 Liqin Yin1†
Liqin Yin1† Lihui Wang
Lihui Wang Zunhan Shi
Zunhan Shi Longhua Liu
Longhua Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 02 March 2022
Sec. Integrative Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.826811
This article is part of the Research Topic Insights in Integrative Physiology: 2021 View all 27 articles
Atherosclerosis is the hallmark of cardiovascular disease (CVD) which is a leading cause of death in type 2 diabetes patients, and glycemic control is not beneficial in reducing the potential risk of CVD. Clinically, it was shown that Thiazolidinediones (TZDs), a class of peroxisome proliferator-activated receptor gamma (PPARγ) agonists, are insulin sensitizers with reducing risk of CVD, while the potential adverse effects, such as weight gain, fluid retention, bone loss, and cardiovascular risk, restricts its use in diabetic treatment. PPARγ, a ligand-activated nuclear receptor, has shown to play a crucial role in anti-atherosclerosis by promoting cholesterol efflux, repressing monocytes infiltrating into the vascular intima under endothelial layer, their transformation into macrophages, and inhibiting vascular smooth muscle cells proliferation as well as migration. The selective activation of subsets of PPARγ targets, such as through PPARγ post-translational modification, is thought to improve the safety profile of PPARγ agonists. Here, this review focuses on the significance of PPARγ activity regulation (selective activation and post-translational modification) in the occurrence, development and treatment of atherosclerosis, and further clarifies the value of PPARγ as a safe therapeutic target for anti-atherosclerosis especially in diabetic treatment.
Atherosclerosis is the hallmark of CVD which is a leading cause of death in type 2 diabetes patients, and glycemic control is not beneficial in reducing the potential risk of CVD (Libby et al., 2016; Vallee et al., 2019; Machado-Oliveira et al., 2020). Atherosclerosis is a disease caused by the combination of high oxidative stress, inflammation (Finn et al., 2012), immune response, lipid deposition, and genetic traits (Falk, 2006; Yu et al., 2013; Johnson, 2017). Atherosclerosis is initiated by a large number of abnormally metabolized lipids including apolipoprotein B-containing lipoproteins (apoB LPs) continuously enter into the vascular intima to trigger an inflammatory response dominated by macrophages in the vascular wall (Chistiakov et al., 2015; Tabas, 2017), promote the migration and proliferation of vascular smooth muscle cells (VSMCs) (Durham et al., 2018), cause the vascular wall thickening and the lumen narrowing, and finally develop atherosclerosis (Wang et al., 2015; Bennett et al., 2016).
PPARγ is a ligand-activated nuclear receptor, that regulates glucose and lipid metabolism, endothelial function, and inflammation (Lehrke and Lazar, 2005; Janani and Ranjitha Kumari, 2015). Due to the different selected promoters and alternative shear modes, the PPARγ gene can transcriptionally generate two PPARγ transcript variants, and translate into two isoforms, PPARγ1 and PPARγ2, with PPARγ2 has 30 more amino acid residues at the N-end (Fajas et al., 1997). PPARγ1 is expressed nearly in all cells, while PPARγ2 is mainly expressed in adipocytes and vascular endothelial cells. Nevertheless, PPARγ2 is a more potent transcription activator (Lehrke and Lazar, 2005; Janani and Ranjitha Kumari, 2015). PPARγ plays a crucial role in anti-atherosclerosis by promoting cholesterol efflux (Ozasa et al., 2011; Tsuboi et al., 2020), inhibiting monocytes infiltrating into the vascular intima under endothelial layer (Namgaladze et al., 2013), and inhibiting their transformation into macrophages (Zhang and Chawla, 2004; Charo, 2007; Oppi et al., 2020), inhibiting VSMCs proliferation and migration (Zhang et al., 2011; Durham et al., 2018). PPARγ has emerged as one of the most promising therapeutic targets for cardiovascular complications, and its synthetic ligands (Lim et al., 2015), such as Thiazolidinediones (TZDs) have also been shown to have anti-atherosclerosis function (Viles-Gonzalez et al., 2004; Nakaya et al., 2009). Although their advantages are recognized, the profiles of numerous adverse effects hinder the continued use of these drugs.
To develop a safer and better treatment of cardiovascular complications targeting PPARγ, novel strategies that preserve the “good” potent insulin sensitization, while reducing or eliminating “bad”-related side effects should be used. These novel strategies may include downstream effectors of PPARγ-mediated insulin sensitization, targeting specific post-translational modification (PTMs) (Wang and Tafuri, 2003; Brunmeir and Xu, 2018) of PPARγ, and selective PPARγ modulators (SPPARMs) (Higgins and Depaoli, 2010; Dunn et al., 2011). PTMs such as phosphorylation (Yin et al., 2006; Choi et al., 2011; Montanari et al., 2020), acetylation (Qiang et al., 2012; Kraakman et al., 2018; Liu L. et al., 2020), ubiquitination (Garin-Shkolnik et al., 2014), and sumoylation (Pascual et al., 2005) are all involved in regulating PPARγ activity. These PTMs of PPARγ could regulate its transcription of downstream genes via changing protein conformation, regulating protein interactions, or altering the affinity between receptors and ligands (van Beekum et al., 2009; Brunmeir and Xu, 2018). PPARγ activation requires ligand recognition and binding to receptor regulatory receptor mediated gene transcription (Lehrke and Lazar, 2005; Janani and Ranjitha Kumari, 2015). The binding site and binding region of the ligand with PPARγ determine the conformation of PPARγ and the subsequent changes in cofactor recruitment (Higgins and Depaoli, 2010; Montanari et al., 2020). The selective PPARγ modulators (SPPARMs) is thought to improve the safety profile of PPARγ agonists avoiding TZDs’ adverse reactions.
In this review, we focus on the significance of PPARγ activity regulation (PTMs and SPPARMs) in the occurrence, development and treatment of atherosclerotic diseases, and further clarifies the value of PPARγ as a therapeutic target for anti-atherosclerosis.
PPARγ has a typical structure of nuclear hormone receptors, including the N-terminal A/B domain, DNA binding domain (DBD) and ligand binding domain (LBD) (Lehrke and Lazar, 2005; Chandra et al., 2008; Janani and Ranjitha Kumari, 2015). Both forms of PPARγ1 and PPARγ2 have a similar structural, except for PPARγ2 containing an N-terminal extension of 28 amino acids (Chandra et al., 2008). PPARγ usually form a heterodimer with Retinoid X Receptor α (RXRαChandra et al., 2008) and binds to PPRE. When the ligand is not bound, PPARγ/RXRα mainly binds to some co-repressors, such as Nuclear Receptor Corepressor (NCoR) or Silencing Mediator of Retinoic Acid and Thyroid Hormone Receptor (SMRT). When the ligand binds to LBD, it will change the conformation of PPARγ, and the co-repressors will be replaced by some co-activators, e.g., cAMP responsive element binding protein (CREBP), PPARγ coactivator-1 (PGC-1), Steroid Receptor Coactivator (SRC), and CBP/P300 (Lehrke and Lazar, 2005; Chandra et al., 2008). The recruited cofactors vary in their transcriptional regulatory target genes, and their transcriptional levels and biological functions will change accordingly.
PPARγ not only participates in fat formation, lipid and glucose metabolism, but also plays an important role on vascular biology and inflammation, and the development of atherosclerosis (Kvandová et al., 2016; Hernandez-Quiles et al., 2021). PPARγ has anti-atherosclerotic effects through the following aspects (Figure 1): (1) PPARγ regulates the expression of cell adhesion molecules, such as inducible nitric oxide synthase (Chen et al., 2001), intracellular cell adhesion molecule-1(ICAM-1), vascular cell adhesion molecule-1(VCAM-1) (Babaev et al., 2005), and inhibit endothelial cells activation and attenuation of monocyte chemoattractant protein 1 (MCP-1), matrix metalloproteinase 9 (MMP9), and metallopeptidase inhibitor 1 (TIMP-1) which induced monocyte migration across endothelial cells (Verrier et al., 2004; Calabrò et al., 2005); (2) PPARγ activates the PPARγ/liver X receptor α (LXR-α) pathway to stimulate the expression of cholesterol efflux-related genes– ATP binding cassette transporter A1 (ABCA1) (Akiyama et al., 2002) and Acy1 Coenzyme A: Cholesterol Acyltransferases (ABCG1) (Sueyoshi et al., 2010; Srivastava, 2011), accelerates the efflux of cholesterol from macrophages (Liu D. et al., 2020; Oppi et al., 2020), therefore inhibits the formation of foam cells (Li et al., 2004; Zhang et al., 2021); (3) PPARγ inhibits the expression of pro-inflammatory factors, such as TNF-α (Zhang et al., 2014), IL-6, IL-18 (Chen et al., 2008), and induces the macrophages differentiation into an anti-inflammatory M2 phenotype (Oppi et al., 2020). And the expression of M2 markers, such as MR, AMAC1, and IL-10 levels correlate positively with the expression of PPARγ (Bouhlel et al., 2007). Therefore, PPARγ can improve the inflammatory response of cardiovascular cells, inhibit plaque formation, and maintain plaques stability. (4) PPARγ inhibits VSMCs proliferation and migration by suppressing TLR4-mediated inflammation (Gu et al., 2019) and ultimately attenuates intimal hyperplasia after carotid injury (Meredith et al., 2009; Osman and Segar, 2016). Meanwhile, PPARγ prevents the degradation of cyclin-dependent kinase inhibitors (CDKIs) and p27 induced by growth factors, and inhibits the formation of cyclin-dependent kinase complex (Cyclin-CDK), thereby inhibits proliferation, migration, and apoptosis (Law et al., 2000; Fu et al., 2001). (5) PPARγ inhibits the expression of MMP-9 and MMP-2 that can decompose collagen and fibers in macrophages (Luo et al., 2007; Reinhold et al., 2020), reduces the fragility of the fiber cap and enhances the stability of the plaque. Therefore, the functional regulation of PPARγ is of very important for the prevention and treatment of atherosclerosis.
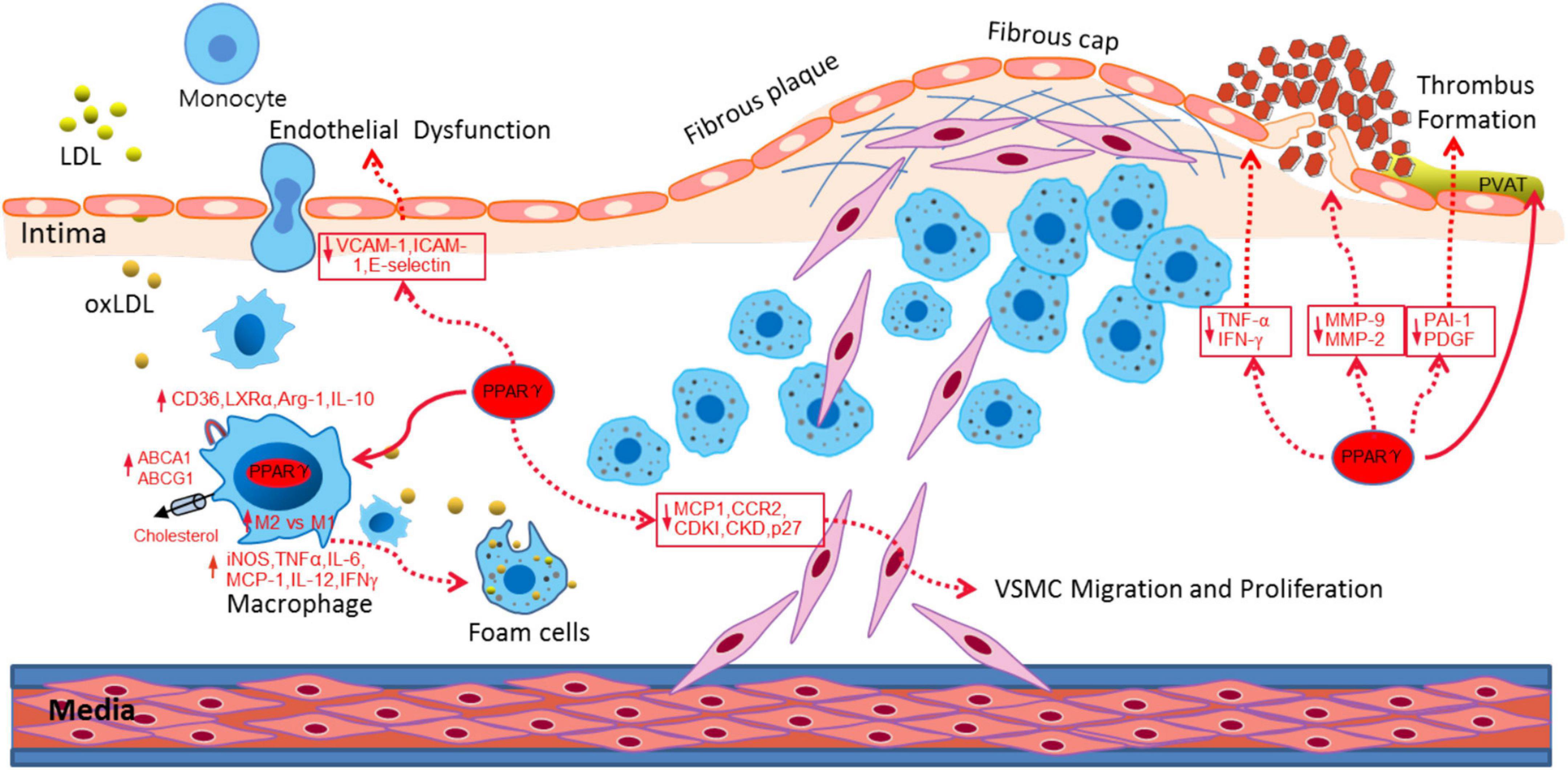
Figure 1. PPARγ attenuated atherosclerosis through different aspects, including alleviating endothelial dysfunction, promoting cholesterol efflux, inducing M1-M2 transition, inhibiting VSMC migration and proliferation and stabilizing the fibrous cap and plaque.
The PPARγ transcriptional activity regulates in diverse ways, including protein expression levels, ligands, and transcriptional cofactors. PTMs of proteins can alter protein conformation, regulate protein interactions, and alter the affinity between receptors and ligands, thus regulating the transcription of downstream genes (Brunmeir and Xu, 2018).
The phosphorylation regulation of PPARγ is one of the main ways to regulate its activity (Montanari et al., 2020). With different stimuli, PPARγ could be phosphorylated at different sites and resulting diverse biological effects (Choi et al., 2011). Cyclin-dependent kinase (CDK) (Choi et al., 2010, 2011; Laghezza et al., 2018) and mitogen-activated protein kinase (MAPK) (Yin et al., 2006; Yang et al., 2013; Ge et al., 2018) are involving in the phosphorylation of PPARγ, and the main sites include Ser273 (245 in isoform 1) (Dias et al., 2020; Hall et al., 2020) and Ser112 (Ser82 in isoform 1) (Figure 2A; Ge et al., 2018). CDK5-mediated phosphorylation of PPARγ S273 results in a decrease in its transcriptional activity and adipogenesis. One of TZDs’ major side effects is due to its activation of PPARγ in adipose tissue, and high-fat diet increased CDK5-mediated of PPARγ phosphorylation, which was negatively associated with TZDs’ insulin sensitization in humans. Meanwhile PPARγ phosphorylation can up-regulate lipid uptake of CD36 and SR-A1 related proteins, inhibit cholesterol efflux ABCA1 and ABCG1 related proteins, induce the expression of TNF-α, IL-1β and other inflammatory factors, and promote the formation of foam cells to accelerate the process of atherosclerosis (Choi et al., 2010, 2011; Banks et al., 2015). PPARγ phosphorylation by CDK5 may contribute to its dissociation with PGC1α and TIF2 coactivators but interaction with SMRT and NCoR corepressors (Dias et al., 2020). NCoR can regulate the phosphorylation of PPARγ on Ser 273 by stabilizing CDK5 (Li et al., 2011). Mice with fat cell NCoR knockout (NCoR–/–) improves glucose tolerance and insulin sensitivity and reduces macrophage infiltration and inflammation (Ribeiro Filho et al., 2019). As from CDK5 studies, compounds can be designed to alter specific PTMs of PPARγ to partly prevent disturbed fat metabolism while retaining anti-diabetic potency. CDK9/CDK7-mediated phosphorylation of S112 can increase the transcriptional activity of PPARγ and promote adipocyte differentiation (Iankova et al., 2006; Li et al., 2017; Ge et al., 2018).
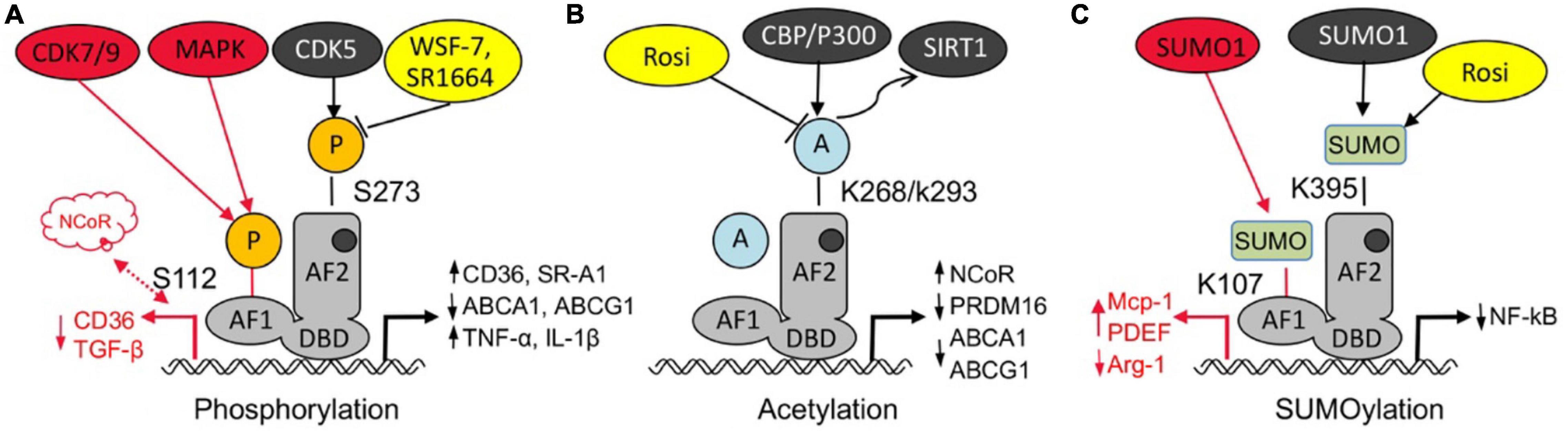
Figure 2. Post-translational modification of PPARγ regulates atherosclerosis. (A) Both phosphorylation of PPARγ at S112 by CDK7/9 or MAPK and phosphorylation of PPARγ at S273 accelerates foam cell formation and atherosclerosis through different signaling pathway, some PPARγ agonist (WSF-7, SR1664) can block cdk5 mediated Ser273 phosphorylation. (B) Acetylation of PPARγ at K268/K293 increases atherosclerosis through upregulating ABCA1, ABCG1, and NcoR but inhibiting PRDM16,while deacetylation of PPARγ at K268 and K293 alleviates atherosclerosis, while PPARγ agonist rosiglitazone (Rosi) could deacetylate PPARγ at K268/K293. (C) Sumoylation of PPARγ at K107 promotes VSMCs proliferation and migration, but sumoylation of PPARγ at K395, such as by Rosi, has anti-inflammation effect.
MAPK can phosphorylate PPARγ in the AF1 region (PPARγ2 Ser112, PPARγ1 Ser82), inhibiting the ligand binding and changing the recruitment of co-factors, and then change the transcriptional activity (Yin et al., 2006; Yang et al., 2013; Ge et al., 2018). MAPK-mediated phosphorylation of PPARγ, which promotes the formation of foam cells by macrophages exposed to ox-LDL (Yin et al., 2006). Growth factors can phosphorylate PPARγ by the MAPK signaling pathway and reduce the transcriptional activity of PPARγ (Yin et al., 2006), such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) (Osman and Segar, 2016). In addition, Choi et al. (2015) recently described the phosphorylation of Y78, is also important for the cytokine and chemokine gene expression’s regulation. PPARγ phosphorylation can alter its transcriptional activity, and the blockage of PPARγ phosphorylation is related to improve insulin sensitization (Choi et al., 2014). However, PPARγ phosphorylation mediated by different enzymes, conformational changes at different sites can cause the recruitment response of different cofactors, its role in atherosclerosis and its mechanism need to be further studied, and also provide ideas for the drug design of PPAR ligand.
Acetylation of PPARγ is a ligand- independent activation of PPARγ. Qiang et al. (2012) showed that five lysine residues (K98, K107, K218, K268, and K293) could be acetylated, of which two K268/K293 could be deacetylated by TZD rosiglitazone (Rosi) via activation of the NAD (Nicotinamide adenine dinucleotide)-dependent deacetylase sirtuin-1 (SIRT1) deacetylase (Figure 2B; Kraakman et al., 2018). PPARγ is acetylated by p300 or CBP (Kim et al., 2006), and it may play an important role in lipid synthesis (Tian et al., 2014). Acetylation of PPARγ at K268/K293 increases atherosclerosis through upregulating ABCA1, ABCG1, and NcoR but inhibiting PRDM16, while deacetylation of PPARγ at K268 and K293 alleviates atherosclerosis.
PPARγ deacetylation on K268 and K293 induces brown remodeling of white adipose tissue and reduces the adverse effects of TZDs while maintaining insulin sensitization (Qiang et al., 2012; Liu L. et al., 2020). Deacetylation of PPARγ can selective regulation the target genes, it could inhibit aP2, Cd36, upregulation genes ucp1 and adipsin on lipid oxidative genes cpt1a (Kraakman et al., 2018). PPARγ deacetylation improves endothelial function with diabetes treatment. The aortic arch lesion size was reduced in 2KR (K268 and K293) LDLr–/– mice, the expression of iNOS, Nox2, and IL-6 in endothelial cells were decreased, while the side effects of TZD, including fluid retention and bone loss were reduced (Liu L. et al., 2020). Deacetylation of PPARγ inhibits the cholesterol efflux PPARγ/LXRα/ABCA1 pathway (Cao et al., 2014; Yang et al., 2015), increased production of proinflammatory M1 macrophages and promotes the development of inflammatory response (Chen et al., 2008, 2010), leading to the onset and development of atherosclerosis. From a mechanistic perspective, deacetylated PPARγ preferentially interacts with PRDM16 and disrupts the binding of the transcriptional corepressor NCoR (Qiang et al., 2012). Therefore, manipulating PPARγ acetylation is a promising therapeutic strategy to anti-atherosclerosis.
PPARγ sumoylation with SUMO1 modification of K107 (K77 in PPARγ1) (Figure 2C), and ubiquitin carrier protein 9 (Ubc9) and PIAS1 (protein inhibitor of activated STAT1) are involved as PPARγ specific E2 binding enzymes and E3 ligases, respectively (Lim et al., 2009). Sumoylation of PPARγ K107 inhibits its transcriptional activity, and is enhanced by the K107 mutation (K107R). PPARγ sumoylation at K107 position strongly inhibited VSMCs proliferation and migration (Katafuchi et al., 2018), and reduced neointimal formation after balloon injury (Lim et al., 2009). Desumoylation at K107 in PPARγ may inhibit serum-stimulated VSMCs proliferation (Lim et al., 2009; Osman and Segar, 2016), might play an important role against atherosclerosis. Moreover, K107R improves insulin sensitivity without body weight gain or adiposity (Wadosky and Willis, 2012; Pourcet et al., 2013). However, some studies have shown that K107 sumoylation plays an important role in the anti-inflammatory response triggered by apoptotic cells, possibly by stabilizing the co-inhibitor NCoR on the target gene (Jennewein et al., 2008; Lu et al., 2013). The sumoylation modification of PPARγ1 inhibits the M2 polarization of macrophages by inhibiting the transcription of Arg-1 (Haschemi et al., 2011). Pascual et al. (2005) shows that the PPAR agonist TZD can exert anti-diabetic and anti-atherosclerotic effects through the NF-kB inflammatory pathway, the first response is associated with TZD-mediated SUMO1 modification of K365 (K395 in PPARγ2) followed by targeted regulation of the PPAR co-cofactor NCoR. The exact biological role of the two modifications in anti-inflammatory responses, especially the potential functional overlap, remains to be determined. Nevertheless, targeting PPARγ sumoylation may provide a novel mechanism for anti-atherosclerosis.
Ubiquitination modification cannot only regulate the proteasome-mediated degradation of target proteins, but also serve as a “scaffold” to recruit other proteins to form signal complexes. PPARγ undergoes conformational changes after binding to the ligand. On the one hand, Makorin RING finger protein 1 (MKRN1) (Kim et al., 2014) and Seven in absentia homolog 2 (SIAH2) (Kilroy et al., 2012) services as PPARγ E3 ligases, targeting PPARγ for proteasomal degradation. Ubiquitination of PPARγ on K184 and K185 inhibits its activity in mature 3T3-L1 adipocytes (Kilroy et al., 2012; Kim et al., 2014). On the other hand, it can also recruit the binding of ubiquitination-related enzymes and induce proteasome-dependent degradation, thereby negatively regulating the transcriptional activity of PPARγ. Rosiglitazone reduces the inflammatory response in diabetic plaques, less ubiquitin, proteasome 20S, TNF-α, and NF-κB, ubiquitin-proteasome activity with diabetic plaque NF-κB-mediated inflammatory response is involved (Marfella et al., 2006). This study strengthens the earlier findings on PPARγ regulation through modulation of its stability. Currently, the use of ubiquitination modifications for the regulation of PPARγ transcriptional activity is still controversial (Watanabe et al., 2015). It has been reported that the ubiquitin-proteasome pathway can mediate the protein renewal of PPARγ (Li et al., 2016), a process that is required for the efficient transcription of its downstream genes, while the ubiquitin activase inhibitor E1 inhibitor, and the proteasome inhibitor MG-132 can cause a decrease in PPARγ transcriptional activity.
PPARγ ligands are generally lipid-derived compounds with natural and synthetic properties, and different ligands have different affinity-binding receptors and activate the receptors. Synthetic ligands TZDs reduce atherosclerosis in certain mouse models (Martens et al., 2002). However, severe side effects associated with TZD use, such as weight gain, fluid retention, bone loss, cardiovascular disk, etc., restricts the use of TZDs (Zinn et al., 2008). In order to maximize the PPAR-mediated insulin sensitization and to minimize the occurrence of related adverse reactions, the concept of “selective PPAR regulator (selective PPARγ modulators, SPPARMs)” was proposed and developed (Higgins and Depaoli, 2010; Camejo, 2016). In contrast to rosiglitazone, SPPRAMs has similar or different PPAR receptor binding sites, or has different affinity or specificity of recruitment receptor cofactors, or a range of target genes where PPAR are biased and selective for regulating transcription. The currently found SPPRAMs that associated with atherosclerosis mainly consists of three types: partial PPARγ agonist, dual PPAR α/γ agonist, and non-agonist PPARγ ligand.
In fact, partial PPARγ agonist is different from the classic TZDs, with rosiglitazone as the “full agonist.” It is generally believed that 20–60% of rosiglitazone efficacy is a partial activator (Table 1), such as GQ-177 (Silva et al., 2016), S 26948 (Carmona et al., 2007), WSF-7 (Zhang et al., 2020) lobeglitazone (Lim et al., 2015), and INT131 (Xie et al., 2017). LDLr–/– mice treated with GQ-177 can significant decrease the VLDL, LDL fractions and increase mean HDL, Glut4 levels, increased the expression of apoA1, CD36, ABCA1, SR-B1, and ABCG5 in hepatic, contrary to rosiglitazone, GQ-177 did not affect fat accumulation and bone mineral density (Silva et al., 2016). It was shown that TZDs facilitated the transport of BM-derived circulating progenitor cells to adipose tissue and their differentiation into multilocular adipose cells (Crossno et al., 2006). Meanwhile, Hu et al. (2021) found RANKL from bone marrow adipose lineage cells promoted osteoclast formation and bone loss. S26948 improves lipid parameters (LDLs, VLDL) and reduces atherosclerotic lesions in ob/ob male C57BL/6 mice (Carmona et al., 2007). WSF-7 upregulated PPARγ-responsive genes, such as adiponectin and Glut4, inhibits PPARγ phosphorylation at Ser273 by obesity and enhances insulin sensitivity in 3T3-L1 Adipocytes (Zhang et al., 2020). Lobeglitazone inhibits the VSMCs proliferation and migration, reduces the vascular cells adhesion, NF-kB p65 translocation, and improves circulating factors related to atherosclerosis, then reduced neointimal formation significantly in balloon injury rat carotid arteries in ApoE–/– mice (Lim et al., 2015; Song et al., 2021). In the presence of pro-inflammatory stimulation, Lobeglitazone effectively inhibited expression of pro-inflammatory gene expression in macrophages and adipocytes (Sohn et al., 2018). The latest study has found that macrophages targeted PPARγ activator Lobeglitazone could rapidly stabilize a coronary artery-sized inflammatory plaque (Song et al., 2021). Both non-clinical and clinical studies have demonstrated that INT131 have the potential to separate insulin-sensitizing actions and undesirable side effects in Patients With Type 2 Diabetes (DePaoli et al., 2014), and it also has the potential to decrease free fatty acids, increase HDL-C (Dunn et al., 2011). However, there is still no relevant study on the effect of INT131 on atherosclerosis.
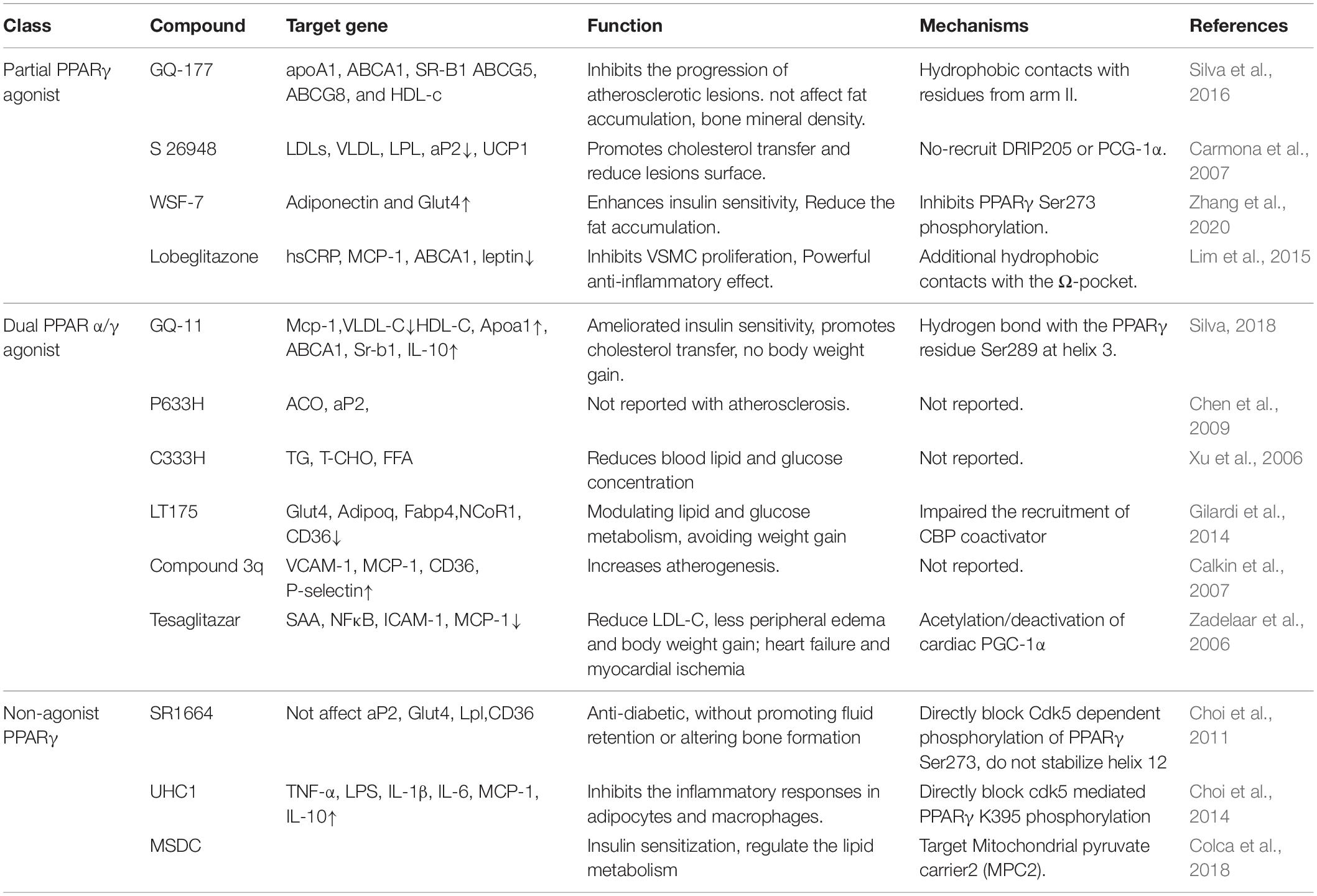
Table 1. Selective PPARγ modulators associated with atherosclerosis.
Full agonist-TZDs forms a key hydrogen bond with the side chain of Y473 on helix 12, mainly interacts with residues from arm I in the ligand binding pocket, to enhance the binding affinity of coactivators/weaken corepressors, inducing transcriptional activation (Brust et al., 2018). Unlike full agonist TZDs, some partial agonists have different binding mode, such as GQ-177 interacts through hydrophobic contacts with residues from arm II (Barros et al., 2010), lobeglitazone makes additional hydrophobic contacts with the Ω-pocket. There are also some partial agonists having similar binding sites to TZDs, but the recruiting cofactors were different. For example, INT131 activates PPARγ, but does not recruit the cofactor MED1 (key factor in regulating adipogenesis) (Lee et al., 2012; Xie et al., 2017), S26948 is unable to recruit DRIP205 or PCG-1α (Key genes in regulating gluconeogenesis), so that it selectively reduces blood glucose without the obvious adverse effects of adipogenesis (Carmona et al., 2007; Sohn et al., 2009). Compared to rosiglitazone, Lobeglitazone strongly blocks the phosphorylation of PPARγ at Ser245, but the pharmacological effects of this translational modification change need further studied (Jang et al., 2018). Although the exact mechanism beyond these effects remains to be determined, partial agonist might represent a new class of therapeutic molecules for the treatment of atherosclerosis.
Dual PPARα/γ agonist has been focusing on the activation of both PPARα and PPARγ, which may provide a wider range of metabolic benefits. Studies have shown that most of the Dual PPARα/γ agonist play an active role in anti-atherosclerosis, such as GQ-11 (Silva, 2018), P633H (Chen et al., 2009), C333H (Xu et al., 2006), LT175 (Gilardi et al., 2014), and Tesaglitazar (Zadelaar et al., 2006; Chira et al., 2007; Table 1). GQ-11 improved insulin sensitivity and enhanced Glut4 expression in the adipose tissue, meanwhile the levels of MCP-1were reduced and the levels of IL-10 were increased. Furthermore, it also upregulation of Apoa1 and ABCA1 gene expression, then reduced triglycerides and VLDL cholesterol and increased HDL cholesterol in LDLr–/– mice (Silva et al., 2018). P633H can be accompanied by the upregulation of ACO and aP2 expression in db/db and KK-Ay mice, and is targeted to regulate PPARα in the liver and PPARγ in adipose tissue, respectively (Chen et al., 2009). C333H efficiently reduced blood lipid and glucose concentration in db/db mice (Xu et al., 2006). LT175 activates PPARγ in adipocytes, increases the expression of PPARγ target gene Glut4 and Adipoq in 3T3-L1 adipocytes and in a mouse model. Moreover, LT175 can also activate PPARα in the liver, trigger triglyceride and fatty acid catabolism, and achieve to eliminate the side effects of some conventional PPAR agonists (Gilardi et al., 2014). However, such effects are not clear in human situation. Tesaglitazar reduces atherosclerosis, reduces macrophage inflammation, number of adhesion monocytes and nuclear factor activity of the vessel wall (Zadelaar et al., 2006). However, some Dual PPARα/γ agonist such as compound 3q may accelerate atherosclerosis, it may related to the increase expression of the vascular endothelial activation and inflammation markers, such as P-selectin, MCP-1, VCAM-1, and CD36, that are also associated with plaque complexity (Calkin et al., 2007). Dual agonists such as Tesaglitazar also showed better insulin sensitization effects as well as the prevention of atherosclerosis progression in clinical studies, but due to adverse side effects, including heart failure and myocardial ischemia, it has been discontinued in phase III clinical trials (Yamaguchi et al., 2014).
The PPARγ agonists shown to increase adipogenesis and body weight, whereas PPARα agonists counteract these effects by decreasing food intake and fat deposits. However, both its PPAR binding mode and its downstream targeting will change accordingly. LT175 impaired the recruitment of CBP coactivator, Tesaglitazar is accounted for by inhibition of both expression and acetylation/deactivation of cardiac PGC1α both in healthy C57BL/6 and diabetic db/db mice. Consistent with other partial agonists, GQ-11 only hydrogen bond with the PPARγ residue Ser289 at helix 3, which could reflect in weak PPARγ agonistic activities, and also interacts and weakly activates PPARα (Silva, 2018). Most of the dual PPARα/γ agonists, although they can improve insulin resistance as the full agonists, and do not have similar weight gain, negative bone effects, but it will appear adverse effects on the urothelial, renal, and cardiovascular system (Kaul et al., 2019; Chen et al., 2021). For the adverse side effects of PPARγ agonists in fluid retention, most studies have shown that this effect was due to increased reabsorption of sodium and water by the renal tubules, but the role of specific renal unit segments and sodium carriers was unclear. PPARγ-induced EGF receptors and non-genomic trans-activation of downstream extracellular signal-modulating kinases (ERKs) may augment sodium reabsorption in the proximal tubule (Beltowski et al., 2013). TZDs-like compounds significantly inhibited PPARγ phosphorylation in Ser112 while telmisartan did not (Kolli et al., 2014). Thus, telmisartan did not have a significant effect on osteoclast differentiation and osteogenesis. Dual PPARα/γ Tesaglitazar activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. It was showed that this cardiac dysfunction was associated with reduced PGC1α expression. These effects are related to competition between PPARα and PPARγ to regulate Ppargc1a gene expression and to reduce cardiac SIRT1 expression (Kalliora et al., 2019). PGC1α is a regulator of mitochondrial function in thermogenic tissues, such as brown fat. Lehman et al. (2000) also found cardiac-specific overexpression of PGC1α in mice lead to uncontrolled mitochondrial proliferation in cardiomyocytes, resulting in loss of sarcomere structure and dilated cardiomyopathy. Inhibiting PGC1α on dual PPARα/γ activation is potentially as a key event that mediates the cardiotoxic effect, which would provide a guide for design of future PPAR agonists.
Non-agonist (Antagonists) PPARγ ligand, also known as PPAR modulators, exhibit high affinity but do not activate PPARγ. The Antagonists consists of two main categories, and one is known as PPARγ phosphorylation inhibitors, such as SR1664 (Cariou et al., 2012; Dias et al., 2020) and UHC1 (Choi et al., 2014). Taken SR1664 as an example, it has basically no transcriptional activation effect on PPARγ, but has a high affinity with PPARγ and belongs to a phosphorylation inhibitor, blocking the CDK5-mediated phosphorylation of PPARγ in Ser273 (Laghezza et al., 2018; Dias et al., 2020). SR1664 did not stimulate lipid accumulation or adipogenesis gene expression (such as aP2, Glut4, Lpl, CD36) in differentiating fat cells. UHC1 blocking CKD5-mediated PPARγ phosphorylation at position K395 in LBD, reducing macrophages inflammatory factor LPS-induced nitric oxide (NO) production both in vitro and in HFD-fed mice (Choi et al., 2014; Ribeiro Filho et al., 2019). Comparison with conventional full agonists, these antagonists do not stabilize helix 12 and display negligible changes in activation, but make unfavorable interactions with F282 on helix 3 (Asteian et al., 2015). Another class of PPAR modulators is relatively special, with structurally TZDs analogs, but with little effect on PPARγ, represent the compound as MSDC. It have been showed a potential therapeutic avenue for treating non-alcoholic steatohepatitis, improve the insulin resistance effect, regulate the lipid metabolism (Colca et al., 2018). However, the current study of atherosclerosis has not been reported. Its target of action was reported as a line Mitochondrial pyruvate carrier 2 (MPC2), which is the mitochondrial target of thiazolidinediones (mTOT) (Vigueira et al., 2017). Although not directly stimulated to PPARγ, but still has the potency of insulin sensitization.
PPARγ plays a crucial role in anti-atherosclerosis, PPARγ-mediated anti-atherosclerosis depends on the basic expression level and activity level of PPARγ. At the same time, PPARγ transcription activity is no longer the only criterion, and the mode of action of the compound and PPARγ is the key to PPARγ activity. Both protein post-translational modification and selective modulators are different modes of PPARγ activity regulation. The mode of action between the compound and PPARγ determines the intensity and breadth of PPARγ-mediated transcriptional activation.
However, there are still many questions to be solved, such as the proteases involved in various post-translational modifications, and the protein interactions and downstream target genes regulated by these modifications are still unclear. In addition to the identified post-translational modifications, novel modification patterns or modification sites are still to be discovered. Different post-translational modifications may also interact and form closely related network, which provides a strong guarantee for the fine control of protein functions. The post-translational modifications of proteins are usually reversible, while the de-modification of PPARγ is relatively lagging behind. Transcriptional alteration of post-translational modifications is an innovative idea of new agonist, and only phosphorylation is partially applied to PPARγ agonist, deacetylation and sumoylation has not been involved. How to selectively activate partial downstream targets of PPARγ to protect from atherosclerosis and relative metabolic diseases as well as reducing adverse effects deserve further investigation.
LY, LW, ZS, XJ, and LL contributed to the literature search. LY, LW, ZS, and LL wrote and revised the manuscript. LL designed the frame of this manuscript. All authors contributed to this manuscript and approved the submitted version.
This work was supported by the Shanghai Frontiers Science Research Base of Exercise and Metabolic Health, the research program of exercise and public health (0831) in Shanghai University of Sport.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akiyama, T. E., Sakai, S., Lambert, G., Nicol, C. J., Matsusue, K., Pimprale, S., et al. (2002). Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol. Cell. Biol. 22, 2607–2619. doi: 10.1128/MCB.22.8.2607-2619.2002
Asteian, A., Blayo, A. L., He, Y., Koenig, M., Shin, Y., Kuruvilla, D. S., et al. (2015). Design, Synthesis, and Biological Evaluation of Indole Biphenylcarboxylic Acids as PPARγ Antagonists. ACS Med. Chem. Lett. 6, 998–1003. doi: 10.1021/acsmedchemlett.5b00218
Babaev, V. R., Yancey, P. G., Ryzhov, S. V., Kon, V., Breyer, M. D., Magnuson, M. A., et al. (2005). Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 25, 1647–1653. doi: 10.1161/01.ATV.0000173413.31789.1a
Banks, A. S., McAllister, F. E., Camporez, J. P., Zushin, P. J., Jurczak, M. J., Laznik-Bogoslavski, D., et al. (2015). An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 517, 391–395. doi: 10.1038/nature13887
Barros, C. D., Amato, A. A., de Oliveira, T. B., Iannini, K. B., Silva, A. L., Silva, T. G., et al. (2010). Synthesis and anti-inflammatory activity of new arylidene-thiazolidine-2,4-diones as PPARgamma ligands. Bioorg. Med. Chem. 18, 3805–3811. doi: 10.1016/j.bmc.2010.04.045
Beltowski, J., Rachanczyk, J., and Wlodarczyk, M. (2013). Thiazolidinedione-induced fluid retention: recent insights into the molecular mechanisms. PPAR Res. 2013:628628. doi: 10.1155/2013/628628
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 118, 692–702. doi: 10.1161/circresaha.115.306361
Bouhlel, M. A., Derudas, B., Rigamonti, E., Dièvart, R., Brozek, J., Haulon, S., et al. (2007). PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143. doi: 10.1016/j.cmet.2007.06.010
Brunmeir, R., and Xu, F. (2018). Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 19:1738. doi: 10.3390/ijms19061738
Brust, R., Shang, J., Fuhrmann, J., Mosure, S. A., Bass, J., Cano, A., et al. (2018). A structural mechanism for directing corepressor-selective inverse agonism of PPARgamma. Nat. Commun. 9:4687. doi: 10.1038/s41467-018-07133-w
Calabrò, P., Samudio, I., Safe, S. H., Willerson, J. T., and Yeh, E. T. (2005). Inhibition of tumor-necrosis-factor-alpha induced endothelial cell activation by a new class of PPAR-gamma agonists. An in vitro study showing receptor-independent effects. J. Vasc. Res. 42, 509–516. doi: 10.1159/000088260
Calkin, A. C., Allen, T. J., Lassila, M., Tikellis, C., Jandeleit-Dahm, K. A., and Thomas, M. C. (2007). Increased atherosclerosis following treatment with a dual PPAR agonist in the ApoE knockout mouse. Atherosclerosis 195, 17–22. doi: 10.1016/j.atherosclerosis.2006.11.021
Camejo, G. (2016). Selective PPAR modulators (SPPARs) may fill the need for treatment of the atherogenic dyslipidemia of insulin resistance and type 2 diabetes: can they reduce the associated cardiac risk? Atherosclerosis 249, 224–225. doi: 10.1016/j.atherosclerosis.2016.03.026
Cao, Q., Rong, S., Repa, J. J., St Clair, R., Parks, J. S., and Mishra, N. (2014). Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 34, 1871–1879. doi: 10.1161/atvbaha.114.303393
Cariou, B., Charbonnel, B., and Staels, B. (2012). Thiazolidinediones and PPARgamma agonists: time for a reassessment. Trends Endocrinol. Metab. 23, 205–215. doi: 10.1016/j.tem.2012.03.001
Carmona, M. C., Louche, K., Lefebvre, B., Pilon, A., Hennuyer, N., and Audinot-Bouchez, V. (2007). S 26948: a new specific peroxisome proliferator activated receptor gamma modulator with potent antidiabetes and antiatherogenic effects. Diabetes 56, 2797–2808. doi: 10.2337/db06-1734
Chandra, V., Huang, P., Hamuro, Y., Raghuram, S., Wang, Y., Burris, T. P., et al. (2008). Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 456, 350–356. doi: 10.1038/nature07413
Charo, I. F. (2007). Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab. 6, 96–98. doi: 10.1016/j.cmet.2007.07.006
Chen, F. L., Yang, Z. H., Liu, Y., Li, L. X., Liang, W. C., Wang, X. C., et al. (2008). Berberine inhibits the expression of TNFalpha, MCP-1, and IL-6 in AcLDL-stimulated macrophages through PPARgamma pathway. Endocrine 33, 331–337. doi: 10.1007/s12020-008-9089-3
Chen, F. L., Yang, Z. H., Wang, X. C., Liu, Y., Yang, Y. H., Li, L. X., et al. (2010). Adipophilin affects the expression of TNF-alpha, MCP-1, and IL-6 in THP-1 macrophages. Mol. Cell. Biochem. 337, 193–199. doi: 10.1007/s11010-009-0299-7
Chen, J., Zhang, X., Millican, R., Sherwood, J., Martin, S., Jo, H., et al. (2021). Recent advances in nanomaterials for therapy and diagnosis for atherosclerosis. Adv. Drug Deliv. Rev. 170, 142–199. doi: 10.1016/j.addr.2021.01.005
Chen, W., Zhou, X. B., Liu, H. Y., Xu, C., Wang, L. L., and Li, S. (2009). P633H, a novel dual agonist at peroxisome proliferator-activated receptors alpha and gamma, with different anti-diabetic effects in db/db and KK-Ay mice. Br. J. Pharmacol. 157, 724–735. doi: 10.1111/j.1476-5381.2009.00231.x
Chen, Z., Ishibashi, S., Perrey, S., Osuga, J., Gotoda, T., Kitamine, T., et al. (2001). Troglitazone inhibits atherosclerosis in apolipoprotein E-knockout mice: pleiotropic effects on CD36 expression and HDL. Arterioscler. Thromb. Vasc. Biol. 21, 372–377. doi: 10.1161/01.atv.21.3.372
Chira, E. C., McMillen, T. S., Wang, S., Haw, A. III, O’Brien, K. D., Wight, T. N., et al. (2007). Tesaglitazar, a dual peroxisome proliferator-activated receptor alpha/gamma agonist, reduces atherosclerosis in female low density lipoprotein receptor deficient mice. Atherosclerosis 195, 100–109. doi: 10.1016/j.atherosclerosis.2006.12.012
Chistiakov, D. A., Orekhov, A. N., and Bobryshev, Y. V. (2015). Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 214, 33–50. doi: 10.1111/apha.12466
Choi, J. H., Banks, A. S., Estall, J. L., Kajimura, S., Bostrom, P., Laznik, D., et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. doi: 10.1038/nature09291
Choi, J. H., Banks, A. S., Kamenecka, T. M., Busby, S. A., Chalmers, M. J., Kumar, N., et al. (2011). Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477–481. doi: 10.1038/nature10383
Choi, S., Jung, J. E., Yang, Y. R., Kim, E. S., Jang, H. J., Kim, E. K., et al. (2015). Novel phosphorylation of PPARgamma ameliorates obesity-induced adipose tissue inflammation and improves insulin sensitivity. Cell Signal. 27, 2488–2495. doi: 10.1016/j.cellsig.2015.09.009
Choi, S. S., Kim, E. S., Koh, M., Lee, S. J., Lim, D., Yang, Y. R., et al. (2014). A novel non-agonist peroxisome proliferator-activated receptor gamma (PPARgamma) ligand UHC1 blocks PPARgamma phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. J. Biol. Chem. 289, 26618–26629. doi: 10.1074/jbc.M114.566794
Colca, J. R., McDonald, W. G., and Adams, W. J. (2018). MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis. Expert Opin. Investig. Drugs 27, 631–636. doi: 10.1080/13543784.2018.1494153
Crossno, J. T. Jr., Majka, S. M., Grazia, T., Gill, R. G., and Klemm, D. J. (2006). Rosiglitazone promotes development of a novel adipocyte population from bone marrow-derived circulating progenitor cells. J. Clin. Invest. 116, 3220–3228. doi: 10.1172/jci28510
DePaoli, A. M., Higgins, L. S., Henry, R. R., Mantzoros, C., and Dunn, F. L. (2014). Can a selective PPARγ modulator improve glycemic control in patients with type 2 diabetes with fewer side effects compared with pioglitazone? Diabetes Care 37, 1918–1923. doi: 10.2337/dc13-2480
Dias, M. M. G., Batista, F. A. H., Tittanegro, T. H., de Oliveira, A. G., Le Maire, A., Torres, F. R., et al. (2020). PPARgamma S273 Phosphorylation Modifies the Dynamics of Coregulator Proteins Recruitment. Front. Endocrinol. 11:561256. doi: 10.3389/fendo.2020.561256
Dunn, F. L., Higgins, L. S., Fredrickson, J., and DePaoli, A. M. INT131-004 study group (2011). Selective modulation of PPARgamma activity can lower plasma glucose without typical thiazolidinedione side-effects in patients with Type 2 diabetes. J. Diabetes Complications 25, 151–158. doi: 10.1016/j.jdiacomp.2010.06.006
Durham, A. L., Speer, M. Y., Scatena, M., Giachelli, C. M., and Shanahan, C. M. (2018). Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 114, 590–600. doi: 10.1093/cvr/cvy010
Fajas, L., Auboeuf, D., Raspé, E., Schoonjans, K., Lefebvre, A. M., Saladin, R., et al. (1997). The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 272, 18779–18789. doi: 10.1074/jbc.272.30.18779
Falk, E. (2006). Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 47, C7–C12. doi: 10.1016/j.jacc.2005.09.068
Finn, A. V., Saeed, O., and Virmani, R. (2012). Macrophage subsets in human atherosclerosis. Circ. Res. 110:e64. doi: 10.1161/circresaha.112.268714
Fu, M., Zhu, X., Wang, Q., Zhang, J., Song, Q., Zheng, H., et al. (2001). Platelet-derived growth factor promotes the expression of peroxisome proliferator-activated receptor gamma in vascular smooth muscle cells by a phosphatidylinositol 3-kinase/Akt signaling pathway. Circ. Res. 89, 1058–1064. doi: 10.1161/hh2301.099642
Garin-Shkolnik, T., Rudich, A., Hotamisligil, G. S., and Rubinstein, M. (2014). FABP4 attenuates PPARgamma and adipogenesis and is inversely correlated with PPARgamma in adipose tissues. Diabetes 63, 900–911. doi: 10.2337/db13-0436
Ge, C., Zhao, G., Li, B., Li, Y., Cawthorn, W. P., MacDougald, O. A., et al. (2018). Genetic inhibition of PPARgamma S112 phosphorylation reduces bone formation and stimulates marrow adipogenesis. Bone 107, 1–9. doi: 10.1016/j.bone.2017.10.023
Gilardi, F., Giudici, M., Mitro, N., Maschi, O., Guerrini, U., Rando, G., et al. (2014). LT175 is a novel PPARalpha/gamma ligand with potent insulin-sensitizing effects and reduced adipogenic properties. J. Biol. Chem. 289, 6908–6920. doi: 10.1074/jbc.M113.506394
Gu, H. F., Li, N., Xu, Z. Q., Hu, L., Li, H., Zhang, R. J., et al. (2019). Chronic Unpredictable Mild Stress Promotes Atherosclerosis via HMGB1/TLR4-Mediated Downregulation of PPARgamma/LXRalpha/ABCA1 in ApoE(-/-) Mice. Front. Physiol. 10:165. doi: 10.3389/fphys.2019.00165
Hall, J. A., Ramachandran, D., Roh, H. C., DiSpirito, J. R., Belchior, T., Zushin, P. H., et al. (2020). Obesity-Linked PPARgamma S273 Phosphorylation Promotes Insulin Resistance through Growth Differentiation Factor 3. Cell Metab. 32, 665–675.e6. doi: 10.1016/j.cmet.2020.08.016
Haschemi, A., Chin, B. Y., Jeitler, M., Esterbauer, H., Wagner, O., Bilban, M., et al. (2011). Carbon monoxide induced PPARγ SUMOylation and UCP2 block inflammatory gene expression in macrophages. PLoS One 6:e26376. doi: 10.1371/journal.pone.0026376
Hernandez-Quiles, M., Broekema, M. F., and Kalkhoven, E. (2021). PPARgamma in Metabolism, Immunity, and Cancer: unified and Diverse Mechanisms of Action. Front. Endocrinol. 12:624112. doi: 10.3389/fendo.2021.624112
Higgins, L. S., and Depaoli, A. M. (2010). Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am. J. Clin. Nutr. 91, 267S–272S. doi: 10.3945/ajcn.2009.28449E
Hu, Y., Li, X., Zhi, X., Cong, W., Huang, B., Chen, H., et al. (2021). RANKL from bone marrow adipose lineage cells promotes osteoclast formation and bone loss. EMBO Rep. 22:e52481. doi: 10.15252/embr.202152481
Iankova, I., Petersen, R. K., Annicotte, J. S., Chavey, C., Hansen, J. B., Kratchmarova, I., et al. (2006). Peroxisome proliferator-activated receptor gamma recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol. 20, 1494–1505. doi: 10.1210/me.2005-0222
Janani, C., and Ranjitha Kumari, B. D. (2015). PPAR gamma gene–a review. Diabetes Metab. Syndr. 9, 46–50. doi: 10.1016/j.dsx.2014.09.015
Jang, J. Y., Bae, H., Lee, Y. J., Choi, Y. I., Kim, H. J., Park, S. B., et al. (2018). Structural Basis for the Enhanced Anti-Diabetic Efficacy of Lobeglitazone on PPARgamma. Sci. Rep. 8:31. doi: 10.1038/s41598-017-18274-1
Jennewein, C., Kuhn, A. M., Schmidt, M. V., Meilladec-Jullig, V., von Knethen, A., Gonzalez, F. J., et al. (2008). Sumoylation of peroxisome proliferator-activated receptor gamma by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from kappaB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 181, 5646–5652. doi: 10.4049/jimmunol.181.8.5646
Johnson, J. L. (2017). Metalloproteinases in atherosclerosis. Eur. J. Pharmacol. 816, 93–106. doi: 10.1016/j.ejphar.2017.09.007
Kalliora, C., Kyriazis, I. D., Oka, S. I., Lieu, M. J., Yue, Y., Area-Gomez, E., et al. (2019). Dual peroxisome-proliferator-activated-receptor-alpha/gamma activation inhibits SIRT1-PGC1alpha axis and causes cardiac dysfunction. JCI Insight 5:e129556. doi: 10.1172/jci.insight.129556
Katafuchi, T., Holland, W. L., Kollipara, R. K., Kittler, R., Mangelsdorf, D. J., and Kliewer, S. A. (2018). PPARgamma-K107 SUMOylation regulates insulin sensitivity but not adiposity in mice. Proc. Natl. Acad. Sci. U. S. A. 115, 12102–12111. doi: 10.1073/pnas.1814522115
Kaul, U., Parmar, D., Manjunath, K., Shah, M., Parmar, K., Patil, K. P., et al. (2019). New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: integrated analysis of the real world evidence. Cardiovasc. Diabetol. 18:80. doi: 10.1186/s12933-019-0884-3
Kilroy, G., Kirk-Ballard, H., Carter, L. E., and Floyd, Z. E. (2012). The ubiquitin ligase Siah2 regulates PPARγ activity in adipocytes. Endocrinology 153, 1206–1218. doi: 10.1210/en.2011-1725
Kim, J. H., Park, K. W., Lee, E. W., Jang, W. S., Seo, J., Shin, S., et al. (2014). Suppression of PPARγ through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ. 21, 594–603. doi: 10.1038/cdd.2013.181
Kim, M. Y., Woo, E. M., Chong, Y. T., Homenko, D. R., and Kraus, W. L. (2006). Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol. Endocrinol. 20, 1479–1493. doi: 10.1210/me.2005-0531
Kolli, V., Stechschulte, L. A., Dowling, A. R., Rahman, S., Czernik, P. J., and Lecka-Czernik, B. (2014). Partial agonist, telmisartan, maintains PPARγ serine 112 phosphorylation, and does not affect osteoblast differentiation and bone mass. PLoS One 9:e96323. doi: 10.1371/journal.pone.0096323
Kraakman, M. J., Liu, Q., Postigo-Fernandez, J., Ji, R., Kon, N., Larrea, D., et al. (2018). PPARgamma deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects. J. Clin. Invest. 128, 2600–2612. doi: 10.1172/JCI98709
Kvandová, M., Majzúnová, M., and Dovinová, I. (2016). The role of PPARgamma in cardiovascular diseases. Physiol. Res. 65, S343–S363. doi: 10.33549/physiolres.933439
Laghezza, A., Piemontese, L., Cerchia, C., Montanari, R., Capelli, D., Giudici, M., et al. (2018). Identification of the First PPARalpha/gamma Dual Agonist Able To Bind to Canonical and Alternative Sites of PPARgamma and To Inhibit Its Cdk5-Mediated Phosphorylation. J. Med. Chem. 61, 8282–8298. doi: 10.1021/acs.jmedchem.8b00835
Law, R. E., Goetze, S., Xi, X. P., Jackson, S., Kawano, Y., Demer, L., et al. (2000). Expression and function of PPARgamma in rat and human vascular smooth muscle cells. Circulation 101, 1311–1318. doi: 10.1161/01.cir.101.11.1311
Lee, D. H., Huang, H., Choi, K., Mantzoros, C., and Kim, Y. B. (2012). Selective PPARγ modulator INT131 normalizes insulin signaling defects and improves bone mass in diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 302, E552–E560. doi: 10.1152/ajpendo.00569.2011
Lehman, J. J., Barger, P. M., Kovacs, A., Saffitz, J. E., Medeiros, D. M., and Kelly, D. P. (2000). Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856. doi: 10.1172/jci10268
Lehrke, M., and Lazar, M. A. (2005). The many faces of PPARgamma. Cell 123, 993–999. doi: 10.1016/j.cell.2005.11.026
Li, A. C., Binder, C. J., Gutierrez, A., Brown, K. K., Plotkin, C. R., Pattison, J. W., et al. (2004). Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J. Clin. Invest. 114, 1564–1576. doi: 10.1172/jci200418730
Li, D., Zhang, L., Xu, L., Liu, L., He, Y., Zhang, Y., et al. (2017). WIP1 phosphatase is a critical regulator of adipogenesis through dephosphorylating PPARγ serine 112. Cell. Mol. Life Sci. 74, 2067–2079. doi: 10.1007/s00018-016-2450-4
Li, J. J., Wang, R., Lama, R., Wang, X., Floyd, Z. E., Park, E. A., et al. (2016). Ubiquitin Ligase NEDD4 Regulates PPARγ Stability and Adipocyte Differentiation in 3T3-L1 Cells. Sci. Rep. 6:38550. doi: 10.1038/srep38550
Li, P., Fan, W., Xu, J., Lu, M., Yamamoto, H., Auwerx, J., et al. (2011). Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 147, 815–826. doi: 10.1016/j.cell.2011.09.050
Libby, P., Bornfeldt, K. E., and Tall, A. R. (2016). Atherosclerosis: successes, Surprises, and Future Challenges. Circ. Res. 118, 531–534. doi: 10.1161/CIRCRESAHA.116.308334
Lim, S., Ahn, B. Y., Chung, S. S., Park, H. S., Cho, B. J., Kim, M., et al. (2009). Effect of a peroxisome proliferator-activated receptor gamma sumoylation mutant on neointimal formation after balloon injury in rats. Atherosclerosis 206, 411–417. doi: 10.1016/j.atherosclerosis.2009.02.031
Lim, S., Lee, K. S., Lee, J. E., Park, H. S., Kim, K. M., Moon, J. H., et al. (2015). Effect of a new PPAR-gamma agonist, lobeglitazone, on neointimal formation after balloon injury in rats and the development of atherosclerosis. Atherosclerosis 243, 107–119. doi: 10.1016/j.atherosclerosis.2015.08.037
Liu, D., Wang, X., Zhang, M., Tian, J., Liu, M., Jin, T., et al. (2020). WISP1 alleviates lipid deposition in macrophages via the PPARgamma/CD36 pathway in the plaque formation of atherosclerosis. J. Cell. Mol. Med. 24, 11729–11741. doi: 10.1111/jcmm.15783
Liu, L., Fan, L., Chan, M., Kraakman, M. J., Yang, J., Fan, Y., et al. (2020). PPARgamma Deacetylation Confers the Antiatherogenic Effect and Improves Endothelial Function in Diabetes Treatment. Diabetes 69, 1793–1803. doi: 10.2337/db20-0217
Lu, Y., Zhou, Q., Shi, Y., Liu, J., Zhong, F., Hao, X., et al. (2013). SUMOylation of PPARγ by rosiglitazone prevents LPS-induced NCoR degradation mediating down regulation of chemokines expression in renal proximal tubular cells. PLoS One 8:e79815. doi: 10.1371/journal.pone.0079815
Luo, F., Wang, Z. H., Du, L. L., and Wang, J. (2007). [Effects of PPAR-gamma agonist and MMP-2 on formation of atherosclerosis plaque in rabbits]. Zhonghua Bing Li Xue Za Zhi 36, 556–557.
Machado-Oliveira, G., Ramos, C., Marques, A. R. A., and Vieira, O. V. (2020). Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 9:2146. doi: 10.3390/cells9102146
Marfella, R., D’Amico, M., Esposito, K., Baldi, A., Di Filippo, C., Siniscalchi, M., et al. (2006). The ubiquitin-proteasome system and inflammatory activity in diabetic atherosclerotic plaques: effects of rosiglitazone treatment. Diabetes 55, 622–632. doi: 10.2337/diabetes.55.03.06.db05-0832
Martens, F. M., Visseren, F. L., Lemay, J., de Koning, E. J., and Rabelink, T. J. (2002). Metabolic and additional vascular effects of thiazolidinediones. Drugs 62, 1463–1480. doi: 10.2165/00003495-200262100-00004
Meredith, D., Panchatcharam, M., Miriyala, S., Tsai, Y. S., Morris, A. J., Maeda, N., et al. (2009). Dominant-negative loss of PPARgamma function enhances smooth muscle cell proliferation, migration, and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 29, 465–471. doi: 10.1161/ATVBAHA.109.184234
Montanari, R., Capelli, D., Yamamoto, K., Awaishima, H., Nishikata, K., Barendregt, A., et al. (2020). Insights into PPARgamma Phosphorylation and Its Inhibition Mechanism. J. Med. Chem. 63, 4811–4823. doi: 10.1021/acs.jmedchem.0c00048
Nakaya, H., Summers, B. D., Nicholson, A. C., Gotto, A. M. Jr., Hajjar, D. P., and Han, J. (2009). Atherosclerosis in LDLR-knockout mice is inhibited, but not reversed, by the PPARgamma ligand pioglitazone. Am. J. Pathol. 174, 2007–2014. doi: 10.2353/ajpath.2009.080611
Namgaladze, D., Kemmerer, M., von Knethen, A., and Brüne, B. (2013). AICAR inhibits PPARγ during monocyte differentiation to attenuate inflammatory responses to atherogenic lipids. Cardiovasc. Res. 98, 479–487. doi: 10.1093/cvr/cvt073
Oppi, S., Nusser-Stein, S., Blyszczuk, P., Wang, X., Jomard, A., Marzolla, V., et al. (2020). Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARgamma signature. Eur. Heart J. 41, 995–1005. doi: 10.1093/eurheartj/ehz667
Osman, I., and Segar, L. (2016). Pioglitazone, a PPARgamma agonist, attenuates PDGF-induced vascular smooth muscle cell proliferation through AMPK-dependent and AMPK-independent inhibition of mTOR/p70S6K and ERK signaling. Biochem. Pharmacol. 101, 54–70. doi: 10.1016/j.bcp.2015.11.026
Ozasa, H., Ayaori, M., Iizuka, M., Terao, Y., Uto-Kondo, H., Yakushiji, E., et al. (2011). Pioglitazone enhances cholesterol efflux from macrophages by increasing ABCA1/ABCG1 expressions via PPARγ/LXRα pathway: findings from in vitro and ex vivo studies. Atherosclerosis 219, 141–150. doi: 10.1016/j.atherosclerosis.2011.07.113
Pascual, G., Fong, A. L., Ogawa, S., Gamliel, A., Li, A. C., Perissi, V., et al. (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763. doi: 10.1038/nature03988
Pourcet, B., Staels, B., and Glineur, C. (2013). PPAR SUMOylation: some useful experimental tips. Methods Mol. Biol. 952, 145–161. doi: 10.1007/978-1-62703-155-4_10
Qiang, L., Wang, L., Kon, N., Zhao, W., Lee, S., Zhang, Y., et al. (2012). Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 150, 620–632. doi: 10.1016/j.cell.2012.06.027
Reinhold, S., Blankesteijn, W. M., and Foulquier, S. (2020). The Interplay of WNT and PPARgamma Signaling in Vascular Calcification. Cells 9:2658. doi: 10.3390/cells9122658
Ribeiro Filho, H. V., Guerra, J. V., Cagliari, R., Batista, F. A. H., Le Maire, A., Oliveira, P. S. L., et al. (2019). Exploring the mechanism of PPARgamma phosphorylation mediated by CDK5. J. Struct. Biol. 207, 317–326. doi: 10.1016/j.jsb.2019.07.007
Silva, J. C. (2018). GQ-11: a new PPAR agonist improves obesity-induced metabolic alterations in LDLr(-/-) mice. Int. J. Obes. 42, 1062–1072.
Silva, J. C., Cesar, F. A., de Oliveira, E. M., Turato, W. M., Tripodi, G. L., Castilho, G., et al. (2016). New PPARgamma partial agonist improves obesity-induced metabolic alterations and atherosclerosis in LDLr(-/-) mice. Pharmacol. Res. 104, 49–60. doi: 10.1016/j.phrs.2015.12.010
Silva, J. C., de Oliveira, E. M., Turato, W. M., Trossini, G. H. G., Maltarollo, V. G., Pitta, M. G. R., et al. (2018). GQ-11: a new PPAR agonist improves obesity-induced metabolic alterations in LDLr(-/-) mice. Int. J. Obes. 42, 1062–1072. doi: 10.1038/s41366-018-0011-7
Sohn, J. H., Kim, J. I., Jeon, Y. G., Park, J., and Kim, J. B. (2018). Effects of Three Thiazolidinediones on Metabolic Regulation and Cold-Induced Thermogenesis. Mol. Cells 41, 900–908. doi: 10.14348/molcells.2018.0294
Sohn, K. A., Cruciani-Guglielmacci, C., Kassis, N., Clement, L., Ouali, F., and Cauzac, M. (2009). S26948, a new specific peroxisome proliferator activated receptor gamma modulator improved in vivo hepatic insulin sensitivity in 48 h lipid infused rats. Eur. J. Pharmacol. 608, 104–111. doi: 10.1016/j.ejphar.2009.02.033
Song, J. W., Nam, H. S., Ahn, J. W., Park, H. S., Kang, D. O., Kim, H. J., et al. (2021). Macrophage targeted theranostic strategy for accurate detection and rapid stabilization of the inflamed high-risk plaque. Theranostics 11, 8874–8893. doi: 10.7150/thno.59759
Srivastava, R. A. (2011). Evaluation of anti-atherosclerotic activities of PPAR-alpha, PPAR-gamma, and LXR agonists in hyperlipidemic atherosclerosis-susceptible F(1)B hamsters. Atherosclerosis 214, 86–93. doi: 10.1016/j.atherosclerosis.2010.10.033
Sueyoshi, S., Mitsumata, M., Kusumi, Y., Niihashi, M., Esumi, M., Yamada, T., et al. (2010). Increased expression of peroxisome proliferator-activated receptor (PPAR)-alpha and PPAR-gamma in human atherosclerosis. Pathol. Res. Pract. 206, 429–438. doi: 10.1016/j.prp.2010.01.010
Tabas, I. (2017). 2016 Russell Ross Memorial Lecture in Vascular Biology: molecular-Cellular Mechanisms in the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 37, 183–189. doi: 10.1161/ATVBAHA.116.308036
Tian, L., Wang, C., Hagen, F. K., Gormley, M., Addya, S., Soccio, R., et al. (2014). Acetylation-defective mutant of Pparγ is associated with decreased lipid synthesis in breast cancer cells. Oncotarget 5, 7303–7315. doi: 10.18632/oncotarget.2371
Tsuboi, T., Lu, R., Yonezawa, T., Watanabe, A., Woo, J. T., Abe-Dohmae, S., et al. (2020). Molecular mechanism for nobiletin to enhance ABCA1/G1 expression in mouse macrophages. Atherosclerosis 297, 32–39. doi: 10.1016/j.atherosclerosis.2020.01.024
Vallee, A., Vallee, J. N., and Lecarpentier, Y. (2019). Metabolic reprogramming in atherosclerosis: opposed interplay between the canonical WNT/beta-catenin pathway and PPARgamma. J. Mol. Cell. Cardiol. 133, 36–46. doi: 10.1016/j.yjmcc.2019.05.024
van Beekum, O., Fleskens, V., and Kalkhoven, E. (2009). Posttranslational modifications of PPAR-gamma: fine-tuning the metabolic master regulator. Obesity 17, 213–219. doi: 10.1038/oby.2008.473
Verrier, E., Wang, L., Wadham, C., Albanese, N., Hahn, C., Gamble, J. R., et al. (2004). PPARgamma agonists ameliorate endothelial cell activation via inhibition of diacylglycerol-protein kinase C signaling pathway: role of diacylglycerol kinase. Circ. Res. 94, 1515–1522. doi: 10.1161/01.Res.0000130527.92537.06
Vigueira, P. A., McCommis, K. S., Hodges, W. T., Schweitzer, G. G., Cole, S. L., Oonthonpan, L., et al. (2017). The beneficial metabolic effects of insulin sensitizers are not attenuated by mitochondrial pyruvate carrier 2 hypomorphism. Exp. Physiol. 102, 985–999. doi: 10.1113/EP086380
Viles-Gonzalez, J. F., Choi, B. G., Fuster, V., and Badimon, J. J. (2004). Peroxisome proliferator-activated receptor ligands in atherosclerosis. Expert Opin. Investig. Drugs 13, 1393–1403. doi: 10.1517/13543784.13.11.1393
Wadosky, K. M., and Willis, M. S. (2012). The story so far: post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 302, H515–H526. doi: 10.1152/ajpheart.00703.2011
Wang, J., Uryga, A. K., Reinhold, J., Figg, N., Baker, L., Finigan, A., et al. (2015). Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 132, 1909–1919. doi: 10.1161/circulationaha.115.016457
Wang, M., and Tafuri, S. (2003). Modulation of PPARgamma activity with pharmaceutical agents: treatment of insulin resistance and atherosclerosis. J. Cell. Biochem. 89, 38–47. doi: 10.1002/jcb.10492
Watanabe, M., Takahashi, H., Saeki, Y., Ozaki, T., Itoh, S., Suzuki, M., et al. (2015). The E3 ubiquitin ligase TRIM23 regulates adipocyte differentiation via stabilization of the adipogenic activator PPARγ. Elife 4:e05615. doi: 10.7554/eLife.05615
Xie, X., Chen, W., Zhang, N., Yuan, M., Xu, C., Zheng, Z., et al. (2017). Selective Tissue Distribution Mediates Tissue-Dependent PPARγ Activation and Insulin Sensitization by INT131, a Selective PPARγ Modulator. Front. Pharmacol. 8:317. doi: 10.3389/fphar.2017.00317
Xu, C., Wang, L. L., Liu, H. Y., Zhou, X. B., Cao, Y. L., and Li, S. (2006). C333H, a novel PPARalpha/gamma dual agonist, has beneficial effects on insulin resistance and lipid metabolism. Acta Pharmacol. Sin. 27, 223–228. doi: 10.1111/j.1745-7254.2006.00263.x
Yamaguchi, K., Wakatsuki, T., Soeki, T., Niki, T., Taketani, Y., Oeduka, H., et al. (2014). Effects of telmisartan on inflammatory cytokines and coronary plaque component as assessed on integrated backscatter intravascular ultrasound in hypertensive patients. Circ. J. 78, 240–247. doi: 10.1253/circj.cj-13-0741
Yang, H., Cheng, J., Song, Z., Li, X., Zhang, Z., Mai, Y., et al. (2013). The anti-adipogenic effect of PGRN on porcine preadipocytes involves ERK1,2 mediated PPARgamma phosphorylation. Mol. Biol. Rep. 40, 6863–6872. doi: 10.1007/s11033-013-2804-z
Yang, X., Yao, H., Chen, Y., Sun, L., Li, Y., Ma, X., et al. (2015). Inhibition of Glutathione Production Induces Macrophage CD36 Expression and Enhances Cellular-oxidized Low Density Lipoprotein (oxLDL) Uptake. J. Biol. Chem. 290, 21788–21799. doi: 10.1074/jbc.M115.654582
Yin, R., Dong, Y. G., and Li, H. L. (2006). PPARgamma phosphorylation mediated by JNK MAPK: a potential role in macrophage-derived foam cell formation. Acta Pharmacol. Sin. 27, 1146–1152. doi: 10.1111/j.1745-7254.2006.00359.x
Yu, X. H., Fu, Y. C., Zhang, D. W., Yin, K., and Tang, C. K. (2013). Foam cells in atherosclerosis. Clin. Chim. Acta 424, 245–252. doi: 10.1016/j.cca.2013.06.006
Zadelaar, A. S., Boesten, L. S., Jukema, J. W., van Vlijmen, B. J., Kooistra, T., Emeis, J. J., et al. (2006). Dual PPARalpha/gamma agonist tesaglitazar reduces atherosclerosis in insulin-resistant and hypercholesterolemic ApoE*3Leiden mice. Arterioscler. Thromb. Vasc. Biol. 26, 2560–2566. doi: 10.1161/01.Atv.0000242904.34700.66
Zhang, L., and Chawla, A. (2004). Role of PPARgamma in macrophage biology and atherosclerosis. Trends Endocrinol. Metab. 15, 500–505. doi: 10.1016/j.tem.2004.10.006
Zhang, L. L., Gao, C. Y., Fang, C. Q., Wang, Y. J., Gao, D., Yao, G. E., et al. (2011). PPARgamma attenuates intimal hyperplasia by inhibiting TLR4-mediated inflammation in vascular smooth muscle cells. Cardiovasc. Res. 92, 484–493. doi: 10.1093/cvr/cvr238
Zhang, Y., Shi, X., Han, J., Peng, W., Fang, Z., Zhou, Y., et al. (2021). Convallatoxin Promotes M2 Macrophage Polarization to Attenuate Atherosclerosis Through PPARgamma-Integrin alphavbeta5 Signaling Pathway. Drug Des. Devel. Ther. 15, 803–812. doi: 10.2147/DDDT.S288728
Zhang, Y., Wang, Y., Li, X., Gu, K., Li, M., Zhang, Y., et al. (2020). WSF-7 Inhibits Obesity-Mediated PPARγ Phosphorylation and Improves Insulin Sensitivity in 3T3-L1 Adipocytes. Biol. Pharm. Bull. 43, 526–532. doi: 10.1248/bpb.b19-00986
Zhang, Y., Yang, X., Bian, F., Wu, P., Xing, S., Xu, G., et al. (2014). TNF-alpha promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: crosstalk between NF-kappaB and PPAR-gamma. J. Mol. Cell. Cardiol. 72, 85–94. doi: 10.1016/j.yjmcc.2014.02.012
Keywords: PPARγ, atherosclerosis, post-translational modifications, selective modulators, cardiovascular disease
Citation: Yin L, Wang L, Shi Z, Ji X and Liu L (2022) The Role of Peroxisome Proliferator-Activated Receptor Gamma and Atherosclerosis: Post-translational Modification and Selective Modulators. Front. Physiol. 13:826811. doi: 10.3389/fphys.2022.826811
Received: 01 December 2021; Accepted: 11 February 2022;
Published: 02 March 2022.
Edited by:
Geoffrey A. Head, Baker Heart and Diabetes Institute, AustraliaReviewed by:
Makoto Makishima, Nihon University, JapanCopyright © 2022 Yin, Wang, Shi, Ji and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Longhua Liu, bGl1bG9uZ2h1YUBzdXMuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.