 Molly E. Streiff
Molly E. Streiff Frank B. Sachse
Frank B. Sachse
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 16 June 2022
Sec. Computational Physiology and Medicine
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.916278
This article is part of the Research Topic Modeling of Cardiovascular Systems View all 9 articles
The intricate regulation of the compartmental Ca2+ concentrations in cardiomyocytes is critical for electrophysiology, excitation-contraction coupling, and other signaling pathways. Research into the complex signaling pathways is motivated by cardiac pathologies including arrhythmia and maladaptive myocyte remodeling, which result from Ca2+ dysregulation. Of interest to this investigation are two types of Ca2+ currents in cardiomyocytes: 1) background Ca2+ entry, i.e., Ca2+ transport across the sarcolemma from the extracellular space into the cytosol, and 2) Ca2+ leak from the sarcoplasmic reticulum (SR) across the SR membrane into the cytosol. Candidates for the ion channels underlying background Ca2+ entry and SR Ca2+ leak channels include members of the mechano-modulated transient receptor potential (TRP) family. We used a mathematical model of a human ventricular myocyte to analyze the individual contributions of background Ca2+ entry and SR Ca2+ leak to the modulation of Ca2+ transients and SR Ca2+ load at rest and during action potentials. Background Ca2+ entry exhibited a positive relationship with both [Ca2+]i and [Ca2+]SR. Modulating SR Ca2+ leak had opposite effects of background Ca2+ entry. Effects of SR Ca2+ leak on Ca2+ were particularly pronounced at lower pacing frequency. In contrast to the pronounced effects of background and leak Ca2+ currents on Ca2+ concentrations, the effects on cellular electrophysiology were marginal. Our studies provide quantitative insights into the differential modulation of compartmental Ca2+ concentrations by the background and leak Ca2+ currents. Furthermore, our studies support the hypothesis that TRP channels play a role in strain-modulation of cardiac contractility. In summary, our investigations shed light on the physiological effects of the background and leak Ca2+ currents and their contribution to the development of disease caused by Ca2+ dysregulation.
Ca2+ concentrations are dynamically controlled in cardiomyocytes by a complex regulatory system comprising ion channels, transporters, exchangers, regulatory proteins, and ion buffers. Intricately regulated levels of Ca2+ concentrations are critical for electrical activity, excitation-contraction coupling, and other signaling pathways. In many cardiac diseases, the delicate balance of Ca2+ cycling is perturbed. Ca2+ dysregulation underlies maladaptive cardiac remodeling. A complete understanding of all components of Ca2+ handling is essential for the development of therapeutic strategies to attenuate cardiac pathologies.
Many ion channels underlying the Ca2+ signaling in cardiomyocytes are well characterized. Ca2+ signaling related to excitation-contraction coupling primarily relies on sarcolemmal Ca2+ entry through voltage-gated L-type Ca2+ channels (LTCC) to trigger Ca2+-induced Ca2+ release from the sarcoplasmic reticulum (SR) through ryanodine receptors (RyR), and, subsequently, Ca2+ extrusion via the sodium-calcium exchanger (NCX) and reuptake into the SR through sarco/endoplasmic Ca2+-ATPase (SERCA). Other Ca2+ currents contribute to the modulation of Ca2+ concentrations in cardiomyocytes as well. Of interest to this study are background Ca2+ entry through the sarcolemma and SR Ca2+ leak. Understanding of the physiological role of these Ca2+ currents is still incomplete.
Beyond Ca2+ transport across the sarcolemma from the extracellular space into the cytosol through LTCC and NCX, sarcolemmal Ca2+ transport comprises background Ca2+ entry. In resting cardiomyocytes, [Ca2+]i is of the order of 100 nM, which indicates the existence of a background Ca2+ entry pathway to balance the Ca2+ efflux of NCX (Eisner et al., 2020). Resting ventricular myocytes depleted of Ca2+ stores with caffeine are able to reload the SR by a mechanism that involves extracellular Ca2+, demonstrating further evidence of background Ca2+ entry (Terracciano and Macleod, 1996). Based on studies measuring background Ca2+ influx in rat ventricular myocytes of the order of 2–5 μmol/L per second (Choi et al., 2000) or 4 μmol/L per second (Sankaranarayanan et al., 2017), the background Ca2+ entry is approximately 10% of the influx through LTCC current (5–10 μmol/L each action potential) at normal heart rates (Eisner et al., 2020). The identity of background Ca2+ flux is still poorly defined, but several channels have been suggested as contributors.
One study identified a Ca2+ entry mechanism that is blocked by the nonspecific agent gadolinium (Gd3+) (Kupittayanant et al., 2006). Connexin hemichannels are candidates for background Ca2+ entry since they can be inhibited by Gd3+ (Stout et al., 2002). While connexin hemichannels primarily form pairs to allow ion fluxes between cells at intercalated discs, some are present as hemichannels in the surface membrane of a single cell (Wang et al., 2012; Leybaert et al., 2017) and may, therefore, provide a route for Ca2+ entry. However, primary candidates for background Ca2+ entry include members of the family of Transient Receptor Potential (TRP) channels, which are also sensitive to Gd3+. The mdx mouse model of muscular dystrophy exhibits [Ca2+]i and [Na+]i overload that can be blocked by Gd3+, and the increase in cation entry has been suggested to involve TRPC channels (Mijares et al., 2014). Myocytes from old mdx mice exhibit increased expression of a putative stretch-activated channel (SAC), TRPC1. Elevated [Ca2+]i levels can also be reduced to [Ca2+]i levels of WT myocytes when exposed to SAC blockers streptomycin or GsMTx-4 (Williams and Allen, 2007; Ward et al., 2008). Upregulated TRPC1 also contributes to increased [Ca2+]i through SAC in hypertrophic myocardium of rats following isoproterenol injection (Chen et al., 2013). Background Ca2+ entry involved in maladaptive cardiac remodeling was more recently shown to critically depend on both TRPC1 and TRPC4 (Camacho Londono et al., 2015, Camacho Londoño et al., 2021). TRPC6 channels have also been shown to modulate cytosolic Ca2+ transients and SR Ca2+ load through sarcolemmal Ca2+ entry (Ahmad et al., 2020). Furthermore, TRPV4 can modulate Ca2+ transients and SR load, and participates in hypoosmotic stress-induced cardiomyocyte Ca2+ entry (Rubinstein et al., 2014; Jones et al., 2019).
Ca2+ transport across the SR membrane from the intracellular store into the cytosol is known as SR Ca2+ leak. During an action potential, activation of RyR clusters triggered by Ca2+ entry through LTCC results in a synchronized release of a large amount of Ca2+ from the SR that forms the Ca2+ transient and leads to cardiomyocyte contraction. Activation of RyR clusters causes Ca2+ sparks. These sparks are an important pathway for SR Ca2+ leak. RyR Ca2+ leak can occur also through a mechanism independent of sparks (Santiago et al., 2010). Further, total SR Ca2+ leak includes a component separate from RyRs (Zima et al., 2010). Many candidates have been suggested as components of this leak, yet it is still ill-defined. A candidate is the inositol 1,4,5-trisphosphate (IP3) receptor (IP3R), which is a Ca2+ release channel expressed at lower densities than RyRs in cardiomyocytes and found upregulated in heart failure (HF) (Go et al., 1995; Ai et al., 2005) and therefore may be a relevant contributor to SR Ca2+ leak (Zima et al., 2010). Members of the TRP family have also been suggested to contribute to SR Ca2+ leak. TRPC1 was found to operate as a SR Ca2+ leak channel in skeletal muscle (Berbey et al., 2009) and more recently in cardiomyocytes (Hu et al., 2020). Additional evidence suggests contribution of TRPC6, TRPM8, TRPP2, and TRPV1 to endoplasmic reticulum Ca2+ leak in various cell types, although characterization is still incomplete for cardiomyocytes (Lemos et al., 2021).
TRP channels constitute primary candidates for explaining background and leak Ca2+ currents through both the sarcolemma and the SR membrane. Interestingly, many members of the TRP family are known to be modulated by stretch (Inoue et al., 2009; Reed et al., 2014; Peyronnet et al., 2016). Cardiomyocyte contractility is known to respond to mechanical stretch in two phases: a rapid and a slow response (Calaghan and White, 1999). The rapid response is the cellular basis for the Frank-Starling Mechanism (FSM). It relies primarily on myofilament overlap and alteration of myofilament Ca2+ sensitivity, and does not involve changes in Ca2+ transients. The slow response has been termed slow force response (SFR) or stress-induced slow increase in contractility (SSC), describing the gradual increase in twitch force corresponding to an increase in [Ca2+]i transients that develops over several minutes when stretch is sustained. Members of the TRP family are likely candidates for mechanotransduction of the SFR.
Though background and leak Ca2+ currents are still poorly defined, they may play important roles in regulating Ca2+ homeostasis and contractility and altered Ca2+ dynamics in cardiac disease. The lack of complete understanding of the identity and mechanics of these channels, in addition to their relatively small amplitude compared to the voltage-gated Ca2+ channels, make them difficult to study in vivo. A computational model provides the advantage to study the currents and their roles in isolation from other cellular mechanisms. In this study, we use a mathematical model of a human ventricular myocyte to analyze the individual contributions of background Ca2+ entry and SR Ca2+ leak to the modulation of Ca2+ transients and SR Ca2+ load. We also assess the effects of the Ca2+ currents on cellular electrophysiology.
We applied a mathematical model of a human ventricular myocyte (Grandi et al., 2010). The model and subsequent analyses were executed in MATLAB (R2020b). The model includes subsarcolemmal and junctional compartments beyond the cytosol compartment. Total background Ca2+ current (ICabk) through the sarcolemma is defined as a summation of junctional (ICabk, junc) and subsarcolemmal (ICabk,sl) components:
where Fjunc = 0.11 and Fsl = 0.89 are constants that determine the fraction of total background current corresponding to the junctional and subsarcolemmal spaces, respectively, Gbkg is the maximum conductance (5.513e-4 A/F) of the channels, Vm is the transmembrane voltage, and ECa,junc and ECa,sl are Nernst potentials corresponding to the junctional and subsarcolemmal spaces, respectively. SR Ca2+ leak (Jleak) describes the Ca2+ flux out of the SR:
where Kleak is the leak constant (5.348e-6/ms), and [Ca2+]SR and [Ca2+]j describe the Ca2+ concentrations in the SR and junctional space, respectively. In this study, we modulated Gbkg and Kleak independently and in conjunction to investigate the effects of altered sarcolemmal Ca2+ entry and altered SR Ca2+ leak, respectively.
A graphical summary of the Ca2+ currents in the cell model is shown in Figure 1. The total Ca2+ current into the junctional (ICa,tot,junc) and subsarcolemmal (ICa,tot,sl) compartments are determined by Eqs 5, 6, respectively:
where ICa describes the L-type Ca2+ current, IpCa describes the sarcolemmal Ca2+ pump current, and INCX describes the NCX current.
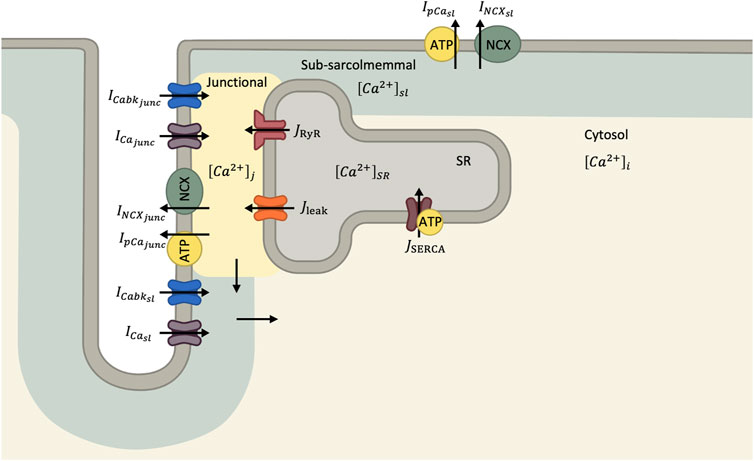
FIGURE 1. Diagram of Ca2+ handling components in human ventricular myocyte model (Grandi et al., 2010). In this mathematical model, the cytosol is divided into junction, sub-sarcolemmal and bulk compartments. Ca2+ currents through the sarcolemma enter both the junctional and sub-sarcolemmal compartments before diffusing to the bulk cytosol.
The rate of change of Ca2+ concentrations in the junctional compartment [Ca2+]j, subsarcolemmal compartment [Ca2+]sl, bulk cytosol [Ca2+]i, and SR [Ca2+]SR are given by:
where Cmem is the membrane capacitance, F is Faraday’s constant, Vjunc is the volume of the junctional compartment, Vsl is the volume of the subsarcolemmal compartment, Vmyo is the volume of the bulk cytosol, and Vsr is the volume of the SR. JCa,juncsl is the rate of Ca2+ flux from junctional to subsarcolemmal compartments and JCa,slmyo is the rate of Ca2+ flux from the subsarcolemmal compartment to bulk cytosol. JRYR describes the SR Ca2+ release and JSERCA describes the SR Ca2+ pump. Ca2+ buffering is described by JCa,B,junction in the junctional compartment, JCa,B,sl in the subsarcolemmal compartment, JCa,B,cytosol in the bulk cytosol, and dCsqnb/dt in the SR. We refer to (Grandi et al., 2010) for more details and the equations describing the mathematical model.
To investigate the effects of altered background Ca2+ entry, we modulated Gbkg from 0 to 300% of its default value, 5.513e-4 A/F. To investigate the effects of altered SR Ca2+ leak, we modulated Kleak from 0 to 300% of its default value, 5.348e-6 ms−1. Simulation durations were 1 min to establish steady state. We analyzed the last beat. We performed sensitivity analyses for the modulation of Gbkg and Kleak independently on measurements of resting Vm, maximum Vm, action potential duration to 90% repolarization (APD90), diastolic [Ca2+]i, systolic [Ca2+]i, [Ca2+]i amplitude, diastolic [Ca2+]SR, systolic [Ca2+]SR, and [Ca2+]SR amplitude. Sensitivity analyses of each measured parameter were then normalized to the measurement of the default model values and fit to the quadratic equation:
where x is the fraction (%) of default Gbkg or Kleak. We evaluated the factor of the quadratic term, A, the linear term, B, and the constant term, C, of this fit for relative comparisons of the effects of modulated background Ca2+ current or modulated SR Ca2+ leak. This analysis was repeated for the model ran at 1, 2, and 3 Hz electrical excitation.
We subsequently performed a dual-sensitivity analysis for the modulations of Gbkg and Kleak to understand how the two Ca2+ currents interact and influence Ca2+ handling in the cardiomyocyte together.
We first examined the changes in Vm, [Ca2+]i, and [Ca2+]SR following modulation of the background Ca2+ entry current, ICabk, at pacing rates of 1, 2 and 3 Hz (Figure 2). Changes to features of the action potential were minimal (Figure 2A). Resting Vm exhibited a positive relationship with % Gbkg (Figure 2B), while maximum depolarization exhibited a negative relationship (Figure 2C) with increasing background Ca2+ current. APD90 showed a positive relationship, where increased background Ca2+ entry lengthens the time for repolarization (Figure 2D). We measured diastolic and systolic values, as well as amplitude of [Ca2+]i transients (Figure 2E). All measurements demonstrate positive relationships with increasing background Ca2+ entry; the increase in systolic [Ca2+]i (Figure 2F) is greater than the increase in diastolic [Ca2+]i (Figure 2G), especially at the slowest pacing rate, so the amplitude increases a considerable amount (Figure 2H). We measured the same parameters of [Ca2+]SR transients, which also demonstrate an increase in amplitude caused by increased background Ca2+ entry (Figures 2I,L). The release was initiated from increased diastolic [Ca2+]SR (Figure 2J) and ended in reduced systolic [Ca2+]SR (Figure 2K). The relationships between background Ca2+ entry and features of the [Ca2+]SR transient were also augmented at the slowest pacing rate.
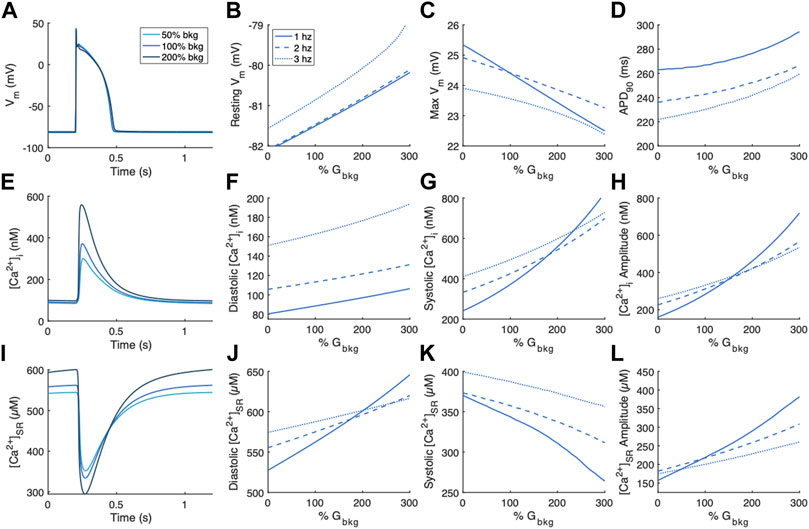
FIGURE 2. Effects of modulating the background Ca2+ entry current, ICabk, on Vm, [Ca2+]i, and [Ca2+]SR. (A) Example action potentials for 50% and 200% Gbkg at 1 Hz pacing. Sensitivity analyses of modulating Gbkg on measurements of (B) resting Vm, (C) maximum Vm, and (D) time to 90% repolarization at 1, 2 and 3 Hz pacing. (E) Example [Ca2+]i transients for 50% and 200% Gbkg at 1 Hz pacing. Sensitivity analyses of modulating Gbkg on measurements of (F) diastolic [Ca2+]i, (G) systolic [Ca2+]i, and (H) [Ca2+]i amplitude at 1, 2 and 3 Hz pacing. (I) Example [Ca2+]SR transients for 50% and 200% Gbkg at 1 Hz pacing. Sensitivity analyses of modulating Gbkg on measurements of (J) diastolic [Ca2+]SR, (K) systolic [Ca2+]SR, and (L) [Ca2+]SR amplitude at 1, 2 and 3 Hz pacing.
Changes following independent modulation of the SR Ca2+ leak current, Jleak, were also examined (Figure 3). The effects of modulating Kleak on Vm were small (Figure 3A). Resting Vm exhibited a slight positive relationship (Figure 3B). Maximum Vm increased with increasing Kleak at 1 Hz pacing but decreased with Kleak at 2 and 3 Hz pacing (Figure 3C). Repolarization time measured as APD90 was unaltered by modulations of Kleak (Figure 3D). Diastolic [Ca2+]i was also relatively unaffected by modulations of Kleak (Figures 3E,F). However systolic [Ca2+]i and thus also [Ca2+]i amplitude decreased with increasing Kleak (Figures 3G,H). The effect was strongest at the slowest pacing frequency of 1 Hz. Alterations in [Ca2+]SR were also strongest at 1 Hz pacing. Diastolic [Ca2+]SR exhibited a negative relationship with Kleak (Figure 3J), while systolic [Ca2+]SR exhibits a positive relationship with Kleak (Figure 3K), both contributing to reduced [Ca2+]SR amplitude with increased Kleak (Figure 3L).
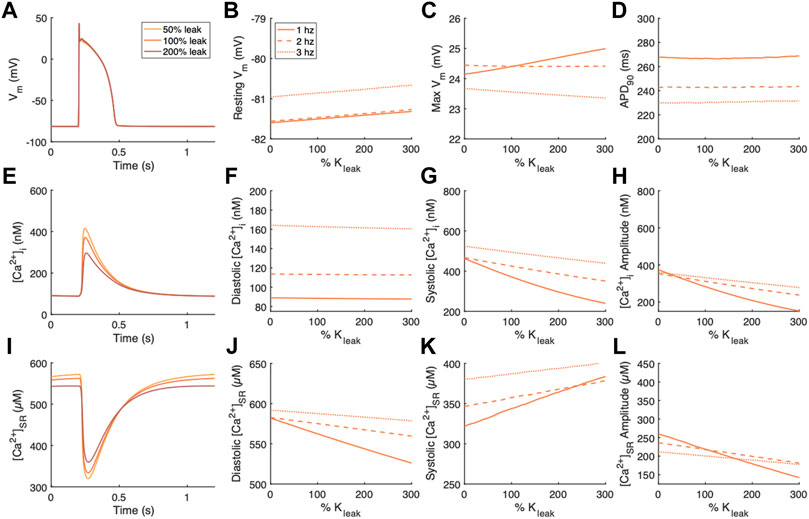
FIGURE 3. Effects of modulating the SR Ca2+ leak flux, Jleak, on Vm, [Ca2+]i, and [Ca2+]SR. (A) Example action potentials for 50% and 200% Kleak at 1 Hz pacing. Sensitivity analyses of modulating Kleak on measurements of (B) resting Vm, (C) maximum Vm, and (D) time to 90% repolarization at 1, 2 and 3 Hz pacing. (E) Example [Ca2+]i transients for 50% and 200% Kleak at 1 Hz pacing. Sensitivity analyses of modulating Kleak on measurements of (F) diastolic [Ca2+]i, (G) systolic [Ca2+]i, and (H) [Ca2+]i amplitude at 1, 2 and 3 Hz pacing. (I) Example [Ca2+]SR transients for 50% and 200% Kleak at 1 Hz pacing. Sensitivity analyses of modulating Kleak on measurements of (J) diastolic [Ca2+]SR, (K) systolic [Ca2+]SR, and (L) [Ca2+]SR amplitude at 1, 2 and 3 Hz pacing.
For comparison of the effects of altered Gbkg or Kleak on features of the action potential, we normalized the measurements of resting Vm, maximum Vm, and APD90 to the default measurements from the model for the given pacing frequency (Figures 4A–F). The relationships of measured vs. modified parameter, both represented as fraction (%) vs. default values, were fit to a 2nd order polynomial model for quantification of the effects (Figures 4G–I). The quadratic term of the fit is negligible for resting Vm and maximum Vm, demonstrating that these measurements exhibit primarily linear relationships with Gbkg and Kleak (Figure 4G). However, the polynomial fit of APD90 to Gbkg has a strong quadratic term, demonstrating a non-linear relationship between APD90 and Gbkg (Figure 4G). Since the relationships of resting Vm and maximum Vm are primarily linear, the linear term of the quadratic polynomial fit demonstrates the sensitivity of the measurements to changes in Gbkg or Kleak (Figure 4H). Both Gbkg and Kleak modulation have a positive relationship with resting Vm, but the changes are negligible for Kleak and minimal for Gbkg, never exceeding an increase greater than 2% of the default model’s resting Vm. Maximum Vm had a strong negative linear relationship with Gbkg for all pacing frequencies, especially at 1 Hz pacing. Maximum Vm had a positive linear relationship with Kleak at 1 Hz pacing but negatives linear relationship for 2 and 3 Hz. The constant of the quadratic polynomial model represents the measurements for the modulated currents set to 0 (Figure 4I). These results revealed that resting Vm was largely unchanged by pacing rate. Maximum Vm was slightly increased with no ICabk and slightly reduced with no Jleak at 1 Hz and slightly increased with no Jleak at 3 Hz. APD90 decreased without ICabk but remained unchanged without Kleak.
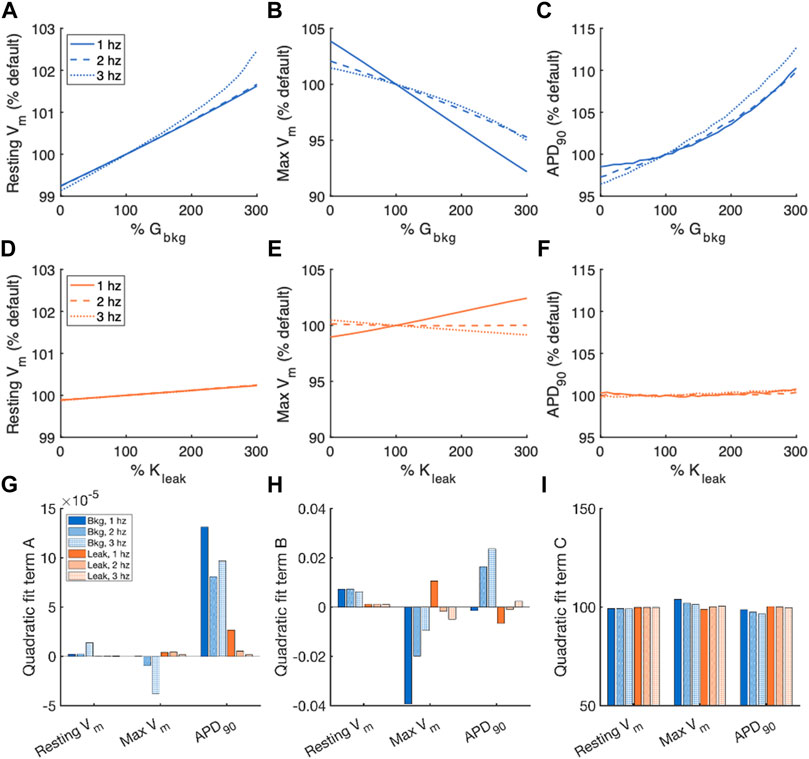
FIGURE 4. Summary of effects of modulating Gbkg or Kleak independently on Vm. Sensitivity analyses of modulating Gbkg on measurements of (A) resting Vm, (B) maximum Vm, and (C) time to 90% repolarization normalized by the default model parameters. Sensitivity analyses of modulating Kleak on measurements of (D) resting Vm, (E) maximum Vm, and (F) time to 90% repolarization normalized by the default model parameters. Summary of the (G) quadratic term, (H) linear term and (I) constant term calculated by quadratic polynomial fits to the normalized sensitivity analyses in (A–H).
Figure 5 contains a summary of [Ca2+]i normalized by measurements of the default model for the sensitivity analyses of Gbkg and Kleak modulation. The relationships of measured parameter vs. modified parameter, both represented as % default values, were fit to a 2nd order polynomial model for quantification of the effects. The quadratic term indicates nonlinearity of the relationship (Figure 5G). The quadratic term of the diastolic [Ca2+]i fits were marginal, indicating a primarily linear relationship. For both Gbkg and Kleak modulations, we noticed a nonlinearity associated with systolic [Ca2+]i and consequently the [Ca2+]i amplitude, with the greatest degree of nonlinearity associated with the lowest pacing frequency. The linear term of the quadratic fit showed a strong linear sensitivity of the measured parameter to changes in Gbkg or Kleak (Figure 5H). The positive sign of this term for Gbkg modulations for each measurement, diastolic, systolic, and amplitude, demonstrates the positive relationship of these parameters with Gbkg, with greater slope of the relationship for systolic and amplitude measurements. The linear term for diastolic [Ca2+]i sensitivity to Kleak was negative but very small, demonstrating that diastolic [Ca2+]i exhibits negligible sensitivity to SR Ca2+ leak. The relationship of systolic [Ca2+]i sensitivity to Kleak was negative, with the largest value for 1 Hz pacing. The same is true for [Ca2+]i transient amplitude. The constant term of the quadratic polynomial fits corresponds to the % default if the modulated channel were 0 (Figure 5I). Elimination of ICabk resulted in a reduction of diastolic [Ca2+]i, systolic [Ca2+]i, and [Ca2+]i amplitude, with the greatest reductions at 1 Hz pacing. Elimination of Jleak did not affect diastolic [Ca2+]i, but results in increased systolic [Ca2+]i and [Ca2+]i amplitude, especially at 1 Hz pacing.
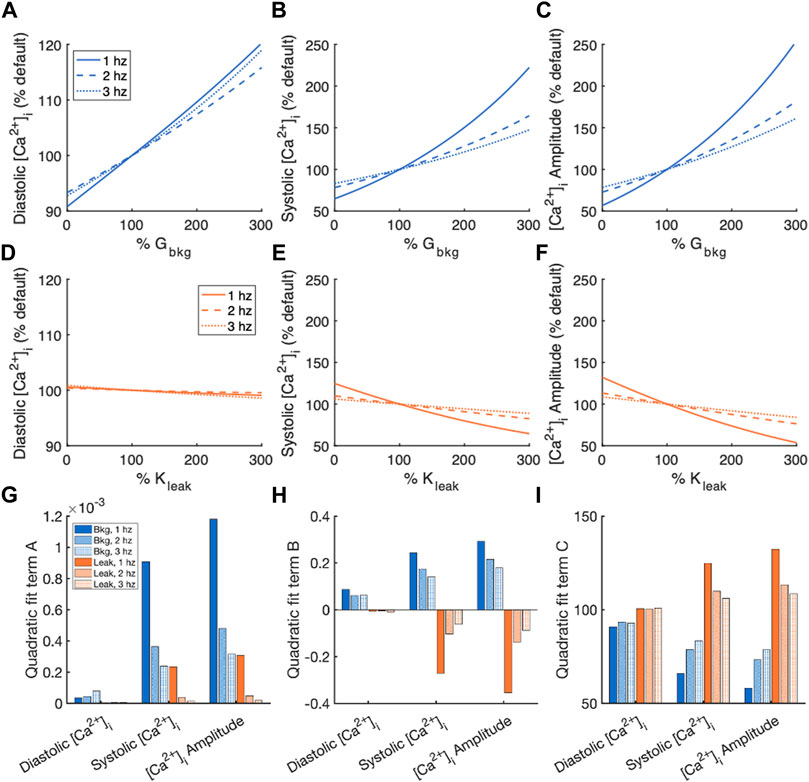
FIGURE 5. Summary of effects of modulating Gbkg or Kleak independently on [Ca2+]i. Sensitivity analyses of modulating Gbkg on measurements of (A) diastolic [Ca2+]i, (B) systolic [Ca2+]i, and (C) [Ca2+]i amplitude normalized by the default model parameters. Sensitivity analyses of modulating Kleak on measurements of (D) diastolic [Ca2+]i, (E) systolic [Ca2+]i, and (F) [Ca2+]i amplitude normalized by the default model parameters. Summary of the (G) quadratic term, (H) linear term and (I) constant term calculated by quadratic polynomial fits to the normalized sensitivity analyses in (A–H).
Measurements of diastolic and systolic [Ca2+]SR and [Ca2+]SR transient amplitude, normalized by measurements of the default model, for the sensitivity analyses of Gbkg and Kleak modulations are summarized in Figure 6. Again, a 2nd order polynomial model for the normalized sensitivity analyses provided information about the relationships. The quadratic term of the fits demonstrates that systolic [Ca2+]SR and [Ca2+]SR transient amplitude both exhibited nonlinearity in the relationship to modulated Gbkg (Figure 6G). The linear term of the relationships demonstrated the best representation sensitivity of the measurements to Gbkg or Kleak modulation (Figure 6H). The sign of this term was opposite for each measured parameter for Gbkg vs. Kleak modulation but similar in amplitude. While the changes to [Ca2+]SR transients all contributed to a positive correlation between Gbkg and [Ca2+]SR transient amplitude, the opposite changes all contribute to a negative correlation between Kleak and [Ca2+]SR transient amplitude. The constant term of the polynomial model, which corresponds to the % of default model measurements if the current is set to 0, showed that in the absence of ICabk, diastolic [Ca2+]SR is lower, systolic [Ca2+]SR is higher, and [Ca2+]SR amplitude is reduced (Figure 6I). In the absence of Jleak, diastolic [Ca2+]SR was slightly elevated, systolic [Ca2+]SR was reduced, and the amplitude of [Ca2+]SR was greater. The effects on all measurements of [Ca2+]SR following modulation of Gbkg or Kleak were stronger at 1 Hz pacing than at faster pacing frequencies.
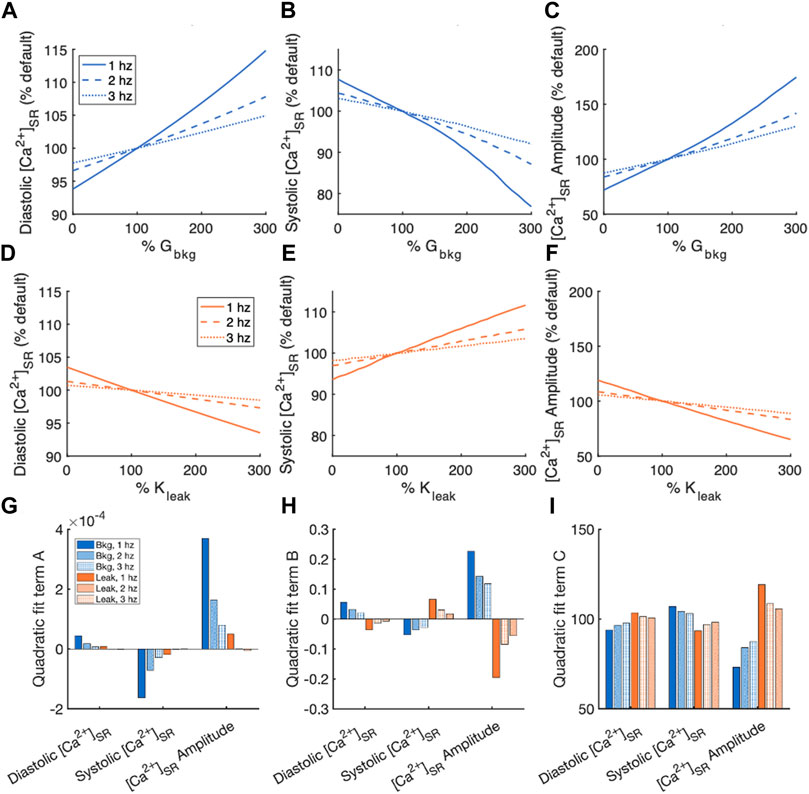
FIGURE 6. Summary of effects of modulating Gbkg or Kleak independently on [Ca2+]SR. Sensitivity analyses of modulating Gbkg on measurements of (A) diastolic [Ca2+]SR, (B) systolic [Ca2+]SR, and (C) [Ca2+]SR amplitude normalized by the default model parameters. Sensitivity analyses of modulating Kleak on measurements of (D) diastolic [Ca2+]SR, (E) systolic [Ca2+]SR, and (F) [Ca2+]SR amplitude normalized by the default model parameters. Summary of the (G) quadratic term, (H) linear term and (I) constant term calculated by quadratic polynomial fits the normalized sensitivity analyses in (A–H).
With a thorough understanding of how Gbkg and Kleak modulations independently affect features of the action potential, [Ca2+]i and [Ca2+]SR transients, we subsequently performed a dual-parameter sensitivity analysis of Gbkg and Kleak together for 1 Hz pacing (Figure 7). Resting Vm is affected minimally by Gbkg, and negligibly by Kleak, apparent by the nearly horizontal contour lines (Figure 7A). Both increasing Gbkg and increasing Kleak resulted in increased Vm, but increasing Gbkg makes a larger contribution. Maximum Vm was modulated in opposite directions by Gbkg and Kleak (Figure 7B). Increasing Gbkg reduces maximum Vm while increasing Kleak increased maximum Vm, but Gbkg is the slightly more dominant effect. APD90 experienced the greatest increase at large increases in Gbkg and small Kleak (Figure 7C). Diastolic [Ca2+]i was primarily affected by a positive relationship to Gbkg, while Kleak appeared to make no contribution (Figure 7D). Interestingly, for all other [Ca2+]i and [Ca2+]SR measurements, the opposing effects of Gbkg and Kleak appeared to balance with a very similar linear relationship (Figures 7E–I). The contour lines of the dual-parameter sensitivity analyses for systolic [Ca2+]i, [Ca2+]i amplitude, diastolic and systolic [Ca2+]SR and [Ca2+]SR amplitude all exhibited strikingly similar slopes.
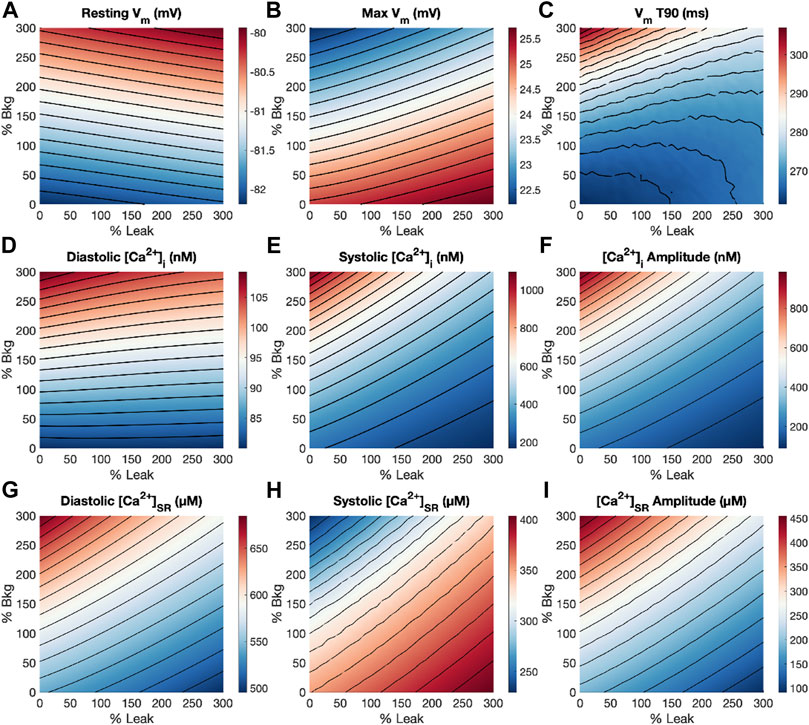
FIGURE 7. Dual-parameter sensitivity analysis of modulating Gbkg and Kleak at 1 Hz pacing. Surface plots with contour lines for measurements of (A) resting Vm, (B) maximum Vm, (C) APD90, (D) diastolic [Ca2+]i, (E) systolic [Ca2+]i, (F) [Ca2+]i amplitude, (G) diastolic [Ca2+]SR, (H) systolic [Ca2+]SR, and (I) [Ca2+]SR amplitude.
If both background and leak currents were modulated along this relationship, measured parameters remained relatively unchanged and only diastolic [Ca2+]i increased. An example of balanced background Ca2+ entry and SR Ca2+ leak demonstrates that a Gbkg value 200% of default and Kleak value of 270% default provided a balanced effect and canceled out the opposing modulations on systolic [Ca2+]i and diastolic [Ca2+]SR load, while diastolic [Ca2+]i was elevated 7.9% (Figure 8).
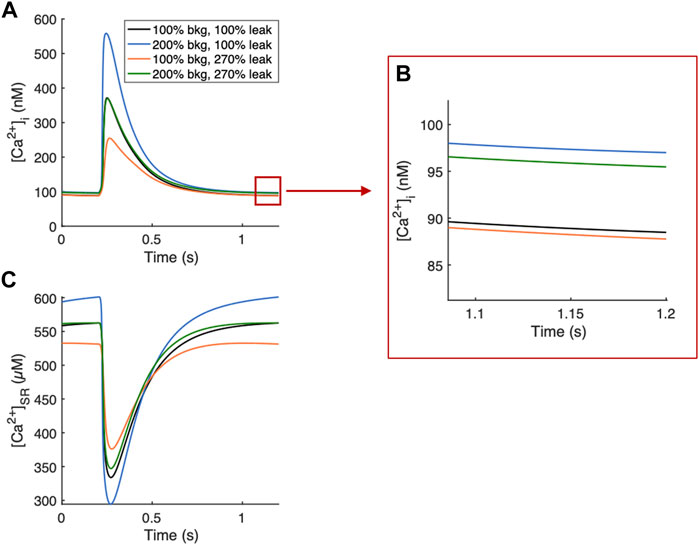
FIGURE 8. Example Ca2+ transients of balanced background Ca2+ entry and SR Ca2+ leak. (A) 200% Gbkg and 270% Kleak balance their opposing modulations on systolic [Ca2+]i while increasing diastolic [Ca2+]i by 7.9%. (B) Zoomed-in region demonstrating 7.9% increase in diastolic [Ca2+]i from default values (black line). (C) 200% Gbkg and 270% Kleak balance opposing effects on SR load.
In this study, we evaluated the contributions of background Ca2+ entry and SR Ca2+ leak to Vm and Ca2+ concentrations in the SR and cytosol in cardiomyocytes in silico. Our investigations shed light on the differential effects of background and leak Ca2+ currents in physiology, and also provide insight into their contributions to disease development due to Ca2+ dysfunction. Below, we discussed background Ca2+ entry as a mechanism to positively modulate Ca2+ entry and SR Ca2+ leak as a critical balancing mechanism to maintain homeostasis.
Our results show that background Ca2+ entry has a positive relationship with diastolic [Ca2+]i and [Ca2+]SR, and the amplitude of their transients (Figure 2). The small increase in diastolic [Ca2+]i is the direct result of an increase in background Ca2+ entry, and the increase in [Ca2+]SR follows as SERCA responds to pump the extra Ca2+ into the SR. The small increase in diastolic [Ca2+]i amplifies Ca2+-induced-Ca2+ release through RyRs, explaining the increased transient amplitudes. This supports the physiological role of background Ca2+ entry in increasing Ca2+ concentrations. These results replicate prior findings for TRP family members suggested as Ca2+ entry channels. For example, we provided evidence for TRPC6 as background Ca2+ entry in neonatal rat ventricular myocytes (Ahmad et al., 2020). Overexpression of TRPC6 contributes to both elevated [Ca2+]i and [Ca2+]SR. TRPC3 and TRPC6 have also both been implicated in the stress-induced slow increase in [Ca2+]i and increased [Ca2+]i transients contributing to the SFR (Seo et al., 2014; Yamaguchi et al., 2017; Yamaguchi et al., 2018). These studies highlight one potential role for Ca2+ entry in strained myocytes. Many of the candidates suggested as background Ca2+ entry channels are known to be modulated by stretch (Inoue et al., 2009; Reed et al., 2014; Peyronnet et al., 2016), so strained myocytes would exhibit an increase in background Ca2+ entry, leading to elevated diastolic levels and larger transients. This mechanism likely contributes to the SFR, increased contractile force following sustained stretch.
SR Ca2+ leak had only a marginal effect on diastolic [Ca2+]i but reduced [Ca2+]SR and the amplitude of Ca2+ transients (Figure 3). In general, modulating SR Ca2+ leak had the opposite effects of background Ca2+ entry, except for the weak effect on diastolic [Ca2+]i. The leaked Ca2+ is removed from the cytosol effectively by NCX, which is why it does not affect free cytosolic levels but reduces SR Ca2+. On its own, this complicates the understanding of the physiological role of SR Ca2+ leak and the purpose of reduced SR load. When considering the dual-parameter sensitivity analysis, it became however evident that while background Ca2+ entry responds to increased needs with increased Ca2+, SR Ca2+ leak likely functions as a critical balancing component to regulate SR stores and maintain Ca2+ homeostasis. The combined effects of increasing both background Ca2+ entry and SR Ca2+ leak exhibit a linear relationship, represented by the contour lines of the dual sensitivity plot for systolic [Ca2+]i, [Ca2+]i amplitude, diastolic and systolic [Ca2+]SR and [Ca2+]SR amplitude (Figure 7). If both background and leak currents are modulated along this relationship, measured parameters remain relatively unchanged and only diastolic [Ca2+]i increases. Examples of balanced background Ca2+ entry and SR Ca2+ leak in Figure 8 demonstrate that modulations in Gbkg and Kleak can be balanced in a way to cancel out the large opposing effects they have on Ca2+ transient amplitudes (Figure 8). This balancing mechanism of SR Ca2+ leak could be critical to prevent Ca2+ overload in the cell. Leak in the form of RyR sparks has been demonstrated as SR load regulator to prevent overload, with a steep dependency on [Ca2+]SR (Shannon et al., 2002). However, large Ca2+ release events through RyR sparks increase sensitivity for arrhythmia (George, 2008). RyR sparks are most prevalent at high [Ca2+]SR, but non-spark RyR leak and non-RyR leak do not appear to exhibit the same steep dependency on [Ca2+]SR and therefore might function differently. Thus, a mechanism of non-RyR leak may be to regulate compartmental Ca2+ before the cells become overloaded, and RyR sparks increase as a more extreme measure.
Like channels involved in background Ca2+ entry in cardiomyocytes, candidates for SR Ca2+ leak also include members of the TRP family and were suggested to be mechano-modulated (Inoue et al., 2009; Reed et al., 2014; Peyronnet et al., 2016). This indicates that the background and leak Ca2+ currents could be modulated in conjunction. Non-RyR leak that can be modulated by stretch may provide a more moderate and steady regulation on a beat-by-beat basis in conjunction with background Ca2+ entry modulated by stretch. Recently we demonstrated that TRPC1 constitutes an SR Ca2+ leak channel, and its overexpression resulted in decreased SR Ca2+ load (Hu et al., 2020). TRPC1 channels are suggested to be modulated by stretch, indicating that the reduction in SR Ca2+ load could be a regulatory mechanism to match increased background Ca2+ entry through, e.g., TRPC6 channels. We speculate that background Ca2+ entry and SR Ca2+ leak fulfill a critical homeostatic function in the modulation of Ca2+ concentrations throughout the cardiomyocyte in response to strain.
Both background Ca2+ entry and SR Ca2+ leak through TRP channels are likely to be modulated by cardiomyocyte strain (Inoue et al., 2009; Reed et al., 2014; Peyronnet et al., 2016). Under chronic pressure overload conditions, strain-modulation of TRPC channels could increase background Ca2+ entry and SR Ca2+ leak, and thus dysregulate Ca2+. Cardiac disease is perpetuated by Ca2+ dysregulation, and a stray from its homeostatic balance. Some of the suggested ion channels for these Ca2+ currents were found to be upregulated in models of cardiac disease, suggesting a role in pathogenesis (Ahmad et al., 2017; Hof et al., 2019). Diastolic [Ca2+]i is elevated in HF causing diastolic dysfunction (Eisner et al., 2020). Elevated background Ca2+ entry could be a contributing factor. Two different models of HF with preserved ejection fraction (HFpEF) display increases in both diastolic and systolic [Ca2+]i (Curl et al., 2018; Rouhana et al., 2019). A hypothesis is that a major difference in Ca2+ handling between HFpEF and HF with reduced ejection fraction (HFrEF) is preserved [Ca2+]SR in HFpEF vs. reduced [Ca2+]SR in HFrEF (Eisner et al., 2020). The decreased SR Ca2+ content contributes largely to the decrease in systolic [Ca2+]i and contractile dysfunction (Bers, 2006). Based on the demonstration of a balancing mechanism between background Ca2+ entry and SR Ca2+ leak in this study, it is reasonable to speculate a difference between maintenance of this balance in HFpEF vs. a stray from this balanced relationship towards overcompensated leak in HFrEF. In addition to reducing SR Ca2+ available for release, causing systolic dysfunction, increased SR Ca2+ leak can be problematic, e.g., triggering arrhythmias and being energetically costly due to increased use of ATP to repump Ca2+ (Bers, 2014). Understanding the balance of background and leak Ca2+ currents in cardiomyocytes and how they affect Ca2+ homeostasis and remodeling in disease will be critical to develop effective drug therapies targeting Ca2+ channels.
In this study, we observed the well-established frequency dependency of Ca2+ transients. Increasing the rate of stimulation increases diastolic [Ca2+]i in isolated myocytes (Frampton et al., 1991; Antoons et al., 2002; Dibb et al., 2007; Horváth et al., 2017; Sankaranarayanan et al., 2017). Background Ca2+ entry and SR Ca2+ leak also both exhibit a frequency effect. The parameters we measured are all more sensitive to modulations of the Ca2+ currents at slower pacing rates than at faster pacing rates (Figures 5, 6). The sensitivity of each measured [Ca2+]i and [Ca2+]SR parameter to Gbkg and Kleak is greatest in amplitude for 1 Hz pacing. An explanation is that at slower pacing rates, the background and leak currents have relatively more time to contribute to the total Ca2+ flux per beat vs. the voltage-gated ion channels that open during the action potential and are closed at rest.
Modulating Kleak had negligible effects on the action potential for any pacing frequency (Figure 4). For the values of Kleak tested, we found that the SR Ca2+ leak flux does not significantly contribute to sarcolemmal electrophysiology. Modulating Gbkg has marginal effects on features of action potentials (Figure 4). While Gbkg positively correlates with increased resting Vm, an increase to 300% Gbkg only resulted in <2% change from basal resting Vm. This minimal change in resting potential is unlikely to be functionally relevant. An increase to 300% Gbkg also reduces maximum depolarization by 7% for 1 Hz pacing. The largest effect is an increase in action potential duration (APD90) by around 10% for maximal Gbkg modulation. It has also been shown that APD prolongation leads to increased Ca2+ (Bouchard et al., 1995), suggesting a positive feedback loop for electrical and Ca2+ signaling. Another important note is that APD increase is known to be inotropic, e.g., in rat ventricular myocytes (Bouchard et al., 1995). This indicates another mechanism for the contribution of background Ca2+ entry to contractility. Conversely, prolonged APD can induce torsades de pointes tachycardia, leading to life-threatening ventricular fibrillation (Roden and Hoffman, 1985; Ravens and Cerbai, 2008).
Mathematical modeling of cellular electrophysiology provides a valuable resource for studying how aspects of cellular physiology interact and affect one another. It provides a means to investigate questions that cannot be easily answered in vivo. However, there are also caveats of mathematical modeling that should be considered. It should be noted that the definitions of ICabk and Jleak used in this model are general simplifications and meant to reproduce poorly defined currents. The equations lack specific gating conditions of the currents. The current equations were not parameterized to match experimental data which is only incompletely characterized in human ventricular myocytes. Instead, the current equations are adjusted such that the model reproduces overall physiological action potentials and calcium transients. This is an important consideration, since the magnitudes of these currents could be largely different in living cells. Thus, interpreting the results of this study should focus on the qualitative trends. As the specific ion channels that contribute to Ca2+ entry and leak are identified and characterized, future work can aim to refine the current definitions and provide detailed current models to replace the general simplifications of ICabk and Jleak.
In a similar way, other ion currents in the model are not fully defined. For example, some K+ channels and isoforms of the Na+/K+-ATPase are modulated by localized Ca2+ concentrations (Tian and Xie, 2008; Weisbrod, 2020). However, the model does not include any Ca2+-dependent terms in the definitions of these currents. The inclusion of these interactions may alter the effects we see on Vm in this study. Future work could address this limitation.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
MS and FS designed the study. MS implemented the modeling, analyzed simulation data and drafted the manuscript. All authors critically revised the manuscript and approved the version to be published.
This work was supported by Nora Eccles Treadwell Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.916278/full#supplementary-material
Ahmad A. A., Streiff M., Hunter C., Hu Q., Sachse F. B. (2017). Physiological and Pathophysiological Role of Transient Receptor Potential Canonical Channels in Cardiac Myocytes. Prog. Biophysics Mol. Biol. 130, 254–263. doi:10.1016/j.pbiomolbio.2017.06.005
Ahmad A. A., Streiff M. E., Hunter C., Sachse F. B. (2020). Modulation of Calcium Transients in Cardiomyocytes by Transient Receptor Potential Canonical 6 Channels. Front. Physiol. 11, 44. doi:10.3389/fphys.2020.00044
Ai X., Curran J. W., Shannon T. R., Bers D. M., Pogwizd S. M. (2005). Ca 2+/Calmodulin-Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca 2+ Leak in Heart Failure. Circulation Res. 97, 1314–1322. doi:10.1161/01.res.0000194329.41863.89
Antoons G., Mubagwa K., Nevelsteen I., Sipido K. R. (2002). Mechanisms Underlying the Frequency Dependence of Contraction and [Ca 2+ ] I Transients in Mouse Ventricular Myocytes. J. Physiology 543, 889–898. doi:10.1113/jphysiol.2002.025619
Berbey C., Weiss N., Legrand C., Allard B. (2009). Transient Receptor Potential Canonical Type 1 (TRPC1) Operates as a Sarcoplasmic Reticulum Calcium Leak Channel in Skeletal Muscle. J. Biol. Chem. 284, 36387–36394. doi:10.1074/jbc.m109.073221
Bers D. M. (2006). Altered Cardiac Myocyte Ca Regulation in Heart Failure. Physiology 21, 380–387. doi:10.1152/physiol.00019.2006
Bers D. M. (2014). Cardiac Sarcoplasmic Reticulum Calcium Leak: Basis and Roles in Cardiac Dysfunction. Annu. Rev. Physiol. 76, 107–127. doi:10.1146/annurev-physiol-020911-153308
Bouchard R. A., Clark R. B., Giles W. R. (1995). Effects of Action Potential Duration on Excitation-Contraction Coupling in Rat Ventricular Myocytes. Circulation Res. 76, 790–801. doi:10.1161/01.res.76.5.790
Calaghan S. C., White E. (1999). The Role of Calcium in the Response of Cardiac Muscle to Stretch. Prog. Biophysics Mol. Biol. 71, 59–90. doi:10.1016/s0079-6107(98)00037-6
Camacho Londoño J. E., Tian Q., Hammer K., Schröder L., Camacho Londoño J., Reil J. C., et al. (2015). A Background Ca2+entry Pathway Mediated by TRPC1/TRPC4 Is Critical for Development of Pathological Cardiac Remodelling. Eur. Heart J. 36, 2257–2266. doi:10.1093/eurheartj/ehv250
Camacho Londoño J. E., Kuryshev V., Zorn M., Saar K., Tian Q., Hübner N., et al. (2021). Transcriptional Signatures Regulated by TRPC1/C4-Mediated Background Ca2+ Entry after Pressure-Overload Induced Cardiac Remodelling. Prog. Biophys. Mol. Biol. 159, 86–104. doi:10.1016/j.pbiomolbio.2020.07.006
Chen M.-S., Xiao J.-H., Wang Y., Xu B.-M., Gao L., Wang J.-L. (2013). Upregulation of TRPC1 Contributes to Contractile Function in Isoproterenol-Induced Hypertrophic Myocardium of Rat. Cell Physiol. Biochem. 32, 951–959. doi:10.1159/000354498
Choi H. S., Trafford A. W., Eisner D. A. (2000). Measurement of Calcium Entry and Exit in Quiescent Rat Ventricular Myocytes. Pflügers Arch. 440, 600–608. doi:10.1007/s004240050011
Curl C. L., Danes V. R., Bell J. R., Raaijmakers A. J. A., Ip W. T. K., Chandramouli C., et al. (2018). Cardiomyocyte Functional Etiology in Heart Failure With Preserved Ejection Fraction Is Distinctive-A New Preclinical Model. J. Am. Heart Assoc. 7, e007451. doi:10.1161/JAHA.117.007451
Dibb K. M., Eisner D. A., Trafford A. W. (2007). Regulation of Systolic [Ca2+]iand Cellular Ca2+flux Balance in Rat Ventricular Myocytes by SR Ca2+, L-type Ca2+current and Diastolic [Ca2+]i. J. Physiol. 585, 579–592. doi:10.1113/jphysiol.2007.141473
Eisner D. A., Caldwell J. L., Trafford A. W., Hutchings D. C. (2020). The Control of Diastolic Calcium in the Heart. Circ. Res. 126, 395–412. doi:10.1161/circresaha.119.315891
Frampton J. E., Orchard C. H., Boyett M. R. (1991). Diastolic, Systolic and Sarcoplasmic Reticulum [Ca2+] during Inotropic Interventions in Isolated Rat Myocytes. J. Physiol. 437, 351–375. doi:10.1113/jphysiol.1991.sp018600
George C. H. (2008). Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure: Mere Observation or Functional Relevance? Cardiovasc Res. 77, 302–314. doi:10.1093/cvr/cvm006
Go L. O., Moschella M. C., Watras J., Handa K. K., Fyfe B. S., Marks A. R. (1995). Differential Regulation of Two Types of Intracellular Calcium Release Channels during End-Stage Heart Failure. J. Clin. Invest. 95, 888–894. doi:10.1172/jci117739
Grandi E., Pasqualini F. S., Bers D. M. (2010). A Novel Computational Model of the Human Ventricular Action Potential and Ca Transient. J. Mol. Cell. Cardiol. 48, 112–121. doi:10.1016/j.yjmcc.2009.09.019
Hof T., Chaigne S., Récalde A., Sallé L., Brette F., Guinamard R. (2019). Transient Receptor Potential Channels in Cardiac Health and Disease. Nat. Rev. Cardiol. 16, 344–360. doi:10.1038/s41569-018-0145-2
Horváth B., Szentandrássy N., Veress R., Almássy J., Magyar J., Bányász T., et al. (2017). Frequency-dependent Effects of Omecamtiv Mecarbil on Cell Shortening of Isolated Canine Ventricular Cardiomyocytes. Naunyn Schmiedeb. Arch. Pharmacol. 390, 1239–1246.
Hu Q., Ahmad A. A., Seidel T., Hunter C., Streiff M., Nikolova L., et al. (2020). Location and Function of Transient Receptor Potential Canonical Channel 1 in Ventricular Myocytes. J. Mol. Cell. Cardiol. 139, 113–123. doi:10.1016/j.yjmcc.2020.01.008
Inoue R., Jian Z., Kawarabayashi Y. (2009). Mechanosensitive TRP Channels in Cardiovascular Pathophysiology. Pharmacol. Ther. 123, 371–385. doi:10.1016/j.pharmthera.2009.05.009
Jones J. L., Peana D., Veteto A. B., Lambert M. D., Nourian Z., Karasseva N. G., et al. (2019). TRPV4 Increases Cardiomyocyte Calcium Cycling and Contractility yet Contributes to Damage in the Aged Heart Following Hypoosmotic Stress. Cardiovasc Res. 115, 46–56. doi:10.1093/cvr/cvy156
Kupittayanant P., Trafford A. W., Díaz M. E., Eisner D. A. (2006). A Mechanism Distinct from the L-type Ca Current or Na-Ca Exchange Contributes to Ca Entry in Rat Ventricular Myocytes. Cell Calcium 39, 417–423. doi:10.1016/j.ceca.2006.01.011
Lemos F. O., Bultynck G., Parys J. B. (2021). A Comprehensive Overview of the Complex World of the Endo- and Sarcoplasmic Reticulum Ca2+-Leak Channels. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1868, 119020. doi:10.1016/j.bbamcr.2021.119020
Leybaert L., Lampe P. D., Dhein S., Kwak B. R., Ferdinandy P., Beyer E. C., et al. (2017). Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 69, 396–478. doi:10.1124/pr.115.012062
Mijares A., Altamirano F., Kolster J., Adams J. A., López J. R. (2014). Age-dependent Changes in Diastolic Ca2+ and Na+ Concentrations in Dystrophic Cardiomyopathy: Role of Ca2+ Entry and IP3. Biochem. Biophysical Res. Commun. 452, 1054–1059. doi:10.1016/j.bbrc.2014.09.045
Peyronnet R., Nerbonne J. M., Kohl P. (2016). Cardiac Mechano-Gated Ion Channels and Arrhythmias. Circ. Res. 118, 311–329. doi:10.1161/circresaha.115.305043
Ravens U., Cerbai E. (2008). Role of Potassium Currents in Cardiac Arrhythmias. Europace 10, 1133–1137. doi:10.1093/europace/eun193
Reed A., Kohl P., Peyronnet R. (2014). Molecular Candidates for Cardiac Stretch-Activated Ion Channels. Glob. Cardiol. Sci. Pract. 2014, 9–25. doi:10.5339/gcsp.2014.19
Roden D. M., Hoffman B. F. (1985). Action Potential Prolongation and Induction of Abnormal Automaticity by Low Quinidine Concentrations in Canine Purkinje Fibers. Relationship to Potassium and Cycle Length. Circ. Res. 56, 857–867. doi:10.1161/01.res.56.6.857
Rouhana S., Farah C., Roy J., Finan A., Rodrigues De Araujo G., Bideaux P., et al. (2019). Early Calcium Handling Imbalance in Pressure Overload-Induced Heart Failure with Nearly Normal Left Ventricular Ejection Fraction. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1865, 230–242. doi:10.1016/j.bbadis.2018.08.005
Rubinstein J., Lasko V. M., Koch S. E., Singh V. P., Carreira V., Robbins N., et al. (2014). Novel Role of Transient Receptor Potential Vanilloid 2 in the Regulation of Cardiac Performance. Am. J. Physiology-Heart Circulatory Physiology 306, H574–H584. doi:10.1152/ajpheart.00854.2013
Sankaranarayanan R., Kistamás K., Greensmith D. J., Venetucci L. A., Eisner D. A. (2017). Systolic [Ca2+ ]i Regulates Diastolic Levels in Rat Ventricular Myocytes. J. Physiol. 595, 5545–5555. doi:10.1113/jp274366
Santiago D. J., Curran J. W., Bers D. M., Lederer W. J., Stern M. D., Ríos E., et al. (2010). Ca Sparks Do Not Explain All Ryanodine Receptor-Mediated SR Ca Leak in Mouse Ventricular Myocytes. Biophysical J. 98, 2111–2120. doi:10.1016/j.bpj.2010.01.042
Seo K., Rainer P. P., Lee D.-i., Hao S., Bedja D., Birnbaumer L., et al. (2014). Hyperactive Adverse Mechanical Stress Responses in Dystrophic Heart Are Coupled to Transient Receptor Potential Canonical 6 and Blocked by cGMP-Protein Kinase G Modulation. Circ. Res. 114, 823–832. doi:10.1161/circresaha.114.302614
Shannon T. R., Ginsburg K. S., Bers D. M. (2002). Quantitative Assessment of the SR Ca 2+ Leak-Load Relationship. Circulation Res. 91, 594–600. doi:10.1161/01.res.0000036914.12686.28
Stout C. E., Costantin J. L., Naus C. C. G., Charles A. C. (2002). Intercellular Calcium Signaling in Astrocytes via ATP Release through Connexin Hemichannels. J. Biol. Chem. 277, 10482–10488. doi:10.1074/jbc.m109902200
Terracciano C. M., Macleod K. T. (1996). Reloading of Ca(2+)-Depleted Sarcoplasmic Reticulum during Rest in guinea Pig Ventricular Myocytes. Am. J. Physiology-Heart Circulatory Physiology 271, H1814–H1822. doi:10.1152/ajpheart.1996.271.5.h1814
Tian J., Xie Z.-j. (2008). The Na-K-ATPase and Calcium-Signaling Microdomains. Physiology 23, 205–211. doi:10.1152/physiol.00008.2008
Wang N., De Bock M., Antoons G., Gadicherla A. K., Bol M., Decrock E., et al. (2012). Connexin Mimetic Peptides Inhibit Cx43 Hemichannel Opening Triggered by Voltage and Intracellular Ca2+ Elevation. Basic Res. Cardiol. 107, 304. doi:10.1007/s00395-012-0304-2
Ward M.-L., Williams I. A., Chu Y., Cooper P. J., Ju Y.-K., Allen D. G. (2008). Stretch-activated Channels in the Heart: Contributions to Length-Dependence and to Cardiomyopathy. Prog. Biophysics Mol. Biol. 97, 232–249. doi:10.1016/j.pbiomolbio.2008.02.009
Weisbrod D. (2020). Small and Intermediate Calcium Activated Potassium Channels in the Heart: Role and Strategies in the Treatment of Cardiovascular Diseases. Front. Physiol. 11, 590534. doi:10.3389/fphys.2020.590534
Williams I. A., Allen D. G. (2007). Intracellular Calcium Handling in Ventricular Myocytes from Mdx Mice. Am. J. Physiology-Heart Circulatory Physiology 292, H846–H855. doi:10.1152/ajpheart.00688.2006
Yamaguchi Y., Iribe G., Nishida M., Naruse K. (2017). Role of TRPC3 and TRPC6 Channels in the Myocardial Response to Stretch: Linking Physiology and Pathophysiology. Prog. Biophysics Mol. Biol. 130, 264–272. doi:10.1016/j.pbiomolbio.2017.06.010
Yamaguchi Y., Iribe G., Kaneko T., Takahashi K., Numaga-Tomita T., Nishida M., et al. (2018). TRPC3 Participates in Angiotensin II Type 1 Receptor-dependent Stress-Induced Slow Increase in Intracellular Ca2+ Concentration in Mouse Cardiomyocytes. J. Physiol. Sci. 68, 153–164. doi:10.1007/s12576-016-0519-3
Keywords: calcium, cardiomyocyte, sarcolemma, sarcoplasmic reticulum, leak
Citation: Streiff ME and Sachse FB (2022) Effects of Sarcolemmal Background Ca2+ Entry and Sarcoplasmic Ca2+ Leak Currents on Electrophysiology and Ca2+ Transients in Human Ventricular Cardiomyocytes: A Computational Comparison. Front. Physiol. 13:916278. doi: 10.3389/fphys.2022.916278
Received: 09 April 2022; Accepted: 27 May 2022;
Published: 16 June 2022.
Edited by:
Rupamanjari Majumder, Max Planck Society, GermanyCopyright © 2022 Streiff and Sachse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank B. Sachse, ZnJhbmsuc2FjaHNlQHV0YWguZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.