 Jaromir Myslivecek
Jaromir Myslivecek- Institute of Physiology, 1st Faculty of Medicine, Charles University, Prague, Czechia
Muscarinic receptors (mAChRs) are typical members of the G protein-coupled receptor (GPCR) family and exist in five subtypes from M1 to M5. Muscarinic receptor subtypes do not sufficiently differ in affinity to orthosteric antagonists or agonists; therefore, the analysis of receptor subtypes is complicated, and misinterpretations can occur. Usually, when researchers mainly specialized in CNS and peripheral functions aim to study mAChR involvement in behavior, learning, spinal locomotor networks, biological rhythms, cardiovascular physiology, bronchoconstriction, gastrointestinal tract functions, schizophrenia, and Parkinson’s disease, they use orthosteric ligands and they do not use allosteric ligands. Moreover, they usually rely on manufacturers’ claims that could be misleading. This review aimed to call the attention of researchers not deeply focused on mAChR pharmacology to this fact. Importantly, limited selective binding is not only a property of mAChRs but is a general attribute of most neurotransmitter receptors. In this review, we want to give an overview of the most common off-targets for established mAChR ligands. In this context, an important point is a mention the tremendous knowledge gap on off-targets for novel compounds compared to very well-established ligands. Therefore, we will summarize reported affinities and give an outline of strategies to investigate the subtype’s function, thereby avoiding ambiguous results. Despite that, the multitargeting nature of drugs acting also on mAChR could be an advantage when treating such diseases as schizophrenia. Antipsychotics are a perfect example of a multitargeting advantage in treatment. A promising strategy is the use of allosteric ligands, although some of these ligands have also been shown to exhibit limited selectivity. Another new direction in the development of muscarinic selective ligands is functionally selective and biased agonists. The possible selective ligands, usually allosteric, will also be listed. To overcome the limited selectivity of orthosteric ligands, the recommended process is to carefully examine the presence of respective subtypes in specific tissues via knockout studies, carefully apply “specific” agonists/antagonists at appropriate concentrations and then calculate the probability of a specific subtype involvement in specific functions. This could help interested researchers aiming to study the central nervous system functions mediated by the muscarinic receptor.
1 Introduction
It is widely accepted that many neurotransmitters bind to different receptor types. As an example, glutamate binds to metabotropic glutamate receptors (mGlu) and to a group of ligand-gated ion channels (N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and kainite receptors). Serotonin binds mainly to G protein-coupled receptors (GPCRs), but 5-HT3 receptors are ligand-gated ion channels. Similarly, acetylcholine (ACh) binds to nicotinic receptors (NRs), which are ligand-gated ion channels, and to muscarinic receptors (mAChRs), which are typical G protein-coupled receptors.
Commonly, less is known about the properties of ligands used in research on the central nervous system (CNS) and peripheral tissue functions. In this review, we will focus on mAChRs. As an example, the recent translational research of human epithelial ovarian carcinoma have described the role of M2 mAChRs in M2 cell growth and survival (Taggi et al., 2022). The authors have used arecaidine propargyl ester as M2 mAChR preferential agonist. However, as it can be deduced from Table 1, arecaidine propargyl ester binds with similar affinity to M2-M4 mAChRs, slightly better that to M1 mAChRs. These conclusions could be therefore misleading.
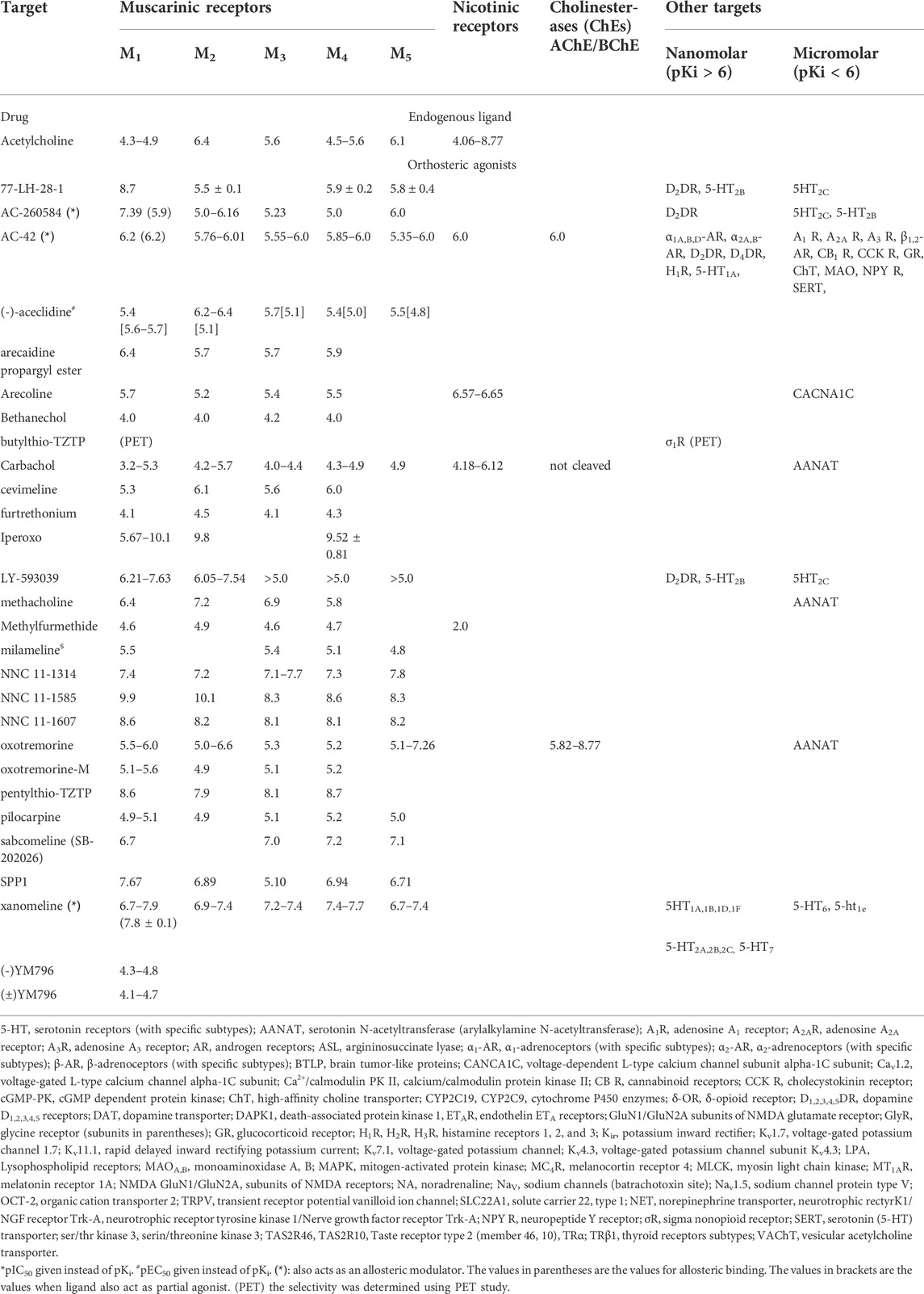
TABLE 1. Effects of orthosteric agonists (+endogenous ligand) on muscarinic receptors and other targets. The compounds are listed alphabetically. The numbers indicate pKi values. Please note that a higher pKi indicates higher affinity. Unavailable data are shown as blank spaces. The affinities of nicotinic receptors are not divided among specific subunits. The affinities of other, noncholinergic targets are divided into nanomolar (pKi>6) and micromolar (pKi<6) groups. The categorization of pKi is compromise between extremely large Table and fast orientation how much are other targets are affected in specific concentration of ligand used (see also examples in Chapter 5). Data are from or adapted from (Pascuzzo et al., 1984; Eglen and Whiting, 1987; Buckley et al., 1989; Buckley et al., 1990; Hudkins et al., 1991; Wess et al., 1991; Bolden et al., 1992; Kashihara et al., 1992; Sowell et al., 1992; Doods et al., 1993; Shen et al., 1993; Stanton et al., 1993; Doods et al., 1994; Ferrari-Dileo et al., 1994; Kleinschroth et al., 1995; Waelbroeck et al., 1996; McKenna et al., 1997; Boess et al., 1998; Bolognesi et al., 1998; Cantı́ et al., 1998; Caulfield and Birdsall, 1998; Lazareno et al., 1998; Choppin et al., 1999; Sánchez and Hyttel, 1999; Eglen and Nahorski, 2000; Kozlowski et al., 2000; Lazareno et al., 2000; Becerra et al., 2001; Dhein et al., 2001; Huang et al., 2001; Lockhart et al., 2001; Apelt et al., 2002; Carlsson et al., 2002; Harvey et al., 2002; Cheng et al., 2002; Lazareno et al., 2002; Böhme et al., 2003; Samochocki et al., 2003; Sur et al., 2003; Jakubik et al., 2004; Kobayashi et al., 2004; Wang et al., 2004; Ghoneim et al., 2006; Spalding et al., 2006; Butini et al., 2008; Langmead et al., 2008; Bridges et al., 2009; Heinrich et al., 2009; Prat et al., 2009; von Coburg et al., 2009; Bridges et al., 2010a; Harada et al., 2010; Hern et al., 2010; Lange et al., 2010; Rook et al., 2010; Sinha et al., 2010; Watt et al., 2011; Daval et al., 2012; Samadi et al., 2012; Sykes et al., 2012; Arunotayanun et al., 2013; Nenasheva et al., 2013; Salmon et al., 2013; Croy et al., 2016; Gaulton et al., 2016; Alexander S. P. et al., 2017; Alexander S. P. H. et al., 2017; Chen et al., 2017; Carr et al., 2018; Hegde et al., 2018; Broad et al., 2019; Myslivecek, 2019; Xu et al., 2019; Olianas et al., 2020; Okimoto et al., 2021) and the IUPHAR/BPS Guide to Pharmacology (www.guidetophamacology.org). For specific ligand references see text. The activity represents the main effect of a specific ligand.
It could be surprising to mention that majority of GPCRs can be activated by multiple ligands (Vass et al., 2018), e.g., 1312 potentially active ligands (searched from ChEMBL database (Gaulton et al., 2016), with Ki<1 µmol/L) exist for M1 mAChRs, of which 930 are active at other GPCRs (i.e., 70%). Similarly, among 1336 potentially active ligands for M2, 974 are active at other GPCRs (i.e., 73%), and the corresponding numbers for M3 mAChRs and M4 mAChRs are 1384 vs. 855 (i.e., 62%) and 466 vs. 387 (i.e., 83%), respectively. Thus, these authors talk about the interactome.
In general, the off-target is defined as targets (proteins or other molecules in the body) other than those for which the drug was meant to bind. This can lead to unexpected side effects that may be both harmful and positive. Learning about the off-target effects of drugs may help in drug development. Alternatively, these drugs are designated as multitargeting drugs.
Muscarinic receptors can provide good examples of ligands exhibiting multitarget properties. Typically, mAChR subtypes do not greatly differ in their affinity to orthosteric antagonists (Farar and Myslivecek, 2016; Valuskova et al., 2018a; Myslivecek, 2019) or agonists (Alexander S. P. et al., 2017), which complicates the analysis determining receptor subtypes involved in the specific function and misinterpretations can occur. Thus, the exact role of a specific muscarinic subtype is an important issue that is usually not precisely considered when studying its functions.
There are five mAChR subtypes (M1-M5). They can be classified into the even-numbered subtypes (M2,M4) that preferentially couple to heterotrimeric Gi proteins and the odd-numbered (M1,3,5) that preferentially couple to heterotrimeric Gq proteins. In detail, odd-numbered mAChRs are more similar in amino acid sequence composition (see Section 6) between each other than this group and the other group (even-numbered mAChRs).
What is important, mAChRs are involved in many functions, such as learning (Fernández De Sevilla et al., 2021), spinal locomotor networks (Mille et al., 2021), locomotion (Ztaou and Amalric, 2019), biological rhythms (Myslivecek et al., 2017), cardiovascular physiology (Saternos et al., 2017), bronchoconstriction (Kistemaker and Gosens, 2015), and gastrointestinal tract functions (Tobin et al., 2009), and they are also involved in pathologies, such as schizophrenia (Dean and Scarr, 2020) and Parkinson’s disease (Ztaou and Amalric, 2019). Therefore, the choice of appropriate ligand(s) is crucial for mAChR subtype determination in the abovementioned functions.
There are several ways how to discriminate between mAChR subtypes. The first is the choice of appropriate ligand(s) with careful competition binding. This is usually a time and source-consuming issue and sometimes it is not possible (e.g., when it is necessary to see the changes in function depending on mAChR subtype). The second one is the use of the procedure as proposed here. The third possibility is the use of a specific ligand (mostly allosteric) whose number and subtype selectivity are limited to date.
The allosteric ligands have been shown to exhibit selectivity for specific mAChR subtypes (Kruse et al., 2013; Kruse et al., 2014; Bock et al., 2018; Coughlin et al., 2019), providing promising therapeutic effects in diseases connected with mAChRs. However, the search for therapeutically useful ligands should also address additional criteria (i.e., security, bioavailability) beyond subtype specificity. As discussed in Section 4, in most cases, orthosteric ligands bind with higher affinity, and allosteric ligands can provide greater receptor selectivity but are not bound with sufficient affinity. On the other hand, not all orthosteric bind with a higher affinity, and not all allosteric ligands may provide greater receptor selectivity. Bitopic (dualsteric) ligands combine allosteric site selectivity with orthosteric high binding affinity.
Another new direction in the development of selective muscarinic ligands involves functionally selective and biased agonists [for detail, see review (Randáková and Jakubík, 2021)]. However, both allosteric ligands and biased agonists are usually not widely known and thus are not used in mAChR functional studies. On the other hand, allosteric ligands can also target multiple receptors; thus, analyses of mAChR functions using these ligands can be complicated. Additionally, very well-established ligands (with a long history of use in MR research, e.g., atropine) are expected to exhibit more additional targets than less established ligands (with a short history of use or only recently discovered), simply because they are better investigated. Another aspect that complicates the determination of appropriate receptor subtypes in specific tissues is the presence of multiple mAChR subtypes in both periphery tissues, e.g., the retina (Ruan et al., 2021) and heart (Myslivecek et al., 2008a; Myslivecek et al., 2008b), and CNS tissues, e.g., the striatum (Felder et al., 2018), hippocampus (Ohno-Shosaku et al., 2003) and other tissues. In the CNS, the presence of multiple mAChR subtypes on the same structure (presynaptic or postsynaptic) thus complicates the identification of target structures. In the brain, some regions have targets being co-expressed, whereas regions have only one target or subtype predominantly expressed. This can be also relevant if a ligand is targeting regions in the brain and only one region is of interest. The number of receptors (Bmax) also plays a role in receptor interactions.
One can argue that it is possible to use antibodies against specific mAChR subtypes; however, antibodies are very often not specific to the declared targets and can reveal the presence of specific subtypes, even when using mAChR subtype-specific knockout (KO) (Pradidarcheep and Michel, 2016).
Here, we focus on the multiple-target binding of widely used muscarinic drugs that are usually considered subtype-specific. We mainly focus on orthosteric ligands because of their broad usage. We also will emphasize the allosteric drugs that have been shown to be subtype-selective.
Usually, but not in all cases, the orthosteric agonists need micromolar concentration to activate the receptors (see Table 1). The concentration of agonists in signaling assays will depend on receptor expression, receptor reserve, and the amplification of the signaling assay. The antagonists can block the receptors with nanomolar concentration. The common problem in the activation/blockade of mAChR is that the ligand is usually used in high concentrations (see examples in Section 5). Thus, we tried to search available databases (PubMed, Web of Science) for all possible molecules that are able to bind to other targets (proteins or other molecules in the body) than mAChRs. The drugs were divided according to the affinity of off-targets in two groups (nanomolar and micromolar). The reader can then compare the concentration used with the target and judge if this off-target is affected or not.
We show here that most mAChR orthosteric ligands that are usually considered subtype-specific can bind to other receptors, which can be important when interpreting the roles of mAChR subtypes in specific events in both periphery and CNS tissues. This review aimed to call the attention of researchers not deeply focused on mAChR pharmacology to this fact. A very simple search of PubMed (accessed on 20 June 2022) “pirenzepine and M1 selective” reached 984 references—i.e., publications in which pirenzepine could be potentially considered as M1 selective, which is far from true (see below). In fact, the number of these publications could be lower as not always pirenzepine is considered as M1 selective (just coincidence can occur). However, in most of these references is pirenzepine considered to be M1 selective. Similarly, a search on “AF-DX 116 and M2 selective” (accessed on 20 June 2022) reached 313 results—thus, these publications potentially consider AF-DX 116 as M2 selective. The data reviewed here, however, do not report this selectivity.
Additionally, it is necessary to stress that multitarget drug interactions can be considered an advantage in drug effects, as they allow drugs to target more receptors and thus activate more signaling pathways (typically involving sigma receptors) (Maurice and Su, 2009; Abatematteo et al., 2021), which can decrease the required dose of a given drug and produce additive effects with the use of one drug.
2 Muscarinic receptor orthosteric agonists
2.1 Established agonists of mAChRs and their off-targets
The endogenous ligand of mAChRs, ACh, also binds to NRs. Other mAChR agonists are able to activate multiple targets, at least including all mAChR subtypes. This can be a problem when one tries to discriminate between effects on specific receptor subtypes. The widely used mAChR agonist carbachol (carbamylcholine, see Table 1) also binds to NRs (Bolchi et al., 2013) and is not hydrolyzable by cholinesterases, which can be both advantageous and disadvantageous when studying associated effects. The presence of NRs on presynaptic or postsynaptic membranes can affect neurotransmission.
In general, mAChR agonists (and partial agonists, see Table 1) are not subtype-selective. Concretely, pilocarpine, bethanechol, oxotremorine, arecoline, oxotremorine-M, xanomeline, cevimeline, methacholine, iperoxo, methylfurmetide, pentylthio-TZTP, sabcomeline, arecaidine propargyl ester, milameline, furtrethonium (furmethid), aceclidine and relatively newly synthesized compounds, such as SPP1, NNC 11-1585, NNC 11-1607, and NNC 11-1314, are nonselective with respect to a single mAChR subtype (Alexander S. P. et al., 2017; Broad et al., 2019), although they can express higher selectivity for some subtypes, as is the case for NNC 11-1585, which is more selective for M1 and M2 mAChRs than for other mAChR subtypes. Some of them, although declared as subtype-selective, do not reveal specific subtype selectivity. For example, oxotremorine, which is usually considered M1-selective, activates all muscarinic subtypes with similar affinity (Jakubík et al., 1997). Similarly, pilocarpine (partial agonist), which is often declared M3-selective, has comparable effects on all muscarinic receptor subtypes (Alexander S. P. et al., 2017). Also, a relatively newly (2011) synthesized drug, LY-593093, is not M1 mAChR selective, as declared but there exists a similar affinity of M1 and M2 mAChRs [pKi = 6.21, 6.05, for M1 and M2 mAChRs, respectively, see (Watt et al., 2011)]. In general, it is plausible to expect that orthosteric mAChR agonists will bind to all mAChR subtypes without specific selectivity to one of the five mAChR subtypes (see Table 1 for mAChR subtype pKis: specific agonists are bound with similar affinity to all subtypes). Some drugs, like (-)YM796, (±)YM796 (Wei et al., 1992), AZD6088, LSN3172176 (Nabulsi et al., 2019), butylthio-TZTP (Farde et al., 1996) are not sufficiently documented with respect to selectivity.
In the following paragraphs, the orthosteric muscarinic agonists will be reviewed with respect to other targets. In the 1970s, methylfurmethide was indicated to also produce some effects on NRs (Ochillo et al., 1977). Another example of such a drug is arecoline, which acts on NRs (Ward et al., 1994; Pei et al., 1998) as well as the Cav1.2 calcium current (So et al., 2015), producing inhibitive effects. mAChR agonists additionally target enzyme activities; thus, methacholine, carbachol, and oxotremorine inhibited the activity of serotonin N-acetyltransferase, which is a melatonin-synthesizing enzyme, in bovine pineal explants (Pujito et al., 1991). Oxotremorine has also been shown to inhibit acetylcholinesterase (AChE) activity (Sowell et al., 1992). Some ligands reveal multiple-target actions. This is the case for xanomeline, which is considered a partial agonist for mAChRs (Alexander S. P. et al., 2017). However, this drug also has properties indicating allosteric binding (Jakubik et al., 2004) and binding to many serotonin receptor (SERT) subtypes (5-HT1A, 5-HT1B, 5-HT1D, 5-HT1F, 5-HT2A-5HT2C, 5-HT6, and 5-HT7) (Watson et al., 1998).
Relatively recently, some new drugs have been synthesized to favor M1 mAChR effects over effects on other receptor subtypes (mainly M3 mAChRs), including AC-42, AC-260584, 77-LH-28-1, and LY-593039 (Heinrich et al., 2009). However, these drugs can also bind to other neurotransmitter receptors. For example, AC-260584 and 77-LH-28-1 bind similarly to M1 mAChRs and D2 dopamine receptors and affect 5-HT2B and 5HT2C receptors (Heinrich et al., 2009). LY-593039 also binds to D2 dopamine receptors, although slightly less than to M1 mAChRs (Heinrich et al., 2009). AC-42 exerts complicated actions on mAChRs and is a typical representative of multitargeting drugs. It acts on M1 mAChR as a full agonist and simultaneously functions as a negative allosteric modulator (NAM, see below for an explanation of this term, (Langmead et al., 2006)). On other mAChR subtypes, it behaves as an atypical agonist (Richards and van Giersbergen, 1995). As assessed by broad pharmacology profiling (the compound was tested against 69 different pharmacological targets - receptors, ion channels, transporters, and enzymes), AC-42 was bound with revealed relatively high affinity to histamine H1 receptors, D2-, and D4 dopamine receptors, 5-HT1A serotonin receptors, and α1A,B,D- and α2A,B-adrenoceptors (Sams et al., 2010). Other targets had a lower affinity to AC-42 than 1 μmol/L.
2.2 Established agonists of GPCRs with mAChRs as off-target
Another aspect of mAChR pharmacology is the existence of the interactome (Vass et al., 2018). Although the identification of other neurotransmitters that act on mAChRs is difficult, some data indicate that adenosine can act as an antagonist of some specific mAChR-mediated events. It has been shown that adenosine acts selectively in opposing mechanisms of depolarization of the rat superior cervical ganglion M-current (Connolly and Stone, 1995). Another report described a profound (isobutylthio)adenosine inhibitive effect on mAChR binding (Pankaskie et al., 1985). Recently, nerve growth factor has been shown to physically interact with M4 mAChRs (Chen et al., 2021), promoting neuroendocrine differentiation in prostate cancer and producing castration resistance. Droxidopa, which is a synthetic precursor of noradrenaline, used in hypotension treatment, acts also as an antagonist with pKi = 7.1 at M1 mAChRs (Croy et al., 2016).
Important findings were obtained with β3-adrenoceptor agonist vibegron. It has been shown (Yamada et al., 2021) to have an inhibitory action on 3H-NMS (N-methylscopolamine, muscarinic antagonist) binding in different tissue with maximal effect in the heart (pIC50 = 6.1 ± 0.16). However, one should consider two facts: first, IC50 is not an optimal indicator of the pharmacodynamic properties of a drug (see Section 6), and second, β3-adrenoceptor are present in the heart (Benes et al., 2012) which can slightly doubt these results.
3 Muscarinic receptor orthosteric antagonists
3.1 Established antagonists of mAChRs and their off-targets
Orthosteric antagonists of mAChRs reveal a much wider spectrum of action targets than agonists. Thus, one should be more cautious in interpreting results obtained with the use of mAChR orthosteric antagonists. Typically, although very often declared by manufacturers as “selective ligands,” the vast majority of antagonists bind with similar affinity to more than one mAChR subtype (see Table 2). We have reviewed these properties recently (Myslivecek, 2019). It can be concluded that only a limited number of ligands (e.g., mamba toxins) are selective for one subtype (see Table 4). However, the usage of toxins is not simple, especially in vivo, it is difficult to make and expensive to use. These toxins also bind allosterically, rather than only orthosterically (Karlsson et al., 2000). Importantly, the selectivity to M3 mAChR was not, according to our knowledge yet proven. The M3 mAChR positive allosteric modulator ASP8302 (Okimoto et al., 2021) revealed a similar increase in activity in M3 as well as in M5 mAChRs (see Table 2).
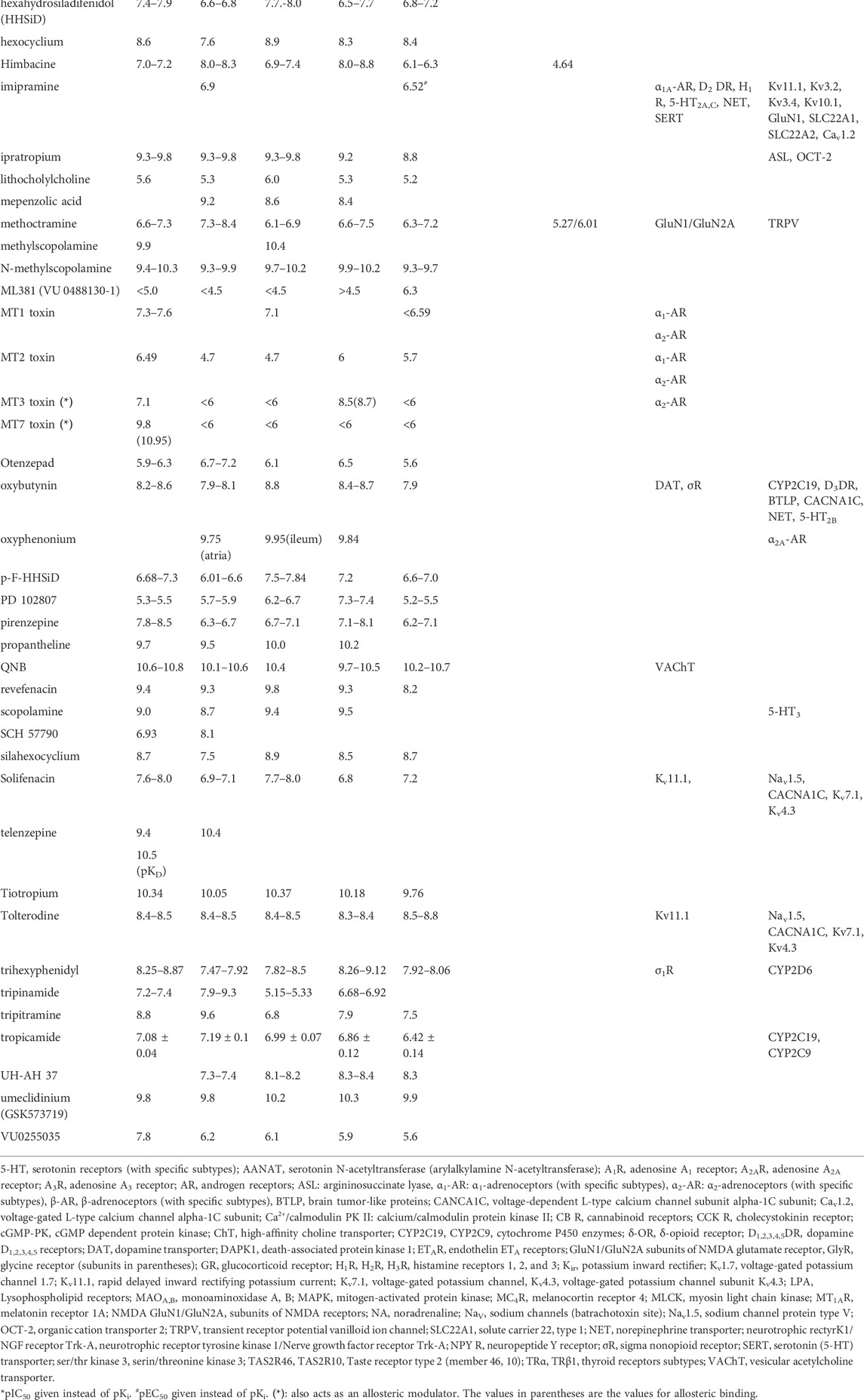
TABLE 2. Effects of orthosteric antagonists on muscarinic receptors and other targets. The compounds are listed alphabetically. The numbers indicate pKi values. Please note that a higher pKi indicates higher affinity. Unavailable data are shown as blank spaces. The affinities of nicotinic receptors are not divided among specific subunits. The affinities of other, noncholinergic targets are divided into nanomolar (pKi>6) and micromolar (pKi<6) groups. The categorization of pKi is compromise between extremely large Table and fast orientation how much are other targets are affected in specific concentration of ligand used (see also examples in Chapter 5). Data are from or adapted from (Pascuzzo et al., 1984; Eglen and Whiting, 1987; Buckley et al., 1989; Buckley et al., 1990; Hudkins et al., 1991; Wess et al., 1991; Bolden et al., 1992; Kashihara et al., 1992; Sowell et al., 1992; Doods et al., 1993; Shen et al., 1993; Stanton et al., 1993; Doods et al., 1994; Ferrari-Dileo et al., 1994; Kleinschroth et al., 1995; Waelbroeck et al., 1996; McKenna et al., 1997; Boess et al., 1998; Bolognesi et al., 1998; Cantı́ et al., 1998; Caulfield and Birdsall, 1998; Lazareno et al., 1998; Choppin et al., 1999; Sánchez and Hyttel, 1999; Eglen and Nahorski, 2000; Kozlowski et al., 2000; Lazareno et al., 2000; Becerra et al., 2001; Dhein et al., 2001; Huang et al., 2001; Lockhart et al., 2001; Apelt et al., 2002; Carlsson et al., 2002; Harvey et al., 2002; Cheng et al., 2002; Lazareno et al., 2002; Böhme et al., 2003; Samochocki et al., 2003; Sur et al., 2003; Jakubik et al., 2004; Kobayashi et al., 2004; Wang et al., 2004; Ghoneim et al., 2006; Spalding et al., 2006; Butini et al., 2008; Langmead et al., 2008; Bridges et al., 2009; Heinrich et al., 2009; Prat et al., 2009; von Coburg et al., 2009; Bridges et al., 2010a; Harada et al., 2010; Hern et al., 2010; Lange et al., 2010; Rook et al., 2010; Sinha et al., 2010; Watt et al., 2011; Daval et al., 2012; Samadi et al., 2012; Sykes et al., 2012; Arunotayanun et al., 2013; Nenasheva et al., 2013; Salmon et al., 2013; Croy et al., 2016; Gaulton et al., 2016; Alexander S. P. et al., 2017; Alexander S. P. H. et al., 2017; Chen et al., 2017; Carr et al., 2018; Hegde et al., 2018; Broad et al., 2019; Myslivecek, 2019; Xu et al., 2019; Olianas et al., 2020; Okimoto et al., 2021) and the IUPHAR/BPS Guide to Pharmacology (www.guidetophamacology.org). For specific ligand references see text. The activity represents the main effect of a specific ligand.
Some examples of “selective antagonists” are as follows (for the affinities of these ligands, see Table 2): pirenzepine is considered an M1 mAChR-selective ligand, but it binds with a similar affinity to M4 mAChRs (Caulfield and Birdsall, 1998; Myslivecek, 2019); methoctramine (and similarly, himbacine, AF-DX 384 and AF-DX 116) (Doods et al., 1994; Caulfield and Birdsall, 1998; Myslivecek, 2019), which is said to preferentially bind to M2 mAChRs, also binds with similar affinity to M4 mAChRs; 4-DAMP, which is known as an M3 mAChR-selective antagonist, reveals a similar affinity to M4 mAChRs (Caulfield and Birdsall, 1998; Myslivecek, 2019); and darifenacin, which is used as an M3 mAChR antagonist in incontinence treatment, antagonizes M5 mAChRs (Myslivecek, 2019). Similar non-selectivity has also been found for other mAChR antagonists. On the other hand, when working in vitro, some antagonists can discriminate between mAChR subtypes when a competitive binding curve is employed; for example, AFDX-384 has a significantly lower affinity for M5 mAChRs (pKi = 6.3) than for other mAChR subtypes (pKi values vary between 7.2 and 9.0). Thus, the proportion of mAChRs detected at concentrations higher than pKi = 6.3 can indicate the amount of M5 mAChRs present [for the affinities of these ligands, see (Myslivecek, 2019)]. Telenzepine, considered M1 mAChR selective, has virtually the same affinity to M1 [pKD = 10.5, (Hern et al., 2010)] as well as to M2 mAChRs [pKi = 10.4, (Nenasheva et al., 2013).
In the following paragraphs, we discuss the binding of mAChR antagonists to other neurotransmitter receptors and target proteins (e.g., ion channels).
Atropine, which is a typical muscarinic antagonist, can also act as an antagonist at the α5 subunit of NRs (although at higher concentrations, pKi = 4.49) (Tomizawa and Yamamoto, 1992) and can block the effects of drugs on α2A- (Carr et al., 2018) and α1D-adrenoceptors (Gaulton et al., 2016) and glycine receptors (all subtypes (Maksay et al., 1999; Yang et al., 2007)). At higher concentrations, it also inhibits solute carrier family 22 member 1 (SLC22A1 (Ahlin et al., 2008)) and serotonin 5-HT1A (Zhuang et al., 1993) and 5-HT2C receptors (Gaulton et al., 2016). In mice, atropine inhibits AChE (Sowell et al., 1992) at very low concentrations (the pKi is 9.15–9.46). Thus, when researchers apply atropine intending to block mAChRs, they should expect some side effects on additional receptors, which can be minor or significant. Moreover, the possible inhibition of AChE may partially artificially diminish the effects of mAChR blockade. Concerning these facts, one should consider replacing atropine with another drug (e.g., aclidinium) when trying to block mAChRs.
Similarly, the mAChR antagonist scopolamine also behaves as an antagonist of serotonin 5-HT3 receptors (Lochner and Thompson, 2016). Another mAChR antagonist, benzatropine, has a wider spectrum of targets including other GPCRs (D3 dopamine receptors, α1A,B,D-adrenoceptors, α2A,B,C-adrenoceptors (more information is available in (Svoboda et al., 2019)), H1 (Kulkarni et al., 2006) and H2 histamine receptors (Svoboda et al., 2019), and SERTs, 5-HT2A,B,C and 5-HT6 (Modell et al., 1989)); other receptors (σR—sigma receptors (Svoboda et al., 2019)); and neurotransmitter transporters (SERT (Zhang et al., 2001), DAT (Runyon and Carroll, 2006), and NET (Newman and Kulkarni, 2002)), and it is also able to inhibit the amino acid transporter SLC6A19 (Cheng et al., 2017).
A next mAChR antagonist, methoctramine, has been proven to inhibit vanilloid receptor (transient receptor potential cation channel, TRPV) function by reducing the single-channel mean opening time and increasing the mean closure time (Mellor et al., 2004). On the other hand, it is necessary to note that the concentration used for this inhibition was relatively high (10 μmol l−1, i.e., 10−5 mol l−1). Another effect of methoctramine lies in GTPase activity, and it also behaves as a direct Gi agonist in mast cells, where it stimulates exocytosis (Chahdi et al., 1998). Additionally, it inhibits cholinesterases (AChE and BChE) (Tumiatti et al., 2003) and produces antagonistic effects on GluN1/GluN2A NMDA receptors (pKIC50 = 6.86, (Saiki et al., 2013)).
Amitriptyline affects mainly (in nanomolar rank) serotonin receptors (5-HT2A, 5-HT2C, 5-HT6, 5-HT7 (Gaulton et al., 2016)), H1R histamine receptors (Gaulton et al., 2016), α1A,B,D-AR, α2A,B,C-AR (Gaulton et al., 2016), D1,2,3,5DR (von Coburg et al., 2009; Gaulton et al., 2016), neurotransmitter transporters SERT, NET, and less DAT (Arunotayanun et al., 2013) and LPA1 lysophospholipid receptors (Olianas et al., 2020). Besides, potassium inward rectifiers (Kir3.2, Kir3.4) are inhibited in micromolar rank (Kobayashi et al., 2004).
Himbacine inhibits AChE, although at micromolar concentrations (Brunhofer et al., 2012). Synthetic analogs of the natural product himbacine can inhibit protease-activated receptor-1 (PAR-1), which is also known as the thrombin receptor (Chackalamannil et al., 2003; Chelliah et al., 2014). However, himbacine itself is devoid of PAR-1 activity.
Darifenacin, which has very good selectivity for M3 mAChRs, surprisingly affects many additional receptors and some channels and proteins. For example, darifenacine inhibits fatty acid-binding protein 4 (Wang et al., 2014), the rapid delayed inward rectifying potassium current (KIR, Kv11.1), the sodium channel protein type V alpha subunit (Nav1.5), voltage-gated potassium channels (IKs, KCNQ1), and voltage-gated potassium channel subunit Kv4.3 (Mirams et al., 2014). Another target of darifenacin is GPCRs, including β1 and β2-adrenoceptors (Beattie et al., 2012) and dopamine D2 receptors (Connolly et al., 2011).
Similar to darifenacine, solifenacin affects the KIR, the Nav1.5/sodium channel protein type V alpha subunit, Kv7.1/IKs, KCNQ1(Kv7.1)/KCNE1(MinK), Kv4.3/IK subunit Kv4.3, and the Cav1.2/voltage-gated L-type calcium channel alpha-1C subunit (Mirams et al., 2014).
Ipratropium can activate the arginine metabolic enzyme argininosuccinate lyase (Hung et al., 2017), which is involved in specific cancer types, including hepatocellular carcinoma. Ipratropium also inhibits the organic cation transporter 2-mediated transport of specific substrates (Belzer et al., 2013).
Tropicamide is a drug usually used as an atropine replacement for pupillary dilatation. It is considered an M4 mAChR antagonist, but all mAChR subtypes reveal a similar affinity to this ligand (Croy et al., 2016). Moreover, this drug also inhibits the cytochrome P450 enzymes CYP2C19 and CYP2C9 (Gaulton et al., 2016). However, CYPs are affected in micromolar rank and thus they are not physiologically significant in that point of view.
Oxybutynin is another example of a drug with multiple targets. In addition to mAChRs, it also inhibits CYP2C19, D3 dopamine receptors, DAT (Gaulton et al., 2016), a group of brain tumor-like proteins (i.e., lethal(3)malignant brain tumor-like protein 1, L3MBTL histone methyl-lysine binding protein 3, lethal(3)malignant brain tumor-like protein 4, MBT domain-containing protein 1) (Kireev et al., 2010), voltage-dependent L-type calcium channel subunit α-1C (Wiśniowska et al., 2012), NET (Gaulton et al., 2016), SERT, 5-HT2B (Gaulton et al., 2016), and sigma nonopioid intracellular receptor 1(Gaulton et al., 2016).
Tolterodine is another drug with multiple targets that binds to Kv11.1/HERG (inward rectifier) (Mirams et al., 2014), the Nav1.5/sodium channel protein type V alpha subunit (Mirams et al., 2014), the Cav1.2/voltage-gated L-type calcium channel alpha-1C subunit (Mirams et al., 2014), the Kv7.1/IK (Mirams et al., 2014), and Kv4.3/IK subunit Kv4.3 (Mirams et al., 2014).
Dicyclomine has a relatively wide spectrum of actions, in nanomolar concentrations, it inhibits 5-HT2A, 5-HT2B, 5-HT2C serotonin receptors, D3DR, σ1R, σ2R, and in micromolar concentrations it inhibits Cav1.2, H2R, 5-HT6, Kv11.1, NaV (for all data see (Gaulton et al., 2016)).
Trihexyphenidyl acts on σ1R in nanomolar rank and on CYPD2 in micromolar rank (Gaulton et al., 2016).
A special example of a drug with multiple targets (reveals orthosteric and allosteric binding) is MT7 toxin, which is obtained from the green mamba. This compound is not only a very selective antagonist of M1 mAChRs that irreversibly blocks but also binds to their allosteric sites with pKi = 10.95 (Fruchart-Gaillard et al., 2006). There are further mamba toxins that bind to muscarinic receptors. MT1 toxin, however, is not specific, it binds similarly to M1 and M4 mAChRs (see Table 2), but also inhibit 3H-prazosin binding in different tissue preparation (in the periphery—vas deferens, in CNS—cerebral binding) indicating it is able to inhibit α1-ARs and α2-ARs (Harvey et al., 2002). It is necessary to mention that inhibition of prazosin binding was not 100% in vas deferens. Similarly, MT2 toxin is not selective and also binds to cerebral (100% inhibition) α1-ARs and α2-ARs (Harvey et al., 2002). MT3 toxin is also not selective (Jolkkonen et al., 1994; Carr et al., 2018), it also binds to the allosteric site on M4 mAChRs (Jolkkonen et al., 1994)and is able to inhibit α2-ARs (Carr et al., 2018).
Another antagonist, (S)-dimetindene is an isomer of histamine antagonist and has a similar ability to antagonize the effects on all mAChRs (Böhme et al., 2003).
Ethopropazine is also able to inhibit cholinesterases—both acetylcholinesterase and butyrylcholinesterase (Brus et al., 2014).
Very often used tritiated ligand for muscarinic receptors, 3H-QNB binds also to vesicular acetylcholine transporter with pKi = 6.24 (Kozaka et al., 2012). However, the pKi for mAChRs is around 10 (see Table 2).
Another radioligand used in mAChR detection, also as PET ligand (Mulholland et al., 1995) is tritiated, radioactively carbonylated, respectively N-methylpiperidyl benzilate (NMPB). This ligand is declared as M2 mAChR selective (Stein et al., 1988). However, no available data on other mAChR subtypes and structure similarity to 4-DAMP (see above) opens the question about the real selectivity of this drug. A similar case comes with clinidium, in which also data on M2 mAChRs are only available (Kovacs et al., 1998).
Although this review summarizes the inability of ligands to bind one mAChR subtype, this obstacle can be overcome. It is possible to use relatively specific ligands under well-defined conditions (concentration, time, and temperature), as described in KO models. Thus, pirenzepine, which is far from a selective ligand (it binds to M1 and M4 mAChRs with similar affinity), has been shown to selectively bind with a specific protocol to M1 mAChRs (Valuskova et al., 2018a).
3.2 Established antagonists of GPCRs with mAChRs as off-target with a special accent to antipsychotic drugs
Some drugs mainly act at other receptors that can antagonize the activation of mAChRs. This was described for the 5-HT1A receptor agonist 7-(dipropylamino)-5,6,7,8-tetrahydronaphthalen-1-ol (8-OH-DPAT) (Fowler et al., 1991; Chidlow and Osborne, 1997). Similarly, the 5-HT4 receptor agonists RS 67333 and RS 67506 also reveal a low affinity to mAChRs (Eglen et al., 1995).
Special attention should be given to antipsychotics. These drugs are a perfect example of multitargeting advantage in treating specific diseases. All drugs listed here can bind not only to “classical” antipsychotic targets like serotonin, dopamine, or histamine receptors but also inhibit muscarinic receptors which are additional, desirable effects that could widen the action of antipsychotics. This is valid for the following drugs. Haloperidol, a typical antipsychotic medication, in addition to its classical effects on dopamine and serotonin receptors, is also able to inhibit the M2 mAChRs, although the affinity is not too high (pKi = 6.62, (Ronsisvalle et al., 1998)). Olanzapine, which is an atypical antipsychotic with a higher affinity for 5-HT2A, SERTs, and D2 dopamine receptors, has also been shown to antagonize the effects of pilocarpine on phosphoinositide hydrolysis, although with lower affinity (Zhang and Bymaster, 1999). In general, many drugs used as antipsychotics (usually targeting dopamine and serotonin receptors), such as chlorpromazine, fluphenazine, levomepromazine, perphenazine [for all these drugs see (Hals et al., 1988)], and blonanserin (Tenjin et al., 2013) have anticholinergic effects and can antagonize the binding of mAChRs. In the first generation of antipsychotics, thioridazine reveals a wide spectrum of effects which also include muscarinic (M1-M5) antagonism in the range pKi = 6-10 (Vass et al., 2018). Clozapine is one of the first atypical antipsychotics. Besides its antagonistic effects on histamine, serotonin, adrenergic, and dopamine receptors, it also reveals positive allosteric effects on M1 mAChRs (Flamez et al., 1994) with high affinity (pKIC50 = 7.9 (Sur et al., 2003), antagonistic effects on M2 mAChRs pKi = 7.32–7.36 (Ohmori et al., 1996), and on M3-M5 mAChRs (Bandyopadhyaya et al., 2012) with pKi = 7-8). This also led to the possibility that muscarinic receptors can be targeted when treating schizophrenia (Foster et al., 2021). In this review, clozapine, xanomeline, and specific allosteric modulators targeting to mAChR subtypes (MK-801 (M4 mAChR selective), BQCA (M1 mAChR selective), VU0238429 (M5 mAChR selective) are discussed. It is necessary to mention that there can also be a possibility that xanomeline act on dopaminergic neurons through its action on M1 and M4 mAChRs (Weiden et al., 2022). Similarly, the effects of MK-801, BQCA, and VU0238429 are the most probably via the mAChRs expressed on dopaminergic/serotoninergic or other neurons (Foster et al., 2021). An important aspect of antipsychotic action has been investigated by (Obara et al., 2019) who determined the inhibitory effects on brain mAChRs in therapeutically achievable concentrations. They found that the pKi values of chlorpromazine (pKi = 5.85–7.55), levomepromazine (pKi = 6.21–7.35), and zotepine (pKi = 6.14–7.04), olanzapine (pKi = 6.37–7.38), and clozapine (pKi = 5.23–6.30) overlapped with their clinically achievable blood concentrations. Importantly, clozapine act as a positive allosteric modulator (see Table 3) with pKIC50 = 7.7–7.9 at M1 mAChRs (Sur et al., 2003; Spalding et al., 2006) and together with this action it also competes with M1 mAChRs [pKi = 7.0–9.01 (Rowley et al., 2001; Bandyopadhyaya et al., 2012)], with M2 mAChRs [pKi = 6.77–7.34 (Gaulton et al., 2016)], with M3 mAChRs [pKi = 7.0–7.77 (Bandyopadhyaya et al., 2012; Gaulton et al., 2016)], with M4 mAChRs [pKi = 7.0–8.2 (Bandyopadhyaya et al., 2012; Gaulton et al., 2016)], and with M5 mAChRs [pKi = 7.0–8.02 (Bandyopadhyaya et al., 2012; Gaulton et al., 2016)]. Another antipsychotic, dosulepin, act with high affinity on histamine receptors (Sánchez and Hyttel, 1999), SERT, and NET(Tatsumi et al., 1997) and, of course, on all mAChR subtypes (Stanton et al., 1993). Thus, it is possible to conclude that mAChRs play important role in antipsychotic action, arising from the effects on mAChR subtypes that are integral to the regulation of DA neural circuits (Weiden et al., 2022).
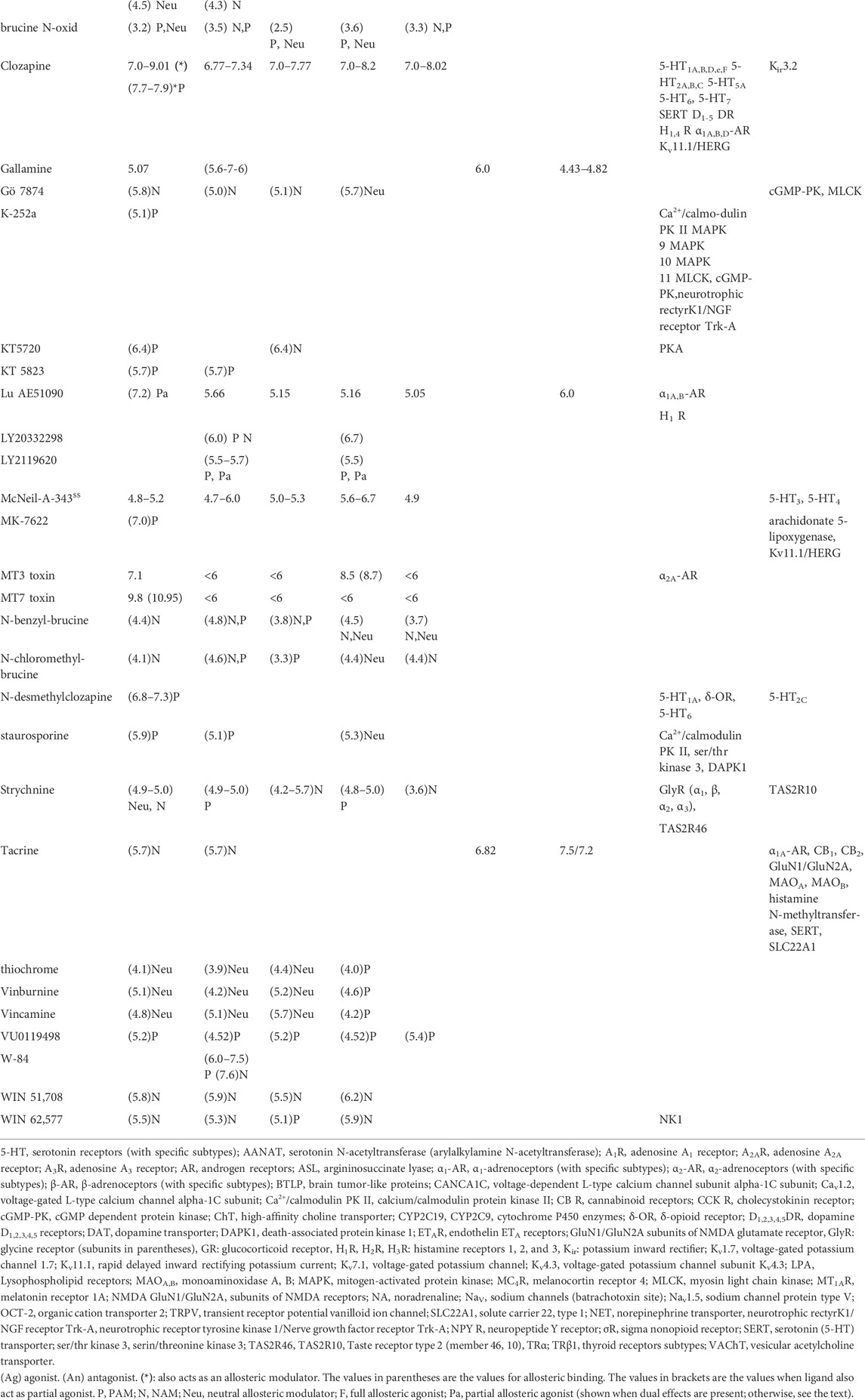
TABLE 3. Effects of allosteric ligands on muscarinic receptors and other targets. The compounds are listed alphabetically. The numbers indicate pKi values. Please note that a higher pKi indicates higher affinity. Unavailable data are shown as blank spaces. The affinities of nicotinic receptors are not divided among specific subunits. The affinities of other, noncholinergic targets are divided into nanomolar (pKi>6) and micromolar (pKi<6) groups. The categorization of pKi is compromise between extremely large Table and fast orientation how much are other targets are affected in specific concentration of ligand used (see also examples in Chapter 5). Data are from or adapted from (Pascuzzo et al., 1984; Eglen and Whiting, 1987; Buckley et al., 1989; Buckley et al., 1990; Hudkins et al., 1991; Wess et al., 1991; Bolden et al., 1992; Kashihara et al., 1992; Sowell et al., 1992; Doods et al., 1993; Shen et al., 1993; Stanton et al., 1993; Doods et al., 1994; Ferrari-Dileo et al., 1994; Kleinschroth et al., 1995; Waelbroeck et al., 1996; McKenna et al., 1997; Boess et al., 1998; Bolognesi et al., 1998; Cantı́ et al., 1998; Caulfield and Birdsall, 1998; Lazareno et al., 1998; Choppin et al., 1999; Sánchez and Hyttel, 1999; Eglen and Nahorski, 2000; Kozlowski et al., 2000; Lazareno et al., 2000; Becerra et al., 2001; Dhein et al., 2001; Huang et al., 2001; Lockhart et al., 2001; Apelt et al., 2002; Carlsson et al., 2002; Harvey et al., 2002; Cheng et al., 2002; Lazareno et al., 2002; Böhme et al., 2003; Samochocki et al., 2003; Sur et al., 2003; Jakubik et al., 2004; Kobayashi et al., 2004; Wang et al., 2004; Ghoneim et al., 2006; Spalding et al., 2006; Butini et al., 2008; Langmead et al., 2008; Bridges et al., 2009; Heinrich et al., 2009; Prat et al., 2009; von Coburg et al., 2009; Bridges et al., 2010a; Harada et al., 2010; Hern et al., 2010; Lange et al., 2010; Rook et al., 2010; Sinha et al., 2010; Watt et al., 2011; Daval et al., 2012; Samadi et al., 2012; Sykes et al., 2012; Arunotayanun et al., 2013; Nenasheva et al., 2013; Salmon et al., 2013; Croy et al., 2016; Gaulton et al., 2016; Alexander S. P. et al., 2017; Alexander S. P. H. et al., 2017; Chen et al., 2017; Carr et al., 2018; Hegde et al., 2018; Broad et al., 2019; Myslivecek, 2019; Xu et al., 2019; Olianas et al., 2020; Okimoto et al., 2021) and the IUPHAR/BPS Guide to Pharmacology (www.guidetophamacology.org). For specific ligand references see text. The activity represents the main effect of a specific ligand.
Imipramine, a tricyclic antidepressant, has a wide range of targets: besides M2 (Kovacs et al., 1998) and M5 mAChRs (Griffith et al., 1984), it binds also to α1A-AR (Yardley et al., 1990), D2 DR (Peters, 2013), H1 R (Peters, 2013), 5-HT2A,C (Peddi et al., 2004), NET, and SERT (Runyon et al., 2001).
Similar multitarget effects can be found (Vass et al., 2018) for astemizole, an H1 histamine receptor antagonist, and cinnarizine was used primarily as an anti-histaminic. All these three drugs were withdrawn from the market because of their side effects. Similarly, econazole - an antifungal agent, primarily acting on TRPM and TRPV channels (Hill et al., 2004) has also anti-muscarinic effects (M1, M3, M4) (Vass et al., 2018).
One should keep it in mind when applying antipsychotics in an experimental condition. As it can be seen one also should be cautious when limiting the effects of antipsychotics on serotonin, dopamine, and histamine receptors only.
An interesting aspect of interneurotransmitter effects has recently been shown—the simple replacement of a flexible alkyl chain with a semirigid aryl or cycloalkyl ring leads to increased mAChR affinity of specific ligands, detrimentally affecting histamine receptor binding (Staszewski et al., 2021). However, these newly synthesized ligands continued to bind to histamine receptors, although with lower affinity.
4 Muscarinic receptor allosteric ligands
The allostery concept was introduced in 1961 by Monod and Jacob and suggests conformational changes in a two-state model [see (Coughlin et al., 2019)]. The search for allosteric mAChRs ligands began in the 1970s (Clark and Mitchelson, 1976) and was further developed in the 1980s.
The general principle of allosteric action lies in the fact that binding to allosteric sites can modulate the activity of orthosteric ligands (Kruse et al., 2014). The common mechanisms of allosteric modulation have been indicated to be generalizable and can be found for many if not all, GPCRs. However, marked structural differences also exist. Mainly among mAChR subtypes, the receptors reveal different sequence homology. While orthosteric site sequence homology is high between all mAChR subtypes, in the extracellular parts of the transmembrane domains and extracellular loops of these receptors, there is a low degree of sequence homology. In summary, the interactions of ligands with receptors can occur as follows (Mohr et al., 2013): 1) positive allosteric modulators (PAMs) increase the affinity or efficacy of an orthosteric ligand or orthosteric agonist–receptor complex, increase the equilibrium binding of the simultaneously bound endogenous or exogenous orthosteric ligand, enhance either the affinity of an orthosteric agonist or lower the activation barrier for the transition from the inactive to the active conformation of the receptor, 2) negative allosteric modulators (NAMs) decrease the affinity or efficacy of an orthosteric ligand or orthosteric agonist–receptor complex, inducing a rightward shift of the concentration-effect curve of the endogenous agonist (as an example), 3) neutral allosteric ligands (NALs) bind to an allosteric site but have no effect on the affinity or efficacy of orthosteric or allosteric ligands, and 4) allosteric agonists activate the receptors themselves.
We can also define some strengths and disadvantages of each mode of mAChR targeting. Orthosteric sites exhibit high affinity to ligands, but mAChR subtype selectivity remains largely unfeasible. Allosteric sites exhibit greater receptor selectivity and promote more physiological response patterns, but there was a problem with low-affinity modulators that have been reported (Melancon et al., 2012b). Recently, some drugs that overcame this obstacle advanced to the preclinical or clinical phase of testing in the treatment of neurological disorders (Moran et al., 2019).
The possible solution to this problem could be bitopic (dualsteric) ligands. Research has been conducted to rationally design hybrid molecules that simultaneously bridge orthosteric and allosteric sites within a single receptor. These compounds have been termed “bitopic,” “dualsteric,” or “multivalent.” These drugs attempt to target allosteric sites to achieve selectivity and orthosteric sites to provide high affinity (Bock et al., 2018).
At the beginning of the 21st century (Disingrini et al., 2006), first combined a nonselective, high-affinity orthosteric agonist (iperoxo) with an M2 receptor-selective allosteric modulator to generate an M2 receptor-selective agonist. However, although bitopic agonists with mAChR subtype selectivity and signaling tendencies have been described, substantial improvements in affinity were not observed [see (Kruse et al., 2014)]. On the other hand, the bitopic mAChR antagonist THRX-160209 has been shown to exhibit both affinity and selectivity for the M2 receptor (Steinfeld et al., 2007b).
The selective ligands (the current situation) are summarized in Table 4. It is necessary to stress that very well-established ligands (with a long history of use in mAChR research, e.g., atropine) are expected to have more additional targets than less established ligands (e.g., allosteric ligands with a short history of use or only recently discovered), simply because they are better investigated. Thus, one should consider Table 4 as the present state of knowledge.
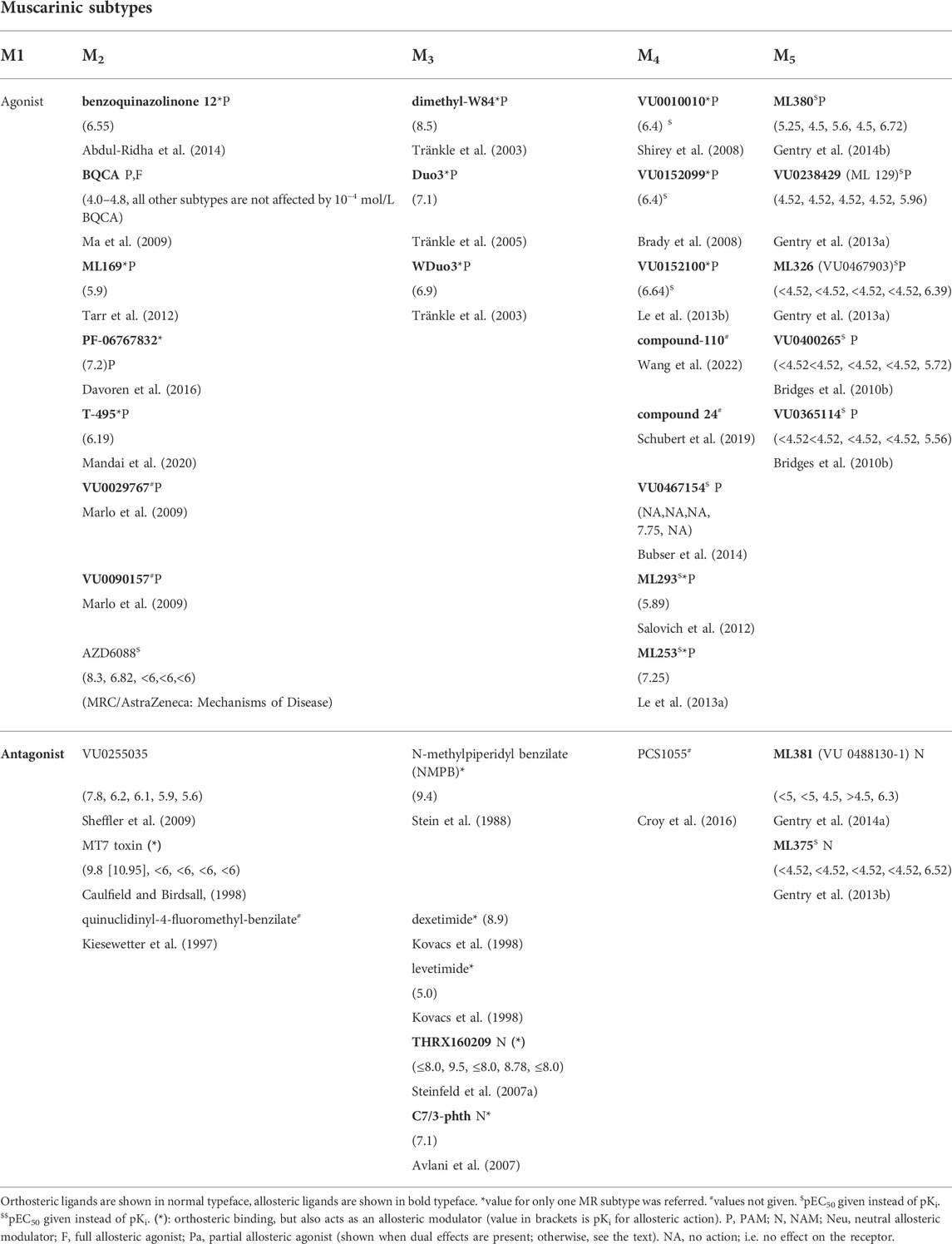
TABLE 4. The list of selective MR ligands. The present state of knowledge. The values in parentheses represent pKi (pKD, respectively) for M1, M2, M3, M4, and M5 MRs). Some orthosteric agonists ((-)YM796, AZD6088, LSN3172176, butylthio-TZTP are not sufficiently documented with respect to selectivity. The same can be stated about allosteric agonist listed in this table (ML169, PF-06767832, T-495) that have been reported as selective but the data on other MR subtypes are missing. VU0029767 and VU0090157 showed selectivity on M1 MRs over other subtypes but values were not reported.
Despite their relatively short history of use, some allosteric ligands have also been shown to target multiple receptors. Following are examples of such allosteric (or bitopic) ligands.
Some of these drugs can act not only on mAChRs but also on other neurotransmitter receptors. This is the case for McNeil-A-343, which acts on all mAChR subtypes as a partial agonist (Alexander S. P. et al., 2017). In addition, it is also considered a bitopic ligand of mAChRs in that it modulates allosteric and orthosteric binding sites (Christopoulos et al., 2014). In addition, this compound can also act as an antagonist of 5-HT4 and 5-HT3 receptors at a range of concentrations overlapping with those relevant to its interaction with mAChRs (Sagrada et al., 1994). An example of a compound with unprecedented selectivity and procognitive potential is Lu AE51090, an allosteric muscarinic M1 mAChR agonist. Although the selectivity to M1 mAChR is really nice (between 36-145× better than to M2-M5 mAChRs), there is still binding to α1A,B-adrenoceptors, and to histamine H1 receptors (pKis between 6.07-6.59, (Sams et al., 2010)). Thus, this compound shows the advantage of multitarget binding in treatment.
Another example of mAChR allosteric ligands acting on other neurotransmitter receptors is as follows. MT3 toxin from the green mamba is considered an M4 mAChR NAM. However, it has also been demonstrated to be an α2A-adrenoceptor antagonist (Carr et al., 2018). The opposite mAChR allosteric ligand situation is seen in amiodarone, which is a classical blocker of voltage-gated potassium channels (Kv1.7) (Bardien-Kruger et al., 2002) and voltage-gated sodium channels (Nav1.5)(Sheldon et al., 1989). Relatively recently, it has also been identified as a PAM of M5 mAChRs (Stahl and Ellis, 2010), but it has an alternative allosteric binding site (Burger et al., 2021). This drug also acts as an M3 mAChR PAM (Stahl et al., 2011). However, this molecule has additional targets, including β-adrenoceptors (Chatelain et al., 1995), thyroid hormone receptors α and β (Carlsson et al., 2002), and other molecules [α2-adrenoceptors, dopamine receptors, 5-HT receptors, and σ nonopioid receptors; see (Gaulton et al., 2016)].
The first pioneer molecules with described allosteric effects on mAChRs were originally targeted toward NRs. This is the case for alcuronium, which is a neuromuscular-blocking drug with negative allosteric effects on mAChRs (Nedoma et al., 1985) that acts as a NAM of M1, M2, M3, and M4 mAChRs. Gallamine (defined as a nondepolarizing muscle relaxant) is a NAM of M2 mAChRs and has a similar affinity to the α1 subunits of NRs, which are inhibited by this drug [e.g., (Psaridi-Linardaki et al., 2003)]. Higher concentrations of gallamine can also inhibit AChE (Butini et al., 2008). Gallamine also exhibits competitive inhibition of M1 mAChR (Daval et al., 2012). Tacrine was first synthesized as a dual inhibitor of butyrylcholine esterase and acetylcholinesterase with the aim to treat Alzheimer’s disease (Fisher, 2008). In addition to these effects (both cholinesterases have a relatively high affinity to tacrine with pKi ≈ 7), this drug also reveals negative allosterism on M1 and M2 mAChRs [pIC50 = 5.7, (Sowell et al., 1992)]. Tacrine also inhibits nicotinic receptor subunit ε (Xu et al., 2019), at a similar concentration as for mAChRs it inhibits α1Α-adrenoceptors (Gaulton et al., 2016), cannabinoid receptors CB1, CB2 (Lange et al., 2010), GluN1/GluN2A/Glutamate NMDA receptor subunits (Rook et al., 2010), histamine N-methyltransferase (Apelt et al., 2002), MAOA and MAOB (Samadi et al., 2012), SERT (McKenna et al., 1997), and SLC22A1 (Chen et al., 2017).
Staurosporine, as another example of a drug with a classical function, is described mainly as a protein kinase inhibitor but is also a PAM of M1 and M2 mAChRs (Lazareno et al., 2000). In addition, it can affect many other molecules, such as calcium calmodulin protein kinase II, serine/threonine kinase 3, and death-associated protein kinase 1 [see (Hall et al., 2009)]. Brucine is a natural alkaloid product with many effects on organisms (Lu et al., 2020), such as antitumor, anti-inflammatory, analgesic effects, and effects on the cardiovascular system and nervous system. This molecule is also a PAM of M1 and M2 mAChRs and a NAM of M3, M4, and M5 mAChRs (Jakubík et al., 1997; Lazareno et al., 1998; Birdsall et al., 1999). WIN 62,577, as another allosteric modulator, is a NAM of M1, M2, M4, and PAM of M3 mAChR that was originally synthesized as an NK1 antagonist (Lazareno et al., 2002). Another allosteric ligand, AC-260584 has also complicated action: it acts as PAM on M1 mAChR (pIC50 = 5.9) and other mAChR subtypes (M2-M4) reveal similar affinity when it acts as a full antagonist. On M5 mAChR it has agonist action and the affinity is also similar (Melancon et al., 2012a). Strychnine, glycine antagonist (Gaulton et al., 2016)—with action on many subunits (Zlotos et al., 2019), can allosterically modulate the muscarinic transmission: positively, negatively, as well as it has a neutral effect on mAChRs (Jakubík et al., 1997) also affects taste receptors (Gaulton et al., 2016).
Another M1 and M4 mAChR PAM, KT5720 (Lazareno et al., 2000), is also a protein kinase A inhibitor (Birdsall et al., 2001). Kinases (cGMP-dependent protein kinase and myosin light chain kinase) are inhibited in mAChRs (M1-M4) action rank by Gö 7874 (Kleinschroth et al., 1995; Lazareno et al., 2000). Another positive allosteric modulator, K-252a, selective to M1 mAChRs (Lazareno et al., 2000), is, however, a potent inhibitor of many kinases in rank (pKi (pIC50, respectively, between 6.44 and 8.74) above the action on M1 mAChRs (pKd = 5.1)—see Table 3. N-desmethylclozapine is a specific M1 mAChR PAM but also has the properties of an atypical agonist of M1-M4 mAChRs (Sur et al., 2003). In addition, N-desmethylclozapine is a potent 5-HT1A (Heusler et al., 2011) partial receptor antagonist, an antagonist of 5-HT2C receptors (Hasuo et al., 2002) and cloned 5-HT6 receptors (Dupuis et al., 2008) and an agonist of δ-opioid receptors (Olianas et al., 2009).
An example of a selective drug (PAM) for M1 mAChRs is MK-7622. The pEC50 = 7.0 (Bertron et al., 2018) seems to be far from the pIC50s (see (Beshore et al., 2018)) for arachidonate 5-lipoxygenase (pIC50 = 5.36) and Kv11.1/HERG (pIC50 = 4.22). With respect to the common criteria for receptor selectivity, it looks good, and one could consider MK-7622 as an M1 selective ligand. This is true but a cautious attitude is recommended concerning possible future discoveries (the drug showing the selectivity at present would be less investigated than notoriously known drugs).
Other examples present the situation in which the activities of ligands depend on the number of mAChRs present in the tissue in which the agonist is used for receptor activation. For example, the M1 mAChR selective modulator BQCA acts solely as a PAM of ACh activity when assayed in a cell line with low M1 mAChR density (Canals et al., 2012), and although signaling pathways are weakly coupled to the M1 mAChR, it acts as both a full allosteric agonist and a PAM in a system with a high M1 mAChR reserve. Another example of such a ligand is LY2033298, which is usually considered an M4-selective agonist and has a more complicated mode of action. It also binds to M2 mAChRs (Valant et al., 2012) and mediates both positive and negative allosteric effects, depending on the orthosteric ligand. More concretely, it is a PAM of oxotremorine-M signaling at M2 mAChRs but a NAM of xanomeline at the same receptor (Valant et al., 2012). Another ligand with mixed pharmacodynamics is LY2119620. LY2119620 acts as a PAM of ACh on M2 mAChRs and M4 mAChRs but also exhibits partial allosteric agonism of M2 and M4 mAChRs (Croy et al., 2014; Schober et al., 2014). Dual effects are seen in W-84 which act both as PAM and NAM on M2 mAChRs (Tränkle et al., 2003). Relatively recently, the allosteric modulator of M3 mAChR was described and introduced in the clinic for the treatment of underactive bladder (Okimoto et al., 2021). This drug acts as PAM, potentiate activation, not only on M3 mAChR but with similar effects on M5 mAChR.
Another characteristic of GPCR allostery is biased agonism, which refers to the abilities of different ligands to stabilize a subset of functionally relevant GPCR conformations, such that different signaling outputs are achieved with the exclusion of others (see (Kruse et al., 2014)). An example of this type of ligand is the compound VU0029767, which potentiates ACh-mediated phospholipase C activity via the M1 mAChR. ACh-induced phospholipase D activation is not, however, affected (Marlo et al., 2009).
Similarly, the development of multitarget drugs with anti-psychotic effects (antagonists on D2 dopamine receptors and 5-HT2A serotonin receptors) with allosteric agonism is a promising strategy how to minimize the side effects of drugs (Szabo et al., 2015).
5 Nonsimple targeting of muscarinic drugs
In this section, three examples of experiments in which orthosteric mAChR agonists/antagonists were used are described. These papers were selected to demonstrate not only the need for selectivity awareness but also the role of the mAChR subtype ratio in the respective tissue. This section aims to show the trickiness of muscarinic ligand use and possible ways to avoid misinterpretations of obtained data. The reader can thereby be aware of the problems associated with mAChR agonist and antagonist specificity.
As an example (Morales-Weil et al., 2020), used different muscarinic antagonists to determine the subtype responsible for long-term GABAergic (inhibitory) potentiation. In a conclusion from their experiments, they declared that M1 mAChRs are responsible for the blockade of long-term inhibitory potentiation at GABA synapses in the hippocampus. The authors used pirenzepine at a concentration (100 nmol l−1, i.e., 10−7mol l−1) with which it was not possible to exclude the blockade of not only M1 mAChRs but also other mAChR subtypes, at least in part. Considering the pKis of pirenzepine for specific mAChR subtypes (Myslivecek, 2019) (M1: pKi = 7.8–8.5, M2: pKi = 6.3–6.7, M3: pKi = 6.7–7.1, M4: pKi = 7.1–8.1, and M5: pKi = 6.2–7.1), it is evident that at 10−7 mol l−1, pirenzepine would block the majority of M1 mAChRs and the majority of M4 mAChRs. We can also assume that approximately one-half of M3 mAChRs were occupied by pirenzepine (pKi = 6.7–7.1), that M5 mAChRs were probably occupied in the same way, and that M2 mAChRs were also partly occupied. Under this condition, focal ACh induced short-term depression of GABAergic transmission, which led to the suggestion that presynaptic M2 mAChRs are responsible for this phenomenon. The authors also used AFDX-116 (10 μmol l−1, i.e., 10−5 mol l−1) as an M2 mAChR antagonist and found no effects of this antagonist on long-term inhibitory potentiation, concluding that M2 mAChRs are not involved in this form of long-term plasticity. However, upon comparing the pKis of AFDX-116 for specific mAChR subtypes (Myslivecek, 2019) (M1: pKi = 5.8–6.9, M2: pKi = 7.1–7.3, M3: pKi = 5.5–6.6, M4: pKi = 6.2–7.0, and M5: pKi = 5.4–6.6), it is apparent that not only M2 mAChRs but also M4 mAChRs and some M1, M3 and M5 mAChRs were inhibited by 10−5 mol l−1 AFDX-116. However, considering that the relative density of M1 mAChRs in the whole hippocampus ranges between 69% (Oki et al., 2005) and 91% (Valuskova et al., 2018a) and is approximately 89% in the CA1 hippocampal area (Valuskova et al., 2018a) and that separate studies indicated that M2 and M4 mAChRs were undetectable/insignificant in the hippocampus and CA1 area (Valuskova et al., 2018a) or that M2 mAChRs represented 31% of receptors (Oki et al., 2005) while M4 mAChRs were undetectable (Oki et al., 2005; Valuskova et al., 2018a), it is possible to conclude that the role of M1 mAChRs is indisputable and that M2 mAChRs are not highly present in hippocampal areas. In summary, it is necessary to consider not only the limited selectivity of mAChR antagonists but also the relative presence (or subtype ratios) of the respective mAChR subtypes.
Another aspect that can lead to problems in results interpretations is the in situ hybridization of mAChR subtype nucleotide transcripts in binding studies of receptor presence in target tissues. In very interesting and fundamental work in the field of cholinergic control of biological rhythms, Dojo et al. (2017) described carbachol-induced phase shifts of Per1 rhythms. The authors used cultured suprachiasmatic nucleus (SCN) slices to overcome problems with intracerebroventricular carbachol treatment. Previously, intracerebroventricular carbachol treatment was shown to cause phase delays during the subjective early night and phase advances in the subjective late night [see (Dojo et al., 2017) for details]. Dojo et al. found a carbachol-induced biphasic effect on the phase shift, which was blocked by atropine but not mecamylamine (nicotinic blocker). Thus, the authors concluded that these phase shifts are mAChR-dependent and used in situ hybridization to detect mAChR subtypes, finding M3 and M4 mAChRs expressed in SCN cells. The first effect that cannot be excluded concerning atropine properties is antagonism at nicotinic receptors (see Section 3.1). Although mecamylamine failed to block the phase shift, it is necessary to stress that mecamylamine blocks only some nicotinic receptors or subunits (α3 with pIC50 = 6.4 and α4 with IC50 = 5.3–6.5) (Alexander S. P. H. et al., 2017), while atropine blocks not only mAChRs but also the α5 subunits of nicotinic receptors and other receptors (see above—α2A- and α1D-adrenoceptors, glycine receptors, and SERTs 5-HT1A and 5-HT2C). Thus, the exclusion of nicotinic receptors cannot be absolute. Additionally, other receptors could be affected by the 100 μmol l−1 (10−4 mol l−1) atropine concentration used in this experiments. α4-containing NRs (which form heteropentameric NRs) have a high affinity for nicotine and are the most frequent receptor subtype in both the rodent and human brains (Weinhart et al., 2021). The α5 subunit does not directly contribute to the agonist-binding site but plays a critical role in the functional properties of the receptor (Weinhart et al., 2021). Concerning mAChR subtypes, by in situ hybridization, the authors found that M3 and M4 mAChRs are expressed in SCN cells. However, no M4 mAChRs were found in the SCN (Valuskova et al., 2018b) in KO animals, and the overall density of mAChRs in the SCN was low. The general problem with the method of using nucleotide sequence detection is that this method localizes specific nucleotide transcripts within a portion or section of tissue but does not detect the presence of target proteins or binding sites.
As the last example, we would like to demonstrate the role of mAChRs in addiction. The study by (Wu et al., 2020) described the role of the rostromedial tegmental nucleus (also known as the tail of the ventral tegmental area) in mAChR-regulated opioid addiction. For the identification of mAChRs involved in opioid modulation of rostromedial tegmental GABAergic neurons, the authors used pilocarpine, 4-DAMP, LY2033298, and tropicamide. They concluded that M3 mAChRs are responsible for the opioid modulation of GABAergic neurons. In detail, pilocarpine (considered an M3 mAChR agonist) inhibited the acquisition of morphine-induced conditional place preference, and 4-DAMP (considered an M3 mAChR antagonist) reversed the inhibitory effect of pilocarpine. 4-DAMP also increased locomotor activity, while pilocarpine partially decreased locomotor activity (when combined with morphine). LY2033298 is usually considered an M4-selective agonist and has a more complicated mode of action—it also binds to M2 mAChRs and mediates both positive and negative allosteric effects, depending on the orthosteric ligand. More concretely, it is a PAM of oxotremorine-M signaling at the M2 mAChR but a NAM of xanomeline at the same receptor. LY2033298 and tropicamide (considered an M4 mAChR antagonist) did not affect the acquisition of morphine-induced conditional place preference or locomotor activity. As discussed above, pilocarpine, which is declared an M3 selective agonist, has comparable effects on all mAChR subtypes, and 4-DAMP, which is known as an M3 mAChR-selective antagonist, reveals similar affinity to M4 mAChRs. Tropicamide is a drug with a similar affinity to all mAChRs and is also far from selective in blocking M4 mAChRs. The exact numbers of M3 and M4 mAChRs in the rostromedial tegmental area are not known. Thus, we can use the numbers of these mAChRs in the midbrain as reference values, although with reservation. According to (Oki et al., 2005), 38% of receptors in the midbrain are M3 mAChRs, and no significant number of M4 mAChRs are present. However, this study (Oki et al., 2005) experienced an important problem: in some cases, the total number of mAChRs when summing the numbers of specific mAChR subtypes exceeded the number of mAChRs in WT animals. Thus, one should be cautious in relying on the respective percentages of specific mAChR subtypes. The most abundant subtype in the midbrain is M2 mAChRs. Thus, it should be concluded that the role of M3 mAChRs in mAChR-regulated opioid addiction is highly probable, but we cannot exclude the roles of other mAChR subtypes (M2 mAChRs).
6 Discussion
We have shown in the previous sections that both agonists and antagonists and some allosteric ligands do not have single target (see Figure 1). Thus, the muscarinic drugs can be considered “multitargeting.”

FIGURE 1. The main receptor targets of muscarinic agonists, antagonists and allosteric ligands. See the legend below.
Importantly, the pharmacological data are not identical, and one should also consider which constant is given. From that point of view, the correct assessment (proper steady-state) of the inhibition constant (Ki) is important information. Half-maximal inhibitory concentration (IC50) is not a good predictor of pharmacodynamics properties. It does not account for many factors in drug action (Lamba and Pesaresi, 2022).
This type of drug action is not specific to mAChRs but is, at least in some cases, an advantage of drugs, allowing them to affect more pathway changes in specific diseases (Abatematteo et al., 2021). Surprisingly, we can find that ligands believed to specifically bind to one neurotransmitter receptor type had much wider activities. For example, dopamine receptor ligands have also a wide spectrum of targets (Myslivecek, 2022). This is just one example of such similar affinities to different neurotransmitter receptors, and many others can be found, which indicates that binding ligands that are believed to be specific to other neurotransmitter receptors are a general property of neurotransmitter receptors. Compared to other cholinergic targets, mAChRs are less selective in their orthosteric binding sites; thus, the selection of drugs for mAChR functions must involve more caution.
The use of mAChR agonists and/or antagonists in research on physiological/pathophysiological functions should thus include caution. When choosing a ligand for mAChR subtype determination, one should consider the limited selectivity. As it can be deduced from ligand structures, the ligands have, in many cases, similar structures. Together with the high degree of amino acid sequence homology, this is the reason for the difficulties in subtype function determination.
This structural similarity is one of the causes of subtype similarity in orthosteric ligands binding (see Figure 2). mAChR subtypes are mainly conserved within seven transmembrane zones - the structure to which ligands are bound (e.g., there is approximately 64% of identity between all mAChR subtypes, i.e., M1-M5 (Peralta et al., 1987; Bonner, 1989). In other words, 64% of amino acids are the same in all mAChRs. Thus, if the identical amino acid residue is present in the same position between mAChR subtypes, then the binding of ligand could be similar to mAChR subtypes. This can be demonstrated by similarities of binding for different substances revealed by crystal structure description (Thal et al., 2016). This high degree of sequence conservation does not exclude that orthosteric ligands can differently bind thanks to differences in tertiary structure. The molecular distinction is present in the third intracellular loop (participating in the G protein interactions) between odd- and even-numbered mAChRs (Caulfield and Birdsall, 1998). The overall amino acid chain identity display 43, 35, and 37% between M4 mAChR and M1, M2, and M3 mAChR, respectively (Peralta et al., 1987). In detail, the comparison of the M1, M2, M3, and M4 tiotropium-bound structures (using crystallization) with the structurally similar ligand QNB, revealed considerable differences around residues D3.32, Y7.39, and Y7.43 (Thal et al., 2016), interacting with an amine group. Similarly, other structure–binding properties are discussed in this review (Thal et al., 2016). Recently, the cryo-electron microscopy structures of M1 and M2 mAChR was described (Maeda et al., 2019), the crystal structure of M5 mAChR (Vuckovic et al., 2019), as well as structural insights into the subtype-selective antagonist binding to the M2 mAChR (Suno et al., 2018).
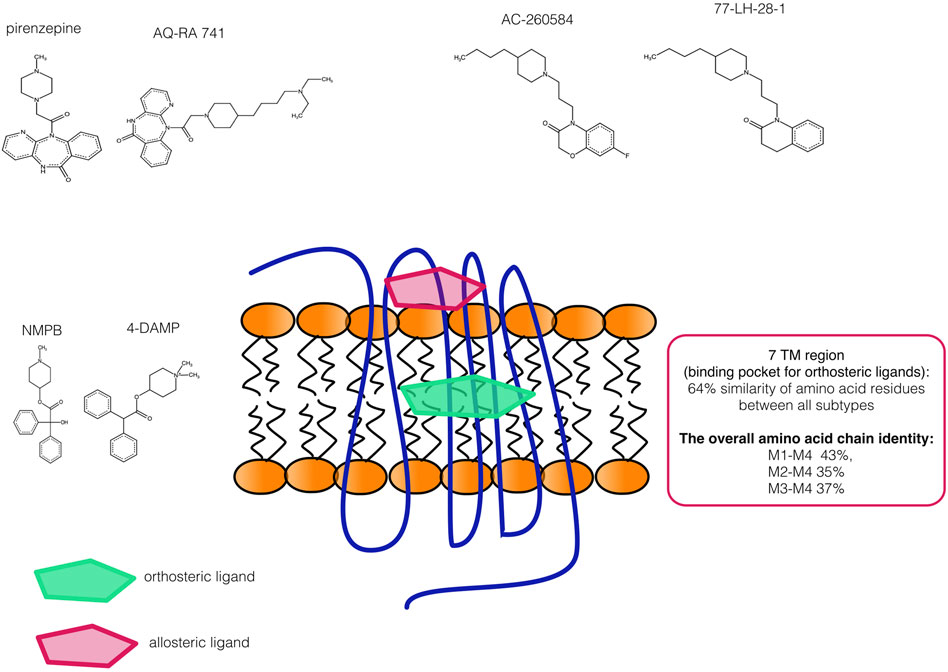
FIGURE 2. The schematic representation of selectivity problem with muscarinic orthosteric ligands. The binding of orthosteric ligands to transmembrane (TM) zones and binding site of allosteric ligands is also sechamatically shown. The similarity in amino acid chain is shown in the frame. Three examples of ligands with similar structure demonstrate that not only amino acid similarity but also ligand structural similarity are the basis of selectivity problems.
In detail, the similarities in orthosteric ligand structures as seen in Figure 2 are one reason for the limited selectivity of ligands. It is possible to demonstrate this by the effort to substitute the part of chains in mAChR ligand structure with other residues (methyl, benzyl, and others) that significantly change the properties of molecules. For examples of these efforts see data on the synthesis of selective ligands (e.g., (Melancon et al., 2012a; Salovich et al., 2012; Gentry et al., 2013a; Gentry et al., 2013b; Le et al., 2013b; Beshore et al., 2018; Nabulsi et al., 2019)). The second reason is the similarities in amino acid residues structure. For example (Suno et al., 2018), have constructed the thermostabilized mutant of M2 mAChRs (S110R) that had a significantly lower affinity for the agonist iperoxo than the wild type, whereas the affinity was unchanged or slightly lower for the inverse agonists (QNB, tiotropium and NMS). There was also difference in binding of antagonists (AF-DX 384 and pirenzepine), versus the inverse agonists with this mutation. This suggest that antagonists bind the M2 mAChR in an allosteric sodium ion–dependent manner, whereas inverse agonists do not. In contrast to these ligands, AFDX-384 had a remarkably higher affinity for the S110R mutant. It is notable that AFDX-384 is bound by M2 mAChR than pirenzepine (see Table 2). Another point is the conformations of critical residues that are needed for the receptor activation (Maeda et al., 2019). These motifs (see (Maeda et al., 2019) for details) are also similar in active conformations of M1 mAChR and M2 mAChR. This suggests that the activation mechanism is shared between M1 and M2 mAChRs, although these receptors are coupled with different G-proteins. (Maeda et al., 2019). Similarly, the amino acid residues, responsible for allosteric binding have been also identified (or more exactly are gradually identified). For example, key residues involved in the activity of BQCA, including Y179 in the second extracellular loop and W4007.35 in TM7, were critical for the activity of all PAMs tested by (Khajehali et al., 2018). This indicates that structurally distinct PAMs share a similar binding site with BQCA. More specifically, an extracellular allosteric site is defined by residues in TM2, TM7 and ECL2 (Khajehali et al., 2018). Similarly (Abdul-Ridha et al., 2014), have optimized the synthesis of benzoquinazolinone 12. They also fully characterized the pharmacology of this drug, finding that its improved potency derived from a 50-fold increase in allosteric site affinity as compared with BQCA, while retaining a similar level of positive cooperativity with acetylcholine.
Important data on the interaction of allosteric compounds with M4 mAChRs were reported by (Wang et al., 2022), where the binding of newly synthesized compound-110, iperoxo, or iperoxo-LY2119620 to M4 mAChR-Gi complex was studied. The authors described different interaction modes and activation mechanisms of M4 mAChR, and the receptor-iperoxo -LY-2119620-Gi cooperativity.
In this review, we tried to include as many ligands as possible for which the relevant off-target were found. The compounds with not clearly defined targets or those that do not target CNS-relevant molecules were excluded.
Most importantly, one should notice that ligands targeting similar (or same structures) reveal also structural similarity. For example, AC-260584 and 77-LH-28-1 (Heinrich et al., 2009) reveal structural similarities (see Figure 2) and can affect D2 DR in nanomolar concentration (see Table 1). In detail, Ki for D2 DR is 0.05, and 0.06, respectively, for AC-260584 and 77-LH-28-1 (Heinrich et al., 2009). Similarly, the structural similarity of atropine and bezatropine could be the reason for nanomolar affinity (pKi = 6.28, and pKi = 7.49) for atropine and benzatropine, respectively) of 5-HT2C serotonin receptors to these ligands (Modell et al., 1989; Gaulton et al., 2016).
To overcome the limited selectivity of orthosteric ligands, it is recommended to carefully study the presence of respective subtypes in specific tissues via KO studies. Two studies conducted on mice with specific deletions of mAChR subtypes can serve as references. The first study by Ito et al., employed KO animals to determine the mAChR subtype numbers in peripheral tissues, such as the salivary glands, lung, heart, stomach, pancreas, bladder, and prostate (Ito et al., 2009). The second study by Oki et al., employed KO animals to investigate the presence of mAChR subtypes in specific areas in the CNS (Oki et al., 2005), including the cerebral cortex, corpus striatum, hippocampus, hypothalamus, thalamus, midbrain, pons-medulla, cerebellum, and spinal cord. Although one may assume that this type of study would provide a clear picture of the presence of mAChR subtypes in specific tissues, a compensatory mechanism also exists—if one subtype is knocked down, the presence of the remaining subtypes in the specific tissue would increase. In fact, an additional problem can also lie in the methodology. (Oki et al., 2005) and (Ito et al., 2009) used direct radioligand binding with hydrophilic ligands in tissue homogenates, which can slightly artificially increase binding. This problem can be overcome by using autoradiography, in which binding can be determined in very small brain areas (Farar and Myslivecek, 2016). Another method reported by (Lebois et al., 2018) employed the detection of mRNA transcript levels expressed as reads per kilobase per million mapped reads. These results are in better agreement with our data obtained using autoradiography.
Thus, the total amount of all mAChR subtypes (i.e., the sum of the density of M1+M2+M3+M4+M5 mAChRs), as reported by (Oki et al., 2005; Ito et al., 2009), could exceed the density of all receptor subtypes determined in WT animals. For example, in the submandibular gland (Ito et al., 2009), 35% M2 mAChRs and 79% M3 mAChRs were found, totaling 114%. It is, therefore, necessary to consider that the major subtype is M3 mAChRs, which are present at more than 70%, and that the second subtype, M2 mAChRs, is present at slightly less than half of the number of M3 mAChRs (i.e., the ratio is approximately 2:1). To present another example of how knocking down a specific subtype can increase the numbers of other subtypes, according to (Oki et al., 2005), 45% of M1 mAChRs, 34% of M2 mAChRs, 19% of M3 mAChRs, and 54% M4 mAChRs are present in the striatum. These numbers total 152% (the total should be 100%). Thus, it is better to count the subtypes according to the M1:M2:M3:M4 ratio, which is 20:22:13:35. According to our measurements using autoradiography (Valuskova et al., 2018a), 37% M1 mAChRs and 46% M4 mAChRs were found, while we did not detect M2 mAChRs. Unfortunately, we did not have M3 (or M5)-KO animals at our disposal. Although the numbers are different, it is evident that the most abundant subtype in the striatum is M4 mAChRs and that a substantial number of M1 mAChRs are present. Recently, we proposed an algorithm for subtype determination (Myslivecek, 2021). Here, we would like to provide the reader with a more detailed procedure for subtype identification. First, as described above, it is necessary to establish a rough ratio of the mAChR subtypes present in the tissue of interest. Let us use the example of the striatum, with an M1:M4 ratio between 36-45:55-64. The second step is the selection of a ligand able to discriminate between M1 mAChRs and M4 mAChRs. This criterion is partly fulfilled by antagonist PD102807 (the orthosteric site does not provide enough selectivity—see Section 3), which has a selectivity for M4 mAChRs over 2 grades of magnitude higher than that for M1 mAChRs (pKi = 7.3–7.4 vs. pKi = 5.3–5.5; see (Myslivecek, 2019)). As mentioned above, to date, no interactions with other targets are known. Thus, the third step, which is the most difficult, is the choice of the antagonist concentration. The pKi defines the concentration at which a ligand binds to 50% of available binding sites. In our case, the pKi = 7.3–7.4 indicates approximately 39.8–50.1 nmol/L. This means that PD 102807 at a concentration of approximately 45 nmol/L will occupy approximately 50% of M4 mAChRs. Virtually no M1 mAChRs will be inhibited at this concentration (Bohme et al., 2002). The affinity of M1 mAChRs for PD 102807 is 72-fold lower than that of M4 mAChRs (Augelli-Szafran et al., 1998). This means that at concentrations approaching 1 μmol/L, all M4 mAChRs will be blocked, but almost no M1 mAChRs will be affected by this drug. However, the affinity of M3 mAChRs for this drug is higher (there is a 10-fold difference—see (Augelli-Szafran et al., 1998)) than that of M2 mAChRs by 38 fold. According to the possible ratios (Oki et al., 2005; Valuskova et al., 2018a), and assuming that the researcher uses PD 102807 at 1 μmol/L, all M4 mAChRs will be blocked, and almost no M1 or M2 mAChRs will be affected by this drug. M3 mAChRs are present at only one-third of the number of M4 mAChRs and have a 10-fold lower affinity for PD 102807. However, discriminating between M3 and M4 mAChRs is impossible, as no drug with an appropriate difference in selectivity is known. Thus, it is necessary to conclude that there is a possibility of minor involvement of M3 mAChRs (with respect to their 10-fold lower affinity for PD 102807 and one-third lower density in comparison to M4 mAChRs).
As additional examples (Oki et al., 2005), identified three mAChR subtypes in the hippocampus (M1-M3) and pons medulla (M2-M4), two mAChR subtypes in the hypothalamus and midbrain (M2-M3), and only M2 mAChRs in the cerebellum and spinal cord. If M1-M3 mAChR subtypes are present in the hippocampus, then it is possible to use pirenzepine, which has approximately one grade of magnitude higher selectivity for M1 mAChRs than M2 and M3 mAChRs. According to (Oki et al., 2005), 69% M1, 30% M2, and 17% M3 mAChRs were present, totaling 116%. The corrected ratio should thus be 59:26:15. On the other hand, we used tritiated pirenzepine for M1 mAChR determination and showed that in M1 mAChR-KO animals (Valuskova et al., 2018a), the binding of 3H-pirenzepine was almost completely abolished in hippocampal areas (dorsal hippocampus, CA1, CA3 area, and dentate gyrus). Examples of antagonists that could be used in specific areas according to the presence of mAChR subtypes are as follows: in the pons-medulla (M2-M4), PD12807 would identify M4 mAChRs, and in the hypothalamus and midbrain (M2-M3), methoctramine would identify M2 mAChRs.
As mentioned above, a new method for identifying mAChR subtypes involved in specific functions involves the use of allosteric ligands (PAMs and NAMs), as recently demonstrated in M4 and M5 mAChR determination as important structures in alcohol-seeking behaviors (Walker et al., 2021). Similarly, some new positron emission tomography (PET) tracers have exhibited promising selectivity (Ozenil et al., 2021).
7 Conclusion
The main conclusion of this review is that the cholinergic system in different peripheral/CNS functions should first be studied by searching for the mAChR subtype affinities of ligands of choice. Although it is sometimes difficult to differentiate between specific subtypes, there are some exceptions in which a ligand can be used as a specific means of mAChR subtype identification (Valuskova et al., 2018a). Another search should determine the respective densities of mAChR subtypes in specific tissues. The combination of these two searches should increase accurate conclusions concerning mAChR subtypes involved in specific functions.
One way to identify the subtypes involved is to use conditional KO animals. However, it is also necessary to keep in mind that targeting one receptor can affect the functions of other receptors. Typically, this is the case for mAChRs vs. adrenoceptors in the heart (Myslivecek et al., 1996; Myslivecek et al., 1998; Myslivecek et al., 2004; Myslivecek et al., 2008c; Benes et al., 2012; Laukova et al., 2014; Tomankova et al., 2015). Moreover, conditional, tissue-specific KO is a better option than classical KO. The finding that muscarinic agonists or antagonists have a multitarget nature is not unique, and one should be aware that this can be true for many GPCR agonists/antagonists.
Importantly, limited binding selectivity for specific neurotransmitter receptors is not a property relevant to only mAChRs but is also a general attribute of most neurotransmitter receptors (see the examples for dopamine receptors in the Discussion).
Author contributions
JM conceptualized, searched references, wrote, and revised the manuscript.
Funding
This research was funded by the Program “Cooperatio” in Pharmaceutical Sciences from Charles University.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abatematteo F. S., Niso M., Contino M., Leopoldo M., Abate C. (2021). Multi-target directed ligands (MTDLs) binding the σ1 receptor as promising therapeutics: State of the art and perspectives. Int. J. Mol. Sci. 22, 6359. doi:10.3390/ijms22126359
Abdul-Ridha A., Lane J. R., Mistry S. N., López L., Sexton P. M., Scammells P. J., et al. (2014). Mechanistic insights into allosteric structure-function relationships at the M1 muscarinic acetylcholine receptor. J. Biol. Chem. 289, 33701–33711. doi:10.1074/jbc.M114.604967
Ahlin G., Karlsson J., Pedersen J. M., Gustavsson L., Larsson R., Matsson P., et al. (2008). Structural requirements for drug inhibition of the liver specific human organic cation transport protein 1. J. Med. Chem. 51, 5932–5942. doi:10.1021/jm8003152
Alexander S. P., Christopoulos A., Davenport A. P., Kelly E., Marrion N. V., Peters J. A., et al. (2017a). The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 174, S17–S129. doi:10.1111/bph.13878
Alexander S. P. H., Peters J. A., Kelly E., Marrion N. V., Faccenda E., Harding S. D., et al. (2017b). The concise guide to pharmacology 2017/18: Ligand‐gated ion channels. Br. J. Pharmacol. 174, S130–S159. doi:10.1111/bph.13879
Apelt J., Ligneau X., Pertz H. H., Arrang J. M., Ganellin C. R., Schwartz J. C., et al. (2002). Development of a new class of nonimidazole histamine H(3) receptor ligands with combined inhibitory histamine N-methyltransferase activity. J. Med. Chem. 45, 1128–1141. doi:10.1021/jm0110845
Arunotayanun W., Dalley J. W., Huang X. P., Setola V., Treble R., Iversen L., et al. (2013). An analysis of the synthetic tryptamines AMT and 5-MeO-DALT: Emerging 'novel psychoactive drugs. Bioorg. Med. Chem. Lett. 23, 3411–3415. doi:10.1016/j.bmcl.2013.03.066
Augelli-Szafran C. E., Jaen J. C., Moreland D. W., Nelson C. B., Penvose-Yi J. R., Schwarz R. D. (1998). Identification and characterization of m4 selective muscarinic antagonists. Bioorg. Med. Chem. Lett. 8, 1991–1996. doi:10.1016/s0960-894x(98)00351-5
Avlani V. A., Gregory K. J., Morton C. J., Parker M. W., Sexton P. M., Christopoulos A. (2007). Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J. Biol. Chem. 282, 25677–25686. doi:10.1074/jbc.M702311200
Bandyopadhyaya A., Rajagopalan D. R., Rath N. P., Herrold A., Rajagopalan R., Napier T. C., et al. (2012). The synthesis and receptor binding affinities of DDD-016, a novel, potential, atypical antipsychotic. MedChemComm 3, 580–583. doi:10.1039/c2md00311b
Bardien-Kruger S., Wulff H., Arieff Z., Brink P., Chandy K. G., Corfield V. (2002). Characterisation of the human voltage-gated potassium channel gene, KCNA7, a candidate gene for inherited cardiac disorders, and its exclusion as cause of progressive familial heart block I (PFHBI). Eur. J. Hum. Genet. 10, 36–43. doi:10.1038/sj.ejhg.5200739
Beattie D., Beer D., Bradley M. E., Bruce I., Charlton S. J., Cuenoud B. M., et al. (2012). An investigation into the structure–activity relationships associated with the systematic modification of the β2-adrenoceptor agonist indacaterol. Bioorg. Med. Chem. Lett. 22, 6280–6285. doi:10.1016/j.bmcl.2012.07.096
Becerra M. A., Herrera M. D., Marhuenda E. (2001). Action of tacrine on muscarinic receptors in rat intestinal smooth muscle. J. Auton. Pharmacol. 21, 113–119. doi:10.1046/j.1365-2680.2001.00213.x
Belzer M., Morales M., Jagadish B., Mash E. A., Wright S. H. (2013). Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J. Pharmacol. Exp. Ther. 346, 300–310. doi:10.1124/jpet.113.203257
Benes J., Novakova M., Rotkova J., Farar V., Kvetnansky R., Riljak V., et al. (2012). Beta3 adrenoceptors substitute the role of M-2 muscarinic receptor in coping with cold stress in the heart: Evidence from M2KO mice. Cell. Mol. Neurobiol. 32, 859–869. doi:10.1007/s10571-011-9781-3
Bertron J. L., Cho H. P., Garcia-Barrantes P. M., Panarese J. D., Salovich J. M., Nance K. D., et al. (2018). The discovery of VU0486846: Steep SAR from a series of M(1) PAMs based on a novel benzomorpholine core. Bioorg. Med. Chem. Lett. 28, 2175–2179. doi:10.1016/j.bmcl.2018.05.009
Beshore D. C., C N. D. M., Chang R. K., Greshock T. J., Ma L., Wittmann M., et al. (2018). MK-7622: A first-in-class M(1) positive allosteric modulator development candidate. ACS Med. Chem. Lett. 9, 652–656. doi:10.1021/acsmedchemlett.8b00095
Birdsall N. J., Farries T., Gharagozloo P., Kobayashi S., Lazareno S., Sugimoto M. (1999). Subtype-selective positive cooperative interactions between brucine analogs and acetylcholine at muscarinic receptors: Functional studies. Mol. Pharmacol. 55, 778–786.
Birdsall N. J., Lazareno S., Popham A., Saldanha J. (2001). Multiple allosteric sites on muscarinic receptors. Life Sci. 68, 2517–2524. doi:10.1016/s0024-3205(01)01047-5
Bock A., Schrage R., Mohr K. (2018). Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 136, 427–437. doi:10.1016/j.neuropharm.2017.09.024
Boess F. G., Riemer C., Bös M., Bentley J., Bourson A., Sleight A. J. (1998). The 5-hydroxytryptamine6 receptor-selective radioligand [3H]Ro 63-0563 labels 5-hydroxytryptamine receptor binding sites in rat and porcine striatum. Mol. Pharmacol. 54, 577–583. doi:10.1124/mol.54.3.577
Bohme T. M., Augelli-Szafran C. E., Hallak H., Pugsley T., Serpa K., Schwarz R. D. (2002). Synthesis and pharmacology of benzoxazines as highly selective antagonists at M(4) muscarinic receptors. J. Med. Chem. 45, 3094–3102. doi:10.1021/jm011116o
Böhme T. M., Keim C., Kreutzmann K., Linder M., Dingermann T., Dannhardt G., et al. (2003). Structure-activity relationships of dimethindene derivatives as new M2-selective muscarinic receptor antagonists. J. Med. Chem. 46, 856–867. doi:10.1021/jm020895l
Bolchi C., Valoti E., Binda M., Fasoli F., Ferrara R., Fumagalli L., et al. (2013). Design, synthesis and binding affinity of acetylcholine carbamoyl analogues. Bioorg. Med. Chem. Lett. 23, 6481–6485. doi:10.1016/j.bmcl.2013.09.023
Bolden C., Cusack B., Richelson E. (1992). Antagonism by antimuscarinic and neuroleptic compounds at the five cloned human muscarinic cholinergic receptors expressed in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 260, 576–580.
Bolognesi M. L., Minarini A., Budriesi R., Cacciaguerra S, Chiarini A., Spampinato S., et al. (1998). Universal template approach to drug design: Polyamines as selective muscarinic receptor antagonists. J. Med. Chem. 41, 4150–4160. doi:10.1021/jm981038d
Bonner T. I. (1989). The molecular basis of muscarinic receptor diversity. Trends Neurosci. 12, 148–151. doi:10.1016/0166-2236(89)90054-4
Brady A. E., Jones C. K., Bridges T. M., Kennedy J. P., Thompson A. D., Heiman J. U., et al. (2008). Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 327, 941–953. doi:10.1124/jpet.108.140350
Bridges T. M., Kennedy J. P., Cho H. P., Breininger M. L., Gentry P. R., Hopkins C. R., et al. (2010b). Chemical lead optimization of a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) lead. Part I: Development of the first highly selective M5 PAM. Bioorg. Med. Chem. Lett. 20, 558–562. doi:10.1016/j.bmcl.2009.11.089
Bridges T. M., Kennedy J. P., Hopkins C. R., Conn P. J., Lindsley C. W. (2010a). Heterobiaryl and heterobiaryl ether derived M5 positive allosteric modulators. Bioorg. Med. Chem. Lett. 20, 5617–5622. doi:10.1016/j.bmcl.2010.08.042
Bridges T. M., Marlo J. E., Niswender C. M., Jones C. K., Jadhav S. B., Gentry P. R., et al. (2009). Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J. Med. Chem. 52, 3445–3448. doi:10.1021/jm900286j
Broad L. M., Sanger H. E., Mogg A. J., Colvin E. M., Zwart R., Evans D. A., et al. (2019). Identification and pharmacological profile of SPP1, a potent, functionally selective and brain penetrant agonist at muscarinic M(1) receptors. Br. J. Pharmacol. 176, 110–126. doi:10.1111/bph.14510
Brunhofer G., Fallarero A., Karlsson D., Batista-Gonzalez A., Shinde P., Gopi Mohan C., et al. (2012). Exploration of natural compounds as sources of new bifunctional scaffolds targeting cholinesterases and beta amyloid aggregation: The case of chelerythrine. Bioorg. Med. Chem. 20, 6669–6679. doi:10.1016/j.bmc.2012.09.040
Brus B., Košak U., Turk S., Pišlar A., Coquelle N., Kos J., et al. (2014). Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 57, 8167–8179. doi:10.1021/jm501195e
Bubser M., Bridges T. M., Dencker D., Gould R. W., Grannan M., Noetzel M. J., et al. (2014). Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem. Neurosci. 5, 920–942. doi:10.1021/cn500128b
Buckley N. J., Bonner T. I., Buckley C. M., Brann M. R. (1989). Antagonist binding properties of five cloned muscarinic receptors expressed in CHO-K1 cells. Mol. Pharmacol. 35, 469–476.
Buckley N. J., Hulme E. C., Birdsall N. J. (1990). Use of clonal cell lines in the analysis of neurotransmitter receptor mechanisms and function. Biochim. Biophys. Acta 1055, 43–53. doi:10.1016/0167-4889(90)90089-v
Burger W. a. C., Gentry P. R., Berizzi A. E., Vuckovic Z., Van Der Westhuizen E. T., Thompson G., et al. (2021). Identification of a novel allosteric site at the M(5) muscarinic acetylcholine receptor. ACS Chem. Neurosci. 12, 3112–3123. doi:10.1021/acschemneuro.1c00383
Butini S., Campiani G., Borriello M., Gemma S., Panico A., Persico M., et al. (2008). Exploiting protein fluctuations at the active-site gorge of human cholinesterases: Further optimization of the design strategy to develop extremely potent inhibitors. J. Med. Chem. 51, 3154–3170. doi:10.1021/jm701253t
Canals M., Lane J. R., Wen A., Scammells P. J., Sexton P. M., Christopoulos A. (2012). A monod-wyman-changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation *. J. Biol. Chem. 287, 650–659. doi:10.1074/jbc.M111.314278
Cantı́ C., Bodas E., Marsal J., Solsona C. (1998). Tacrine and physostigmine block nicotinic receptors in Xenopus oocytes injected with Torpedo electroplaque membranes. Eur. J. Pharmacol. 363, 197–202. doi:10.1016/s0014-2999(98)00793-6
Carlsson B., Singh B. N., Temciuc M., Nilsson S., Li Y.-L., Mellin C., et al. (2002). Synthesis and preliminary characterization of a novel antiarrhythmic compound (KB130015) with an improved toxicity profile compared with amiodarone. J. Med. Chem. 45, 623–630. doi:10.1021/jm001126+
Carr B. J., Mihara K., Ramachandran R., Saifeddine M., Nathanson N. M., Stell W. K., et al. (2018). Myopia-inhibiting concentrations of muscarinic receptor antagonists block Activation of Alpha2A-adrenoceptors in vitro. Invest. Ophthalmol. Vis. Sci. 59, 2778–2791. doi:10.1167/iovs.17-22562
Caulfield M. P., Birdsall N. J. (1998). International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 50, 279–290.
Chackalamannil S., Ahn H. S., Xia Y., Doller D., Foster C. (2003). Potent non-peptide thrombin receptor antagonists. Curr. Med. Chem. Cardiovasc. Hematol. Agents 1, 37–45. doi:10.2174/1568016033356706
Chahdi A., Daeffler L., Gies J. P., Landry Y. (1998). Drugs interacting with G protein alpha subunits: Selectivity and perspectives. Fundam. Clin. Pharmacol. 12, 121–132. doi:10.1111/j.1472-8206.1998.tb00932.x
Chatelain P., Meysmans L., Matteazzi J. R., Beaufort P., Clinet M. (1995). Interaction of the antiarrhythmic agents SR 33589 and amiodarone with the beta-adrenoceptor and adenylate cyclase in rat heart. Br. J. Pharmacol. 116, 1949–1956. doi:10.1111/j.1476-5381.1995.tb16397.x
Chelliah M. V., Chackalamannil S., Xia Y., Greenlee W. J., Ahn H. S., Kurowski S., et al. (2014). Himbacine-derived thrombin receptor antagonists: c7-aminomethyl and c9a-hydroxy analogues of vorapaxar. ACS Med. Chem. Lett. 5, 183–187. doi:10.1021/ml400452v
Chen E. C., Khuri N., Liang X., Stecula A., Chien H. C., Yee S. W., et al. (2017). Discovery of competitive and noncompetitive ligands of the organic cation transporter 1 (OCT1; SLC22A1). J. Med. Chem. 60, 2685–2696. doi:10.1021/acs.jmedchem.6b01317
Chen W.-Y., Wen Y.-C., Lin S.-R., Yeh H.-L., Jiang K.-C., Chen W.-H., et al. (2021). Nerve growth factor interacts with CHRM4 and promotes neuroendocrine differentiation of prostate cancer and castration resistance. Commun. Biol. 4, 22. doi:10.1038/s42003-020-01549-1
Cheng K., Khurana S., Chen Y., Kennedy R. H., Zimniak P., Raufman J. P. (2002). Lithocholylcholine, a bile acid/acetylcholine hybrid, is a muscarinic receptor antagonist. J. Pharmacol. Exp. Ther. 303, 29–35. doi:10.1124/jpet.102.036376
Cheng Q., Shah N., Bröer A., Fairweather S., Jiang Y., Schmoll D., et al. (2017). Identification of novel inhibitors of the amino acid transporter B(0) AT1 (SLC6A19), a potential target to induce protein restriction and to treat type 2 diabetes. Br. J. Pharmacol. 174, 468–482. doi:10.1111/bph.13711
Chidlow G., Osborne N. N. (1997). Antagonism of muscarinic receptors in the rabbit iris-ciliary body by 8-OH-DPAT and other 5-HT1A receptor agonists. J. Neural Transm. 104, 1015–1025. doi:10.1007/BF01273315
Choppin A., Stepan G. J., Loury D. N., Watson N., Eglen R. M. (1999). Characterization of the muscarinic receptor in isolated uterus of sham operated and ovariectomized rats. Br. J. Pharmacol. 127, 1551–1558. doi:10.1038/sj.bjp.0702696
Christopoulos A., Changeux J.-P., Catterall W. A., Fabbro D., Burris T. P., Cidlowski J. A., et al. (2014). International union of basic and clinical pharmacology. XC. Multisite pharmacology: Recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol. Rev. 66, 918–947. doi:10.1124/pr.114.008862
Clark A. L., Mitchelson F. (1976). The inhibitory effect of gallamine on muscarinic receptors. Br. J. Pharmacol. 58, 323–331. doi:10.1111/j.1476-5381.1976.tb07708.x
Connolly G. P., Stone T. W. (1995). Adenosine selectively depresses muscarinic compared with non-muscarinic receptor mediated depolarisation of the rat superior cervical ganglion. Gen. Pharmacol. 26, 865–873. doi:10.1016/0306-3623(94)00257-n
Connolly S., Alcaraz L., Bailey A., Cadogan E., Christie J., Cook A. R., et al. (2011). Design-driven LO: The discovery of new ultra long acting dibasic β2-adrenoceptor agonists. Bioorg. Med. Chem. Lett. 21, 4612–4616. doi:10.1016/j.bmcl.2011.05.097
Coughlin Q., Hopper A. T., Blanco M.-J., Tirunagaru V., Robichaud A. J., Doller D. (2019). Allosteric modalities for membrane-bound receptors: Insights from drug hunting for brain diseases. J. Med. Chem. 62, 5979–6002. doi:10.1021/acs.jmedchem.8b01651
Croy C. H., Chan W. Y., Castetter A. M., Watt M. L., Quets A. T., Felder C. C. (2016). Characterization of PCS1055, a novel muscarinic M4 receptor antagonist. Eur. J. Pharmacol. 782, 70–76. doi:10.1016/j.ejphar.2016.04.022
Croy C. H., Schober D. A., Xiao H., Quets A., Christopoulos A., Felder C. C. (2014). Characterization of the novel positive allosteric modulator, LY2119620, at the muscarinic M2 and M4 receptors. Mol. Pharmacol. 86, 106–115. doi:10.1124/mol.114.091751
Daval S. B., Valant C., Bonnet D., Kellenberger E., Hibert M., Galzi J.-L., et al. (2012). Fluorescent derivatives of AC-42 to probe bitopic orthosteric/allosteric binding mechanisms on muscarinic M1 receptors. J. Med. Chem. 55, 2125–2143. doi:10.1021/jm201348t
Davoren J. E., Lee C. W., Garnsey M., Brodney M. A., Cordes J., Dlugolenski K., et al. (2016). Discovery of the potent and selective M1 PAM-agonist N-[(3R, 4S)-3-Hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1, 3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): Evaluation of efficacy and cholinergic side effects. J. Med. Chem. 59, 6313–6328. doi:10.1021/acs.jmedchem.6b00544
Dean B., Scarr E. (2020). Muscarinic M1 and M4 receptors: Hypothesis driven drug development for schizophrenia. Psychiatry Res. 288, 112989. doi:10.1016/j.psychres.2020.112989
Dhein S., Van Koppen C. J., Brodde O. E. (2001). Muscarinic receptors in the mammalian heart. Pharmacol. Res. 44, 161–182. doi:10.1006/phrs.2001.0835
Disingrini T., Muth M., Dallanoce C., Barocelli E., Bertoni S., Kellershohn K., et al. (2006). Design, synthesis, and action of oxotremorine-related hybrid-type Allosteric modulators of muscarinic acetylcholine receptors. J. Med. Chem. 49, 366–372. doi:10.1021/jm050769s
Dojo K., Yamaguchi Y., Fustin J.-M., Doi M., Kobayashi M., Okamura H. (2017). Carbachol induces phase-dependent phase shifts of Per1 transcription rhythms in cultured suprachiasmatic nucleus slices. J. Biol. Rhythms 32, 101–108. doi:10.1177/0748730417691205
Doods H. N., Entzeroth M., Ziegler H., Mayer N., Holzer P. (1994). Pharmacological profile of selective muscarinic receptor antagonists on Guinea-pig ileal smooth muscle. Eur. J. Pharmacol. 253, 275–281. doi:10.1016/0014-2999(94)90202-x
Doods H. N., Willim K. D., Boddeke H. W., Entzeroth M. (1993). Characterization of muscarinic receptors in Guinea-pig uterus. Eur. J. Pharmacol. 250, 223–230. doi:10.1016/0014-2999(93)90385-u
Dupuis D. S., Mannoury La Cour C., Chaput C., Verrièle L., Lavielle G., Millan M. J. (2008). Actions of novel agonists, antagonists and antipsychotic agents at recombinant rat 5-HT6 receptors: A comparative study of coupling to G alpha s. Eur. J. Pharmacol. 588, 170–177. doi:10.1016/j.ejphar.2008.04.039
Eglen R. M., Bonhaus D. W., Johnson L. G., Leung E., Clark R. D. (1995). Pharmacological characterization of two novel and potent 5-HT4 receptor agonists, RS 67333 and RS 67506, in vitro and in vivo. Br. J. Pharmacol. 115, 1387–1392. doi:10.1111/j.1476-5381.1995.tb16628.x
Eglen R. M., Nahorski S. R. (2000). The muscarinic M(5) receptor: A silent or emerging subtype? Br. J. Pharmacol. 130, 13–21. doi:10.1038/sj.bjp.0703276
Eglen R. M., Whiting R. L. (1987). Competitive and non-competitive antagonism exhibited by 'selective' antagonists at atrial and ileal muscarinic receptor subtypes. Br. J. Pharmacol. 90, 701–707. doi:10.1111/j.1476-5381.1987.tb11223.x
Farar V., Myslivecek J. (2016). “Autoradiography assessment of muscarinic receptors in the central nervous system,” in Muscarinic receptor: From structure to animal models. Editors J. Myslivecek, and J. Jakubik (New York: Springer), 159–180.
Farde L., Suhara T., Halldin C., Nybäck H., Nakashima Y., Swahn C. G., et al. (1996). PET study of the M1-agonists [11C]xanomeline and [11C]butylthio-TZTP in monkey and man. Dementia 7, 187–195. doi:10.1159/000106877
Felder C. C., Goldsmith P. J., Jackson K., Sanger H. E., Evans D. A., Mogg A. J., et al. (2018). Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology 136, 449–458. doi:10.1016/j.neuropharm.2018.01.028
Fernández De Sevilla D., Núñez A., Buño W. (2021). Muscarinic receptors, from synaptic plasticity to its role in network activity. Neuroscience 456, 60–70. doi:10.1016/j.neuroscience.2020.04.005
Ferrari-Dileo G., Waelbroeck M., Mash D. C., Flynn D. D. (1994). Selective labeling and localization of the M4 (m4) muscarinic receptor subtype. Mol. Pharmacol. 46, 1028–1035.
Fisher A. (2008). Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer's disease. Neurotherapeutics 5, 433–442. doi:10.1016/j.nurt.2008.05.002
Flamez A., De Backer J.-P., Wilczak N., Vauquelin G., De Keyser J. (1994). [3H]Clozapine is not a suitable radioligand for the labelling of D4 dopamine receptors in postmortem human brain. Neurosci. Lett. 175, 17–20. doi:10.1016/0304-3940(94)91067-7
Foster D. J., Bryant Z. K., Conn P. J. (2021). Targeting muscarinic receptors to treat schizophrenia. Behav. Brain Res. 405, 113201. doi:10.1016/j.bbr.2021.113201
Fowler C. J., Ahlgren P. C., O'neill C. (1991). Antagonism by 8-hydroxy-2(di-n-propylamino)tetraline and other serotonin agonists of muscarinic M1-type receptors coupled to inositol phospholipid breakdown in human IMR-32 and SK-N-MC neuroblastoma cells. Life Sci. 48, 959–967. doi:10.1016/0024-3205(91)90361-e
Fruchart-Gaillard C., Mourier G., Marquer C., Ménez A., Servent D. (2006). Identification of various allosteric interaction sites on M1 muscarinic receptor using 125I-Met35-Oxidized muscarinic toxin 7. Mol. Pharmacol. 69, 1641–1651. doi:10.1124/mol.105.020883
Gaulton A., Hersey A., Nowotka M., Bento A. P., Chambers J., Mendez D., et al. (2016). The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954. doi:10.1093/nar/gkw1074
Gentry P. R., Bridges T. M., Lamsal A., Vinson P. N., Smith E., Chase P., et al. (2013a). Discovery of ML326: The first sub-micromolar, selective M5 PAM. Bioorg. Med. Chem. Lett. 23, 2996–3000. doi:10.1016/j.bmcl.2013.03.032
Gentry P. R., Kokubo M., Bridges T. M., Cho H. P., Smith E., Chase P., et al. (2014a). Discovery, synthesis and characterization of a highly muscarinic acetylcholine receptor (mAChR)-selective M5-orthosteric antagonist, VU0488130 (ML381): A novel molecular probe. ChemMedChem 9, 1677–1682. doi:10.1002/cmdc.201402051
Gentry P. R., Kokubo M., Bridges T. M., Kett N. R., Harp J. M., Cho H. P., et al. (2013b). Discovery of the first M5-selective and CNS penetrant negative allosteric modulator (NAM) of a muscarinic acetylcholine receptor: (S)-9b-(4-chlorophenyl)-1-(3, 4-difluorobenzoyl)-2, 3-dihydro-1H-imidazo[2, 1-a]isoindol-5(9bH)-one (ML375). J. Med. Chem. 56, 9351–9355. doi:10.1021/jm4013246
Gentry P. R., Kokubo M., Bridges T. M., Noetzel M. J., Cho H. P., Lamsal A., et al. (2014b). Development of a highly potent, novel M5 positive allosteric modulator (PAM) demonstrating CNS exposure: 1-((1H-indazol-5-yl)sulfoneyl)-N-ethyl-N-(2-(trifluoromethyl)benzyl)piperidine-4-carboxamide (ML380). J. Med. Chem. 57, 7804–7810. doi:10.1021/jm500995y
Ghoneim O. M., Legere J. A., Golbraikh A., Tropsha A., Booth R. G. (2006). Novel ligands for the human histamine H1 receptor: Synthesis, pharmacology, and comparative molecular field analysis studies of 2-dimethylamino-5-(6)-phenyl-1, 2, 3, 4-tetrahydronaphthalenes. Bioorg. Med. Chem. 14, 6640–6658. doi:10.1016/j.bmc.2006.05.077
Griffith R. C., Gentile R. J., Robichaud R. C., Frankenheim J. (1984). cis-1, 3, 4, 6, 7, 11b-Hexahydro-2-methyl-7-phenyl-2H-pyrazino[2, 1-a] isoquinoline: a new atypical antidepressant. J. Med. Chem. 27, 995–1003. doi:10.1021/jm00374a011
Hall M. D., Salam N. K., Hellawell J. L., Fales H. M., Kensler C. B., Ludwig J. A., et al. (2009). Synthesis, activity, and pharmacophore development for isatin-β-thiosemicarbazones with selective activity toward multidrug-resistant cells. J. Med. Chem. 52, 3191–3204. doi:10.1021/jm800861c
Hals P. A., Hall H., Dahl S. G. (1988). Muscarinic cholinergic and histamine H1 receptor binding of phenothiazine drug metabolites. Life Sci. 43, 405–412. doi:10.1016/0024-3205(88)90519-x
Harada T., Fushimi K., Kato A., Ito Y., Nishijima S., Sugaya K., et al. (2010). Demonstration of muscarinic and nicotinic receptor binding activities of distigmine to treat detrusor underactivity. Biol. Pharm. Bull. 33, 653–658. doi:10.1248/bpb.33.653
Harvey A. L., Kornisiuk E., Bradley K. N., Cerveñansky C., Durán R., Adrover M., et al. (2002). Effects of muscarinic toxins MT1 and MT2 from green mamba on different muscarinic cholinoceptors. Neurochem. Res. 27, 1543–1554. doi:10.1023/a:1021660708187
Hasuo H., Matsuoka T., Akasu T. (2002). Activation of presynaptic 5-hydroxytryptamine 2A receptors facilitates excitatory synaptic transmission via protein kinase C in the dorsolateral septal nucleus. J. Neurosci. 22, 7509–7517. doi:10.1523/jneurosci.22-17-07509.2002
Hegde S. S., Pulido-Rios M. T., Luttmann M. A., Foley J. J., Hunsberger G. E., Steinfeld T., et al. (2018). Pharmacological properties of revefenacin (TD-4208), a novel, nebulized long-acting, and lung selective muscarinic antagonist, at human recombinant muscarinic receptors and in rat, Guinea pig, and human isolated airway tissues. Pharmacol. Res. Perspect. 6, e00400. doi:10.1002/prp2.400
Heinrich J. N., Butera J. A., Carrick T., Kramer A., Kowal D., Lock T., et al. (2009). Pharmacological comparison of muscarinic ligands: Historical versus more recent muscarinic M1-preferring receptor agonists. Eur. J. Pharmacol. 605, 53–56. doi:10.1016/j.ejphar.2008.12.044
Hern J. A., Baig A. H., Mashanov G. I., Birdsall B., Corrie J. E., Lazareno S., et al. (2010). Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. U. S. A. 107, 2693–2698. doi:10.1073/pnas.0907915107
Heusler P., Bruins Slot L., Tourette A., Tardif S., Cussac D. (2011). The clozapine metabolite N-desmethylclozapine displays variable activity in diverse functional assays at human dopamine D₂ and serotonin 5-HT₁A receptors. Eur. J. Pharmacol. 669, 51–58. doi:10.1016/j.ejphar.2011.07.031
Hill K., Mcnulty S., Randall A. D. (2004). Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn. Schmiedeb. Arch. Pharmacol. 370, 227–237. doi:10.1007/s00210-004-0981-y
Huang F., Buchwald P., Browne C. E., Farag H. H., Wu W. M., Ji F., et al. (2001). Receptor binding studies of soft anticholinergic agents. AAPS PharmSci 3, E30. doi:10.1208/ps030430
Hudkins R. L., Dehaven-Hudkins D. L., Stubbins J. F. (1991). Muscarinic receptor binding profile of para-substituted caramiphen analogues. J. Med. Chem. 34, 2984–2989. doi:10.1021/jm00114a005
Hung Y. H., Huang H. L., Chen W. C., Yen M. C., Cho C. Y., Weng T. Y., et al. (2017). Argininosuccinate lyase interacts with cyclin A2 in cytoplasm and modulates growth of liver tumor cells. Oncol. Rep. 37, 969–978. doi:10.3892/or.2016.5334
Ito Y., Oyunzul L., Seki M., Fujino Oki T., Matsui M., Yamada S. (2009). Quantitative analysis of the loss of muscarinic receptors in various peripheral tissues in M1-M5 receptor single knockout mice. Br. J. Pharmacol. 156, 1147–1153. doi:10.1111/j.1476-5381.2009.00113.x
Jakubík J., Bacáková L., El-Fakahany E. E., Tucek S. (1997). Positive cooperativity of acetylcholine and other agonists with allosteric ligands on muscarinic acetylcholine receptors. Mol. Pharmacol. 52, 172–179. doi:10.1124/mol.52.1.172
Jakubik J., Tucek S., El-Fakahany E. E. (2004). Role of receptor protein and membrane lipids in xanomeline wash-resistant binding to muscarinic M1 receptors. J. Pharmacol. Exp. Ther. 308, 105–110. doi:10.1124/jpet.103.058594
Jolkkonen M., Van Giersbergen P. L., Hellman U., Wernstedt C., Karlsson E. (1994). A toxin from the green mamba dendroaspis angusticeps: Amino acid sequence and selectivity for muscarinic m4 receptors. FEBS Lett. 352, 91–94. doi:10.1016/0014-5793(94)00933-3
Karlsson E., Jolkkonen M., Mulugeta E., Onali P., Adem A. (2000). Snake toxins with high selectivity for subtypes of muscarinic acetylcholine receptors. Biochimie 82, 793–806. doi:10.1016/s0300-9084(00)01176-7
Kashihara K., Varga E. V., Waite S. L., Roeske W. R., Yamamura H. I. (1992). Cloning of the rat M3, M4 and M5 muscarinic acetylcholine receptor genes by the polymerase chain reaction (PCR) and the pharmacological characterization of the expressed genes. Life Sci. 51, 955–971. doi:10.1016/0024-3205(92)90403-c
Khajehali E., Valant C., Jörg M., Tobin A. B., Conn P. J., Lindsley C. W., et al. (2018). Probing the binding site of novel selective positive allosteric modulators at the M1 muscarinic acetylcholine receptor. Biochem. Pharmacol. 154, 243–254. doi:10.1016/j.bcp.2018.05.009
Kiesewetter D. O., Carson R. E., Jagoda E. M., Endres C. J., Der M. G., Herscovitch P., et al. (1997). In vivo muscarinic binding selectivity of (R, S)- and (R, R)-[18F]-fluoromethyl QNB. Bioorg. Med. Chem. 5, 1555–1567. doi:10.1016/s0968-0896(97)00100-4
Kireev D., Wigle T. J., Norris-Drouin J., Herold J. M., Janzen W. P., Frye S. V. (2010). Identification of non-peptide malignant brain tumor (MBT) repeat antagonists by virtual screening of commercially available compounds. J. Med. Chem. 53, 7625–7631. doi:10.1021/jm1007374
Kistemaker L. E. M., Gosens R. (2015). Acetylcholine beyond bronchoconstriction: Roles in inflammation and remodeling. Trends Pharmacol. Sci. 36, 164–171. doi:10.1016/j.tips.2014.11.005
Kleinschroth J., Hartenstein J., Rudolph C., Schächtele C. (1995). Novel indolocarbazole protein kinase c inhibitors with improved biochemical and physicochemical properties. Bioorg. Med. Chem. Lett. 5, 55–60. doi:10.1016/0960-894x(94)00458-r
Kobayashi T., Washiyama K., Ikeda K. (2004). Inhibition of G protein-activated inwardly rectifying K+ channels by various antidepressant drugs. Neuropsychopharmacology 29, 1841–1851. doi:10.1038/sj.npp.1300484
Kovacs I., Yamamura H. I., Waite S. L., Varga E. V., Roeske W. R. (1998). Pharmacological comparison of the cloned human and rat M2 muscarinic receptor genes expressed in the murine fibroblast (B82) cell line. J. Pharmacol. Exp. Ther. 284, 500–507.
Kozaka T., Uno I., Kitamura Y., Miwa D., Ogawa K., Shiba K. (2012). Syntheses and in vitro evaluation of decalinvesamicol analogues as potential imaging probes for vesicular acetylcholine transporter (VAChT). Bioorg. Med. Chem. 20, 4936–4941. doi:10.1016/j.bmc.2012.06.040
Kozlowski J. A., Lowe D. B., Guzik H. S., Zhou G., Ruperto V. B., Duffy R. A., et al. (2000). Diphenyl sulfoxides as selective antagonists of the muscarinic M2 receptor. Bioorg. Med. Chem. Lett. 10, 2255–2257. doi:10.1016/s0960-894x(00)00438-8
Kruse A. C., Kobilka B. K., Gautam D., Sexton P. M., Christopoulos A., Wess J. (2014). Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nat. Rev. Drug Discov. 13, 549–560. doi:10.1038/nrd4295
Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., et al. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106. doi:10.1038/nature12735
Kulkarni S. S., Kopajtic T. A., Katz J. L., Newman A. H. (2006). Comparative structure-activity relationships of benztropine analogues at the dopamine transporter and histamine H(1) receptors. Bioorg. Med. Chem. 14, 3625–3634. doi:10.1016/j.bmc.2006.01.017
Lamba D., Pesaresi A. (2022). Kinetic modeling of time-dependent enzyme inhibition by pre-steady-state analysis of progress curves: The case study of the anti-alzheimer’s drug galantamine. Int. J. Mol. Sci. 23, 5072. doi:10.3390/ijms23095072
Lange J. H., Coolen H. K., Van Der Neut M. A., Borst A. J., Stork B., Verveer P. C., et al. (2010). Design, synthesis, biological properties, and molecular modeling investigations of novel tacrine derivatives with a combination of acetylcholinesterase inhibition and cannabinoid CB1 receptor antagonism. J. Med. Chem. 53, 1338–1346. doi:10.1021/jm901614b
Langmead C. J., Austin N. E., Branch C. L., Brown J. T., Buchanan K. A., Davies C. H., et al. (2008). Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br. J. Pharmacol. 154, 1104–1115. doi:10.1038/bjp.2008.152
Langmead C. J., Fry V. A., Forbes I. T., Branch C. L., Christopoulos A., Wood M. D., et al. (2006). Probing the molecular mechanism of interaction between 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine (AC-42) and the muscarinic M(1) receptor: Direct pharmacological evidence that AC-42 is an allosteric agonist. Mol. Pharmacol. 69, 236–246. doi:10.1124/mol.105.017814
Laukova M., Tillinger A., Novakova M., Krizanova O., Kvetnansky R., Myslivecek J. (2014). Repeated immobilization stress increases expression of β3-adrenoceptor in the left ventricle and atrium of the rat heart. Stress Health 30, 301–309. doi:10.1002/smi.2515
Lazareno S., Gharagozloo P., Kuonen D., Popham A., Birdsall N. J. (1998). Subtype-selective positive cooperative interactions between brucine analogues and acetylcholine at muscarinic receptors: Radioligand binding studies. Mol. Pharmacol. 53, 573–589. doi:10.1124/mol.53.3.573
Lazareno S., Popham A., Birdsall N. J. M. (2000). Allosteric interactions of staurosporine and other indolocarbazoles withN-[methyl-3H]scopolamine and acetylcholine at muscarinic receptor subtypes: Identification of a second allosteric site. Mol. Pharmacol. 58, 194–207. doi:10.1124/mol.58.1.194
Lazareno S., Popham A., Birdsall N. J. M. (2002). Analogs of WIN 62, 577 define a second allosteric site on muscarinic receptors. Mol. Pharmacol. 62, 1492–1505. doi:10.1124/mol.62.6.1492
Le U., Melancon B. J., Bridges T. M., Vinson P. N., Utley T. J., Lamsal A., et al. (2013a). Discovery of a selective M4 positive allosteric modulator based on the 3-amino-thieno[2, 3-b]pyridine-2-carboxamide scaffold: Development of ML253, a potent and brain penetrant compound that is active in a preclinical model of schizophrenia. Bioorg. Med. Chem. Lett. 23, 346–350. doi:10.1016/j.bmcl.2012.10.073
Le U., Melancon B. J., Bridges T. M., Vinson P. N., Utley T. J., Lamsal A., et al. (2013b). Discovery of a selective M₄ positive allosteric modulator based on the 3-amino-thieno[2, 3-b]pyridine-2-carboxamide scaffold: Development of ML253, a potent and brain penetrant compound that is active in a preclinical model of schizophrenia. Bioorg. Med. Chem. Lett. 23, 346–350. doi:10.1016/j.bmcl.2012.10.073
Lebois E. P., Thorn C., Edgerton J. R., Popiolek M., Xi S. (2018). Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer's disease. Neuropharmacology 136, 362–373. doi:10.1016/j.neuropharm.2017.11.018
Lochner M., Thompson A. J. (2016). The muscarinic antagonists scopolamine and atropine are competitive antagonists at 5-HT3 receptors. Neuropharmacology 108, 220–228. doi:10.1016/j.neuropharm.2016.04.027
Lockhart B., Closier M., Howard K., Steward C., Lestage P. (2001). Differential inhibition of [3H]-oxotremorine-M and [3H]-quinuclinidyl benzilate binding to muscarinic receptors in rat brain membranes with acetylcholinesterase inhibitors. Naunyn. Schmiedeb. Arch. Pharmacol. 363, 429–438. doi:10.1007/s002100000382
Lu L., Huang R., Wu Y., Jin J.-M., Chen H.-Z., Zhang L.-J., et al. (2020). Brucine: A review of phytochemistry, pharmacology, and toxicology. Front. Pharmacol. 11, 377. doi:10.3389/fphar.2020.00377
Ma L., Seager M. A., Wittmann M., Jacobson M., Bickel D., Burno M., et al. (2009). Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U. S. A. 106, 15950–15955. doi:10.1073/pnas.0900903106
Maeda S., Qu Q., Robertson M. J., Skiniotis G., Kobilka B. K. (2019). Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364, 552–557. doi:10.1126/science.aaw5188
Maksay G., Laube B., Betz H. (1999). Selective blocking effects of tropisetron and atropine on recombinant glycine receptors. J. Neurochem. 73, 802–806. doi:10.1046/j.1471-4159.1999.0730802.x
Mandai T., Sako Y., Kurimoto E., Shimizu Y., Nakamura M., Fushimi M., et al. (2020). T-495, a novel low cooperative M1 receptor positive allosteric modulator, improves memory deficits associated with cholinergic dysfunction and is characterized by low gastrointestinal side effect risk. Pharmacol. Res. Perspect. 8, e00560. doi:10.1002/prp2.560
Marlo J. E., Niswender C. M., Days E. L., Bridges T. M., Xiang Y., Rodriguez A. L., et al. (2009). Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol. 75, 577–588. doi:10.1124/mol.108.052886
Maurice T., Su T.-P. (2009). The pharmacology of sigma-1 receptors. Pharmacol. Ther. 124, 195–206. doi:10.1016/j.pharmthera.2009.07.001
McKenna M. T., Proctor G. R., Young L. C., Harvey A. L. (1997). Novel tacrine analogues for potential use against Alzheimer's disease: Potent and selective acetylcholinesterase inhibitors and 5-HT uptake inhibitors. J. Med. Chem. 40, 3516–3523. doi:10.1021/jm970150t
Melancon B. J., Gogliotti R. D., Tarr J. C., Saleh S. A., Chauder B. A., Lebois E. P., et al. (2012a). Continued optimization of the MLPCN probe ML071 into highly potent agonists of the hM1 muscarinic acetylcholine receptor. Bioorg. Med. Chem. Lett. 22, 3467–3472. doi:10.1016/j.bmcl.2012.03.088
Melancon B. J., Hopkins C. R., Wood M. R., Emmitte K. A., Niswender C. M., Christopoulos A., et al. (2012b). Allosteric modulation of seven transmembrane spanning receptors: Theory, practice, and opportunities for central nervous system drug discovery. J. Med. Chem. 55, 1445–1464. doi:10.1021/jm201139r
Mellor I. R., Ogilvie J., Pluteanu F., Clothier R. H., Parker T. L., Rosini M., et al. (2004). Methoctramine analogues inhibit responses to capsaicin and protons in rat dorsal root ganglion neurons. Eur. J. Pharmacol. 505, 37–50. doi:10.1016/j.ejphar.2004.10.005
Mille T., Quilgars C., Cazalets J.-R., Bertrand S. S. (2021). Acetylcholine and spinal locomotor networks: The insider. Physiol. Rep. 9, e14736. doi:10.14814/phy2.14736
Mirams G. R., Davies M. R., Brough S. J., Bridgland-Taylor M. H., Cui Y., Gavaghan D. J., et al. (2014). Prediction of Thorough QT study results using action potential simulations based on ion channel screens. J. Pharmacol. Toxicol. Methods 70, 246–254. doi:10.1016/j.vascn.2014.07.002
Modell J. G., Tandon R., Beresford T. P. (1989). Dopaminergic activity of the antimuscarinic antiparkinsonian agents. J. Clin. Psychopharmacol. 9, 347–351. doi:10.1097/00004714-198910000-00006
Mohr K., Schmitz J., Schrage R., Tränkle C., Holzgrabe U. (2013). Molecular alliance—from orthosteric and allosteric ligands to dualsteric/bitopic agonists at G protein coupled receptors. Angew. Chem. Int. Ed. Engl. 52, 508–516. doi:10.1002/anie.201205315
Morales-Weil K., Moreno M., Ahumada J., Arriagada J., Fuentealba P., Bonansco C., et al. (2020). Priming of GABAergic long-term potentiation by muscarinic receptors. Neuroscience 428, 242–251. doi:10.1016/j.neuroscience.2019.12.033
Moran S. P., Maksymetz J., Conn P. J. (2019). Targeting muscarinic acetylcholine receptors for the treatment of psychiatric and neurological disorders. Trends Pharmacol. Sci. 40, 1006–1020. doi:10.1016/j.tips.2019.10.007
Mulholland G. K., Kilbourn M. R., Sherman P., Carey J. E., Frey K. A., Koeppe R. A., et al. (1995). Synthesis, in vivo biodistribution and dosimetry of [11C]N-methylpiperidyl benzilate ([11C]NMPB), a muscarinic acetylcholine receptor antagonist. Nucl. Med. Biol. 22, 13–17. doi:10.1016/0969-8051(94)00082-u
Myslivecek J. (2022). Dopamine and dopamine-related ligands can bind not only to dopamine receptors. Life (Basel). 12 (5), 606. doi:10.3390/life12050606
Myslivecek J., Farar V., Valuskova P. (2017). M(4) muscarinic receptors and locomotor activity regulation. Physiol. Res. 66, S443–s455. doi:10.33549/physiolres.933796
Myslivecek J., Klein M., Novakova M., Ricny J. (2008a). The detection of the non-M-2 muscarinic receptor subtype in the rat heart atria and ventricles. Naunyn. Schmiedeb. Arch. Pharmacol. 378, 103–116. doi:10.1007/s00210-008-0285-8
Myslivecek J., Lisa V., Trojan S., Tucek S. (1998). Heterologous regulation of muscarinic and beta-adrenergic receptors in rat cardiomyocytes in culture. Life Sci. 63, 1169–1182. doi:10.1016/s0024-3205(98)00378-6
Myslivecek J. (2019). “M4 muscarinic receptors – structure, ligands, detection and function,” in Acetylcholine receptors in health and disease. Editor A. E. Gupta (New York: Nova Science Publishers), 41–68.
Myslivecek J., Novakova M., Klein M. (2008b). Receptor subtype abundance as a tool for effective intracellular signalling. Cardiovasc. Hematol. Disord. Drug Targets 8, 66–79. doi:10.2174/187152908783884939
Myslivecek J., Ricny J., Palkovits M., Kvetnansky R. (2004). The effects of short-term immobilization stress on muscarinic receptors, β-adrenoceptors, and adenylyl cyclase in different heart regions. Ann. N. Y. Acad. Sci. 1018, 315–322. doi:10.1196/annals.1296.038
Myslivecek J. (2021). Social isolation: How can the effects on the cholinergic system Be isolated? Front. Pharmacol. 12, 716460. doi:10.3389/fphar.2021.716460
Myslivecek J., Tillinger A., Novakova M., Kvetnansky R., Kvetnansky R., Aguilera G., et al. (2008c). Regulation of adrenoceptor and muscarinic receptor gene expression after single and repeated stress. Stress, Neurotransmitters, Hormones Neuroendocrine Genet. Mech. 1148, 367–376.
Myslivecek J., Trojan S., Tucek S. (1996). Biphasic changes in the density of muscarinic and beta-adrenergic receptors in cardiac atria of rats treated with diisopropylfluorophosphate. Life Sci. 58, 2423–2430. doi:10.1016/0024-3205(96)00246-9
Nabulsi N. B., Holden D., Zheng M. Q., Bois F., Lin S. F., Najafzadeh S., et al. (2019). Evaluation of (11)C-LSN3172176 as a novel PET tracer for imaging M(1) muscarinic acetylcholine receptors in nonhuman primates. J. Nucl. Med. 60, 1147–1153. doi:10.2967/jnumed.118.222034
Nedoma J., Dorofeeva N. A., Tucek S., Shelkovnikov S. A., Danilov A. F. (1985). Interaction of the neuromuscular blocking drugs alcuronium, decamethonium, gallamine, pancuronium, ritebronium, tercuronium and d-tubocurarine with muscarinic acetylcholine receptors in the heart and ileum. Naunyn. Schmiedeb. Arch. Pharmacol. 329, 176–181. doi:10.1007/BF00501209
Nenasheva T. A., Neary M., Mashanov G. I., Birdsall N. J., Breckenridge R. A., Molloy J. E. (2013). Abundance, distribution, mobility and oligomeric state of M₂ muscarinic acetylcholine receptors in live cardiac muscle. J. Mol. Cell. Cardiol. 57, 129–136. doi:10.1016/j.yjmcc.2013.01.009
Newman A. H., Kulkarni S. (2002). Probes for the dopamine transporter: New leads toward a cocaine-abuse therapeutic-A focus on analogues of benztropine and rimcazole. Med. Res. Rev. 22, 429–464. doi:10.1002/med.10014
Obara K., Horiguchi S., Shimada T., Ikarashi T., Yamaki F., Matsuo K., et al. (2019). Characterization of binding of antipsychotics to muscarinic receptors using mouse cerebral cortex. J. Pharmacol. Sci. 140, 197–200. doi:10.1016/j.jphs.2019.05.006
Ochillo R. F., Kau S. T., Sastry B. V. (1977). Activities of 5-methylfurfuryltrimethylammonium iodide (5-methylfurmethide) at nicotinic receptors. Pharmacol. Res. Commun. 9, 719–727. doi:10.1016/s0031-6989(77)80063-5
Ohmori J., Maeno K., Hidaka K., Nakato K., Matsumoto M., Tada S., et al. (1996). Dopamine D3 and D4 receptor antagonists: Synthesis and Structure−Activity relationships of (S)-(+)-N-(1-Benzyl-3-pyrrolidinyl)-5-chloro-4- [(cyclopropylcarbonyl)amino]-2-methoxybenzamide (YM-43611) and related compounds. J. Med. Chem. 39, 2764–2772. doi:10.1021/jm9601720
Ohno-Shosaku T., Matsui M., Fukudome Y., Shosaku J., Tsubokawa H., Taketo M. M., et al. (2003). Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur. J. Neurosci. 18, 109–116. doi:10.1046/j.1460-9568.2003.02732.x
Oki T., Takagi Y., Inagaki S., Taketo M. M., Manabe T., Matsui M., et al. (2005). Quantitative analysis of binding parameters of [3H]N-methylscopolamine in central nervous system of muscarinic acetylcholine receptor knockout mice. Mol. Brain Res. 133, 6–11. doi:10.1016/j.molbrainres.2004.09.012
Okimoto R., Ino K., Ishizu K., Takamatsu H., Sakamoto K., Yuyama H., et al. (2021). Potentiation of muscarinic M(3) receptor activation through a new allosteric site with a novel positive allosteric modulator ASP8302. J. Pharmacol. Exp. Ther. 379, 64–73. doi:10.1124/jpet.121.000709
Olianas M. C., Dedoni S., Ambu R., Onali P. (2009). Agonist activity of N-desmethylclozapine at delta-opioid receptors of human frontal cortex. Eur. J. Pharmacol. 607, 96–101. doi:10.1016/j.ejphar.2009.02.025
Olianas M. C., Dedoni S., Onali P. (2020). Antidepressants induce profibrotic responses via the lysophosphatidic acid receptor LPA(1). Eur. J. Pharmacol. 873, 172963. doi:10.1016/j.ejphar.2020.172963
Ozenil M., Aronow J., Millard M., Langer T., Wadsak W., Hacker M., et al. (2021). Update on PET tracer development for muscarinic acetylcholine receptors. Pharmaceuticals 14, 530. doi:10.3390/ph14060530
Pankaskie M. C., Kachur J. F., Itoh T., Gordon R. K., Chiang P. K. (1985). Inhibition of muscarinic receptor binding and acetylcholine-induced contraction of Guinea pig ileum by analogues of 5'-(isobutylthio)adenosine. J. Med. Chem. 28, 1117–1119. doi:10.1021/jm00146a027
Pascuzzo G. J., Akaike A., Maleque M. A., Shaw K. P., Aronstam R. S., Rickett D. L., et al. (1984). The nature of the interactions of pyridostigmine with the nicotinic acetylcholine receptor-ionic channel complex. I. Agonist, desensitizing, and binding properties. Mol. Pharmacol. 25, 92–101.
Peddi S., Roth B. L., Glennon R. A., Westkaemper R. B. (2004). Structural determinants for high 5-HT(2A) receptor affinity of spiro[9, 10-dihydroanthracene]-9, 3(')-pyrrolidine (SpAMDA). Bioorg. Med. Chem. Lett. 14, 2279–2283. doi:10.1016/j.bmcl.2004.02.014
Pei X.-F., Gupta T. H., Badio B., Padgett W. L., Daly J. W. (1998). 6β-Acetoxynortropane: A potent muscarinic agonist with apparent selectivity toward M2-receptors. J. Med. Chem. 41, 2047–2055. doi:10.1021/jm9705115
Peralta E. G., Ashkenazi A., Winslow J. W., Smith D. H., Ramachandran J., Capon D. J. (1987). Distinct primary structures, ligand-binding properties and tissue-specific expression of four human muscarinic acetylcholine receptors. Embo J. 6, 3923–3929. doi:10.1002/j.1460-2075.1987.tb02733.x
Peters J. U. (2013). Polypharmacology - foe or friend? J. Med. Chem. 56, 8955–8971. doi:10.1021/jm400856t
Pradidarcheep W., Michel M. C. (2016). “Use of antibodies in the research on muscarinic receptor subtypes,” in Muscarinic receptor: From structure to animal models. Editors J. Myslivecek, and J. Jakubik (New York, NY: Springer New York), 83–94.
Prat M., Fernández D., Buil M. A., Crespo M. I., Casals G., Ferrer M., et al. (2009). Discovery of novel quaternary ammonium derivatives of (3r)-quinuclidinol esters as potent and long-acting muscarinic antagonists with potential for minimal systemic exposure after inhaled administration: Identification of (3r)-3-{[hydroxy(di-2-thienyl)acetyl]oxy}-1-(3-phenoxypropyl)-1-azoniabicyclo[2.2.2]octane bromide (aclidinium bromide). J. Med. Chem. 52, 5076–5092. doi:10.1021/jm900132z
Psaridi-Linardaki L., Mamalaki A., Tzartos S., J. (2003). Future therapeutic strategies in autoimmune myasthenia gravis. Ann. N. Y. Acad. Sci. 998, 539–548. doi:10.1196/annals.1254.071
Pujito P. P., Govitrapong P., Ebadi M. (1991). Inhibitory actions of muscarinic cholinergic receptor agonists on serotonin N-acetyltransferase in bovine pineal explants in culture. Neurochem. Res. 16, 885–889. doi:10.1007/BF00965537
Randáková A., Jakubík J. (2021). Functionally selective and biased agonists of muscarinic receptors. Pharmacol. Res. 169, 105641. doi:10.1016/j.phrs.2021.105641
Richards M. H., van Giersbergen P. L. (1995). Human muscarinic receptors expressed in A9L and CHO cells: Activation by full and partial agonists. Br. J. Pharmacol. 114, 1241–1249. doi:10.1111/j.1476-5381.1995.tb13339.x
Ronsisvalle G., Marrazzo A., Prezzavento O., Pasquinucci L., Vittorio F., Pittalà V., et al. (1998). (+)-cis-N-ethyleneamino-N-normetazocine derivatives. Novel and selective sigma ligands with antagonist properties. J. Med. Chem. 41, 1574–1580. doi:10.1021/jm970333f
Rook Y., Schmidtke K. U., Gaube F., Schepmann D., Wünsch B., Heilmann J., et al. (2010). Bivalent beta-carbolines as potential multitarget anti-Alzheimer agents. J. Med. Chem. 53, 3611–3617. doi:10.1021/jm1000024
Rowley M., Bristow L. J., Hutson P. H. (2001). Current and novel approaches to the drug treatment of schizophrenia. J. Med. Chem. 44, 477–501. doi:10.1021/jm0002432
Ruan Y., Patzak A., Pfeiffer N., Gericke A. (2021). Muscarinic acetylcholine receptors in the retina—therapeutic implications. Int. J. Mol. Sci. 22, 4989. doi:10.3390/ijms22094989
Runyon S. P., Carroll F. I. (2006). Dopamine transporter ligands: Recent developments and therapeutic potential. Curr. Top. Med. Chem. 6, 1825–1843. doi:10.2174/156802606778249775
Runyon S. P., Savage J. E., Taroua M., Roth B. L., Glennon R. A., Westkaemper R. B. (2001). Influence of chain length and N-alkylation on the selective serotonin receptor ligand 9-(aminomethyl)-9, 10-dihydroanthracene. Bioorg. Med. Chem. Lett. 11, 655–658. doi:10.1016/s0960-894x(01)00023-3
Sagrada A., Schiavi G. B., Cereda E., Ladinsky H. (1994). Antagonistic properties of McNeil-A-343 at 5-HT4 and 5-HT3 receptors. Br. J. Pharmacol. 113, 711–716. doi:10.1111/j.1476-5381.1994.tb17051.x
Saiki R., Yoshizawa Y., Minarini A., Milelli A., Marchetti C., Tumiatti V., et al. (2013). In vitro and in vivo evaluation of polymethylene tetraamine derivatives as NMDA receptor channel blockers. Bioorg. Med. Chem. Lett. 23, 3901–3904. doi:10.1016/j.bmcl.2013.04.063
Salmon M., Luttmann M. A., Foley J. J., Buckley P. T., Schmidt D. B., Burman M., et al. (2013). Pharmacological characterization of GSK573719 (umeclidinium): A novel, long-acting, inhaled antagonist of the muscarinic cholinergic receptors for treatment of pulmonary diseases. J. Pharmacol. Exp. Ther. 345, 260–270. doi:10.1124/jpet.112.202051
Salovich J. M., Vinson P. N., Sheffler D. J., Lamsal A., Utley T. J., Blobaum A. L., et al. (2012). Discovery of N-(4-methoxy-7-methylbenzo[d]thiazol-2-yl)isonicatinamide, ML293, as a novel, selective and brain penetrant positive allosteric modulator of the muscarinic 4 (M4) receptor. Bioorg. Med. Chem. Lett. 22, 5084–5088. doi:10.1016/j.bmcl.2012.05.109
Samadi A., De Los Ríos C., Bolea I., Chioua M., Iriepa I., Moraleda I., et al. (2012). Multipotent MAO and cholinesterase inhibitors for the treatment of Alzheimer's disease: Synthesis, pharmacological analysis and molecular modeling of heterocyclic substituted alkyl and cycloalkyl propargyl amine. Eur. J. Med. Chem. 52, 251–262. doi:10.1016/j.ejmech.2012.03.022
Samochocki M., Höffle A., Fehrenbacher A., Jostock R., Ludwig J., Christner C., et al. (2003). Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 305, 1024–1036. doi:10.1124/jpet.102.045773
Sams A. G., Hentzer M., Mikkelsen G. K., Larsen K., Bundgaard C., Plath N., et al. (2010). Discovery of N-1-[3-(3-oxo-2, 3-dihydrobenzo[1, 4]oxazin-4-yl)propyl]piperidin-4-yl-2-phenylacetamide (Lu AE51090): An allosteric muscarinic M1 receptor agonist with unprecedented selectivity and procognitive potential. J. Med. Chem. 53, 6386–6397.
Sánchez C., Hyttel J. (1999). Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell. Mol. Neurobiol. 19, 467–489. doi:10.1023/a:1006986824213
Saternos H. C., Almarghalani D. A., Gibson H. M., Meqdad M. A., Antypas R. B., Lingireddy A., et al. (2017). Distribution and function of the muscarinic receptor subtypes in the cardiovascular system. Physiol. Genomics 50, 1–9. doi:10.1152/physiolgenomics.00062.2017
Schober D. A., Croy C. H., Xiao H., Christopoulos A., Felder C. C. (2014). Development of a radioligand, [3H]LY2119620, to probe the human M2 and M4 muscarinic receptor allosteric binding sites. Mol. Pharmacol. 86, 116–123. doi:10.1124/mol.114.091785
Schubert J. W., Harrison S. T., Mulhearn J., Gomez R., Tynebor R., Jones K., et al. (2019). Discovery, optimization, and biological characterization of 2, 3, 6-trisubstituted pyridine-containing M4 positive allosteric modulators. ChemMedChem 14, 943–951. doi:10.1002/cmdc.201900088
Sheffler D. J., Williams R., Bridges T. M., Xiang Z., Kane A. S., Byun N. E., et al. (2009). A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol. Pharmacol. 76, 356–368. doi:10.1124/mol.109.056531
Sheldon R. S., Hill R. J., Cannon N. J., Duff H. J. (1989). Amiodarone: Biochemical evidence for binding to a receptor for class I drugs associated with the rat cardiac sodium channel. Circ. Res. 65, 477–482. doi:10.1161/01.res.65.2.477
Shen Y., Monsma F. J., Metcalf M. A., Jose P. A., Hamblin M. W., Sibley D. R. (1993). Molecular cloning and expression of a 5-hydroxytryptamine7 serotonin receptor subtype. J. Biol. Chem. 268, 18200–18204. doi:10.1016/s0021-9258(17)46830-x
Shirey J. K., Xiang Z., Orton D., Brady A. E., Johnson K. A., Williams R., et al. (2008). An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat. Chem. Biol. 4, 42–50. doi:10.1038/nchembio.2007.55
Sinha S., Gupta S., Malhotra S., Krishna N. S., Meru A. V., Babu V., et al. (2010). AE9C90CB: A novel, bladder-selective muscarinic receptor antagonist for the treatment of overactive bladder. Br. J. Pharmacol. 160, 1119–1127. doi:10.1111/j.1476-5381.2010.00752.x
So E. C., Huang Y. M., Hsing C. H., Liao Y. K., Wu S. N. (2015). Arecoline inhibits intermediate-conductance calcium-activated potassium channels in human glioblastoma cell lines. Eur. J. Pharmacol. 758, 177–187. doi:10.1016/j.ejphar.2015.03.065
Sowell J. W., Tang Y., Valli M. J., Chapman J. M., Usher L. A., Vaughan C. M., et al. (1992). Synthesis and cholinergic properties of bis[[(dimethylamino)methyl]furanyl] analogs of ranitidine. J. Med. Chem. 35, 1102–1108. doi:10.1021/jm00084a015
Spalding T. A., Ma J. N., Ott T. R., Friberg M., Bajpai A., Bradley S. R., et al. (2006). Structural requirements of transmembrane domain 3 for activation by the M1 muscarinic receptor agonists AC-42, AC-260584, clozapine, and N-desmethylclozapine: Evidence for three distinct modes of receptor activation. Mol. Pharmacol. 70, 1974–1983. doi:10.1124/mol.106.024901
Stahl E., Ellis J. (2010). Novel allosteric effects of amiodarone at the muscarinic M5 receptor. J. Pharmacol. Exp. Ther. 334, 214–222. doi:10.1124/jpet.109.165316
Stahl E., Elmslie G., Ellis J. (2011). Allosteric modulation of the M₃ muscarinic receptor by amiodarone and N-ethylamiodarone: Application of the four-ligand allosteric two-state model. Mol. Pharmacol. 80, 378–388. doi:10.1124/mol.111.072991
Stanton T., Bolden-Watson C., Cusack B., Richelson E. (1993). Antagonism of the five cloned human muscarinic cholinergic receptors expressed in CHO-K1 cells by antidepressants and antihistaminics. Biochem. Pharmacol. 45, 2352–2354. doi:10.1016/0006-2952(93)90211-e
Staszewski M., Nelic D., Jonczyk J., Dubiel M., Frank A., Stark H., et al. (2021). Guanidine derivatives: How simple structural modification of histamine H3R antagonists has led to the discovery of potent muscarinic m2r/m4r antagonists. ACS Chem. Neurosci. 12, 2503–2519. doi:10.1021/acschemneuro.1c00237
Stein R., Pinkas-Kramarski R., Sokolovsky M. (1988). Cloned M1 muscarinic receptors mediate both adenylate cyclase inhibition and phosphoinositide turnover. Embo J. 7, 3031–3035. doi:10.1002/j.1460-2075.1988.tb03167.x
Steinfeld T., Mammen M., Smith J. a. M., Wilson R. D., Jasper J. R. (2007b). A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol. Pharmacol. 72, 291–302. doi:10.1124/mol.106.033746
Steinfeld T., Mammen M., Smith J. A., Wilson R. D., Jasper J. R. (2007a). A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Mol. Pharmacol. 72, 291–302. doi:10.1124/mol.106.033746
Suno R., Lee S., Maeda S., Yasuda S., Yamashita K., Hirata K., et al. (2018). Structural insights into the subtype-selective antagonist binding to the M(2) muscarinic receptor. Nat. Chem. Biol. 14, 1150–1158. doi:10.1038/s41589-018-0152-y
Sur C., Mallorga P. J., Wittmann M., Jacobson M. A., Pascarella D., Williams J. B., et al. (2003). N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc. Natl. Acad. Sci. 100, 13674–13679. doi:10.1073/pnas.1835612100
Svoboda D. L., Saddler T., Auerbach S. S. (2019). “An overview of national toxicology program’s toxicogenomic applications: DrugMatrix and ToxFX,” in Advances in computational toxicology: Methodologies and applications in regulatory science. Editor H. Hong (Cham: Springer International Publishing), 141–157.
Sykes D. A., Dowling M. R., Leighton-Davies J., Kent T. C., Fawcett L., Renard E., et al. (2012). The Influence of receptor kinetics on the onset and duration of action and the therapeutic index of NVA237 and tiotropium. J. Pharmacol. Exp. Ther. 343, 520–528. doi:10.1124/jpet.112.194456
Szabo M., Lim H. D., Klein Herenbrink C., Christopoulos A., Lane J. R., Capuano B. (2015). Proof of concept study for designed multiple ligands targeting the dopamine D2, serotonin 5-HT2A, and muscarinic M1 acetylcholine receptors. J. Med. Chem. 58, 1550–1555. doi:10.1021/jm5013243
Taggi M., Kovacevic A., Capponi C., Falcinelli M., Cacciamani V., Vicini E., et al. (2022). The activation of M2 muscarinic receptor inhibits cell growth and survival in human epithelial ovarian carcinoma. J. Cell. Biochem. doi:10.1002/jcb.30303
Tarr J. C., Turlington M. L., Reid P. R., Utley T. J., Sheffler D. J., Cho H. P., et al. (2012). Targeting selective activation of M(1) for the treatment of Alzheimer's disease: Further chemical optimization and pharmacological characterization of the M(1) positive allosteric modulator ML169. ACS Chem. Neurosci. 3, 884–895. doi:10.1021/cn300068s
Tatsumi M., Groshan K., Blakely R. D., Richelson E. (1997). Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 340, 249–258. doi:10.1016/s0014-2999(97)01393-9
Tenjin T., Miyamoto S., Ninomiya Y., Kitajima R., Ogino S., Miyake N., et al. (2013). Profile of blonanserin for the treatment of schizophrenia. Neuropsychiatr. Dis. Treat. 9, 587–594. doi:10.2147/NDT.S34433
Thal D. M., Sun B., Feng D., Nawaratne V., Leach K., Felder C. C., et al. (2016). Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531, 335–340. doi:10.1038/nature17188
Tobin G., Giglio D., Lundgren O. (2009). Muscarinic receptor subtypes in the alimentary tract. J. Physiol. Pharmacol. 60, 3–21.
Tomankova H., Valuskova P., Varejkova E., Rotkova J., Benes J., Myslivecek J. (2015). The M 2 muscarinic receptors are essential for signaling in the heart left ventricle during restraint stress in mice. Stress 18, 208–220. doi:10.3109/10253890.2015.1007345
Tomizawa M., Yamamoto I. (1992). Binding of nicotinoids and the related compounds to the insect nicotinic acetyicholine receptor. ournal Pesticide Sci. 17, 231–236.
Tränkle C., Dittmann A., Schulz U., Weyand O., Buller S., Jöhren K., et al. (2005). Atypical muscarinic allosteric modulation: Cooperativity between modulators and their atypical binding topology in muscarinic M2 and M2/M5 chimeric receptors. Mol. Pharmacol. 68, 1597–1610. doi:10.1124/mol.105.017707
Tränkle C., Weyand O., Voigtländer U., Mynett A., Lazareno S., Birdsall N. J., et al. (2003). Interactions of orthosteric and allosteric ligands with [3H]dimethyl-W84 at the common allosteric site of muscarinic M2 receptors. Mol. Pharmacol. 64, 180–190. doi:10.1124/mol.64.1.180
Tumiatti V., Rosini M., Bartolini M., Cavalli A., Marucci G., Andrisano V., et al. (2003). Structure−Activity relationships of acetylcholinesterase noncovalent inhibitors based on a polyamine backbone. 2. Role of the substituents on the phenyl ring and nitrogen atoms of caproctamine. J. Med. Chem. 46, 954–966. doi:10.1021/jm021055+
Valant C., Felder C. C., Sexton P. M., Christopoulos A. (2012). Probe dependence in the allosteric modulation of a G protein-coupled receptor: Implications for detection and validation of allosteric ligand effects. Mol. Pharmacol. 81, 41–52. doi:10.1124/mol.111.074872
Valuskova P., Farar V., Forczek S., Krizova I., Myslivecek J. (2018a). Autoradiography of 3H-pirenzepine and 3H-AFDX-384 in mouse brain regions: Possible insights into M1, M2, and M4 muscarinic receptors distribution. Front. Pharmacol. 9, 124. doi:10.3389/fphar.2018.00124
Valuskova P., Forczek S. T., Farar V., Myslivecek J. (2018b). The deletion of M4 muscarinic receptors increases motor activity in females in the dark phase. Brain Behav. 8, e01057. doi:10.1002/brb3.1057
Vass M., Kooistra A. J., Yang D., Stevens R. C., Wang M.-W., De Graaf C. (2018). Chemical diversity in the G protein-coupled receptor superfamily. Trends Pharmacol. Sci. 39, 494–512. doi:10.1016/j.tips.2018.02.004
von Coburg Y., Kottke T., Weizel L., Ligneau X., Stark H. (2009). Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg. Med. Chem. Lett. 19, 538–542. doi:10.1016/j.bmcl.2008.09.012
Vuckovic Z., Gentry P. R., Berizzi A. E., Hirata K., Varghese S., Thompson G., et al. (2019). Crystal structure of the M5 muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. U. S. A. 116, 26001–26007. doi:10.1073/pnas.1914446116
Waelbroeck M., De Neef P., Domenach V., Vandermeers-Piret M. C., Vandermeers A. (1996). Binding of the labelled muscarinic toxin 125I-MT1 to rat brain muscarinic M1 receptors. Eur. J. Pharmacol. 305, 187–192. doi:10.1016/0014-2999(96)00136-7
Walker L. C., Huckstep K. L., Chen N. A., Hand L. J., Lindsley C. W., Langmead C. J., et al. (2021). Muscarinic M4 and M5 receptors in the ventral subiculum differentially modulate alcohol seeking versus consumption in male alcohol-preferring rats. Br. J. Pharmacol. 178, 3730–3746. doi:10.1111/bph.15513
Wang J., Wu M., Chen Z., Wu L., Wang T., Cao D., et al. (2022). The unconventional activation of the muscarinic acetylcholine receptor M4R by diverse ligands. Nat. Commun. 13, 2855. doi:10.1038/s41467-022-30595-y
Wang Y., Law W. K., Hu J. S., Lin H. Q., Ip T. M., Wan D. C. (2014). Discovery of FDA-approved drugs as inhibitors of fatty acid binding protein 4 using molecular docking screening. J. Chem. Inf. Model. 54, 3046–3050. doi:10.1021/ci500503b
Wang Z., Shi H., Wang H. (2004). Functional M3 muscarinic acetylcholine receptors in mammalian hearts. Br. J. Pharmacol. 142, 395–408. doi:10.1038/sj.bjp.0705787
Ward J. S., Merritt L., Bymaster F. P., Calligaro D. O. (1994). Isoarecolones and arecolones: Selective central nicotinic agonists that cross the blood-brain barrier. Bioorg. Med. Chem. Lett. 4, 573–578. doi:10.1016/s0960-894x(01)80157-8
Watson J., Brough S., Coldwell M. C., Gager T., Ho M., Hunter A. J., et al. (1998). Functional effects of the muscarinic receptor agonist, xanomeline, at 5-HT1 and 5-HT2 receptors. Br. J. Pharmacol. 125, 1413–1420. doi:10.1038/sj.bjp.0702201
Watt M. L., Schober D. A., Hitchcock S., Liu B., Chesterfield A. K., Mckinzie D., et al. (2011). Pharmacological characterization of LY593093, an M1 muscarinic acetylcholine receptor-selective partial orthosteric agonist. J. Pharmacol. Exp. Ther. 338, 622–632. doi:10.1124/jpet.111.182063
Wei H. B., Roeske W. R., Lai J., Wanibuchi F., Hidaka K., Usuda S., et al. (1992). Pharmacological characterization of a novel muscarinic partial agonist, YM796, in transfected cells expressing the m1 or m2 muscarinic receptor gene. Life Sci. 50, 355–363. doi:10.1016/0024-3205(92)90437-t
Weiden P. J., Yohn S., Felder C. C. (2022). Understanding why muscarinic receptor agonists have antipsychotic properties. CNS Spectr. 27, 249. doi:10.1017/s1092852922000608
Weinhart C. G., Wifling D., Schmidt M. F., Neu E., Höring C., Clark T., et al. (2021). Dibenzodiazepinone-type muscarinic receptor antagonists conjugated to basic peptides: Impact of the linker moiety and unnatural amino acids on M2R selectivity. Eur. J. Med. Chem. 213, 113159. doi:10.1016/j.ejmech.2021.113159
Wess J., Lambrecht G., Mutschler E., Brann M. R., Dörje F. (1991). Selectivity profile of the novel muscarinic antagonist UH-AH 37 determined by the use of cloned receptors and isolated tissue preparations. Br. J. Pharmacol. 102, 246–250. doi:10.1111/j.1476-5381.1991.tb12161.x
Wiśniowska B., Mendyk A., Fijorek K., Glinka A., Polak S. (2012). Predictive model for L-type channel inhibition: Multichannel block in QT prolongation risk assessment. J. Appl. Toxicol. 32, 858–866. doi:10.1002/jat.2784
Wu J., Li X., Zhou P., Li X. (2020). M3 but not M4 muscarinic receptors in the rostromedial tegmental nucleus are involved in the acquisition of morphine-induced conditioned place preference. Eur. J. Pharmacol. 882, 173274. doi:10.1016/j.ejphar.2020.173274
Xu M., Peng Y., Zhu L., Wang S., Ji J., Rakesh K. P. (2019). Triazole derivatives as inhibitors of Alzheimer's disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 180, 656–672. doi:10.1016/j.ejmech.2019.07.059
Yamada S., Chimoto J., Shiho M., Okura T., Morikawa K., Kagota S., et al. (2021). Muscarinic receptor binding activity in rat tissues by vibegron and prediction of its receptor occupancy levels in the human bladder. Int. J. Urol. 28, 1298–1303. doi:10.1111/iju.14696
Yang Z., Ney A., Cromer B. A., Ng H. L., Parker M. W., Lynch J. W. (2007). Tropisetron modulation of the glycine receptor: Femtomolar potentiation and a molecular determinant of inhibition. J. Neurochem. 100, 758–769. doi:10.1111/j.1471-4159.2006.04242.x
Yardley J. P., Husbands G. E., Stack G., Butch J., Bicksler J., Moyer J. A., et al. (1990). 2-Phenyl-2-(1-hydroxycycloalkyl)ethylamine derivatives: Synthesis and antidepressant activity. J. Med. Chem. 33, 2899–2905. doi:10.1021/jm00172a035
Zhang W., Bymaster F. P. (1999). The in vivo effects of olanzapine and other antipsychotic agents on receptor occupancy and antagonism of dopamine D1, D2, D3, 5HT2A and muscarinic receptors. Psychopharmacology 141, 267–278. doi:10.1007/s002130050834
Zhang Y., Joseph D. B., Bowen W. D., Flippen-Anderson J. L., Dersch C. M., Rothman R. B., et al. (2001). Synthesis and biological evaluation of tropane-like 1-{2-[Bis(4-fluorophenyl)methoxy]ethyl}-4-(3-phenylpropyl)piperazine (GBR 12909) analogues. J. Med. Chem. 44, 3937–3945. doi:10.1021/jm0101592
Zhuang Z. P., Kung M. P., Kung H. F. (1993). Synthesis of (R, S)-trans-8-hydroxy-2-[N-n-propyl-N-(3'-iodo-2'-propenyl)amino]tetralin (trans 8-OH-PIPAT): A new 5-HT1A receptor ligand. J. Med. Chem. 36, 3161–3165. doi:10.1021/jm00073a016
Zlotos D. P., Mohsen A. M. Y., Mandour Y. M., Marzouk M. A., Breitinger U., Villmann C., et al. (2019). 11-Aminostrychnine and N-(Strychnine-11-yl)propionamide: Synthesis, configuration, and pharmacological evaluation at Glycine receptors. J. Nat. Prod. 82, 2332–2336. doi:10.1021/acs.jnatprod.9b00180
Keywords: muscarinic receptors, multitarget, muscarinic agonist, muscarinic antagonist, allosteric, orthosteric
Citation: Myslivecek J (2022) Multitargeting nature of muscarinic orthosteric agonists and antagonists. Front. Physiol. 13:974160. doi: 10.3389/fphys.2022.974160
Received: 20 June 2022; Accepted: 01 August 2022;
Published: 06 September 2022.
Edited by:
George Grant, University of Aberdeen, United KingdomReviewed by:
David Thal, Faculty of Pharmacy and Pharmaceutical Sciences, Monash University, AustraliaYair Ben Chaim, Open University of Israel, Israel
Copyright © 2022 Myslivecek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaromir Myslivecek, am15c0BsZjEuY3VuaS5jeg==