellular carcinoma: From mechanism to treatment. Life Sci. 268, 119007, doi:10.1016/j.lfs.2020.119007
Jiang C., Wen Y., Kuroda K., Hannon K., Rudnicki M., Kuang S. (2014). Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis. Model. Mech. 7, 997, 1004. doi:10.1242/dmm.015917
Joanisse S., Nederveen J. P., Snijders T., McKay B. R., Parise G. (2017). Skeletal muscle regeneration, repair and remodelling in aging: The importance of muscle stem cells and vascularization. Gerontology 63, 91–100. doi:10.1159/000450922
Kamath B. M., Bauer R. C., Loomes K. M., Chao G., Gerfen J., Hutchinson A., et al. (2012). NOTCH2 mutations in Alagille syndrome. J. Med. Genet. 49, 138–144. doi:10.1136/jmedgenet-2011-100544
Kanisicak O., Mendez J. J., Yamamoto S., Yamamoto M., Goldhamer D. J. (2009). Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD., Dev. Biol. 332, 131–141. doi:10.1016/j.ydbio.2009.05.554
Kann A. P., Hung M., Krauss R. S. (2021). Cell–cell contact and signaling in the muscle stem cell niche. Curr. Opin. Cell Biol. 73, 78–83. doi:10.1016/j.ceb.2021.06.003
Kitamoto T., Hanaoka K. (2010). Notch3 null mutation in mice causes muscle hyperplasia by repetitive muscle regeneration. Stem Cells 28, 2205–2216. doi:10.1002/stem.547
Kornegay J. N. (2017). The golden retriever model of Duchenne muscular dystrophy. Skelet. Muscle 7, 9, doi:10.1186/s13395-017-0124-z
Kottlors M., Kirschner J. (2010). Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 340, 541–548. doi:10.1007/s00441-010-0976-6
Kuang S., Kuroda K., Le Grand F., Rudnicki M. A. (2007). Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010. doi:10.1016/j.cell.2007.03.044
Kuroda K., Tani S., Tamura K., Minoguchi S., Kurooka H., Honjo T. (1999). Delta-induced notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis*. J. Biol. Chem. 274, 7238–7244. doi:10.1074/jbc.274.11.7238
Law P., Bertorini T., Goodwin T., Chen M., Fang Q., Li H.-J., et al. (1990). Dystrophin production induced by myoblast transfer therapy in Duchenne muscular dystrophy. Lancet 336, 114–115. doi:10.1016/0140-6736(90)91628-n
Li C., Vargas-Franco D., Saha M., Davis R. M., Manko K. A., Draper I., et al. (2021). Megf10 deficiency impairs skeletal muscle stem cell migration and muscle regeneration. FEBS Open Bio 11, 114–123. doi:10.1002/2211-5463.13031
Lin S., Shen H., Jin B., Gu Y., Chen Z., Cao C., et al. (2013). Brief report: Blockade of Notch signaling in muscle stem cells causes muscular dystrophic phenotype and impaired muscle regeneration. Stem Cells 31, 823–828. doi:10.1002/stem.1319
Liu Q., Deng J., Qiu Y., Gao J., Li J., Guan L., et al. (2021). Non-coding RNA basis of muscle atrophy. Mol. Ther. Nucleic Acids 26, 1066–1078. doi:10.1016/j.omtn.2021.10.010
Logan C. V., Lucke B., Pottinger C., Abdelhamed Z. A., Parry D. A., Szymanska K., et al. (2011). Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat. Genet. 43, 1189–1192. doi:10.1038/ng.995
Lovering R. M., Porter N. C., Bloch R. J. (2005). The muscular dystrophies: From genes to therapies. Phys. Ther. 85, 1372–1388. doi:10.1093/ptj/85.12.1372
Low S., Barnes J. L., Zammit P. S., Beauchamp J. R. (2018). Delta-like 4 activates notch 3 to regulate self-renewal in skeletal muscle stem cells. Stem Cells 36, 458–466. doi:10.1002/stem.2757
Lundkvist J., Zhu S., Hansson E. M., Schweinhardt P., Miao Q., Beatus P., et al. (2005). Mice carrying a R142C Notch 3 knock-in mutation do not develop a CADASIL-like phenotype. Genesis 41, 13–22. doi:10.1002/gene.20091
Luo D., Renault V. M., Rando T. A. (2005). The regulation of Notch signaling in muscle stem cell activation and postnatal myogenesis. Semin. Cell Dev. Biol. 16, 612–622. doi:10.1016/j.semcdb.2005.07.002
Mah J. K. (2016). Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr. Dis. Treat. 12, 1795–1807. doi:10.2147/NDT.S93873
Mara A., Schroeder J., Holley S. A. (2008). Two deltaC splice-variants have distinct signaling abilities during somitogenesis and midline patterning. Dev. Biol. 318, 126–132. doi:10.1016/j.ydbio.2008.03.009
McIntyre B., Asahara T., Alev C. (2020). Overview of basic mechanisms of Notch signaling in development and disease BT - Notch signaling in embryology and cancer: Molecular biology of Notch signaling. J. Reichrath, and S. Reichrath, eds. (Cham: Springer International Publishing), pp. 9–27. doi:10.1007/978-3-030-36422-9_2
Mohamadian M., Rastegar M., Pasamanesh N., Ghadiri A., Ghandil P., Naseri M. (2022). Clinical and molecular spectrum of muscular dystrophies (MDs) with intellectual disability (ID): A comprehensive overview. J. Mol. Neurosci. 72, 9–23. doi:10.1007/s12031-021-01933-4
Moore G., Annett S., McClements L., Robson T. (2020). Top notch targeting strategies in cancer: A detailed overview of recent insights and current perspectives. Cells 9. 1503. doi:10.3390/cells9061503
Morgan J. E., Zammit P. S. (2010). Direct effects of the pathogenic mutation on satellite cell function in muscular dystrophy. Exp. Cell Res. 316, 3100–3108. doi:10.1016/j.yexcr.2010.05.014
Motohashi N., Shimizu-Motohashi Y., Roberts T. C., Aoki Y. (2019). Potential therapies using myogenic stem cells combined with bio-engineering approaches for treatment of muscular dystrophies. Cells 8, 1066, doi:10.3390/cells8091066
Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V., Tajbakhsh S. (2012). A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30, 243–252. doi:10.1002/stem.775
Mu X., Tang Y., Lu A., Takayama K., Usas A., Wang B., et al. (2015). The role of Notch signaling in muscle progenitor cell depletion and the rapid onset of histopathology in muscular dystrophy. Hum. Mol. Genet. 24, 2923–2937. doi:10.1093/hmg/ddv055
Nagata Y., Kiyono T., Okamura K., Goto Y. I., Matsuo M., Ikemoto-Uezumi M., et al. (2017). Interleukin-1beta (IL-1β)-induced Notch ligand Jagged1 suppresses mitogenic action of IL-1β on human dystrophic myogenic cells. PLoS One. 12, e0188821, doi:10.1371/journal.pone.0188821
Nakamura A., Fueki N., Shiba N., Motoki H., Miyazaki D., Nishizawa H., et al. (2016). Deletion of exons 3-9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 61, 663–667. doi:10.1038/jhg.2016.28
Nandagopal N., Santat L. A., LeBon L., Sprinzak D., Bronner M. E., Elowitz M. B. (2018). Dynamic ligand discrimination in the notch signaling pathway. Cell 172, 869–880. doi:10.1016/j.cell.2018.01.002e19
Ogasawara M., Iida A., Kumutpongpanich T., Ozaki A., Oya Y., Konishi H., et al. (2020). CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol. Commun. 8, 204. doi:10.1186/s40478-020-01084-4
Pandey A., Niknejad N., Jafar-Nejad H. (2021). Multifaceted regulation of Notch signaling by glycosylation. Glycobiology 31, 8–28. doi:10.1093/glycob/cwaa049
Parker M. H., Loretz C., Tyler A. E., Duddy W. J., Hall J. K., Olwin B. B., et al. (2012). Activation of Notch signaling during ex vivo expansion maintains donor muscle cell engraftment. Stem Cells 30, 2212–2220. doi:10.1002/stem.1181
Parker M. H., Tapscott S. J. (2013). Expanding donor muscle-derived cells for transplantation. Curr. Protoc. Stem Cell Biol. Chapter 2, Unit 2C.4. doi:10.1002/9780470151808.sc02c04s25Chapter 2, Unit-2C.4
Plantié E., Migocka-Patrzałek M., Daczewska M., Jagla K. (2015). Model organisms in the fight against muscular dystrophy: Lessons from drosophila and zebrafish. Molecules 20, 6237–6253. doi:10.3390/molecules20046237
Preuße K., Tveriakhina L., Schuster-Gossler K., Gaspar C., Rosa A. I., Henrique D., et al. (2015). Context-dependent functional divergence of the notch ligands DLL1 and DLL4 in vivo. PLoS Genet. 11, e1005328. doi:10.1371/journal.pgen.1005328
Ricotti V., Mandy W. P. L., Scoto M., Pane M., Deconinck N., Messina S., et al. (2016). Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev. Med. Child. Neurol. 58, 77–84. doi:10.1111/dmcn.12922
Rocheteau P., Gayraud-Morel B., Siegl-Cachedenier I., Blasco M. A., Tajbakhsh S. (2012). A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 148, 112–125. doi:10.1016/j.cell.2011.11.049
Sacco A., Mourkioti F., Tran R., Choi J., Llewellyn M., Kraft P., et al. (2010). Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143, 1059–1071. doi:10.1016/j.cell.2010.11.039
Safaee H., Bakooshli M. A., Davoudi S., Cheng R. Y., Martowirogo A. J., Li E. W., et al. (2017). Tethered jagged-1 synergizes with culture substrate stiffness to modulate notch-induced myogenic progenitor differentiation. Cell. Mol. Bioeng. 10, 501–513. doi:10.1007/s12195-017-0506-7
Saha M., Mitsuhashi S., Jones M. D., Manko K., Reddy H. M., Bruels C. C., et al. (2017). Consequences of MEGF10 deficiency on myoblast function and Notch1 interactions. Hum. Mol. Genet. 26, 2984–3000. doi:10.1093/hmg/ddx189
Sakai H., Fukuda S., Nakamura M., Uezumi A., Noguchi Y.-T., Sato T., et al. (2017). Notch ligands regulate the muscle stem-like state ex vivo but are not sufficient for retaining regenerative capacity. PLoS One 12, e0177516. doi:10.1371/journal.pone.0177516
Sato C., Zhao G., Ilagan M. X. G. (2012). An overview of notch signaling in adult tissue renewal and maintenance. Curr. Alzheimer Res. 9, 227–240. doi:10.2174/156720512799361600
Servián-Morilla E., Takeuchi H., Lee T. V., Clarimon J., Mavillard F., Area-Gómez E., et al. (2016). A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol. Med. 8, 1289–1309. doi:10.15252/emmm.201505815
Shayevitz C., Cohen O. S., Faraone S. V., Glatt S. J. (2012). A re-review of the association between the NOTCH4 locus and schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 477–483. doi:10.1002/ajmg.b.32050
Shimizu-Motohashi Y., Komaki H., Motohashi N., Takeda S., Yokota T., Aoki Y. (2019). Restoring dystrophin expression in Duchenne muscular dystrophy: Current status of therapeutic approaches. J. Pers. Med. 9, E1. doi:10.3390/jpm9010001
Siebel C., Lendahl U. (2017). Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294. doi:10.1152/physrev.00005.2017
Sousa-Victor P., García-Prat L., Muñoz-Cánoves P. (2022). Control of satellite cell function in muscle regeneration and its disruption in ageing. Nat. Rev. Mol. Cell Biol. 23, 204–226. doi:10.1038/s41580-021-00421-2
Starosta A., Konieczny P. (2021). Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 78, 4867–4891. doi:10.1007/s00018-021-03821-x
Sun C., Shen L., Zhang Z., Xie X. (2020). Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes (Basel). 11.doi:10.3390/genes11080837
Takeuchi H., Schneider M., Williamson D. B., Ito A., Takeuchi M., Handford P. A., et al. (2018). Two novel protein O-glucosyltransferases that modify sites distinct from POGLUT1 and affect Notch trafficking and signaling. Proc. Natl. Acad. Sci. U. S. A. 115, E8395–E8402. doi:10.1073/pnas.1804005115
Tedesco F. S., Dellavalle A., Diaz-Manera J., Messina G., Cossu G. (2010). Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 120, 11–19. doi:10.1172/JCI40373
Tokuoka H., Imae R., Nakashima H., Manya H., Masuda C., Hoshino S., et al. (2022). CDP-ribitol prodrug treatment ameliorates ISPD-deficient muscular dystrophy mouse model. Nat. Commun. 13, 1847. doi:10.1038/s41467-022-29473-4
Tominari T., Aoki Y. (2022). Clinical development of novel therapies for Duchenne muscular dystrophy—current and future. Neurol. Clin. Neurosci., 1–8. doi:10.1111/ncn3.12656
Turk R., Sterrenburg E., de Meijer E. J., van Ommen G. J., den Dunnen J. T., 't Hoen P. A. (2005). Muscle regeneration in dystrophin-deficient mdx mice studied by gene expression profiling. BMC Genomics. 6, 98, doi:10.1186/1471-2164-6-98
Vargas-Franco D., Kalra R., Draper I., Pacak C. A., Asakura A., Kang P. B. (2022). The Notch signaling pathway in skeletal muscle health and disease. Muscle Nerve 2022, 1–15. doi:10.1002/mus.27684
Venugopal V., Pavlakis S. (2022). Duchenne muscular dystrophy. (Treasure Island, FL: StatPearls Publishing)
Verma M., Asakura Y., Murakonda B. S. R., Pengo T., Latroche C., Chazaud B., et al. (2018). Muscle satellite cell cross-talk with a vascular niche maintains quiescence via VEGF and notch signaling. Cell Stem Cell 23, 530–543. doi:10.1016/j.stem.2018.09.007e9
Vieira N. M., Elvers I., Alexander M. S., Moreira Y. B., Eran A., Gomes J. P., et al. (2015). Jagged 1 rescues the Duchenne muscular dystrophy phenotype. Cell 163, 1204–1213. doi:10.1016/j.cell.2015.10.049
Waldrop M. A., Gumienny F., El Husayni S., Frank D. E., Weiss R. B., Flanigan K. M. (2018). Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul. Disord. 28, 116–121. doi:10.1016/j.nmd.2017.11.007
Wen Y., Bi P., Liu W., Asakura A., Keller C., Kuang S. (2012). Constitutive Notch activation upregulates Pax7 and promotes the self-renewal of skeletal muscle satellite cells. Mol. Cell. Biol. 32, 2300–2311. doi:10.1128/MCB.06753-11
Wilschut K. J., Ling V. B., Bernstein H. S. (2012). Concise review: Stem cell therapy for muscular dystrophies. Stem Cells Transl. Med. 1, 833–842. doi:10.5966/sctm.2012-0071
Yao S., Chen Z., Yu Y., Zhang N., Jiang H., Zhang G., et al. (2021). Current pharmacological strategies for Duchenne muscular dystrophy. Front. Cell Dev. Biol. 9, 689533, doi:10.3389/fcell.2021.689533
Yartseva V., Goldstein L. D., Rodman J., Kates L., Chen M. Z., Chen Y.-J. J., et al. (2020). Heterogeneity of satellite cells implicates DELTA1/NOTCH2 signaling in self-renewal. Cell Rep. 30, 1491–1503. doi:10.1016/j.celrep.2019.12.100
Yucel N., Chang A. C., Day J. W., Rosenthal N., Blau H. M. (2018). Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 3, 4, doi:10.1038/s41536-018-0045-4
Zhang Y., Lahmann I., Baum K., Shimojo H., Mourikis P., Wolf J., et al. (2021). Author Correction: Oscillations of Delta-like1 regulate the balance between differentiation and maintenance of muscle stem cells. Nat. Commun. 12, 1766, doi:10.1038/s41467-021-22144-w
Zhang Y., Xie Z.-Y., Guo X.-T., Xiao X.-H., Xiong L.-X. (2019). Notch and breast cancer metastasis: Current knowledge, new sights and targeted therapy. Oncol. Lett. 18, 2743–2755. doi:10.3892/ol.2019.10653
Zhou X., Xiao Y., Mao Z., Huang J., Geng Q., Wang W., et al. (2015). Soluble Jagged-1 inhibits restenosis of vein graft by attenuating Notch signaling. Microvasc. Res. 100, 9–16. doi:10.1016/j.mvr.2015.01.009
Zhu W., Cheng Y.-S., Xu M., Farkhondeh A., Beers J., Zou J., et al. (2021). Generation of Alagille syndrome derived induced pluripotent stem cell line carrying heterozygous mutation in the JAGGED-1 gene at splicing site (Chr20: 10, 629, 709C>A) before exon 11. Stem Cell Res. 53, 102366, doi:10.1016/j.scr.2021.102366
Keywords: notch, muscular dystrophy, muscle regeneration, satellite cell, muscle stem cell
Citation: Den Hartog L and Asakura A (2022) Implications of notch signaling in duchenne muscular dystrophy. Front. Physiol. 13:984373. doi: 10.3389/fphys.2022.984373
Received: 01 July 2022; Accepted: 05 September 2022;
Published: 27 September 2022.
Edited by:
Norio Fukuda, Jikei University School of Medicine, JapanReviewed by:
Boel De Paepe, Ghent University, BelgiumCopyright © 2022 Den Hartog and Asakura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Asakura, YXNha3VyYUB1bW4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.