
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Physiol. , 08 June 2022
Sec. Red Blood Cell Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.907444
This article is part of the Research Topic Rising Stars in Red Blood Cell Physiology: 2022 View all 6 articles
 Alkmini T. Anastasiadi1*
Alkmini T. Anastasiadi1* Vassilis L. Tzounakas1
Vassilis L. Tzounakas1 Monika Dzieciatkowska2Vasiliki-Zoi Arvaniti1
Monika Dzieciatkowska2Vasiliki-Zoi Arvaniti1 Effie G. Papageorgiou3
Effie G. Papageorgiou3 Issidora S. Papassideri1Konstantinos Stamoulis4
Issidora S. Papassideri1Konstantinos Stamoulis4 Angelo D’Alessandro2Anastasios G. Kriebardis3
Angelo D’Alessandro2Anastasios G. Kriebardis3 Marianna H. Antonelou1*
Marianna H. Antonelou1*The broad spectrum of beta-thalassemia (βThal) mutations may result in mild reduction (β++), severe reduction (β+) or complete absence (β0) of beta-globin synthesis. βThal heterozygotes eligible for blood donation are “good storers” in terms of red blood cell (RBC) fragility, proteostasis and redox parameters of storage lesion. However, it has not been examined if heterogeneity in genetic backgrounds among βThal-trait donors affects their RBC storability profile. For this purpose, a paired analysis of physiological and omics parameters was performed in freshly drawn blood and CPD/SAGM-stored RBCs donated by eligible volunteers of β++ (N = 4), β+ (N = 9) and β0 (N = 2) mutation-based phenotypes. Compared to β+, β++ RBCs were characterized by significantly lower RDW and HbA2 but higher hematocrit, MCV and NADPH levels in vivo. Moreover, they had lower levels of reactive oxygen species and markers of oxidative stress, already from baseline. Interestingly, their lower myosin and arginase membrane levels were accompanied by increased cellular fragility and arginine values. Proteostasis markers (proteasomal activity and/or chaperoning-protein membrane-binding) seem to be also diminished in β++ as opposed to the other two phenotypic groups. Overall, despite the low number of samples in the sub-cohorts, it seems that the second level of genetic variability among the group of βThal-trait donors is reflected not only in the physiological features of RBCs in vivo, but almost equally in their storability profiles. Mutations that only slightly affect the globin chain equilibrium direct RBCs towards phenotypes closer to the average control, at least in terms of fragility indices and proteostatic dynamics.
The storability profile of red blood cells (RBCs) seems to be highly dependent on intrinsic donor characteristics. Both genetic and environmental factors have been studied during the last decade in the context of donor variation effects upon storage and transfusion therapy. Donor’s sex (Szczesny-Malysiak et al., 2021), ethnicity (Kanias et al., 2017) and glucose-6-phosphate dehydrogenase (G6PD) activity (Tzounakas et al., 2016; Francis et al., 2020), as well as lifestyle aspects, such as smoking (Stefanoni et al., 2020) and caffeine or alcohol consumption (D'Alessandro et al., 2020a; D'Alessandro et al., 2020b) have been proven to affect differentially the storage and/or post-transfusion efficacy of donated RBCs. The same is true for RBCs with distinct hemoglobin (Hb) variants. For instance, stored RBCs from donors with elevated glycosylated Hb present increased susceptibility to lysis, phosphatidylserine (PS) externalization and non-reversible shape modifications (Li et al., 2022). Moreover, polymorphisms in HbA2 gene are associated with a reduced hemoglobin increment (Roubinian et al., 2022), and sickle cell trait with increased storage hemolysis and removal in animal models of transfusion (Osei-Hwedieh et al., 2016).
RBCs from beta-thalassemia minor (βThal+) eligible donors have been extensively studied lately with respect to their physiological, metabolic and proteomic profiles during storage. These cells seem to possess an intrinsic resistance to both spontaneous and induced lysis (Tzounakas et al., 2022), as well as an array of metabolic and proteomic features indicative of advantageous control of oxidative and proteotoxic stresses (Anastasiadi et al., 2021b; Tzounakas et al., 2021; Tzounakas et al., 2022). Regarding their post-transfusion aspects, stored βThal+ RBCs demonstrate resilience against lysis following exposure to plasma at body temperature, along with a trend for increased recovery post transfusion in mice recipients (Anastasiadi et al., 2021a). These superior post-storage phenotypes have been found linked to βThal+-specific variations in baseline or storage parameters, such as cell fragilities, cytoskeleton composition, or urate (Anastasiadi et al., 2021a; Anastasiadi et al., 2022).
Besides divergence of specific donor cohorts from the average control, a range of within-group variation is also anticipated. In the case of beta-thalassemia the highly heterogenous genetic setting of mutations and related polymorphisms is translated to a broad spectrum of clinical and cellular phenotypes (Giardine et al., 2021) with variable prevalence within distinct national settings. More specifically, in Greece, mutations leading to severe reduction in ß-globin synthesis (β+) represent almost 50% of the reported thalassemia alleles (Boussiou et al., 2008) followed by mutations resulting in null synthesis or slight reduction of ß-chains. Therefore, the cellular effects of each mutation might lead to a different storability phenotype. Having this in mind, the aim of the present study was to examine the innate variation of hematological, physiological, metabolic and protein parameters in freshly drawn and stored RBCs from a group of βThal+ donors stratified by the degree of ß-globin synthesis imposed by the affected allele.
Venous blood from fifteen regular βThal+ blood donors was collected into EDTA and citrate vacutainer tubes. The same subjects donated blood to prepare and store RBC units in citrate-phosphate-dextrose (CPD)/saline-adenine-glucose-mannitol (SAGM), for 42 days at 4°C. βThal+ trait was confirmed by Hb electrophoresis and molecular identification of mutations (IVS I-1, IVS I-6, IVS I-110, IVS II-1 and IVS II-745). The samples were subsequently categorized as β++ (n = 4), β+ (n = 9) and β0 (n = 2) according to the impact that the mutations have upon the ß-globin synthesis (from slight -β++- to severe -β0- reduction in ß-globin levels). The RBC units were sampled every week under aseptic conditions. The study was approved by the Ethics Committee of the Department of Biology, School of Science, NKUA and investigations were carried out upon donor consent, in accordance with the principles of the Declaration of Helsinki.
BC-3000 PLUS, MINDRAY Celltac E, MEK-7222 Κ, NIHON KOHDEN automatic blood cell counters were used for complete blood count through double measurements to achieve maximum reliability, while the automatic analysers Hitachi 902, AVL Series Electrolyte Analyzer 9,180 and Elecsys Systems Analyzer (Roche Diagnostics, Risch-Rotkreuz, Switzerland) were used for the biochemical analysis of triglycerides, lipoproteins, iron (Fe), electrolytes and ferritin.
Storage hemolysis was calculated via spectrophotometry using Harboe’s method (Harboe, 1959) followed by Allen’s correction. For assessment of osmotic hemolysis, the samples were exposed to ascending concentrations of NaCl and then the mean corpuscular fragility (MCF) index (i.e., %NaCl at 50% hemolysis) was calculated. Mechanical hemolysis was estimated following rocking of RBCs with stainless steel beads for 1 h and measurement of the Hb released in the supernatant compared to non-rocked counterparts (Tzounakas et al., 2022).
The intracellular accumulation of reactive oxygen species (ROS) was measured via fluorometry (BIORAD Hercules, CA, United States) by using the membrane permeable and redox-sensitive probe 5-(and-6)-chloromethyl-2′,7′-dichloro-dihydro-fluoresceindiacetate, acetyl ester (CM-H2DCFDA; Invitrogen, Molecular Probes, Eugene, OR, United States). This assay was performed with or without prior oxidative stimulation of RBCs by diamide (2 mM) or phenylhydrazine (PHZ; 100 μM) for 45 min at 37°C. Fluorometry was also used for the determination of caspase-like (CASP-like), chymotrypsin-like (CH-like) and trypsin-like (TR-like) proteasome activities in cytosol and membrane fractions. For this purpose, 120–200 μg of protein samples were incubated with the fluorogenic substrates Suc-Leu-Leu-Val-Tyr-aminomethylcoumarin (AMC) (CH-like), z-Leu-Leu-Glu-AMC (CASP-like), and Boc-Leu-Arg-Arg-AMC (TR-like) for 1.30 h (CH-like) or 3 h (CASP- and TR-like) at 37°C in the dark (Anastasiadi et al., 2021b). All substrates were produced from Enzo Life Sciences (New York, NY, United States). Fluorescent units were normalized to protein levels to reach a quantitative result.
For the metabolomics analysis, 100 μl of stored RBCs (or 20 μl of plasma/supernatants) were collected on a weekly basis, extracted at 1:6 (or 1:25) dilution in methanol:acetonitrile:water (5:3:2) and analyzed by UHPLC-MS (Ultimate 3000 RSLC-Q Exactive, Thermo Fisher), as previously described (D'Alessandro et al., 2019; Nemkov et al., 2019). Sample extracts (10 μl) were loaded onto a Kinetex XB-C18 column (150 mm × 2.1 mm × 1.7 μm—Phenomenex, Torrance, CA, United States). A 5-min gradient from 5 to 95% B (phase A: water +0.1% formic acid and B: acetonitrile +0.1% formic acid) eluted metabolites into a Q Exactive system (Thermo, Bremen, Germany), scanning in full MS mode or performing acquisition independent fragmentation (MS/MS analysis—5 min method) at 70,000 resolution in the 60–900 m/z range, 4 kV spray voltage, 15 sheath gas, and five auxiliary gas, operated in negative and then positive ion mode (separate runs). Metabolite assignment was performed against an in-house standard library, as reported (Nemkov et al., 2017), through the freely available software Maven (Princeton University, United States). No data pre-processing (neither normalization nor log-transformation) was performed. Proteomics analysis was performed on isolated membranes of early- and late-stored RBCs (n = 12 at each time point; n = 2 for β++, n = 8 for β+, n = 2 for β0; obtained by hypotonic lysis). Samples (200 ng) were loaded onto individual Evotips (desalting) and were subsequently washed (20 μl 0.1% formic acid), followed by the addition of 0.1% formic acid to keep the Evotips wet. The Evosep One system was coupled to a timsTOF Pro mass spectrometer (Bruker Daltonics, Bremen, Germany). Data were collected over a m/z range of 100–1700 for MS and MS/MS on the timsTOF Pro instrument using an accumulation and ramp time of 100 ms. PEAKS studio (Version X+, Bioinformatics Solutions, Waterloo, ON, United States) was used for post-processing. The relative protein levels were normalized on the total amount of proteins.
All physiological experiments were performed in triplicate. Statistical analysis was performed by using the statistical package SPSS Version 22.0 (IBM Hellas, Athens, Greece, administered by NKUA). Between-group differences in freshly drawn blood (in vivo comparison) were assessed by independent t-test. Repeated measures ANOVA with Bonferroni-like adjustments for multiple comparisons was used for the evaluation of time-course and between groups differences in stored RBC units. Significance was accepted at p < 0.05. Due to the low number of samples analyzed by proteomics methods (distinguished by dashed boxes throughout the figures in order to highlight their mostly qualitative assessment) or those falling into the β0 category (Figure 4) all selected parameters satisfied the criteria of both statistical significance (<0.05) and fold change (>1.25), with the exception of Piezo-1 that satisfies only the first criterion.
At first, we focused on probable differences between β++ (slight reduction in ß-globin) and β+ (severe reduction but not null synthesis of ß-globin chains) samples. In freshly drawn blood, β++ heterozygotes presented a trend toward higher hematocrit and mean corpuscular volume (MCV), but significantly lower red cell distribution width (RDW) and HbA2 (Figure 1A). Moving on to storage, while similar levels of spontaneous hemolysis were observed in β++ and β+ units (e.g., late storage: 7.42 ± 2.10 vs. 5.83 ± 2.81 mg Hb/dL, β++ vs. β+, p = 0.294), β++ RBCs presented increased osmotic fragility and sporadically increased mechanical fragility throughout storage, with both differences evident in freshly drawn blood as well (Figure 1B). At the same time, different levels of myosin-9 and piezo-1 proteins (that play a role in RBC deformability and volume regulation) were also detected in those groups (Figure 1B).
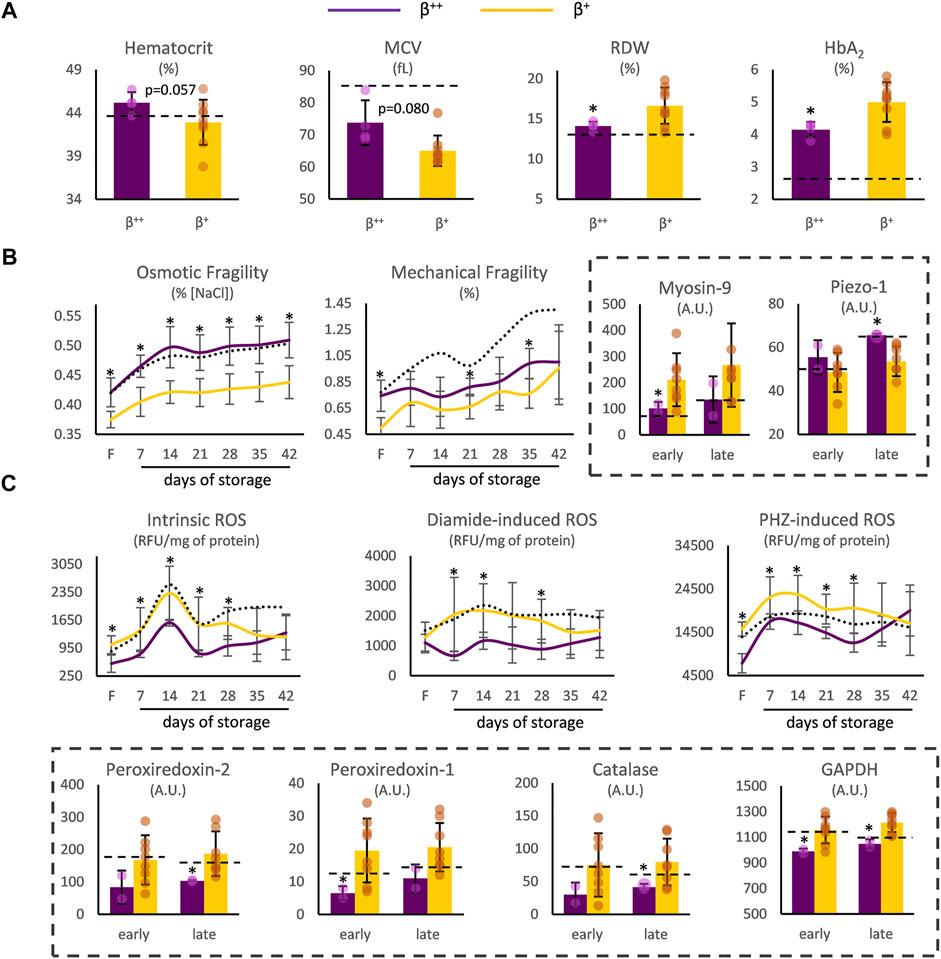
FIGURE 1. RBC indices and physiology differences between β++ and β+ heterozygotes. (A) Hematological indices in freshly drawn blood. (B) Cellular fragility and (C) redox parameters before and during storage. Values for average controls are shown by dashed lines. Proteomic parameters are shown in dashed boxes (n = 2 vs. 8, β++ vs. β+). (*) p < 0.05. F: freshly drawn blood; A.U, arbitrary units; ROS: reactive oxygen species; RFU: relative fluorescence units; PHZ: phenylhydrazine; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
Concerning biochemical and proteomic features that vary as a function of the oxidative burden of RBCs, β++ RBCs presented lower intracellular levels of ROS when compared to β+, either intrinsic or induced by thiol- and hemoglobin-oxidizing agents, from the beginning until the middle of the storage period (Figure 1C). Interestingly, intrinsic, and phenylhydrazine-induced ROS already differed at baseline. The binding of redox-related proteins on the membrane was also distinct: members of the peroxiredoxin family, along with catalase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were less evident in the isolated β++ membranes (Figure 1C). It should be noted that no differences arose regarding membrane protein carbonylation (e.g., protein carbonylation index day 21: 36.73 ± 7.04 vs. 33.43 ± 9.12, β++ vs. β+, p > 0.05) and extracellular antioxidant capacity (e.g., total antioxidant capacity day 35: 428 ± 93 vs. 354 ± 64 μM Fe2+, β++ vs. β+, p > 0.05).
Proteostasis was also affected by the degree of ß-globin synthesis. In the cytosol of β++, CASP- and TR-like (but not CH-like) activities were significantly reduced before (for TR-like activity at baseline p = 0.057) and during storage (Figure 2A) compared to the β+ values. With regards to the membrane, all three proteasomal activities presented lower levels in β++ vs. β+ at both early and late storage. This finding was accompanied by a trend for lower binding of the b5, b1 and b2 proteasome subunits (where the three different proteolytic specificities of the proteasome are located) in the membrane, especially at late storage, in β++ RBCs (Figure 2A). In parallel, heat shock proteins and components of the chaperoning T-complex also exhibited lower levels in the membranes of β++ RBCs in comparison to β+ (Figure 2B).
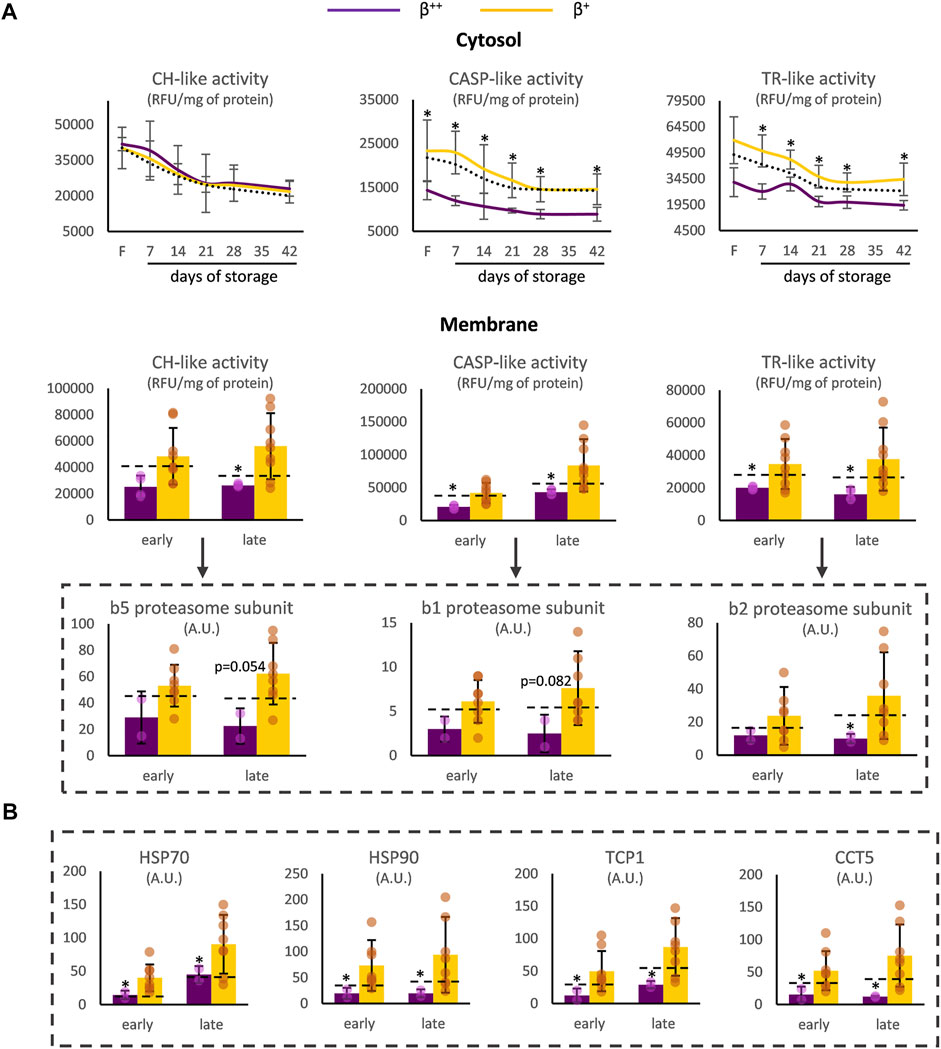
FIGURE 2. Proteostasis differences between β++ and β+ heterozygotes. (A) Proteasome activity in the cytosol and the membrane and binding of proteasome activity subunits on the membrane. (B) Binding of chaperoning proteins on the membrane. Values for average controls are shown by dashed lines. Proteomic parameters are shown in dashed boxes (n = 2 vs. 8, β++ vs. β+). (*) p < 0.05. F: freshly drawn blood; CH-like: chymotrypsin-like, CASP-like: caspase-like, TR-like: trypsin-like proteasome activities; RFU: relative fluorescence units; A.U, arbitrary units; HSP: heat shock protein; TCP1: T-complex protein one; CCT: T-complex subunit.
Moreover, the RBCs of β++ donors demonstrated increased levels of G6PD activity and of the relevant metabolites NADPH and pyridoxal in vivo (Figure 3A). Both before and throughout storage, l-arginine stood out by presenting elevated values in β++ vs. β+ RBCs, probably in agreement with the lower membrane binding levels of arginase-1 (Figure 3B). Another metabolite that was differentially affected by the mutation’s severity was dihydrothymine. Either throughout storage (intracellularly) or in the last 2 weeks of it (extracellularly) lower levels of dihydrothymine were detected in the group of β++ versus β+ (Figure 3C). Finally, two metabolites implicated in the biosynthesis of glycerophospholipids, namely choline and sphingosine-1-phosphate (S1P), also showed lower levels in the same subgroup throughout the storage period (Figure 3D). All other metabolites tested presented similar levels between the two groups (e.g., urate day 7: 17 × 106±6 × 106 vs. 13 × 106±5 × 106 A.U., β++ vs. β+).
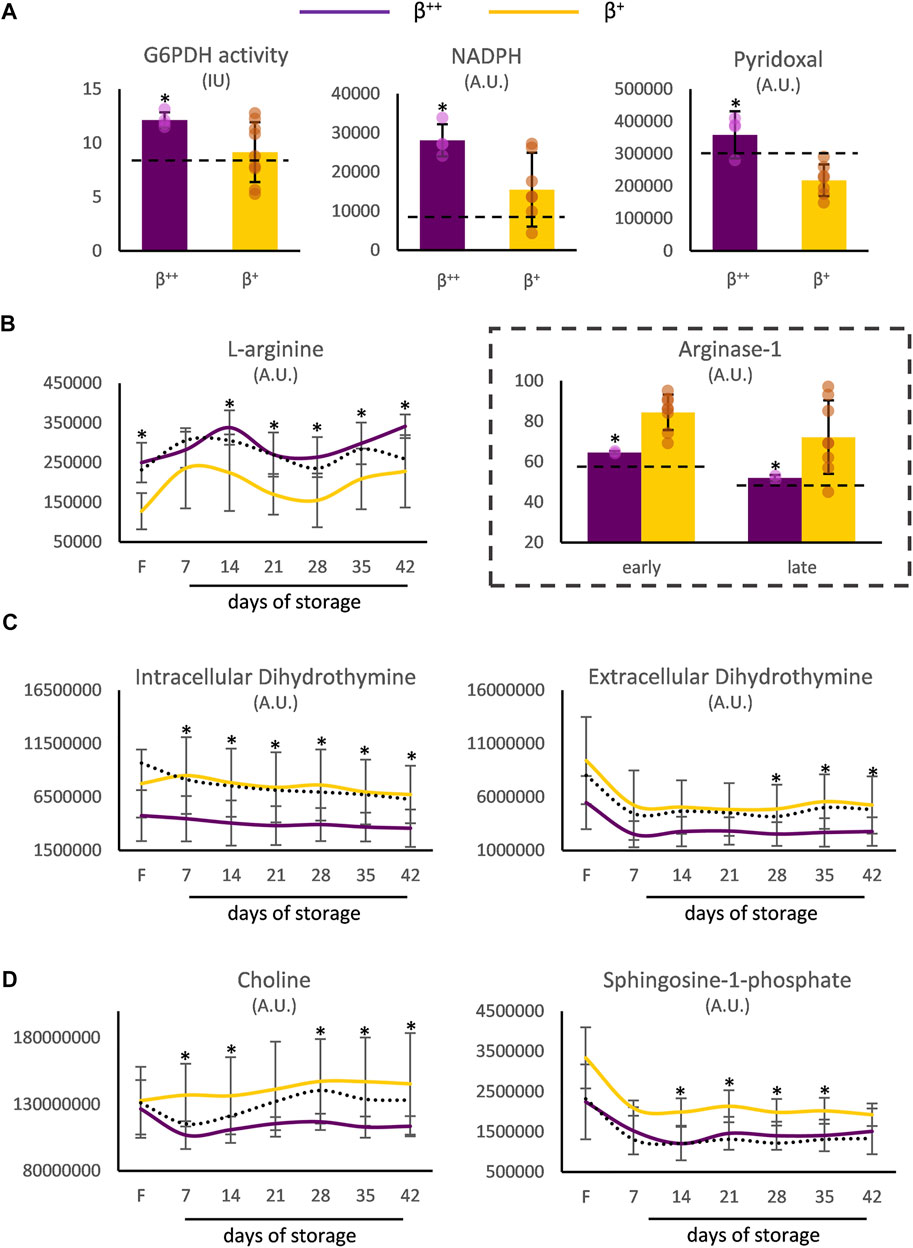
FIGURE 3. Metabolism differences between β++ and β+ heterozygotes. (A) In vivo differences in reducing and antioxidant powers. Differences in freshly drawn and stored red blood cells in (B) arginine, (C) dihydrothymine and (D) metabolites implicated in the biosynthesis of glycerophospholipids. Values for average controls are shown by dashed lines. Proteomic parameters are shown in dashed boxes (n = 2 vs. 8, β++ vs. β+). (*) p < 0.05. F: freshly drawn blood; G6PDH: glucose-6-phosphate dehydrogenase; IU: international units; NADPH: nicotinamide adenine dinucleotide phosphate; A.U. arbitrary units.
Despite the low number of β0 donors, that renders comparisons with the other groups mainly qualitative, the β0 (null ß-globin synthesis) subjects were characterized by higher levels of HbA2 but lower levels of ferritin, transferrin and osmotic fragility compared to β++ or β+ donors in vivo (Figure 4A). In terms of metabolism, β0 RBCs presented a downregulated amino acid metabolism sporadically during storage, as in the case of l-arginine (lower levels than in β+) and l-tryptophan (lower levels than in β++). Nucleotides such as IMP, carboxylic acids and glutathione-related metabolites were also decreased in β0, mainly throughout storage and against both other donor groups. Additionally, molecules related to fatty acid and carnitine metabolism were found significantly lower in the same group, especially when compared to β+ RBCs in specific storage periods (Figure 4A). Lastly, some proteins also exhibited distinct profiles in the β0 RBC membrane (Figure 4B), including several proteasome subunits that along with GAPDH were upregulated during late or early storage, respectively, when compared to β++. On the other hand, the presence of annexin A7 and peroxiredoxin-1 was minor in the membrane of late-stored β0 in comparison to β+. Another protein that differed among the three groups was carnitine O-palmitoyltransferase 1, the early levels of which gradually declined from the β++ to the β0 status (Figure 4B).
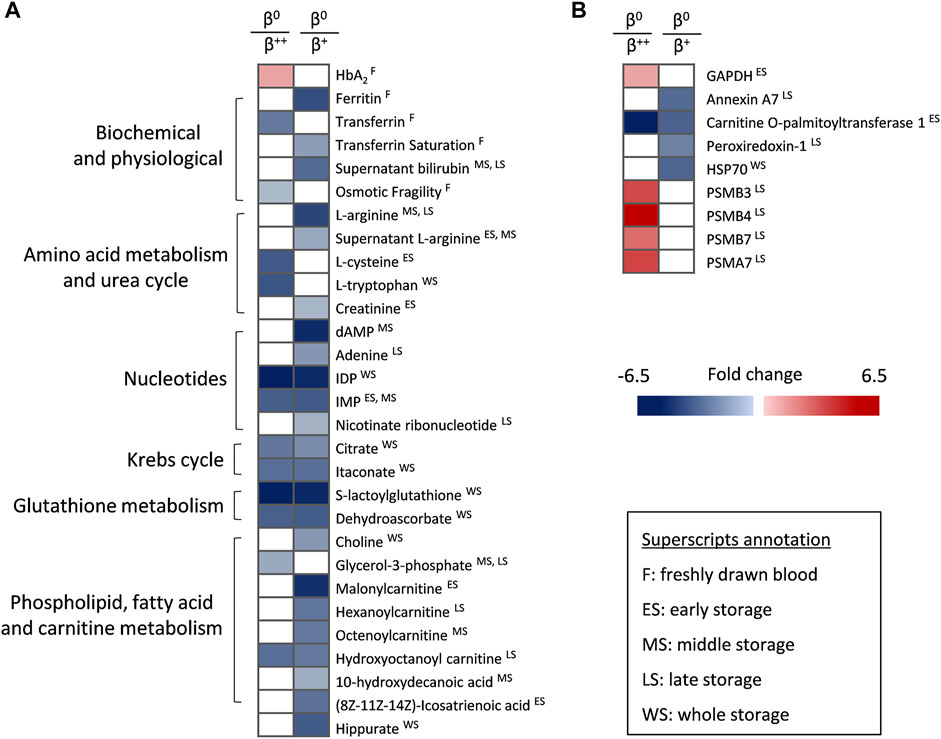
FIGURE 4. Statistically significant differences between β++ or β+ heterozygotes and β0. (A) Biochemical, physiological, metabolic and (B) proteomic differences. All differences shown satisfy the criterion of 1.25 fold. n = 2 vs. 8 vs. 2, β++ vs. β+ vs. β0. GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HSP: heat shock protein; PSM: proteasome subunit.
As previously shown, RBCs from βThal+ donors can effectively cope with storage lesion, especially with regards to the storage-related hemolysis stress, maintaining at the same time enhanced proteostasis capacity and uric acid-related intracellular and extracellular antioxidant power. Hereby, we report a variation within the group of heterozygotes, with β++ donors exhibiting worse RBC fragility indices but lower oxidative burden and proteasome activity than β+. It was surprising to also find some β0 subjects within the cohort of eligible donors, who showed unique nucleotide and amino acid metabolism features. A rather expected variation was found in hematological and biochemical data among the three sub-groups (β++, β+ and β0) in vivo.
Silent mutations, which lie behind the β++ phenotype, lead to just a small imbalance of the α-/β-globin synthesis ratio (Cao and Galanello, 2010), therefore, less excess of α-chains is present in cells. It is established that the accumulation of unpaired α-globin chains promotes the generation of ROS, thus, modifying the redox equilibrium of the cell and leading to oxidative damages (Olivieri, 1999). At the same time, RBCs in beta-thalassemia trait seem to possess an advantageous genetic regulation of antioxidant enzymes, leading to upregulated expression of peroxiredoxin-2 and superoxide dismutase (Teran et al., 2020). Our cohort of beta-thalassemia carriers, in contrast to previously studied groups (Selek et al., 2007), also exhibits higher than average extracellular antioxidant capacity, as well as superior intracellular equilibrium of redox metabolites (e.g., increased urate, decreased s-allantoin) (Tzounakas et al., 2022), irrespectively to the variable degree of beta-globin synthesis imposed by the heterozygous state of β++ and β+ underlying mutations. It is tempting to hypothesize that the slight excess of α-globin chains in β++ vs. β+ subjects, along with the boosted antioxidant system observed in both subgroups, altogether provide the first with an advantage regarding the control of ROS accumulation. The above hypothesis is also supported by the susceptibility to PHZ- (and thus, to oxidized-Hb) induced ROS production: while overall similar to the control (when all mutations analyzed as a group), when stratified in the currently examined subgroups β+ RBCs present sporadically higher but β++ lower susceptibility to ROS elevation (e.g., day 28: 20,079 ± 5,267 vs. 16,165 ± 4,529 vs. 12,058 ± 1,946 RFU/mg of protein, β+ vs. control vs. β++, p < 0.05). Moreover, while the storage levels of ROS in the β+ subgroup followed the general variation pattern observed in heterozygous versus control samples, namely lower levels at late-storage (Tzounakas et al., 2022), the levels of β++ RBCs were inferior throughout the storage period (e.g., day 14: 2,321 ± 678 vs. 2,539 ± 789 vs. 1,584 ± 83 RFU/mg of protein, β+ vs. control vs. β++, p > 0.05 only for β+ vs. control). In this context, it would be very interesting to study the redox equilibrium of RBCs from the minority group of β0 eligible donors during the storage period.
Following binding to the RBC cytoskeletal network, the free α-globin chains tend to autoxidize and cause massive membrane oxidative damage (Ficarra et al., 2009). The enhanced proteostatic system of β+ and β0 cells, observed through the elevated levels of proteasomal activity and/or the increased binding of chaperone and proteasome proteins to the membrane, seems capable to ameliorate the detrimental effects of oxidative stress upon the membrane. Indeed, we found no difference between the levels of membrane protein carbonylation between the distinct sub-groups. The proteasome machinery is abundant in the beta-thalassemic precursor cells to decongest the cell from the free α-globin chains (Khandros et al., 2012; Rivella, 2012), while in mature RBCs the same supramolecular complex appears to be involved in the degradation of oxidized Hb (Abi Habib et al., 2020). Regarding the latter, it has been indicated that several chaperones are also involved in α-globin detoxification (Khandros et al., 2012). The simultaneous recruitment of antioxidant cytosolic proteins such as peroxiredoxin-2 (Cho et al., 2014) in the membrane of β+ RBCs (which is considered a response to local oxidative stress) (Rocha et al., 2009), might also assist in the overall membrane protection, especially considering the remarkable crosstalk between the redox and proteostasis networks in βThal+ stored RBCs (Anastasiadi et al., 2021b). It should not be omitted that peroxiredoxin-2 competes with hemichromes for Band 3 binding and can prevent the latter’s clustering and formation of senescence neo-antigen (Bayer et al., 2016). Nonetheless, excess of hemichromes, as shown in RBCs of βThal mice models can displace peroxiredoxin-2 from the membrane, highlighting the excessive oxidative challenges (Matte et al., 2010). Thus, there might be a fine line regarding the recruitment of peroxiredoxin-2 to the membrane under different levels of oxidative stress. In our case, the increased oxidative stress of β+ RBCs, that is additionally burdened during the stressful storage period, seems not to be translated to extreme membrane damage and subsequent cell lysis, since the βThal+ sub-groups presented similar spontaneous hemolysis levels. In striking contrast, in beta-thalassemia intermedia and major RBCs, the severe defects of the membrane components, caused by the precipitation of the unstable α-globin chains, contribute to hemolysis (Romanello et al., 2018). The increased binding of the cytosolic GAPDH to the membranes of β0 and β+ RBCs further highlights the oxidative stress imposed by the unmatched α-globin chains, since the relocation of GAPDH is observed under pro-oxidant conditions during storage and triggers glucose consumption through the pentose phosphate pathway to supply RBCs with reducing power (Reisz et al., 2016).
It has been previously shown that the α-globin related oxidation of membrane and skeletal proteins in thalassemic RBCs affects their mechanical stability (Schrier and Mohandas, 1992). The augmented proteostasis at the membrane level along with the observed variation in the membrane association of myosin proteoforms and piezo-1 protein could take the credit for the superior fragility of β+ versus β++ RBCs. Indeed, non-muscle myosin IIA plays a significant role in the control of RBC shape and deformability (Smith et al., 2018), while piezo-1 is involved in the regulation of cell volume (Svetina et al., 2019). Moreover, it has been shown that the membrane localization of peroxiredoxin-2 promotes K+ efflux through activation of the Gardos channel (Low et al., 2008), benefitting the cell in terms of osmotic stress tolerance. To further support this, elevated RBC osmotic fragility has been previously shown in Gardos-knockout mice (Grgic et al., 2009).
Only a few differences were detected in the levels of RBC metabolites between the three βThal+ subgroups, revealing that the metabolism of RBCs is rather compact in the thalassemia trait. Freshly drawn RBCs from β++ subjects presented a redox equilibrium advantage compared to the β+ counterparts, since they were enriched in NADPH −the driving force of several antioxidant pathways in RBCs− and the redox-related pyridoxal (Jain and Lim, 2001), a finding that is in line with the lower levels of ROS accumulation observed in this subgroup. Arginine was found in gradually lower levels from the β++ towards the β0 stored RBCs. It is known that arginine metabolism is dysregulated in thalassemia (Jain and Lim, 2001), while increased arginase-1 expression (currently observed in β+ versus β++) and activity, in parallel with low l-arginine levels have been linked to oxidative stress and hemolysis (Morris et al., 2017; Contreras-Zentella et al., 2019). In this context, the arginase-1 enriched β+ RBCs also presented increased intrinsic oxidative burden when compared to β++. During aging-related oxidative stress, arginase-1 is elevated (at least in animal models (Pandya et al., 2019)), thus the successively higher α-globin chain accumulation in the βThal+ subgroups might play a role in the differential arginine metabolism. Another metabolite that distinguished β++ from β+ was dihydrothymine. Interestingly, this pyrimidine metabolite that has been associated with the beneficial osmotic stability of βThal+ RBCs in previous studies (Anastasiadi et al., 2022) is currently found increased in the least fragile group. Nonetheless, the mechanistic basis (if any) underlying these observations needs further examination. Of note, the elevated levels of spingosine-1-phosphate in β+ compared to β++ stored RBCs, a lysophospholipid related to RBC energy metabolism regulation (Sun et al., 2016) and transfusion biology (Selim et al., 2011), predispose for favorable storability profile under hypoxic storage conditions. To support this, a previous study on G6PD deficient donors demonstrated significant correlations between baseline spingosine-1-phosphate levels and quality characteristics of stored RBCs (Reisz et al., 2017). Finally, β0 RBCs presented decreased metabolites of the carboxylic acid, nucleotide, fatty acid and glutathione pathways when compared to the other two subgroups. RBCs from beta-thalassemic patients are characterized by decreased glutathione levels (Kalpravidh et al., 2013) and, therefore, sustained oxidative stress that can be ameliorated by inhibitors of glutathione efflux transporters (Muanprasat et al., 2013). In the same context, serum from beta-thalassemic subjects demonstrates down-regulated fatty acid metabolism (Musharraf et al., 2017), whereas carboxylic acids and metabolites of the arginine and glutathione routes were found slightly or significantly downregulated in the lungs of murine models of beta-thalassemia (Buehler et al., 2021). According to these findings, it appears that the metabolism features of β0 RBCs are closer to the thalassemia disease profile, even though the currently studied β0 heterozygotes were considered eligible blood donors. Again, the metabolic profile of donated RBCs from the β0 subgroup deserves further examination by studies in bigger cohorts.
Overall, while presenting some solid characteristics that differentiate them from control RBCs, including the highly important superior end-of-storage hemolysis and antioxidant arsenal (such as urate), βThal+ stored RBCs also exhibit an inside-group variation. It is plausible to suspect that this variation is α-globin excess-dependent, since milder mutations lead to phenotypes closer to the average control, while severe mutations tend to phenotypic features that are closer (at some level) to the disease state. One limitation of this study that does not allow drawing of broad hypotheses is the low number of donors enrolled, especially in the group of β0 subjects, and the small number of samples used for proteomics analyses. However, this is the first time that the special features of the βThal+ donor subgroups are reported in the research field of RBC transfusion and donor variation effects. In fact, it would be really interesting and helpful to design large-scale studies, with βThal+ subgroups exhibiting wider genetic heterogeneity. Such targeted research could give us the ability to draw more sound conclusions regarding the relation between the magnitude of the thalassemia imprint on RBCs and their storage quality metrics.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by The Ethics Committee of the Department of Biology, School of Science, NKUA. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
VT and MA designed the study. AA, VT, and V-ZA performed the physiological experiments. MD performed the proteomic analyses. AD’A analysed the metabolomic data. AA and VT analysed the results and prepared the figures. KS and AK were responsible for the sample acquisition and the preparation of RBC units. AA, VT, and MA wrote the first draft of the manuscript. MD, V-ZA, EP, IP, KS, AD’A, and AK drafted the manuscript and contributed to the final version. All authors contributed to the article and approved the submitted version.
This project has received funding from the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Innovation (GSRI), under grant agreement No 2032.
Though unrelated to the contents of this manuscript, AD’A declares that he is a founder of Omix Technologies Inc and Altis Biosciences LLC and a consultant for Hemanext Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the blood donation volunteers and the NKUA students Dimitrios Karadimas (MSc) and Christos Cristogeorgos (BSc) who contributed to a part of the physiological experiments. M. S. Jacovides Hellas S.A. is specially acknowledged for the kind offer of the LTRC blood bags. We are very grateful toward the post graduate program “Pedagogy through innovative Technologies and Biomedical approaches” for supporting this research.
Abi Habib J., De Plaen E., Stroobant V., Zivkovic D., Bousquet M.-P., Guillaume B., et al. (2020). Efficiency of the Four Proteasome Subtypes to Degrade Ubiquitinated or Oxidized Proteins. Sci. Rep. 10 (1), 15765. doi:10.1038/s41598-020-71550-5
Anastasiadi A. T., Arvaniti V.-Z., Paronis E. C., Kostomitsopoulos N. G., Stamoulis K., Papassideri I. S., et al. (2022). Corpuscular Fragility and Metabolic Aspects of Freshly Drawn Beta-Thalassemia Minor RBCs Impact Their Physiology and Performance Post Transfusion: A Triangular Correlation Analysis In Vitro and In Vivo. Biomedicines 10, 530. doi:10.3390/biomedicines10030530
Anastasiadi A. T., Paronis E. C., Arvaniti V.-Z., Velentzas A. D., Apostolidou A. C., Balafas E. G., et al. (2021a). The Post-Storage Performance of RBCs from Beta-Thalassemia Trait Donors Is Related to Their Storability Profile. Ijms 22 (22), 12281. doi:10.3390/ijms222212281
Anastasiadi A. T., Tzounakas V. L., Arvaniti V.-Z., Dzieciatkowska M., Stamoulis K., Lekka M. E., et al. (2021b). Red Blood Cell Proteasome in Beta-Thalassemia Trait: Topology of Activity and Networking in Blood Bank Conditions. Membranes 11 (9), 716. doi:10.3390/membranes11090716
Bayer S. B., Low F. M., Hampton M. B., Winterbourn C. C. (2016). Interactions Between Peroxiredoxin 2, Hemichrome and the Erythrocyte Membrane. Free Radic. Res. 50 (12), 1329–1339. doi:10.1080/10715762.2016.1241995
Boussiou M., Karababa P., Sinopoulou K., Tsaftaridis P., Plata E., Loutradi-Anagnostou A. (2008). The Molecular Heterogeneity of β-Thalassemia in Greece. Blood Cells, Mol. Dis. 40 (3), 317–319. doi:10.1016/j.bcmd.2007.11.003
Buehler P. W., Swindle D., Pak D. I., Fini M. A., Hassell K., Nuss R., et al. (2021). Murine Models of Sickle Cell Disease and Beta‐Thalassemia Demonstrate Pulmonary Hypertension with Distinctive Features. Pulm. Circ. 11 (4), 1–12. doi:10.1177/20458940211055996
Cao A., Galanello R. (2010). Beta-Thalassemia. Genet. Med. 12 (2), 61–76. doi:10.1097/GIM.0b013e3181cd68ed
Cho C.-S., Yoon H. J., Kim J. Y., Woo H. A., Rhee S. G. (2014). Circadian Rhythm of Hyperoxidized Peroxiredoxin II Is Determined by Hemoglobin Autoxidation and the 20S Proteasome in Red Blood Cells. Proc. Natl. Acad. Sci. U.S.A. 111 (33), 12043–12048. doi:10.1073/pnas.1401100111
Contreras-Zentella M. L., Sánchez-Sevilla L., Suárez-Cuenca J. A., Olguín-Martínez M., Alatriste-Contreras M. G., García-García N., et al. (2019). The Role of Oxidant Stress and Gender in the Erythrocyte Arginine Metabolism and Ammonia Management in Patients with Type 2 Diabetes. PLoS One 14 (7), e0219481. doi:10.1371/journal.pone.0219481
D'Alessandro A., Fu X., Reisz J. A., Kanias T., Page G. P., Stone M., et al. (2020a). Stored RBC Metabolism as a Function of Caffeine Levels. Transfusion 60 (6), 1197–1211. doi:10.1111/trf.15813
D'Alessandro A., Fu X., Reisz J. A., Stone M., Kleinman S., Zimring J. C., et al. (2020b). Ethyl Glucuronide, a Marker of Alcohol Consumption, Correlates with Metabolic Markers of Oxidant Stress but Not with Hemolysis in Stored Red Blood Cells from Healthy Blood Donors. Transfusion 60 (6), 1183–1196. doi:10.1111/trf.15811
D’Alessandro A., Reisz J. A., Zhang Y., Gehrke S., Alexander K., Kanias T., et al. (2019). Effects of Aged Stored Autologous Red Blood Cells on Human Plasma Metabolome. Blood Adv. 3 (6), 884–896. doi:10.1182/bloodadvances.2018029629
Ficarra S., Tellone E., Giardina B., Scatena R., Russo A., Misiti F., et al. (2009). Derangement of Erythrocytic AE1 in Beta-Thalassemia by Caspase 3: Pathogenic Mechanisms and Implications in Red Blood Cell Senescence. J. Membr. Biol. 228 (1), 43–49. doi:10.1007/s00232-009-9157-5
Francis R. O., D’Alessandro A., Eisenberger A., Soffing M., Yeh R., Coronel E., et al. (2020). Donor Glucose-6-Phosphate Dehydrogenase Deficiency Decreases Blood Quality for Transfusion. J. Clin. Invest 130 (5), 2270–2285. doi:10.1172/JCI133530
Giardine B. M., Joly P., Pissard S., Wajcman H., K. Chui D. H., Hardison R. C., et al. (2021). Clinically Relevant Updates of the HbVar Database of Human Hemoglobin Variants and Thalassemia Mutations. Nucleic Acids Res. 49 (D1), D1192–D1196. doi:10.1093/nar/gkaa959
Grgic I., Kaistha B. P., Paschen S., Kaistha A., Busch C., Si H., et al. (2009). Disruption of the Gardos Channel (KCa3.1) in Mice Causes Subtle Erythrocyte Macrocytosis and Progressive Splenomegaly. Pflugers Arch. - Eur. J. Physiol. 458 (2), 291–302. doi:10.1007/s00424-008-0619-x
Harboe M. (1959). A Method for Determination of Hemoglobin in Plasma by Near-Ultraviolet Spectrophotometry. Scand. J. Clin. Laboratory Investigation 11, 66–70. doi:10.3109/00365515909060410
Jain S. K., Lim G. (2001). Pyridoxine and Pyridoxamine Inhibits Superoxide Radicals and Prevents Lipid Peroxidation, Protein Glycosylation, and (Na+ + K+)-ATPase Activity Reduction in High Glucose-Treated Human Erythrocytes. Free Radic. Biol. Med. 30 (3), 232–237. doi:10.1016/s0891-5849(00)00462-7
Kalpravidh R. W., Tangjaidee T., Hatairaktham S., Charoensakdi R., Panichkul N., Siritanaratkul N., et al. (20132013). Glutathione Redox System Inβ-Thalassemia/Hb E Patients. Sci. World J. 2013, 1–7. doi:10.1155/2013/543973
Kanias T., Lanteri M. C., Page G. P., Guo Y., Endres S. M., Stone M., et al. (2017). Ethnicity, Sex, and Age Are Determinants of Red Blood Cell Storage and Stress Hemolysis: Results of the REDS-III RBC-Omics Study. Blood Adv. 1 (15), 1132–1141. doi:10.1182/bloodadvances.2017004820
Khandros E., Thom C. S., D'Souza J., Weiss M. J. (2012). Integrated Protein Quality-Control Pathways Regulate Free α-Globin in Murine β-Thalassemia. Blood 119 (22), 5265–5275. doi:10.1182/blood-2011-12-397729
Li H., Fang K., Peng H., He L., Wang Y. (2022). The Relationship Between Glycosylated Hemoglobin Level and Red Blood Cell Storage Lesion in Blood Donors. Transfusion 62 (3), 663–674. doi:10.1111/trf.16815
Low F. M., Hampton M. B., Winterbourn C. C. (2008). Peroxiredoxin 2 and Peroxide Metabolism in the Erythrocyte. Antioxidants Redox Signal. 10 (9), 1621–1630. doi:10.1089/ars.2008.2081
Matte A., Low P. S., Turrini F., Bertoldi M., Campanella M. E., Spano D., et al. (2010). Peroxiredoxin-2 Expression Is Increased in β-Thalassemic Mouse Red Cells but Is Displaced from the Membrane as a Marker of Oxidative Stress. Free Radic. Biol. Med. 49 (3), 457–466. doi:10.1016/j.freeradbiomed.2010.05.003
Morris C. R., Hamilton-Reeves J., Martindale R. G., Sarav M., Ochoa Gautier J. B. (2017). Acquired Amino Acid Deficiencies: A Focus on Arginine and Glutamine. Nutr. Clin. Pract. 32, 30S–47S. doi:10.1177/0884533617691250
Muanprasat C., Wongborisuth C., Pathomthongtaweechai N., Satitsri S., Hongeng S. (2013). Protection against Oxidative Stress in Beta Thalassemia/hemoglobin E Erythrocytes by Inhibitors of Glutathione Efflux Transporters. PLoS One 8 (1), e55685. doi:10.1371/journal.pone.0055685
Musharraf S. G., Iqbal A., Ansari S. H., Parveen S., Khan I. A., Siddiqui A. J. (2017). β-Thalassemia Patients Revealed a Significant Change of Untargeted Metabolites in Comparison to Healthy Individuals. Sci. Rep. 7, 42249. doi:10.1038/srep42249
Nemkov T., Hansen K. C., D'Alessandro A. (2017). A Three-Minute Method for High-Throughput Quantitative Metabolomics and Quantitative Tracing Experiments of Central Carbon and Nitrogen Pathways. Rapid Commun. Mass Spectrom. 31 (8), 663–673. doi:10.1002/rcm.7834
Nemkov T., Reisz J. A., Gehrke S., Hansen K. C., D’Alessandro A. (2019). High-Throughput Metabolomics: Isocratic and Gradient Mass Spectrometry-Based Methods. Methods Mol. Biol. 1978, 13–26. doi:10.1007/978-1-4939-9236-2_2
Olivieri N. F. (1999). The β-Thalassemias. N. Engl. J. Med. 341 (2), 99–109. doi:10.1056/NEJM199907083410207
Osei-Hwedieh D. O., Kanias T., Croix C. S., Jessup M., Xiong Z., Sinchar D., et al. (2016). Sickle Cell Trait Increases Red Blood Cell Storage Hemolysis and Post-Transfusion Clearance in Mice. EBioMedicine 11, 239–248. doi:10.1016/j.ebiom.2016.08.006
Pandya C. D., Lee B., Toque H. A., Mendhe B., Bragg R. T., Pandya B., et al. (2019). Age-Dependent Oxidative Stress Elevates Arginase 1 and Uncoupled Nitric Oxide Synthesis in Skeletal Muscle of Aged Mice. Oxidative Med. Cell. Longev. 2019, 1–9. doi:10.1155/2019/1704650
Reisz J. A., Tzounakas V. L., Nemkov T., Voulgaridou A. I., Papassideri I. S., Kriebardis A. G., et al. (2017). Metabolic Linkage and Correlations to Storage Capacity in Erythrocytes from Glucose 6-Phosphate Dehydrogenase-Deficient Donors. Front. Med. 4, 248. doi:10.3389/fmed.2017.00248
Reisz J. A., Wither M. J., Dzieciatkowska M., Nemkov T., Issaian A., Yoshida T., et al. (2016). Oxidative Modifications of Glyceraldehyde 3-Phosphate Dehydrogenase Regulate Metabolic Reprogramming of Stored Red Blood Cells. Blood 128 (12), e32–e42. doi:10.1182/blood-2016-05-714816
Rivella S. (2012). Do Not Super-Excess Me! Blood 119 (22), 5064–5065. doi:10.1182/blood-2012-04-418590
Rocha S., Costa E., Coimbra S., Nascimento H., Catarino C., Rocha-Pereira P., et al. (2009). Linkage of Cytosolic Peroxiredoxin 2 to Erythrocyte Membrane Imposed by Hydrogen Peroxide-Induced Oxidative Stress. Blood Cells, Mol. Dis. 43 (1), 68–73. doi:10.1016/j.bcmd.2009.03.002
Romanello K. S., Teixeira K. K. L., Silva J. P. M. O., Nagamatsu S. T., Bezerra M. A. C., Domingos I. F., et al. (2018). Global Analysis of Erythroid Cells Redox Status Reveals the Involvement of Prdx1 and Prdx2 in the Severity of Beta Thalassemia. PLoS One 13 (12), e0208316. doi:10.1371/journal.pone.0208316
Roubinian N. H., Reese S. E., Qiao H., Plimier C., Fang F., Page G. P., et al. (2022). Donor Genetic and Nongenetic Factors Affecting Red Blood Cell Transfusion Effectiveness. JCI Insight 7 (1). doi:10.1172/jci.insight.152598
Schrier S., Mohandas N. (1992). Globin-Chain Specificity of Oxidation-Induced Changes in Red Blood Cell Membrane Properties. Blood 79 (6), 1586–1592. doi:10.1182/blood.v79.6.1586.1586
Selek S., Aslan M., Horoz M., Gur M., Erel O. (2007). Oxidative Status and Serum PON1 Activity in Beta-Thalassemia Minor. Clin. Biochem. 40 (5-6), 287–291. doi:10.1016/j.clinbiochem.2006.10.028
Selim S., Sunkara M., Salous A. K., Leung S. W., Berdyshev E. V., Bailey A., et al. (2011). Plasma Levels of Sphingosine 1-Phosphate Are Strongly Correlated with Haematocrit, but Variably Restored by Red Blood Cell Transfusions. Clin. Sci. (Lond) 121 (12), 565–572. doi:10.1042/CS20110236
Smith A. S., Nowak R. B., Zhou S., Giannetto M., Gokhin D. S., Papoin J., et al. (2018). Myosin IIA Interacts with the Spectrin-Actin Membrane Skeleton to Control Red Blood Cell Membrane Curvature and Deformability. Proc. Natl. Acad. Sci. U.S.A. 115 (19), E4377–E4385. doi:10.1073/pnas.1718285115
Stefanoni D., Fu X., Reisz J. A., Kanias T., Nemkov T., Page G. P., et al. (2020). Nicotine Exposure Increases Markers of Oxidant Stress in Stored Red Blood Cells from Healthy Donor Volunteers. Transfusion 60 (6), 1160–1174. doi:10.1111/trf.15812
Sun K., Zhang Y., D’Alessandro A., Nemkov T., Song A., Wu H., et al. (2016). Sphingosine-1-Phosphate Promotes Erythrocyte Glycolysis and Oxygen Release for Adaptation to High-Altitude Hypoxia. Nat. Commun. 7, 12086. doi:10.1038/ncomms12086
Svetina S., Švelc Kebe T., Božič B. (2019). A Model of Piezo1-Based Regulation of Red Blood Cell Volume. Biophysical J. 116 (1), 151–164. doi:10.1016/j.bpj.2018.11.3130
Szczesny-Malysiak E., Mohaissen T., Bulat K., Kaczmarska M., Wajda A., Marzec K. M. (2021). Sex-Dependent Membranopathy in Stored Human Red Blood Cells. Haematol. 106 (10), 2779–2782. doi:10.3324/haematol.2021.278895
Terán M. M., Mónaco M. E., Lazarte S. S., Haro C., Ledesma Achem E., Asensio N. A., et al. (2020). Genetic Regulation of Redox Balance in β-Thalassemia Trait. Hemoglobin 44 (2), 122–127. doi:10.1080/03630269.2020.1765794
Tzounakas V. L., Anastasiadi A. T., Dzieciatkowska M., Karadimas D. G., Stamoulis K., Papassideri I. S., et al. (2021). Proteome of Stored RBC Membrane and Vesicles from Heterozygous Beta Thalassemia Donors. Ijms 22 (7), 3369. doi:10.3390/ijms22073369
Tzounakas V. L., Anastasiadi A. T., Stefanoni D., Cendali F., Bertolone L., Gamboni F., et al. (2022). Beta Thalassemia Minor Is a Beneficial Determinant of Red Blood Cell Storage Lesion. Haematol. 107 (1), 112–125. doi:10.3324/haematol.2020.273946
Tzounakas V. L., Kriebardis A. G., Georgatzakou H. T., Foudoulaki-Paparizos L. E., Dzieciatkowska M., Wither M. J., et al. (2016). Glucose 6-Phosphate Dehydrogenase Deficient Subjects May Be Better “Storers” Than Donors of Red Blood Cells. Free Radic. Biol. Med. 96, 152–165. doi:10.1016/j.freeradbiomed.2016.04.005
Keywords: genetic variability, storage lesion, beta-thalassemia trait, red blood cells, omics
Citation: Anastasiadi AT, Tzounakas VL, Dzieciatkowska M, Arvaniti V-Z, Papageorgiou EG, Papassideri IS, Stamoulis K, D’Alessandro A, Kriebardis AG and Antonelou MH (2022) Innate Variability in Physiological and Omics Aspects of the Beta Thalassemia Trait-Specific Donor Variation Effects. Front. Physiol. 13:907444. doi: 10.3389/fphys.2022.907444
Received: 29 March 2022; Accepted: 02 May 2022;
Published: 08 June 2022.
Edited by:
Giampaolo Minetti, University of Pavia, ItalyReviewed by:
Egarit Noulsri, Mahidol University, ThailandCopyright © 2022 Anastasiadi, Tzounakas, Dzieciatkowska, Arvaniti, Papageorgiou, Papassideri, Stamoulis, D’Alessandro, Kriebardis and Antonelou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alkmini T. Anastasiadi, YWxrYW5hc3RAYmlvbC51b2EuZ3I=; Marianna H. Antonelou, bWFudG9uQGJpb2wudW9hLmdy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.