 Yiru Wang
Yiru Wang Siyang Liu
Siyang Liu Qingquan Liu
Qingquan Liu Yongman Lv
Yongman Lv
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Physiol., 04 March 2022
Sec. Renal Physiology and Pathophysiology
Volume 13 - 2022 | https://doi.org/10.3389/fphys.2022.826686
Acute kidney injury (AKI) is a common disorder in critically ill hospitalized patients. Its main pathological feature is the activation of the sympathetic nervous system and the renin-angiotensin system (RAS). This disease shows a high fatality rate. The reason is that only renal replacement therapy and supportive care can reduce the impact of the disease, but those measures cannot significantly improve the mortality. This review focused on a generalization of the interaction between acute kidney injury and the central nervous system (CNS). It was found that the CNS further contributes to kidney injury by regulating sympathetic outflow and oxidative stress in response to activation of the RAS and increased pro-inflammatory factors. Experimental studies suggested that inhibiting sympathetic activity and RAS activation in the CNS and blocking oxidative stress could effectively reduce the damage caused by AKI. Therefore, it is of significant interest to specify the mechanism on how the CNS affects AKI, as we could use such mechanism as a target for clinical interventions to further reduce the mortality and improve the complications of AKI.
Systematic Review Registration: [www.ClinicalTrials.gov], identifier [registration number].
Acute kidney injury (AKI) is a complex syndrome marked by sudden decrease in glomerular filtration rate (GFR). Generally, the disease shows a slight and sustained elevation in serum creatinine concentration, oliguria or anuric renal failure (Levey and James, 2017). According to the anatomical location of primary cause, the etiologies of AKI are roughly classified into three categories: pre-renal, intra-renal, and post-renal origin (Kellum et al., 2013). The hall marks of AKI pathophysiology include renal microvasculature injury (Maringer and Sims-Lucas, 2016), maladaptive repair (Ferenbach and Bonventre, 2015) and activation of neurohumoral mechanisms, like sympathetic nervous system (SNS), renin–angiotensin system (RAS), systemic inflammatory response and oxidative stress (Grisk, 2020; Grivei et al., 2020; Legrand and Bokoch, 2021). Under normal conditions, interaction between nervous system and kidney is reasonable to maintain a normal physiological balance. Activation of central nervous system (CNS) and RAS promotes the maintenance of renal homeostasis osmolality, sodium balance and renal blood flow by regulating the secretion of vasopressin and modulating efferent renal sympathetic nerve activity (ERSNA) to induce renin secretion and decrease urinary sodium excretion (Tanaka and Okusa, 2020). Under pathological conditions, such as hypertension, AKI and chronic kidney disease (CKD), the interaction between the nervous system and the kidney could be out of balance. Such imbalance further leads to loss of normal homeostasis. Damaged kidneys usually involve acute activation of the SNS, which in turn may further accelerate the loss of renal function. These adaptive changes eventually become a deleterious factor for the progression of kidney damage (Zhang et al., 2018; Cao et al., 2020).
At present, clinical treatment of AKI was primarily symptomatic and supportive treatment, including dialysis, renal replacement therapy (RRT), and fluid administration according to personal circumstances. There was no specific therapy for rapid recovery from AKI. That was the reason why the mortality of AKI remained high. Current AKI therapy primarily focused on dysfunction, cell death, oxidative stress and inflammation of renal tubule epitheliums (Aparicio-Trejo et al., 2020). In recent years, researchers on AKI therapy targets devoted their efforts in fields concerning the neuroimmune system (Inoue et al., 2016). The activation of the CNS caused by AKI has been shown to relate to more severe renal impairment (Campese and Kogosov, 1995). Furthermore, AKI has deleterious effects on the CNS (e.g., uremic encephalopathy and stroke) and this disease can result in higher mortality, lower quality of life and higher health care costs (Nongnuch et al., 2014). Therefore, modulating the activation of CNS in AKI might be a new and promising therapeutic target. The purposes of this review were to describe the mechanisms of altered CNS pathways due to AKI and to consider the potential therapeutic implications.
Kidney is densely under control by afferent renal sensory nerve and efferent renal sympathetic nerve that run in the outer layer of renal artery and around kidney (Tellez et al., 2013). There is interaction between the renal afferent and efferent nerves. The pathway is mediated by norepinephrine (NE), which increases and decreases afferent renal nerve activity (ARNA) by activating renal α1-and α2-adrenoceptors, respectively (Kopp et al., 2011). The renorenal reflex is a negative feedback loop to maintain renal function in a physiological state, consisting of a combination of renal afferent and efferent nerves (Larsen et al., 2014). ERSNA increases renin secretion rate and renal tubular sodium reabsorption, and it reduces renal blood flow while increasing ARNA (Colindres et al., 1980). Activation of ARNA triggers inhibitory renorenal reflex and decreases ERSNA. Those activities would lead to diuresis and minimize sodium retention (Kopp et al., 2007; Ma et al., 2008; Johns et al., 2011).
Previous study showed that ARNA has a predominantly inhibitory effect in normal rats (Colindres et al., 1980). Renal afferent sensory nerves are activated by renal pelvic pressure as well as stimuli such as NE, producing an inhibition on ERSNA to minimize sodium retention (Kopp et al., 2011). This response to renal afferent sensory nerves is modulated by sodium intake. The mechanisms include increased activation of angiotensin II type 1 (AT1) receptors and α2 adrenergic receptors under low sodium diet conditions, thereby inhibiting ARNA, and increased activation of endothelin-B receptors under high sodium diet conditions, thereby enhancing the responsiveness of afferent renal sensory nerves (Kopp, 2015). In summary, in healthy individuals, afferent sensory nerve, efferent sympathetic nerve and the kidney work together to keep body sodium and water balance by inhibitory renorenal reflex pathway in response to varying sodium intake.
Acute kidney injury is defined as an abrupt decrease in renal function resulting from a combination of multiple factors (Makris and Spanou, 2016). Renal ischemia-reperfusion injury (IRI) is one of the most common causes of AKI in hospitalized patients. Animal studies have shown that the inhibitory renorenal reflex pathway is impaired in the IRI model, and that the absence of the inhibitory renorenal reflex leads to an inappropriate increase in ERSNA (Kopp and Buckley-Bleiler, 1989). Increased ERSNA stimulates the release of renin from the juxtaglomerular cell, resulting in increased production of angiotensin II (Ang II), which in turn triggers renal vasoconstriction (Naito et al., 2008). Renal vasoconstriction leads to a decrease in renal blood flow and GFR, thereby aggravating ischemic renal damage (Stefanska et al., 2016). In addition, Ang II also stimulates aldosterone release, which strongly enhances sodium and water reabsorption in the distal tubule and the collecting duct, leading to sodium and water retention (Satou et al., 2020).
Increased ERSNA leads to increased release of plasma NE (Fujii et al., 2003). In recent years, NE was found to be associated with inflammation and progressive tissue injury after AKI. Increased ERSNA during ischemia and post-reperfusion renal venous NE overflow plays a significant role in the development of ischemic AKI. A study of rat IRI model showed that plasma NE levels were significantly elevated after AKI and the levels were differed by sex. More specifically, male rats had a more pronounced increase than females did the degree of renal histological damage was also significantly different, which correlated with NE levels (Tanaka et al., 2013). Tyrosine hydroxylase (TH) is the rate-limiting enzyme for NE synthesis. Other studies also found increased expression of TH in IRI model (Cronin et al., 1978; Bellomo and Giantomasso, 2001). In addition, a study showed that renal NE levels remained elevated until day 16 after unilateral renal ischemia-reperfusion, whereas renal denervation significantly reduced renal NE expression and attenuated apoptosis and tubular injury. The study also revealed that denervated kidneys had a significant reduction in GFR after NE treatment as compared with intact kidneys (Kim and Padanilam, 2015). Early experimental studies showed that renal arterial infusion of norepinephrine could induce ischemic AKI in rats by causing renal vasoconstriction (Conger et al., 1991). In conclusion, evidence from experimental studies suggested that renal sympathetic activation might play a major role in the development and progression of AKI.
As an important sensory organ, the kidney has abundant baroreceptors and chemoreceptors, as well as a large number of afferent sensory innervation. It communicates with the CNS via afferent sensory nerves (Webb and Brody, 1987). Renal baroreceptors sense alterations in renal perfusion and intrarenal pressure, and renal chemoreceptors receive stimuli from ischemic metabolites or uremic toxins (Koepke and DiBona, 1985). Renal afferent nerves have been shown to project directly to regions of the CNS such as the lateral hypothalamic area, the paraventricular nucleus (PVN), and indirectly to other areas of the hypothalamus (Calaresu and Ciriello, 1981). When the renal afferent nerve receives adverse stimulation due to the increase of adenosine and other metabolites caused by ischemia, ERSNA will reflexively increase (Katholi et al., 1984). Related studies have shown that renal injury activates renal afferent sensory nerve signals, which activate norepinephrine neurons in the posterior hypothalamus, lateral hypothalamus and the locus coeruleus (LC) (Campese and Kogosov, 1995). Notably, among brainstem noradrenergic neurons, the LC is the largest source of norepinephrine production (Chandler, 2016). In contrast, Selective renal afferent block inhibit the increase in NE levels in the posterior hypothalamus and the LC by cutting the dorsal root of T10-L2 without damaging the efferent sympathetic nerve (Campese and Kogosov, 1995). The findings further demonstrated that renal afferent nerve would activate the CNS and enhance sympathetic activity.
The hypothalamus plays a crucial role in the regulation of fluid homeostasis, with the PVN being a crucial brain region in the hypothalamus (Badoer, 2010). The PVN is located near the third ventricle and is an essential integrated site involved in endocrine and neural control. PVN consists of two types of neurons, magnocellular neurons and parvocellular neurons. The magnocellular neurons project to the posterior pituitary gland and release arginine vasopressin (AVP), which are involved in the regulation of fluid homeostasis (Swanson and Sawchenko, 1983), while the parvocellular neurons play a prominent role in the regulation of sympathetic nerve activity (Shafton et al., 1998).
It has been confirmed that during stimulation of the afferent renal nerve, the firing frequency of magnocellular neurons in the PVN increases (Ciriello, 1998). Stimulation of magnocellular neurons in the PVN by the afferent renal nerve projects to the pituitary gland, resulting in AVP release upon afferent renal nerve activation. Electrical stimulation of the afferent renal nerve increases the firing rate of PVN neurons that project directly to the rostral ventrolateral medulla (RVLM). It suggested that stimulation of afferent renal sensory nerves might lead to activation of neurons within the PVN (Nishi et al., 2017). The RVLM is a decisive nucleus in sympathetic regulation. The majority of renal sympathetic premotor neurons are located in the RVLM the most represented neurons are catecholamine synthesizing cells in the cell population of the RVLM (Ding et al., 1993). Studies have confirmed that electrical stimulation of renal afferent nerve causes the activation of TH-positive containing neurons in the RVLM and non-TH neurons in the NTS (Nishi et al., 2017).
Different neurons in the PVN play different roles, with pre-sympathetic neurons mediating sympathetic excitation and GABAergic or NO-producing interneurons mediating sympathetic inhibition (Affleck et al., 2012). Treatment of ischemic AKI rats with intravenous GABA inhibited the enhancement of ERSNA during ischemia and the increase of NE overflow after reperfusion (Kobuchi et al., 2009). There are two known GABA receptor subtypes: GABAA and GABAB. The GABAA receptors work by coupling to the ligand-gated Cl–channel, and the GABAB receptors are the archetype of heterodimeric G protein-coupled receptors (Johnston, 1996). Further studies demonstrated that the protective effect of intravenous GABA on AKI is mediated by inhibition of central sympathetic outflow, and the prevention of the AKI long-term kidney dysfunction is achieved through activation of GABAB rather than GABAA receptors (Kobuchi et al., 2015). Furthermore, removal of the common renal nerve, which includes afferent sensory nerves and efferent sympathetic nerves, increased GABAergic neuronal activity in the PVN (Chen et al., 2016). This suggested that the improvement in GABAergic neurotransmission in the PVN is partly due to the absence of afferent renal sensory nerve.
The above experimental studies allowed us to reasonably speculate that AKI stimulates PVN pre-sympathetic neuronal activity through activation of afferent renal nerves in processes including ischemia and metabolites. The stimulus then activates catecholaminergic neurons in the RVLM and enhances renal sympathetic activity. These would cause renal vasoconstriction, decrease GFR, and exacerbate renal tissue injury (Figure 1). However, additional exploration of the underlying mechanisms was still needed.
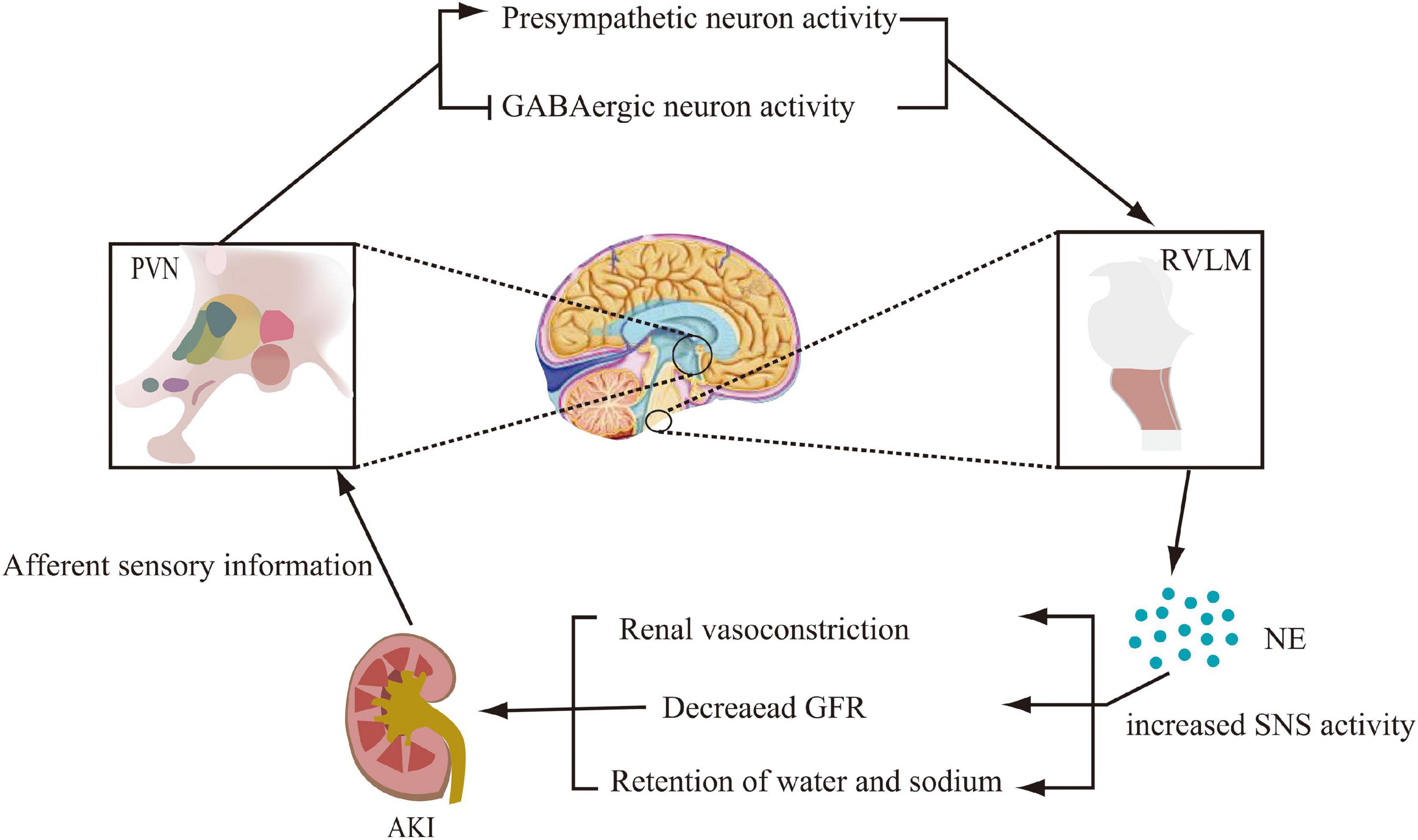
Figure 1. Modification of the central nervous system pathways involved in sympathetic activation during acute kidney injury (AKI). Activation of renal afferent sensory nerves, which project to the paraventricular nucleus (PVN) of the hypothalamus, promotes pre-sympathetic neuronal activity and inhibits GABAergic neuronal activity. By projecting from the PVN to norepinephrinergic neurons in the rostral ventrolateral medulla (RVLM), norepinephrine (NE) levels are increased, thereby promoting sympathetic nervous system (SNS) activity, which further leads to renal vasoconstriction, decreased glomerular filtration rate (GFR), and sodium and water retention.
Since activation of the renin-angiotensin system (RAS) is strongly up regulated in AKI, it is particularly important to understand the mechanisms involved. Previous studies have shown that activation of the intrarenal RAS is mediated by overexpression of the brain RAS, which in turn activates the SNS (Campese et al., 2002). Under physiological conditions, Ang II regulates the RAS through negative feedback and inhibits renin release directly (Johns et al., 1990). However, in acute kidney injury, Ang II plays an indirect role in promoting the release of renin through sympathetic activation, thus forming a positive feedback pathway (Hering and Winklewski, 2017). Ischemic AKI mice showed increased brain TH synthesis and increased renal NE levels, indicating increased central sympathetic drive and enhanced ERSNA (Cao et al., 2017). PVN and RVLM are the sites of response that regulates ERSNA in renal IRI, and Ang II in this region acts via the oxidative stress pathway (Seifi et al., 2014). Ang II stimulates the AT1 receptors of PVN and RVLM to activate intracellular pathways, thereby stimulating NAD(P)H oxidase to mediate reactive oxygen species (ROS) synthesis to increase sympathetic outflow (Gao et al., 2004). Ang II was found to bind to the AT 1 receptor in the PVN to activate c-Src, which then modulates the effects of Ang II on sympathetic activity by activating NAD(P)H oxidase and subsequent superoxide anion production in the PVN. Microinjection of Ang II into the PVN increased c-Src activity, NAD(P)H oxidase activity and superoxide anion levels in the PVN of normal rats (Li and Pan, 2005; Han et al., 2011). Central sympathetic excitation during ischemic AKI is clearly dependent on the central RAS and ROS. In the ischemic AKI model, inhibition of central RAS/ROS with central RAS inhibitors or antioxidants prevented intrarenal RAS activation, decreased renal NE and reduced IRI-induced renal injury by approximately 60% (Cao et al., 2017), which demonstrated the importance of the central RAS/ROS activation pathway in AKI. Interestingly, the study also found that capsaicin treatment interrupted renal afferent innervation or renal denervation, inhibited brain-renal RAS axis activation and improved renal injury, implying that afferent renal sensory nerves play an important role in central sympathetic nerve excitation and renal injury. In addition, Ang II also exerts a regulatory effect on GABAergic neurons in the PVN. Ang II activates presynaptic AT-1 receptors, which increase the firing of PVN-output neurons projecting to the spinal cord and RVLM by inhibiting GABA release (Li and Pan, 2005). ROS, particularly superoxide anion, is closely associated with presynaptic modulatory signaling by Ang II on GABAergic synaptic inputs to pre-sympathetic neurons of the PVN, mediated by Gi/o proteins (Chen and Pan, 2007). In conclusion, the brain-renal RAS axis is an important part of the mechanism of renal injury caused by AKI, but its complete mechanism remains unclear.
Acute kidney injury is a systemic inflammatory response process. It involves activation of pro-inflammatory cytokines and chemokines and infiltration of neutrophils and macrophages (Bonventre and Zuk, 2004). Those processes could further exacerbate kidney injury and distant organ damage (Doi and Rabb, 2016). In a mouse model of renal ischemia, there was evidence of varying activation of numerous circulating cytokines, including KC, G-CSF, IL-6, IL-1β, and IL-12 (Grams and Rabb, 2012). Systemic administration of IL-1β increased TH mRNA expression in brainstem noradrenergic nuclei and increased central NE synthesis, thereby affecting sympathetic efferent activity (Sirivelu et al., 2012). Moreover, renal IRI increased the permeability of the blood-brain barrier (BBB), induced neuronal and microglia damage, and increased levels of pro-inflammatory cytokines including G-CSF, IL-1β in the brain (Liu et al., 2008). The increase in pro-inflammatory cytokines such as TNF-α and IL-1β in PVN activates RAS and ROS in the brain, increasing sympathetic activity (Kang et al., 2008; Shi et al., 2011). Ginsenoside has been shown to have anti-inflammatory and antioxidant effects, and, in AKI model induced by glycerol, the use of ginsenoside attenuated AKI-induced kidney and brain oxidative stress damage and decreased malondialdehyde (MDA) level, a marker of oxidative stress (Mao et al., 2020). Moreover, the use of the central ROS inhibitor tempol attenuated levels of oxidative stress and inflammation as well as renal tissue damage following renal IRI (Cao et al., 2017). The findings further demonstrated the protective effect of blocking central oxidative stress on the kidney. In addition, both systemic and centrally administered AT1 receptor blockers, losartan, might have beneficial effects on renal function by reducing serum creatinine and BUN levels (Liu et al., 2008; Sharifi et al., 2019). The main mechanism of its renoprotective effects might be that anti-inflammatory and antioxidant work by blocking the proliferation and activation of leukocytes and chemokines, which were thought to be directly stimulated by Ang II (Vaziri et al., 2007; Crowley and Rudemiller, 2017).
The central action of IL-1β is mediated through the nuclear factor-kappa B (NF-κB) pathway (Nadjar et al., 2003). IL-1β binds to its receptor and undergoes a phosphorylation cascade that translocates NF-κB to the nucleus (Nadjar et al., 2005). In addition, NF-κB activation is facilitated by NAD(P)H oxidase-derived ROS, which are induced by proinflammatory cytokines (Bonizzi et al., 1999; Gloire et al., 2006). An animal study demonstrated that AKI in rats could lead to upregulation of NF-κB and its downstream cyclic oxygenase-2 (COX2)/ prostaglandin E2 (PGE2) expression in brain tissue (Zhang et al., 2014). ROS stimulates COX2 and PGE2 activity via the NF-κB pathway, which in turn stimulates the synthesis of pro-inflammatory cytokines, angiotensinogen and AT1 receptor (Chen and Pan, 2007; Kang et al., 2008). Thus, by acting on the CNS, cytokines and RAS promote each other, creating a positive feedback loop. NF-κB activation promotes inflammatory factor production and NE synthesis in the PVN, while PVN-targeted injection of the NF-κB inhibitor, PDTC, attenuates NF-κB expression and sympathetic nerve activity (Wang et al., 2019). Thus, it could be inferred that central pro-inflammatory factor production due to AKI in turn affects tissue damage therein by increasing central sympathetic activity output, and that inhibition of the CNS inflammation might be a new therapeutic direction (Figure 2).
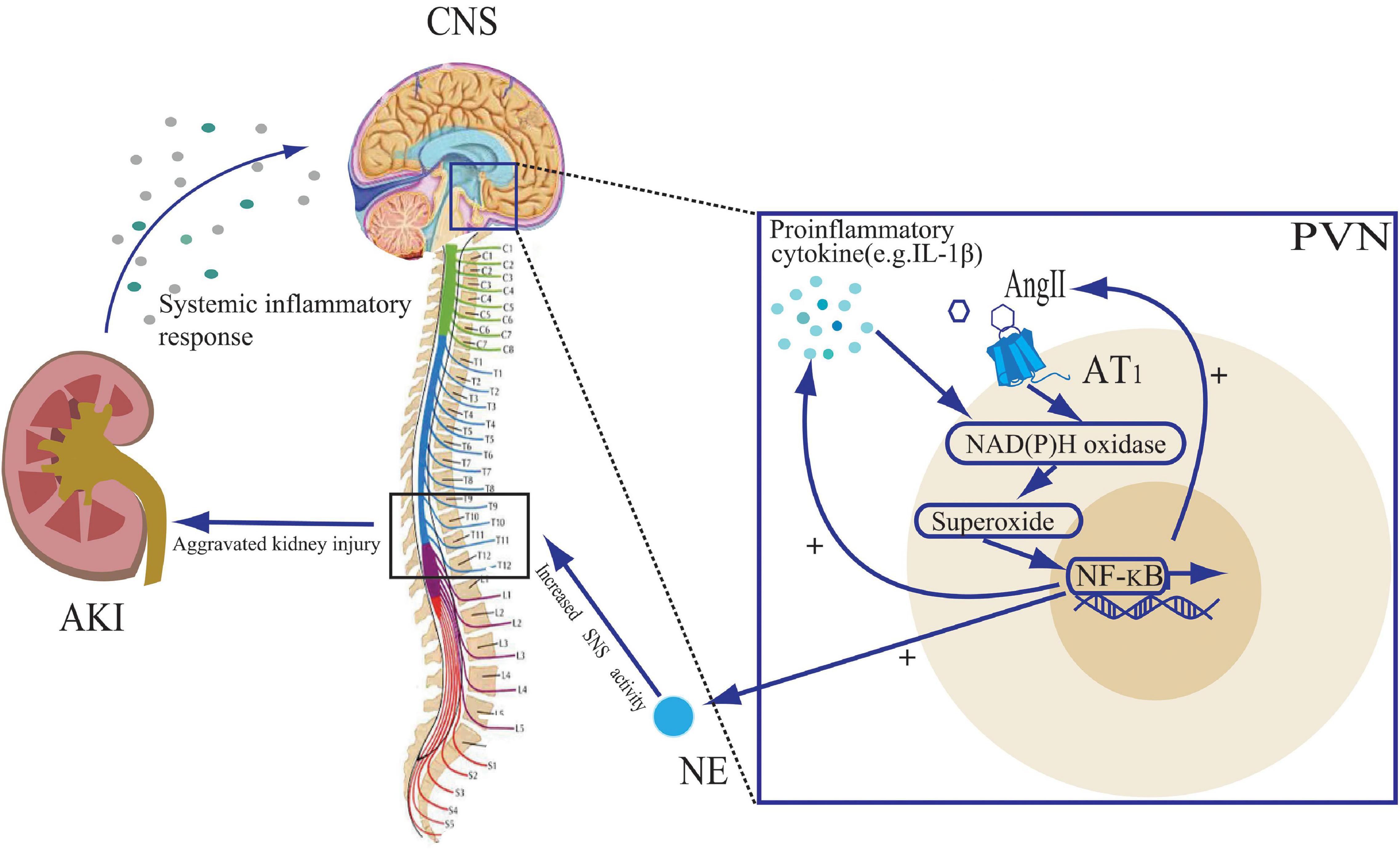
Figure 2. The role of proinflammatory cytokines and angiotensin II (Ang II) in the paraventricular nuclear (PVN) during acute kidney injury (AKI). The systemic inflammatory response to AKI leads to an increase in central nervous system (CNS) pro-inflammatory cytokines. Both Ang II and pro-inflammatory cytokines stimulate NAD(P)H oxidase activity and activate the NF-κB signaling pathway, leading to further cytokine synthesis and Ang II binding to the Ang II type 1 (AT1) receptor, creating positive feedback loops. The activation of the NF-κB signaling pathway also leads to an increase in norepinephrine (NE) release, promoting sympathetic nervous system (SNS) activity, further exacerbating kidney injury.
The high mortality rate from AKI is mainly due to extra-renal organ dysfunction (Kelly, 2006). The CNS is more commonly affected by such disease. Many potential uremic toxins, including guanidine-based compounds that regulate nitric oxide synthase (e.g., creatinine, guanidine, and methyl guanidine), have been associated with uremic encephalopathy (Vanholder et al., 2003). Symptoms of encephalopathy are usually more pronounced and progress more rapidly in AKI patients than in patients with CKD and end-stage renal disease (De Deyn et al., 1992). This might be explained by inadequate adaptation time of the uremic toxins accumulated after AKI. A clinical study showed that patients who recovered from AKI had a higher incidence of stroke and mortality than those who did not develop AKI (hazard ratio:1.25) (Wu et al., 2014). Some relevance has been described in animal models of AKI. An experimental study demonstrated that there were reduced dopamine turnover in the striatum, midbrain and hypothalamus of AKI rats, with a reduction in spontaneous motor activity (Adachi et al., 2001). In a mouse model of systemic inflammation, increases in circulating inflammatory cytokines (IL-1β, TNF-α) altered the transcriptional proteins of NF-κB, CCL2, and IL-1β in the hypothalamus and hippocampus (Skelly et al., 2013). In addition, these changes in mice IRI model were shown to result in altered brain function, including a significant increase in fixation neuronal cells in the CA1 region of the hippocampus and an increase in the number of activated microglia and a reduction in its activity (Liu et al., 2008). Interestingly, neuronal sequestration and activated glial cells in the brain were not significantly increased in the corresponding animal model of acute liver injury (Liu et al., 2008), suggesting that this effect is specific to AKI and not just connected with systemic inflammation following organ injuries. A recent study suggested that AKI might lead to hippocampal apoptosis and electrophysiological damage, increased BBB permeability, and memory loss through inflammatory mediators, and that uremia might lead to neuronal necrosis (Firouzjaei et al., 2019).
In addition, it has been shown that renal IRI could lead to oxidative stress in the kidney and brain, and the use of seaweed extract with antioxidant activity could significantly reduce MDA levels in the kidney and hippocampus of rats after IRI to reduce oxidative damage in the kidney and brain (Yu et al., 2016). There were also gender differences in the effects of AKI on brain damage, with the female rats having milder brain damage and MDA levels as compared with the male ones. The differences might be resulted from the protective effect of estrogen by increasing antioxidant enzymes and decreasing inducible NO synthase (iNOS) activity (Azarkish et al., 2021). The level of MDA was significantly decreased in AKI rats treated with losartan intervention. The hyperactivation of AT1 receptor by brain Ang II during acute kidney injury has been previously described, and this hyperactivation might precede the release of proinflammatory or inflammatory cytokines in the brain (Saavedra et al., 2011). It appeared that brain inflammation caused by peripheral or central inflammation is closely related to AT1 receptor function. In addition, brain MDA levels were significantly reduced in rats with AKI by losartan intervention (Sharifi et al., 2019). Possibly due to the attenuation of neuroinflammation and oxidative stress, losartan-treated rats attenuated AKI-induced memory deficits and impairments (Sharifi et al., 2019). A large US prospective cohort study showed that angiotensin receptor blocker administration could reduce cognitive impairment and Alzheimer’s disease progression (Li et al., 2010). Taken together, these findings suggested that AKI might affect brain function through the production of some toxic metabolites, and through the inflammatory response resulting in BBB destruction and by stimulating inflammation and oxidative stress in the brain. Those processes would lead to modifications in neuronal protein transcription and cell activation.
So far, there were only limited number of measures for treatment of AKI, most of which were supportive therapy and RRT (Vaara et al., 2020). But the mortality rate for AKI patients remained disturbingly high. Targeted treatment developed from the mechanism on how the CNS affects AKI might be a new strategy to improve the effectiveness of AKI treatment and to reduce the occurrence of complications. A retrospective study of 46,253 patients who developed AKI during hospitalization suggested that the use of angiotensin-converting enzyme inhibitor (ACEI) or angiotensin receptor blocker (ARB) might reduce mortality in patients with AKI for two years. This finding could be a potential benefit for AKI patients, but complications of kidney diseases should be carefully monitored in practice (Brar et al., 2018). Similarly, the benefits of ARB have been demonstrated in animal studies. Losartan treatment altered the expression of genes associated with inflammation and oxidative stress in AKI rats, suppressed significant increases in urea and creatinine levels, and reduced tubular structural damage and renal cell apoptosis (Wu et al., 2019). As previously mentioned, activation of the intrarenal RAS in AKI is caused by activation of the central RAS (Campese et al., 2002). Peripherally administered AT1 receptor blockers, such as losartan, are able to enter the brain and cause AT1 receptor blockade in the CNS, such as the PVN (Wang et al., 2003), thus providing additional therapeutic benefit. In addition, intracerebroventricular administration of losartan to block AT1 receptor and tempol to block central oxidative stress in AKI mice both reduced RAS activation, ROS and NE synthesis in the brain, improving brain inflammation, renal injury and dysfunction (Liu et al., 2008). In summary, we could use the blockade of the central RAS and ROS activation, and the control of central inflammation to reduce the damage to the kidney and other organs caused by AKI. Moreover, it was known that AKI could lead to reduced activity of GABAergic neurons in the PVN, and that intravenous GABA treatment or lateral ventricular injection of GABA agonists might reduce renal injury in rats by inhibiting central nervous excitability (Kobuchi et al., 2015). Those findings could also bring new perspective to the treatment of AKI.
The activation of the CNS due to AKI stimulates the SNS and RAS, exacerbating AKI-induced renal impairment. Therefore, AKI could in turn contribute to the CNS pathology. At present, there were no specific drugs for treatment of AKI. RRT was the only way to minimize the damage, but the mortality for this approach remained high. Recent studies have improved our understanding on the interaction between the CNS and the kidney, with AKI modulating the CNS through a weakening of sympathetic inhibition and a worsening of sympathetic excitation. The end result of this modulation is an increase in central sympathetic outflow, which exacerbates renal injury. Considering the high morbidity and mortality of AKI, there was a requirement to further investigate the mechanisms by which AKI interacts with the nervous system and to develop targeted interventions to reduce the threat of AKI to individuals. By shifting our focus from peripheral to CNS mechanisms, it would help further reduce AKI mortality through use of existing drugs, development of new drugs and application of other treatments with promising perspectives. The safety of drug use, however, must still be taken into account.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
QL and YL: conceptualization. YW: methodology and writing—original draft preparation. YW and SL: writing—editing. YL: visualization. QL: supervision. All authors have read and agreed to the published version of the manuscript.
This study was financially supported by the foundation from the National Natural Science Foundation of China (No. 81800609).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adachi, N., Lei, B., Deshpande, G., Seyfried, F. J., Shimizu, I., Nagaro, T., et al. (2001). Uraemia suppresses central dopaminergic metabolism and impairs motor activity in rats. Intensive Care Med. 27, 1655–1660. doi: 10.1007/s001340101067
Affleck, V. S., Coote, J. H., and Pyner, S. (2012). The projection and synaptic organisation of NTS afferent connections with presympathetic neurons, GABA and nNOS neurons in the paraventricular nucleus of the hypothalamus. Neuroscience 219, 48–61. doi: 10.1016/j.neuroscience.2012.05.070
Aparicio-Trejo, O. E., Avila-Rojas, S. H., Tapia, E., Rojas-Morales, P., Leon-Contreras, J. C., Martinez-Klimova, E., et al. (2020). Chronic impairment of mitochondrial bioenergetics and beta-oxidation promotes experimental AKI-to-CKD transition induced by folic acid. Free Radic. Biol. Med. 154, 18–32. doi: 10.1016/j.freeradbiomed.2020.04.016
Azarkish, F., Armin, F., Parvar, A. A. A., and Dehghani, A. (2021). The influence of renal ischemia-reperfusion injury on remote organs: the histological brain changes in male and female rats. Brain Circ. 7, 194–200. doi: 10.4103/bc.bc_3_21
Badoer, E. (2010). Role of the hypothalamic PVN in the regulation of renal sympathetic nerve activity and blood flow during hyperthermia and in heart failure. Am. J. Physiol. Renal. Physiol. 298, F839–F846. doi: 10.1152/ajprenal.00734.2009
Bellomo, R., and Giantomasso, D. D. (2001). Noradrenaline and the kidney: friends or foes? Crit. Care 5, 294–298. doi: 10.1186/cc1052
Bonizzi, G., Piette, J., Schoonbroodt, S., Greimers, R., Havard, L., Merville, M. P., et al. (1999). Reactive oxygen intermediate-dependent NF-kappaB activation by interleukin-1beta requires 5-lipoxygenase or NADPH oxidase activity. Mol. Cell. Biol. 19, 1950–1960. doi: 10.1128/mcb.19.3.1950
Bonventre, J. V., and Zuk, A. (2004). Ischemic acute renal failure: an inflammatory disease? Kidney Int. 66, 480–485. doi: 10.1111/j.1523-1755.2004.761_2.x
Brar, S., Ye, F., James, M. T., Hemmelgarn, B., Klarenbach, S., Pannu, N., et al. (2018). Association of angiotensin-converting enzyme inhibitor or angiotensin receptor blocker use with outcomes after acute kidney injury. JAMA Intern. Med. 178, 1681–1690. doi: 10.1001/jamainternmed.2018.4749
Calaresu, F. R., and Ciriello, J. (1981). Renal afferent nerves affect discharge rate of medullary and hypothalamic single units in the cat. J. Auton. Nerv. Syst. 3, 311–320. doi: 10.1016/0165-1838(81)90072-2
Campese, V. M., and Kogosov, E. (1995). Renal afferent denervation prevents hypertension in rats with chronic renal failure. Hypertension 25(4 Pt 2), 878–882. doi: 10.1161/01.hyp.25.4.878
Campese, V. M., Ye, S., and Zhong, H. (2002). Downregulation of neuronal nitric oxide synthase and interleukin-1beta mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension 39(2 Pt 2), 519–524. doi: 10.1161/hy0202.102815
Cao, W., Li, A., Li, J., Wu, C., Cui, S., Zhou, Z., et al. (2017). Reno-cerebral reflex activates the renin-angiotensin system, promoting oxidative stress and renal damage after ischemia-reperfusion injury. Antioxid. Redox Signal. 27, 415–432. doi: 10.1089/ars.2016.6827
Cao, W., Wu, L., Zhang, X., Zhou, J., Wang, J., Yang, Z., et al. (2020). Sympathetic overactivity in CKD disrupts buffering of neurotransmission by endothelium-derived hyperpolarizing factor and enhances vasoconstriction. J. Am. Soc. Nephrol. 31, 2312–2325. doi: 10.1681/ASN.2020030234
Chandler, D. J. (2016). Evidence for a specialized role of the locus coeruleus noradrenergic system in cortical circuitries and behavioral operations. Brain Res. 1641(Pt B), 197–206. doi: 10.1016/j.brainres.2015.11.022
Chen, H. H., Cheng, P. W., Ho, W. Y., Lu, P. J., Lai, C. C., Tseng, Y. M., et al. (2016). Renal denervation improves the baroreflex and GABA system in chronic kidney disease-induced hypertension. Sci. Rep. 6:38447. doi: 10.1038/srep38447
Chen, Q., and Pan, H. L. (2007). Signaling mechanisms of angiotensin II-induced attenuation of GABAergic input to hypothalamic presympathetic neurons. J. Neurophysiol. 97, 3279–3287. doi: 10.1152/jn.01329.2006
Ciriello, J. (1998). Afferent renal inputs to paraventricular nucleus vasopressin and oxytocin neurosecretory neurons. Am. J. Physiol. 275, R1745–R1754. doi: 10.1152/ajpregu.1998.275.6.R1745
Colindres, R. E., Spielman, W. S., Moss, N. G., Harrington, W. W., and Gottschalk, C. W. (1980). Functional evidence for renorenal reflexes in the rat. Am. J. Physiol. 239, F265–F270. doi: 10.1152/ajprenal.1980.239.3.F265
Conger, J. D., Robinette, J. B., and Hammond, W. S. (1991). Differences in vascular reactivity in models of ischemic acute renal failure. Kidney Int. 39, 1087–1097. doi: 10.1038/ki.1991.138
Cronin, R. E., Erickson, A. M., de Torrente, A., McDonald, K. M., and Schrier, R. W. (1978). Norepinephrine-induced acute renal failure: a reversible ischemic model of acute renal failure. Kidney Int. 14, 187–190. doi: 10.1038/ki.1978.106
Crowley, S. D., and Rudemiller, N. P. (2017). Immunologic effects of the renin-angiotensin system. J. Am. Soc. Nephrol. 28, 1350–1361. doi: 10.1681/ASN.2016101066
De Deyn, P. P., Saxena, V. K., Abts, H., Borggreve, F., D’Hooge, R., Marescau, B., et al. (1992). Clinical and pathophysiological aspects of neurological complications in renal failure. Acta Neurol. Belg. 92, 191–206.
Ding, Z. Q., Li, Y. W., Wesselingh, S. L., and Blessing, W. W. (1993). Transneuronal labelling of neurons in rabbit brain after injection of herpes simplex virus type 1 into the renal nerve. J. Auton. Nerv. Syst. 42, 23–31. doi: 10.1016/0165-1838(93)90338-u
Doi, K., and Rabb, H. (2016). Impact of acute kidney injury on distant organ function: recent findings and potential therapeutic targets. Kidney Int. 89, 555–564. doi: 10.1016/j.kint.2015.11.019
Ferenbach, D. A., and Bonventre, J. V. (2015). Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 11, 264–276. doi: 10.1038/nrneph.2015.3
Firouzjaei, M. A., Haghani, M., and Shid Moosavi, S. M. (2019). Renal ischemia/reperfusion induced learning and memory deficit in the rat: insights into underlying molecular and cellular mechanisms. Brain Res. 1719, 263–273. doi: 10.1016/j.brainres.2019.05.018
Fujii, T., Kurata, H., Takaoka, M., Muraoka, T., Fujisawa, Y., Shokoji, T., et al. (2003). The role of renal sympathetic nervous system in the pathogenesis of ischemic acute renal failure. Eur. J. Pharmacol. 481, 241–248. doi: 10.1016/j.ejphar.2003.09.036
Gao, L., Wang, W., Li, Y. L., Schultz, H. D., Liu, D., Cornish, K. G., et al. (2004). Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ. Res. 95, 937–944. doi: 10.1161/01.RES.0000146676.04359.64
Gloire, G., Legrand-Poels, S., and Piette, J. (2006). NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72, 1493–1505. doi: 10.1016/j.bcp.2006.04.011
Grams, M. E., and Rabb, H. (2012). The distant organ effects of acute kidney injury. Kidney Int. 81, 942–948. doi: 10.1038/ki.2011.241
Grisk, O. (2020). The sympathetic nervous system in acute kidney injury. Acta Physiol. (Oxf.) 228:e13404. doi: 10.1111/apha.13404
Grivei, A., Giuliani, K. T. K., Wang, X., Ungerer, J., Francis, L., Hepburn, K., et al. (2020). Oxidative stress and inflammasome activation in human rhabdomyolysis-induced acute kidney injury. Free Radic. Biol. Med. 160, 690–695. doi: 10.1016/j.freeradbiomed.2020.09.011
Han, Y., Yuan, N., Zhang, S. J., Gao, J., Shi, Z., Zhou, Y. B., et al. (2011). c-Src in paraventricular nucleus modulates sympathetic activity and cardiac sympathetic afferent reflex in renovascular hypertensive rats. Pflugers Arch. 461, 437–446. doi: 10.1007/s00424-011-0932-7
Hering, D., and Winklewski, P. J. (2017). Autonomic nervous system in acute kidney injury. Clin. Exp. Pharmacol. Physiol. 44, 162–171. doi: 10.1111/1440-1681.12694
Inoue, T., Abe, C., Sung, S. S., Moscalu, S., Jankowski, J., Huang, L., et al. (2016). Vagus nerve stimulation mediates protection from kidney ischemia-reperfusion injury through alpha7nAChR+ splenocytes. J. Clin. Invest. 126, 1939–1952. doi: 10.1172/JCI83658
Johns, D. W., Peach, M. J., Gomez, R. A., Inagami, T., and Carey, R. M. (1990). Angiotensin II regulates renin gene expression. Am. J. Physiol. 259(6 Pt 2), F882–F887. doi: 10.1152/ajprenal.1990.259.6.F882
Johns, E. J., Kopp, U. C., and DiBona, G. F. (2011). Neural control of renal function. Compr. Physiol. 1, 731–767. doi: 10.1002/cphy.c100043
Johnston, G. A. (1996). GABAA receptor pharmacology. Pharmacol. Ther. 69, 173–198. doi: 10.1016/0163-7258(95)02043-8
Kang, Y. M., Ma, Y., Elks, C., Zheng, J. P., Yang, Z. M., and Francis, J. (2008). Cross-talk between cytokines and renin-angiotensin in hypothalamic paraventricular nucleus in heart failure: role of nuclear factor-kappaB. Cardiovasc. Res. 79, 671–678. doi: 10.1093/cvr/cvn119
Katholi, R. E., Whitlow, P. L., Hageman, G. R., and Woods, W. T. (1984). Intrarenal adenosine produces hypertension by activating the sympathetic nervous system via the renal nerves in the dog. J. Hypertens. 2, 349–359.
Kellum, J. A., Lameire, N., and Group, K. A. G. W. (2013). Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit. Care 17:204. doi: 10.1186/cc11454
Kelly, K. J. (2006). Acute renal failure: much more than a kidney disease. Semin. Nephrol. 26, 105–113. doi: 10.1016/j.semnephrol.2005.09.003
Kim, J., and Padanilam, B. J. (2015). Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 87, 350–358. doi: 10.1038/ki.2014.300
Kobuchi, S., Shintani, T., Sugiura, T., Tanaka, R., Suzuki, R., Tsutsui, H., et al. (2009). Renoprotective effects of gamma-aminobutyric acid on ischemia/reperfusion-induced renal injury in rats. Eur. J. Pharmacol. 623, 113–118. doi: 10.1016/j.ejphar.2009.09.023
Kobuchi, S., Tanaka, R., Shintani, T., Suzuki, R., Tsutsui, H., Ohkita, M., et al. (2015). Mechanisms underlying the renoprotective effect of GABA against ischaemia/reperfusion-induced renal injury in rats. Clin. Exp. Pharmacol. Physiol. 42, 278–286. doi: 10.1111/1440-1681.12350
Kopp, U. C. (2015). Role of renal sensory nerves in physiological and pathophysiological conditions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R79–R95. doi: 10.1152/ajpregu.00351.2014
Kopp, U. C., and Buckley-Bleiler, R. L. (1989). Impaired renorenal reflexes in two-kidney, one clip hypertensive rats. Hypertension 14, 445–452. doi: 10.1161/01.hyp.14.4.445
Kopp, U. C., Cicha, M. Z., and Smith, L. A. (2007). Activation of endothelin-a receptors contributes to angiotensin-induced suppression of renal sensory nerve activation. Hypertension 49, 141–147. doi: 10.1161/01.HYP.0000249634.46212.7b
Kopp, U. C., Cicha, M. Z., Smith, L. A., Ruohonen, S., Scheinin, M., Fritz, N., et al. (2011). Dietary sodium modulates the interaction between efferent and afferent renal nerve activity by altering activation of α2-adrenoceptors on renal sensory nerves. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R298–R310. doi: 10.1152/ajpregu.00469.2010
Larsen, R., Thorp, A., and Schlaich, M. (2014). Regulation of the sympathetic nervous system by the kidney. Curr. Opin. Nephrol. Hypertens. 23, 61–68. doi: 10.1097/01.mnh.0000437610.65287.db
Legrand, M., and Bokoch, M. P. (2021). The Yin and Yang of the renin-angiotensin-aldosterone system in acute kidney injury. Am. J. Respir. Crit. Care Med. 203, 1053–1055. doi: 10.1164/rccm.202012-4419ED
Levey, A. S., and James, M. T. (2017). Acute kidney injury. Ann. Intern. Med. 167, ITC66–ITC80. doi: 10.7326/AITC201711070
Li, D. P., and Pan, H. L. (2005). Angiotensin II attenuates synaptic GABA release and excites paraventricular-rostral ventrolateral medulla output neurons. J. Pharmacol. Exp. Ther. 313, 1035–1045. doi: 10.1124/jpet.104.082495
Li, N. C., Lee, A., Whitmer, R. A., Kivipelto, M., Lawler, E., Kazis, L. E., et al. (2010). Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 340, b5465. doi: 10.1136/bmj.b5465
Liu, M., Liang, Y., Chigurupati, S., Lathia, J. D., Pletnikov, M., Sun, Z., et al. (2008). Acute kidney injury leads to inflammation and functional changes in the brain. J. Am. Soc. Nephrol. 19, 1360–1370. doi: 10.1681/ASN.2007080901
Ma, M. C., Huang, H. S., Chen, Y. S., and Lee, S. H. (2008). Mechanosensitive N-methyl-D-aspartate receptors contribute to sensory activation in the rat renal pelvis. Hypertension 52, 938–944. doi: 10.1161/HYPERTENSIONAHA.108.114116
Makris, K., and Spanou, L. (2016). Acute kidney injury: definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 37, 85–98.
Mao, H., Jiang, C., Xu, L., Chen, D., Liu, H., Xu, Y., et al. (2020). Ginsenoside protects against AKI via activation of HIF-1α and VEGF-A in the kidney-brain axis. Int. J. Mol. Med. 45, 939–946. doi: 10.3892/ijmm.2020.4466
Maringer, K., and Sims-Lucas, S. (2016). The multifaceted role of the renal microvasculature during acute kidney injury. Pediatr. Nephrol. 31, 1231–1240. doi: 10.1007/s00467-015-3231-2
Nadjar, A., Bluthé, R. M., May, M. J., Dantzer, R., and Parnet, P. (2005). Inactivation of the cerebral NFkappaB pathway inhibits interleukin-1beta-induced sickness behavior and c-Fos expression in various brain nuclei. Neuropsychopharmacology 30, 1492–1499. doi: 10.1038/sj.npp.1300755
Nadjar, A., Combe, C., Layé, S., Tridon, V., Dantzer, R., Amédée, T., et al. (2003). Nuclear factor kappaB nuclear translocation as a crucial marker of brain response to interleukin-1. A study in rat and interleukin-1 type I deficient mouse. J. Neurochem. 87, 1024–1036. doi: 10.1046/j.1471-4159.2003.02097.x
Naito, K., Anzai, T., Yoshikawa, T., Anzai, A., Kaneko, H., Kohno, T., et al. (2008). Impact of chronic kidney disease on postinfarction inflammation, oxidative stress, and left ventricular remodeling. J. Card. Fail. 14, 831–838. doi: 10.1016/j.cardfail.2008.07.233
Nishi, E. E., Martins, B. S., Milanez, M. I., Lopes, N. R., de Melo, J. F. Jr., Pontes, R. B., et al. (2017). Stimulation of renal afferent fibers leads to activation of catecholaminergic and non-catecholaminergic neurons in the medulla oblongata. Auton. Neurosci. 204, 48–56. doi: 10.1016/j.autneu.2017.01.003
Nongnuch, A., Panorchan, K., and Davenport, A. (2014). Brain-kidney crosstalk. Crit. Care 18:225. doi: 10.1186/cc13907
Saavedra, J. M., Sanchez-Lemus, E., and Benicky, J. (2011). Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications. Psychoneuroendocrinology 36, 1–18. doi: 10.1016/j.psyneuen.2010.10.001
Satou, R., Cypress, M. W., Woods, T. C., Katsurada, A., Dugas, C. M., Fonseca, V. A., et al. (2020). Blockade of sodium-glucose cotransporter 2 suppresses high glucose-induced angiotensinogen augmentation in renal proximal tubular cells. Am. J. Physiol. Renal. Physiol. 318, F67–F75. doi: 10.1152/ajprenal.00402.2019
Seifi, B., Kadkhodaee, M., Bakhshi, E., Ranjbaran, M., Ahghari, P., and Rastegar, T. (2014). Enhancement of renal oxidative stress by injection of angiotensin II into the paraventricular nucleus in renal ischemia-reperfusion injury. Can. J. Physiol. Pharmacol. 92, 752–757. doi: 10.1139/cjpp-2014-0108
Shafton, A. D., Ryan, A., and Badoer, E. (1998). Neurons in the hypothalamic paraventricular nucleus send collaterals to the spinal cord and to the rostral ventrolateral medulla in the rat. Brain Res. 801, 239–243. doi: 10.1016/s0006-8993(98)00587-3
Sharifi, F., Reisi, P., and Malek, M. (2019). Angiotensin 1 receptor antagonist attenuates acute kidney injury-induced cognitive impairment and synaptic plasticity via modulating hippocampal oxidative stress. Life Sci. 234:116775. doi: 10.1016/j.lfs.2019.116775
Shi, Z., Gan, X. B., Fan, Z. D., Zhang, F., Zhou, Y. B., Gao, X. Y., et al. (2011). Inflammatory cytokines in paraventricular nucleus modulate sympathetic activity and cardiac sympathetic afferent reflex in rats. Acta Physiol. (Oxf.) 203, 289–297. doi: 10.1111/j.1748-1716.2011.02313.x
Sirivelu, M. P., MohanKumar, P. S., and MohanKumar, S. M. (2012). Differential effects of systemic interleukin-1beta on gene expression in brainstem noradrenergic nuclei. Life Sci. 90, 77–81. doi: 10.1016/j.lfs.2011.10.006
Skelly, D. T., Hennessy, E., Dansereau, M. A., and Cunningham, C. (2013). A systematic analysis of the peripheral and CNS effects of systemic LPS, IL-1β, [corrected] TNF-α and IL-6 challenges in C57BL/6 mice. PLoS One 8:e69123. doi: 10.1371/journal.pone.0069123
Stefanska, A., Eng, D., Kaverina, N., Pippin, J. W., Gross, K. W., Duffield, J. S., et al. (2016). Cells of renin lineage express hypoxia inducible factor 2α following experimental ureteral obstruction. BMC Nephrol. 17:5. doi: 10.1186/s12882-015-0216-0
Swanson, L. W., and Sawchenko, P. E. (1983). Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu. Rev. Neurosci. 6, 269–324. doi: 10.1146/annurev.ne.06.030183.001413
Tanaka, R., Tsutsui, H., Ohkita, M., Takaoka, M., Yukimura, T., and Matsumura, Y. (2013). Sex differences in ischemia/reperfusion-induced acute kidney injury are dependent on the renal sympathetic nervous system. Eur. J. Pharmacol. 714, 397–404. doi: 10.1016/j.ejphar.2013.07.008
Tanaka, S., and Okusa, M. D. (2020). Crosstalk between the nervous system and the kidney. Kidney Int. 97, 466–476. doi: 10.1016/j.kint.2019.10.032
Tellez, A., Rousselle, S., Palmieri, T., Rate, W. R. T., Wicks, J., Degrange, A., et al. (2013). Renal artery nerve distribution and density in the porcine model: biologic implications for the development of radiofrequency ablation therapies. Transl. Res. 162, 381–389. doi: 10.1016/j.trsl.2013.07.002
Vaara, S. T., Ostermann, M., Selander, T., Bitker, L., Schneider, A., Poli, E., et al. (2020). Protocol and statistical analysis plan for the REstricted fluid therapy VERsus standard trEatment in acute kidney injury-REVERSE-AKI randomized controlled pilot trial. Acta Anaesthesiol. Scand. 64, 831–838. doi: 10.1111/aas.13557
Vanholder, R., De Smet, R., Glorieux, G., Argiles, A., Baurmeister, U., Brunet, P., et al. (2003). Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 63, 1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x
Vaziri, N. D., Bai, Y., Ni, Z., Quiroz, Y., Pandian, R., and Rodriguez-Iturbe, B. (2007). Intra-renal angiotensin II/AT1 receptor, oxidative stress, inflammation, and progressive injury in renal mass reduction. J. Pharmacol. Exp. Ther. 323, 85–93. doi: 10.1124/jpet.107.123638
Wang, J. M., Tan, J., and Leenen, F. H. (2003). Central nervous system blockade by peripheral administration of AT1 receptor blockers. J. Cardiovasc. Pharmacol. 41, 593–599. doi: 10.1097/00005344-200304000-00012
Wang, Y., Hu, H., Yin, J., Shi, Y., Tan, J., Zheng, L., et al. (2019). TLR4 participates in sympathetic hyperactivity Post-MI in the PVN by regulating NF-κB pathway and ROS production. Redox Biol. 24:101186. doi: 10.1016/j.redox.2019.101186
Webb, R. L., and Brody, M. J. (1987). Functional identification of the central projections of afferent renal nerves. Clin. Exp. Hypertens. A 9(Suppl. 1), 47–57. doi: 10.3109/10641968709160163
Wu, V. C., Wu, P. C., Wu, C. H., Huang, T. M., Chang, C. H., Tsai, P. R., et al. (2014). The impact of acute kidney injury on the long-term risk of stroke. J. Am. Heart Assoc. 3:e000933. doi: 10.1161/jaha.114.000933
Wu, Y., Peng, W., Wei, R., Zhou, Y., Fang, M., Liu, S., et al. (2019). Rat mRNA expression profiles associated with inhibition of ischemic acute kidney injury by losartan. Biosci. Rep. 39:BSR20181774. doi: 10.1042/BSR20181774
Yu, F., Liang, H., and Xin, S. (2016). Renal ischemia reperfusion causes brain Hippocampus oxidative damage and inhibition effect. Afr. J. Tradit. Complement. Altern. Med. 13, 61–66. doi: 10.21010/ajtcam.v13i5.9
Zhang, N., Cheng, G. Y., Liu, X. Z., and Zhang, F. J. (2014). Expression of Bcl-2 and NF-kappaB in brain tissue after acute renal ischemia-reperfusion in rats. Asian Pac. J. Trop. Med. 7, 386–389. doi: 10.1016/S1995-7645(14)60061-4
Keywords: central nervous system, acute kidney injury, paraventricular hypothalamic, sympathetic nervous system, neuromodulation
Citation: Wang Y, Liu S, Liu Q and Lv Y (2022) The Interaction of Central Nervous System and Acute Kidney Injury: Pathophysiology and Clinical Perspectives. Front. Physiol. 13:826686. doi: 10.3389/fphys.2022.826686
Received: 01 December 2021; Accepted: 03 February 2022;
Published: 04 March 2022.
Edited by:
Adriana Castello Costa Girardi, University of São Paulo, BrazilReviewed by:
Olaf Grisk, Städtisches Klinikum Brandenburg GmbH, GermanyCopyright © 2022 Wang, Liu, Liu and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingquan Liu, cXFsaXV0akAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.