
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 19 August 2022
Sec. Gastrointestinal Sciences
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.896951
This article is part of the Research Topic Gastrointestinal Tract Barrier Damage in Health and in Inflammatory Diseases View all 7 articles
 Isabel A. Jimenez1,2
Isabel A. Jimenez1,2 Allison P. Stilin1Kanako Morohaku1,3
Allison P. Stilin1Kanako Morohaku1,3 Mahmoud H. Hussein1
Mahmoud H. Hussein1 Prasanthi P. Koganti1
Prasanthi P. Koganti1 Vimal Selvaraj1*
Vimal Selvaraj1*In human patients and animal models of ulcerative colitis (UC), upregulation of the mitochondrial translocator protein (TSPO) in the colon is consistent with inflammation. Although the molecular function for TSPO remains unclear, it has been investigated as a therapeutic target for ameliorating UC pathology. In this study, we examined the susceptibility of Tspo gene-deleted (Tspo−/−) mice to insults as provided by the dextran sodium sulfate (DSS)-induced acute UC model. Our results show that UC clinical signs and pathology were severely exacerbated in Tspo−/− mice compared to control Tspofl/fl cohorts. Histopathology showed extensive inflammation and epithelial loss in Tspo−/− mice that caused an aggravated disease. Colonic gene expression in UC uncovered an etiology linked to precipitous loss of epithelial integrity and disproportionate mast cell activation assessed by tryptase levels in Tspo−/− colons. Evaluation of baseline homeostatic shifts in Tspo−/− colons revealed gene expression changes noted in elevated epithelial Cdx2, mast cell Cd36 and Mcp6, with general indicators of lower proliferation capacity and elevated mitochondrial fatty acid oxidation. These findings demonstrate that intact physiological TSPO function serves to limit inflammation in acute UC, and provide a systemic basis for investigating TSPO-targeting mechanistic therapeutics.
Overexpression of the mitochondrial translocator protein (TSPO) in the colon is a pathological feature observed in human ulcerative colitis (UC) (Ostuni et al., 2010), a form of inflammatory bowel disease (IBD). Consistent with human pathology, rodent models of UC have confirmed that TSPO upregulation in the colon is characteristic of UC (Ostuni et al., 2010). This finding is supported by studies investigating TSPO-binding drugs as radioligands for positron emission tomography (PET)-based diagnostic imaging; it has been concluded that TSPO detection using these probes provides a non-invasive method to evaluate the level and localization of colon inflammation (Bernards et al., 2015; Kurtys et al., 2017). High expression of TSPO in chronic UC has also been linked to an increased risk of colorectal cancers (Katz et al., 1988, 1990). Association between TSPO and inflammation is not only restricted to observations made in the colon; TSPO upregulation has been reported in conditions that induce neuroinflammation (Airas et al., 2015), cardiovascular inflammation (Fujimura et al., 2008), and rheumatoid arthritis (Narayan et al., 2018). Indicative of an active function and/or etiology for TSPO involvement in these pathologies, preclinical models have demonstrated that a variety of TSPO-binding drugs are efficacious as therapeutics in ameliorating inflammation. Protective effects of TSPO-targeting therapies have been reported for disorders such as multiple sclerosis (Daugherty et al., 2013), brain injury after intracerebral hemorrhage (Li et al., 2017), diabetic neuropathy (Giatti et al., 2009), autoimmune arthritis (Waterfield et al., 1999), and Alzheimer’s disease (Barron et al., 2013). As the etiology and pathophysiology of UC remains to be fully defined, TSPO has been considered as a therapeutic target and investigated for reducing both stress and colon inflammation.
Of direct relevance to therapeutic efficacy in UC, the TSPO-binding drug ONO-2952 (1-[(1S)-1-(4-chloro-2-methoxyphenyl)-5-fluoro-1,9-dihydrospiro [β-carboline-4,1’- cycloprop ane]-2(3H)-yl]ethenone) has been under development as a treatment for diarrhea-predominant irritable bowel syndrome (IBS-D) (Ono Pharma United States Inc.; NCT01844180, ClinicalTrials.gov). IBS-D patients have significantly higher levels of pro-inflammatory cytokines indicative of gut inflammation (Rana et al., 2012). ONO-2952 showed high selectivity in binding to TSPO, and could promote anti-stress effects by inducing a variety of protective responses (Mitsui, 2012; Mitsui et al., 2015). In preclinical rat models, ONO-2952 attenuated stress-induced defecation and rectal hyperalgesia (Kawahara et al., 2018). In the Phase 2 trial, ONO-2952 showed evidence for clinical efficacy with promising trends supporting its consideration as a potential treatment (Whitehead et al., 2017). Similarly, flunitrazepam, albeit a non-selective TSPO-binding drug, could protect the intestinal mucosa against DSS-induced damage (Ostuni et al., 2010). However, actions of other TSPO-binding drugs have not been consistent; PK11195 and Ro5-4864 were observed to enhance initial severity of colonic erosion, but accelerate recovery (Ostuni et al., 2010). Because the molecular function of TSPO remains to be elucidated (Selvaraj and Stocco, 2015; Selvaraj and Tu, 2016), TSPO-binding drugs cannot be confirmed as agonists or antagonists based on current understanding of target effects, potentially accounting for these differences in TSPO-targeting therapeutic efficacy.
First identified as a “receptor” showing high affinity binding to diazepam and its derivative 4’chlorodiazepam (Braestrup and Squires, 1977), the protein sequence of TSPO is highly conserved across microbes, plants and animals (Selvaraj and Stocco, 2015). Although pharmacology linked to TSPO-binding has indicated a variety of potential functions (Gavish et al., 1999), development and utilization of TSPO gene-deleted (Tspo−/−) models have ruled out its involvement in key purported pharmacological functions such as induction of de novo steroidogenesis (Tu et al., 2014a; Banati et al., 2014; Morohaku et al., 2014b; Wang et al., 2016), mitochondrial permeability transition (Šileikyte et al., 2014), and heme biosynthesis (Zhao et al., 2016). Phenotypic observations in Tspo−/− models point to TSPO’s functional involvement in lipid/energy metabolism (Leduc et al., 2011; Thompson et al., 2013; Tu et al., 2016; Liu et al., 2017; Kim et al., 2019; Koganti and Selvaraj, 2020). While such metabolic modulation might be relevant to certain TSPO over-expressing colon cancers (Katz et al., 1990; Maaser et al., 2005; Königsrainer et al., 2007), the precise mechanism of TSPO function in this context remains to be defined (Selvaraj and Stocco, 2015; Betlazar et al., 2020).
Given the considerable relevance of TSPO in the context of etiology, pathogenesis and therapeutic interventions for UC, we investigated Tspo−/− mice as a loss-of-function preclinical model to study the relevance of TSPO in homeostasis and TSPO-mediated mechanisms in UC. Such genetic studies provide an opportunity to delineate gene function and distinguish this from the non-specific actions prominently seen with different TSPO-binding drugs (Bordet et al., 2007; Li et al., 2008; Luc Do Rego et al., 2015; Tu et al., 2015; Singh et al., 2020). Our results identify a clear protective function for TSPO expression in colitis, deficiency of which results in exacerbated colonic inflammatory pathology.
Generation and validation of Tspo−/− mice has been previously described (Morohaku et al., 2014a; Tu et al., 2014b; Zhao et al., 2016). Breeding colonies for Tspofl/fl and Tspo−/− mice were propagated in the C57BL/6 background. Both male and female mice (Mus musculus) (8–10 weeks of age) were used in this study. Animals were maintained in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals. All experiments reported in this manuscript were approved by The Institutional Animal Care and Use Committee of Cornell University.
Detection of TSPO in Western blots was performed as previously described (Morohaku et al., 2013). In brief, colon samples were homogenized using a bead beater (Biospec Products), boiled in Laemmli sample buffer (Laemmli, 1970), and total protein was quantified using a bicinchoninic acid (BCA) colorimetric assay. Twenty micrograms of protein was then separated by SDS-PAGE, transferred to PVDF membranes and immunoblotted for the presence of TSPO. In brief, membranes were blocked using 5% non-fat dry milk in tris buffered saline containing 0.2% Tween 20 (TBST) and incubated with rabbit anti-TSPO monoclonal antibody (1:1,000; Abcam, EPR5384) and control mouse anti-actin affinity purified polyclonal antibody (1:1,000; LI-COR). Detection was performed by incubation with IRDye® 800 conjugated goat anti-rabbit IgG and IRDye® 700 conjugated goat anti-mouse IgG secondary antibodies followed by imaging using a laser fluorescence scanner (LI-COR) for simultaneous detection of the two emission wavelengths in the same blot.
Induction of acute UC was performed using a dextran sodium sulfate (DSS) model (Okayasu et al., 1990). This model involved the oral administration of DSS (36–50 kDa, MP Biomedicals) that compromises mucosal barrier function in treated mice. For the experimental treatment groups, Tspofl/fl and Tspo−/− mice received DSS dissolved in autoclaved drinking water to a 2.5% solution (w/v) in negative pressure bottles ad libitum for 7 days. For the control groups, Tspofl/fl and Tspo−/− mice continued to receive regular drinking water for the same time period. For both the treatment and control groups of both genotypes, phenotypic recordings were performed daily. This included body weight measurements, stool consistency recordings, and scoring the phenotypic severity. Criteria used for stool scoring system were: 0—Normal appearance, 1—Observation of soft feces or pink color in anus, 2—Mild rectal bleeding, or blood observed only in feces, 3—Moderate rectal bleeding, 4—Severe rectal bleeding. Criteria used for posture scoring system were: 0—Normal appearance, 1—Slight hunched posturing, 2—Moderate hunching, reduced mobility, 3—Severe hunching, very little mobility. Criteria used for scoring phenotypic severity were: 1—observation of soft feces or pink color in anus, indicative of irritation, 2—blood observed in feces, or mild rectal bleeding, 3—moderate rectal bleeding, 4—severe rectal bleeding. The phenotypic clinical scoring data was utilized to generate an average stool score and average posturing score for each group. For sample collection, mice were euthanized for tissue collection for histopathology at the end of the 7-day treatment period; separate experimental runs were performed for collections at 0 (baseline, prior to any treatment), 4- and 7-day treatment periods for gene expression analyses.
At the end of each study, colons (the complete colorectal region) were collected after euthanasia and flushed with ice-cold phosphate-buffered saline from proximal to distal to remove fecal matter. Their length was measured using digital calipers. Colons were then dissected along the long axis to open up the lumen for gross pathological examination of the mucosa (hyperemia, petechial hemorrhage, punctate to broad-based ulcers of various sizes). Tissues were then either fixed for histopathology, or snap frozen in liquid nitrogen and held at -80°C before further processing for gene expression assays.
Full lengths of the colons were prepared for fixation by rolling from distal to proximal as a “Swiss roll” with the mucosa facing inside, a previously described method to examine the full length of tissue in sections (Whittem et al., 2010). The rolled tissue was fixed in place with 4% formaldehyde in phosphate buffered saline for 48 h. Tissues were then transferred to 70% ethanol for 24 h and taken through steps of dehydration and embedded in paraffin. Sections (4 µm thick) were cut using a microtome, and hematoxylin and eosin staining was performed to visualize tissue morphology using standard histological methods, also previously described (Morohaku et al., 2013; 2014a). Slides were studied under a light microscope (DM1000-LED, Leica), and images were acquired using a HD camera (ICC50HD, Leica); full slides were also scanned at ×40 magnification using the Aperio CS2 Scanscope (Leica). Image analysis was performed to quantify proximal distance of ulcerated tissue showing epithelial loss. In brief, lengthwise measurements of colonic epithelial pathology (from distal to proximal) and total colon length were made from scanned histopathology images after calibration using Adobe Illustrator. These values were used to generate a ratio of affected to total colon length for comparison between the groups.
After deparaffinization and rehydration (as above), sections were subjected to antigen retrieval using 0.01 M citrate buffer. Non-specific binding was blocked using 5% normal goat serum, then samples were incubated with anti-TSPO antibody (1∶200; Abcam, EPR5384) in 1% BSA in PBS overnight at 4°C. After incubation, slides were washed in PBS and incubated with pHRP-conjugated anti-rabbit secondary antibody, and processed using the DAB chemistry to visualize positive staining as previously described (Morohaku et al., 2013). To highlight morphology, slides were counterstained for a weak hematoxylin background. Images were acquired as described for histopathology.
Total RNA was extracted from the colon (distal 1.5 cm) from Tspo−/− and Tspofl/fl treatments and controls using TRIzolTM Reagent (Life Technologies), after tissue disruption (Bead beater, Biospec). Extracted total RNA was further purified using lithium chloride (Viennois et al., 2013), as we confirmed that DSS inhibited reverse transcription for qPCR amplification of cDNA from tissues as previously described (Kerr et al., 2012). In brief, 0.1 volume of 8M LiCl was added to total RNA, and incubated at −20°C for 2 h. After incubation, samples were immediately centrifuged for 30 min to pellet total RNA at 14,000 x g at 4°C. After discarding the supernatant, the pellet was washed with 70% ethanol and air dried before resuspension in RNAse-free water. RNA concentration was quantified using a spectrophotometer (NanoPhotometer® Pearl, Implen). Reverse transcription of 1 µg of total RNA was carried out using MultiscribeTM reverse transcriptase (Thermo Fischer Scientific). Validated TaqMan™ gene expression assays (Applied Biosystems) and intron-spanning primers for SYBR® green detection were used for quantitative PCR to estimate levels of Krt18, Cd34, Mcp6, Cd11c, F4/80, Mcp1, Tnfα, Lgr5, Ascl2, Cdh1, Ndrg2, cMyc, Ccnd1, Cdx2, Socs3, Cpt1a, Acadm, Acadl, Hadha, Cd36, FpnI, Hmox, and Nrf2. All expressions were normalized to an internal control gene Gapdh or Tbp. Primer probe sets and sequences are provided in Supplementary Table S1. Relative fold-change between Tspo−/− and Tspofl/fl mice was calculated using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Means for the posturing and stool scores were compared between genotypes using a Student’s t-test. Colon length and daily body weight changes were compared using repeated measures ANOVA and individual timepoints were compared using Student’s two-tailed t-tests. Histopathological colon length was analyzed by first calculating a ratio of affected to normal colonic tissue for each mouse after 7 days of DSS treatment, and comparing the ratios from Tspo−/− and Tspofl/fl groups using a two-proportions z-test. Gene expression differences between Tspo−/− and Tspofl/fl groups at the different timepoints were compared using Student’s two-tailed t-tests. For all analyses p < 0.05 was considered significant.
Deletion of TSPO was confirmed in Tspo−/− mice used in these studies using both Western blots and immunolocalization in the colon (Figure 1); expression of TSPO was observed in the epithelium, with noted absence in submucosal and muscular layers. In Tspo−/− mice, clinical signs and pathology after DSS-induced UC were significantly more severe compared to Tspofl/fl mice. Tspo−/− mice with UC exhibited significantly worse external clinical signs (phenotypic scores: posture and stool) compared to Tspofl/fl mice (Figures 2A,B). There was a significantly higher degree of rectal bleeding and diarrhea, decreased movement, and hunched posturing observed in Tspo−/− mice. Body weight loss, a well-studied indicator of disease severity in DSS-induced UC, was significantly higher from days 4 through 7 in Tspo−/− mice compared to Tspofl/fl mice (Figure 2C). On postmortem gross pathology, Tspo−/− mice exhibited subjectively more petechiae, ecchymoses, and ulceration of the colonic mucosa compared to Tspofl/fl mice (Figure 2D). Tspo−/− mice also exhibited significantly greater colonic shortening, indicative of the severity of colitis, compared to Tspofl/fl mice (Figure 2E). There was no significant difference in colon lengths between male and female mice within their respective cohorts.
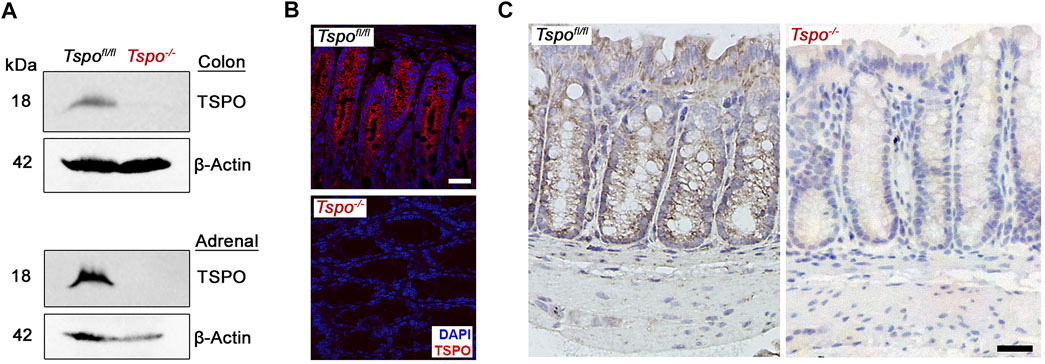
FIGURE 1. TSPO expression in Tspofl/fl and Tspo−/− colons. (A) Western blot showing specific recognition of TSPO in Tspofl/fl but not in Tspo−/− colons; adrenals that show high expression of TSPO are presented for comparison. In the same lanes, β-actin detection was used to indicate protein loading (control). (B–C) Immunofluorescence and immunohistochemistry images showing TSPO expression in Tspofl/fl but not in Tspo−/− colons. Expression of TSPO can be observed localized to the mucosal layer but not in the submucosal or muscular layers of the colon wall. (Scale bar 30 µm).
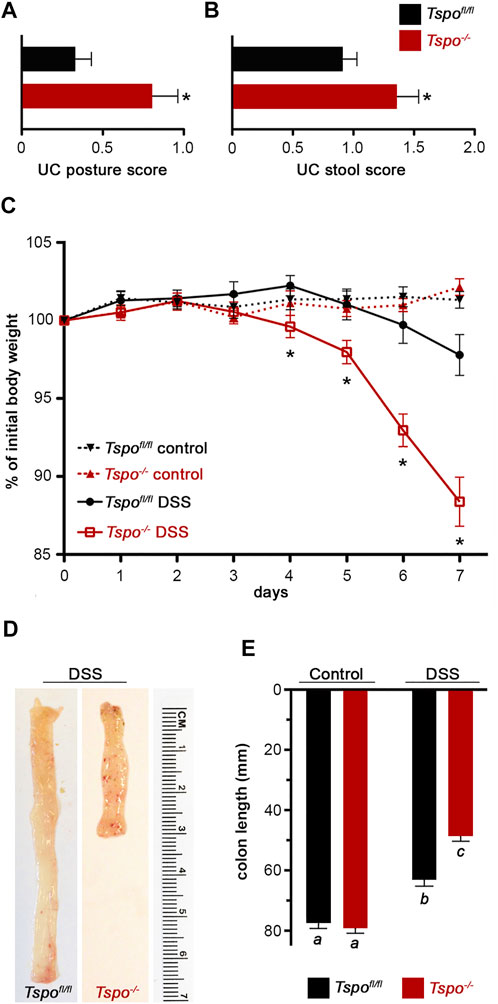
FIGURE 2. Exacerbated clinical signs and gross pathology of UC in Tspo−/− mice. (A) Severity of UC assessed by posturing scoring, days 5–7 of DSS in Tspo−/− and Tspofl/fl mice (n = 17 per group; p = 0.0053). Scoring: 0, normal behavior; 1, mild hunched posture; 2, hunched posture and reduced movement. (B) Severity of UC assessed by stool score, days 3–7 of DSS in Tspo−/− and Tspofl/fl mice (n = 17 per group; p = 0.03). Scoring: 0, normal stool; 1, soft feces, pink anus; 2, bloody feces, mild rectal bleeding; 3, moderate rectal bleeding; 4, diarrhea, severe rectal bleeding. (C) Body weight changes during UC in Tspo−/− mice indicated a significantly higher weight loss compared to Tspofl/fl mice. Temporal changes over the 7-day DSS treatment graphed as percentage of initial body weight in Tspo−/− and Tspofl/fl mice (n = 21 per group; ANOVA p=<0.0001). Body weights at Day 4 (p = 0.0328), Day 5 (p = 0.0193), Day 6 (p < 0.0001), and Day 7 (p < 0.0001), were significantly different. (D) Representative images of colons from Tspofl/fl and Tspo−/− mice after DSS-induced colitis, showing differences in length and the presence of petechiation (red hemorrhagic spots) on the mucosal surface indicative of inflammatory severity. (E) Average colon lengths which correlate with severity of UC was significantly shorter in Tspo−/− mice than Tspofl/fl mice after 7 days of DSS treatment (n = 16–21 per group; p < 0.0001).
Histopathological lesions were consistently more severe after DSS-induced UC in Tspo−/− mice compared to Tspofl/fl mice. Extremely severe colonic pathologies were noted with UC in Tspo−/− mice compared to Tspofl/fl mice (Figure 3A). These included diffuse edema, immune infiltrations, erosion and ulceration of the epithelium, and loss of crypt and goblet cells; progression of ulceration occurred continuously from distal to proximal and correlated with severity of disease. In evaluating the proximal extent of epithelial damage as an indicator of disease severity between the two genotypes, lengthwise measurements of colonic epithelial damage (from distal to proximal) indicated a substantial increase in ulcerative pathology in Tspo−/− mice compared to Tspofl/fl mice (Figure 3B). Based on the ratio of affected:total colon length as an indicator of disease severity, Tspo−/− mice demonstrated a much more proximal extent of epithelial damage (ratio = 0.44), while colonic pathology was limited to the distal colon in Tspofl/fl mice (ratio = 0.17). Based on evaluation of Krt18 gene expression as a quantitative indicator of colonic epithelial cells, Tspo−/− mice showed significantly more severe epithelial loss (4-fold) following DSS-induced UC treatment compared to Tspofl/fl mice (Figure 3C). This finding indicated a substantial structural loss of epithelial barrier integrity in Tspo−/− mice compared to Tspofl/fl mice, potentially increasing microbial infiltration and exacerbating associated pathologies.
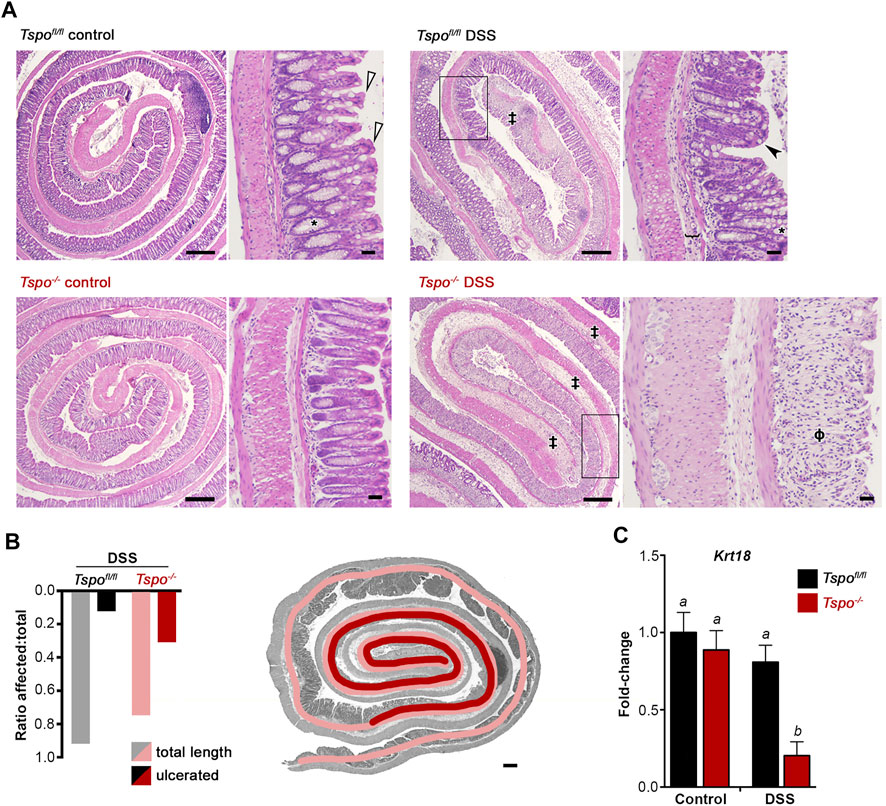
FIGURE 3. Histopathology and epithelial loss indicate severe inflammation with UC in Tspo−/− mice. (A) Histopathological images showing colon rolls (clockwise: inside distal to outside proximal), from control colons and 7-day DSS-induced UC colons. Representative images are shown for both Tspo−/− and Tspofl/fl colons. Control Tspo−/− and Tspofl/fl colons were histologically indistinguishable. Normal morphology consists of basally-located goblet cells (*), a narrow submucosa and thin muscularis mucosa, cuboidal to columnar mature absorptive epithelial cells (white arrowhead). In both UC cohorts, the pathology was most severe at the distal colon, consistent with the pathogenesis of UC. DSS-treated Tspofl/fl animals exhibited segmental moderate UC, with the distal colon demonstrating moderate edema, loss of crypt and goblet cells, moderate to severe inflammation, and erosion and ulceration of the colonic epithelium (‡). In the mid-colon (black box), Tspofl/fl colons showed less severe pathology, characterized by mild submucosal edema (bracket), and mild mononuclear inflammation. The mucosa displays shortened, immature epithelial cells (black arrowhead). Partial loss of the colonic crypts is present, as well as goblet cell hyperplasia and altered distribution of goblet cells towards the apex of the crypts. DSS-treated Tspo−/− animals developed more diffuse UC, affecting the entirety of the distal colon and extending into the mid-colon (black box) and proximal colon. These animals exhibited severe, diffuse colonic edema, extensive crypt and goblet cell loss (ϕ), muscular hypertrophy, epithelial ulceration and erosion, and severe diffuse mononuclear inflammation. (Low magnification scale bar: 300 μm; high magnification scale bar: 30 µm). (B) In quantitating the regional degree of colonic epithelial ulceration and erosion due to UC, the ratio of affected:total colon length demonstrated significantly greater damage in Tspo−/− colons than Tspofl/fl colons (0.44 vs. 0.17 respectively; p = 0.0039, Z = 2.885, n = 9 per group). Representative histopathology image with an overlay of color-coded measurement lines is shown to demonstrate this quantification of disease extent. (C) Expression of Krt18, an intestinal epithelial marker, in the distal colon of Tspo−/− and Tspofl/fl mice at baseline and at day-7 of DSS-induced UC; significant down-regulation of Krt18 indicative of epithelial loss is observed only with UC in Tspo−/− colons (n = 7 per group; p = 0.0011).
The involvement of immune cells in DSS-induced colitis between Tspo−/− and Tspofl/fl mice was evaluated via a variety of markers. At baseline, expression of Cd34, a marker of mature mast cells, was significantly elevated in Tspo−/− mice compared to Tspofl/fl mice, perhaps indicating a larger colonic mast cell population (Figure 4A). However, Cd34 levels were not different between Tspo−/− and Tspofl/fl colons during the course of DSS-induced UC. Mast cell tryptase (Mcp6) levels were elevated at both baseline and after DSS-induced colitis in Tspo−/− mice compared to Tspofl/fl mice (Figure 4B), indicative of increased activation of mast cells in Tspo−/− colons. Expression of Cd11c, a marker of macrophages and dendritic cells, was increased at day-4 of UC colitis in Tspo−/− compared to Tspofl/fl colons (Figure 4C). This was also indicative of differences in Cd11c expression across different UC timepoints within each genotype, in that Tspo−/− colons had an earlier peak in Cd11c expression, with significant up-regulation at day-4 compared to day-0 (p = 0.0132). In contrast, Tspofl/fl colons showed no significant difference between baseline and day-4 levels of Cd11c (p = 0.0706). Expression of F4/80, the macrophage surface adhesion protein, as well as Mcp1, indicative of macrophage activation, and Tnfα, a pro-inflammatory cytokine, progressively increased throughout DSS-induced colitis, with no significant difference in levels between Tspo−/− and Tspofl/fl colons (Figures 4D–F).
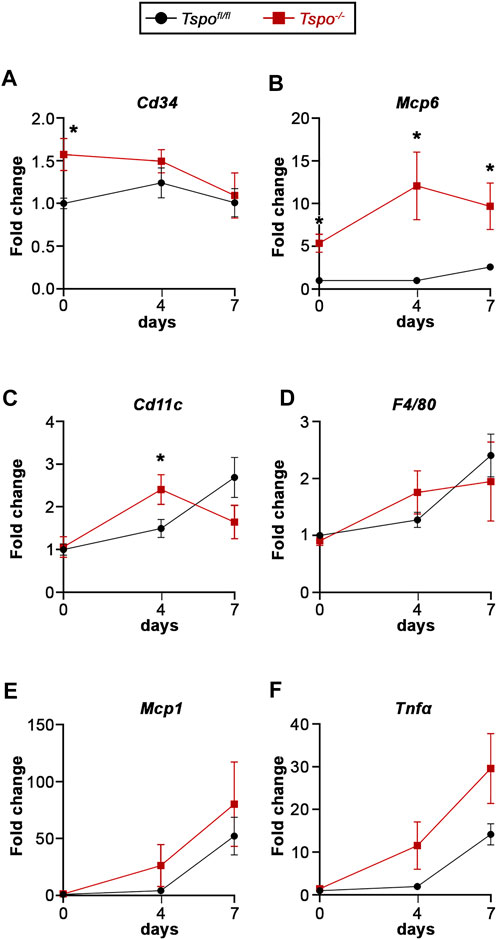
FIGURE 4. Gene expression indicative of inflammation with UC in Tspo−/− and Tspofl/fl mice. (A) Expression of Cd34, a surface marker for mast cells, was significantly higher at baseline (day-0) in Tspo−/− colons compared to Tspofl/fl colons (p = 0.0077), but not at the later UC timepoints. (B) Expression of Mcp6, a mast cell tryptase, was significantly up-regulated at baseline (p = 0.0010) and at all UC timepoints in Tspo−/− colons compared to Tspofl/fl colons (p ≤ 0.0111). (C) Expression of Cd11c, a dendritic cell transmembrane protein, showed significant up-regulation at day-4 of UC in Tspo−/− colons compared to Tspofl/fl colons (p < 0.0392). (D) Expression of F4/80, a surface adhesion protein also observed in macrophages, appeared up-regulated in UC but not significantly different between the two genotypes. (E) Expression of Mcp1, a macrophage activation marker, appeared up-regulated in UC, but not significantly different between the two genotypes. (F) Expression of Tnfα, a mast cell- and macrophage-released proinflammatory cytokine, was up-regulated with progression of UC, with significant increases between baseline and day-7 in both Tspo−/− (p = 0.0089) and Tspofl/fl (p = 0.0002) colons. Although there was a trend showing relative upregulation in Tspo−/− colons compared to Tspofl/fl colons at day-4 (p = 0.0652) and day-7 (p = 0.0638), it did not reach statistical significance. For all gene expression changes, n = 5-7 per group; significance indicated as *p < 0.05.
Additional genes were examined to evaluate baseline differences in expression between Tspo−/− and Tspofl/fl colons. There was no significant difference in expression of Leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5, a receptor seen in epithelial stem cells) and Achaete-scute-like 2 (Ascl2, a transcription factor that manages stem cell fate), suggesting that undifferentiated intestinal stem cell populations are similar between Tspo−/− and Tspofl/fl colons (Figure 5A). Expression of Cadherin 1 (Cdh1, an epithelial tight junctional protein) and N-myc downstream regulated gene 2 (Ndrg2, a regulator of gut epithelial permeability) also remained unchanged between Tspo−/− and Tspofl/fl colons, suggesting no obvious perturbations to the setup of epithelial integrity (Figure 5B). Of the proliferation markers, expression of cellular myelocytomatosis (cMyc, a cell proliferation indicator) was not different between Tspo−/− and Tspofl/fl colons; however, expression of Cyclin D1 (Ccnd1, relevant to cell cycle progression) was significantly decreased in Tspo−/− colons. This suggested that the rate of intestinal epithelial turnover could be modestly decreased in Tspo−/− colons (Figure 5C). Expression of Caudal-type homeobox 2 (Cdx2, an epithelium-restricted transcription factor in the intestine), known to be responsible for intestinal identity and regulating immune cell infiltration, was significantly up-regulated in Tspo−/− compared to Tspofl/fl colons (Figure 4D). Expression of Suppressor of cytokine signaling 3 (Socs3, a regulator of cytokine signaling and inflammation), albeit not different between Tspo−/− and Tspofl/fl colons, showed a trend of increased baseline expression in Tspo−/− colons (Figure 5D).
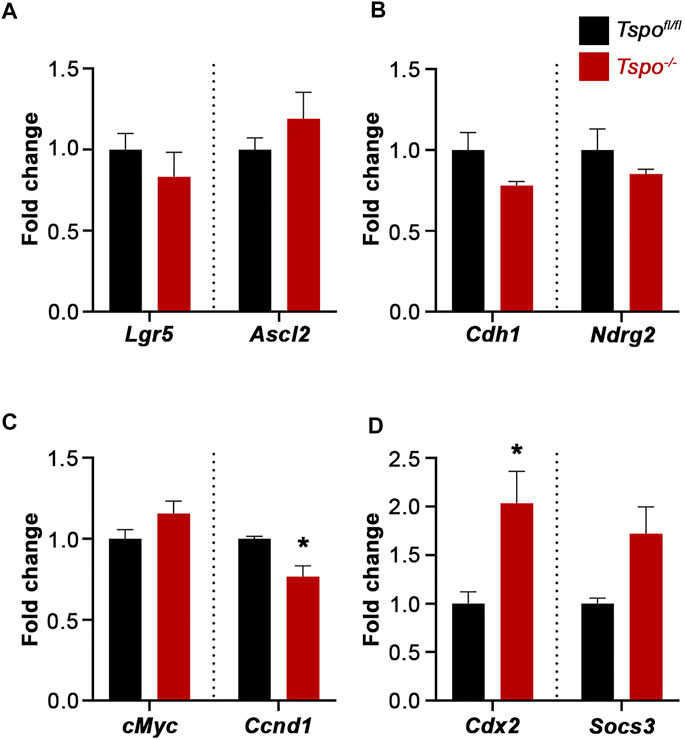
FIGURE 5. Baseline gene expression evaluating colonic epithelial functions in Tspo−/− mice. (A) Expression of Lgr5 and Ascl2, indicative of intestinal stem cell identity, showed no difference between Tspo−/− and Tspofl/fl colons. (B) Expression of Cdh1 and Ndrg2, indicative of adhesion/epithelial integrity, showed no difference between Tspo−/− and Tspofl/fl colons. (C) Expression of cMyc and Ccnd1, Wnt targets indicative of proliferation showed divergent effects; cMyc showed no difference between Tspo−/− and Tspofl/fl colons, whereas Ccnd1 was significantly downregulated in Tspo−/− colons (p = 0.0280). (D) Expression of Cdx2, transcription factor associated with intestinal epithelial development/identity, was significantly higher in Tspo−/− colons compared to Tspofl/fl colons (p = 0.044). Expression of Socs3, a regulator of cytokine signaling in inflammation, was higher on an average in Tspo−/− colons compared to Tspofl/fl colons, but did not reach statistical significance (p = 0.0625). Significance indicated as *p < 0.05.
In examining genes associated with an intestinal oxidative stress response: Cd36 (involved in the uptake of oxidized lipids), Ferroportin 1 (Fpn1, regulates iron homeostasis and the redox state), Heme oxygenase 1 (Hmox1, catalyzes degradation of heme and a sensitive indicator of oxidative stress), and Nuclear factor erythroid 2-related factor 2 (Nrf2, a transcriptional regulator of antioxidant and cellular protective genes) were not significantly affected in baseline Tspo−/− compared to Tspofl/fl colons (Figure 6A). Expression of genes associated with mitochondrial fatty acid oxidation indicated that Acyl-CoA dehydrogenase medium chain (Acadm, catalyzes the initial step in the mitochondrial β-oxidation of medium chain fatty acids), was significantly increased in Tspo−/− compared to Tspofl/fl colons (Figure 6B). Other genes involved in this pathway such as Carnitine palmitoyl transferase (Cpt1a, essential for transport of long-chain fatty acids into the mitochondria for fatty acid oxidation), Acyl-CoA dehydrogenase long-chain (Acadl, catalyzes the initial step in the mitochondrial β-oxidation of long chain fatty acids) and Hydroxyacyl-CoA dehydrogenase subunit α (Hadha, catalyzes the last three steps in the mitochondrial β-oxidation of the long-chain fatty acids) were not significantly different between Tspo−/− and Tspofl/fl colons (Figure 6B).
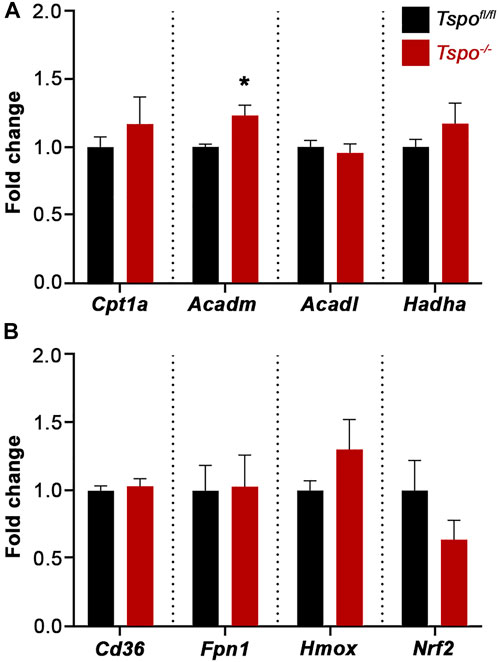
FIGURE 6. Baseline gene expression evaluating regulators of fatty acid oxidation and oxidative stress in Tspo−/− colons. (A) Expression of elements that indicate and/or regulate oxidative stress such as Cd36, Fpn1, Hmox, and Nrf2, showed no difference between Tspo−/− and Tspofl/fl colons. (B) Expression of Acadm, a mitochondrial enzyme essential to metabolize medium chain fatty acids, was significantly higher in Tspo−/− colons compared to Tspofl/fl colons (p = 0.0425). Expression of other elements associated with mitochondrial fatty acid β-oxidation such as Cpt1a, Acadl, and Hadha, showed no difference between Tspo−/− and Tspofl/fl colons. Significance indicated as *p < 0.05.
In using Tspo−/− mice to investigate the mechanism and test the foundational value of TSPO-targeting pharmacological agents in UC, we uncover a functional link that corroborates TSPO involvement in the pathological basis of the disease. The DSS-induced colitis model has been shown to primarily induce epithelial barrier disruption, with inflammation only secondary to loss of epithelial integrity (Cooper et al., 1993; Laroui et al., 2012). This model yields colonic pathology that closely mimics that of human patients with UC (Wirtz et al., 2007). In this study, TSPO deficiency substantially exacerbated the inflammatory pathology associated with DSS-induced colitis. Although the impact of local versus global TSPO loss-of-function remains difficult to delineate, the pathologic findings provide conclusive evidence that effects specific to TSPO can influence inflammatory injury in colon. Physiologic TSPO function appears to offer a significant level of protection against colonic insults that might induce inflammation and injury. As TSPO expression is observed both in the intestinal epithelium and immune cells, we believe the exacerbated UC in Tspo−/− mice results from a combination of vulnerability in the epithelial barrier and dysregulation in specific immune responses.
The exacerbated UC phenotype in Tspo−/− mice became obvious as our pilot experiments to generate a colon cancer model using DSS and azoxymethane (Thaker et al., 2012) resulted in complete mortality in the Tspo−/− group during the second cycle of treatment (unpublished observations). During the 7-day DSS treatment in this study, we observed progressively aggravated clinical signs and body weight loss in Tspo−/− mice compared to Tspofl/fl controls. The aggravated response in Tspo−/− mice was consistent with known pathological parameters of severity such as progressive, continuous ulceration of the colonic epithelium, edema, and shortened colon length (Okayasu et al., 1990). The severity of UC pathology Tspo−/− mice was also indicative of a degree of unrecoverable loss of intestinal structure.
As TSPO is expressed in the colonic epithelial cells, we first considered the possibility that specific loss of function in this cell type might be linked to a local predisposition to injury in Tspo−/− colons. Our finding that Krt18, the type I intermediate filament associated with the colonic epithelium, significantly decreases with UC in Tspo−/− mice was a clear indication of higher levels of colonic epithelial loss compared to Tspofl/fl cohorts. Epithelial denudation has been correlated to the severity of DSS-induced UC in human patients (Corfe et al., 2015), and in our study, indicated the increased severity of UC pathology in Tspo−/− mice. In addition to serving as a marker, mutations in Krt18 that disrupt the intermediate filament networks have also been shown to functionally increase epithelial permeability in human colonocytes (Zupancic et al., 2014). Keratin downregulation has also shown to dramatically increase susceptibility to cell death mediated by inflammatory cytokines such as TNF⍺ (Caulin et al., 2000). Supporting our gene expression results, substantial epithelial loss could be visualized in histopathology of Tspo−/− colons. Excessive inflammation seen with UC in Tspo−/− mice compared to Tspofl/fl cohorts could be a consequence of amplified disruption of the epithelial barrier.
High-affinity benzodiazepine binding sites indicative of TSPO expression have also been identified in mast cells (Miller et al., 1988). Rats deficient in mast cells fail to develop clinical signs of DSS-induced colitis, indicating a crucial role for mast cells in UC pathogenesis (Araki et al., 2000). Moreover, it has been demonstrated that mast cell activation, identified by tryptase levels, has an essential proinflammatory role in triggering DSS-induced UC (Hamilton et al., 2011). Mast cell tryptase can also act as a trigger for other mediators of inflammation (Compton et al., 1998; Huang et al., 1998), potentially contributing to UC pathology. Functionally, different drugs that bind TSPO have been shown to have varying effects on mast cell degranulation (Miller et al., 1988; Suzuki-Nishimura et al., 1989; Sano et al., 1990; Yousefi et al., 2013). Investigating whether Tspo deletion affected the mast cell response, we identified significant upregulation of murine Mcp6 (which is similar to the human mast cell hTryptase-β) in Tspo−/− colons. This can be considered an activation response, as Cd34 expression indicative of mature mast cells (Drew et al., 2002), albeit increased in Tspo−/− colons, was not significantly different between subsequent (day-4 and day-7) timepoints between the two genotypes. It has been shown that Mcp6 deficiency (Hamilton et al., 2011), or tryptase inhibition (Tremaine et al., 2002; Liu et al., 2021), could reduce UC pathology and intestinal fibrosis. In parallel, degranulation of mast cells has also been associated with the release of proinflammatory TNF⍺ (Gordon and Galli, 1991; Bischoff et al., 1999). We observed higher levels of Tnfα expression during UC in Tspo−/− colons, although this difference was not statistically significant. In forms of mast cell driven colitis, TNF⍺-driven effects have been demonstrated to be crucial for UC pathology (Rijnierse et al., 2006). It has also been shown that TNFα and other proinflammatory cytokines could persist in the colon at high levels for at least 14 days after DSS withdrawal (Yan et al., 2009). Therefore, protracted effects on inflammation could also be anticipated in Tspo−/− mice driven by both the inflammatory milieu and the degree of injury sustained.
We also observed that Cd11c levels peaked at day-4 of UC in Tspo−/− colons, which was significantly different and earlier than that measured for UC in Tspofl/fl colons. Cd11c is expressed on the surface of monocytes, macrophages, and dendritic cells in the intestinal mucosa; these cells are important players in immune recognition and tolerance to the gut microbiota (Bain and Mowat, 2014). Increased infiltration of CD11c+ macrophages has been demonstrated to trigger mucosal injury in UC (Bernardo et al., 2018; Fuke et al., 2018), while reduction in CD11c+ macrophages ameliorated mucosal pathology (Arnold et al., 2015). Earlier up-regulation of Cd11c in Tspo−/− animals may reflect and/or influence the rapid onset and severity of colonic inflammation. In addition, CD11c+ phagocytes may also increase gut mucosal epithelial permeability (Ren et al., 2017), further contributing to colonic pathology through increased bacterial translocation across the gut barrier.
The upregulation of baseline colonic Cdx2 expression in Tspo−/− mice indicates that TSPO deficiency contributed to a homeostatic shift, perhaps compensating for decreased epithelial barrier function. Cdx2 gene-deleted heterozygous (Cdx2+/−) mice showed hypersensitivity to DSS-induced acute UC and increased permeability of the colonic epithelium (Calon et al., 2007). Cdx2 is known to control cell-cell adhesion and extracellular matrix composition/interactions (Lorentz et al., 1997); baseline Cdx2 upregulation may serve as a compensatory mechanism to offset increased vulnerability to epithelial permeability in Tspo−/− mice. High expression of TNFα, through NF-κB and p38 MAPK pathways, could downregulate Cdx2 (Coskun et al., 2014), suggesting that there might be significant cross-talk between epithelium-specific and different immune pathways.
There also exists cross-talk between Cdx2 and Wnt signaling, regulating the proliferative compartment of normal intestinal crypts (Guo et al., 2010). Our finding that the Wnt target Ccnd1 was decreased in Tspo−/− colons might indicate a lower capacity for epithelial proliferation and turnover, potentially affecting regeneration after ulcerative injury. However, based on similar Lgr5 and Ascl2 levels between Tspofl/fl and Tspo−/− colons, stem cell populations and adhesion molecule expression were not altered in Tspo−/− colons. Although Cd34 expression also believed to be associated with these adult stem cells (Stzepourginski et al., 2017), are higher at baseline in Tspo−/− colons, the differences are in all probability linked to mature mast cell numbers (consistent with higher baseline Mcp6 expression levels). Although stem cell populations are similar, given the progressive severity of UC pathology Tspo−/− mice, it is plausible that TSPO deficiency might also negatively impact epithelial recovery and functional restoration in the colon. This speculation is supported by pharmacology that TSPO binding drugs, PK11195 and Ro5-4864 could accelerate recovery after DSS-induced UC (Ostuni et al., 2010). Although the extent of TSPO-mediated action remains unclear, positive and negative outcomes in DSS-induced UC have been reported for relatively low-affinity TSPO binding molecules such as curcumin (Li et al., 2013; Liu et al., 2013; Guo et al., 2022) and retinoic acid (Oehlers et al., 2012; Li et al., 2013; Rampal et al., 2021).
As recent developments on TSPO function have indicated possible roles in regulation of mitochondrial fatty acid oxidation (Tu et al., 2016) and oxidative stress responses (Gatliff et al., 2014), we examined genes associated with these functions in Tspo−/− colons. The finding that Acadm is up-regulated in Tspo−/− colons is consistent with evidence that there exist subtle metabolic shifts with TSPO loss-of-function. In tissues active in lipid metabolism that show very high expression of TSPO, we have previously reported a functional up-regulation of mitochondrial fatty acid oxidation, and associated increase in gene expression of Cpt1a, Acadm, Acadl, and Hadha (Tu et al., 2016). Increases in Cpt1a, Acadl and Hadha were not observed in Tspo−/− colons; therefore, it is conceivable that the present study’s use of entire colons for gene expression assays, as opposed to just TSPO-expressing epithelial layers or mast cells, could have diluted any cell-type specific phenotypes. With regard to oxidative stress, direct evidence previously demonstrated that TSPO could stabilize mitochondrial architecture during inflammatory stress in colonic cells (Issop et al., 2016). We have previously observed that TSPO involvement in oxidative stress is cell-type dependent (Tu et al., 2016; Zhao et al., 2016). Although we did not observe any baseline perturbations in gene expression associated with oxidative stress responses in Tspo−/− colons, baseline expression may not reflect active inflammatory cell states associated with UC.
In summary, despite lack of a clear structure-function relationship for the different TSPO-mediated effects that are described in the literature (Selvaraj et al., 2015; Selvaraj and Stocco, 2015), and mechanisms that delineate TSPO pharmacology (Gavish et al., 1999; Gavish and Veenman, 2018), our findings in this Tspo−/− model demonstrates a protective effect for TSPO on the colonic epithelium and confirms that there might be potential for therapeutic interventions with ligands that enhance TSPO function. For example, the TSPO-binding benzodiazepine derivatives Ro5-4864 (4'-chlorodiazepam) and flunitrazepam, which were both shown to quickly suppress mast cell activation in vivo and in vitro (Yousefi et al., 2013), may be considered to be TSPO agonists. Understandably, similar actions albeit with differing efficacies, occur for these same drugs in the in vivo DSS-induced colitis model (Ostuni et al., 2010). The hypothesis that TSPO likely provides a dual therapeutic effect, involving both epithelial barrier-specific functions and immune-cell mediated pathways, is an attractive mechanism that merits more detailed investigation. Fundamentally, we uncover that TSPO deletion/loss-of-function, albeit inconsequential at a resting/healthy state, makes mice more vulnerable to insults. Future studies that dissect the precise molecular function of TSPO might help describe its physiological and pharmacological significance in UC and other inflammatory disease models.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee, Cornell University.
Conceptualization: VS; Methodology: IJ, AS, KM, MH, PK, and VS; Formal analysis: IJ; Investigation: IJ, AS, KM, MH, PK, and VS; Data curation: IJ; Resources: VS; Writing—original draft: IJ.; Writing—review editing: VS; Supervision: VS; Project administration: VS; Funding acquisition: IJ, VS All authors contributed to the article and approved the submitted version.
This work was supported by startup funds from the College of Agriculture and Life Sciences, Cornell University (to VS), a grant from the National Institutes of Health DK110059 (to VS), and a seed grant from the Center for Vertebrate Genomics at Cornell University. Undergraduate research was supported by a National Science Foundation Biology Research Fellowship for Underrepresented minorities at Cornell #0933921 (to IJ), and the Office of Academic Diversity Initiatives (OADI)—Biology Research Scholars Program (to IJ).
The authors thank Amy H. Zhao and Lan N. Tu (former graduate students in our group), for all the leadership and enthusiastic support provided to facilitate undergraduate research in the Selvaraj Laboratory of Integrative Physiology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2022.896951/full#supplementary-material
Airas L., Rissanen E., Rinne J. O. (2015). Imaging neuroinflammation in multiple sclerosis using TSPO-PET. Clin. Transl. Imaging 3, 461–473. doi:10.1007/s40336-015-0147-6
Araki Y., Andoh A., Fujiyama Y., Bamba T. (2000). Development of dextran sulphate sodium-induced experimental colitis is suppressed in genetically mast cell-deficient Ws/Ws rats. Clin. Exp. Immunol. 119, 264–269. doi:10.1046/J.1365-2249.2000.01094.X
Arnold I. C., Mathisen S., Schulthess J., Danne C., Hegazy A. N., Powrie F. (2015). CD11c+ monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. 92 (9), 352–363. doi:10.1038/mi.2015.65
Bain C. C., Mowat A. M. I. (2014). The monocyte-macrophage axis in the intestine. Cell. Immunol. 291, 41–48. doi:10.1016/J.CELLIMM.2014.03.012
Banati R. B., Middleton R. J., Chan R., Hatty C. R., Wai-Ying Kam W., Quin C., et al. (2014). Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 5, 5452. doi:10.1038/ncomms6452
Barron A. M., Garcia-Segura L. M., Caruso D., Jayaraman A., Lee J. W., Melcangi R. C., et al. (2013). Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 33, 8891–8897. doi:10.1523/JNEUROSCI.1350-13.2013
Bernardo D., Marin A. C., Fernández-Tomé S., Montalban-Arques A., Carrasco A., Tristán E., et al. (2018). Human intestinal pro-inflammatory CD11c high CCR2 + CX3CR1 + macrophages, but not their tolerogenic CD11c - CCR2 - CX3CR1 - counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. 11, 1114–1126. doi:10.1038/S41385-018-0030-7
Bernards N., Pottier G., Thézé B., Dollé F., Boisgard R. (2015). In vivo evaluation of inflammatory bowel disease with the aid of μPET and the translocator protein 18 kDa radioligand [18F]DPA-714. Mol. imaging Biol. 17, 67–75. doi:10.1007/S11307-014-0765-9
Betlazar C., Middleton R. J., Banati R., Liu G.-J. (2020). The translocator protein (TSPO) in mitochondrial bioenergetics and immune processes. Cells 9, 512. doi:10.3390/cells9020512
Bischoff S. C., Lorentz A., Schwengberg S., Weier G., Raab R., Manns M. P. (1999). Mast cells are an important cellular source of tumour necrosis factor in human intestinal tissue. Gut 44, 643–652. doi:10.1136/gut.44.5.643
Bordet T., Buisson B., Michaud M., Drouot C., Galéa P., Delaage P., et al. (2007). Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 322, 709–720. doi:10.1124/jpet.107.123000
Braestrup C., Squires R. F. (1977). Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc. Natl. Acad. Sci. U. S. A. 74, 3805–3809. doi:10.1073/pnas.74.9.3805
Calon A., Gross I., Lhermitte B., Martin E., Beck F., Duclos B., et al. (2007). Different effects of the Cdx1 and Cdx2 homeobox genes in a murine model of intestinal inflammation. Gut 56, 1688–1695. doi:10.1136/GUT.2007.125542
Caulin C., Ware C. F., Magin T. M., Oshima R. G. (2000). Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J. Cell Biol. 149, 17–22. doi:10.1083/JCB.149.1.17
Compton S. J., Cairns J. A., Holgate S. T., Walls A. F. (1998). The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: Tryptase induces expression of mRNA for IL-1 beta and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J. Immunol. 161, 1939–1946.
Cooper H. S., Murthy S. N. S., Shah R. S., Sedergran D. J. (1993). Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 69, 238–249. doi:10.1016/s0021-5198(19)41298-5
Corfe B. M., Majumdar D., Assadsangabi A., Marsh A. M. R., Cross S. S., Connolly J. B., et al. (2015). Inflammation decreases keratin level in ulcerative colitis; inadequate restoration associates with increased risk of colitis-associated cancer. BMJ Open Gastroenterol. 2, e000024. doi:10.1136/BMJGAST-2014-000024
Coskun M., Olsen A. K., Bzorek M., Holck S., Engel U. H., Nielsen O. H., et al. (2014). Involvement of CDX2 in the cross talk between TNF-α and Wnt signaling pathway in the colon cancer cell line Caco-2. Carcinogenesis 35, 1185–1192. doi:10.1093/CARCIN/BGU037
Daugherty D. J., Selvaraj V., Chechneva O. V., Liu X. B., Pleasure D. E., Deng W. (2013). A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol. Med. 5, 891–903. doi:10.1002/emmm.201202124
Drew E., Merkens H., Chelliah S., Doyonnas R., McNagny K. M. (2002). CD34 is a specific marker of mature murine mast cells. Exp. Hematol. 30, 1211–1218. doi:10.1016/S0301-472X(02)00890-1
Fujimura Y., Hwang P. M., Trout H., Kozloff L., Imaizumi M., Innis R. B., et al. (2008). Increased peripheral benzodiazepine receptors in arterial plaque of patients with atherosclerosis: An autoradiographic study with [3H]PK 11195. Atherosclerosis, 201:108–111. doi:10.1016/j.atherosclerosis.2008.02.032
Fuke N., Takagi T., Higashimura Y., Tsuji T., Umeda R., Mizushima K., et al. (2018). Lactobacillus brevis KB290 with vitamin A ameliorates murine intestinal inflammation associated with the increase of CD11c+ macrophage/cd103- dendritic cell ratio. Inflamm. Bowel Dis. 24, 317–331. doi:10.1093/IBD/IZX003
Gatliff J., East D., Crosby J., Abeti R., Harvey R., Craigen W., et al. (2014). TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 10, 2279–2296. doi:10.4161/15548627.2014.991665
Gavish M., Bachman I., Shoukrun R., Katz Y., Veenman L., Weisinger G., et al. (1999). Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 51, 629–650.
Gavish M., Veenman L. (2018). Regulation of mitochondrial, cellular, and organismal functions by TSPO. Adv. Pharmacol., 82:103–136. doi:10.1016/bs.apha.2017.09.004
Giatti S., Pesaresi M., Cavaletti G., Bianchi R., Carozzi V., Lombardi R., et al. (2009). Neuroprotective effects of a ligand of translocator protein-18kDa (Ro5-4864) in experimental diabetic neuropathy. Neuroscience 164, 520–529. doi:10.1016/j.neuroscience.2009.08.005
Gordon J. R., Galli S. J. (1991). Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J. Exp. Med. 174, 103–107. doi:10.1084/jem.174.1.103
Guo J., Zhang Y. Y., Sun M., Xu L. f. (2022). Therapeutic potential of curcumin in a rat model of dextran sulfate sodium-induced ulcerative colitis by regulating the balance of treg/Th17 cells. Inflammation. doi:10.1007/S10753-022-01678-1
Guo R. J., Funakoshi S., Lee H. H., Kong J., Lynch J. P. (2010). The intestine-specific transcription factor Cdx2 inhibits β-catenin/TCF transcriptional activity by disrupting the β-catenin–TCF protein complex. Carcinogenesis 31, 159–166. doi:10.1093/CARCIN/BGP213
Hamilton M. J., Sinnamon M. J., Lyng G. D., Glickman J. N., Wang X., Xing W., et al. (2011). Essential role for mast cell tryptase in acute experimental colitis. Proc. Natl. Acad. Sci. U. S. A. 108, 290–295. doi:10.1073/pnas.1005758108
Huang C., Friend D. S., Qiu W.-T., Wong G. W., Morales G., Hunt J., et al. (1998). Induction of a selective and persistent extravasation of neutrophils into the peritoneal cavity by tryptase mouse mast cell protease 6. J. Immunol. 160, 1910–1919.
Issop L., Ostuni M. A., Lee S., Laforge M., Péranzi G., Rustin P., et al. (2016). Translocator protein-mediated stabilization of mitochondrial architecture during inflammation stress in colonic cells. PLoS One 11, e0152919. doi:10.1371/JOURNAL.PONE.0152919
Katz Y., Eitan A., Amiri Z., Gavish M. (1988). Dramatic increase in peripheral benzodiazepine binding sites in human colonic adenocarcinoma as compared to normal colon. Eur. J. Pharmacol. 148, 483–484. doi:10.1016/0014-2999(88)90135-5
Katz Y., Ettari A., Gavish M. (1990). Increase in peripheral benzodiazepine binding sites in colonic adenocarcinoma. Oncology 47, 139–142. doi:10.1159/000226806
Kawahara Y., Mitsui K., Niwa T., Morimoto N., Kawaharada S., Katsumata S. (2018). Translocator protein 18kDa antagonist ameliorates stress-induced stool abnormality and abdominal pain in rodent stress models. Neurogastroenterol. Motil. 30, e13425. doi:10.1111/NMO.13425
Kerr T. A., Ciorba M. A., Matsumoto H., Davis V. R. T., Luo J., Kennedy S., et al. (2012). Dextran sodium sulfate inhibition of real-time polymerase chain reaction amplification: A poly-A purification solution. Inflamm. Bowel Dis. 18, 344–348. doi:10.1002/ibd.21763
Kim S., Kim N., Park S., Jeon Y., Lee J., Yoo S. J., et al. (2019). Tanycytic TSPO inhibition induces lipophagy to regulate lipid metabolism and improve energy balance. Autophagy 16, 1200–1220. doi:10.1080/15548627.2019.1659616
Koganti P. P., Selvaraj V. (2020). Lack of adrenal TSPO/PBR expression in hamsters reinforces correlation to triglyceride metabolism. J. Endocrinol. 247, 1–10. doi:10.1530/JOE-20-0189
Königsrainer I., Vogel U. F., Beckert S., Sotlar K., Coerper S., Braun A., et al. (2007). Increased translocator protein (TSPO) mRNA levels in colon but not in rectum carcinoma. Eur. Surg. Res. 39, 359–363. doi:10.1159/000106380
Kurtys E., Doorduin J., Eisel U. L. M., Dierckx R. A. J. O., de Vries E. F. J. (2017). Evaluating [11C]PBR28 PET for monitoring gut and brain inflammation in a rat model of chemically induced colitis. Mol. Imaging Biol. 19, 68–76. doi:10.1007/s11307-016-0979-0
Laemmli U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi:10.1038/227680a0
Laroui H., Ingersoll S. A., Liu H. C., Baker M. T., Ayyadurai S., Charania M. A., et al. (2012). Dextran sodium sulfate (dss) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS One 7, e32084. doi:10.1371/journal.pone.0032084
Leduc M. S., Hageman R. S., Verdugo R. A., Tsaih S. W., Walsh K., Churchill G. A., et al. (2011). Integration of QTL and bioinformatic tools to identify candidate genes for triglycerides in mice. J. Lipid Res. 52, 1672–1682. doi:10.1194/jlr.M011130
Li F., Xia Y., Meiler J., Ferguson-Miller S. (2013). Characterization and modeling of the oligomeric state and ligand binding behavior of purified translocator protein 18 kDa from Rhodobacter sphaeroides. Biochemistry 52, 5884–5899. doi:10.1021/BI400431T/SUPPL_FILE/BI400431T_SI_001
Li L., Chen T., Stanton J. D., Sueyoshi T., Negishi M., Wang H. (2008). The peripheral benzodiazepine receptor ligand 1-(2-chlorophenyl-methylpropyl)-3-isoquinoline-carboxamide is a novel antagonist of human constitutive androstane receptor. Mol. Pharmacol. 74, 443–453. doi:10.1124/mol.108.046656
Li M., Ren H., Sheth K. N., Shi F. D., Liu Q. (2017). A TSPO ligand attenuates brain injury after intracerebral hemorrhage. FASEB J. 31, 3278–3287. doi:10.1096/fj.201601377RR
Liu B., Yang M. Q., Yu T. Y., Yin Y. Y., Liu Y., Wang X. D., et al. (2021). Mast cell tryptase promotes inflammatory bowel disease-induced intestinal fibrosis. Inflamm. Bowel Dis. 27, 242–255. doi:10.1093/IBD/IZAA125
Liu G. J., Middleton R. J., Kam W. W. Y., Chin D. Y., Hatty C. R., Chan R. H. Y., et al. (2017). Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 16, 436–447. doi:10.1080/15384101.2017.1281477
Liu L., Liu Y. L., Liu G. X., Chen X., Yang K., Yang Y. X., et al. (2013). Curcumin ameliorates dextran sulfate sodium-induced experimental colitis by blocking STAT3 signaling pathway. Int. Immunopharmacol. 17, 314–320. doi:10.1016/J.INTIMP.2013.06.020
Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi:10.1006/meth.2001.1262
Lorentz O., Duluc I., De Arcangelis A., Simon-Assmann P., Kedinger M., Freund J. N. (1997). Key role of the Cdx2 homeobox gene in extracellular matrix–mediated intestinal cell differentiation. J. Cell Biol. 139, 1553–1565. doi:10.1083/JCB.139.6.1553
Luc Do Rego J., Vaudry D., Vaudry H. (2015). The non-benzodiazepine anxiolytic drug etifoxine causes a rapid, receptor-independent stimulation of neurosteroid biosynthesis. PLoS One 10, e0120473. doi:10.1371/journal.pone.0120473
Maaser K., Grabowski P., Oezdem Y., Krahn A., Heine B., Stein H., et al. (2005). Up-regulation of the peripheral benzodiazepine receptor during human colorectal carcinogenesis and tumor spread. Clin. Cancer Res. 11, 1751–1756. doi:10.1158/1078-0432.CCR-04-1955
Miller L. G., Lee-Paritz A., Greenblatt D. J., Theoharides T. C. (1988). High-affinity benzodiazepine binding sites on rat peritoneal mast cells and rbl-1 cells: Binding characteristics and effects on granule secretion. Pharmacology 36, 52–60. doi:10.1159/000138346
Mitsui K., Niwa T., Kawahara Y., Morimoto N., Ohmoto K., Kato M., et al. (2015). Anti-stress effects of ONO-2952, a novel translocator protein 18 kDa antagonist, in rats. Neuropharmacology 99, 51–66. doi:10.1016/J.NEUROPHARM.2015.07.011
Mitsui K., Sasamura T., Katsumata S., Niwa T., Kawahara Y., Morimoto N., et al. (2012). Tu1370 effects of ono-2952, a novel translocator protein 18kda antagonist, on stress-induced rectal hyperalgesia and defecation in rats. Gastroenterology 142, S813–S814. doi:10.1016/s0016-5085(12)63164-3
Morohaku K., Pelton S. H., Daugherty D. J., Butler W. R., Deng W., Selvaraj V. (2014a). Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 155, 89–97. doi:10.1210/en.2013-1556
Morohaku K., Pelton S. H., Daugherty D. J., Butler W. R., Deng W., Selvaraj V. (2014b). Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 155, 89–97. doi:10.1210/en.2013-1556
Morohaku K., Phuong N. S., Selvaraj V. (2013). Developmental expression of translocator protein/peripheral benzodiazepine receptor in reproductive tissues. PLoS One 8, e74509. doi:10.1371/journal.pone.0074509
Narayan N., Owen D. R., Mandhair H., Smyth E., Carlucci F., Saleem A., et al. (2018). Translocator protein as an imaging marker of macrophage and stromal activation in rheumatoid arthritis pannus. J. Nucl. Med. 59, 1125–1132. doi:10.2967/jnumed.117.202200
Oehlers S. H., Flores M. V., Hall C. J., Crosier K. E., Crosier P. S. (2012). Retinoic acid suppresses intestinal mucus production and exacerbates experimental enterocolitis. Dis. Model. Mech. 5, 457–467. doi:10.1242/dmm.009365
Okayasu I., Hatakeyama S., Yamada M., Ohkusa T., Inagaki Y., Nakaya R. (1990). A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702. doi:10.1016/0016-5085(90)90290-H
Ostuni M. A., Issop L., Péranzi G., Walker F., Fasseu M., Elbim C., et al. (2010). Overexpression of translocator protein in inflammatory bowel disease: Potential diagnostic and treatment value. Inflamm. Bowel Dis. 16, 1476–1487. doi:10.1002/ibd.21250
Rampal R., Wari N., Singh A. K., Das U., Bopanna S., Gupta V., et al. (2021). Retinoic acid is elevated in the mucosa of patients with active ulcerative colitis and displays a proinflammatory role by augmenting IL-17 and IFNγ production. Inflamm. Bowel Dis. 27, 74–83. doi:10.1093/IBD/IZAA121
Rana S. V., Sharma S., Sinha S. K., Parsad K. K., Malik A., Singh K. (2012). Pro-inflammatory and anti-inflammatory cytokine response in diarrhoea-predominant irritable bowel syndrome patients. Trop. Gastroenterol. 33, 251–256. doi:10.7869/tg.2012.66
Ren Y. J., Lei Z., Bai T., Yu H. L., Li Y., Qian W., et al. (2017). Transfer of CD11c+ lamina propria mononuclear phagocytes from post-infectious irritable bowel syndrome causes mucosal barrier dysfunction and visceral hypersensitivity in recipient mice. Int. J. Mol. Med. 39, 1555–1563. doi:10.3892/IJMM.2017.2966
Rijnierse A., Koster A. S., Nijkamp F. P., Kraneveld A. D. (2006). TNF-α is crucial for the development of mast cell-dependent colitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G969–G976. doi:10.1152/ajpgi.00146.2006
Sano T., Uchida M. K., Suzuki-Nishimura T. (1990). The effects of clozapine on rat mast cells are different from those of benzodiazepines. Gen. Pharmacol. 21, 559–561. doi:10.1016/0306-3623(90)90715-X
Selvaraj V., Stocco D. M. (2015). The changing landscape in translocator protein (TSPO) function. Trends Endocrinol. Metab. 26, 341–348. doi:10.1016/j.tem.2015.02.007
Selvaraj V., Stocco D. M., Tu L. N. (2015). Minireview: Translocator protein (TSPO) and steroidogenesis: A reappraisal. Mol. Endocrinol. 29, 490–501. doi:10.1210/me.2015-1033
Selvaraj V., Tu L. N. (2016). Current status and future perspectives: TSPO in steroid neuroendocrinology. J. Endocrinol. 231, R1–R30. doi:10.1530/JOE-16-0241
Šileikyte J., Blachly-Dyson E., Sewell R., Carpi A., Menabò R., Di Lisa F., et al. (2014). Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (translocator protein of 18 kDa (TSPO)). J. Biol. Chem. 289, 13769–13781. doi:10.1074/jbc.M114.549634
Singh A., Dashnyam M., Chim B., Escobar T. M., Dulcey A. E., Hu X., et al. (2020). Anxiolytic drug FGIN-1-27 ameliorates autoimmunity by metabolic reprogramming of pathogenic Th17 cells. Sci. Rep. 10, 3766. doi:10.1038/s41598-020-60610-5
Stzepourginski I., Nigro G., Jacob J. M., Dulauroy S., Sansonetti P. J., Eberl G., et al. (2017). CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc. Natl. Acad. Sci. U. S. A. 114, E506–E513. doi:10.1073/PNAS.1620059114/SUPPL_FILE/PNAS.201620059SI
Suzuki-Nishimura T., Sano T., Uchida M. K. (1989). Effects of benzodiazepines on serotonin release from rat mast cells. Eur. J. Pharmacol. 167, 75–85. doi:10.1016/0014-2999(89)90749-8
Thaker A. I., Shaker A., Suprada Rao M., Ciorba M. A. (2012). Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS). J. Vis. Exp., 11:4100. doi:10.3791/4100
Thompson M. M., Manning H. C., Ellacott K. L. (2013). Translocator protein 18 kDa (TSPO) is regulated in white and Brown adipose tissue by obesity. PLoS One 8, e79980. doi:10.1371/journal.pone.0079980
Tremaine W. J., Brzezinski A., Katz J. A., Wolf D. C., Fleming T. J., Mordenti J., et al. (2002). Treatment of mildly to moderately active ulcerative colitis with a tryptase inhibitor (APC 2059): An open-label pilot study. Aliment. Pharmacol. Ther. 16, 407–413. doi:10.1046/J.1365-2036.2002.01194.X
Tu L. N., Morohaku K., Manna P. R., Pelton S. H., Butler W. R., Stocco D. M., et al. (2014a). Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 289, 27444–27454. doi:10.1074/jbc.M114.578286
Tu L. N., Morohaku K., Manna P. R., Pelton S. H., Butler W. R., Stocco D. M., et al. (2014b). Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 289, 27444–27454. doi:10.1074/jbc.M114.578286
Tu L. N., Zhao A. H., Hussein M., Stocco D. M., Selvaraj V. (2016). Translocator protein (TSPO) affects mitochondrial fatty acid oxidation in steroidogenic cells. Endocrinology 157, 1110–1121. doi:10.1210/en.2015-1795
Tu L. N., Zhao A. H., Stocco D. M., Selvaraj V. (2015). PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology 156, 1033–1039. doi:10.1210/en.2014-1707
Viennois E., Chen F., Laroui H., Baker M. T., Merlin D. (2013). Dextran sodium sulfate inhibits the activities of both polymerase and reverse transcriptase: Lithium chloride purification, a rapid and efficient technique to purify RNA. BMC Res. Notes 6, 360. doi:10.1186/1756-0500-6-360
Wang H., Zhai K., Xue Y., Yang J., Yang Q., Fu Y., et al. (2016). Global deletion of TSPO does not affect the viability and gene expression profile. PLoS One 11, e0167307. doi:10.1371/journal.pone.0167307
Waterfield J. D., McGeer E. G., McGeer P. L. (1999). The peripheral benzodiazepine receptor ligand PK 11195 inhibits arthritis in the MRL-lpr mouse model. Rheumatology 38, 1068–1073. doi:10.1093/rheumatology/38.11.1068
Whitehead W. E., Duffy K., Sharpe J., Nabata T., Bruce M. (2017). Randomised clinical trial: Exploratory phase 2 study of ONO-2952 in diarrhoea-predominant irritable bowel syndrome. Aliment. Pharmacol. Ther. 45, 14–26. doi:10.1111/apt.13839
Whittem C. G., Williams A. D., Williams C. S. (2010). Murine colitis modeling using dextran sulfate sodium (DSS). J. Vis. Exp., 35:1652. doi:10.3791/1652
Wirtz S., Neufert C., Weigmann B., Neurath M. F. (2007). Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2, 541–546. doi:10.1038/nprot.2007.41
Yan Y., Kolachala V., Dalmasso G., Nguyen H., Laroui H., Sitaraman S. V., et al. (2009). Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One 4, e6073. doi:10.1371/journal.pone.0006073
Yousefi O. S., Wilhelm T., Maschke-Neuß K., Kuhny M., Martin C., Molderings G. J., et al. (2013). The 1, 4-benzodiazepine Ro5-4864 (4-chlorodiazepam) suppresses multiple pro-inflammatory mast cell effector functions. Cell Commun. Signal. 11, 13. doi:10.1186/1478-811X-11-13
Zhao A. H., Tu L. N., Mukai C., Sirivelu M. P., Pillai V. V., Morohaku K., et al. (2016). Mitochondrial translocator protein (TSPO) function is not essential for heme biosynthesis. J. Biol. Chem. 291, 1591–1603. doi:10.1074/jbc.M115.686360
Keywords: colon, inflammation, mast cell, mitochondria, intestinal epithelium
Citation: Jimenez IA, Stilin AP, Morohaku K, Hussein MH, Koganti PP and Selvaraj V (2022) Mitochondrial translocator protein deficiency exacerbates pathology in acute experimental ulcerative colitis. Front. Physiol. 13:896951. doi: 10.3389/fphys.2022.896951
Received: 15 March 2022; Accepted: 26 July 2022;
Published: 19 August 2022.
Edited by:
Daniele Maria-Ferreira, Pelé Pequeno Príncipe Research Institute, BrazilReviewed by:
Mariano A. Ostuni, Université Paris Cité, FranceCopyright © 2022 Jimenez, Stilin, Morohaku, Hussein, Koganti and Selvaraj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vimal Selvaraj, dnM4OEBjb3JuZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.