 Shengyuan Wang
Shengyuan Wang Hongyan Li5
Hongyan Li5 Minghao Yuan
Minghao Yuan Zhiyou Cai
Zhiyou Cai- 1Chongqing Medical University, Chongqing, China
- 2Department of Neurology, Chongqing General Hospital, University of Chinese Academy of Sciences, Chongqing, China
- 3Department of Neurology, Chongqing School, University of Chinese Academy of Sciences, Chongqing, China
- 4Chongqing Key Laboratory of Neurodegenerative Diseases, Chongqing, China
- 5Department of Neurology, The Affiliated Hospital of Southwest Medical University, Sichuan, China
Adenosine monophosphate-activated protein kinase (AMPK) is a significant energy sensor in the maintenance of cellular energy homeostasis. Autophagy is a highly conserved catabolic process that involves an intracellular degradation system in which cytoplasmic components, such as protein aggregates, organelles, and other macromolecules, are directed to the lysosome through the self-degradative process to maintain cellular homeostasis. Given the triggered autophagy process in various situations including the nutrient deficit, AMPK is potentially linked with different stages of autophagy. Above all, AMPK increases ULK1 activity by directly phosphorylating Ser467, Ser555, Thr574, and Ser637 at least four sites, which increases the recruitment of autophagy-relevant proteins (ATG proteins) to the membrane domains which affects autophagy at the initiation stage. Secondly, AMPK inhibits VPS34 complexes that do not contain pro-autophagic factors and are thus involved in isolation membrane forming processes, by direct phosphorylation of VPS34 on Thr163 and Ser165. After phosphorylation, AMPK can govern autophagosome formation through recruiting downstream autophagy-related proteins to the autophagosome formation site. Finally, the AMPK-SIRT1 signaling pathway can be activated by upregulating the transcription of autophagy-related genes, thereby enhancing autophagosome-lysosome fusion. This review provides an introduction to the role of AMPK in different stages of autophagy.
Introduction
Adenosine monophosphate-activated protein kinase (AMPK), a serine/threonine kinase in eukaryotes, plays a crucial role in regulating cell energy balance in eukaryotes (Chen et al., 2018). When the ADP/ATP or AMP/ATP ratio is increased, AMPK is activated and triggers a series of physiological regulation processes (Mancini and Salt, 2018). Activation of AMPK increases the rate of catabolic (ATP-generating) pathways and compatibly decreases ATP usage. In addition, AMPK regulates cellular energy balance as well as whole-body energy metabolism (Liang et al., 2021; Wu et al., 2021). Dysregulation of energy balance plays an important role in the onset of many diseases, such as type 2 diabetes, obesity, and cancer (Hoffman et al., 2020). AMPK plays a central regulatory role in energy homeostasis, making it an important target for drugs to prevent and/or treat metabolic diseases (Suh et al., 2017).
Among eukaryotes, autophagy, a highly conserved and ubiquitous physiological process, is the intracellular catabolic pathway (de Pablos et al., 2019). Although autophagy is active to a certain extent at the physiological level, nutrient deficiency or energy deficiency is the main driving factor inducing autophagy in the physiological process (Wang et al., 2021). Autophagy is a lysosomal-mediated degradation system that has been highly conserved during evolution. Autophagy circulates cell contents and removes accumulated proteins, damaged organelles, and invading pathogens (such as bacteria and viruses) for the maintenance of normal cell function and intracellular homeostasis (Zhang et al., 2015; Newman et al., 2017). The process of autophagy is continuous, including sequential membrane reconstitution (Zhao et al., 2016; Miyamoto, 2019). This review describes the important role of AMPK in autophagy and metabolic regulation. We will discuss how AMPK activity is energy produced in the process of intracellular material synthesis and the catabolism pathway of macromolecules and explain how AMPK mediates signaling pathways by directly or indirectly regulating autophagy-related proteins and the activity of autophagy regulatory factors to explain the mechanism of AMPK regulation of autophagy.
AMPK: Structures and activation mechanism
AMPK, a serine/threonine kinase, plays a crucial role in regulating cellular energy balance across eukaryotes. When the intracellular RATIO of AMP/ATP increases relative to ATP, AMPK is activated and targets a series of subsequent physiological processes (Ke et al., 2018). Activation of AMPK increases the rate of catabolic pathways, resulting in increased ATP production and a corresponding decrease in ATP use efficiency (Bonanno et al., 2019). AMPK also regulates whole-body energy metabolism in addition to maintaining intracellular energy balance (Seabright et al., 2020). AMPK plays a crucial role in the steady-state energy imbalance (Kishton Rigel et al., 2016), and the energy balance is one of the important inducing factors of many diseases, such as type 2 diabetes, obesity, and cancer, so the important role of AMPK in steady-state energy form to prevent and/or treat many metabolic diseases (including cancer) is an important drug target (Faubert et al., 2015).
AMPK is a highly dynamic trimer consisting of α, β and γ subunits, with the β subunit sandwiched between the α and γ subunits (Hardie et al., 2016). The α, β and γ subunits are subdivided into different subtypes: the α and β subunits contain two subtypes (α1, α2 and β1, β2), and the γ subunit can be divided into three subtypes (γ1, γ2 and γ3). Combinations of all subtypes are possible, resulting in 12 different AMPK complexes (Carling, 2017). The N-terminus of the α subunit contains the catalytic domain of serine/threonine-protein kinase and residues of Thr172, whose phosphorylation plays an important role in AMPK activation. The C-terminus of the α subunit contains domains necessary for interaction with the β and γ subunits. Amino acids 312–335 of the α subunit have domains responsible for autoinhibitory activity, and the β subunit is responsible for the interaction between glycogen and AMPK (Herzig and Shaw, 2018). The specific action of each subunit is due to its different properties, among which the N-terminus of the β subunit contains CBM (carbohydrate-binding module) dedicated to AMPK targeting glycogen. In addition, the C-terminal domain of the β subunit (amino acids 186–270) stabilizes the interaction between the α and γ subunits (Figure 1). In addition, the γ subunit contains four CBS (cythionine β-synthase) domains, which are involved in regulating the sulfur conversion pathway in vivo (Oligschlaeger et al., 2015). AMP, ADP, and ATP bind to the γ subunit of AMPK to catalyze structural changes in the α-subunit and regulate phosphorylation of upstream kinase and Thr172, thereby activating AMPK (Lin and Hardie, 2018; Lyons and Roche, 2018). Each binding site requires a tandem CBS pair to form a functional unit called the Bateman domain (CBS1+CBS2 = Bateman domain 1, CBS3+CBS4 = Bateman domain 2). There are three sites (CBS1, 3, 4) for binding AMP, ADP, and ATP-associated adenine nucleotides in the core structure of the mammalian AMPK complex. CBS1, 3, and four have their own functions. However, in CBS2, the Asp residues bound to ribosomes were replaced by Arg, while the nucleotide binding of CBS2 was not observed in the heterotrimer structure (Jeon, 2016).
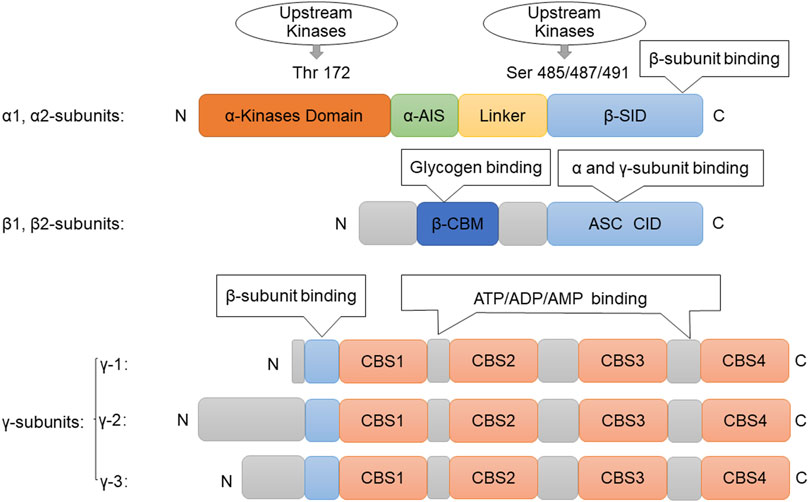
FIGURE 1. Structure of AMPK. Picture of the components of the AMPK subunits. The catalytic α-subunit can be phosphorylated at Thr172by upstream kinases including, LKB1, CaMKKβ, and TAK1, leading to enzyme activation. The β-subunit contains a glycogen-binding domain (GBD). The γ-subunits contain four nucleotide-binding modules (CBS domains), which can synergistically bind AMP, ADP, and ATP. Abbreviations: AMPK, adenosine monophosphate-activated protein kinase; ATP, Adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; ASC, association with Snf1 complex domain; AIS, autoinhibitory sequence; CBM, carbohydrate-binding module; CBS, cystathionine-β-synthase domain; Thr, threonine; Ser, serine; GBD, glycogen bind domain; SID, subunit interacting domain.
AMPK is activated by binding of AMP through three distinct mechanisms in the canonical mechanism. Specifically, AMP first stimulates the phosphorylation of Thr172 by directly activating upstream kinases (Kim et al., 2020). Thr172 can be phosphorylated by several different upstream kinases, which establishes the main mechanism by which AMPK activity is regulated in the short term (Song et al., 2021). Different studies have suggested that LBK1 (liver kinase B1) kinase and MAPKKK family member TAK/MAP3K7 (transforming growth factor beta-activated kinase 1/mitogen-activated kinase 7) can respond to various signals by phosphorylating Thr172 (Fritzen et al., 2016). It has also been shown that Thr172 can be phosphorylated by CAMKK2 (calcium/calmodulin-dependent protein kinase 2) kinase in response to changes in calcium flux. Second, AMP can be allosterically regulated to make AMPK more attractive to its upstream kinase (Jang et al., 2018). Finally, AMP also protects Thr172 from phosphatase activity, leaving Thr172 phosphorylated and increasing AMPK activity (Hardie et al., 2012). AMPK can increase ATP levels in the body through two pathways. First, it enhances energy catabolic processes such as glucose metabolism, lipid oxidation, mitochondrial biogenesis, and autophagy. It can also be achieved by reducing energy anabolic processes, such as lipid synthesis, glycogen storage, gluconeogenesis, and protein synthesis.
Autophagy biogenesis
Eukaryotes are highly conserved in their autophagy process, which is a degradation and recycling process (Ren and Zhang, 2018). It is possible that autophagy can create a cellular milieu that supports survival in some manner. The specific way is through the degradation of organelles, proteins and macromolecules to achieve the recycling of intracellular substances (Zhong et al., 2016). Depending on how the cytoplasmic material is transported into the lysosomal lumen, autophagy is classified into three main categories in mammalian cells: microautophagy, macroautophagy and chaperone-mediated autophagy (CMA) (Lamming and Bar-Peled, 2019). The three autophagy mechanisms are different (Cuomo et al., 2019). Macroautophagy occurs by fusion of autophagosomes and lysosomes, while microautophagy occurs by lysosomes directly wrapping and degrading the cell contents, while chaperone-mediated autophagy occurs by recognition of cytoplasmic proteins by molecular chaperones through special modules and then binding to special receptors on the lysosome membrane to enter the lysosome and be degraded (Mizushima and Komatsu, 2011; Martinez et al., 2016). All three types of autophagy can achieve lysosomal-mediated degradation and cycling of cytoplasmic components, including organelles, through different mechanisms (Yang and Klionsky, 2010; Lamming and Bar-Peled, 2019; Boukhalfa et al., 2021). Among them, macroautophagy is the most common type, and its biogenic process can be triggered by starvation, hypoxia, ER stress or other stimuli, thus involving a cascade of signaling and recruitment events (Corona and Jackson, 2018; Hu et al., 2021; Zeng and Jiang, 2021).
Once macrophages are activated, they specifically degrade damaged or excess organelles in the cell into metabolites for the biosynthesis process or energy generation in the cell so that other cells can survive and maintain the normal growth of the body (Parzych and Klionsky, 2014; Icard et al., 2021). Macroautophagy is generally protective for the body (Levine et al., 2011); however, excessive degradation may also cause damage to the body. Accordingly, the disruption of autophagy is associated with several human pathologies, such as lung, liver, and heart disease, neurodegeneration, myopathies, cancer, aging, and metabolic diseases (Kaur et al., 2021). Under the induction of the ULK1/2 complex, the nucleation of phagocytic groups initiates a series of subsequent biogenic processes of macroautophagy (Jayaraj et al., 2020; Karabiyik and Rubinsztein, 2021). The ATG12-ATG5-ATG16L1 complex, the class III PtdIns3K complex, LC3-II and ATG9 can assist phagocytic mass extension. Eventually, the cell component is enveloped by an expansive bilayer to form an autophagosome, and LC3-II divides from the outer membrane of the autophagosome (Nieto-Torres et al., 2021). Under certain conditions, autophagosomes fuse with endosomes to form amphisomes (Ho et al., 2017). In general, the outer membrane of autophagosomes fuses with lysosomes to form autophagolysosomes, and then the contents of autophagolysosomes are degraded and exported to the cytoplasm for cell reuse (Kimmelman and White, 2017).
Regulation of autophagy by AMPK
AMPK plays a critical role in cellular energy metabolism and can act directly on the metabolism of proteins or indirectly affect the expression of genes related to the regulation of a variety of metabolic processes (Martin-Montalvo et al., 2013; Cordero et al., 2018). There are a number of downstream targets inhibited by AMPK, acetyl-CoA carboxylase (ACC), mammalian target of rapamycin (mTOR), fatty acid synthase (FAS), PPARγ coactivator 1α (PGC1α), and glycerol phosphate acyltransferase (GPAT), which are involved in fatty acid synthesis, protein synthesis, glycerolipid synthesis, and mitochondrial function. In this way, the energy balance is maintained, and the internal environment is kept in balance. Autophagy is a protective mechanism by which cells adapt to a variety of different types of stress, which is regulated by AMPK (Mihaylova and Shaw, 2011; Jia et al., 2020). By specifically phosphorylating autophagy-related protein complexes, AMPK promotes autophagy at multiple levels of autophagy regulation (Blagih et al., 2012).
Autophagy is a lysosome/vacuole-dependent catabolic pathway in eukaryotes that is evolutionarily conserved (Lahiri et al., 2019). Nutrient deprivation or energy scarcity induces autophagy in a physiological manner (Yi et al., 2018). In organisms as diverse as yeast and humans, this intrinsic property remains unchanged. Thus, the involvement of AMPK in the regulation of this process seems logical (Tamargo-Gomez and MarinoAMPK, 2018). Autophagy begins with the formation of bowl-shaped membranes in the cytoplasm that expand into double-membrane vesicles called autophagosomes. Autophagosomes sequester cytoplasmic cargo and travel along the microtubule network until they merge with lysosomes (Mehrpour et al., 2010). In addition to the degradation of autophagosome cargo and its inner membrane, autophagosome-lysosome fusion also promotes the degradation of the autophagosome membrane. After degradation takes place, the resulting biomolecules are recycled back into the cytoplasm, where they are used to synthesize new biomolecules (Glick et al., 2010). ATG (autophagy-related proteins) are evolutionarily conserved genes/proteins that are necessary for the autophagy pathway. Originally described in yeast, these proteins are required for autophagosome formation, maturation, transport, or degradation, taking part in the various steps of autophagic degradation (Figure 2).
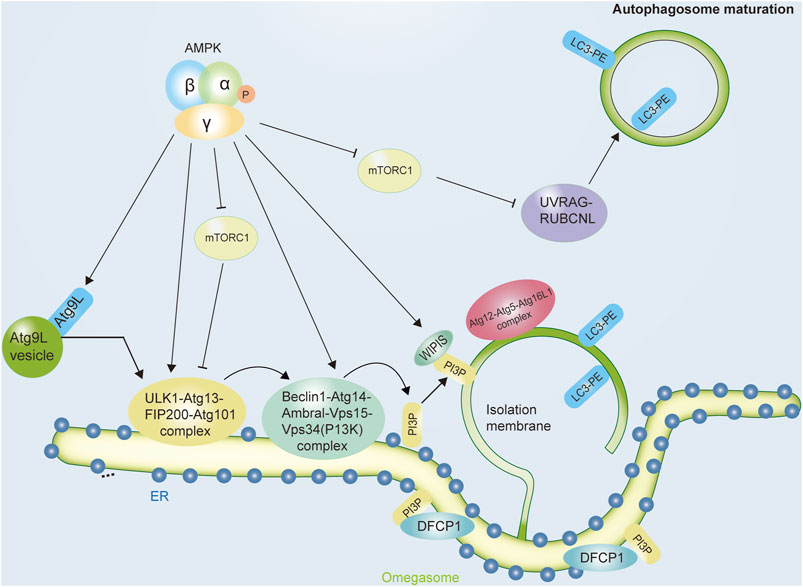
FIGURE 2. The role of AMPK in initiating the autophagic process. Autophagy, a lysosome-mediated degradation system, starts with the initiation and nucleation of a double membrane vesicle, known as the isolation membrane (or phagophore), in the cytoplasm. Autophagy occurs in a stepwise manner involving sequential membrane reshaping processes. Autophagy induction is initiated by the ULK1 complex, which contains ULK1, ATG13, FIP200, and ATG101. The activity of this protein complex is regulated by AMPK, which both inhibits the mTORC1 complex and activates the activity of ULK1 complexes. In mammalian cells, another complex ClassIII PI3K complex formed by VPS34, Beclin1, ATG14, AMBRA1, and other subunits act coordinately with the ULK1 complex to initiate autophagosome nucleation. AMPK can increase the pro-autophagic function of this complex and enhance its formation. Abbreviations: AMPK, adenosine monophosphate-activated protein kinase; ATG, autophagy-related protein; DFCP1, double FYVE-containing protein one; ER, endoplasmic reticulum; FIP200, focal adhesion kinase family interacting protein of 200kDa; LC3, microtubule-associated protein 1 A/1B-light chain three; PE, phosphatidylethanolamine; PI3P complex, the class III phosphatidylinositol (PtdIns) 3-kinase complex; mTOR, mechanistic target of rapamycin; mTORC1, mechanistic target of rapamycin complex one; ULK1, Unc-51-like kinase one; UVRAG, ultraviolet radiation resistance genes; WIPI proteins, WD-repeat protein interacting with phosphoinoside.
AMPK facilitates the initiation of autophagy
During autophagy induction, ULK1 and VPS34 are the major components that drive autophagy initiation. The activation of AMPK induces autophagy via two different mechanisms: negative regulation of the mammalian target of rapamycin (mTOR) protein kinase complex and activation of ULK1 (Unc-51-Like Kinase 1, a mammalian ortholog of Atg1) by direct phosphorylation. ULK1 is a protein complex formed by interacting with ATG13, RB1CC1/FIP200, and ATG101. Even though there are many signaling pathways that can influence autophagy, two of the most important ones are AMPK and mTOR (mechanistic/mammalian target of rapamycin). AMPK positively regulates autophagic activity. At least four sites on ULK1 are phosphorylated by AMPK to stimulate its activity. To regulate the activity of the ULK complex, AMPK antagonizes mTORC1 (Kim et al., 2015; Park et al., 2016). There are two protein complexes formed by mTOR, mTORC1 and mTORC2 (Egan et al., 2011; Gonzalez et al., 2020). Autophagy regulation is insignificantly influenced by mTORC2 (Schmitz et al., 2021). In contrast, a mammalian cell’s main autophagy suppressor is mTORC1 (Schmitz et al., 2021). As mTORC1 is normally activated when energy levels are high, cellular amino acid content is high, or growth factors are stimulated, these conditions are damaging to autophagy (Cohen and Hall, 2009). Due to their antagonistic roles, it is known that AMPK increases autophagic degradation by inhibiting mTORC1 activity (Rabinowitz and White, 2010), and the activity of mTORC1 is a molecular pathway that is closely related to that of AMPK (Tamargo-Gomez and MarinoAMPK, 2018; Yoo and Jung, 2018). When sufficient energy/growth factors or amino acids were present, mTORC1 inhibited autophagy by inhibiting phosphorylation of ATG13, thereby reducing the activity of the ULK1 complex and thus the rate of autophagosome formation. Similarly, ULK1 itself is a direct target of mTORC1, so mTORC1 inhibits the autophagic process by targeting ULK1 and ATG13 (Kim et al., 2011; Huang et al., 2020).
According to AMPK, a variety of pro-autophagic stimuli negatively regulates ULK1 complex activity, whereas mTORC1 positively regulates ULK1 complex activity (Zhao and Zhang, 2016). AMPK increases ULK1 activity by directly phosphorylating Ser555, Thr574, Ser467, and Ser637. The formation of autophagosomes is enhanced by increasing the recruitment of autophagy-relevant proteins to membrane domains. Furthermore, AMPK inhibits ULK1 inhibition by both inhibiting mTORC1 and blocking its inhibitory activity. First, in addition to activating TSC2(Tuberous sclerosis complex 2) by phosphorylating its Thr1227 and Ser1345 residues, AMPK also negatively influences the activity of mTORC1 by promoting the assembly of the heterodimer between TSC1 and TSC2 (Huang and Manning, 2008; Egan et al., 2011; Pengo et al., 2017). Second, inhibition of mTORC1 by AMPK is brought about by direct phosphorylation of the Ser722 and Ser792 residues in RAPTOR (Regulatory-associated protein of mTOR) (Johnson et al., 2013).
AMPK regulates the activity of Class III PI3K complexes as well (Kahn et al., 2005). It is also through phosphorylation in different Class III PI3K subunits that AMPK regulates autophagy, it also modulates their affinity for other complex components, including VPS34 itself (Kim et al., 2013a). Therefore, AMPK controls the relative abundance of various VPS34-containing complexes, which in turn regulates cellular energy levels through vesicle trafficking (Yang et al., 2011). Furthermore, divergent regulation of Vps34 complexes by AMPK occurs in the context of nutrient stress and autophagy (Kim et al., 2013a; Yoon et al., 2016). Studies have shown that AMPK regulates the balance of Class III PI3K complexes in this regard (Kim et al., 2013b). Beclin1’s binding to VPS34 and ATG14 is increased when AMPK phosphorylates Thr388 in Beclin1, the higher autophagy activity is promoted upon glucose withdrawal than in a wild-type cell (Zhao et al., 2015). A similar effect can be seen when AMPK phosphorylates mouse Beclin1 at Ser-91 and Ser-94 under nutritional stress (Stjepanovic et al., 2017).
Additionally, it interacts with different Class III PI3K complex components, AMPK can influence their composition by phosphorylating other proteins that are essential for the formation and stability of VPS34-containing complexes (Russell et al., 2013). It has also been demonstrated that AMPK-mediated phosphorylation of an ATG14 L/VPS34 scaffolding protein, Thr32 on PAQR3 (progestin and adipo-Q receptors member 3), or that of Thr50 on the VPS34 associated protein RACK1 (Receptor for activated C kinase 1) enhances stability and pro-autophagic activity of Class III PI3K complexes (Russell et al., 2013; Park et al., 2016). In parallel with the activation of VPS34, it is also phosphorylated in a variety of components of the pro-autophagic complexes, AMPK, which is generally pro-autophagic, suppresses the activity of late autophagy PI3KC3–C2 by phosphorylating Thr163 and Ser165 in VPS34. Therefore, in conditions that promote autophagy, by activating AMPK, pro-autophagic VPS34 complexes are activated while other Class III PI3K complexes involved in autophagy-independent processes are inhibited (Xu et al., 2016).
AMPK promotes autophagosome biogenesis
Phagophore expansion is thought to be responsible for autophagosome formation; that is, in autophagosomes, a sheet of membrane equal to the size of a complete autophagosome does not separate from the endomembrane system, but simply curls up (Grasso et al., 2018). The formation of autophagosomes is thought to be influenced by almost every compartment of the membrane, according to numerous lines of evidence, counting the mitochondria, ER, plasma membrane, and Golgi apparatus (Kimura et al., 2007). It appears that autophagosome generation may be mediated by at least two mechanisms (Yang and Klionsky, 2010; Kageyama et al., 2021). In yeast, membranes are believed to be delivered from various organelles, and Another system uses an omega-shaped membrane structure, called an omegasome, which is derived from phosphatidylinositol-3-phosphate (PtdIns3P)-enriched ER subdomains. Autophagosomes of yeast are formed from a single preautophagosomal structure (PAS), where most Atg proteins are recruited (Xie and Klionsky, 2007). There is a hierarchy of recruitment of the Atg protein to the PAS in yeast based on genetic studies (Polson et al., 2010). Upstream of Atg1 are the Vps34 lipid kinase complex and the Atg1 protein kinase complex (Reggiori and Klionsky, 2005). PAS expansion into a phagophore and autophagosome closure are dependent on Atg proteins downstream of these initiating complexes (Galluzzi and Green, 2019).
On the phagophore surface, Atg12 and Atg5 are covalently linked, triggering autophagosome formation (Nishimura et al., 2017). Atg16 and a second cytosolic Atg12-Atg5 conjugate are drawn to this compartment, where they begin to redistribute and focus primarily on the outside lipid bilayer. As Atg12-Atg5-Atg16 form larger oligomers, they determine phagophore elongation and curvature through the delivery of Atg8-containing membranes. After the autophagosome has been formed, Atg12 and Atg16 dissolve from this structure, while Atg4 releases the lipid bilayer-bound Atg8 to the cytosol by proteolytically cleaving it (Kawabata and Yoshimori, 2020). When the coating is uncoated, the autophagosome fuses with the lysosome. AMPK-MTORC1-ULK1/2 signaling axis regulates autophagosome formation in response to amino acid availability and energy balance in cells. It has been suggested that AMPK plays a role in the maturation of autophagosomes. Through UVRAG phosphorylation, AMPK can inhibit mTORC1 signaling, suppressing autophagosome maturation (Dunlop and Tee, 2013).
AMPK regulates autophagosome-lysosome fusion
The next stage of autophagy is the fusion of the outer membrane of autophagosomes and lysosomes to form autolysosomes and finally transport the materials encapsulated in autophagosomes to the lysosomal lumen (Mauthe et al., 2018). Autolysosomes degrade the inner membrane of the autophagosome and its encapsulated substances (Zhou et al., 2013). Moreover, the formation of autophagosomes requires a specific period. Studies have shown that only closed autophagosomes can fuse with lysosomes to form autolysosomes. However, how is the closure of autophagosomes regulated? In mammals, a defect in the ATG-conjugation system results in the accumulation of unclosed autophagosomes, implying that it is likely to function in the elongation and closure of autophagosomes and is important for the transition of the isolation membrane into the autophagosome (Nieto-Torres et al., 2021). It also participates in the maturation of autophagosomes, it was shown that the ATG conjugation system (consisting of ATG3, ATG7, and ATG5) is essential for degrading inner autophagic membranes; however, it is not required for autophagosome-lysosome fusion. In contrast, according to another study, cell lines that were knocked out of the entire ATG8 protein family showed that Although LC3 and GABARAP proteins do not participate in autophagosome formation, they play an essential role during lysosome-autophagosome fusion (Itakura et al., 2012).
Late endosomes and lysosomes are predominantly found in the perinuclear region, while autophagosomes form at random in the cytoplasm (Savini et al., 2019). Thus, once autophagosomes have been produced, they must be delivered to the perinuclear region. Dynein-dynactin motor complexes move cargo to the perinuclear region, while most kinesins drive cargo toward the periphery of the cell with their plus-end directed motor proteins (Yamamoto and Noda, 2020). Autophagosomes are transported minus-end-directed, given that lysosomes are found in perinuclear regions, and indeed, live imaging reveals mature autophagosomes traveling along microtubule tracks to lysosomes. By microinjecting antibodies against LC3, autophagosome movement is inhibited, suggesting LC3 plays a role during autophagosome movement. Furthermore, Autophagosome positioning requires plus-end-directed transport mediated by kinesin because the depletion of KIF5B blocks autophagy and causes clustering of autophagosomes in the perinuclear space (Lorincz and Juhasz, 2020). It is interesting to note that lysosome localization affects the rate of autophagosome fusion. Thus, it is crucial for fusion to coordinate autophagosome and lysosome transport. However, how do autophagosomes and lysosomes connect with microtubules? Rab7 is a small GTPase that acts as a molecular switch and, presumably, Connects autophagosomes and microtubule motors through FYCO1(FYVE and coiled-coil domain-containing 1) in late autophagosomes, thereby kinesin-driven movement towards the cell periphery is mediated (Nakamura and Yoshimori, 2017). The Rab7 protein also functions in the reverse direction by facilitating the transport of autophagosomes, autolysosomes, and lysosomes to the perinuclear region through its interaction with Rab-interacting lysosomal protein (RILP), dynein and the cholesterol sensor ORP1 L (also known as OSBPL1A) (Feng et al., 2013). Actin filaments utilize the myosin family of motor proteins for moving intracellular charges similarly to microtubules (Zhao et al., 2021).
Autophagosome fused with the endocytic system is the result of its arrival at its destination. However, since both processes seem to occur almost simultaneously, it is difficult to distinguish movement from fusion. Currently, In order to understand this process, we need to understand intracellular membrane trafficking in general, a particular focus is on three protein families: membrane-tethering complexes, Rab GTPases, and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) (Tian et al., 2020). As Rab proteins localize to specific membranes, they recruit tethering complexes that act as bridges between compartments intended for fusion. In turn, these tethering complexes enable SNARE proteins to drive the fusion of opposing lipid bilayers. By binding to LC3 and STX17–SNAP29, EPG5 binds to late endosomes/lysosomes along with Rab7 and VAMP-8; In this way, the trans-SNARE complex can be assembled for fusion more easily (Wang et al., 2016). Unlike EPG5, ATG14 L binds to STX17 and STX17-SNAP29 SNARE complexes on autophagosomes, but not to STX17–SNAP29-V AMP8 RQabc SNARE complexes, suggesting that ATG14 L appears to act earlier than EPG5. An adaptor protein, PLEKHM1, interacts with Rab7, LC3 proteins, HOPS–SNARE complexes, and GABARAP proteins to facilitate fusion between autophagosomes and lysosomes. It is still unclear how EPG5, ATG14 L, and the HOPS complex function as tethering factors. The O-GlcNAcylated SNAP-29, which is generated by O-linked N-acetylglucosamine transferase (OGT), has a reduced affinity for its SNARE partners. By decreasing levels of O-GlcNAcylated SNAP-29, starvation suppresses this modification, and SNAP-29-containing trans-SNARE complexes are assembled, stimulating autophagy.
There is robust evidence that AMPK plays a crucial role in autophagosome-lysosome fusion (Figure 3), and the activation of AMPK can positively regulate lysosomal biogenesis and function (Carroll and Dunlop, 2017). In mammals, some studies have shown that the coactivator-associated arginine methyltransferase 1 (CARM1) plays a crucial role in autophagy. A genome-wide analysis reveals that CARM1 acts as a transcriptional coactivator by interacting with transcription factor EB (TFEB) (Huang et al., 2019; Fang et al., 2021). An AMPK–SKP2–CARM1 signaling axis is responsible for regulating autophagy induction after nutrient starvation by CARM1-dependent histone arginine methylation, a crucial nuclear event in autophagy. Under nutrient-rich conditions, CARM1 stability is regulated by the SKP2-containing SCF (SKP1-cullin1-F-box protein) E3 ubiquitin ligase in the nucleus, but not in the cytoplasm (Shin et al., 2016). In addition, this study suggests that nutrient deprivation causes nucleus FOXO3a to undergo AMPK-dependent phosphorylation, which in turn inhibits transcription of the SKP2 gene. CARM1 is upregulated as a result of this repression, and it acts as a coactivator of TFEB in regulating lysosome function. As a result of glucose starvation, a lysosomal AMPK-activating complex is formed composed of AMPK and, V-ATPase, liver kinase B1 (LKB1), aldolase, AXIN, and Ragulator-RAG (Holland et al., 2020). AMPK is effectively activated by lysosomal N-myristoylation and DNA-dependent protein kinase (DNA-PK)-mediated phosphorylation of its γ-subunit. In contrast to mTORC1, AMPK inhibits diverse anabolic pathways while promoting lipolysis, glycolysis, and macroautophagy. Growth factors and cellular energy status are also involved in microtubule-mediated lysosomal trafficking. AMPK is activated by resveratrol when it interacts with its direct target SIRT1. Resveratrol has been shown to protect against Parkinson’s disease by enhancing autophagy in various models (Moors et al., 2017). It is a well known phenomenon that exercise increased the brain’s lysosome and autophagy levels. Exercise also promotes the nuclear translocation of TFEB in the cortex, increasing the transcription of genes related to autophagy and lysosomes. Fang Lin’s team discovered that exercise activated autolysosomal function and the gene transcription regulated by TFEB in the cerebral cortex through AMPK-SIRT1-TFEB pathway (Huang et al., 2019). Moreover, it has been shown that lysosomal function is activated by AMPK inhibition of mTORC1, and mTORC1 localization to lysosomes is not directly related to its role in regulating lysosomal function. In addition, as a result of mTORC1 suppression, TFEB activation is necessary, but not sufficient to activate lysosomes (Carroll and Dunlop, 2017). The expression of autophagy and lysosome genes is repressed by BRD4 in a study by Sakamaki et al. This inhibition is mitigated by AMPK-SIRT1 signaling during nutrient deficiency, thus allowing autophagy activation. By blocking BRD4 function, autophagy and lysosomal function are enhanced, as well as protein aggregate degradation is promoted (Sakamaki et al., 2017).
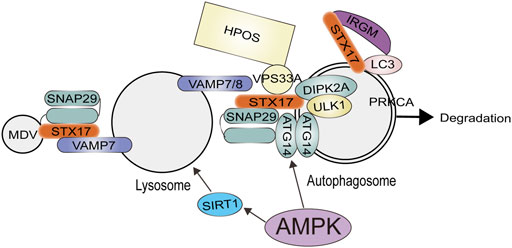
FIGURE 3. The role of AMPK for Autophagosome-lysosome fusion of autophagosomes with lysosomes involves the concerted action of RABs, tethers, and the SNARE complex. AMPK can positively regulate lysosomal biogenesis and function. AMPK activates ATG14 thereby promoting the fusion of the autophagosome and lysosome. Abbreviations: ATG14, autophagy-related protein14; DIPK2A, divergent protein kinase domain 2A; HOPS, homotypic fusion and protein sorting; IRGM, immunity related GTPase M; MDV, mitochondrial-derived vesicle; PRKCA, Protein Kinase C Alpha; SNAP29, synaptosomal associated protein 29; STX17, syntaxin 17; VAMP7, vesicle associated membrane protein seven; VPS33A, vacuolar protein sorting 33 homolog A; ULK1, unc-51 like.
Conclusion and perspectives
In higher eukaryotes, the regulation of autophagy has become increasingly complex, and multiple signal cascades are related to key autophagy factors. However, AMPK may still be the main molecular autophagy inducer that counteracts the activity of mTORC1, and mTORC1 has also been the main molecular autophagy inhibitor during the evolutionary process. AMPK inhibits anabolic activities that are activated due to a lack of energy or nutrients and switches off cell growth. While regulating the function of autophagy, AMPK’s role in adapting cell metabolism to energy supply is also conserved in various organisms from yeast to mammals.
As discussed in this review, AMPK plays a key role in the regulation of autophagy, coupled with various processes for which autophagic activity is beneficial, suggesting a potential mechanistic involvement of autophagy for some of the positive effects of AMPK activation. Future studies aimed at dissecting the precise molecular mechanisms by which AMPK exerts its wide variety of beneficial effects on human health will shed more light on these questions.
Author contributions
SW, MY, and HF wrote and revised the paper. HL draw the figures. ZC provided financial support.
Funding
This work was supported by the Construction Project of Capacity Improvement Plan for Chongqing Municipal Health Commission affiliated unit (2019NLTS001)-ZS03174, operating grant to Chongqing Key Laboratory of Neurodegenerative Diseases (1000013), Chongqing Talent Project (2000062), Overseas Students entrepreneurial fund (2000079), and Plan for High-level Talent Introduction (2000055).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Blagih J., Krawczyk C. M., Jones R. G. (2012). LKB1 and AMPK: Central regulators of lymphocyte metabolism and function. Immunol. Rev. 249 (1), 59–71. doi:10.1111/j.1600-065X.2012.01157.x
Bonanno L., Zulato E., Pavan A., Attili I., Pasello G., Conte P., et al. (2019). LKB1 and tumor metabolism: The interplay of immune and angiogenic microenvironment in lung cancer. Int. J. Mol. Sci. 20 (8), E1874. doi:10.3390/ijms20081874
Boukhalfa A., Roccio F., Dupont N., Codogno P., Morel E. (2021). When the autophagy protein ATG16L1 met the ciliary protein IFT20. Autophagy 17 (7), 1791–1793. doi:10.1080/15548627.2021.1935004
Carling D. (2017). AMPK signalling in health and disease. Curr. Opin. Cell Biol. 45, 31–37. doi:10.1016/j.ceb.2017.01.005
Carroll B., Dunlop E. A. (2017). The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 474 (9), 1453–1466. doi:10.1042/bcj20160780
Chen X., Li X., Zhang W., He J., Xu B., Lei B., et al. (2018). Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism 83, 256–270. doi:10.1016/j.metabol.2018.03.004
Cohen A., Hall M. N. (2009). An amino acid shuffle activates mTORC1. Cell 136 (3), 399–400. doi:10.1016/j.cell.2009.01.021
Cordero M. D., Williams M. R., Ryffel B. (2018). AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol. Metab. 29 (1), 8–17. doi:10.1016/j.tem.2017.10.009
Corona A. K., Jackson W. T. (2018). Finding the middle ground for autophagic fusion requirements. Trends Cell Biol. 28 (11), 869–881. doi:10.1016/j.tcb.2018.07.001
Cuomo F., Altucci L., Cobellis G. (2019). Autophagy function and dysfunction: Potential drugs as anti-cancer therapy. Cancers (Basel) 11 (10), E1465. doi:10.3390/cancers11101465
de Pablos R. M., Espinosa-Oliva A. M., Hornedo-Ortega R., Cano M., Arguelles S. (2019). Hydroxytyrosol protects from aging process via AMPK and autophagy; a review of its effects on cancer, metabolic syndrome, osteoporosis, immune-mediated and neurodegenerative diseases. Pharmacol. Res. 143, 58–72. doi:10.1016/j.phrs.2019.03.005
Dunlop E. A., Tee A. R. (2013). The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem. Soc. Trans. 41 (4), 939–943. doi:10.1042/BST20130030
Egan D., Kim J., Shaw R. J., Guan K. L. (2011). The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7 (6), 643–644. doi:10.4161/auto.7.6.15123
Fang S., Wan X., Zou X., Sun S., Hao X., Liang C., et al. (2021). Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway. Cell Death Dis. 12 (1), 88. doi:10.1038/s41419-020-03357-1
Faubert B., Vincent E. E., Poffenberger M. C., Jones R. G. (2015). The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 356 (2), 165–170. doi:10.1016/j.canlet.2014.01.018
Feng Y., He D., Yao Z., Klionsky D. J. (2013). The machinery of macroautophagy. Cell Res. 24 (1), 24–41. doi:10.1038/cr.2013.168
Fritzen A. M., Frosig C., Jeppesen J., Jensen T. E., Lundsgaard A. M., Serup A. K., et al. (2016). Role of AMPK in regulation of LC3 lipidation as a marker of autophagy in skeletal muscle. Cell. Signal. 28 (6), 663–674. doi:10.1016/j.cellsig.2016.03.005
Galluzzi L., Green D. R. (2019). Autophagy-independent functions of the autophagy machinery. Cell 177 (7), 1682–1699. doi:10.1016/j.cell.2019.05.026
Glick D., Barth S., Macleod K. F. (2010). Autophagy: Cellular and molecular mechanisms. J. Pathol. 221 (1), 3–12. doi:10.1002/path.2697
Gonzalez A., Hall M. N., Lin S. C., Hardie D. G. (2020). AMPK and TOR: The yin and yang of cellular nutrient sensing and growth control. Cell Metab. 31 (3), 472–492. doi:10.1016/j.cmet.2020.01.015
Grasso D., Renna F. J., Vaccaro M. I. (2018). Initial steps in mammalian autophagosome biogenesis. Front. Cell Dev. Biol. 6, 146. doi:10.3389/fcell.2018.00146
Hardie D. G., Ross F. A., Hawley S. A. (2012). Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13 (4), 251–262. doi:10.1038/nrm3311
Hardie D. G., Schaffer B. E., Brunet A. (2016). Ampk: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26 (3), 190–201. doi:10.1016/j.tcb.2015.10.013
Herzig S., Shaw R. J. (2018). Ampk: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19 (2), 121–135. doi:10.1038/nrm.2017.95
Ho T. T., Warr M. R., Adelman E. R., Lansinger O. M., Flach J., Verovskaya E. V., et al. (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543 (7644), 205–210. doi:10.1038/nature21388
Hoffman N., Whitfield J., Janzen N., Belhaj M., Galic S., Murray-Segal L., et al. (2020). Genetic loss of AMPK-glycogen binding destabilises AMPK and disrupts metabolism. Mol. Metab. 41, 101048. doi:10.1016/j.molmet.2020.101048
Holland L. K. K., Nielsen I. O., Maeda K., Jaattela M. (2020). SnapShot: Lysosomal functions. Cell 181 (3), 748–e1. doi:10.1016/j.cell.2020.03.043
Hu J., Ding X., Tian S., Chu Y., Liu Z., Li Y., et al. (2021). TRIM39 deficiency inhibits tumor progression and autophagic flux in colorectal cancer via suppressing the activity of Rab7. Cell Death Dis. 12 (4), 391. doi:10.1038/s41419-021-03670-3
Huang J., Manning B. D. (2008). The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 412 (2), 179–190. doi:10.1042/BJ20080281
Huang J., Wang X., Zhu Y., Li Z., Zhu Y. T., Wu J. C., et al. (2019). Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci. Ther. 25 (6), 796–807. doi:10.1111/cns.13114
Huang Y., Li S., Jia Z., Zhao W., Zhou C., Zhang R., et al. (2020). Transient receptor potential melastatin 8 (TRPM8) channel regulates proliferation and migration of breast cancer cells by activating the AMPK-ULK1 pathway to enhance basal autophagy. Front. Oncol. 10, 573127. doi:10.3389/fonc.2020.573127
Icard P., Coquerel A., Wu Z., Gligorov J., Fuks D., Fournel L., et al. (2021). Understanding the central role of citrate in the metabolism of cancer cells and tumors: An update. Int. J. Mol. Sci. 22 (12), 6587. doi:10.3390/ijms22126587
Itakura E., Kishi-Itakura C., Mizushima N. (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151 (6), 1256–1269. doi:10.1016/j.cell.2012.11.001
Jang M., Park R., Kim H., Namkoong S., Jo D., Huh Y. H., et al. (2018). AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 8 (1), 12637. doi:10.1038/s41598-018-30977-7
Jayaraj R. L., Beiram R., Azimullah S., Mf N. M., Ojha S. K., Adem A., et al. (2020). Valeric acid protects dopaminergic neurons by suppressing oxidative stress, neuroinflammation and modulating autophagy pathways. Int. J. Mol. Sci. 21 (20), E7670. doi:10.3390/ijms21207670
Jeon S. M. (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48 (7), e245. doi:10.1038/emm.2016.81
Jia J., Bissa B., Brecht L., Allers L., Choi S. W., Gu Y., et al. (2020). AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol. Cell 77 (5), 951–969. doi:10.1016/j.molcel.2019.12.028
Johnson S. C., Rabinovitch P. S., Kaeberlein M. (2013). mTOR is a key modulator of ageing and age-related disease. Nature 493 (7432), 338–345. doi:10.1038/nature11861
Kageyama S., Gudmundsson S. R., Sou Y. S., Ichimura Y., Tamura N., Kazuno S., et al. (2021). p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 12 (1), 16. doi:10.1038/s41467-020-20185-1
Kahn B. B., Alquier T., Carling D., Hardie D. G. (2005). AMP-Activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1 (1), 15–25. doi:10.1016/j.cmet.2004.12.003
Karabiyik C., Rubinsztein D. C. (2021). AMPK-activated ULK1 phosphorylates PIKFYVE to drive formation of PtdIns5P-containing autophagosomes during glucose starvation. Autophagy 17 (11), 3877–3878. doi:10.1080/15548627.2021.1961409
Kaur J., Goldsmith J., Tankka A., Bustamante Eguiguren S., Gimenez A. A., Vick L., et al. (2021). Atg32-dependent mitophagy sustains spermidine and nitric oxide required for heat-stress tolerance in Saccharomyces cerevisiae. J. Cell Sci. 134 (11), jcs253781. doi:10.1242/jcs.253781
Kawabata T., Yoshimori T. (2020). Autophagosome biogenesis and human health. Cell Discov. 6 (1), 33. doi:10.1038/s41421-020-0166-y
Ke R., Xu Q., Li C., Luo L., Huang D. (2018). Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 42 (4), 384–392. doi:10.1002/cbin.10915
Kim J., Kim Y. C., Fang C., Russell R. C., Kim J. H., Fan W., et al. (2013). Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152 (1-2), 290–303. doi:10.1016/j.cell.2012.12.016
Kim J., Kim Young C., Fang C., Russell Ryan C., Kim Jeong H., Fan W., et al. (2013). Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152 (1-2), 290–303. doi:10.1016/j.cell.2012.12.016
Kim J., Kundu M., Viollet B., Guan K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13 (2), 132–141. doi:10.1038/ncb2152
Kim M. J., Min Y., Son J., Kim J. Y., Lee J. S., Kim D. H., et al. (2020). AMPKα1 regulates lung and breast cancer progression by regulating TLR4-mediated TRAF6-BECN1 signaling Axis. Cancers (Basel) 12 (11), E3289. doi:10.3390/cancers12113289
Kim Y. M., Jung C. H., Seo M., Kim E. K., Park J. M., Bae S. S., et al. (2015). mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 57 (2), 207–218. doi:10.1016/j.molcel.2014.11.013
Kimmelman A. C., White E. (2017). Autophagy and tumor metabolism. Cell Metab. 25 (5), 1037–1043. doi:10.1016/j.cmet.2017.04.004
Kimura S., Noda T., Yoshimori T. (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3 (5), 452–460. doi:10.4161/auto.4451
Kishton Rigel J., Barnes Carson E., Nichols Amanda G., Cohen S., Gerriets Valerie A., Siska Peter J., et al. (2016). AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 23 (4), 649–662. doi:10.1016/j.cmet.2016.03.008
Lahiri V., Hawkins W. D., Klionsky D. J. (2019). Watch what you (self-) eat: Autophagic mechanisms that modulate metabolism. Cell Metab. 29 (4), 803–826. doi:10.1016/j.cmet.2019.03.003
Lamming D. W., Bar-Peled L. (2019). Lysosome: The metabolic signaling hub. Traffic (Copenhagen, Den. 20 (1), 27–38. doi:10.1111/tra.12617
Levine B., Mizushima N., Virgin H. W. (2011). Autophagy in immunity and inflammation. Nature 469 (7330), 323–335. doi:10.1038/nature09782
Liang J., Zhang H., Zeng Z., Wu L., Zhang Y., Guo Y., et al. (2021). Lifelong aerobic exercise alleviates sarcopenia by activating autophagy and inhibiting protein degradation via the AMPK/PGC-1α signaling pathway. Metabolites 11 (5), 323. doi:10.3390/metabo11050323
Lin S-C., Hardie D. G. (2018). Ampk: Sensing glucose as well as cellular energy status. Cell Metab. 27 (2), 299–313. doi:10.1016/j.cmet.2017.10.009
Lorincz P., Juhasz G. (2020). Autophagosome-lysosome fusion. J. Mol. Biol. 432 (8), 2462–2482. doi:10.1016/j.jmb.2019.10.028
Lyons C. L., Roche H. M. (2018). Nutritional modulation of AMPK-impact upon metabolic-inflammation. Int. J. Mol. Sci. 19 (10), E3092. doi:10.3390/ijms19103092
Mancini S. J., Salt I. P. (2018). Investigating the role of AMPK in inflammation. Methods Mol. Biol. 1732, 307–319. doi:10.1007/978-1-4939-7598-3_20
Martin-Montalvo A., Mercken E. M., Mitchell S. J., Palacios H. H., Mote P. L., Scheibye-Knudsen M., et al. (2013). Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192. doi:10.1038/ncomms3192
Martinez J., Cunha L. D., Park S., Yang M., Lu Q., Orchard R., et al. (2016). Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533 (7601), 115–119. doi:10.1038/nature17950
Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K-J., et al. (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14 (8), 1435–1455. doi:10.1080/15548627.2018.1474314
Mehrpour M., Esclatine A., Beau I., Codogno P. (2010). Overview of macroautophagy regulation in mammalian cells. Cell Res. 20 (7), 748–762. doi:10.1038/cr.2010.82
Mihaylova M. M., Shaw R. J. (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13 (9), 1016–1023. doi:10.1038/ncb2329
Miyamoto S. (2019). Autophagy and cardiac aging. Cell Death Differ. 26 (4), 653–664. doi:10.1038/s41418-019-0286-9
Mizushima N., Komatsu M. (2011). Autophagy: Renovation of cells and tissues. Cell 147 (4), 728–741. doi:10.1016/j.cell.2011.10.026
Moors T., Hoozemans J., Ingrassia A., Beccari T., Parnetti L., Chartier-Harlin M., et al. (2017). Therapeutic potential of autophagy-enhancing agents in Parkinson's disease. Mol. Neurodegener. 12 (1), 11. doi:10.1186/s13024-017-0154-3
Nakamura S., Yoshimori T. (2017). New insights into autophagosome-lysosome fusion. J. Cell Sci. 130 (7), 1209–1216. doi:10.1242/jcs.196352
Newman A. C., Kemp A. J., Drabsch Y., Behrends C., Wilkinson S. (2017). Autophagy acts through TRAF3 and RELB to regulate gene expression via antagonism of SMAD proteins. Nat. Commun. 8 (1), 1537. doi:10.1038/s41467-017-00859-z
Nieto-Torres J. L., Encalada S. E., Hansen M. (2021). LC3B phosphorylation: autophagosome's ticket for a ride toward the cell nucleus. Autophagy 17 (10), 3266–3268. doi:10.1080/15548627.2021.1961073
Nishimura T., Tamura N., Kono N., Shimanaka Y., Arai H., Yamamoto H., et al. (2017). Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 36 (12), 1719–1735. doi:10.15252/embj.201695189
Oligschlaeger Y., Miglianico M., Chanda D., Scholz R., Thali R. F., Tuerk R., et al. (2015). The recruitment of AMP-activated protein kinase to glycogen is regulated by autophosphorylation. J. Biol. Chem. 290 (18), 11715–11728. doi:10.1074/jbc.M114.633271
Park J. M., Jung C. H., Seo M., Otto N. M., Grunwald D., Kim K. H., et al. (2016). The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 12 (3), 547–564. doi:10.1080/15548627.2016.1140293
Parzych K. R., Klionsky D. J. (2014). An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 20 (3), 460–473. doi:10.1089/ars.2013.5371
Pengo N., Agrotis A., Prak K., Jones J., Ketteler R. (2017). A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat. Commun. 8 (1), 294. doi:10.1038/s41467-017-00303-2
Polson H. E., de Lartigue J., Rigden D. J., Reedijk M., Urbe S., Clague M. J., et al. (2010). Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6 (4), 506–522. doi:10.4161/auto.6.4.11863
Rabinowitz J. D., White E. (2010)., 330. New York, NY), 1344–1348. doi:10.1126/science.1193497Autophagy and metabolismScience6009
Reggiori F., Klionsky D. J. (2005). Autophagosomes: Biogenesis from scratch? Curr. Opin. Cell Biol. 17 (4), 415–422. doi:10.1016/j.ceb.2005.06.007
Ren J., Zhang Y. (2018). Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol. Sci. 39 (12), 1064–1076. doi:10.1016/j.tips.2018.10.005
Russell R. C., Tian Y., Yuan H., Park H. W., Chang Y. Y., Kim J., et al. (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15 (7), 741–750. doi:10.1038/ncb2757
Sakamaki J. I., Wilkinson S., Hahn M., Tasdemir N., O'Prey J., Clark W., et al. (2017). Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell 66 (4), 517–532. doi:10.1016/j.molcel.2017.04.027
Savini M., Zhao Q., Wang M. C. (2019). Lysosomes: Signaling hubs for metabolic sensing and longevity. Trends Cell Biol. 29 (11), 876–887. doi:10.1016/j.tcb.2019.08.008
Schmitz K., Cox J., Esser L. M., Voss M., Sander K., Loffler A., et al. (2021). An essential role of the autophagy activating kinase ULK1 in snRNP biogenesis. Nucleic Acids Res. 49 (11), 6437–6455. doi:10.1093/nar/gkab452
Seabright A. P., Fine N. H. F., Barlow J. P., Lord S. O., Musa I., Gray A., et al. (2020). AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 34 (5), 6284–6301. doi:10.1096/fj.201903051R
Shin H. J., Kim H., Oh S., Lee J. G., Kee M., Ko H. J., et al. (2016). AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534 (7608), 553–557. doi:10.1038/nature18014
Song S., Bao S., Zhang C., Zhang J., Lv J., Li X., et al. (2021). Stimulation of AMPK prevents diabetes-induced photoreceptor cell degeneration. Oxid. Med. Cell. Longev. 2021, 5587340–5587415. doi:10.1155/2021/5587340
Stjepanovic G., Baskaran S., Lin M. G., Hurley J. H. (2017). Vps34 kinase domain dynamics regulate the autophagic PI 3-kinase complex. Mol. Cell 67 (3), 528–534. doi:10.1016/j.molcel.2017.07.003
Suh K. S., Chon S., Jung W. W., Choi E. M. (2017). Magnolol protects pancreatic β-cells against methylglyoxal-induced cellular dysfunction. Chem. Biol. Interact. 277, 101–109. doi:10.1016/j.cbi.2017.09.014
Tamargo-Gomez I., MarinoAMPK G. (2018). Ampk: Regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci. 19 (12), E3812. doi:10.3390/ijms19123812
Tian X., Teng J., Chen J. (2020). New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 17 (10), 2680–2688. doi:10.1080/15548627.2020.1823124
Wang B., Shi Y., Chen J., Shao Z., Ni L., Lin Y., et al. (2021). High glucose suppresses autophagy through the AMPK pathway while it induces autophagy via oxidative stress in chondrocytes. Cell Death Dis. 12 (6), 506. doi:10.1038/s41419-021-03791-9
Wang Y., Li L., Hou C., Lai Y., Long J., Liu J., et al. (2016). SNARE-mediated membrane fusion in autophagy. Semin. Cell Dev. Biol. 60, 97–104. doi:10.1016/j.semcdb.2016.07.009
Wu W. K., Ivanova E. A., Orekhov A. N. (2021). Gut microbiome: A possible common therapeutic target for treatment of atherosclerosis and cancer. Semin. Cancer Biol. 70, 85–97. doi:10.1016/j.semcancer.2020.06.017
Xie Z., Klionsky D. J. (2007). Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 9 (10), 1102–1109. doi:10.1038/ncb1007-1102
Xu D. Q., Wang Z., Wang C. Y., Zhang D. Y., Wan H. D., Zhao Z. L., et al. (2016). PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. Embo J. 35 (5), 496–514. doi:10.15252/embj.201592864
Yamamoto Y-h., Noda T. (2020). Autophagosome formation in relation to the endoplasmic reticulum. J. Biomed. Sci. 27 (1), 97. doi:10.1186/s12929-020-00691-6
Yang Y., Atasoy D., Su H. H., Sternson S. M. (2011). Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146 (6), 992–1003. doi:10.1016/j.cell.2011.07.039
Yang Z., Klionsky D. J. (2010). Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22 (2), 124–131. doi:10.1016/j.ceb.2009.11.014
Yi C., Tong J. J., Yu L. (2018). Mitochondria: The hub of energy deprivation-induced autophagy. Autophagy 14 (6), 1084–1085. doi:10.1080/15548627.2017.1382785
Yoo S. M., Jung Y. K. (2018). A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 41 (1), 18–26. doi:10.14348/molcells.2018.2277
Yoon M. S., Son K., Arauz E., Han J. M., Kim S., Chen J. (2016). Leucyl-tRNA synthetase activates Vps34 in amino acid-sensing mTORC1 signaling. Cell Rep. 16 (6), 1510–1517. doi:10.1016/j.celrep.2016.07.008
Zeng Y., Jiang L. (2021). A unique COPII population in plant autophagy. Autophagy 17 (7), 1785–1787. doi:10.1080/15548627.2021.1933298
Zhang H., Chang J. T., Guo B., Hansen M., Jia K., Kovács A. L., et al. (2015). Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 11 (1), 9–27. doi:10.1080/15548627.2014.1003478
Zhao B., Qiang L., Joseph J., Kalyanaraman B., Viollet B., He Y. Y. (2016). Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis. 3 (1), 82–87. doi:10.1016/j.gendis.2015.12.002
Zhao Y., Wang Q., Qiu G., Zhou S., Jing Z., Wang J., et al. (2015). RACK1 promotes autophagy by enhancing the Atg14L-beclin 1-vps34-vps15 complex formation upon phosphorylation by AMPK. Cell Rep. 13 (7), 1407–1417. doi:10.1016/j.celrep.2015.10.011
Zhao Y. G., Codogno P., Zhang H. (2021). Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 22 (11), 733–750. doi:10.1038/s41580-021-00392-4
Zhao Y. G., Zhang H. (2016). ULK1 cycling: The ups and downs of the autophagy response. J. Cell Biol. 215 (6), 757–759. doi:10.1083/jcb.201611177
Zhong Z., Sanchez-Lopez E., Karin M. (2016). Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 166 (2), 288–298. doi:10.1016/j.cell.2016.05.051
Keywords: AMPK, autophagy autophagy, autophagosome autophagosome, lysosome, mTOR
Citation: Wang S, Li H, Yuan M, Fan H and Cai Z (2022) Role of AMPK in autophagy. Front. Physiol. 13:1015500. doi: 10.3389/fphys.2022.1015500
Received: 09 August 2022; Accepted: 10 November 2022;
Published: 25 November 2022.
Edited by:
Chun Yang, Nanjing Medical University, ChinaReviewed by:
Eric Wauson, Des Moines University, United StatesCarlos Guillén, Complutense University, Spain
Lingsha Ju, University of Florida, United States
Copyright © 2022 Wang, Li, Yuan, Fan and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyou Cai, Y2FpemhpeW91QHVjYXMuYWMuY24=