 Kenya Takahashi
Kenya Takahashi Yuki Tamura
Yuki Tamura Yu Kitaoka
Yu Kitaoka Yutaka Matsunaga1
Yutaka Matsunaga1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 30 June 2022
Sec. Mitochondrial Research
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.920034
This article is part of the Research TopicNew Insights into the Role of Mitochondria in Muscle PathophysiologyView all 5 articles
Recent evidence has shown that mitochondrial respiratory function contributes to exercise performance and metabolic health. Given that lactate is considered a potential signaling molecule that induces mitochondrial adaptations, we tested the hypothesis that lactate would change mitochondrial respiratory function in skeletal muscle. Male ICR mice (8 weeks old) received intraperitoneal injection of PBS or sodium lactate (1 g/kg BW) 5 days a week for 4 weeks. Mitochondria were isolated from freshly excised gastrocnemius muscle using differential centrifugation and were used for all analyses. Lactate administration significantly enhanced pyruvate + malate- and glutamate + malate-induced (complex I-driven) state 3 (maximal/ATP synthesis-coupled) respiration, but not state 2 (basal/proton conductance) respiration. In contrast, lactate administration significantly decreased succinate + rotenone-induced (complex II-driven) state 3 and 2 respiration. No significant differences were observed in malate + octanoyl-l-carnitine-induced state 3 or 2 respiration. The enzymatic activity of complex I was tended to increase and those of complexes I + III and IV were significantly increased after lactate administration. No differences were observed in the activities of complexes II or II + III. Moreover, lactate administration increased the protein content of NDUFS4, a subunit of complex I, but not those of the other components. The present findings suggest that lactate alters mitochondrial respiratory function in skeletal muscle.
Mitochondria are fundamental cellular components that underpin skeletal muscle function and, consequently, contribute to physical well-being and athletic performance (Joyner and Coyle, 2008; Nunnari and Suomalainen, 2012). Earlier studies focused mainly on changes in mitochondrial enzyme activity, such as citrate synthase (CS), as a biomarker of mitochondrial content in skeletal muscle (Larsen et al., 2015). Changes in mitochondrial respiratory function were presumed to be proportional to mitochondrial content (Hoppeler et al., 1987). However, subsequent studies reported decreased mitochondrial respiration without changes in mitochondrial enzyme activity in patients with type 2 diabetes (Mogensen et al., 2007; Phielix et al., 2008). Another study reported that, after a period of training, subjects with higher mitochondrial respiration resulted in greater exercise performance despite no significant increase in mitochondrial enzyme activity (Granata et al., 2016). These studies suggest that mitochondrial respiratory function can be dissociated from mitochondrial content and may be relevant to physical fitness and exercise performance. Thus, elucidating factors that alters mitochondrial respiratory function would contribute to establishing effective strategy for improving athletic performance and physical health.
Lactate is not merely an end product of glycolysis, but also a metabolic intermediate that serves as an oxidizable substrate and gluconeogenic precursor (Gladden, 2004; Brooks, 2009, 2018). In recent years, lactate has attracted considerable attention as a potential signaling molecule that induces physiological adaptations (Brooks, 2020; Brooks et al., 2021). Intriguingly, a previous study demonstrated that incubation of L6 cells with lactate increased the mitochondrial protein content (Hashimoto et al., 2007). Additionally, we previously reported that lactate administration enhanced mitochondrial enzyme activity in mouse skeletal muscle (Takahashi et al., 2019b; Takahashi et al., 2020b). Other researchers previously reported the improved mitochondrial respiration in skeletal muscle following high-intensity training (Granata et al., 2016), during which the circulating lactate level increases. These observations led us to hypothesize that lactate would possess a potential to alter mitochondrial respiratory function in skeletal muscle.
In this study, mitochondria isolated from the gastrocnemius muscle of mice administered with lactate for 4 weeks were suspended in four different media (pyruvate + malate, glutamate + malate, succinate + rotenone, and malate + octanoyl-l-carnitine) in the presence (state 3, maximal/ATP synthesis-coupled) or absence (state 2, basal/proton conductance) of ADP. Moreover, we evaluated mitochondrial enzyme activities and protein contents in the isolated mitochondria to better understand the mechanisms underlying lactate-induced mitochondrial functional adaptations.
Male ICR mice (8 weeks old; CLEA Japan, Tokyo, Japan) were used in this study. Mice were housed in an air-conditioned room at 22°C with a 12 h light-dark cycle (dark 7:00 a.m. to 7:00 p.m.). All mice were provided with standard chow (MF diet, Oriental Yeast, Tokyo, Japan) and water ad libitum during the experimental period. All experiments were approved by the Animal Experimental Committee of The University of Tokyo (approval number: 27–14).
The animals were randomly divided into control (CON; n = 10) and lactate (LAC; n = 9) groups. They received intraperitoneal injections of phosphate-buffered saline (PBS) or sodium lactate (1 g/kg of body weight) 5 days a week for 4 weeks. The administration was performed at the same time of day (10:00 a.m.) when animals were active. We previously reported that the method of administration and volume of lactate increased blood lactate concentration to an upper physiological level (∼20 mM), and returned blood lactate to basal level 3 h after the injection (Kitaoka et al., 2016; Takahashi et al., 2019b). Twenty-4 hours after the last administration, the gastrocnemius muscles were taken and used for mitochondrial analysis.
Mitochondria were isolated by differential centrifugation as previously described (Tomiya et al., 2019; Wakabayashi et al., 2020). Briefly, freshly excised gastrocnemius muscle was immediately placed in ice-cold buffer (PBS, 10 mM EDTA, pH 7.4), minced with scissors, supplemented with 0.025% trypsin, and then incubated on ice for 5 min. After tissue suspensions were centrifuged at 200 × g for 5 min at 4°C, the supernatants were discarded. Tissue precipitate was homogenized using a Tenbroeck tissue grinder (Wheaton, NJ, United States ) with 10 strokes in a buffer (50 mM MOPS, 100 mM KCl, 1 mM EGTA, 5 mM MgSO4, and 2.0 g/L BSA, pH 7.1). The homogenates were centrifuged at 500 × g for 10 min at 4°C to collect supernatants containing mitochondria. The supernatants were re-centrifuged at 10,000 × g for 10 min at 4°C to obtain the mitochondrial pellets. The mitochondrial pellets were washed with buffer (50 mM MOPS, 100 mM KCl, 1 mM EGTA, and 5 mM MgSO4) and resuspended in buffer (105 mM potassium-MES, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, and 2.5 g/L BSA, pH 7.2). The protein concentration was determined using the BCA protein assay (TaKaRa BIO INC., Shiga, Japan). For the respiratory function assays, the protein concentration of the mitochondrial solution was adjusted to 2.0 mg/ml.
Mitochondrial OCR was measured as previously described, with minor modifications (Kitaoka et al., 2019; Tomiya et al., 2019; Tamura et al., 2020; Wakabayashi et al., 2020). Briefly, freshly isolated mitochondria (20 µg) were incubated in buffer (105 mM potassium-MES, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, and 2.5 g/L BSA, pH 7.2). Mitochondrial respiration was initiated by addition of the respiration buffers described below. Mitochondrial OCR was measured in an oxygen-monitoring 96-well microplate (OP96C, PreScan Precision Sensing, Regensburg, Germany) using a Tecan Spark multimode plate reader (Spark 20M, Tecan, Männedorf, Switzerland) (excitation: 540 nm; emission: 650 nm). Relative fluorescence changes were measured per minute and normalized to CS activity. Data without normalization are shown in Supplementary Figure S2. We previously reported that the mitochondrial preparation protocol did not increase the OCR after adding 10 mM cytochrome c (Kitaoka et al., 2019; Tomiya et al., 2019; Wakabayashi et al., 2020), suggesting that integrity of the mitochondrial outer membrane was not disturbed.
Pyruvate + malate-induced (complex I-driven) state 3 (PM3) and 2 (PM2) respiration was measured by the addition of a PM3 (5 mM potassium pyruvate, 1.5 mM malic acid, 2.5 mM ADP, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) and a PM2 (5 mM potassium pyruvate, 1.5 mM malic acid, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) buffer, respectively.
Glutamate + malate-induced (complex I-driven) state 3 (GM3) and 2 (GM2) respiration was measured by the addition of a GM3 (10 mM glutamic acid, 1.5 mM malic acid, 2.5 mM ADP, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) and a GM2 (10 mM glutamic acid, 1.5 mM malic acid, 2.5 mM ADP, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) buffer, respectively.
Succinate + rotenone-induced (complex II-driven) state 3 (S3) and 2 (S2) respiration was measured by the addition of an S3 (10 mM sodium succinate, 1 mM rotenone, 2.5 mM ADP, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) and an S2 (10 mM sodium succinate, 1 mM rotenone, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) buffer, respectively
Malate + octanoyl-l-carnitine-induced state 3 (MOc3) and 2 (MOc2) respiration was measured by the addition of an MOc3 (10 mM octanoyl-l-carnitine, 1.5 mM malic acid, 2.5 mM ADP, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) and an MOc2 (10 mM octanoyl-l-carnitine, 1.5 mM malic acid, 10 mM Tris, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 2.5 g/L BSA, pH 7.2) buffer, respectively.
Mitochondrial enzyme activity was measured as previously described, with minor modifications (Takahashi et al., 2019b; Wakabayashi et al., 2020). Electron transport chain enzyme activity data were normalized to CS activity. Data without normalization are shown in Supplementary Figure S3.
Mitochondrial aliquots were mixed with a reaction mixture (100 μM DTNB, 300 μM acetyl-CoA, and 50 μM oxaloacetate) in a 96-well plate. The changes in absorbance were determined at 412 nm/min. We confirmed that CS activity in the isolated mitochondria did not significantly differ between the two groups (CON: 1.00 ± 0.12; LAC: 0.96 ± 0.19, relative to CON) (Supplementary Figure S1A).
Mitochondrial aliquots were mixed with a reaction mixture (1 M Tris, 5 mM EDTA, 450 µM NADH, and 100 µM acetoacetyl-CoA, pH 7.0) in a 96-well microplate. The absorbance changes at 340 nm/min were determined.
Mitochondrial aliquots were mixed with a reaction mixture (100 μM NADH, 60 μM ubiquinone, 300 μM KCN, and 3 mg/ml BSA) in a 96-well plate. The changes in absorbance were determined at 340 nm/min. Rotenone-sensitive enzyme activity (i.e., rotenone absence − rotenone presence) was regarded as complex I enzyme activity.
Mitochondrial aliquots were mixed with a reaction mixture (200 μM NADH, 50 μM cytochrome c, 300 μM KCN, and 1.0 mg/ml BSA) in a 96-well plate. The changes in absorbance were determined at 550 nm/min.
Mitochondrial aliquots were mixed with a reaction mixture [20 mM succinate, 0.015% (wt/vol) 2,6-dichlorophenolindophenol, 12.5 μM decylubiquinone, 300 μM KCN, and 1 mg/ml BSA] in a 96-well plate. The changes in absorbance were determined at 600 nm/min.
Mitochondrial aliquots were mixed with a reaction mixture (10 mM succinate, 50 μM cytochrome c, and 300 μM KCN) in a 96-well plate. The changes in absorbance were determined at 550 nm/min.
Mitochondrial aliquots were mixed with a reaction mixture (50 μM cytochrome c reduced with dithiothreitol) in a 96-well plate. The changes in absorbance were determined at 550 nm/min.
Mitochondrial pellets were suspended in ice-cold radioimmunoprecipitation assay buffer (25 mM Tris–HCl, pH 7.6, 150 mM NaCl, and 1% NP-40) and mixed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (199–16,132, Fujifilm Wako Pure Chemical, Osaka, Japan). Equal amounts of protein were loaded onto SDS-PAGE gels (10–15%) and separated by electrophoresis. After proteins were transferred to polyvinylidene difluoride (PVDF) membranes, western blotting was performed as described previously (Takahashi et al., 2020a; Takahashi et al., 2020b). The primary and secondary antibodies used in this study are described below. Blots were scanned and quantified using a ChemiDoc XRS (Bio-Rad Laboratories, Hercules, CA, United States ) system and Quantity One software (version 4.5.2; Bio-Rad), respectively. The respiratory chain protein content was normalized to the CS content. Data without normalization are shown in Supplementary Figure S4. We confirmed that CS protein content in the isolated mitochondria did not significantly differ between the two groups (CON: 1.00 ± 0.08; LAC: 1.01 ± 0.13, relative to CON) (Supplementary Figure S1B).
The primary antibodies used in this study were as follows: MitoProfile Total OXPHOS Rodent WB Antibody Cocktail [NADH dehydrogenase (ubiquinone) 1β subcomplex 8 (NDUFB8), succinate dehydrogenase complex subunit B (SDHB), ubiquinol-cytochrome c reductase core protein II (UQCRC2), ATP synthase, H+ transporting, mitochondrial F1 complex, α-subunit (ATP5A), ab110413, Abcam], NADH dehydrogenase (ubiquinone) iron-sulfur protein 4 (NDUFS4; ab139178, Abcam), cytochrome c oxidase subunit IV (COX IV; ab14744, Abcam), and CS (ab129095; Abcam). The following secondary antibodies were used in the present study: rabbit anti-goat IgG (H&L) (#A102PT; American Qualex, San Clemente, CA, United States ) and mouse anti-goat IgG (H&L) (#A106PU; American Qualex).
Mitochondrial supercomplex assembly was evaluated by two-dimensional PAGE, as described previously (Wakabayashi et al., 2020). Mitochondrial specimens (50 µg of protein) were suspended in 5% digitonin [digitonin/protein ratio: 8 (g/g); BN 2006, Thermo Fisher Scientific] and NativePAGE™ 4X Sample Buffer (BN20032, Thermo Fisher Scientific). After incubation on ice for 30 min, the suspensions were centrifuged at 20,000 × g for 15 min at 4°C. The supernatants were mixed with NativePAGE™ 5% G-250 Sample Additive (BN 2004, Thermo Fisher Scientific). Samples were loaded on 4–15% gradient gels (4,561,096, Bio-Rad) and were separated by blue native PAGE (BN-PAGE) using native PAGE buffer (BN 2001, Thermo Fisher Scientific). Proteins were then separated by SDS-PAGE and transferred to PVDF membranes. Western blotting was performed using anti-oxidative phosphorylation antibodies (MitoProfile Total OXPHOS Rodent WB Antibody Cocktail; ab110413, Abcam), as described above.
All data are expressed as mean ± standard deviation (SD). Comparisons between two groups were made using the unpaired Student’s t-test. Correlations between two variables were studied using least-squares linear regression followed by the Pearson’s correlation coefficient test. All statistical analyses were performed using the GraphPad Prism software (Ver. 9.0, Macintosh, GraphPad Software, La Jolla, CA). Statistical significance was defined as p < 0.05. To minimize type II errors, all the results within the range 0.05 ≤ p ≤ 0.10 are shown.
To elucidate whether lactate administration enhances mitochondrial respiratory function, we measured the mitochondrial OCR under state 3 and 2 conditions. The LAC group exhibited significantly higher PM3 (p < 0.05; Figure 1A) and GM3 (p < 0.05; Figure 1B) than those of the CON group. In contrast to the complex I-driven OCR, S3 and S2 were significantly lower in the LAC group than those in the CON group (p < 0.05; Figure 1C). No differences were observed between the two groups in PM2 (Figure 1A), GM2 (Figure 1B), MOc3, and MOc2 (Figure 1D). Together, these observations suggest that lactate alters mitochondrial respiratory function.
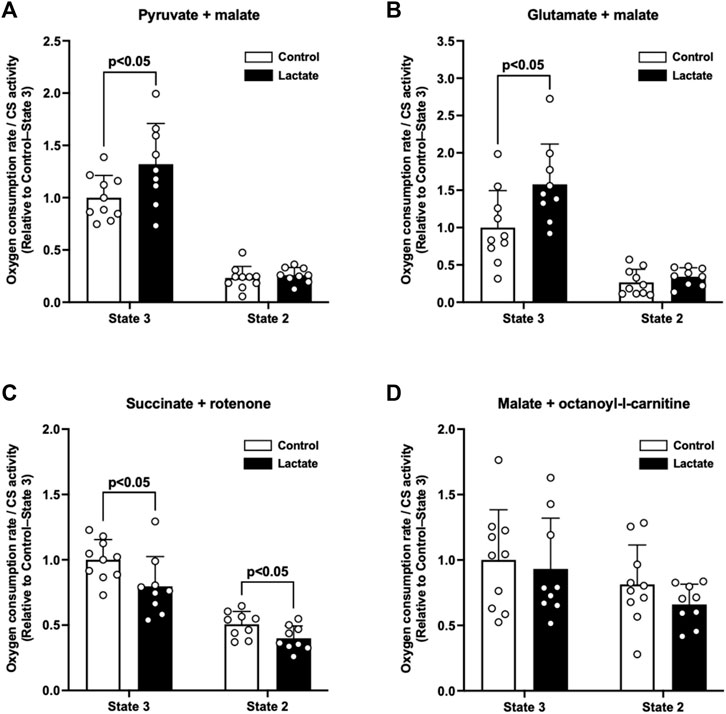
FIGURE 1. Oxygen consumption rate (OCR) in isolated mitochondria. (A) Pyruvate + malate-induced (complex I-driven) OCR. (B) Glutamate + malate-induced (complex I-driven) OCR. (C) Succinate + rotenone-induced (complex II-driven) OCR. (D) Malate + octanoyl-l-carnitine-induced OCR. Data are expressed as mean ± SD and are normalized to citrate synthase (CS) activity. Control group: n = 10, Lactate group: n = 9. Unpaired Student’s t-test was used for statistical evaluation.
To clarify the mechanisms underlying the changes in mitochondrial respiration, we evaluated enzymatic activities of the electron transport chain (complexes I, I + III, II, II + III, and IV). Complex I activity tended to be higher in the LAC group than that in the CON group (p = 0.06; Figure 2). Additionally, the activities of complexes I + III and IV were significantly higher in the LAC group than those in the CON group (p < 0.05; Figure 2). No significant differences were observed in the activities of complexes II and II + III (Figure 2). Moreover, complex I activity positively correlated with PM3 (r = 0.67, p < 0.01; Figure 3A) and GM3 (r = 0.77, p < 0.01; Figure 3B). No significant correlation between the activity of complex II and S3 was found (Figure 3C). Overall, it is likely that the increase in PM3 and GM3 respiration results from the enhanced complex I capacity. However, decline in S2and S3 respiration are not likely to be explained by complex II capacity.
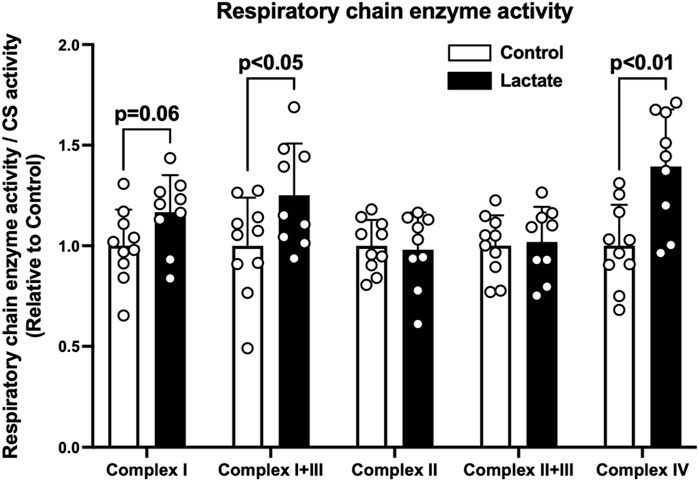
FIGURE 2. Mitochondrial respiratory chain enzyme activity. Data are expressed as mean ± SD and are normalized to citrate synthase (CS) activity. Control group: n = 10, Lactate group: n = 9. Unpaired Student’s t-test was used for statistical evaluation.
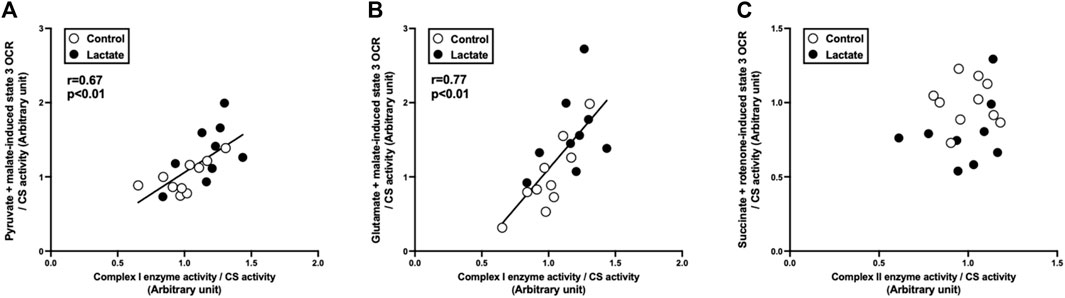
FIGURE 3. Correlation between state 3 oxygen consumption rate (OCR) and respiratory chain enzyme activity. (A) pyruvate + malate-induced (complex I-driven) state 3 OCR against complex I enzyme activity. (B) glutamate + malate-induced (complex I-driven) state 3 OCR against complex I enzyme activity. (C) succinate + rotenone-induced (complex II-driven) state 3 OCR against complex II enzyme activity. Data are expressed as mean ± SD. Control group: n = 10, Lactate group: n = 9. The correlation was studied using least-squares linear regressions, followed by the Pearson’s correlation coefficient test.
To further explore the mechanisms whereby mitochondrial respiration and electron transport chain enzyme activity were altered, we assessed the mitochondrial protein content and supercomplex assembly. First, we measured the protein content of the electron transport chain components. The protein content of NDUFS4, a subunit of complex I, was significantly higher in the LAC group than that in the CON group (Figure 4A). No significant differences were observed in protein content of NDUFB8, SDHB, UQCRC2, MTCO1, COXIV, and ATP5A (Figure 4A). These observations suggest that the increased NDUFS4 protein content may contributed to the enhanced complex I enzyme activity. Emerging evidence suggests that the mitochondrial supercomplex organization contributes to mitochondrial respiration (Ikeda et al., 2013; Greggio et al., 2017). Next, we evaluated mitochondrial supercomplex assembly using two-dimensional PAGE (BN-PAGE followed by SDS-PAGE). No differences in supercomplex assembly were observed between the CON and LAC groups (Figure 4B), suggesting that lactate administration changed mitochondrial respiration and electron transport enzyme activity without altering mitochondrial supercomplex assembly.
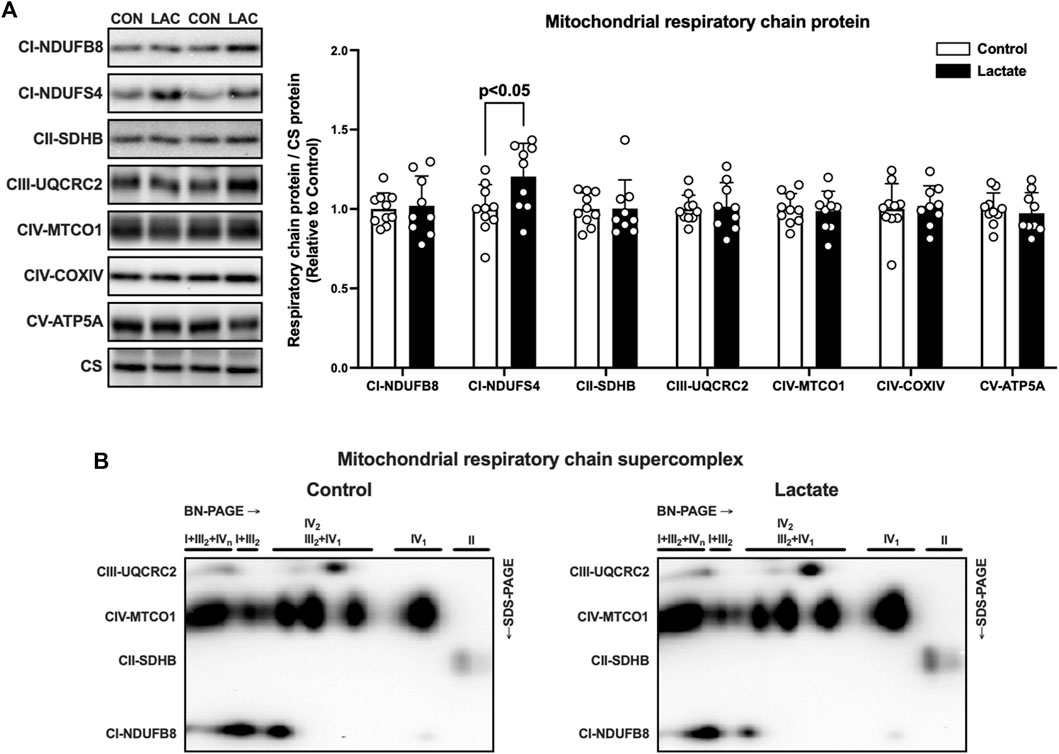
FIGURE 4. The protein content of respiratory chain components (A) and supercomplex assembly (B) in isolated mitochondria. Data are expressed as mean ± SD and are normalized to citrate synthase (CS) protein. Control group: n = 10, Lactate group: n = 9. Unpaired Student’s t-test was used for statistical evaluation.
Research has shown that not only mitochondrial content but also mitochondrial respiratory function is associated with exercise performance and metabolic disorders (Phielix et al., 2008; Granata et al., 2016). Although we and others previously reported that lactate increases mitochondrial content biomarkers, such as enzyme activity and protein content, in L6 cells (Hashimoto et al., 2007) and mouse skeletal muscle (Takahashi et al., 2019b; Takahashi et al., 2020b), whether lactate alters mitochondrial respiratory function remains to be elucidated. In the current study, we showed that 4 weeks of lactate administration enhanced PM3 and GM3 respiration, and decreased S3 and S2 respiration. These novel findings suggest that lactate changes mitochondrial respiratory function in skeletal muscle.
Although mitochondrial respiratory capacity is typically proportional to mitochondrial content (Hoppeler, Hudlicka and Uhlmann 1987; Jacobs et al., 2013), a dissociation between these two variables has been observed in mice (Rowe et al., 2013) and humans (Jacobs and Lundby 2013; Montero et al., 2015; Granata et al., 2016; Meinild Lundby et al., 2018). A previous study reported that sprint interval training, but not traditional endurance training, enhanced mitochondria-specific respiration in skeletal muscle without changes in mitochondrial content (Granata et al., 2016), although most studies have reported the increased mitochondrial content after high-intensity training (Gibala et al., 2006; Burgomaster et al., 2008; Jacobs et al., 2013). In contrast, traditional endurance training decreased intrinsic mitochondrial respiration in skeletal muscle, albeit, with an increase in mitochondrial content (Montero et al., 2015; Meinild Lundby et al., 2018). Based on these observations, it is believed that high training intensity contributes to the improvement of mitochondrial respiration rather than mitochondrial content (Bishop, Granata and Eynon 2014; Granata, Jamnick and Bishop 2018; Bishop et al., 2019). We have previously reported that the method of administration and volume of lactate used in this study increased the blood lactate concentration similar to a level that could be reached during high-intensity exercise (∼20 mM) (Kitaoka et al., 2016; Takahashi et al., 2019b). In the current study, we observed that 4 weeks of lactate administration enhanced PM3 and GM3 respiration. Taken together, improved mitochondrial respiration in skeletal muscle following high-intensity training may result, in part, from lactate production during exercise.
In the present study, we evaluated substrate- and complex-dependent mitochondrial respiratory function using different substrate combinations. Substrate combinations of pyruvate + malate and glutamate + malate stimulate dehydrogenases to produce NADH, which provides electrons for complex I. Flux through pyruvate dehydrogenase (PDH), a gateway enzyme for the entry of pyruvate into the tricarboxylic acid cycle, has been suggested to be a rate-limiting factor for pyruvate oxidation in the presence of malate (Bookelman et al., 1978; Lemieux, Tardif and Blier 2010). In the current study, complex I-driven state 3 respiration, irrespective of substrate combination (pyruvate + malate or glutamate + malate), was positively correlated with complex I enzyme activity. These observations suggest that, when mitochondrial respiration is maximally activated with adequate complex I substrates and ADP, NADH consumption by complex I appears to be a key step for mitochondrial oxygen consumption and subsequent ATP production.
In line with our previous observation that Ndufs4 mRNA level in mouse skeletal muscle increased 3 h after a single lactate administration (Kitaoka et al., 2016), we presently found that NDUFS4 protein level in isolate mitochondria increased after 4 weeks of lactate administration. In NDUFS4 deficiency, complex I fails to assemble properly or is unstable (Petruzzella et al., 2001), resulting in reduced complex I activity and complex I-driven respiration (Kruse et al., 2008; Ingraham et al., 2009). This suggests that NDUFS4 plays a key role in complex I function. Thus, the present observations of elevated PM3 and GM3 respiration associated with complex I enzyme activity may be explained, at least in part, by an increase in NDUFS4 protein levels.
Contrary to complex I-driven state 3 respiration, lactate administration diminished S3 and S2 respiration without changes in complex II enzyme activity or component protein (SDHB) level. These observations suggest that FADH2 production and consumption by complex II (SDH) do not explain the decline in S3 and S2 respiration. One possible explanation is inhibition of the dicarboxylate carrier that accounts for the uptake of succinate by isolated mitochondria. Previous studies have reported that the rate of uptake in mitochondria as catalyzed by the dicarboxylate carrier of dicarboxylates was inhibited in the presence of other intermediate metabolites (van Dam and Tsou 1968; Palmieri et al., 1971). Future studies are required to clarify whether lactate administration changes mitochondrial intermediary metabolism, which impairs mitochondrial succinate uptake and succinate-fueled respiration.
The observation of increased PM3 and GM3 respiration with decreased S3 respiration indicates that possible metabolic remodeling in favor of complex I substrates relative to complex II substrates occurred. In the skeletal muscle of streptozotocin-induced type 1 diabetic rats, complex I-driven respiration has increased despite no difference in complex I + II-driven respiration, where the substrate control ratio for succinate and complex II protein content are reduced (Larsen et al., 2015). Other studies have shown that electron flow through complex II increases in complex I-deficient mice (Alam et al., 2015; Terburgh et al., 2019). These observations suggest that complexes I and II reciprocally compensate for deteriorated complex functions. Importantly, another study reported that resistance training increased complex I-driven respiration together with complex I protein abundance, while the substrate control ratio for succinate was reduced even though complex II protein levels remained unchanged (Porter et al., 2015). This suggests that augmented electron transfer through complex I may result in diminished complex II electron transfer. Thus, the observation of reduced S3 respiration is likely due to improved complex I function rather than a decline in complex II abundance or function.
NADH and FADH2 generated in the course of fatty acid oxidation are substrates for complex I and the electron-transferring flavoprotein complex. In the present study, although we observed a tendency to increase complex I enzyme activity and a significant increase in complex I + III enzyme activity after lactate administration, neither MOc3 nor MOc2 respiration was changed. While both pyruvate and fatty acids are converted to acetyl-CoA and NADH, the maximal flux through PDH to produce these metabolites from pyruvate is much higher than the reaction of fatty acid oxidation (Petrick and Holloway 2019). These observations suggest that NADH production through fatty acid oxidation did not reach the maximal capacity of complex I to consume NADH, and that reactions other than NADH consumption are key events for fatty acid-induced mitochondrial respiration. We previously reported that high-intensity training enhanced palmitate-induced oxygen consumption with the increased activity of ß-HAD, through which NADH is yielded, in the isolated mitochondria of rat skeletal muscle (Hoshino et al., 2013). In the present study, ß-HAD activity did not change after lactate administration (CON: 1.00 ± 0.16; LAC: 0.99 ± 0.10, relative to CON) (Supplementary Figure S1C). Collectively, it appears that increasing NADH production through ß-HAD reaction is likely to be a key step for fatty acid-induced mitochondrial respiration.
The conversion of lactate to pyruvate and subsequent pyruvate oxidation in the tricarboxylic acid cycle yield NADH, which is then consumed by complex I. Previous studies have reported an elevated lactate concentration in the circulation and tissues of NDUFS4 deficient mice (Kruse et al., 2008; Ingraham et al., 2009; Jin et al., 2014; Saito et al., 2019). Additionally, humans with a mutation of NDUFS4 genes, including patients with Leigh syndrome, have repeatedly shown an increased blood lactate level (Budde et al., 2000; Loeffen et al., 2000; Petruzzella et al., 2001; Iuso et al., 2006; Assouline et al., 2012). Without proper function of complex I due in part to loss of NDUFS4, pyruvate cannot be oxidized in mitochondria, but instead, converted to lactate to regenerate NAD+ (Ingraham et al., 2009). These suggest that decline in complex I capacity cause lactate accumulation; on the contrary, increasing complex I capacity enhances lactate oxidation. Therefore, it is speculated that lactate enhances complex I capacity, presumably enabling the mitochondria to preferentially oxidize lactate in the condition that abundant lactate is available. In support of this notion we previously observed that, after 3 weeks of lactate administration with endurance training, lactate removal from the blood circulation in mice was enhanced to a greater extent than with endurance training alone (Takahashi et al., 2019b).
The molecular mechanisms whereby lactate administration induces mitochondrial adaptation remain unclear. Growing evidence has shown that lactate regulates cell signaling and adaptation (Brooks 2020; Brooks et al., 2021). Previous studies have reported that lactate activates cell signaling by binding to hydroxycarboxylic acid receptor 1 (Ahmed et al., 2010; Ohno et al., 2018). Another study reported that lactate facilitated adipose secretion of TGF-β, which subsequently enhanced mRNA expression of peroxisome proliferator-activated receptor γ coactivator 1-α (Pgc-1α), a master regulator of mitochondrial adaptation (Wu et al., 1999), in mouse skeletal muscles (Takahashi et al., 2019a), suggesting that inter-organ communication activated by lactate possibly contributed to the current findings. Other researchers have identified a lactate-responsive form of PGC1 (LRPGC1), which has been reported to enhance lactate consumption by enhancing estrogen-related receptor γ-mediated transcription in mouse hepatic and renal cells (Tanida, Matsuda and Tanaka 2020). Furthermore, a previous study demonstrated metabolic regulation of gene expression by histone lactylation in cancer cells (Zhang et al., 2019). Lastly, it should be noted that exposure to high lactate levels can change the catabolic fate of pyruvate in skeletal muscle and other cells. Because of mass action restrictions, relatively little, if any, pyruvate is reduced to lactate, which instead needs to be oxidized by mitochondria. This relatively high substrate pressure can cause a reduced mitochondrial environment with increased NADH/NAD+ and ATP/ADP ratios, increased protonmotive force and, related, increased likelihood of reactive oxygen species generation, which is considered to trigger gene expressions, leading to enhanced oxidative activity and capacity (Hashimoto et al., 2007; Brooks 2020). Whether these previous findings explain the current observations of altered mitochondrial function should be clarified in future studies.
The present study examined whether lactate administration alters mitochondrial respiratory function in skeletal muscle. Using isolated mitochondria, we demonstrated that lactate administration increased pyruvate + malate- and glutamate + malate-induced (complex I-driven) mitochondrial respiration, with a tendency to increase complex I enzyme activity and a significant increase in complex I component protein. Additionally, lactate administration decreased succinate + rotenone-induced (complex II-driven) mitochondrial respiration, despite no changes in complex II enzyme activity or component proteins. These findings suggest that lactate alters mitochondrial respiratory function, and that exercise training-induced increases in mitochondrial respiration may result, in part, from lactate production during exercise.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Animal Experimental Committee of The University of Tokyo.
KT, YT, YK, and HH conceived and designed research. KT, YT, and YM performed experiments; KT analyzed data. All authors contributed to the interpretation of the results and in drafting and revising the content of the manuscript. All authors read and approved the final manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers 20H04071 (to YK and HH) and 21K21249 (to KT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.920034/full#supplementary-material
Ahmed K., Tunaru S., Tang C., Müller M., Gille A., Sassmann A., et al. (2010). An Autocrine Lactate Loop Mediates Insulin-dependent Inhibition of Lipolysis through GPR81. Cell. Metab. 11, 311–319. doi:10.1016/j.cmet.2010.02.012
Alam M. T., Manjeri G. R., Rodenburg R. J., Smeitink J. A. M., Notebaart R. A., Huynen M., et al. (2015). Skeletal Muscle Mitochondria of NDUFS4−/− Mice Display Normal Maximal Pyruvate Oxidation and ATP Production. Biochimica Biophysica Acta (BBA) - Bioenergetics 1847, 526–533. doi:10.1016/j.bbabio.2015.02.006
Assouline Z., Jambou M., Rio M., Bole-Feysot C., de Lonlay P., Barnerias C., et al. (2012). A Constant and Similar Assembly Defect of Mitochondrial Respiratory Chain Complex I Allows Rapid Identification of NDUFS4 Mutations in Patients with Leigh Syndrome. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1822, 1062–1069. doi:10.1016/j.bbadis.2012.01.013
Bishop D. J., Botella J., Genders A. J., Lee M. J.-C., Saner N. J., Kuang J., et al. (2019). High-Intensity Exercise and Mitochondrial Biogenesis: Current Controversies and Future Research Directions. Physiology 34, 56–70. doi:10.1152/physiol.00038.2018
Bishop D. J., Granata C., Eynon N. (2014). Can We Optimise the Exercise Training Prescription to Maximise Improvements in Mitochondria Function and Content? Biochimica Biophysica Acta (BBA) - General Subj. 1840, 1266–1275. doi:10.1016/j.bbagen.2013.10.012
Bookelman H., Trijbels J. M. F., Sengers R. C. A., Janssen A. J. M., Veerkamp J. H., Stadhouders A. M. (1978). Pyruvate Oxidation in Rat and Human Skeletal Muscle Mitochondria. Biochem. Med. 20, 395–403. doi:10.1016/0006-2944(78)90089-3
Brooks G. A. (202010145). Lactate as a Fulcrum of Metabolism. Redox Biol. doi:10.1016/j.redox.2020.101454
Brooks G. A., Arevalo J. A., Osmond A. D., Leija R. G., Curl C. C., Tovar A. P. (2021). Lactate in Contemporary Biology: a Phoenix Risen. J. Physiol. doi:10.1113/jp280955
Budde S. M. S., van den Heuvel L. P. W. J., Janssen A. J., Smeets R. J. P., Buskens C. A. F., DeMeirleir L., et al. (2000). Combined Enzymatic Complex I and III Deficiency Associated with Mutations in the Nuclear Encoded NDUFS4 Gene. Biochem. Biophysical Res. Commun. 275, 63–68. doi:10.1006/bbrc.2000.3257
Burgomaster K. A., Howarth K. R., Phillips S. M., Rakobowchuk M., Macdonald M. J., McGee S. L., et al. (2008). Similar Metabolic Adaptations during Exercise after Low Volume Sprint Interval and Traditional Endurance Training in Humans. J. Physiol. 586, 151–160. doi:10.1113/jphysiol.2007.142109
Gibala M. J., Little J. P., van Essen M., Wilkin G. P., Burgomaster K. A., Safdar A., et al. (2006). Short-term Sprint Intervalversustraditional Endurance Training: Similar Initial Adaptations in Human Skeletal Muscle and Exercise Performance. J. Physiol. 575, 901–911. doi:10.1113/jphysiol.2006.112094
Gladden L. B. (2004). Lactate Metabolism: A New Paradigm for the Third Millennium. J. Physiol. 558, 5–30.
Granata C., Jamnick N. A., Bishop D. J. (2018). Training-Induced Changes in Mitochondrial Content and Respiratory Function in Human Skeletal Muscle. Sports Med. 48, 1809–1828. doi:10.1007/s40279-018-0936-y
Granata C., Oliveira R. S. F., Little J. P., Renner K., Bishop D. J. (2016). Training Intensity Modulates Changes in PGC‐1α and P53 Protein Content and Mitochondrial Respiration, but Not Markers of Mitochondrial Content in Human Skeletal Muscle. FASEB J. 30, 959–970. doi:10.1096/fj.15-276907
Greggio C., Jha P., Kulkarni S. S., Lagarrigue S., Broskey N. T., Boutant M., et al. (2017). Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell. Metab. 25, 301–311. doi:10.1016/j.cmet.2016.11.004
Hashimoto T., Hussien R., Oommen S., Gohil K., Brooks G. A. (2007). Lactate Sensitive Transcription Factor Network in L6 Cells: Activation of MCT1 and Mitochondrial Biogenesis. FASEB J. 21, 2602–2612. doi:10.1096/fj.07-8174com
Hoppeler H., Hudlicka O., Uhlmann E. (1987). Relationship between Mitochondria and Oxygen Consumption in Isolated Cat Muscles. J. Physiol. 385, 661–675. doi:10.1113/jphysiol.1987.sp016513
Hoshino D., Yoshida Y., Kitaoka Y., Hatta H., Bonen A. (2013). High-intensity Interval Training Increases Intrinsic Rates of Mitochondrial Fatty Acid Oxidation in Rat Red and White Skeletal Muscle. Appl. Physiol. Nutr. Metab. 38, 326–333. doi:10.1139/apnm-2012-0257
Ikeda K., Shiba S., Horie-Inoue K., Shimokata K., Inoue S. (2013). A Stabilizing Factor for Mitochondrial Respiratory Supercomplex Assembly Regulates Energy Metabolism in Muscle. Nat. Commun. 4, 2147. doi:10.1038/ncomms3147
Ingraham C. A., Burwell L. S., Skalska J., Brookes P. S., Howell R. L., Sheu S.-S., et al. (2009). NDUFS4: Creation of a Mouse Model Mimicking a Complex I Disorder. Mitochondrion 9, 204–210. doi:10.1016/j.mito.2009.02.001
Iuso A., Scacco S., Piccoli C., Bellomo F., Petruzzella V., Trentadue R., et al. (2006). Dysfunctions of Cellular Oxidative Metabolism in Patients with Mutations in the NDUFS1 and NDUFS4 Genes of Complex I. J. Biol. Chem. 281, 10374–10380. doi:10.1074/jbc.m513387200
Jacobs R. A., Flück D., Bonne T. C., Bürgi S., Christensen P. M., Toigo M., et al. (20131985). Improvements in Exercise Performance with High-Intensity Interval Training Coincide with an Increase in Skeletal Muscle Mitochondrial Content and Function. J. Appl. Physiology 115, 785–793. doi:10.1152/japplphysiol.00445.2013
Jacobs R. A., Lundby C. (20131985). Mitochondria Express Enhanced Quality as Well as Quantity in Association with Aerobic Fitness across Recreationally Active Individuals up to Elite Athletes. J. Appl. Physiology 114, 344–350. doi:10.1152/japplphysiol.01081.2012
Jin Z., Wei W., Yang M., Du Y., Wan Y. (2014). Mitochondrial Complex I Activity Suppresses Inflammation and Enhances Bone Resorption by Shifting Macrophage-Osteoclast Polarization. Cell. Metab. 20, 483–498. doi:10.1016/j.cmet.2014.07.011
Joyner M. J., Coyle E. F. (2008). Endurance Exercise Performance: The Physiology of Champions. J. Physiol. 586, 35–44.
Kitaoka Y., Takeda K., Tamura Y., Hatta H. (2016). Lactate Administration Increases mRNA Expression of PGC-1α and UCP3 in Mouse Skeletal Muscle. Appl. Physiol. Nutr. Metab. 41, 695–698. doi:10.1139/apnm-2016-0016
Kitaoka Y., Tamura Y., Takahashi K., Takeda K., Takemasa T., Hatta H. (2019). Effects of Nrf2 Deficiency on Mitochondrial Oxidative Stress in Aged Skeletal Muscle. Physiol. Rep. 7, e13998. doi:10.14814/phy2.13998
Kruse S. E., Watt W. C., Marcinek D. J., Kapur R. P., Schenkman K. A., Palmiter R. D. (2008). Mice with Mitochondrial Complex I Deficiency Develop a Fatal Encephalomyopathy. Cell. Metab. 7, 312–320. doi:10.1016/j.cmet.2008.02.004
Larsen S., Scheede-Bergdahl C., Whitesell T., Boushel R., Bergdahl A. (2015). Increased Intrinsic Mitochondrial Respiratory Capacity in Skeletal Muscle from Rats with Streptozotocin-Induced Hyperglycemia. Physiol. Rep. 3, e12467. doi:10.14814/phy2.12467
Lemieux H., Tardif J.-C., Blier P. U. (2010). Thermal Sensitivity of Oxidative Phosphorylation in Rat Heart Mitochondria: Does Pyruvate Dehydrogenase Dictate the Response to Temperature? J. Therm. Biol. 35, 105–111. doi:10.1016/j.jtherbio.2009.12.003
Loeffen J. L. C. M., Smeitink J. A. M., Trijbels J. M. F., Janssen A. J. M., Triepels R. H., Sengers R. C. A., et al. (2000). Isolated Complex I Deficiency in Children: Clinical, Biochemical and Genetic Aspects. Hum. Mutat. 15, 123–134. doi:10.1002/(sici)1098-1004(200002)15:2<123:aid-humu1>3.0.co;2-p
Meinild Lundby A. K., Jacobs R. A., Gehrig S., de Leur J., Hauser M., Bonne T. C., et al. (2018). Exercise Training Increases Skeletal Muscle Mitochondrial Volume Density by Enlargement of Existing Mitochondria and Not De Novo Biogenesis. Acta Physiol. (Oxf) 222. doi:10.1111/apha.12905
Mogensen M., Sahlin K., Fernström M., Glintborg D., Vind B. F., Beck-Nielsen H., et al. (2007). Mitochondrial Respiration is Decreased in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes 56, 1592–1599.
Montero D., Cathomen A., Jacobs R. A., Flück D., de Leur J., Keiser S., et al. (2015). Haematological rather Than Skeletal Muscle Adaptations Contribute to the Increase in Peak Oxygen Uptake Induced by Moderate Endurance Training. J. Physiol. 593, 4677–4688. doi:10.1113/jp270250
Ohno Y., Oyama A., Kaneko H., Egawa T., Yokoyama S., Sugiura T., et al. (2018). Lactate Increases Myotube Diameter via Activation of MEK/ERK Pathway in C2C12 Cells. Acta Physiol. 223, e13042. doi:10.1111/apha.13042
Palmieri F., Prezioso G., Quagliariello E., Klingenberg M. (1971). Kinetic Study of the Dicarboxylate Carrier in Rat Liver Mitochondria. Eur. J. Biochem. 22, 66–74. doi:10.1111/j.1432-1033.1971.tb01515.x
Petrick H. L., Holloway G. P. (2019). The Regulation of Mitochondrial Substrate Utilization during Acute Exercise. Curr. Opin. Physiology 10, 75–80. doi:10.1016/j.cophys.2019.04.021
Petruzzella V., Vergari R., Puzziferri I., Boffoli D., Lamantea E., Zeviani M., et al. (2001). A Nonsense Mutation in the NDUFS4 Gene Encoding the 18 kDa (AQDQ) Subunit of Complex I Abolishes Assembly and Activity of the Complex in a Patient with Leigh-like Syndrome. Hum. Mol. Genet. 10, 529–535. doi:10.1093/hmg/10.5.529
Phielix E., Schrauwen-Hinderling V. B., Mensink M., Lenaers E., Meex R., Hoeks J., et al. (2008). Lower Intrinsic ADP-Stimulated Mitochondrial Respiration Underlies In Vivo Mitochondrial Dysfunction in Muscle of Male Type 2 Diabetic Patients. Diabetes 57, 2943–2949. doi:10.2337/db08-0391
Porter C., Reidy P. T., Bhattarai N., Sidossis L. S., Rasmussen B. B. (2015). Resistance Exercise Training Alters Mitochondrial Function in Human Skeletal Muscle. Med. Sci. Sports Exerc 47, 1922–1931. doi:10.1249/mss.0000000000000605
Rowe G. C., Patten I. S., Zsengeller Z. K., El-Khoury R., Okutsu M., Bampoh S., et al. (2013). Disconnecting Mitochondrial Content from Respiratory Chain Capacity in PGC-1-Deficient Skeletal Muscle. Cell. Rep. 3, 1449–1456. doi:10.1016/j.celrep.2013.04.023
Saito S., Takahashi Y., Ohki A., Shintani Y., Higuchi T. (2019). Early Detection of Elevated Lactate Levels in a Mitochondrial Disease Model Using Chemical Exchange Saturation Transfer (CEST) and Magnetic Resonance Spectroscopy (MRS) at 7T-MRI. Radiol. Phys. Technol. 12, 46–54. doi:10.1007/s12194-018-0490-1
Takahashi H., Alves C. R. R., Stanford K. I., Middelbeek R. J. W., Nigro P., Ryan R. E., et al. (2019a). TGF-β2 Is an Exercise-Induced Adipokine that Regulates Glucose and Fatty Acid Metabolism. Nat. Metab. 1, 291–303. doi:10.1038/s42255-018-0030-7
Takahashi K., Kitaoka Y., Matsunaga Y., Hatta H. (2020a). Effect of Post-exercise Lactate Administration on Glycogen Repletion and Signaling Activation in Different Types of Mouse Skeletal Muscle. Curr. Res. Physiol. 3, 34–43. doi:10.1016/j.crphys.2020.07.002
Takahashi K., Kitaoka Y., Matsunaga Y., Hatta H. (2019b). Effects of Lactate Administration on Mitochondrial Enzyme Activity and Monocarboxylate Transporters in Mouse Skeletal Muscle. Physiol. Rep. 7, e14224. doi:10.14814/phy2.14224
Takahashi K., Kitaoka Y., Yamamoto K., Matsunaga Y., Hatta H. (2020b). Oral Lactate Administration Additively Enhances Endurance Training-Induced Increase in Cytochrome C Oxidase Activity in Mouse Soleus Muscle. Nutrients 12, 770. doi:10.3390/nu12030770
Tamura Y., Kouzaki K., Kotani T., Nakazato K. (2020). Electrically Stimulated Contractile Activity-Induced Transcriptomic Responses and Metabolic Remodeling in C2C12 Myotubes: Twitch vs. Tetanic Contractions. Am. J. Physiology-Cell Physiology 319, C1029–c1044. doi:10.1152/ajpcell.00494.2019
Tanida T., Matsuda K. I., Tanaka M. (2020)). Metabolic System for Lactic Acid via LRPGC1/ERRγ Signaling Pathway. Faseb J. 34 (10), 13239–13256. doi:10.1096/fj.202000492R
Terburgh K., Lindeque Z., Mason S., van der Westhuizen F., Louw R. (2019). Metabolomics of Ndufs4−/− Skeletal Muscle: Adaptive Mechanisms Converge at the Ubiquinone-Cycle. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1865, 98–106. doi:10.1016/j.bbadis.2018.10.034
Tomiya S., Tamura Y., Kouzaki K., Kotani T., Wakabayashi Y., Noda M., et al. (2019). Cast Immobilization of Hindlimb Upregulates Sarcolipin Expression in Atrophied Skeletal Muscles and Increases Thermogenesis in C57BL/6J Mice. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 317, R649–r661. doi:10.1152/ajpregu.00118.2019
van Dam K., Tsou C. S. (1968). Accumulation of Substrates by Mitochondria. Biochimica Biophysica Acta (BBA) - Bioenergetics 162, 301–309. doi:10.1016/0005-2728(68)90116-3
Wakabayashi Y., Tamura Y., Kouzaki K., Kikuchi N., Hiranuma K., Menuki K., et al. (2020). Acetaldehyde Dehydrogenase 2 Deficiency Increases Mitochondrial Reactive Oxygen Species Emission and Induces Mitochondrial Protease Omi/HtrA2 in Skeletal Muscle. Am. J. Physiology-Regulatory, Integr. Comp. Physiology 318, R677–r690. doi:10.1152/ajpregu.00089.2019
Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., et al. (1999). Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell. 98, 115–124. doi:10.1016/s0092-8674(00)80611-x
Keywords: lactate, mitochondria, oxygen consumption rate, supercomplex, skeletal muscle
Citation: Takahashi K, Tamura Y, Kitaoka Y, Matsunaga Y and Hatta H (2022) Effects of Lactate Administration on Mitochondrial Respiratory Function in Mouse Skeletal Muscle. Front. Physiol. 13:920034. doi: 10.3389/fphys.2022.920034
Received: 14 April 2022; Accepted: 09 June 2022;
Published: 30 June 2022.
Edited by:
Damien Roussel, Université Claude Bernard Lyon 1, FranceReviewed by:
Charles Affourtit, University of Plymouth, United KingdomCopyright © 2022 Takahashi, Tamura, Kitaoka, Matsunaga and Hatta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideo Hatta, aGF0dGFAaWRhdGVuLmMudS10b2t5by5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.