
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 23 June 2022
Sec. Developmental Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.899149
This article is part of the Research TopicWomen in Developmental Physiology: 2022View all 7 articles
 Huiling Hu1†Shuoping Zhang2†Jing Guo2†Fei Meng2Xueqin Chen1Fei Gong1,2Guangxiu Lu1,2
Huiling Hu1†Shuoping Zhang2†Jing Guo2†Fei Meng2Xueqin Chen1Fei Gong1,2Guangxiu Lu1,2 Wei Zheng1,2*Ge Lin1,2*
Wei Zheng1,2*Ge Lin1,2*Zygotic cleavage failure (ZCF) is a severe, early type of embryonic arrest in which zygotes cannot complete the first cleavage. Although mutations in BTG4 and CHEK1 have been identified as genetic causes of ZCF, these genes only explain a small population of ZCF cases. Thus, the underlying genetic causes for other affected individuals need to be identified. Here, we identified three TRIP13 missense variants responsible for ZCF in two patients and showed that they followed a recessive inheritance pattern. All three variants resulted in obvious changes in hydrogen bonding and consistent increase in DNA damage. Additionally, transcriptomic sequencing of oocytes and arrested embryos containing these variants suggested a greater number of differentially expressed transcripts in germinal vesicle (GV) oocytes than in 1-cell embryos. Vital genes for energy metabolism and cell cycle procession were widely and markedly downregulated, while DNA repair-related genes were significantly upregulated in both GV oocytes and 1-cell embryos of patients. These findings highlight a critical role of TRIP13 in meiosis and mitosis, as well as expand the genetic and phenotypic spectra of TR1P13 variants with respect to female infertility, especially in relation to ZCF.
Infertility affects approximately 48 million females worldwide and is routinely treated using in vitro fertilization (IVF) or intracytoplasmic sperm injection (ICSI) (Mascarenhas et al., 2012; Chambers et al., 2021). However, some females experience recurrent IVF/ICSI failure because of oocyte meiotic arrest (OMA), total fertilization failure, and early embryonic arrest (EEA), and EEA is the most common abnormality (Betts and Madan, 2008; Sang et al., 2021).
In the first cytokinesis of the zygote, the pronuclear envelopes break down and maternal and paternal chromosomes align at the metaphase plate mediated by the bipolar mitotic spindle (Li et al., 2013). Metaphase chromosomes are arranged on the equatorial plate, forming mitotic checkpoint complexes (MCCs); only when each spindle microtubule is properly connected to the centromere can the MCC be separated and anaphase be initiated (Lara-Gonzalez et al., 2012; Faesen et al., 2017). Subsequently, the zygotes are converted from the 1-cell to the 2-cell stage and then undergo consecutive occurrences of cytokinesis, producing six to eight blastomeres on day 3 of cultivation (Niakan et al., 2012).
Zygotic cleavage failure (ZCF) is a unique early type of EEA in which successfully fertilized zygotes cannot complete the first cleavage and remain arrested in the 1-cell stage. We defined this phenotype as a Mendelian disease for the first time in 2020, and identified variants in the B-cell translocation gene-4 (BTG4) as a genetic cause for maternal mRNA decay and ZCF (Zheng et al., 2020). In 2021, dominant variants in checkpoint kinase 1 (CHEK1) were proven to cause G2/M transition failure and ZCF owing to overactivation of kinase activity (Chen et al., 2021; Zhang et al., 2021). However, known variants in BTG4 and CHEK1 only account for 20% of the ZCF population (Zhang et al., 2021), and the underlying genetic causes for other affected individuals remain largely unknown.
Thyroid hormone receptor interaction protein 13 (TRIP13) functions as an important regulatory molecule of MCC dissociation and chromosome recombination in meiosis (Li and Schimenti, 2007; Wang et al., 2014). Female mice lacking TRIP13 display a severely impaired ovarian reserve (Li and Schimenti, 2007), and biallelic pathogenic variants of TRIP13 reportedly cause OMA and female infertility (Zhang et al., 2020). However, it is unknown whether there is any other phenotype related to female infertility, thus meriting further investigation.
Here, we report two newly discovered TRIP13 variants (c.1141G > A, p. Glu381Lys and c.1258A > G, p. Lys420Glu), and one recurrent previously reported TRIP13 variant (c.77A > G, p. His26Arg), resulting in phenotypic variability. This study suggests a novel, possible genetic pathogenesis of ZCF and extends the spectrum of phenotypes for TRIP13 variants.
Infertile patients diagnosed with ZCF were recruited from the Reproductive and Genetic Hospital of CITIC-XIANGYA. All blood samples, immature germinal vesicle (GV) oocytes, and arrested embryos were donated for the study after written informed consent from the participants.
Genomic DNA was extracted from peripheral blood samples using a QIAamp DNA Blood Kit (Qiagen, 51106, Germany) following the manufacturer’s protocol. Exome capture and sequencing were performed using xGen Exome Research Panel v1.0 and the Illumina HiSeq 2500 platform. Single nucleotide variants (SNVs) and small insertions and deletions (indels) were generated with the Genome Analysis Toolkit (GATK) pipeline and annotated with ANNOVAR. Variants were filtered, as previously described (Zheng et al., 2020), using the following criteria: 1) variants with minor allele frequencies (<1% in the Genome Aggregation Database); 2) exonic nonsynonymous or splice site variants, or coding insertions or deletions (indels); 3) mRNA and/or proteins that were highly or specifically expressed in oocytes; 4) coexistence in at least two probands, but absence in fertile populations.
Wild-type (WT) and TRIP13 variants were assessed using the SWISS-MODEL software (https://swissmodel.expasy.org) based on the 6f0x.1.C.pdb template. Variants were mapped to the atomic model using PyMol (http://www.pymol.org). Evolutionary conservation analysis was performed with DNAMAN software.
HeLa cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/high glucose (Hyclone, SH3002201) with 10% fetal bovine serum (Hyclone, SH30070) in a humidified incubator at 37°C with 5% CO2. Transient transfections were performed using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) according to the manufacturer’s instructions. Approximately 48 h after transfection, cells were fixed for immunofluorescence.
HeLa cells were fixed in 4% paraformaldehyde for 20 min at room temperature and permeated with 0.1% Triton X-100 for 15 min. After blocking for 1 h with 4% bovine serum albumin (A600332-0100, Sangon Biotech), samples were incubated with primary anti-FLAG (1:500 dilution, 701629, Invitrogen), anti-γH2Ax (1:500 dilution, 9718S, Cell signaling technology) and corresponding donkey anti-mouse Ig (H + G) (1:1000 dilution, A21203, Alexa Flour) antibodies. Hoechst 33342 (Invitrogen, H3570, 1:1000 dilution) was used to stain the nucleus. Four groups of cells were imaged using the same magnification on the same day to compensate for experimental error.
Cells were lysed with RIPA lysis buffer (Strong) (P0013E-1, Beyotime) and proteins denatured with 4× sodium dodecyl sulphate (SDS) loading buffer (NP0007, Invitrogen). The proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE Gel Kit, CW0022M, CWBIO), then transferred to a PVDF membrane. After blocking with 5% non-fat milk in Tris-HCl buffer solution with Tween-20 (TBST), the PVDF membrane was incubated with primary antibodies overnight at 4 °C (anti-FLAG, 1:1000 dilution, 8146S, Cell Signaling; and anti-GAPDH, 1:1000 dilution, AF0006, Beyotime). After washing three times with TBST, membranes were incubated with goat anti-mouse IgG (1:4000 dilution, A0216, Beyotime) to detect the primary antibodies.
For RNA sequencing (RNA-seq), the following samples were collected: 1) seven GV oocytes from patient 1 and ten GV oocytes from a control patient, from whom several embryos were collected for transfer during assisted reproductive technology, and 2) three 1-cell stage arrested embryos each from patient 1 and patient 2, and seven control 1-cell embryos of which previously described (Leng et al., 2019). RNA-seq libraries were prepared using the SMARTSeq2 protocol, sequenced using the BGISEQ500 platform (BGI-Shenzhen), and the data analyzed as previously described (Leng et al., 2019; Sha et al., 2020). Gene expression levels were normalized as transcripts per million (TPM). Transcripts whose levels increased or decreased by more than 2-fold were considered as differentially expressed.
Patient 1 from a consanguineous family, was 29 years old and diagnosed with primary infertility for unexplained reasons at 25 years of age. Her parents were first cousins. During her previous two ICSI attempts, 43 oocytes were retrieved, of which 25 were arrested at the GV or MI stage, and the other 18 mature MII oocytes formed 7 zygotes. Six zygotes remained at the 1-cell stage. Patient 2 was 36 years old, and experienced primary infertility for six years, despite a normal menstrual cycle and no endocrine disorders. In her previous three ICSI attempts, 6 of 13 oocytes retrieved were mature, of which 3 fertilized oocytes formed; however, they were arrested at the 1-cell stage (Table 1 and Figure 1B).
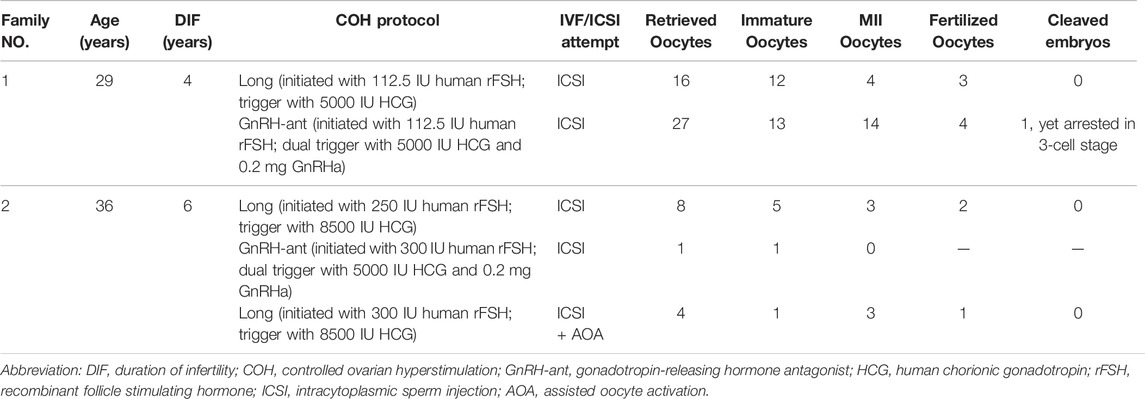
TABLE 1. Oocyte and embryo characteristics of ICSI attempts for the affected individuals.
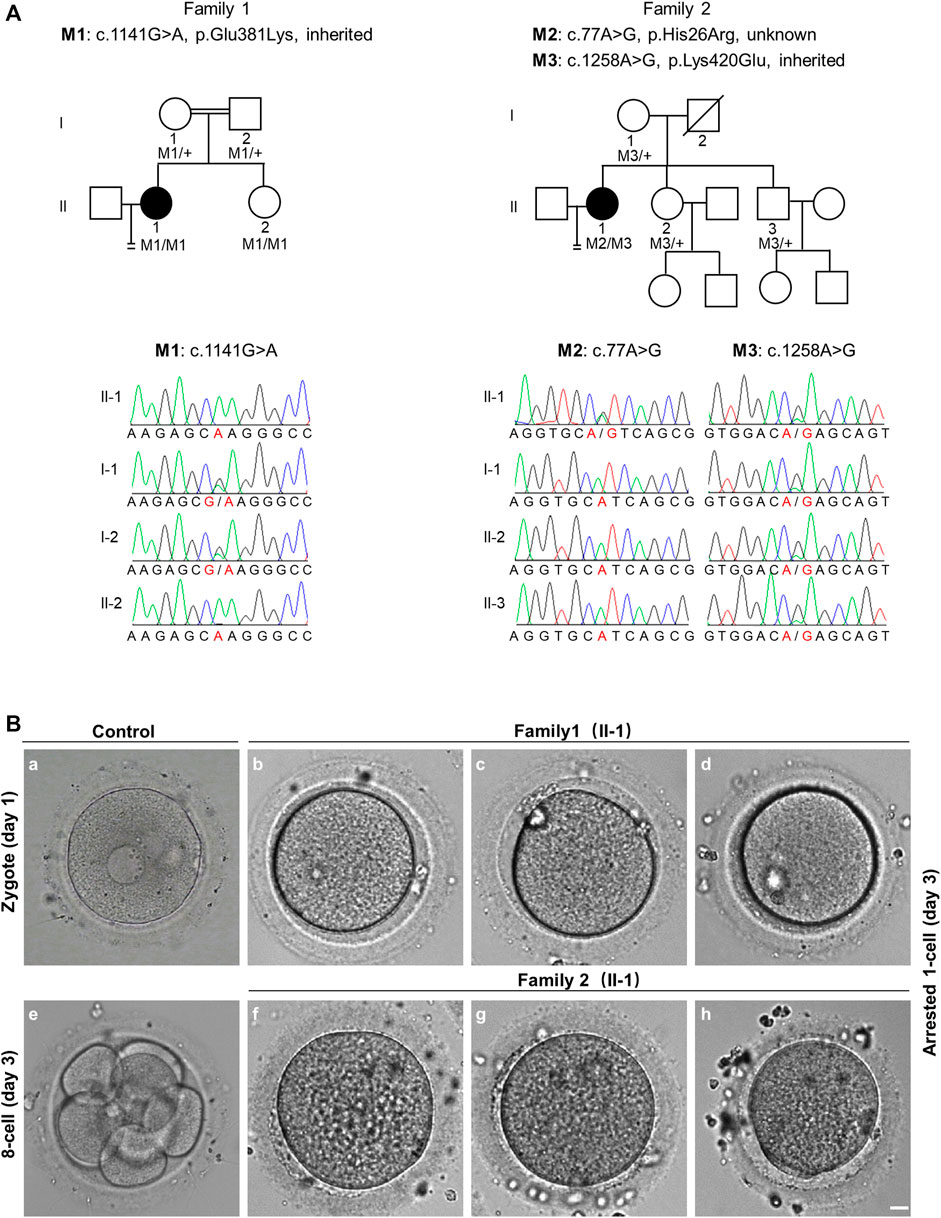
FIGURE 1. Pedigree-based identification of TRIP13 variants and the zygotic cleavage failure (ZCF) phenotype. (A) Genetic analysis of two patients from different families affected by ZCF. Black circles indicate the affected individuals. Sanger sequencing confirmation is shown below the pedigrees. (B) Morphology of a control embryo and arrested embryos from both patients on day 3. All six arrested 1-cell embryos were from two-pronucleus fertilized oocytes. Scale bar = 10 μm.
Since patient 1 is from a consanguineous family, the homozygous variant was considered a priority. After data filtering, only one rare homozygous variant c.1141G > A, p. Glu381Lys in TRIP13 was identified. This variant was inherited from her parents, and her sister carries the same homozygous variant; however, her fertility is not known owing to no sexual history. Patient 2 had the following compound heterozygous variants: c.77A > G, p. His26Arg and c.1258A > G, p. Lys420Glu. The c.1258A > G variant was inherited from her mother, and her fertile sister and brother also harbor this variant. However, the source of the c.77A > G variant is unknown owing to the unavailability of DNA sample from her father (Figure 1A and Table 2).
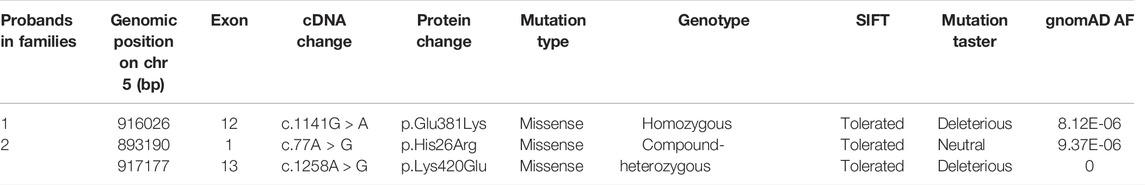
TABLE 2. Overview of the TRIP13 variants observed in two families.
All three TRIP13 variant residues herein are located in both sides of the ATPase domain; variant p. His26Arg is in the N-terminus and the other two variants are in the C-terminus. The variants p. Glu381Lys and p. Lys420Glu are highly conserved at the amino acid level among six different species, whereas the variant p. His26Arg is not conserved in mouse (Figure 2A). None of the three variants had any obvious effect on the TRIP13 protein structure, but they did result in a loss of hydrogen bonding (as p.Glu381Lys) or the formation of new hydrogen bonds (as p.His26Arg and p.Lys420Glu) with nearby amino acids, based on the constructed three-dimensional structure (Figure 2B).
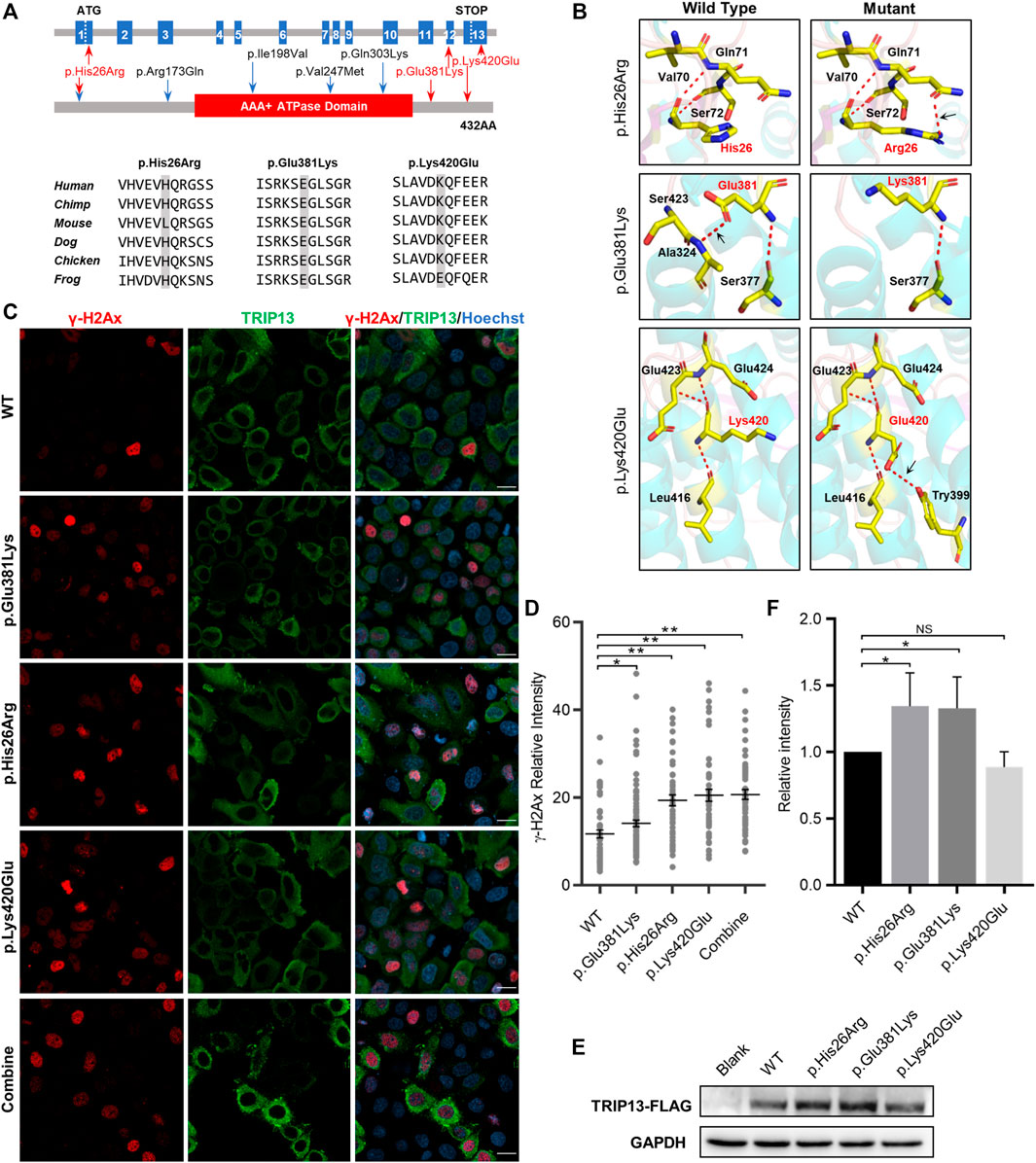
FIGURE 2. Effects of TRIP13 variants on predicted conformation, location of TRIP13 protein and DNA damage level in HeLa cells. (A) Localization of variants in the genome (top) and in a schematic of the TRIP13 protein structure (middle), and conservation of mutated amino acids in the variants in six species (bottom). The red arrows indicate novel variants identified herein; blue arrows indicate previously reported variants. (B) Protein conformation predictions of TRIP13 variants. Red dashed lines represent hydrogen bonds and black arrows indicate hydrogen bond changes. (C) Effects of TRIP13 variants on cellular localization of TRIP13 protein and DNA damage level in transfected HeLa cells. “combine” indicated co-transfected the p. Glu381Lys and p.Lys420Glu plasmids into the HeLa cells. Scale bar = 20 μm. (D) Statistical analysis of γH2Ax intensity shown in panel (C). Data are the mean ± SD. (E) Effects of TRIP13 variants on TRIP13 protein levels in transfected HeLa cells. (F) Statistical analysis of TRIP13 protein abundance shown in panel (E). The relative protein intensities of FLAG/GAPDH in the WT group were set as 1. Data are the mean ± SD.
We examined the subcellular localization of TRIP13 and DNA damage level by immunofluorescence and its abundance by western blotting in transfected HeLa cells. Both the wild-type and mutant TRIP13 proteins were localized in the cytoplasm, with no obvious difference (Figure 2C). However, all mutant TRIP13 caused the γH2Ax (a classical DNA damage marker) signals accumulation (Figures 2C,D). However, compared with wild-type TRIP13 protein abundance, the variants p. Glu381Lys and p. His26Arg showed a significant increase in TRIP13 levels, whereas variant p.Lys420Glu showed a decrease trend (Figures 2E,F).
Principal component analysis revealed an appreciable distinction among the controls and patients (Figure 3A). There were 7,005 and 3,111 differentially expressed transcripts (>2-fold change) in GV oocytes and 1-cell stage embryos of patients with TRIP13 variants, respectively (Supplementary Tables S1, S2). More transcripts were downregulated than upregulated in the GV oocytes of patient than in the controls (ndownregulated vs. nupregulated = 4,462 vs. 2,543; 1.7-fold difference); however, a similar number of transcripts were downregulated and upregulated in 1-cell stage embryos of patients compared with the controls (ndownregulated vs. nupregulated = 1,628 vs. 1,483) (Figure 3B). Additionally, more transcripts with a high abundance (TPM ≥ 5) were observed in the upregulated transcripts than in the downregulated transcripts, especially in the GV oocytes (Figure 3C).
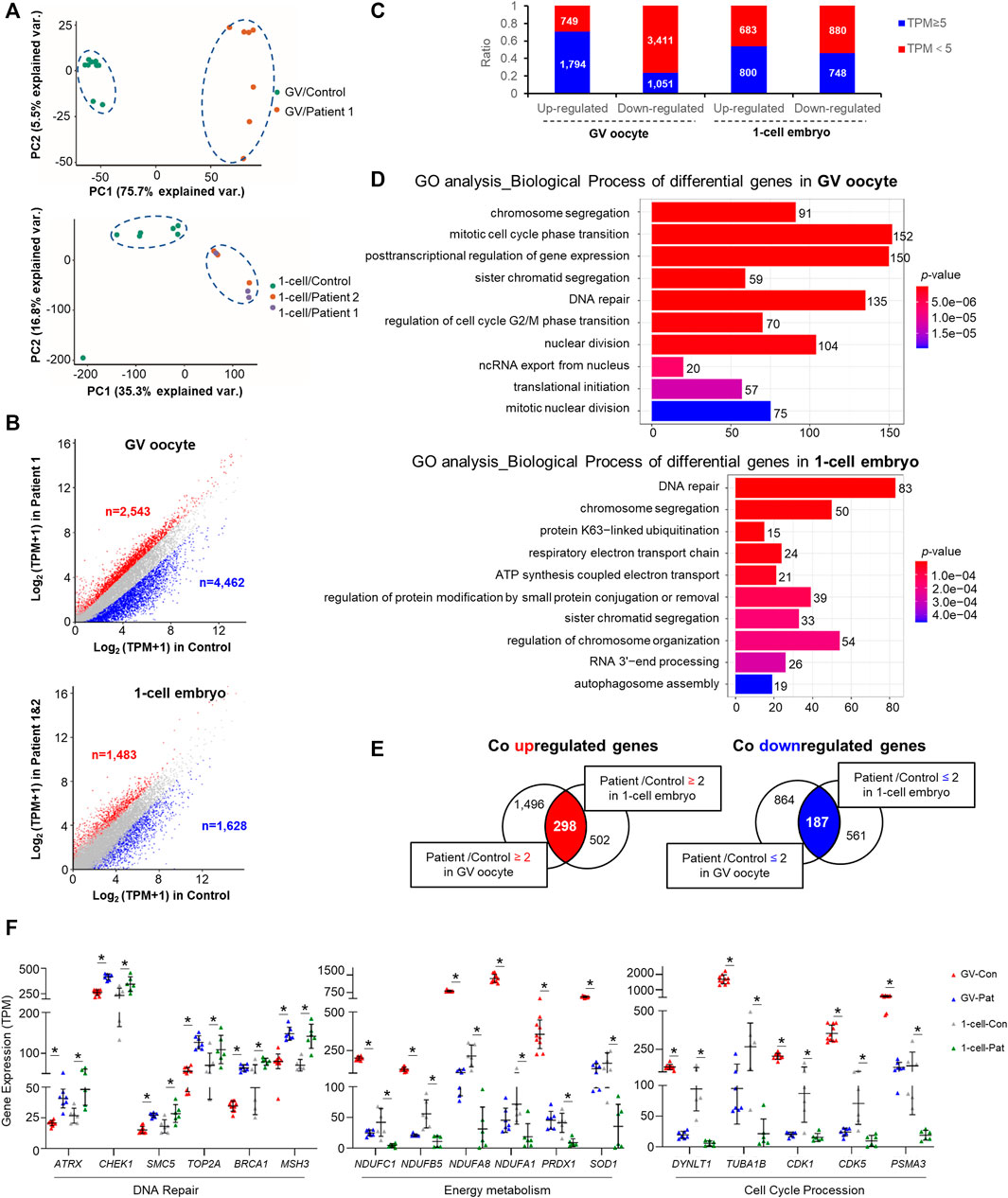
FIGURE 3. Changes in oocyte and embryo transcriptomes in the TRIP13 variants. (A) Principal component analysis (PCA) of gene expression in germinal vesicle (GV) oocytes and 1-cell embryos of TRIP13 variants and controls. (B) Scatter plot comparing the number of upregulated and downregulated gene transcripts between the TRIP13 variant in patient and control GV oocytes (upper) or 1-cell embryos (lower). Transcripts whose levels increased or decreased by more than 2-fold in the TRIP13 variant patients are highlighted in red or blue, respectively. (C) Ratio of the number of differentially expressed transcripts based on transcripts per million (TPM) prevalence in GV oocytes and 1-cell embryos. (D) Enriched top 10 biological processes of differentially expressed transcripts in GV oocytes and 1-cell embryos obtained via Gene Ontology (GO) analysis. Transcript numbers for each biological process are provided on the right. (E) Venn diagrams showing the shared upregulated (red region) or downregulated (blue region) transcripts between the control and GV oocytes (left) or 1-cell embryos (right). (F) Scatter plots of mRNA expression of selected transcripts related to three functions in the TRIP13 variants and control groups. Data are the mean ± SD. *p < 0.01, Student’s t-test.
Gene Ontology analysis revealed that the differentially expressed transcripts in GV oocytes and 1-cell embryos were consistently enriched in chromosome segregation and DNA repair process as the top 10 enriched biological processes (BPs) (Figure 3D). The other BPs were cell cycle phase transition, translation initiation, and nucleus division in GV oocyte and protein ubiquitination/modification, energy metabolism, and mRNA decay in 1-cell embryo. Based on the differences in biological events between GV oocytes and the 1-cell stage embryos, we investigated the commonalities between the two groups of regulated genes, identifying 298 co-upregulated and 187 co-downregulated transcripts among the high abundance differentially expressed genes (Figure 3E). Among the co-differentially expressed transcripts, we found that several genes responsible for DNA repair were highly expressed, whereas those involved in energy metabolism or cell cycle procession were significantly decreased in GV oocytes and 1-cell embryos of patients (Figure 3F). These results indicate that TRIP13 may repair DNA damage and maintain energy support to promote cell cycle progression during human oocyte maturation and mitosis initiation.
TRIP13 is a cell cycle regulator involved in both meiosis and mitosis (Li and Schimenti, 2007; Roig et al., 2010; Wang et al., 2014; Ma and Poon, 2016; Zhang et al., 2020). In this study, we report three different missense variants in TRIP13 that are responsible for human ZCF, which followed a recessive inheritance pattern.
Zhang et al. (2020) identified five variants in TRIP13 responsible for OMA and reported that two independent individuals who experienced OMA carried c.77A > G homozygous variant (Zhang et al., 2020). However, here, variants c.77A > G and c.1258A > G constituted a compound heterozygous pattern, and this patient exhibited a milder abnormality, with half the retrieved oocytes being mature. This phenotypic heterogeneity may suggest that the increase in TRIP13 protein abundance caused by c.77A > G was partially neutralized by the decrease in TRIP13 protein abundance caused by c.1258A > G (Figure 2F). However, because of a scarcity of human oocytes and embryos, the exact protein abundance in oocytes or embryos is largely unknown.
Yost et al. (2017) reported that the homozygous nonsense variant c.1060C > T (p.Arg354*) and the splicing c.673-1G > C pathogenic variant are both related to cause Wilms tumors (Yost et al., 2017). The differentially expressed transcripts in GV oocytes and 1-cell embryos were consistently enriched in chromosome segregation (Figure 3D), interestingly, chromosome segregation error is also the pathogenesis for Wilms tumors. However, both patients in our study suffered from infertility with no other diseases. The two distinct diseases, Wilms tumors and ZCF, might be caused by the state of the mutant protein. The Wilms tumor-associated variants caused the complete loss of the full-length protein, whereas the missense variants in the present study only affected the protein abundance.
Double strand breaks (DSBs) arise from endogenous and exogenous DNA damaging stimulation, persistent DSB could activate cell cycle checkpoints and resulted in cell cycle arrest (Mehta and Haber, 2014). Cells generally repair the DSB via homology-directed repair (HDR) or non-homologous end joining (NHEJ) to maintain the integrity of genomes (Chapman et al., 2012; Scully et al., 2019). TRIP13 directly activates DNA-dependent protein kinase catalytic subunit (DNA-PKcs) to activate the NHEJ pathway (Banerjee et al., 2014). We found the mutant TRIP13 plasmid transfection cause a more γH2Ax signals accumulation in HeLa cell (Figure 2D), additionally, the DNA repair pathway related genes significantly upregulated and cell cycle procession related genes significantly downregulated in both GV oocytes and 1-cell embryos of TRIP13 mutant patients (Figure 3F), we suggested the mutant TRIP13 may reduce ability to repair DNA damage. However, because of a scarcity of human oocytes and embryos, the exact γH2Ax signals in oocytes or embryos cannot been visualized.
Global transcriptional silencing is a highly conserved event from fully grown oocytes to zygotic genome activation (ZGA) at 4–8 cell embryo stage (Yan et al., 2013; Sha et al., 2020). Thus, the GV oocyte is the highest point of maternal mRNA accumulation, followed mainly by mRNA decay until ZGA. We found a large number of differentially expressed transcripts in the GV oocyte phase, especially a high proportion of downregulated transcripts (Figure 3B), indicating that the effect of TRIP13 variants was initiated during oocyte growth. Indeed, Trip13 knockout mice exhibit total oocyte ovarian reserve loss, whereas Trip13 hypomorphic variant mice still exhibit some growing follicles (Li and Schimenti, 2007; Pacheco et al., 2015). However, the ovarian reserve was not obviously affected in the patients in this study, which may be explained by the degree of TRIP13 function loss.
Among the co-differentially expressed transcripts, CHEK1 transcript levels were significantly increased in both oocytes and the 1-cell stage embryos of patients. CHEK1 encodes a serine/threonine-protein kinase, which is required for cell cycle control. Dominant variants in CHEK1 result in G2/M transition arrest of the zygote (Chen et al., 2021; Zhang et al., 2021). Here, we established a relationship between the two cell cycle regulators TRIP13 and CHEK1. However, whether zygote arrest can be rescued by CHEK1 inhibitor treatment needs further investigation.
In summary, our study expanded the mutation and phenotype spectrum of TRIP13 and suggested that human TRIP13 biallelic variants can cause cell cycle progress abnormalities that result in female infertility characterized by human ZCF. These findings should facilitate genetic diagnosis of ZCF and guide the development of precise therapy in the future for clinical female infertility.
The datasets presented in this article are not readily available because of local data protection laws. However, raw data is available from the corresponding authors on reasonable request.
The studies involving human participants were reviewed and approved by the Ethics Committee of the Reproductive and Genetic Hospital of CITIC-XIANGYA (reference LL-SC-2017-009 and LL-SC-2021-002). The patients/participants provided their written informed consent to participate in this study.
WZ and GL designed the study; WZ and HH analyzed the data and wrote the draft of this manuscript; WZ, HH, JG, FM, and XC performed the research; SZ, G-XL, and FG performed the clinical work; GL and WZ editing and improving the manuscript.
This work was supported by the National Natural Science Foundation of China (82001633 and 81901473), China Postdoctoral Science Foundation (2020M682575 and 2021T140198), Fundamental Research Funds for the Central Universities of Central South University (2021zzts0927), and the Scientific Research Foundation of Reproductive and Genetic Hospital of CITIC XIANGYA (YNXM-202202).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.899149/full#supplementary-material
Banerjee R., Russo N., Liu M., Basrur V., Bellile E., Palanisamy N., et al. (2014). TRIP13 Promotes Error-Prone Nonhomologous End Joining and Induces Chemoresistance in Head and Neck Cancer. Nat. Commun. 5, 4527. doi:10.1038/ncomms5527
Betts D. H., Madan P. (2008). Permanent Embryo Arrest: Molecular and Cellular Concepts. Mol. Hum. Reprod. 14 (8), 445–453. doi:10.1093/molehr/gan035
Chambers G. M., Dyer S., Zegers-Hochschild F., de Mouzon J., Ishihara O., Banker M., et al. (2021). International Committee for Monitoring Assisted Reproductive Technologies World Report: Assisted Reproductive Technology, 2014. Hum. Reprod. 36 (11), 2921–2934. doi:10.1093/humrep/deab198
Chapman J. R., Taylor M. R. G., Boulton S. J. (2012). Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 47 (4), 497–510. doi:10.1016/j.molcel.2012.07.029
Chen B., Guo J., Wang T., Lee Q., Ming J., Ding F., et al. (2021). Maternal Heterozygous Mutation in CHEK1 Leads to Mitotic Arrest in Human Zygotes. Protein Cell 13, 148–154. doi:10.1007/s13238-021-00844-9
Faesen A. C., Thanasoula M., Maffini S., Breit C., Müller F., van Gerwen S., et al. (2017). Basis of Catalytic Assembly of the Mitotic Checkpoint Complex. Nature 542 (7642), 498–502. doi:10.1038/nature21384
Lara-Gonzalez P., Westhorpe F. G., Taylor S. S. (2012). The Spindle Assembly Checkpoint. Curr. Biol. 22 (22), R966–R980. doi:10.1016/j.cub.2012.10.006
Leng L., Sun J., Huang J., Gong F., Yang L., Zhang S., et al. (2019). Single-Cell Transcriptome Analysis of Uniparental Embryos Reveals Parent-Of-Origin Effects on Human Preimplantation Development. Cell Stem Cell 25 (5), 697–712. doi:10.1016/j.stem.2019.09.004
Li X., Schimenti J. C. (2007). Mouse Pachytene Checkpoint 2 (Trip13) Is Required for Completing Meiotic Recombination but Not Synapsis. PLoS Genet. 3 (8), e130. doi:10.1371/journal.pgen.0030130
Li L., Lu X., Dean J. (2013). The Maternal to Zygotic Transition in Mammals. Mol. Aspects Med. 34 (5), 919–938. doi:10.1016/j.mam.2013.01.003
Ma H. T., Poon R. Y. C. (2016). TRIP13 Regulates Both the Activation and Inactivation of the Spindle-Assembly Checkpoint. Cell Rep. 14 (5), 1086–1099. doi:10.1016/j.celrep.2016.01.001
Mascarenhas M. N., Flaxman S. R., Boerma T., Vanderpoel S., Stevens G. A. (2012). National, Regional, and Global Trends in Infertility Prevalence since 1990: a Systematic Analysis of 277 Health Surveys. PLoS Med. 9 (12), e1001356. doi:10.1371/journal.pmed.1001356
Mehta A., Haber J. E. (2014). Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 6 (9), a016428. doi:10.1101/cshperspect.a016428
Niakan K. K., Han J., Pedersen R. A., Simon C., Pera R. A. R. (2012). Human Pre-implantation Embryo Development. Development 139 (5), 829–841. doi:10.1242/dev.060426
Pacheco S., Marcet-Ortega M., Lange J., Jasin M., Keeney S., Roig I. (2015). The ATM Signaling Cascade Promotes Recombination-dependent Pachytene Arrest in Mouse Spermatocytes. PLoS Genet. 11 (3), e1005017. doi:10.1371/journal.pgen.1005017
Roig I., Dowdle J. A., Toth A., de Rooij D. G., Jasin M., Keeney S. (2010). Mouse TRIP13/PCH2 Is Required for Recombination and Normal Higher-Order Chromosome Structure during Meiosis. PLoS Genet. 6 (8), e1001062. doi:10.1371/journal.pgen.1001062
Sang Q., Zhou Z., Mu J., Wang L. (2021). Genetic Factors as Potential Molecular Markers of Human Oocyte and Embryo Quality. J. Assist. Reprod. Genet. 38 (5), 993–1002. doi:10.1007/s10815-021-02196-z
Scully R., Panday A., Elango R., Willis N. A. (2019). DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 20 (11), 698–714. doi:10.1038/s41580-019-0152-0
Sha Q.-Q., Zheng W., Wu Y.-W., Li S., Guo L., Zhang S., et al. (2020). Dynamics and Clinical Relevance of Maternal mRNA Clearance during the Oocyte-To-Embryo Transition in Humans. Nat. Commun. 11 (1), 4917. doi:10.1038/s41467-020-18680-6
Wang K., Sturt-Gillespie B., Hittle J. C., Macdonald D., Chan G. K., Yen T. J., et al. (2014). Thyroid Hormone Receptor Interacting Protein 13 (TRIP13) AAA-ATPase Is a Novel Mitotic Checkpoint-Silencing Protein. J. Biol. Chem. 289 (34), 23928–23937. doi:10.1074/jbc.M114.585315
Yan L., Yang M., Guo H., Yang L., Wu J., Li R., et al. (2013). Single-cell RNA-Seq Profiling of Human Preimplantation Embryos and Embryonic Stem Cells. Nat. Struct. Mol. Biol. 20 (9), 1131–1139. doi:10.1038/nsmb.2660
Yost S., de Wolf B., Hanks S., Zachariou A., Marcozzi C., Clarke M., et al. (2017). Biallelic TRIP13 Mutations Predispose to Wilms Tumor and Chromosome Missegregation. Nat. Genet. 49 (7), 1148–1151. doi:10.1038/ng.3883
Zhang Z., Li B., Fu J., Li R., Diao F., Li C., et al. (2020). Bi-allelic Missense Pathogenic Variants in TRIP13 Cause Female Infertility Characterized by Oocyte Maturation Arrest. Am. J. Hum. Genet. 107 (1), 15–23. doi:10.1016/j.ajhg.2020.05.001
Zhang H., Chen T., Wu K., Hou Z., Zhao S., Zhang C., et al. (2021). Dominant Mutations in CHK1 Cause Pronuclear Fusion Failure and Zygote Arrest that Can Be Rescued by CHK1 Inhibitor. Cell Res. 31 (7), 814–817. doi:10.1038/s41422-021-00507-8
Keywords: female infertility, variants, TRIP13, zygotic cleavage failure, DNA damage
Citation: Hu H, Zhang S, Guo J, Meng F, Chen X, Gong F, Lu G, Zheng W and Lin G (2022) Identification of Novel Variants of Thyroid Hormone Receptor Interaction Protein 13 That Cause Female Infertility Characterized by Zygotic Cleavage Failure. Front. Physiol. 13:899149. doi: 10.3389/fphys.2022.899149
Received: 18 March 2022; Accepted: 07 June 2022;
Published: 23 June 2022.
Edited by:
Warren W. Burggren, University of North Texas, United StatesReviewed by:
Ying Wai Chan, The University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2022 Hu, Zhang, Guo, Meng, Chen, Gong, Lu, Zheng and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zheng, dXN0Y3p3QDE2My5jb20=; Ge Lin, bGluZ2dmQGhvdG1haWwuY29t
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.