 Alexandra K. Turvey
Alexandra K. Turvey Gabriella A. Horvath
Gabriella A. Horvath André R. O. Cavalcanti
André R. O. Cavalcanti- 1Department of Biology, Pomona College, Claremont, CA, United States
- 2Division of Biochemical Genetics, Department of Pediatrics, University of British Columbia, BC Children’s Hospital, Vancouver, BC, Canada
- 3Adult Metabolic Diseases Clinic, Vancouver General Hospital, Vancouver, BC, Canada
The Aminoacyl-tRNA Synthetases (aaRSs) are an evolutionarily ancient family of enzymes that catalyze the esterification reaction linking a transfer RNA (tRNA) with its cognate amino acid matching the anticodon triplet of the tRNA. Proper functioning of the aaRSs to create aminoacylated (or “charged”) tRNAs is required for efficient and accurate protein synthesis. Beyond their basic canonical function in protein biosynthesis, aaRSs have a surprisingly diverse array of non-canonical functions that are actively being defined. The human genome contains 37 genes that encode unique aaRS proteins. To date, 56 human genetic diseases caused by damaging variants in aaRS genes have been described: 46 are autosomal recessive biallelic disorders and 10 are autosomal dominant monoallelic disorders. Our appreciation of human diseases caused by damaging genetic variants in the aaRSs has been greatly accelerated by the advent of next-generation sequencing, with 89% of these gene discoveries made since 2010. In addition to these genetic disorders of the aaRSs, anti-synthetase syndrome (ASSD) is a rare autoimmune inflammatory myopathy that involves the production of autoantibodies that disrupt aaRS proteins. This review provides an overview of the basic biology of aaRS proteins and describes the rapidly growing list of human diseases known to be caused by genetic variants or autoimmune targeting that affect both the canonical and non-canonical functions of these essential proteins.
1 Introduction
The Central Dogma of molecular biology explains the flow of genetic information in a biological system from DNA through RNA to proteins (Crick, 1970). The Aminoacyl-tRNA Synthetases (aaRSs)—a family of enzymes present in all eukaryotes, archaea, and bacteria—link the worlds of nucleic acids and proteins and are key for the faithful translation of the genetic code (Kaiser et al., 2020). aaRSs catalyze the esterification that links a transfer RNA (tRNA) with its cognate amino acid matching the anticodon triplet of the tRNA (Ibba and Söll, 2000; Rubio Gomez and Ibba, 2020). aaRSs are evolutionarily ancient, emerging during the time of the last universal common ancestor (LUCA), and are distributed across all branches of life.
Given their central role in human biology, it is unsurprising that genetic variants that disrupt aaRS protein structure and function cause disease. Damaging variants in aaRSs have now been linked to over 50 human diseases (Figure 1A). Notably 2/3 of these newly recognized diseases have only been described in the past decade since next-generation sequencing technologies have become more widely available. This review will provide an overview of the basic biology of aaRS proteins and will describe the rapidly growing list of human diseases known to be caused by variants that affect both the canonical and non-canonical functions of these essential proteins. We anticipate this review will be of value to clinicians who care for patients with diseases related to aaRS function, and scientists interested in the links between the aaRSs and disease.
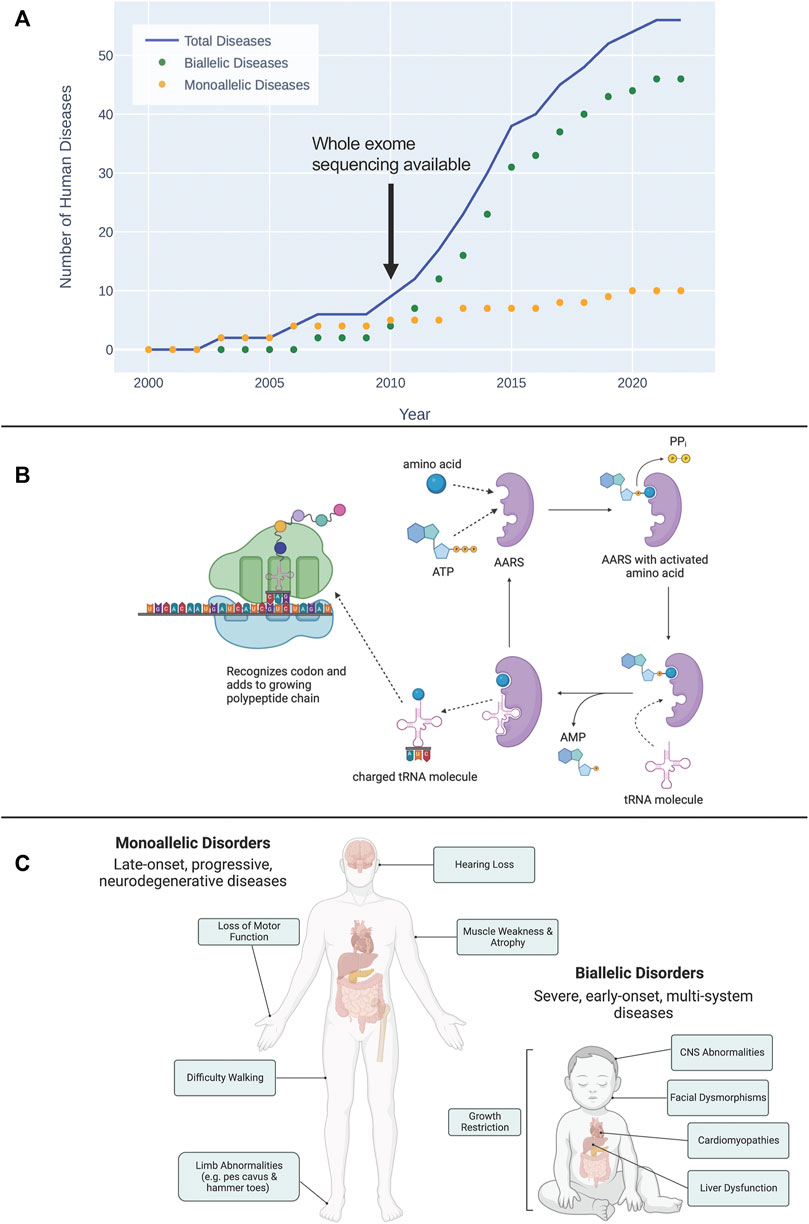
FIGURE 1. (A) The timeline of discovery of biallelic and monoallelic human diseases caused by damaging aaRS variants since 2000. (B) Overview of the esterification reaction, catalyzed by aaRS proteins, that charges a tRNA molecule with its cognate amino acid. This charged tRNA then travels to a translating ribosome where it recognizes a codon and adds its amino acid to the growing polypeptide chain. (C) General characterization of symptoms associated with diseases caused by monoallelic and biallelic pathogenic aaRS variants. Diseases caused by monoallelic variants are typically late-onset, progressive, neurodegenerative disorders. Damaging biallelic variants cause severe, early-onset, multi-system disorders.
2 Overview of basic biological functions of the aaRSs
2.1 Canonical function
The synthesis of functional proteins relies on the accurate transfer of genetic information from DNA, through mRNA, to protein. To generate new proteins, DNA is first transcribed to mRNA using the complementarity of nucleotide bases. The mRNA is then translated into a protein by the ribosome. The ribosome reads the mRNA in groups of triplet codons with each codon corresponding to one amino acid.
Once the ribosome reads a codon, elongation factors bring a tRNA containing the three bases complementary to a codon (anticodon) and charged with the correct amino acid. The amino acid delivered by the tRNA is then added to the growing polypeptide chain following the order of codons specified by the mRNA. This elongating polypeptide eventually becomes a functional protein within the cell.
The sequence of amino acids in a protein is defined by what amino acid is brought to the ribosome by the tRNA cognate to each codon. The essential step in this process involves linking the appropriate amino acid with its matching tRNA. This is the reaction catalyzed by the aaRSs, the esterification that links each tRNA molecule with its cognate amino acid creating an aminoacylated, or “charged” tRNA (Figure 1B). In addition to the aminoacylation functions, during evolution some tRNA synthetases added an editing function to remove the wrong amino acid from its cognate tRNA (Ling et al., 2009).
With few exceptions, all living organisms contain genes for the 20 aaRSs, one for each amino acid. The degeneracy of the genetic code means that there are more codons than amino acids. Therefore, the aaRS for an amino acid can recognize several tRNAs cognate to that amino acid. Eukaryotes contain genes for cytoplasmic and mitochondrial aaRSs (and in plants, chloroplastic aaRSs). All of these proteins are nuclear encoded and synthesized in the cytoplasm with the organellar aaRSs being imported to their final destinations following translation.
In humans and other higher eukaryotes, 9 of the aaRS proteins (ArgRS, AspRS, GlnRS, GluRS, IleRS, LeuRS, LysRS, MetRS, and ProRS) bind together to form the cytoplasmic multi-tRNA synthetase complex (MSC) (Kerjan et al., 1994; Havrylenko and Mirande, 2015; Khan et al., 2022). While the exact cellular function of the MSC is still unknown (Cui et al., 2021), it has been proposed that the MSC may act to enhance translation efficiency by channeling charged tRNAs to the A-site of a protein-synthesizing ribosome (Kyriacou and Deutscher, 2008; Khan et al., 2020; Sissler, 2021).
2.2 Non-canonical functions
During evolution, most eukaryotic cytoplasmic aaRSs gained noncatalytic domains not found in their respective orthologs in bacteria or archaea (Guo et al., 2010a; 2010b; Yao and Fox, 2020). These additions correlate with the progressive complexity of eukaryotes. In most cases, these additional domains are dispensable for aminoacylation or editing, suggesting a role beyond the ‘housekeeping’ function of aaRSs in protein translation. aaRSs have been recognized to have a surprisingly diverse array of non-canonical (or ‘moonlighting’) functions involved in gene expression regulation, RNA splicing, tumorigenesis, angiogenesis, and the immune responses (Ivanov et al., 2000; Smirnova et al., 2012). While it is beyond the scope of this review to address all non-canonical functions, the well-characterized ‘moonlighting’ human tyrosyl tRNA synthetase (TyrRS) serves as an illustrative example. TyrRS has no cell signaling capacity as a full molecule but when split by proteolysis each fragment can act as a cytokine (Wakasugi and Schimmel, 1999). For example, mini-TyrRS, the N-terminal domain of TyrRS, is released by endothelial cells and exhibits angiogenic and leukocyte chemoattractant properties (Wakasugi et al., 2002).
Our understanding of these non-canonical functions of aaRSs is growing rapidly. As we try to understand how pathogenic genetic changes in the aaRS genes cause human disease, it is essential that we look beyond the canonical aminoacylation role of aaRSs and consider how alterations in non-canonical functions may also contribute to pathophysiology (Guo et al., 2010b).
3 aaRS nomenclature and the exceptions
aaRS nomenclature is somewhat complex as it integrates functional classes, subcellular localization, and gene names. Here we clarify and summarize this nomenclature. First, aaRS proteins are divided into two classes based on their specific structural and functional properties (Cusack et al., 1990; Eriani et al., 1990). Class I aaRSs contain two highly conserved sequence motifs (‘KMSKS’ and ‘HIGH’) that are part of the larger conserved Rossman fold domain (Eriani et al., 1990; Shepard et al., 1992). Class II aaRSs, on the other hand, are less conserved than the Class I enzymes and contain a unique alpha-beta fold in their catalytic domains (Bullwinkle and Ibba, 2014; Smith and Hartman, 2015). Although the two aaRS classes are evolutionarily and structurally very different, the overall chemistry of the tRNA aminoacylation reaction is similar in both—an example of convergent evolution (Arnez and Moras, 1997).
The naming convention for genes encoding aaRSs is as follows: 1) gene names begin with the one-letter symbol for the amino acid the aaRS recognizes (e.g., A for alanine; Y for tyrosine); 2) this is followed by ARS1 or ARS2 depending on whether the aaRS is located in the cytoplasm or mitochondria, respectively. For example, LARS2 specifies leucyl-tRNA synthetase 2, which charges a tRNA molecule with leucine in the mitochondria of the cell.
While the single-letter amino acid code is used for aaRS gene names, the 3-letter amino acid code is generally used as a prefix to refer to the protein product. For example, IleRS refers to isoleucyl-tRNA synthetase.
The majority of genes encoding aaRSs follow the naming convention described above, but there are a few exceptions. EPRS1 encodes a bifunctional glutamyl-prolyl-tRNA synthetase that has the capacity to charge tRNA molecules with either glutamic acid or proline in the cytoplasm (Cerini et al., 1991; Jin et al., 2021). The mitochondrial glutaminyl-tRNA synthetase is not encoded for by a separate gene in mammalian species, and it has instead been proposed that an indirect pathway allows for the synthesis of GlnRS in mammalian mitochondria (Nagao et al., 2009). Both KARS1 and GARS1 encode for synthetases that dually-localize to both the mitochondria and cytoplasm (Yao and Fox, 2013). Finally, the cytoplasmic phenylalanine-tRNA synthetase consists of a heterodimer of two protein subunits: FARSA encodes the catalytic alpha subunit while FARSB encodes the regulatory beta subunit (Rodova et al., 1999).
In total, human cells contain 37 genes that encode unique aaRS proteins. Of these, 18 encode cytoplasmic aaRSs (2 of these genes encode cytoplasmic PheRS), 17 encode mitochondrial synthetases, and two genes encode proteins that will exist in both locations (Wei et al., 2019).
4 Human diseases associated with genetic variants in aaRS-encoding genes
Next-generation sequencing (NGS) technology has transformed our ability to make genetic diagnoses. Since the first successful application of NGS for gene identification in 2010 (Ng et al., 2010), the discovery of human diseases caused by pathogenic genetic variants has rapidly increased (Bamshad et al., 2019). It is anticipated that this number will continue to grow with improvements in both sequencing technologies and bioinformatic tools to pinpoint pathogenic variants (Schuler et al., 2022). Throughout this review we will use the term ‘variant’ to describe a change in the germline DNA sequence, as it has been recommended to replace the terms ‘mutation’ and ‘polymorphism’ with the term ‘variant’ (Richards et al., 2015).
To date, 56 human diseases caused by damaging variants in aaRS genes have been described (see Figure 1A). Emphasizing the diagnostic impact of NGS, 89% of these gene discoveries were made since 2010. Of these 56 diseases, 46 are autosomal recessive and are caused by damaging biallelic variants, while the remaining 10 are autosomal dominant and are caused by damaging monoallelic variants (Figure 1C). Biallelic disease occurs when there is a pathogenic variant on both alleles of a given gene, whereas a monoallelic disease is caused by a pathogenic variant affecting one of the two alleles. Biallelic disease can follow two possible inheritance patterns: 1) the same damaging variant occurs on both alleles (homozygous inheritance); and 2) unique damaging variants occur on each allele (compound heterozygous inheritance). The 56 human diseases linked to genetic variation in the aaRSs have been shown to span all three of these possible inheritance patterns (see Table 1).
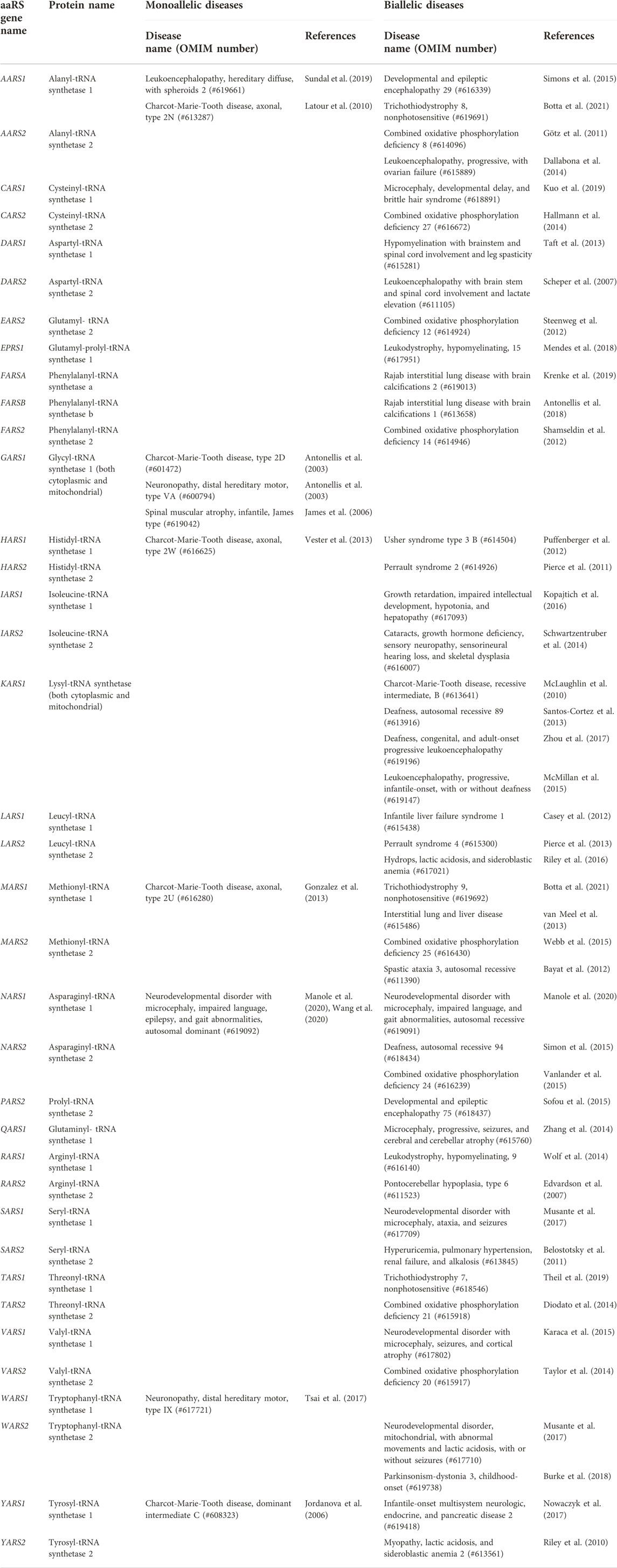
TABLE 1. Complete list of each aaRS-encoding gene and every human disease in the Online Mendelian Inheritance in Man resource (OMIM - https://www.omim.org) caused by damaging monoallelic and biallelic variants in that gene.
4.1 Monoallelic diseases
To date, 10 monoallelic diseases arising from autosomal dominant variants in seven aaRS genes (NARS1, HARS1, GARS1, AARS1, MARS1, WARS1, YARS1) have been identified (see Table 1). Interestingly, all of these monoallelic conditions occur in cytoplasmic AARS-encoding genes.
4.1.1 Charcot-marie-tooth disease
The most common monoallelic condition associated with AARS variants is Charcot-Marie-Tooth (CMT) disease. CMT is a clinically and genetically heterogeneous neurodegenerative disorder that affects the peripheral nervous system in roughly 1 in 2,500 individuals (Skre, 1974; Blocquel et al., 2019). Symptoms typically arise in early adulthood, manifesting as the progressive loss of motor and sensory functions. Clinical features include progressive weakness and atrophy in distal muscles leading to motor impairment, areflexia, limb abnormalities (esp. foot deformities), and a range of sensory loss (Skre, 1974; Rossor et al., 2013; Bansagi et al., 2015; Wei et al., 2019). There are currently no curative options for CMT. Treatment is supportive and focuses on maximizing function through physical and occupational therapies, orthopedic devices such as braces, and sometimes orthopedic surgery. Pain relief medications are used for CMT patients who experience severe pain. However, as the genetics of CMT are defined, therapies addressing the underlying molecular dysfunction are being developed (Pisciotta et al., 2021).
CMT is divided into various subtypes, with demyelinating Type 1 CMT (CMT1) and axonal Type 2 CMT (CMT2) containing the majority of cases. Demyelinating Type 1 CMT occurs from breakdown of the myelin sheath of nerves, while Type 2 axonal CMT occurs from direct damage to the axons of nerves (Harding and Thomas, 1980; Bird, 1993; Teunissen et al., 2003). Intermediate CMT has features of both Type 1 and Type 2.
While over 90 genes have been linked to the pathogenesis of CMT, aaRS-encoding genes constitute the largest gene family connected with this disorder (Bansagi et al., 2015; Blocquel et al., 2019). So far, variants in seven cytoplasmic aaRS genes have been established to cause CMT (YARS1, MARS1, KARS1, WARS1, AARS1, GARS1, HARS1). All of these genes except YARS1 and KARS1 are associated with monoallelic axonal CMT (CMT2). YARS1 causes dominant intermediate C CMT. KARS1 is an outlier in that it causes autosomal recessive CMT, and specifically the intermediate B subtype.
CMT is specifically associated with monoallelic pathogenic variants in cytoplasmic aaRSs, indicating a special sensitivity of the peripheral nervous system to this type of genetic change. Given the estimate that only ∼20% of the CMT-related variants affect canonical catalytic function (Datt and Sharma, 2014), the simple loss of aminoacylation activity is not a prerequisite for disease (Storkebaum et al., 2009; Froelich and First, 2011; Niehues et al., 2016; Zhang et al., 2021). Much work has been done to try to understand how monoallelic variants in cytoplasmic aaRSs cause disease in an autosomal dominant fashion and here we will highlight some key themes. Because the aaRS proteins are essential for protein synthesis in every cell, the challenge has been to determine if the pathogenic variants result in disease because they impair general protein synthesis or whether the disease causing aaRS variants become toxic to normal cellular function. The general experimental strategy has been to over express the wild-type version of the aaRS in an animal model of CMT disease (e.g., WT GlyRS in the dominant mouse models of CMT2D (Motley et al., 2011)). This experimental strategy can then test the hypothesis that if the disease is caused by a loss of function, then the overexpression would rescue the disease phenotype. In general, these types of genetic engineering studies in various model organisms (i.e., mice, flies, worms, and fish) have confirmed the dominant toxicity of pathogenic aaRS variants known to cause CMT (reviewed in (Wei et al., 2019)).
In recent studies, a complementary pair of papers by Zuko et al. and Spaulding et al. expanded our understanding of disease mechanisms in CMT (Mellado and Willis, 2021; Spaulding et al., 2021; Zuko et al., 2021). The investigators found that a disease causing GlyRS variant bound tRNAGly but failed to release it. This sequestration likely reduced the cellular tRNAGly pool, leading to insufficient tRNAGly supply to the ribosome. Indeed, they observed ribosome stalling at glycine codons and chronic activation of the damaging integrated stress response (ISR) in affected motor neurons through the sensor kinase GCN2. Importantly, these complementary studies identified two strategies with potential therapeutic benefit: 1) overexpression of tRNAGly to rescue protein synthesis, avoiding ISR activation and the ensuing peripheral neuropathy; or 2) inhibiting GCN2 to avoid activation of the neurotoxic ISR. Currently, the mechanism(s) linking aaRS variants to human CMT remains an area of intense investigation with the ultimate goal of developing treatments that will prevent neurodegeneration in patients born with aaRS variants that cause CMT.
4.1.2 Other monoallelic neurological disorders
While CMT is the most common monoallelic condition associated with aaRS variants, several other related neurological conditions are caused by damaging aaRS variants. It is helpful to appreciate that borders between these disease definitions are rather ‘porous’ and that these additional monoallelic aaRS-related conditions share features with CMT and with each other.
Distal hereditary motor neuronopathy (dHMN) is a pure motor neuropathy characterized by progressive distal muscle weakness and muscular atrophy without sensory impairment. Pathogenic variants in both WARS1 and GARS1 have been associated with dHMN (Antonellis et al., 2003; Tsai et al., 2017).
Neurodevelopmental disorder with microcephaly, impaired language, epilepsy, and gait abnormalities (designated NEDMILEG) is reported to be caused by de novo heterozygous variants in the NARS1 gene (Manole et al., 2020). Notably, bi-allelic variants in NARS1 also cause a similar neurodevelopmental disease. The mechanism of disease for the de novo heterozygous variants was suggested to be toxic gain-of-function, while the bi-allelic recessive variants were thought to cause disease through partial loss-of-function.
James type of infantile spinal muscular atrophy (SMAJI) is a severe neuromuscular disorder with symptoms beginning in the first weeks or months of life. Several unrelated children with SMAJI have been found to have de novo heterozygous variants in GARS1 (James et al., 2006; Eskuri et al., 2012; Forrester et al., 2020; Markovitz et al., 2020).
4.2 Biallelic diseases
Biallelic diseases arising from the disruption of both alleles of genes encoding the aaRSs cause severe, early-onset disorders affecting multiple organ systems. Biallelic disease can be caused by homozygous or compound heterozygous variants.
Autosomal recessive aaRS deficiencies represent a rapidly growing group of severe inherited diseases (Figure 1A) involving multiple organ systems and currently without curative treatment options. Fuchs et al. (2019) recently analyzed symptoms across aaRS biallelic disorders and found that the most common features of these disorders are: central nervous system (CNS) abnormalities, growth restriction, liver dysfunction, and facial dysmorphisms (Fuchs et al., 2019).
Current treatment options for autosomal recessive aaRS deficiencies are very limited. However, functional studies on variants that cause biallelic disease have demonstrated a reduction of the relevant aaRS protein level and/or decreases in aminoacylation enzymatic activity (Meyer-Schuman and Antonellis, 2017; Kok et al., 2021). Importantly, these patients still have some intrinsic aminoacylation activity. It is hypothesized that deficiencies in the aaRS enzymes may result in the inability to supply sufficient charged tRNAs to support protein synthesis, especially during periods of increased demand, such as rapid growth and infections (Kok et al., 2022). Knowledge of this disease mechanism led Kok et al. to trial a personalized intervention in four patients based on oral supplementation with the cognate amino acid matching the patients’ aaRS deficiency (e.g., the patient with biallelic IARS1 variants received high doses of oral l-isoleucine) (Kok et al., 2021). This amino acid supplementation was well-tolerated and safe, and showed encouraging results in terms of improvements in growth, development, and ability to cope with intercurrent infections. It is anticipated that this result will encourage more trials in additional patients to more formally assess the safety and efficacy of this treatment approach which targets the underlying aminoacylation defect in patients with autosomal recessive aaRS deficiencies.
Biallelic aaRS diseases are multi-system disorders with significant cross-over between phenotypes. It is likely that more clarity will emerge around the clinical phenotypes as more patients and more genetic diagnoses are described. Indeed, Fuchs et al. emphasized the importance of deep phenotyping of patients with aaRS-related diseases and reporting all clinical features, so the full extent of the phenotypes can be appreciated (Fuchs et al., 2019). For simplicity we have divided this section into biallelic disorders affecting mitochondrial or cytoplasmic aaRSs.
4.2.1 Biallelic disorders affecting mitochondrial aaRSs
All aaRS proteins are nuclear-encoded enzymes. After their translation in the cytosol, the mitochondrial aaRSs must be imported into the mitochondrial matrix to perform their canonical role of charging mitochondrial genome-encoded tRNA molecules with their cognate amino acids. Essential cellular processes rely on available ATP, the cellular energy currency, which is generated by oxidative phosphorylation that takes place in the five respiratory complexes in the mitochondria (Sissler et al., 2017; González-Serrano et al., 2019). AaRS proteins play a role in mitochondrial oxidative phosphorylation because accurate translation of the 13 mitochondrial-encoded proteins involved in oxidative phosphorylation and ATP production requires properly functioning mitochondrial aaRS proteins (Fine et al., 2019).
Combined oxidative phosphorylation deficiency (COXPD) is a unifying umbrella term describing a large group of multisystem disorders caused by defects in the mitochondrial oxidative phosphorylation system. Currently more than 50 different types of COXPD have been described, each caused by damaging variants in genes critical to the integrity of mitochondrial oxidative phosphorylation. The most common biallelic disease category associated with mitochondrial aaRSs is COXPD. To date, eight mitochondrial aaRS-encoding genes have been found to cause various forms of COXPD (MARS2, CARS2, EARS2, VARS2, TARS2, AARS2, FARS2, NARS2). All forms of this COXPD caused by pathogenic aaRS variants cause damage to the CNS (which requires an abundant and constant energy supply) (Moulinier et al., 2017; Sissler et al., 2017; Zheng et al., 2022). Other common features include liver disease, visual impairment, and microcephaly. COXPD8, which arises from variants in the AARS2 gene, has been shown to cause lethal cardiomyopathy (Götz et al., 2011; Taylor et al., 2014).
Beyond the umbrella term of COXPD, when viewed in aggregate, biallelic disorders of the mitochondrial aaRSs predominantly cause disease of the central nervous system (i.e. leukoencephalopathies, epilepsy, developmental delay, intellectual disability, sensorial neural hearing loss). However, other organs systems are also affected, manifesting as liver disease, myopathies, and ovarian failure (Meyer-Schuman and Antonellis, 2017). Hence, not all biallelic disorders of the mitochondrial aaRSs cause disease that falls cleanly under the broad COXPD phenotype. For example, damaging biallelic variants in HARS2 and LARS2 cause Perrault syndrome, an autosomal recessive disorder characterized by sensorineural deafness in both males and females, and ovarian dysgenesis in affected females (Pierce et al., 2011; Pierce et al., 2013). While there remains much to learn about these disorders, the fact that biallelic defects in mitochondrial aaRS enzymes do not all lead to identical phenotypes suggests that the underlying disease mechanisms might involve alterations in non-canonical ‘moonlighting’ function rather than solely defects in aminoacylation (Roux et al., 2021).
4.2.2 Biallelic disorders affecting cytosolic aaRSs
Biallelic variants in the cytosolic aaRSs predominantly cause neurological disease, notably leukodystrophies, leukoencephalopathies, and other neurodevelopmental disorders. Leukodystrophies are genetic disorders affecting the white matter of the CNS with or without peripheral nervous system involvement. Genetic leukoencephalopathies refer to related neurological conditions with significant white matter abnormalities that do not meet criteria for inclusion as a leukodystrophy (Vanderver et al., 2015). Leukodystrophies and leukoencephalopathies both profoundly impact the CNS, causing abnormalities and degeneration of cerebral white matter (Kaye and Moser, 2004; Parikh et al., 2015; Tang et al., 2019). Biallelic cytosolic aaRS variants are known to cause both leukoencephalopathies (linked to KARS1) and leukodystrophies (RARS1, EPRS1). Interestingly, a damaging monoallelic variant in the AARS1 gene has also been found to cause leukoencephalopathy in two members of an affected Swedish family (Sundal et al., 2019). Other neurodevelopmental disorders have been linked to three biallelic variants (VARS1, SARS1, NARS1) and one monoallelic variant (NARS1) in cytosolic aaRS genes. Symptoms of these disorders include intellectual disability, delayed language development and ability to walk, microcephaly, movement disorders, and in some cases seizures (Hübers et al., 2020). There is clear cross-over between the manifestations of the disorders caused by biallelic mitochondrial and cytosolic aaRS variants which will only be resolved through the sequencing and careful phenotyping of more affected individuals.
5 Anti-synthetase syndrome
Autoimmune diseases are the result of the body inappropriately mounting an immune response against itself. In addition to the genetic disorders of the aaRSs, there is also an acquired autoimmune condition affecting aaRSs, called Anti-Synthetase Syndrome (ASSD). ASSD is a rare condition that involves the production of autoantibodies that bind with, and mount a response against, aaRS proteins (Kron and Härtlein, 2013; Galindo-Feria et al., 2022). ASSD is an idiopathic inflammatory myopathy with organ complications beyond the muscles, including interstitial lung disease (Mahler et al., 2014). ASSD has features that overlap with dermatomyositis and polymyositis (Lepreux et al., 2018). The formal diagnostic criteria for ASSD are based on the presence of anti-aminoacyl tRNA synthetase antibodies along with major (interstitial lung disease and/or polymyositis or dermatomyositis) and minor (arthritis, Raynaud’s phenomenon, mechanic’s hands) criteria (Solomon et al., 2011; Witt et al., 2016). The most commonly recognized autoantigen is HisRS (recognized by anti-Jo-1 autoantibodies), but to date, autoantibodies targeting eight aaRSs (HisRS, ThrRS, AlaRS, GlyRS, IleRS, AsnRS, PheRS, TyrRS) have been linked to ASSD (Ascherman, 2015; Galindo-Feria et al., 2022). A combination of immunosuppressive agents is used to treat ASSD (Witt et al., 2016). While our understanding of the pathophysiology of ASSD remains incomplete, autoimmune targeting of the aaRSs may trigger their non-canonical immune functions to activate the innate and adaptive immunity (Gallay et al., 2018).
6 Discussion and future directions
The recent rapid advances in defining the role of aaRSs in human disease opens many avenues for life changing improvements in diagnosis and targeted treatment. The ability to provide a complete genetic diagnosis for individuals with monoallelic or biallelic aaRS diseases is transformative in many ways—new treatments can be explored based on the genetic findings (i.e., personalized medicine), new potential medical issues or risks can be anticipated and avoided, and accurate genetic counselling can be provided for the patient and their extended family. Ultimately, our current understanding of aaRSs in health and disease represents a powerful integration of knowledge that has emerged through the study of evolutionary science, basic biochemistry, and clinical medicine.
Author contributions
Conceptualization (AT, GH, AC), Writing-Original Draft (AT), Writing-Review & Editing (AT, GH, AC), Supervision (AC).
Funding
We acknowledge funding from the Pomona College Summer Undergraduate Research Program (to AT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Antonellis A., Ellsworth R. E., Sambuughin N., Puls I., Abel A., Lee-Lin S.-Q., et al. (2003). Glycyl tRNA synthetase mutations in charcot-marie-tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 72, 1293–1299. doi:10.1086/375039
Antonellis A., Oprescu S. N., Griffin L. B., Heider A., Amalfitano A., Innis J. W. (2018). Compound heterozygosity for loss-of-function FARSB variants in a patient with classic features of recessive aminoacyl-tRNA synthetase-related disease. Hum. Mutat. 39, 834–840. doi:10.1002/humu.23424
Arnez J. G., Moras D. (1997). Structural and functional considerations of the aminoacylation reaction. Trends biochem. Sci. 22, 211–216. doi:10.1016/s0968-0004(97)01052-9
Ascherman D. P. (2015). Role of jo-1 in the immunopathogenesis of the anti-synthetase syndrome. Curr. Rheumatol. Rep. 17, 56. doi:10.1007/s11926-015-0532-1
Bamshad M. J., Nickerson D. A., Chong J. X. (2019). Mendelian gene discovery: Fast and furious with No end in sight. Am. J. Hum. Genet. 105, 448–455. doi:10.1016/j.ajhg.2019.07.011
Bansagi B., Antoniadi T., Burton-Jones S., Murphy S. M., McHugh J., Alexander M., et al. (2015). Genotype/phenotype correlations in AARS-related neuropathy in a cohort of patients from the United Kingdom and Ireland. J. Neurol. 262, 1899–1908. doi:10.1007/s00415-015-7778-4
Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., et al. (2012). Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 10, e1001288. doi:10.1371/journal.pbio.1001288
Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., et al. (2011). Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 88, 193–200. doi:10.1016/j.ajhg.2010.12.010
Bird T. D. (1993). “Charcot-marie-tooth hereditary neuropathy overview,” in GeneReviews®. Editors M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Beanet al. (Seattle, WA: University of Washington, Seattle).
Blocquel D., Sun L., Matuszek Z., Li S., Weber T., Kuhle B., et al. (2019). CMT disease severity correlates with mutation-induced open conformation of histidyl-tRNA synthetase, not aminoacylation loss, in patient cells. Proc. Natl. Acad. Sci. U. S. A. 116, 19440–19448. doi:10.1073/pnas.1908288116
Botta E., Theil A. F., Raams A., Caligiuri G., Giachetti S., Bione S., et al. (2021). Protein instability associated with AARS1 and MARS1 mutations causes trichothiodystrophy. Hum. Mol. Genet. 30, 1711–1720. doi:10.1093/hmg/ddab123
Bullwinkle T. J., Ibba M. (2014). Emergence and evolution. Top. Curr. Chem. 344, 43–87. doi:10.1007/128_2013_423
Burke E. a., Frucht S. j., Thompson K., Wolfe L. a., Yokoyama T., Bertoni M., et al. (2018). Biallelic mutations in mitochondrial tryptophanyl-tRNA synthetase cause Levodopa-responsive infantile-onset Parkinsonism. Clin. Genet. 93, 712–718. doi:10.1111/cge.13172
Casey J. P., McGettigan P., Lynam-Lennon N., McDermott M., Regan R., Conroy J., et al. (2012). Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol. Genet. Metab. 106, 351–358. doi:10.1016/j.ymgme.2012.04.017
Cerini C., Kerjan P., Astier M., Gratecos D., Mirande M., Sémériva M. (1991). A component of the multisynthetase complex is a multifunctional aminoacyl-tRNA synthetase. EMBO J. 10, 4267–4277. doi:10.1002/j.1460-2075.1991.tb05005.x
Cui H., Kapur M., Diedrich J. K., Yates J. R., Ackerman S. L., Schimmel P. (2021). Regulation of ex-translational activities is the primary function of the multi-tRNA synthetase complex. Nucleic Acids Res. 49, 3603–3616. doi:10.1093/nar/gkaa1183
Cusack S., Berthet-Colominas C., Härtlein M., Nassar N., Leberman R. (1990). A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 Å. Nature 347, 249–255. doi:10.1038/347249a0
Dallabona C., Diodato D., Kevelam S. H., Haack T. B., Wong L.-J., Salomons G. S., et al. (2014). Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 82, 2063–2071. doi:10.1212/WNL.0000000000000497
Datt M., Sharma A. (2014). Evolutionary and structural annotation of disease-associated mutations in human aminoacyl-tRNA synthetases. BMC Genomics 15, 1063. doi:10.1186/1471-2164-15-1063
Diodato D., Melchionda L., Haack T. B., Dallabona C., Baruffini E., Donnini C., et al. (2014). VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat. 35, 983–989. doi:10.1002/humu.22590
Edvardson S., Shaag A., Kolesnikova O., Gomori J. M., Tarassov I., Einbinder T., et al. (2007). Deleterious mutation in the mitochondrial arginyl–transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 81, 857–862. doi:10.1086/521227
Eriani G., Delarue M., Poch O., Gangloff J., Moras D. (1990). Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347, 203–206. doi:10.1038/347203a0
Eskuri J. M., Stanley C. M., Moore S. A., Mathews K. D. (2012). Infantile onset CMT2D/dSMA V in monozygotic twins due to a mutation in the anticodon-binding domain of GARS. J. Peripher. Nerv. Syst. 17, 132–134. doi:10.1111/j.1529-8027.2012.00370.x
Fine A. S., Nemeth C. L., Kaufman M. L., Fatemi A. (2019). Mitochondrial aminoacyl-tRNA synthetase disorders: An emerging group of developmental disorders of myelination. J. Neurodev. Disord. 11, 29. doi:10.1186/s11689-019-9292-y
Forrester N., Rattihalli R., Horvath R., Maggi L., Manzur A., Fuller G., et al. (2020). Clinical and genetic features in a series of eight unrelated patients with neuropathy due to glycyl-tRNA synthetase ( GARS) variants. J. Neuromuscul. Dis. 7, 137–143. doi:10.3233/JND-200472
Froelich C. A., First E. A. (2011). Dominant Intermediate Charcot-Marie-Tooth disorder is not due to a catalytic defect in tyrosyl-tRNA synthetase. Biochemistry 50, 7132–7145. doi:10.1021/bi200989h
Fuchs S. A., Schene I. F., Kok G., Jansen J. M., Nikkels P. G. J., van Gassen K. L. I., et al. (2019). Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet. Med. 21, 319–330. doi:10.1038/s41436-018-0048-y
Galindo-Feria A. S., Notarnicola A., Lundberg I. E., Horuluoglu B. (2022). Aminoacyl-tRNA synthetases: On anti-synthetase syndrome and beyond. Front. Immunol. 13. doi:10.3389/fimmu.2022.866087
Gallay L., Gayed C., Hervier B. (2018). Antisynthetase syndrome pathogenesis: Knowledge and uncertainties. Curr. Opin. Rheumatol. 30, 664–673. doi:10.1097/BOR.0000000000000555
Gonzalez M., McLaughlin H., Houlden H., Guo M., Yo-Tsen L., Hadjivassilious M., et al. (2013). Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry 84, 1247–1249. doi:10.1136/jnnp-2013-305049
González-Serrano L. E., Chihade J. W., Sissler M. (2019). When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 294, 5309–5320. doi:10.1074/jbc.REV118.002953
Götz A., Tyynismaa H., Euro L., Ellonen P., Hyötyläinen T., Ojala T., et al. (2011). Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 88, 635–642. doi:10.1016/j.ajhg.2011.04.006
Guo M., Schimmel P., Yang X.-L. (2010a). Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett. 584, 434–442. doi:10.1016/j.febslet.2009.11.064
Guo M., Yang X.-L., Schimmel P. (2010b). New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 11, 668–674. doi:10.1038/nrm2956
Hallmann K., Zsurka G., Moskau-Hartmann S., Kirschner J., Korinthenberg R., Ruppert A.-K., et al. (2014). A homozygous splice-site mutation in CARS2 is associated with progressive myoclonic epilepsy. Neurology 83, 2183–2187. doi:10.1212/WNL.0000000000001055
Harding A. E., Thomas P. K. (1980). The clinical features of hereditary motor and sensory neuropathy types I and ii. Brain 103, 259–280. doi:10.1093/brain/103.2.259
Havrylenko S., Mirande M. (2015). Aminoacyl-tRNA synthetase complexes in evolution. Int. J. Mol. Sci. 16, 6571–6594. doi:10.3390/ijms16036571
Hübers A., Huppertz H.-J., Wortmann S. B., Kassubek J. (2020). Mutation of the WARS2 gene as the cause of a severe hyperkinetic movement disorder. Mov. Disord. Clin. Pract. 7, 88–90. doi:10.1002/mdc3.12855
Ibba M., Söll D. (2000). Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 69, 617–650. doi:10.1146/annurev.biochem.69.1.617
Ivanov K. A., Moor N. A., Lavrik O. I. (2000). Non-canonical functions of aminoacyl-tRNA synthetases. Biochemistry. 65, 888–897.
James P. A., Cader M. Z., Muntoni F., Childs A.-M., Crow Y. J., Talbot K. (2006). Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology 67, 1710–1712. doi:10.1212/01.wnl.0000242619.52335.bc
Jin D., Wek S. A., Kudlapur N. T., Cantara W. A., Bakhtina M., Wek R. C., et al. (2021). Disease-associated mutations in a bifunctional aminoacyl-tRNA synthetase gene elicit the integrated stress response. J. Biol. Chem. 297, 101203. doi:10.1016/j.jbc.2021.101203
Jordanova A., Irobi J., Thomas F. P., Van Dijck P., Meerschaert K., Dewil M., et al. (2006). Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 38, 197–202. doi:10.1038/ng1727
Kaiser F., Krautwurst S., Salentin S., Haupt V. J., Leberecht C., Bittrich S., et al. (2020). The structural basis of the genetic code: Amino acid recognition by aminoacyl-tRNA synthetases. Sci. Rep. 10, 12647. doi:10.1038/s41598-020-69100-0
Karaca E., Harel T., Pehlivan D., Jhangiani S. N., Gambin T., Coban Akdemir Z., et al. (2015). Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88, 499–513. doi:10.1016/j.neuron.2015.09.048
Kaye E. M., Moser H. (2004). Where has all the white matter gone?: Unraveling the mysteries of leukoencephalopathies. Neurology 62, 1464–1465. doi:10.1212/WNL.62.9.1464
Kerjan P., Cerini C., Sémériva M., Mirande M. (1994). The multienzyme complex containing nine aminoacyl-tRNA synthetases is ubiquitous from Drosophila to mammals. Biochim. Biophys. Acta 1199, 293–297. doi:10.1016/0304-4165(94)90009-4
Khan K., Baleanu-Gogonea C., Willard B., Gogonea V., Fox P. L. (2020). 3-Dimensional architecture of the human multi-tRNA synthetase complex. Nucleic Acids Res. 48, 8740–8754. doi:10.1093/nar/gkaa569
Khan K., Gogonea V., Fox P. L. (2022). Aminoacyl-tRNA synthetases of the multi-tRNA synthetase complex and their role in tumorigenesis. Transl. Oncol. 19, 101392. doi:10.1016/j.tranon.2022.101392
Kok G., Tseng L., Schene I. F., Dijsselhof M. E., Salomons G., Mendes M. I., et al. (2021). Treatment of ARS deficiencies with specific amino acids. Genet. Med. 23, 2202–2207. doi:10.1038/s41436-021-01249-z
Kok G., van Karnebeek C. D. M., Fuchs S. A. (2022). Response to Shen et al. Genet. Med. 24, 506–507. doi:10.1016/j.gim.2021.09.022
Kopajtich R., Murayama K., Janecke A. R., Haack T. B., Breuer M., Knisely A. S., et al. (2016). Biallelic IARS mutations cause growth retardation with prenatal onset, intellectual disability, muscular hypotonia, and infantile hepatopathy. Am. J. Hum. Genet. 99, 414–422. doi:10.1016/j.ajhg.2016.05.027
Krenke K., Szczałuba K., Bielecka T., Rydzanicz M., Lange J., Koppolu A., et al. (2019). FARSA mutations mimic phenylalanyl-tRNA synthetase deficiency caused by FARSB defects. Clin. Genet. 96, 468–472. doi:10.1111/cge.13614
Kron M., Härtlein M. (2013). Aminoacyl-tRNA synthetases and disease. Madame Curie Bioscience Database [Internet]
Kuo M. E., Theil A. F., Kievit A., Malicdan M. C., Introne W. J., Christian T., et al. (2019). Cysteinyl-tRNA synthetase mutations cause a multi-system, recessive disease that includes microcephaly, developmental delay, and brittle hair and nails. Am. J. Hum. Genet. 104, 520–529. doi:10.1016/j.ajhg.2019.01.006
Kyriacou S. V., Deutscher M. P. (2008). An important role for the multienzyme aminoacyl-tRNA synthetase complex in mammalian translation and cell growth. Mol. Cell 29, 419–427. doi:10.1016/j.molcel.2007.11.038
Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., et al. (2010). A major determinant for binding and aminoacylation of tRNAala in cytoplasmic alanyl-tRNA synthetase is mutated in dominant axonal charcot-marie-tooth disease. Am. J. Hum. Genet. 86, 77–82. doi:10.1016/j.ajhg.2009.12.005
Lepreux S., Hainfellner J. A., Vital A. (2018). Idiopathic inflammatory myopathies overlapping with systemic diseases. Clin. Neuropathol. 37, 6–15. doi:10.5414/NP301077
Ling J., Reynolds N., Ibba M. (2009). Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 63, 61–78. doi:10.1146/annurev.micro.091208.073210
Mahler M., Miller F. W., Fritzler M. J. (2014). Idiopathic inflammatory myopathies and the anti-synthetase syndrome: A comprehensive review. Autoimmun. Rev. 13, 367–371. doi:10.1016/j.autrev.2014.01.022
Manole A., Efthymiou S., O’Connor E., Mendes M. I., Jennings M., Maroofian R., et al. (2020). De novo and Bi-allelic pathogenic variants in NARS1 cause neurodevelopmental delay due to toxic gain-of-function and partial loss-of-function effects. Am. J. Hum. Genet. 107, 311–324. doi:10.1016/j.ajhg.2020.06.016
Markovitz R., Ghosh R., Kuo M. E., Hong W., Lim J., Bernes S., et al. (2020). GARS-related disease in infantile spinal muscular atrophy: Implications for diagnosis and treatment. Am. J. Med. Genet. A 182, 1167–1176. doi:10.1002/ajmg.a.61544
McLaughlin H. M., Sakaguchi R., Liu C., Igarashi T., Pehlivan D., Chu K., et al. (2010). Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 87, 560–566. doi:10.1016/j.ajhg.2010.09.008
McMillan H. J., Humphreys P., Smith A., Schwartzentruber J., Chakraborty P., Bulman D. E., et al. (2015). Congenital visual impairment and progressive microcephaly due to lysyl–transfer ribonucleic acid (RNA) synthetase (KARS) mutations: The expanding phenotype of aminoacyl–transfer RNA synthetase mutations in human disease. J. Child. Neurol. 30, 1037–1043. doi:10.1177/0883073814553272
Mellado W., Willis D. E. (2021). Stressing out translation. Science 373, 1089–1090. doi:10.1126/science.abk3261
Mendes M. I., Salazar M. G., Guerrero K., Thiffault I., Salomons G. S., Gauquelin L., et al. (2018). Bi-Allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 102, 676–684. doi:10.1016/j.ajhg.2018.02.011
Meyer-Schuman R., Antonellis A. (2017). Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet. 26, R114-R127–R127. doi:10.1093/hmg/ddx231
Motley W. W., Seburn K. L., Nawaz M. H., Miers K. E., Cheng J., Antonellis A., et al. (2011). Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet. 7, e1002399. doi:10.1371/journal.pgen.1002399
Moulinier L., Ripp R., Castillo G., Poch O., Sissler M. (2017). MiSynPat: An integrated knowledge base linking clinical, genetic, and structural data for disease-causing mutations in human mitochondrial aminoacyl-tRNA synthetases. Hum. Mutat. 38, 1316–1324. doi:10.1002/humu.23277
Musante L., Püttmann L., Kahrizi K., Garshasbi M., Hu H., Stehr H., et al. (2017). Mutations of the aminoacyl-tRNA-synthetases SARS and WARS2 are implicated in the etiology of autosomal recessive intellectual disability. Hum. Mutat. 38, 621–636. doi:10.1002/humu.23205
Nagao A., Suzuki T., Katoh T., Sakaguchi Y., Suzuki T. (2009). Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc. Natl. Acad. Sci. U. S. A. 106, 16209–16214. doi:10.1073/pnas.0907602106
Ng S. B., Buckingham K. J., Lee C., Bigham A. W., Tabor H. K., Dent K. M., et al. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35. doi:10.1038/ng.499
Niehues S., Bussmann J., Steffes G., Erdmann I., Köhrer C., Sun L., et al. (2016). Corrigendum: Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat. Commun. 7, 10497. doi:10.1038/ncomms10497
Nowaczyk M. J. M., Huang L., Tarnopolsky M., Schwartzentruber J., Majewski J., Bulman D. E., et al. (2017). A novel multisystem disease associated with recessive mutations in the tyrosyl-tRNA synthetase (YARS) gene. Am. J. Med. Genet. A 173, 126–134. doi:10.1002/ajmg.a.37973
Parikh S., Bernard G., Leventer R. J., van der Knaap M. S., van Hove J., Pizzino A., et al. (2015). A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol. Genet. Metab. 114, 501–515. doi:10.1016/j.ymgme.2014.12.434
Pierce S. B., Chisholm K. M., Lynch E. D., Lee M. K., Walsh T., Opitz J. M., et al. (2011). Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. U. S. A. 108, 6543–6548. doi:10.1073/pnas.1103471108
Pierce S. B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M. K., Malach D., et al. (2013). Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 92, 614–620. doi:10.1016/j.ajhg.2013.03.007
Pisciotta C., Saveri P., Pareyson D. (2021). Challenges in treating charcot-marie-tooth disease and related neuropathies: Current management and future perspectives. Brain Sci. 11, 1447. doi:10.3390/brainsci11111447
Puffenberger E. G., Jinks R. N., Sougnez C., Cibulskis K., Willert R. A., Achilly N. P., et al. (2012). Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLOS ONE 7, e28936. doi:10.1371/journal.pone.0028936
Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Riley L. G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., et al. (2010). Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 87, 52–59. doi:10.1016/j.ajhg.2010.06.001
Riley L. G., Rudinger-Thirion J., Schmitz-Abe K., Thorburn D. R., Davis R. L., Teo J., et al. (2016). “LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure,” in JIMD reports, volume 28 JIMD reports. Editors E. Morava, M. Baumgartner, M. Patterson, S. Rahman, J. Zschocke, and V. Peters (Berlin, Heidelberg: Springer), 49–57. doi:10.1007/8904_2015_515
Rodova M., Ankilova V., Safro M. G. (1999). Human phenylalanyl-tRNA synthetase: Cloning, characterization of the deduced amino acid sequences in terms of the structural domains and coordinately regulated expression of the α and β subunits in chronic myeloid leukemia cells. Biochem. Biophys. Res. Commun. 255, 765–773. doi:10.1006/bbrc.1999.0141
Rossor A. M., Polke J. M., Houlden H., Reilly M. M. (2013). Clinical implications of genetic advances in Charcot–Marie–Tooth disease. Nat. Rev. Neurol. 9, 562–571. doi:10.1038/nrneurol.2013.179
Roux C.-J., Barcia G., Schiff M., Sissler M., Levy R., Dangouloff-Ros V., et al. (2021). Phenotypic diversity of brain MRI patterns in mitochondrial aminoacyl-tRNA synthetase mutations. Mol. Genet. Metab. 133, 222–229. doi:10.1016/j.ymgme.2021.04.004
Rubio Gomez M. A., Ibba M. (2020). Aminoacyl-tRNA synthetases. RNA 26, 910–936. doi:10.1261/rna.071720.119
Santos-Cortez R. L. P., Lee K., Azeem Z., Antonellis P. J., Pollock L. M., Khan S., et al. (2013). Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am. J. Hum. Genet. 93, 132–140. doi:10.1016/j.ajhg.2013.05.018
Scheper G. C., van der Klok T., van Andel R. J., van Berkel C. G. M., Sissler M., Smet J., et al. (2007). Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 39, 534–539. doi:10.1038/ng2013
Schuler B. A., Nelson E. T., Koziura M., Cogan J. D., Hamid R., Phillips J. A. (2022). Lessons learned: Next-generation sequencing applied to undiagnosed genetic diseases. J. Clin. Invest. 132, e154942. doi:10.1172/JCI154942
Schwartzentruber J., Buhas D., Majewski J., Sasarman F., Papillon-Cavanagh S., Thiffaut I., et al. (2014). Mutation in the nuclear-encoded mitochondrial isoleucyl–tRNA synthetase IARS2 in patients with cataracts, growth hormone deficiency with short stature, partial sensorineural deafness, and peripheral neuropathy or with leigh syndrome. Hum. Mutat. 35, 1285–1289. doi:10.1002/humu.22629
Shamseldin H. E., Alshammari M., Al-Sheddi T., Salih M. A., Alkhalidi H., Kentab A., et al. (2012). Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 49, 234–241. doi:10.1136/jmedgenet-2012-100836
Shepard A., Shiba K., Schimmel P. (1992). RNA binding determinant in some class I tRNA synthetases identified by alignment-guided mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 89, 9964–9968. doi:10.1073/pnas.89.20.9964
Simon M., Richard E. M., Wang X., Shahzad M., Huang V. H., Qaiser T. A., et al. (2015). Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and leigh syndrome. PLoS Genet. 11, e1005097. doi:10.1371/journal.pgen.1005097
Simons C., Griffin L. B., Helman G., Golas G., Pizzino A., Bloom M., et al. (2015). Loss-of-Function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet. 96, 675–681. doi:10.1016/j.ajhg.2015.02.012
Sissler M. (2021). Decoding the impact of disease-causing mutations in an essential aminoacyl-tRNA synthetase. J. Biol. Chem. 297, 101386. doi:10.1016/j.jbc.2021.101386
Sissler M., González-Serrano L. E., Westhof E. (2017). Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol. Med. 23, 693–708. doi:10.1016/j.molmed.2017.06.002
Skre H. (1974). Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 6, 98–118. doi:10.1111/j.1399-0004.1974.tb00638.x
Smirnova E. V., Lakunina V. A., Tarassov I., Krasheninnikov I. A., Kamenski P. A. (2012). Noncanonical functions of aminoacyl-tRNA synthetases. Biochemistry. 77, 15–25. doi:10.1134/S0006297912010026
Smith T. F., Hartman H. (2015). The evolution of Class II Aminoacyl-tRNA synthetases and the first code. FEBS Lett. 589, 3499–3507. doi:10.1016/j.febslet.2015.10.006
Sofou K., Kollberg G., Holmström M., Dávila M., Darin N., Gustafsson C. M., et al. (2015). Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genomic Med. 3, 59–68. doi:10.1002/mgg3.115
Solomon J., Swigris J. J., Brown K. K. (2011). Myositis-related interstitial lung disease and antisynthetase syndrome. J. Bras. Pneumol. 37, 100–109. doi:10.1590/s1806-37132011000100015
Spaulding E. L., Hines T. J., Bais P., Tadenev A. L. D., Schneider R., Jewett D., et al. (2021). The integrated stress response contributes to tRNA synthetase-associated peripheral neuropathy. Science 373, 1156–1161. doi:10.1126/science.abb3414
Steenweg M. E., Ghezzi D., Haack T., Abbink T. E. M., Martinelli D., van Berkel C. G. M., et al. (2012). Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain 135, 1387–1394. doi:10.1093/brain/aws070
Storkebaum E., Leitão-Gonçalves R., Godenschwege T., Nangle L., Mejia M., Bosmans I., et al. (2009). Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc. Natl. Acad. Sci. U. S. A. 106, 11782–11787. doi:10.1073/pnas.0905339106
Sundal C., Carmona S., Yhr M., Almström O., Ljungberg M., Hardy J., et al. (2019). An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol. Commun. 7, 188. doi:10.1186/s40478-019-0843-y
Taft R. J., Vanderver A., Leventer R. J., Damiani S. A., Simons C., Grimmond S. M., et al. (2013). Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 92, 774–780. doi:10.1016/j.ajhg.2013.04.006
Tang Y., Qin Q., Xing Y., Guo D., Di L., Jia J. (2019). AARS2 leukoencephalopathy: A new variant of mitochondrial encephalomyopathy. Mol. Genet. Genomic Med. 7, e00582. doi:10.1002/mgg3.582
Taylor R. W., Pyle A., Griffin H., Blakely E. L., Duff J., He L., et al. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77. doi:10.1001/jama.2014.7184
Teunissen L. L., Notermans N. C., Franssen H., van Engelen B. G. M., Baas F., Wokke J. H. J. (2003). Disease course of charcot-marie-tooth disease type 2: A 5-year follow-up study. Arch. Neurol. 60, 823–828. doi:10.1001/archneur.60.6.823
Theil A. F., Botta E., Raams A., Smith D. E. C., Mendes M. I., Caligiuri G., et al. (2019). Bi-Allelic TARS mutations are associated with brittle hair phenotype. Am. J. Hum. Genet. 105, 434–440. doi:10.1016/j.ajhg.2019.06.017
Tsai P.-C., Soong B.-W., Mademan I., Huang Y.-H., Liu C.-R., Hsiao C.-T., et al. (2017). A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain 140, 1252–1266. doi:10.1093/brain/awx058
van Meel E., Wegner D. J., Cliften P., Willing M. C., White F. V., Kornfeld S., et al. (2013). Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet. 14, 106. doi:10.1186/1471-2350-14-106
Vanderver A., Prust M., Tonduti D., Mochel F., Hussey H. M., Helman G., et al. (2015). Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 114, 494–500. doi:10.1016/j.ymgme.2015.01.006
Vanlander A. V., Menten B., Smet J., De Meirleir L., Sante T., De Paepe B., et al. (2015). Two siblings with homozygous pathogenic splice-site variant in mitochondrial asparaginyl–tRNA synthetase (NARS2). Hum. Mutat. 36, 222–231. doi:10.1002/humu.22728
Vester A., Velez-Ruiz G., McLaughlin H. M., Program N. C. S., Lupski J. R., Talbot K., et al. (2013). A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat. 34, 191–199. doi:10.1002/humu.22210
Wakasugi K., Schimmel P. (1999). Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 284, 147–151. doi:10.1126/science.284.5411.147
Wakasugi K., Slike B. M., Hood J., Ewalt K. L., Cheresh D. A., Schimmel P. (2002). Induction of angiogenesis by a fragment of human tyrosyl-tRNA synthetase. J. Biol. Chem. 277, 20124–20126. doi:10.1074/jbc.C200126200
Wang L., Li Z., Sievert D., Smith D. E. C., Mendes M. I., Chen D. Y., et al. (2020). Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat. Commun. 11, 4038. doi:10.1038/s41467-020-17454-4
Webb B. D., Wheeler P. G., Hagen J. J., Cohen N., Linderman M. D., Diaz G. A., et al. (2015). Novel, compound heterozygous, single-nucleotide variants in MARS2 associated with developmental delay, poor growth, and sensorineural hearing loss. Hum. Mutat. 36, 587–592. doi:10.1002/humu.22781
Wei N., Zhang Q., Yang X.-L. (2019). Neurodegenerative Charcot–Marie–Tooth disease as a case study to decipher novel functions of aminoacyl-tRNA synthetases. J. Biol. Chem. 294, 5321–5339. doi:10.1074/jbc.REV118.002955
Witt L. J., Curran J. J., Strek M. E. (2016). The diagnosis and treatment of antisynthetase syndrome. Clin. Pulm. Med. 23, 218–226. doi:10.1097/CPM.0000000000000171
Wolf N. I., Salomons G. S., Rodenburg R. J., Pouwels P. J. W., Schieving J. H., Derks T. G. J., et al. (2014). Mutations in RARS cause hypomyelination. Ann. Neurol. 76, 134–139. doi:10.1002/ana.24167
Yao P., Fox P. L. (2020). Aminoacyl-tRNA synthetases in cell signaling. Enzymes 48, 243–275. doi:10.1016/bs.enz.2020.04.002
Yao P., Fox P. L. (2013). Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 5, 332–343. doi:10.1002/emmm.201100626
Zhang H., Zhou Z.-W., Sun L. (2021). Aminoacyl-tRNA synthetases in charcot-marie-tooth disease: A gain or a loss? J. Neurochem. 157, 351–369. doi:10.1111/jnc.15249
Zhang X., Ling J., Barcia G., Jing L., Wu J., Barry B. J., et al. (2014). Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Genet. 94, 547–558. doi:10.1016/j.ajhg.2014.03.003
Zheng W.-Q., Pedersen S. V., Thompson K., Bellacchio E., French C. E., Munro B., et al. (2022). Elucidating the molecular mechanisms associated with TARS2-related mitochondrial disease. Hum. Mol. Genet. 31, 523–534. doi:10.1093/hmg/ddab257
Zhou X.-L., He L.-X., Yu L.-J., Wang Y., Wang X.-J., Wang E.-D., et al. (2017). Mutations in KARS cause early-onset hearing loss and leukoencephalopathy: Potential pathogenic mechanism. Hum. Mutat. 38, 1740–1750. doi:10.1002/humu.23335
Keywords: aminoacyl-tRNA synthetases (aaRSs), genetic diseases, human health, charcot-marie-tooth (CMT) disease, rare disease
Citation: Turvey AK, Horvath GA and Cavalcanti ARO (2022) Aminoacyl-tRNA synthetases in human health and disease. Front. Physiol. 13:1029218. doi: 10.3389/fphys.2022.1029218
Received: 26 August 2022; Accepted: 04 October 2022;
Published: 18 October 2022.
Edited by:
Haissi Cui, University of Toronto, CanadaReviewed by:
Joseph Chihade, Carleton College, United StatesCopyright © 2022 Turvey, Horvath and Cavalcanti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra K. Turvey, YXRhYjIwMjBAbXltYWlsLnBvbW9uYS5lZHU=