 Elidie Beard
Elidie Beard Sylvain Lengacher
Sylvain Lengacher Sara Dias
Sara Dias Charles Finsterwald
Charles Finsterwald
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 11 January 2022
Sec. Integrative Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.825816
This article is part of the Research TopicThe Role of Astrocyte Signaling Pathways in Ageing-induced Neurodegenerative PathologiesView all 8 articles
Astrocytes play key roles in the regulation of brain energy metabolism, which has a major impact on brain functions, including memory, neuroprotection, resistance to oxidative stress and homeostatic tone. Energy demands of the brain are very large, as they continuously account for 20–25% of the whole body’s energy consumption. Energy supply of the brain is tightly linked to neuronal activity, providing the origin of the signals detected by the widely used functional brain imaging techniques such as functional magnetic resonance imaging and positron emission tomography. In particular, neuroenergetic coupling is regulated by astrocytes through glutamate uptake that triggers astrocytic aerobic glycolysis and leads to glucose uptake and lactate release, a mechanism known as the Astrocyte Neuron Lactate Shuttle. Other neurotransmitters such as noradrenaline and Vasoactive Intestinal Peptide mobilize glycogen, the reserve for glucose exclusively localized in astrocytes, also resulting in lactate release. Lactate is then transferred to neurons where it is used, after conversion to pyruvate, as a rapid energy substrate, and also as a signal that modulates neuronal excitability, homeostasis, and the expression of survival and plasticity genes. Importantly, glycolysis in astrocytes and more generally cerebral glucose metabolism progressively deteriorate in aging and age-associated neurodegenerative diseases such as Alzheimer’s disease. This decreased glycolysis actually represents a common feature of several neurological pathologies. Here, we review the critical role of astrocytes in the regulation of brain energy metabolism, and how dysregulation of astrocyte-mediated metabolic pathways is involved in brain hypometabolism. Further, we summarize recent efforts at preclinical and clinical stages to target brain hypometabolism for the development of new therapeutic interventions in age-related neurodegenerative diseases.
The brain requires high amounts of energy to function. As a result, 20–25% of the energy consumed by the human body is dedicated to cerebral functions, although the brain only represents 2% of the total body mass. Maintenance and restoration of neuronal ion gradients and synaptic transmission, as well as uptake and recycling of neurotransmitters are the major contributors to these energy demands (Riveros et al., 1986; Wong-Riley, 1989; Attwell and Laughlin, 2001; Alle et al., 2009; Hyder et al., 2013; Magistretti and Allaman, 2016; Yu et al., 2018). Glucose is the main energy substrate in the adult brain. However, other sources of energy can be used under particular circumstances, such as ketone bodies that are consumed during development and fasting, and lactate that can be preferentially used during periods of intense physical activity (Owen et al., 1967; Nehlig, 2004; Rasmussen et al., 2011; Chowdhury et al., 2014; Magistretti and Allaman, 2018). Importantly, when plasma lactate concentrations rise, central nervous system (CNS) lactate levels also increase, which is correlated with decreased glucose uptake, and indicating a preferential utilization of lactate over glucose as brain energy source (Smith et al., 2003).
Several neurodegenerative diseases linked to aging are characterized by a decrease in the consumption of energy by the brain in specific regions. These include, among others, Alzheimer’s disease (AD), Parkinson’s disease (PD), Frontotemporal dementia (FTD), Amyotrophic lateral sclerosis (ALS), depression and certain neurodevelopmental disorders. Hypometabolism also occurs in physiological aging, a fact that may participate in the vulnerability of the nervous system to pathological states of aging. Decreased energy availability for neurons results in neurodegeneration, cognitive impairment, as well as abnormalities in neuronal function and excitability (Muddapu et al., 2020). Astrocytes, a type of glial cells in the brain, support essential functions such as maintenance of neurotransmitter pools, trophic support, metabolism, synaptic formation and plasticity, myelin sheath formation, injury healing, and immune surveillance (Burda et al., 2016; Manninen et al., 2020). They are key in regulating neurometabolic and neurovascular couplings, thereby linking neuronal activity to brain energy consumption. In particular, astrocytes respond to neuronal activity by taking up glutamate at the synapse, which triggers aerobic glycolysis and lactate secretion. Then, lactate can be used by neurons as preferred energy source upon activity, as formulated by the astrocyte-neuron lactate shuttle (ANLS) model (Pellerin and Magistretti, 1994; Belanger et al., 2011; Magistretti and Allaman, 2018). Astrocytes also modulate activity-dependent vasodilation through nitric oxide-mediated pathways (Bonvento et al., 2002).
In this review, we discuss the different roles played by astrocytes in the control of brain energy metabolism and homeostasis, and how these pathways are affected in aging and hypometabolic neurodegenerative diseases such as AD. Further, we review the current therapeutic strategies from in vitro, in vivo, and clinical evidence that aim at restoring brain energy deficits in neuropathologies with metabolic dysfunctions.
Under normal homeostatic conditions, the supply and demand of energy are tightly coupled. For instance, cerebral blood flow (CBF) and glucose utilization increase in response to neuronal activity through processes known as neurovascular and neurometabolic couplings (Belanger et al., 2011). These processes constitute the bases of functional brain imaging techniques, among which positron emission tomography (PET) that allows determination of CBF, cerebral metabolic rate of glucose consumption (CMRglc), cerebral metabolic rate of oxygen consumption CMRO2, as well as functional magnetic resonance imaging (fMRI) that measures brain oxygenation and blood volume (Magistretti and Pellerin, 1996; Raichle and Mintun, 2006; Figley and Stroman, 2011; Roumes et al., 2021).
Astrocytes have unique cytoarchitectural features that ideally position them to sense their surrounding environment and dynamically respond to extracellular changes (Belanger et al., 2011). They possess numerous processes that form highly organized anatomical domains interconnected through functional networks via gap junctions. Some of these processes closely ensheath synapses, whereas others are in contact with brain capillaries (Iadecola and Nedergaard, 2007; Oberheim et al., 2009; Mathiisen et al., 2010). At the synapse level, astrocytes’ perisynaptic processes express glutamate transporters that can sense changes in neuronal activity, while at the vasculature level, luminal surface of their endfeet that is in contact with vascular endothelium express glucose transporter 1 (GLUT1) (Patching, 2017), that will allow facilitated diffusion of glucose into astrocytes, to supply energy upon neuronal activity. Finally, astrocytes can release vasoactive substances to act on brain glucose supply depending on neuronal activity state (Belanger et al., 2011; MacVicar and Newman, 2015).
Neurons and astrocytes possess distinct metabolic profiles. In the presence of oxygen, neurons process glucose in an oxidative way to yield ATP through mitochondrial activity, while glucose entering astrocytes preferentially undergoes glycolysis to produce pyruvate and lactate (Magistretti and Allaman, 2015; Supplie et al., 2017). Astrocytes specifically express glycolytic enzymes, which make them utilize 80% of the glucose through glycolysis. In neurons, glycolytic enzymes, such as 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) and pyruvate dehydrogenase kinase 4 (PDK4) are inhibited, which make them highly phosphorylative cells (Magistretti and Allaman, 2018). Furthermore, astrocytes preferentially express lactate dehydrogenase 5 (LDH5), which favors the conversion of pyruvate into lactate, while neurons exclusively express lactate dehydrogenase 1 (LDH1) that favors conversion of lactate into pyruvate (Bittar et al., 1996). Astrocytes also have higher NADH to NAD+ ratio than neurons, which favors the reduction of pyruvate into lactate (Mongeon et al., 2016). Interestingly, inhibiting mitochondrial activity specifically in astrocytes did not have any phenotypic effect in mice (Supplie et al., 2017). In contrast, enhancing glycolysis in neurons led to dramatic decrease in glucose utilization in the pentose phosphate pathway, increased oxidative stress and apoptosis (Herrero-Mendez et al., 2009). These studies highlight the cellular specificity of distinct metabolic pathways in the brain with astrocytes being predominantly glycolytic, while neurons are oxidative.
As formulated by the ANLS, glutamate is taken up by astrocytes and recycled through the glutamate-glutamine cycle (Bak et al., 2006; McKenna, 2007). This process, which is mediated by astrocytic Na+-dependent glutamate transporters, leads to increases in cytosolic Na+ that activates Na+/K+ ATPase, thereby increasing ATP consumption (Magistretti and Chatton, 2005) and stimulation of GLUT1 activity (Porras et al., 2008). In turn, glycolysis is activated and results in enhanced glucose uptake in astrocytes and release of lactate toward neurons (Pellerin and Magistretti, 2012). Under resting conditions, astrocytes release 85% of the glucose they consume in the form of lactate (Bolanos et al., 1994). An in vivo study using two photon microscopy and lactate fluorescence resonance energy transfer (FRET) nanosensors confirmed lactate gradient between astrocytes and neurons (Machler et al., 2016). Another level of astrocyte-neuron metabolic coupling is through the activity-dependent production of NH4+ in neurons that, upon transfer to astrocytes, favors astrocytic glycolysis. Thus, in neurons, conversion of glutamine into glutamate by phosphate activated glutaminase (PAG) leads to the production of NH4+, which can be transferred to astrocytes through transporters and K+ channels (Kelly and Rose, 2010). In astrocytes, NH4+ can enter the mitochondria and acidifies mitochondrial matrix, which in turn inhibits mitochondrial incorporation of pyruvate that depends on the H+-coupled mitochondrial pyruvate carrier (MPC) (Herzig et al., 2012; Lerchundi et al., 2015).
Lactate is a metabolic end-product that cannot directly be used and requires its conversion into pyruvate to serve as energy and carbon source to the tricarboxylic acid (TCA) cycle (Barros et al., 2020). One of the advantages of producing lactate that is not readily consumed is to allow its distribution and exchanges between lactate producing and lactate consuming cells (Brooks, 2018). Importantly, lactate also serves as a signaling molecule that modulates mechanisms underlying synaptic plasticity and memory consolidation through the regulation of plasticity genes expression (Suzuki et al., 2011; Yang et al., 2014; Margineanu et al., 2018). Neuroprotective effects of lactate have been demonstrated in various types of brain damages, including ischemic (Schurr et al., 2001; Smith et al., 2003), excitotoxic and mechanical insults (Ros et al., 2001; Cureton et al., 2010). The transfer of lactate between cells is specific and controlled by monocarboxylate transporters (MCTs). There are different types of MCTs that are differentially expressed between producing and receiving cells and have different affinities for lactate. For instance, neurons exclusively express high-affinity MCT2, while astrocytes express lower-affinity MCT1 and MCT4 (Roosterman and Cottrell, 2020). Some studies shown that MCT2 expression in neurons is co-localized with glutamate receptors at the postsynaptic membranes of fast acting excitatory synapses, further supporting the intracellular signaling roles of lactate (Bergersen et al., 2001, 2005). Since lactate is co-transported through MCTs with H+, regulation of pH is essential for the transport of lactate (Bosshart et al., 2019). Lactate symbiosis between astrocytes and neurons is also well demonstrated through the role of energy sensor AMP-activated protein kinase (AMPK). Thus, intracerebral levels of lactate were found to be decreased in AMPK-deficient mice, which was concomitant with decreased glycolysis, oxidative phosphorylation and neuronal survival (Muraleedharan et al., 2020). Mechanistically, phosphorylation of AMPK in astrocytes was found to destabilize thioredoxin-interaction protein (TXNIP), which led to the translocation of GLUT1 at the plasma membrane, glucose uptake and lactate production that in turn provided neuroprotection in a non-cell-autonomous manner (Muraleedharan et al., 2020). The lactate signaling may also occur through the activation of the lactate responsive-G-protein-coupled receptor 81 (GPR81) (Lauritzen et al., 2014; Morland et al., 2015). Activation of GPR81 triggers Gi-mediated pathway that in turn inhibits Adenylate cyclase (AC), resulting in a decrease in cyclic AMP (cAMP) levels and changes in numerous intracellular mechanisms (Ahmed et al., 2009, 2010).
In a key study by Suzuki et al. (2011), transfer of lactate from astrocytes to neurons was found to be critical to mediate synaptic plasticity and memory consolidation. Pharmacological inhibition or genetic targeting of MCT2 irreversibly impairs long-term memory in mice (Newman et al., 2011; Suzuki et al., 2011). Long-term memory impairment could be reversed in MCT4-deficient mice by intrahippocampal administration of lactate, but not glucose (Suzuki et al., 2011). These results indicate that the neuronal uptake of lactate is important for the establishment of long-term memories. Further, degradation of glycogen, which, in the brain, is exclusively localized in astrocytes, is required for memory formation (Newman et al., 2011; Suzuki et al., 2011). Interestingly, exercise-mediated lactate increase was shown to enhance lactate levels in the hippocampus and to be beneficial for memory in mice (El Hayek et al., 2019). Activation of astrocytic, but not neuronal, β2-adrenergic receptors led to lactate production that mediated memory formation (Gao et al., 2016; Dong et al., 2017). Furthermore, lactate mediates neuroprotective effects following traumatic brain injury (TBI) (Alvarez et al., 2014; Zhou et al., 2018), hypoxia (Schurr et al., 1997), cerebral ischemia, and glutamate-mediated excitotoxicity (Bliss et al., 2004; Berthet et al., 2009; Jourdain et al., 2016) and was found to promote adult hippocampal neurogenesis (Lev-Vachnish et al., 2019). Interestingly, lactate has a dual impact on NMDA receptors. With low glutamate, lactate stimulates NMDA receptor signaling, resulting in plasticity gene induction and memory consolidation. However, in excitotoxic conditions with high glutamate, lactate decreases NMDA receptor-mediated signaling, thereby preventing glutamate-induced neuronal death (Jourdain et al., 2018). Recent evidence in vivo indicates that lactate is preferred to glucose as an energy substrate in active neurons, and that lactate metabolism shapes neuronal activity through KATP channels (Karagiannis et al., 2021). An important study finally showed that the effect of circulating glucose on neuronal depolarization was exclusively mediated by astrocyte-mediated lactate release, providing strong evidence for the role of ANLS in vivo (Sada et al., 2015).
Aging leads to many physiological changes in body functioning, including cerebral and cognitive functions such as decreased working, spatial and episodic memory (Mattay et al., 2006; Glisky, 2007). With age, aerobic glycolysis and consumption of glucose were found to be severely decreased in the brain, particularly in the temporal, parietal and frontal lobes, and motor cortex (Goyal et al., 2017). This is accompanied in normal aging by the degeneration of brain structures, leading to loss in brain weight and volume (Dekaban, 1978; Fox and Schott, 2004), cortical thickness in the prefrontal cortex, medial temporal lobe and hippocampus (Sowell et al., 2003; Salat et al., 2004), gray matter atrophy, disruptions of white matter integrity (Bi et al., 2021), and synaptic density (Masliah et al., 1993). These non-pathological changes contribute to age-related cognitive decline in elderly subjects (Resnick et al., 2003; Yang et al., 2015; Bender et al., 2016).
Advances in neuroimaging techniques, such as MRI and PET allow to investigate the dynamic brain changes with aging in vivo. For instance, brain network connectome, which is assessed through diffusion MRI tractography efficiency, was found to decline with age in specific brain regions such as the hippocampus, thalamus, and frontal and parietal cortices (Bi et al., 2021). Glucose hypometabolism was observed with aging in the anterior cingulate cortex, several parts of the orbital and frontal gyrus and in the thalamus (Bi et al., 2021). By combining these two measurements, the study revealed a close coupling between age-dependent decreased brain network connectome and hypometabolism in specific brain regions that include frontal and temporal lobes, cingulate gyrus, hippocampus and hypothalamus (Gong et al., 2009; Bi et al., 2021). Importantly, several studies showed that glucose hypometabolism is due to a decrease of brain aerobic glycolysis as measured by the difference between glucose and oxygen consumptions (Goyal et al., 2017; Hipkiss, 2019; Tang, 2020; Yan et al., 2020). Both animal models and human studies showed that aging is characterized by a decreased aerobic glycolysis in astrocytes (Goyal et al., 2017) and mitochondrial oxidative phosphorylation in neurons (Boumezbeur et al., 2010; Jiang and Cadenas, 2014). It has been proposed that pathological neurons first exhibit mitochondrial dysfunction and compensatory increase in oxidative phosphorylation that results in a competition for a limited energetic resource, i.e., astrocyte-derived lactate, as the fuel of oxidative phosphorylation (Demetrius and Driver, 2015). This competition for energetic resource leads to deleterious consequences on initially healthy neurons in the vicinity of neurons with mitochondrial dysfunction, thereby spreading neurodegeneration and development of the pathological state, from normal aging to neurodegeneration (Demetrius et al., 2014). At the cellular level, impaired glucose uptake is correlated with a decrease in the expression and membrane translocation of the insulin-sensitive neuronal glucose transporters, GLUT3 and GLUT4, which influence neuronal survival in the rat brain (Jiang et al., 2013). Decrease of microvascular endothelium GLUT1 was also observed in the hypometabolic rat brain (Jiang et al., 2013). The disruption in glucose metabolism due to the loss of glucose transporters is closely associated with synaptic dysfunction and renders neurons vulnerable to degeneration.
Alzheimer’s disease, the most prevalent cause of age-associated dementia, is a progressive neurodegenerative disease with biochemical, metabolic and physiological changes that impact memory, thinking and behavior. In addition to the historical description of the pathology that include β amyloid plaques and hyperphosphorylated tau in the brain, it is characterized by clear mitochondrial and metabolic impairments (Butterfield and Halliwell, 2019). Hence, AD can be considered as a metabolic disease with impairment in mitochondrial bioenergetics, as well as glucose brain import and metabolism (Zulfiqar et al., 2019). Brain glucose hypometabolism appears early in the genesis of the pathology and is frequently present before the onset of clinically measurable symptoms (Costantini et al., 2008; Cunnane et al., 2011). For instance, numerous studies have highlighted reduced regional activity-dependent glucose uptake and utilization in AD using 18F-fluorodeoxyglucose (FDG) PET (Ferreira et al., 2010; Demetrius and Driver, 2013; Tomi et al., 2013; Demetrius et al., 2014; Fu and Jhamandas, 2014; Yin et al., 2016; Weise et al., 2018). These decreases are mostly observed in the parieto-temporal and posterior cingulate cortices and extended to the frontal areas while disease advances, whereas primary motor and visual cortices are less severely affected, and cerebellum, thalamus and basal ganglia are relatively spared (Friedland et al., 1985; Koss et al., 1985; Minoshima et al., 1997). In AD, degeneration occurs in the locus coeruleus (LC) depending on the disease progression (Chan-Palay and Asan, 1989; Rub et al., 2001; Wilson et al., 2013; Arendt et al., 2015; Peterson and Li, 2018). Noradrenaline (NA), which is released from the LC, activates cellular response in astrocytes that trigger increase in Ca2+ and cAMP, resulting in numerous cellular responses including enhanced aerobic glycolysis (Arendt et al., 2015; Vardjan et al., 2018). Therefore, early destruction of the LC may contribute, at least in part, to the impaired glucose metabolism in AD (Moore and Bloom, 1979). Another study reported a reduction of several glycolysis intermediates in the cerebrospinal fluid (CSF) of AD patients compared with controls (Bergau et al., 2019). In postmortem AD brains, dysregulation of nutrient transporters was observed, with a decrease of neuronal GLUT3 and astrocytic GLUT1 (Simpson et al., 1994; Harr et al., 1995; Mooradian et al., 1997). A similar reduction in GLUT1 and lactate transporters has been reported in culture of astrocytes from AD mouse model (Merlini et al., 2011). Postmortem studies of AD brains also revealed alterations in glycolytic enzymes activity, glucose utilization and amino acid metabolism (Marcus and Freedman, 1997; Palmer, 1999; Butterfield and Halliwell, 2019). Several genes involved in energy regulation were downregulated in AD patients and mouse model of AD (Liang et al., 2008). AD symptoms essentially never occur without glucose hypometabolism, and the extent of these metabolic changes are strongly correlated with the severity of clinical symptoms (Woo et al., 2010; Thomas et al., 2015). Of relevance, mitochondrial dysfunction, which is associated with age-related neurodegeneration, is also particularly important in AD (Beal, 2005; Yao and Brinton, 2011). In line with the decreased glycolysis in AD brains, an interesting recent study showed that levels of lactate were reduced in the CSF of patients with AD, although no correlation were found between CSF lactate and amyloid levels (Bonomi et al., 2021). Recently, physical exercise was found to have beneficial effects in AD through the improvement of brain glucose metabolism. Thus, aerobic exercise leads to the maintenance of brain glucose uptake in mild AD patients (Robinson et al., 2018), and to the protection against hypometabolism in brain regions particularly vulnerable in AD (Dougherty et al., 2017).
In humans, Apolipoprotein E (APOE) exists in three different isoforms: APOE2, APOE3 and APOE4. Homozygous and heterozygous carriers of APOE4 have respectively 12 fold and 2–3 fold times increased risk of developing late-stage AD than APOE2 or 3 carriers (Belloy et al., 2019). Depending on the ethnicity, 10–25% of the population is carrier of APOE4, which makes it the most prevalent genetic risk factor for AD. APOE4 has been clearly linked to brain hypometabolism, which was shown to precede neurodegeneration by years in APOE4(+) patients (Farrer et al., 1997; Raber et al., 2004; Reiman et al., 2004). APOE is a major cholesterol carrier involved in lipid metabolism. In the brain, APOE is primarily produced by astrocytes and regulates lipids delivery to neurons that are necessary for their structural maintenance, as well as injury repair (Xu et al., 1996, 2006; Mahley and Rall, 2000; Bu, 2009). In conditions of stress or injury, APOE can also be expressed by neurons (Mahley et al., 2006). Several studies have shown strong association between APOE4, metabolic genes expression and cerebral glucose uptake in human brains (Jagust et al., 2012; Carbonell et al., 2016; Wu et al., 2018). Mouse models carrying the APOE4 human allele also have reduced metabolic gene expression and cerebral glucose uptake compared to APOE3 expressing models (Alata et al., 2015; Lin et al., 2017; Williams et al., 2020). At the cellular level, APOE4-expressing astrocytes exhibit altered glycolysis, glucose uptake and lactate secretion (Wu et al., 2018; Williams et al., 2020). Interestingly, lactate transferred from astrocytes to neurons is used for the synthesis of lipid droplets in neurons, which in turn are transported back to astrocytes through carriers that include fatty acid transport proteins (FATPs) and apolipoproteins neurodegeneration (Liu et al., 2017). Expression of APOE4 impairs this transport of lipid droplets between neurons and astrocytes, which in turn promotes neurodegeneration (Liu et al., 2017).
Brain insulin resistance is also believed to contribute to metabolic dysfunctions in AD (Rivera et al., 2005). Thus, a growing body of epidemiological and molecular evidence indicates an overlap in risk, comorbidity, and pathophysiological mechanisms across Type 2 diabetes (T2D), mild cognitive impairment (MCI), AD and other types of dementia such as vascular dementia, Lewy body dementia (LBD) and FTD (Arnold et al., 2018). Studies also indicate that T2D patients are at increased risk of developing MCI or AD (Arnold et al., 2018). While insulin resistance is a central feature of T2D, research from the past few years has also shown that it is present in the brains of patients with dementia, even in the absence of T2D (De Felice et al., 2009; Zhao and Townsend, 2009; El Khoury et al., 2014). Moreover, cerebral levels of insulin and insulin receptor (IR) are lower in the brain of AD patients, and evidence for insulin signaling impairment in post-mortem brain tissue of AD patients and in animal models of AD has been shown (Steen et al., 2005; Chiu et al., 2008; Talbot et al., 2012). Insulin and insulin-like growth factors (IGFs) regulate key neuronal functions such as survival, energy metabolism and synaptic plasticity (Hoyer, 2002). Interestingly, insulin-mediated signaling pathways are impacted by APOE4 through the reduction of the expression of insulin receptor substrate 1 (IRS1) and Akt pathway in both mouse models and human brain tissue (Ong et al., 2014; Keeney et al., 2015), and the sequestration of IR in endosomes in an age-dependent manner (Zhao et al., 2017).
Human and animal studies have shown that dysregulation of insulin function contributes to aging and to the development of neurodegenerative diseases (Craft and Watson, 2004). In this context, impaired glucose utilization, mitochondrial dysfunction, reduced ATP production, and energy shortage in AD led to the hypothesis that these abnormalities could be mediated, at least in part, by desensitization of IR in the brain (Hoyer, 2002; Craft and Watson, 2004; de la Monte, 2009). Several preclinical studies have highlighted the impact of insulin dysregulation in models of cognition. In mice, intracerebroventricular injection of streptozotocin was found to reduce brain glucose metabolism, mitochondrial function, IR activity and spatial learning and memory (Duelli et al., 1994; Hoyer et al., 2000; de la Monte and Wands, 2006). Experimental induction of brain insulin resistance and insulin deficiency in mice causes AD-like neurodegeneration and cognitive impairment (Lester-Coll et al., 2006). In the brain, both neurons and astrocytes are impacted by insulin signaling. In neurons, insulin signaling modulates the expression of GABA, NMDA and AMPA receptors, catecholamine release, and glucose uptake via GLUT3. In astrocytes, insulin enhances glycogen storage, stimulates glucose uptake via GLUT1 and modulates inflammatory response (Heni et al., 2011; Arnold et al., 2018). Interestingly, activation of insulin-mediated pathways was downregulated in astrocytes in response to elevated chronic insulin levels, but not in neurons (Clarke et al., 1984). These cellular differences could have implications in the effects of T2D and insulin resistance on the function of different brain cell types.
In AD brains, reactive astrocytes are preferentially located in the vicinity of amyloid plaques, where they exhibit abnormal morphology (Rodriguez et al., 2009; Acosta et al., 2017; Liddelow et al., 2017). In the early stage of the disease, activated astrocytes have neuroprotective action by internalizing and degrading amyloid plaques, while upon progression of the disease, deposit of amyloid plaques leads to astrocytic death that in turn contribute to further development of the pathology (Nagele et al., 2004). Regarding the consequences of hypometabolic state in the brain, a study showed that amyloid plaques impair glucose uptake by interfering with exocytosis-dependent GLUT3 membrane expression (Uemura and Greenlee, 2001; Prapong et al., 2002). Several reports have described some adaptations of the astrocytic metabolism to amyloid plaques in vitro, with alterations of glycolysis and mitochondrial activity (Allaman et al., 2010; Oksanen et al., 2017; van Gijsel-Bonnello et al., 2017; Carter et al., 2019) and the activation of several intracellular cascades leading to inflammation, oxidative stress and calcium dysregulation (De Strooper and Karran, 2016).
Considering the hypometabolic state and the emerging consideration of insulin signaling in AD, a number of therapeutic strategies targeting insulin-mediated pathways have been considered in order to restore brain energy metabolism (Kellar and Craft, 2020). These approaches include the use of insulin sensitizer agents or intranasal insulin to restore insulin signaling in AD, as well as antidiabetic drugs such as Metformin and Glucagon-like peptide-1 receptor (GLP-1R) agonists.
First, intranasal insulin has been developed with the objective to efficiently deliver insulin directly into the brain without changing peripheral levels that could cause insulin resistance (Born et al., 2002). Insulin has been known for many years to positively modulate brain glucose utilization (Havrankova et al., 1978; Bingham et al., 2002; Taouis and Torres-Aleman, 2019). In animal models of AD, intranasal insulin was found to reduce cerebral oxidative stress, tau phosphorylation and amyloid load, and improves cognitive functions (Barone et al., 2019) (see Table 1). In humans, intranasal insulin has shown promising clinical data in MCI and AD (Kellar and Craft, 2020) (see Table 2). For instance, a pilot trial reported improvement of cognition in healthy volunteers after intranasal insulin administration (Benedict et al., 2004, 2007). A subsequent study confirmed positive effect of intranasal insulin in patients with MCI or mild AD (Reger et al., 2008a,b). Further study on over 100 patients with MCI or mild to moderate AD reported some preservation of cognition and function, and higher cerebral glucose utilization assessed by FDG PET, although no changes were observed in AD biomarkers (Craft et al., 2012; Claxton et al., 2013). These results led to the establishment of a larger Phase 2 and 3 studies that have enrolled nearly 300 people with MCI or early-stage AD. In this trial, treatment with intranasal insulin showed positive impact on primary outcome ADAS-Cog12 memory assessment at 12 and 18 months in a patient’s cohort that has used one of the two devices used for insulin delivery, while another patient’s cohort that has used another delivery device failed to benefit from the treatment (Craft et al., 2020). Other series of clinical studies have evaluated long-acting insulin analog Detemir and showed that treatment of 50 MCI or AD patients led to memory improvement in APOE4 carriers, but worsened memory in non-carriers (Claxton et al., 2013). Another study showed that insulin, but not long-acting analog Detemir, increased memory and preserved volume in several brain regions (Craft et al., 2017). A small trial examining the effect of the rapid acting insulin analog Glulisine in patients with MCI or middle-stage AD failed to show any acute impact in cognition (Rosenbloom et al., 2021). Nasal insulin has also been tested in other neurodegenerative diseases, showing for example improved clinical outcome in PD severity (Novak et al., 2019).
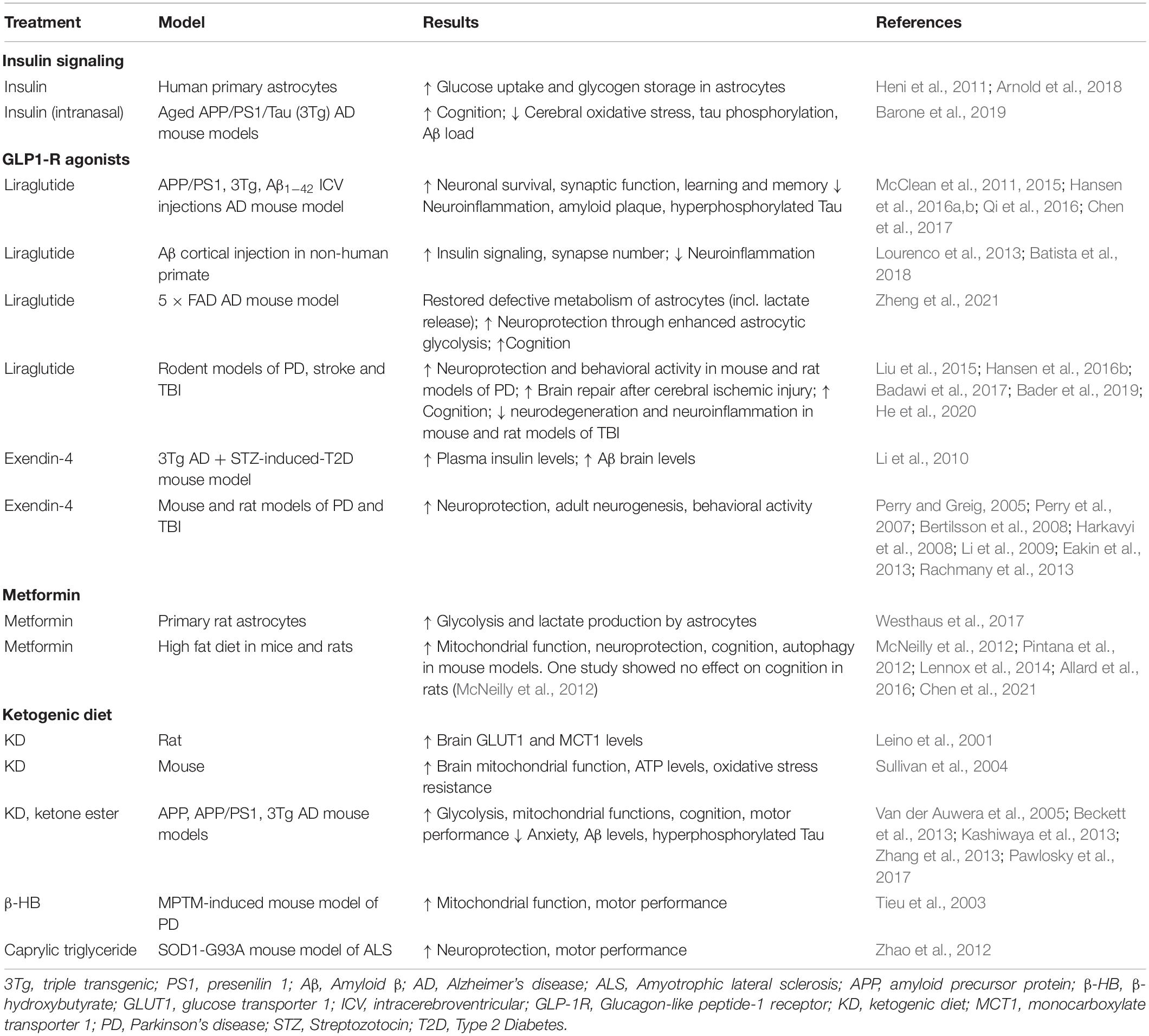
Table 1. Preclinical evidence of compounds targeting brain energy metabolism.
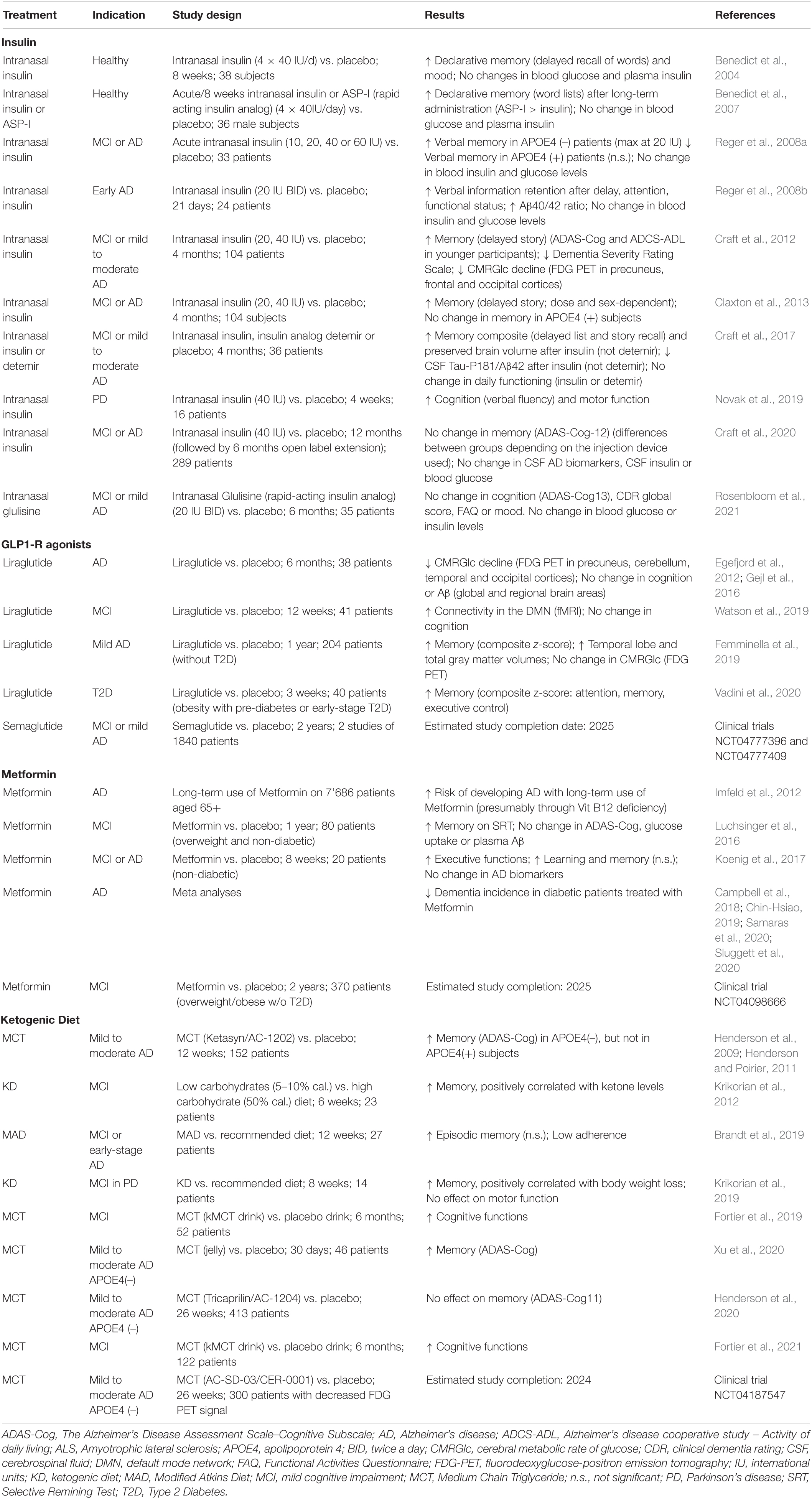
Table 2. Clinical evidence of compounds targeting brain energy metabolism in neurodegenerative diseases.
Another therapeutic strategy aiming at restoring brain metabolism by targeting insulin-related pathway is the use of Glucagon-Like Peptide-1 Receptor (GLP-1R) agonists. GLP-1 is an incretin hormone derived from proglucagon and secreted by the small intestine in response to food intake. GLP-1R is expressed in pancreatic β-cells, kidney, heart, and CNS (Yildirim Simsir et al., 2018). Activation of GLP-1R leads to insulin release by β-cells, which in turn stimulates glucose uptake. While GLP-1 is well known for its action in the regulation of peripheral metabolism, it was also shown to play key roles in CNS functions. For instance, GLP-1 is secreted by neurons in the nucleus tractus solitarius (NTS), which results in anorexic effect and transmit vagal motor information to the pancreas (Yildirim Simsir et al., 2018). Other studies showed that overexpression of GLP-1R in the rat hippocampus improves learning, memory and neuroprotection (During et al., 2003), while transgenic mice lacking GLP-1R have deficits in learning, synaptic plasticity and cognition (Abbas et al., 2009). Interestingly, effects of GLP-1 on energy balance were found to be mediated by astrocytes (Reiner et al., 2016). Thus, large number of astrocytes in the NTS respond to GLP-1R agonists by intracellular calcium and cAMP signaling, while blocking NTS astrocytes activity attenuated GLP-1R agonist effects on food intake in rats (Reiner et al., 2016). These data suggest that astrocytes play a role in the effects of GLP-1 in the brain (Cui et al., 2021).
Preclinical and clinical evidence indicate therapeutic potential for some of the GLP-1R agonists that are commonly used for the treatment of diabetes and obesity (Yildirim Simsir et al., 2018) (see Tables 1, 2, respectively). They include Liraglutide (Novo Nordisk), Semaglutide (Novo Nordisk) and Exendin-4, also known as Exenatide (AstraZeneca). First, in rodent models of AD, the GLP-1 analog Liraglutide was shown to promote neuronal survival, increase synaptic function, reduce neuroinflammation, amyloid plaque and hyperphosphorylated Tau, and support learning and memory (McClean et al., 2011, 2015; Hansen et al., 2016a; Qi et al., 2016; Chen et al., 2017; Holscher, 2018). In non-human primates, Liraglutide improved insulin signaling, reduced inflammation and restored synapse number that were caused by the cortical injections of β amyloid (Lourenco et al., 2013; Batista et al., 2018). Interestingly, a recent study using mouse model of AD showed that Liraglutide has a specific impact on astrocytes (Zheng et al., 2021). Treatment of AD mouse-derived astrocytes with Liraglutide resulted in a neuroprotective action and restored defective metabolic pathways, including lactate secretion (Zheng et al., 2021). Liraglutide also showed positive effects in preclinical models of other neurological diseases, including PD (Liu et al., 2015; Hansen et al., 2016b; Badawi et al., 2017), stroke (He et al., 2020) and TBI (Bader et al., 2019). The other GLP-1R agonist Exendin-4 also exhibited neuroprotective effects in a mouse model of AD (Li et al., 2010) and PD (Perry and Greig, 2005; Perry et al., 2007; Bertilsson et al., 2008; Harkavyi et al., 2008; Li et al., 2009; Eakin et al., 2013; Rachmany et al., 2013).
In humans, a pilot study has first tested the effects of 6-month Liraglutide treatment in 38 patients with AD (Egefjord et al., 2012). Liraglutide led to a clear increase in brain glucose utilization, as revealed by FDG PET (Femminella and Edison, 2014; Gejl et al., 2016). Another pilot trial was done with Liraglutide on 41 middle- to late-aged individuals with elevated blood glucose or diabetes and cognitive complaints. Primary outcome, which included fMRI before and after treatment, revealed improved connectivity within the default mode network (DMN), a system that is defective in AD (Watson et al., 2019). A study in 40 obese subjects with pre-diabetes or newly diagnosed T2D treated for 3 weeks with Liraglutide indicated an increase in short term memory (Vadini et al., 2020). Most recent Phase IIb study (ELAD study) on 204 patients with mild AD and no diabetes failed to see benefit of Liraglutide on primary outcome of FDG PET, but revealed improved in cognition, as well as temporal lobe and total brain gray matter volumes (Femminella et al., 2019). Semaglutide, another GLP-1R agonist, is set to be tested in two large Phase 3 studies (EVOKE and EVOKE Plus; NCT04777396 and NCT04777409). These trials each plan to enroll 1’840 patients with MCI or middle-stage dementia for a duration of 2 years. The clinical protocol is based on the post hoc data analyses from three clinical studies on T2D showing a 53% reduced risk of developing dementia in people who received Semaglutide or Liraglutide. Liraglutide, Semaglutide and Exendin-4 are also all currently being tested in clinical studies for PD.
Repositioning of Metformin, one of the most used medication for the treatment of T2D, has been another strategy to improve brain energy deficits in neurodegenerative diseases. Metformin acts through the activation of AMPK, an important regulator of glucose homeostasis. Activation of AMPK decreases gluconeogenesis, lowers blood glucose and restores insulin sensitivity. Metformin has been shown to reduce inflammation and oxidative stress, and to promote neurogenesis (Wang et al., 2012; Rotermund et al., 2018; Bharath et al., 2020). Interestingly, pleiotropic effects of Metformin have been hypothesized to be mediated, at least in part, by the increased circulating concentrations of lactate produced by AMPK-mediated glucose uptake, and its use as a direct energy source in various organs (Giaccari et al., 2021). In this context, Metformin was found to directly enhance glycolysis and production of lactate by astrocytes in the brain (Westhaus et al., 2017). Metformin was found to have positive impact in cognition in high fat diet mouse models (Pintana et al., 2012; Lennox et al., 2014; Allard et al., 2016), but others found no effect of Metformin in a rat model of high fat diet-induced cognitive deficit (McNeilly et al., 2012). In a mouse model of AD, Metformin attenuated Tau aggregates and amyloid load by ameliorating microglial autophagy (Chen et al., 2021) (Table 1). Interestingly, Metformin was found to repress the expression of Thioredoxin-interacting protein (Txnip), an inhibitor of Thioredoxin, through AMPK-mediated pathway (Chai et al., 2012). Repression of Txnip, which has been shown to improve glucose utilization and uptake (Parikh et al., 2007), is postulated to participate, at least in part, in the effects of Metformin on glucose metabolism.
In humans, several clinical studies have been assessing the impact of Metformin in MCI and AD (Table 2). First, a pilot study in 80 overweight non-diabetic people with MCI indicated that administration of Metformin for a duration of 1 year led to better performance on the Selective Reminding Test (SRT) of memory, but did not change other outcomes including ADAs-Cog, glucose uptake and plasma amyloid levels (Luchsinger et al., 2016). A smaller trial in 20 non-diabetic people with MCI or AD showed that Metformin improved executive functions, led to a trend toward better learning and memory, but did not change AD biomarkers (Koenig et al., 2017). However, contrasting study showed that long-term use of Metformin could also increase the risk of developing AD, presumably through its effect on Vitamin B12 deficiency, which is a cause of dementia (Imfeld et al., 2012). A multicentric Phase 2/3 trial that investigates the effect of Metformin on memory of 370 overweight or obese people with MCI is currently ongoing (NCT04098666). Interestingly, clinical meta-analyses showed that cognitive impairment, as well as dementia incidence, were significantly reduced in diabetic patients that were treated with Metformin (Campbell et al., 2018; Chin-Hsiao, 2019; Samaras et al., 2020; Sluggett et al., 2020).
Ketogenesis is a physiological mechanism whereby ketone bodies (acetoacetate, β-hydroxybutyrate and acetone) are produced by the liver in response to fasting, exercise or reduced carbohydrate availability (Puchalska and Crawford, 2017). Ketone bodies that are released in the circulation can be used by extra-hepatic tissues as alternative energy sources. In the mitochondria of energy-consuming cells, ketone bodies are converted in acetyl-CoA and incorporated in the TCA cycle. The brain can adapt to the utilization of ketone bodies for up to 70% of its energy requirements (Veech, 2004). In humans, the classical ketogenic diet (KD) consists in a 4 to 1 ratio of fats to proteins and carbohydrates, which physiologically mimics the fasting state and leads to hepatic ketogenesis. Although safe and efficiently enhancing levels of circulating ketone bodies, KD usually lacks long-term adherence. Alternative diets consist in the less strict Modified Atkins Diet (MAD) or supplementation to the normal diet with exogenous ketogenic agents such as medium-chain triglycerides (MCTs).
Ketogenic diet is commonly used for the treatment of refractory epilepsy (D’Andrea Meira et al., 2019), and has been proposed as a therapeutic strategy to restore energy deficit in neuropathologies with metabolic dysfunction. Interestingly, transport of ketone bodies is directly affected by ketogenic diet through the upregulation of MCT1 and GLUT1 in the brain (Leino et al., 2001). Preclinical evidence showed promising results for the neuroprotective properties of KD (Paoli et al., 2014) (Table 1). For instance, KD in mice improved mitochondrial function, decreased oxidative stress, and increased ATP cerebral concentrations (Sullivan et al., 2004). In several transgenic mouse models of AD, KD was found to improve glycolysis, mitochondrial function and cognition, while reducing oxidative stress and amyloid deposition (Van der Auwera et al., 2005; Beckett et al., 2013; Kashiwaya et al., 2013; Zhang et al., 2013; Pawlosky et al., 2017). KD also improved metabolism, mitochondrial activity and motor functions in mouse models of PD (Tieu et al., 2003) and ALS (Zhao et al., 2012).
In humans, the impact of KD in neurodegenerative diseases has been tested in different clinical settings (Dewsbury et al., 2021) (Table 2). First, pilot study showed that 12-week KD in MCI or early-stage AD patients led to non-significant trend of memory improvement (Brandt et al., 2019). In a group of 23 patients with MCI, low carbohydrate diet was found to improve learning and memory performance compared to high carbohydrate diet (Krikorian et al., 2012). In a pilot study on 14 participants with MCI in PD, 8-week long KD enhanced cognitive performance (Krikorian et al., 2019). A larger multicentric study with 152 AD patients showed that 12-week-long administration of MCT Ketasyn/AC-1202 (Cerecin), improved memory performance in APOE4(–), but not in APOE4(+) patients (Henderson et al., 2009; Henderson and Poirier, 2011). Another pilot study showed that treatment with MCTs in APOE4(–) AD patients for 30 days improved memory performance (Xu et al., 2020). However, no significant differences were observed in subsequent trial on 413 AD patients after 26-week long intervention with MCT Tricaprilin/AC-1204 (Cerecin) (Henderson et al., 2020). Despite these contrasting results, Phase 3 trial with Cerecin’s MCT AC-SD-03/CER-0001 has been registered and plans to enroll 300 people with mild to moderate AD that exhibit decreased FDG PET signal and have APOE4(–) genotype (NCT04187547). Finally, a clinical study that assessed the metabolic effect of MCT-based regimen kMCT-ONS (Nestlé Health Science) in 52 people with MCI reported increase in plasma and brain ketones, while brain glucose uptake did not differ (Fortier et al., 2019). A larger 6-month trial in 122 MCI patients showed that treatment with kMCT-ONS diet led to improvements in memory tests, executive function and language (Fortier et al., 2021).
Brain hypometabolism is one of the first homeostatic dysregulation that occurs in age-related neurodegenerative diseases. Aging, APOE4 and insulin resistance are among the key factors that lead to brain hypometabolism. Hypometabolic changes are characterized by decreases in brain glucose uptake, expression of glucose transporters, astrocytic aerobic glycolysis, lactate release, neuronal mitochondrial function and increased oxidative stress (Figure 1). This homeostatic imbalance results in an energy gap, which renders neurons more vulnerable to a variety of insults and also decreases network connectivity (de la Torre, 2008). While brain hypometabolism occurs in physiological aging, it represents a significant contributing factor to a number of neurodegenerative diseases such as AD (Thomas et al., 2015; Butterfield and Halliwell, 2019), ALS (Lee et al., 2012; Vandoorne et al., 2018), depression (Rajkowska and Stockmeier, 2013), multiple sclerosis (Saab et al., 2016), migraine (Gross et al., 2019), epilepsy (de Melo et al., 2021), TBI (Carteron et al., 2018), retinal degeneration (Ait-Ali et al., 2015), stroke (Berthet et al., 2012), or spinal cord injury (SCI) (Babetto et al., 2020; Li et al., 2020).
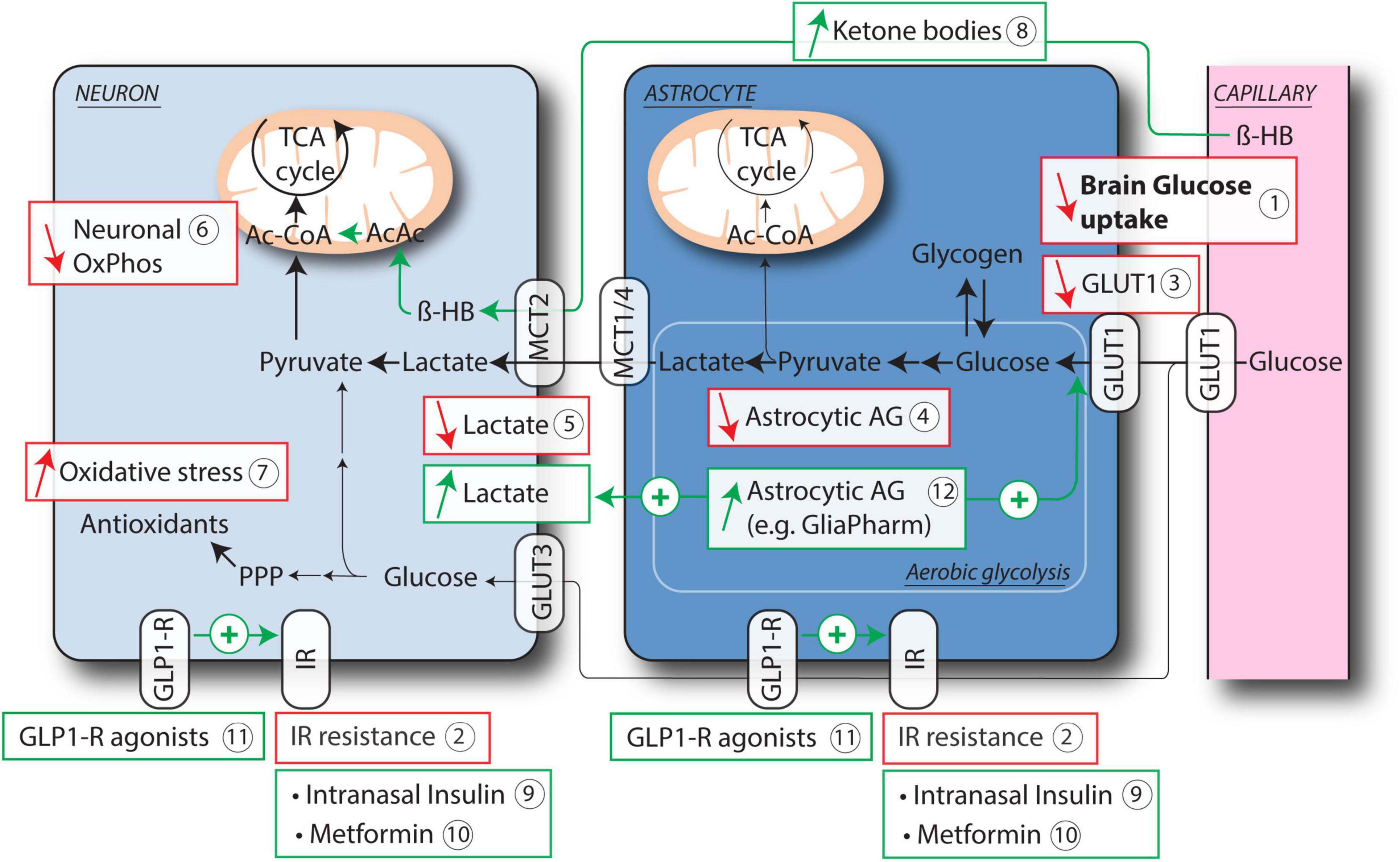
Figure 1. Age-related astrocytic and neuronal deficits leading to brain hypometabolism and current therapeutic strategies. Brain glucose hypometabolism is a hallmark of aging and neurodegeneration, as shown in particular by FDG PET studies (1). Resistance to Insulin has been proposed to account, at least in part, for this hypometabolism (2). Astrocytes and neurons both express insulin receptor (IR). Other key features of brain hypometabolism include reduced expression of GLUT1 on astrocytes and endothelial cells (3), decreased aerobic glycolysis (AG) in astrocytes (4) and consequent impaired release of lactate (5), reduced mitochondrial activity in neurons (6) and increased oxidative stress (7). Therapeutic strategies that aim at restoring brain energy metabolism include the use of ketone bodies as alternative energy source for neuronal mitochondrial OxPhos, either through ketogenic diet or medium chain triglycerides (8), targeting IR resistance either directly with intranasal Insulin (9) or Metformin (10), or via activation of GLP-1R (11). Another specific therapeutic approach consists in improving astrocytic AG (12), which results in increased glucose uptake and lactate release by astrocytes. Ac-CoA, acetyl-CoA; AcAc, acetoacetate; β-HB, β-hydroxybutyrate; GLP1-R, glucagon-like peptide 1 receptor; GLUT1, 3, glucose transporter 1, 3; IR, insulin receptor; MCT1, 2, 4, monocarboxylate transporter 1, 2, 4; OxPhos, oxidative phosphorylation; PPP, pentose phosphate pathway; TCA cycle, tricarboxylic acid cycle.
To target brain hypometabolism, several different therapeutic approaches that have shown promising results are presented in this review. For instance, intranasal insulin, which increases brain glucose uptake, was shown to improve cognition in AD and MCI patients. GLP-1R agonists and Metformin also improved glucose utilization and cognitive function in AD mouse models and patients. Ketogenic diet, another therapeutic strategy that aims at providing alternative source of energy to neurons, improves metabolic functions and cognition in preclinical models and human AD and MCI patients. These approaches have shown promising results, but lack selectivity to brain pathways. More targeted metabolic approaches constitute future avenues of development to tackle hypometabolic neurological diseases. Among these innovative approaches, our strategy at GliaPharm aims at specifically improving aerobic glycolysis in astrocytes, which results in the activation of the ANLS, increase in brain glucose uptake and release of lactate that is used by neurons as preferential energy source. This approach led to promising results in the impact on brain energy metabolism and neuroprotection in vitro and in different preclinical models.
The increasing amount of evidence linking brain aging, neurological diseases and hypometabolism has therefore opened avenue for innovative therapeutic strategies, either through non-specific drug repurposing or targeted approaches to improve brain metabolism. These approaches could have disease-modifying impact in the management of the brain energy crisis in a number of neurological diseases.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
EB, SL, SD, and CF were employed by company GliaPharm SA. PM is an advisor for GliaPharm SA.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbas, T., Faivre, E., and Holscher, C. (2009). Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: interaction between type 2 diabetes and Alzheimer’s disease. Behav. Brain Res. 205, 265–271. doi: 10.1016/j.bbr.2009.06.035
Acosta, C., Anderson, H. D., and Anderson, C. M. (2017). Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 95, 2430–2447.
Ahmed, K., Tunaru, S., and Offermanns, S. (2009). GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 30, 557–562. doi: 10.1016/j.tips.2009.09.001
Ahmed, K., Tunaru, S., Tang, C., Muller, M., Gille, A., Sassmann, A., et al. (2010). An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 11, 311–319. doi: 10.1016/j.cmet.2010.02.012
Ait-Ali, N., Fridlich, R., Millet-Puel, G., Clerin, E., Delalande, F., Jaillard, C., et al. (2015). Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 161, 817–832. doi: 10.1016/j.cell.2015.03.023
Alata, W., Ye, Y., St-Amour, I., Vandal, M., and Calon, F. (2015). Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab. 35, 86–94. doi: 10.1038/jcbfm.2014.172
Allaman, I., Gavillet, M., Belanger, M., Laroche, T., Viertl, D., Lashuel, H. A., et al. (2010). Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J. Neurosci. 30, 3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010
Allard, J. S., Perez, E. J., Fukui, K., Carpenter, P., Ingram, D. K., and De Cabo, R. (2016). Prolonged metformin treatment leads to reduced transcription of Nrf2 and neurotrophic factors without cognitive impairment in older C57BL/6J mice. Behav. Brain Res. 301, 1–9. doi: 10.1016/j.bbr.2015.12.012
Alle, H., Roth, A., and Geiger, J. R. (2009). Energy-efficient action potentials in hippocampal mossy fibers. Science 325, 1405–1408. doi: 10.1126/science.1174331
Alvarez, Z., Castano, O., Castells, A. A., Mateos-Timoneda, M. A., Planell, J. A., Engel, E., et al. (2014). Neurogenesis and vascularization of the damaged brain using a lactate-releasing biomimetic scaffold. Biomaterials 35, 4769–4781. doi: 10.1016/j.biomaterials.2014.02.051
Arendt, T., Bruckner, M. K., Morawski, M., Jager, C., and Gertz, H. J. (2015). Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol. Commun. 3:10. doi: 10.1186/s40478-015-0187-1
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H. Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. doi: 10.1038/nrneurol.2017.185
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Babetto, E., Wong, K. M., and Beirowski, B. (2020). A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 23, 1215–1228.
Badawi, G. A., Abd El Fattah, M. A., Zaki, H. F., and El Sayed, M. I. (2017). Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 25, 369–382. doi: 10.1007/s10787-017-0331-6
Bader, M., Li, Y., Tweedie, D., Shlobin, N. A., Bernstein, A., Rubovitch, V., et al. (2019). Neuroprotective effects and treatment potential of incretin mimetics in a murine model of mild traumatic brain injury. Front. Cell Dev. Biol. 7:356. doi: 10.3389/fcell.2019.00356
Bak, L. K., Schousboe, A., and Waagepetersen, H. S. (2006). The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98, 641–653. doi: 10.1111/j.1471-4159.2006.03913.x
Barone, E., Tramutola, A., Triani, F., Calcagnini, S., Di Domenico, F., Ripoli, C., et al. (2019). Biliverdin reductase-a mediates the beneficial effects of intranasal insulin in Alzheimer disease. Mol. Neurobiol. 56, 2922–2943. doi: 10.1007/s12035-018-1231-5
Barros, L. F., San Martin, A., Ruminot, I., Sandoval, P. Y., Baeza-Lehnert, F., Arce-Molina, R., et al. (2020). Fluid brain glycolysis: limits, speed, location, moonlighting, and the fates of glycogen and lactate. Neurochem. Res. 45, 1328–1334. doi: 10.1007/s11064-020-03005-2
Batista, A. F., Forny-Germano, L., Clarke, J. R., Lyra, E. S. N. M., Brito-Moreira, J., Boehnke, S. E., et al. (2018). The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 245, 85–100. doi: 10.1002/path.5056
Beal, M. F. (2005). Oxidative damage as an early marker of Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 26, 585–586. doi: 10.1016/j.neurobiolaging.2004.09.022
Beckett, T. L., Studzinski, C. M., Keller, J. N., Paul Murphy, M., and Niedowicz, D. M. (2013). A ketogenic diet improves motor performance but does not affect beta-amyloid levels in a mouse model of Alzheimer’s disease. Brain Res. 1505, 61–67. doi: 10.1016/j.brainres.2013.01.046
Belanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Belloy, M. E., Napolioni, V., and Greicius, M. D. (2019). A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron 101, 820–838. doi: 10.1016/j.neuron.2019.01.056
Bender, A. R., Prindle, J. J., Brandmaier, A. M., and Raz, N. (2016). White matter and memory in healthy adults: coupled changes over two years. Neuroimage 131, 193–204. doi: 10.1016/j.neuroimage.2015.10.085
Benedict, C., Hallschmid, M., Hatke, A., Schultes, B., Fehm, H. L., Born, J., et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334.
Benedict, C., Hallschmid, M., Schmitz, K., Schultes, B., Ratter, F., Fehm, H. L., et al. (2007). Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32, 239–243. doi: 10.1038/sj.npp.1301193
Bergau, N., Maul, S., Rujescu, D., Simm, A., and Navarrete Santos, A. (2019). Reduction of glycolysis intermediate concentrations in the cerebrospinal fluid of Alzheimer’s disease patients. Front. Neurosci. 13:871. doi: 10.3389/fnins.2019.00871
Bergersen, L., Waerhaug, O., Helm, J., Thomas, M., Laake, P., Davies, A. J., et al. (2001). A novel postsynaptic density protein: the monocarboxylate transporter MCT2 is co-localized with delta-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp. Brain Res. 136, 523–534. doi: 10.1007/s002210000600
Bergersen, L. H., Magistretti, P. J., and Pellerin, L. (2005). Selective postsynaptic co-localization of MCT2 with AMPA receptor GluR2/3 subunits at excitatory synapses exhibiting AMPA receptor trafficking. Cereb. Cortex 15, 361–370. doi: 10.1093/cercor/bhh138
Berthet, C., Castillo, X., Magistretti, P. J., and Hirt, L. (2012). New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 34, 329–335.
Berthet, C., Lei, H., Thevenet, J., Gruetter, R., Magistretti, P. J., and Hirt, L. (2009). Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. 29, 1780–1789. doi: 10.1038/jcbfm.2009.97
Bertilsson, G., Patrone, C., Zachrisson, O., Andersson, A., Dannaeus, K., Heidrich, J., et al. (2008). Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 86, 326–338. doi: 10.1002/jnr.21483
Bharath, L. P., Agrawal, M., Mccambridge, G., Nicholas, D. A., Hasturk, H., Liu, J., et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55.e6. doi: 10.1016/j.cmet.2020.04.015
Bi, Q., Wang, W., Niu, N., Li, H., Wang, Y., Huang, W., et al. (2021). Relationship between the disrupted topological efficiency of the structural brain connectome and glucose hypometabolism in normal aging. Neuroimage 226:117591. doi: 10.1016/j.neuroimage.2020.117591
Bingham, E. M., Hopkins, D., Smith, D., Pernet, A., Hallett, W., Reed, L., et al. (2002). The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes 51, 3384–3390. doi: 10.2337/diabetes.51.12.3384
Bittar, P. G., Charnay, Y., Pellerin, L., Bouras, C., and Magistretti, P. J. (1996). Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Metab. 16, 1079–1089. doi: 10.1097/00004647-199611000-00001
Bliss, T. M., Ip, M., Cheng, E., Minami, M., Pellerin, L., Magistretti, P., et al. (2004). Dual-gene, dual-cell type therapy against an excitotoxic insult by bolstering neuroenergetics. J. Neurosci. 24, 6202–6208. doi: 10.1523/JNEUROSCI.0805-04.2004
Bolanos, J. P., Peuchen, S., Heales, S. J., Land, J. M., and Clark, J. B. (1994). Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J. Neurochem. 63, 910–916. doi: 10.1046/j.1471-4159.1994.63030910.x
Bonomi, C. G., De Lucia, V., Mascolo, A. P., Assogna, M., Motta, C., Scaricamazza, E., et al. (2021). Brain energy metabolism and neurodegeneration: hints from CSF lactate levels in dementias. Neurobiol. Aging 105, 333–339. doi: 10.1016/j.neurobiolaging.2021.05.011
Bonvento, G., Sibson, N., and Pellerin, L. (2002). Does glutamate image your thoughts? Trends Neurosci. 25, 359–364. doi: 10.1016/s0166-2236(02)02168-9
Born, J., Lange, T., Kern, W., Mcgregor, G. P., Bickel, U., and Fehm, H. L. (2002). Sniffing neuropeptides: a transnasal approach to the human brain. Nat. Neurosci. 5, 514–516. doi: 10.1038/nn849
Bosshart, P. D., Kalbermatter, D., Bonetti, S., and Fotiadis, D. (2019). Mechanistic basis of L-lactate transport in the SLC16 solute carrier family. Nat. Commun. 10:2649. doi: 10.1038/s41467-019-10566-6
Boumezbeur, F., Mason, G. F., De Graaf, R. A., Behar, K. L., Cline, G. W., Shulman, G. I., et al. (2010). Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J. Cereb. Blood Flow Metab. 30, 211–221. doi: 10.1038/jcbfm.2009.197
Brandt, J., Buchholz, A., Henry-Barron, B., Vizthum, D., Avramopoulos, D., and Cervenka, M. C. (2019). Preliminary report on the feasibility and efficacy of the modified atkins diet for treatment of mild cognitive impairment and early Alzheimer’s disease. J. Alzheimers Dis. 68, 969–981. doi: 10.3233/JAD-180995
Brooks, G. A. (2018). The science and translation of lactate shuttle theory. Cell Metab. 27, 757–785. doi: 10.1016/j.cmet.2018.03.008
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Burda, J. E., Bernstein, A. M., and Sofroniew, M. V. (2016). Astrocyte roles in traumatic brain injury. Exp Neurol 275(Pt 3), 305–315. doi: 10.1016/j.expneurol.2015.03.020
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Campbell, J. M., Stephenson, M. D., De Courten, B., Chapman, I., Bellman, S. M., and Aromataris, E. (2018). Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J. Alzheimers Dis. 65, 1225–1236. doi: 10.3233/JAD-180263
Carbonell, F., Zijdenbos, A. P., Mclaren, D. G., Iturria-Medina, Y., Bedell, B. J., and Alzheimer’s Disease Neuroimaging Initiative. (2016). Modulation of glucose metabolism and metabolic connectivity by beta-amyloid. J. Cereb. Blood Flow Metab. 36, 2058–2071. doi: 10.1177/0271678X16654492
Carter, S. F., Chiotis, K., Nordberg, A., and Rodriguez-Vieitez, E. (2019). Longitudinal association between astrocyte function and glucose metabolism in autosomal dominant Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 46, 348–356. doi: 10.1007/s00259-018-4217-7
Carteron, L., Solari, D., Patet, C., Quintard, H., Miroz, J. P., Bloch, J., et al. (2018). Hypertonic lactate to improve cerebral perfusion and glucose availability after acute brain injury. Crit. Care Med. 46, 1649–1655. doi: 10.1097/CCM.0000000000003274
Chai, T. F., Hong, S. Y., He, H., Zheng, L., Hagen, T., Luo, Y., et al. (2012). A potential mechanism of metformin-mediated regulation of glucose homeostasis: inhibition of Thioredoxin-interacting protein (Txnip) gene expression. Cell Signal. 24, 1700–1705. doi: 10.1016/j.cellsig.2012.04.017
Chan-Palay, V., and Asan, E. (1989). Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J. Comp. Neurol. 287, 373–392. doi: 10.1002/cne.902870308
Chen, S., Sun, J., Zhao, G., Guo, A., Chen, Y., Fu, R., et al. (2017). Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem. Res. 42, 2326–2335. doi: 10.1007/s11064-017-2250-8
Chen, Y., Zhao, S., Fan, Z., Li, Z., Zhu, Y., Shen, T., et al. (2021). Metformin attenuates plaque-associated tau pathology and reduces amyloid-beta burden in APP/PS1 mice. Alzheimers Res. Ther. 13:40. doi: 10.1186/s13195-020-00761-9
Chin-Hsiao, T. (2019). Metformin and the risk of dementia in type 2 diabetes patients. Aging Dis. 10, 37–48.
Chiu, S. L., Chen, C. M., and Cline, H. T. (2008). Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58, 708–719. doi: 10.1016/j.neuron.2008.04.014
Chowdhury, G. M., Jiang, L., Rothman, D. L., and Behar, K. L. (2014). The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. J. Cereb. Blood Flow Metab. 34, 1233–1242. doi: 10.1038/jcbfm.2014.77
Clarke, D. W., Boyd, F. T. Jr., Kappy, M. S., and Raizada, M. K. (1984). Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 259, 11672–11675.
Claxton, A., Baker, L. D., Wilkinson, C. W., Trittschuh, E. H., Chapman, D., Watson, G. S., et al. (2013). Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J. Alzheimers Dis. 35, 789–797. doi: 10.3233/JAD-122308
Costantini, L. C., Barr, L. J., Vogel, J. L., and Henderson, S. T. (2008). Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 9(Suppl. 2):S16. doi: 10.1186/1471-2202-9-S2-S16
Craft, S., Baker, L. D., Montine, T. J., Minoshima, S., Watson, G. S., Claxton, A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38.
Craft, S., Claxton, A., Baker, L. D., Hanson, A. J., Cholerton, B., Trittschuh, E. H., et al. (2017). Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: a pilot clinical trial. J. Alzheimers Dis. 57, 1325–1334. doi: 10.3233/JAD-161256
Craft, S., Raman, R., Chow, T. W., Rafii, M. S., Sun, C. K., Rissman, R. A., et al. (2020). Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: a randomized clinical trial. JAMA Neurol. 77, 1099–1109. doi: 10.1001/jamaneurol.2020.1840
Craft, S., and Watson, G. S. (2004). Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 3, 169–178. doi: 10.1016/S1474-4422(04)00681-7
Cui, Q. N., Stein, L. M., Fortin, S. M., and Hayes, M. R. (2021). The role of glia in the physiology and pharmacology of glucagon-like peptide-1: implications for obesity, diabetes, neurodegeneration and glaucoma. Br. J. Pharmacol. doi: 10.1111/bph.15683
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., et al. (2011). Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27, 3–20. doi: 10.1016/j.nut.2010.07.021
Cureton, E. L., Kwan, R. O., Dozier, K. C., Sadjadi, J., Pal, J. D., and Victorino, G. P. (2010). A different view of lactate in trauma patients: protecting the injured brain. J. Surg. Res. 159, 468–473. doi: 10.1016/j.jss.2009.04.020
D’Andrea Meira, I., Romao, T. T., Pires do Prado, H. J., Kruger, L. T., Pires, M. E. P., and Da Conceicao, P. O. (2019). Ketogenic diet and epilepsy: what we know so far. Front. Neurosci. 13:5. doi: 10.3389/fnins.2019.00005
De Felice, F. G., Vieira, M. N., Bomfim, T. R., Decker, H., Velasco, P. T., Lambert, M. P., et al. (2009). Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 1971–1976. doi: 10.1073/pnas.0809158106
de la Monte, S. M., and Wands, J. R. (2006). Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 9, 167–181. doi: 10.3233/jad-2006-9209
de la Torre, J. C. (2008). Pathophysiology of neuronal energy crisis in Alzheimer’s disease. Neurodegener. Dis. 5, 126–132. doi: 10.1159/000113681
de Melo, I. S., Pacheco, A. L. D., Dos Santos, Y. M. O., Figueiredo, L. M., Nicacio, D., Cardoso-Sousa, L., et al. (2021). Modulation of glucose availability and effects of hypo- and hyperglycemia on status epilepticus: what we do not know yet? Mol. Neurobiol. 58, 505–519. doi: 10.1007/s12035-020-02133-8
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615.
Dekaban, A. S. (1978). Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann. Neurol. 4, 345–356. doi: 10.1002/ana.410040410
Demetrius, L. A., and Driver, J. (2013). Alzheimer’s as a metabolic disease. Biogerontology 14, 641–649. doi: 10.1007/s10522-013-9479-7
Demetrius, L. A., and Driver, J. A. (2015). Preventing Alzheimer’s disease by means of natural selection. J. R. Soc. Interface 12:20140919. doi: 10.1098/rsif.2014.0919
Demetrius, L. A., Magistretti, P. J., and Pellerin, L. (2014). Alzheimer’s disease: the amyloid hypothesis and the inverse warburg effect. Front Physiol. 5:522. doi: 10.3389/fphys.2014.00522
Dewsbury, L. S., Lim, C. K., and Steiner, G. Z. (2021). The efficacy of ketogenic therapies in the clinical management of people with neurodegenerative disease: a systematic review. Adv. Nutr. 12, 1571–1593. doi: 10.1093/advances/nmaa180
Dong, J. H., Wang, Y. J., Cui, M., Wang, X. J., Zheng, W. S., Ma, M. L., et al. (2017). Adaptive activation of a stress response pathway improves learning and memory through gs and beta-arrestin-1-regulated lactate metabolism. Biol. Psychiatry 81, 654–670. doi: 10.1016/j.biopsych.2016.09.025
Dougherty, R. J., Schultz, S. A., Kirby, T. K., Boots, E. A., Oh, J. M., Edwards, D., et al. (2017). Moderate physical activity is associated with cerebral glucose metabolism in adults at risk for Alzheimer’s disease. J. Alzheimers Dis. 58, 1089–1097. doi: 10.3233/JAD-161067
Duelli, R., Schrock, H., Kuschinsky, W., and Hoyer, S. (1994). Intracerebroventricular injection of streptozotocin induces discrete local changes in cerebral glucose utilization in rats. Int. J. Dev. Neurosci. 12, 737–743. doi: 10.1016/0736-5748(94)90053-1
During, M. J., Cao, L., Zuzga, D. S., Francis, J. S., Fitzsimons, H. L., Jiao, X., et al. (2003). Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 9, 1173–1179. doi: 10.1038/nm919
Eakin, K., Li, Y., Chiang, Y. H., Hoffer, B. J., Rosenheim, H., Greig, N. H., et al. (2013). Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS One 8:e82016. doi: 10.1371/journal.pone.0082016
Egefjord, L., Gejl, M., Moller, A., Braendgaard, H., Gottrup, H., Antropova, O., et al. (2012). Effects of liraglutide on neurodegeneration, blood flow and cognition in Alzheimer’s disease - protocol for a controlled, randomized double-blinded trial. Dan. Med. J. 59:A4519.
El Hayek, L., Khalifeh, M., Zibara, V., Abi Assaad, R., Emmanuel, N., Karnib, N., et al. (2019). Lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor (BDNF). J. Neurosci. 39, 2369–2382. doi: 10.1523/JNEUROSCI.1661-18.2019
El Khoury, N. B., Gratuze, M., Papon, M. A., Bretteville, A., and Planel, E. (2014). Insulin dysfunction and Tau pathology. Front. Cell Neurosci. 8:22. doi: 10.3389/fncel.2014.00022
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA 278, 1349–1356.
Femminella, G. D., and Edison, P. (2014). Evaluation of neuroprotective effect of glucagon-like peptide 1 analogs using neuroimaging. Alzheimers Dement. 10, S55–S61. doi: 10.1016/j.jalz.2013.12.012
Femminella, G. D., Frangou, E., Love, S. B., Busza, G., Holmes, C., Ritchie, C., et al. (2019). Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials 20:191.
Ferreira, I. L., Resende, R., Ferreiro, E., Rego, A. C., and Pereira, C. F. (2010). Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets 11, 1193–1206. doi: 10.2174/1389450111007011193
Figley, C. R., and Stroman, P. W. (2011). The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur. J. Neurosci. 33, 577–588. doi: 10.1111/j.1460-9568.2010.07584.x
Fortier, M., Castellano, C. A., Croteau, E., Langlois, F., Bocti, C., St-Pierre, V., et al. (2019). A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 15, 625–634. doi: 10.1016/j.jalz.2018.12.017
Fortier, M., Castellano, C. A., St-Pierre, V., Myette-Cote, E., Langlois, F., Roy, M., et al. (2021). A ketogenic drink improves cognition in mild cognitive impairment: results of a 6-month RCT. Alzheimers Dement. 17, 543–552. doi: 10.1002/alz.12206
Fox, N. C., and Schott, J. M. (2004). Imaging cerebral atrophy: normal ageing to Alzheimer’s disease. Lancet 363, 392–394. doi: 10.1016/S0140-6736(04)15441-X
Friedland, R. P., Brun, A., and Budinger, T. F. (1985). Pathological and positron emission tomographic correlations in Alzheimer’s disease. Lancet 1:228. doi: 10.1016/s0140-6736(85)92074-4
Fu, W., and Jhamandas, J. H. (2014). Role of astrocytic glycolytic metabolism in Alzheimer’s disease pathogenesis. Biogerontology 15, 579–586. doi: 10.1007/s10522-014-9525-0
Gao, V., Suzuki, A., Magistretti, P. J., Lengacher, S., Pollonini, G., Steinman, M. Q., et al. (2016). Astrocytic beta2-adrenergic receptors mediate hippocampal long-term memory consolidation. Proc. Natl. Acad. Sci. U.S.A. 113, 8526–8531. doi: 10.1073/pnas.1605063113
Gejl, M., Gjedde, A., Egefjord, L., Moller, A., Hansen, S. B., Vang, K., et al. (2016). In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 8:108. doi: 10.3389/fnagi.2016.00108
Giaccari, A., Solini, A., Frontoni, S., and Del Prato, S. (2021). Metformin benefits: another example for alternative energy substrate mechanism? Diabetes Care 44, 647–654. doi: 10.2337/dc20-1964
Glisky, E. L. (2007). “Changes in cognitive function in human aging,” in Brain Aging: Models, Methods, and Mechanisms, ed. D. R. Riddle (Boca Raton, FL: CRC Press).
Gong, G., Rosa-Neto, P., Carbonell, F., Chen, Z. J., He, Y., and Evans, A. C. (2009). Age- and gender-related differences in the cortical anatomical network. J. Neurosci. 29, 15684–15693. doi: 10.1523/JNEUROSCI.2308-09.2009
Goyal, M. S., Vlassenko, A. G., Blazey, T. M., Su, Y., Couture, L. E., Durbin, T. J., et al. (2017). Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26, 353–360.e3. doi: 10.1016/j.cmet.2017.07.010
Gross, E. C., Lisicki, M., Fischer, D., Sandor, P. S., and Schoenen, J. (2019). The metabolic face of migraine - from pathophysiology to treatment. Nat. Rev. Neurol. 15, 627–643. doi: 10.1038/s41582-019-0255-4
Hansen, H. H., Barkholt, P., Fabricius, K., Jelsing, J., Terwel, D., Pyke, C., et al. (2016a). The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 1634, 158–170. doi: 10.1016/j.brainres.2015.12.052
Hansen, H. H., Fabricius, K., Barkholt, P., Mikkelsen, J. D., Jelsing, J., Pyke, C., et al. (2016b). Characterization of liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, in rat partial and full nigral 6-hydroxydopamine lesion models of Parkinson’s disease. Brain Res. 1646, 354–365. doi: 10.1016/j.brainres.2016.05.038
Harkavyi, A., Abuirmeileh, A., Lever, R., Kingsbury, A. E., Biggs, C. S., and Whitton, P. S. (2008). Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J. Neuroinflammation 5:19. doi: 10.1186/1742-2094-5-19
Harr, S. D., Simonian, N. A., and Hyman, B. T. (1995). Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J. Neuropathol. Exp. Neurol. 54, 38–41.
Havrankova, J., Schmechel, D., Roth, J., and Brownstein, M. (1978). Identification of insulin in rat brain. Proc. Natl. Acad. Sci. U.S.A. 75, 5737–5741.
He, W., Wang, H., Zhao, C., Tian, X., Li, L., and Wang, H. (2020). Role of liraglutide in brain repair promotion through Sirt1-mediated mitochondrial improvement in stroke. J. Cell Physiol. 235, 2986–3001. doi: 10.1002/jcp.29204
Henderson, S. T., Morimoto, B. H., Cummings, J. L., Farlow, M. R., and Walker, J. (2020). A placebo-controlled, parallel-group, randomized clinical trial of AC-1204 in mild-to-moderate Alzheimer’s disease. J. Alzheimers Dis. 75, 547–557. doi: 10.3233/JAD-191302
Henderson, S. T., and Poirier, J. (2011). Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimer’s disease; a randomized, double-blind, placebo-controlled study. BMC Med. Genet. 12:137. doi: 10.1186/1471-2350-12-137
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. (Lond.). 6:31. doi: 10.1186/1743-7075-6-31
Heni, M., Hennige, A. M., Peter, A., Siegel-Axel, D., Ordelheide, A. M., Krebs, N., et al. (2011). Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One 6:e21594. doi: 10.1371/journal.pone.0021594
Herrero-Mendez, A., Almeida, A., Fernandez, E., Maestre, C., Moncada, S., and Bolanos, J. P. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–752. doi: 10.1038/ncb1881
Herzig, S., Raemy, E., Montessuit, S., Veuthey, J. L., Zamboni, N., Westermann, B., et al. (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96. doi: 10.1126/science.1218530
Hipkiss, A. R. (2019). Aging, Alzheimer’s disease and dysfunctional glycolysis; similar effects of too much and too little. Aging Dis. 10, 1328–1331. doi: 10.14336/AD.2019.0611
Holscher, C. (2018). Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology 136, 251–259. doi: 10.1016/j.neuropharm.2018.01.040
Hoyer, S. (2002). The aging brain. Changes in the neuronal insulin/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini-review. J. Neural Transm. (Vienna) 109, 991–1002. doi: 10.1007/s007020200082
Hoyer, S., Lee, S. K., Loffler, T., and Schliebs, R. (2000). Inhibition of the neuronal insulin receptor. An in vivo model for sporadic Alzheimer disease? Ann. N. Y. Acad. Sci. 920, 256–258. doi: 10.1111/j.1749-6632.2000.tb06932.x
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. U.S.A. 110, 3549–3554. doi: 10.1073/pnas.1214912110
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376. doi: 10.1038/nn2003
Imfeld, P., Bodmer, M., Jick, S. S., and Meier, C. R. (2012). Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case-control study. J. Am. Geriatr. Soc. 60, 916–921. doi: 10.1111/j.1532-5415.2012.03916.x
Jagust, W. J., Landau, S. M., and Alzheimer’s Disease Neuroimaging Initiative. (2012). Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J. Neurosci. 32, 18227–18233. doi: 10.1523/JNEUROSCI.3266-12.2012
Jiang, T., and Cadenas, E. (2014). Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell 13, 1059–1067. doi: 10.1111/acel.12268
Jiang, T., Yin, F., Yao, J., Brinton, R. D., and Cadenas, E. (2013). Lipoic acid restores age-associated impairment of brain energy metabolism through the modulation of Akt/JNK signaling and PGC1alpha transcriptional pathway. Aging Cell 12, 1021–1031. doi: 10.1111/acel.12127
Jourdain, P., Allaman, I., Rothenfusser, K., Fiumelli, H., Marquet, P., and Magistretti, P. J. (2016). L-Lactate protects neurons against excitotoxicity: implication of an ATP-mediated signaling cascade. Sci. Rep. 6:21250. doi: 10.1038/srep21250
Jourdain, P., Rothenfusser, K., Ben-Adiba, C., Allaman, I., Marquet, P., and Magistretti, P. J. (2018). Dual action of L-Lactate on the activity of NR2B-containing NMDA receptors: from potentiation to neuroprotection. Sci. Rep. 8:13472. doi: 10.1038/s41598-018-31534-y
Karagiannis, A., Gallopin, T., Lacroix, A., Plaisier, F., Piquet, J., Geoffroy, H., et al. (2021). Lactate is an energy substrate for rodent cortical neurons and enhances their firing activity. Elife 10:e71424. doi: 10.7554/eLife.71424
Kashiwaya, Y., Bergman, C., Lee, J. H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Keeney, J. T., Ibrahimi, S., and Zhao, L. (2015). Human ApoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: evidence of the mechanism of neuroprotection by ApoE2 and implications for Alzheimer’s disease prevention and early intervention. J. Alzheimers Dis. 48, 411–424. doi: 10.3233/JAD-150348
Kellar, D., and Craft, S. (2020). Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 19, 758–766. doi: 10.1016/S1474-4422(20)30231-3
Kelly, T., and Rose, C. R. (2010). Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J. Neurochem. 115, 1123–1136. doi: 10.1111/j.1471-4159.2010.07009.x
Koenig, A. M., Mechanic-Hamilton, D., Xie, S. X., Combs, M. F., Cappola, A. R., Xie, L., et al. (2017). Effects of the Insulin Sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Disord. 31, 107–113. doi: 10.1097/WAD.0000000000000202
Koss, E., Friedland, R. P., Ober, B. A., and Jagust, W. J. (1985). Differences in lateral hemispheric asymmetries of glucose utilization between early- and late-onset Alzheimer-type dementia. Am J. Psychiatry 142, 638–640. doi: 10.1176/ajp.142.5.638
Krikorian, R., Shidler, M. D., Dangelo, K., Couch, S. C., Benoit, S. C., and Clegg, D. J. (2012). Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 33, e419–e427. doi: 10.1016/j.neurobiolaging.2010.10.006
Krikorian, R., Shidler, M. D., Summer, S. S., Sullivan, P. G., Duker, A. P., Isaacson, R. S., et al. (2019). Nutritional ketosis for mild cognitive impairment in Parkinson’s disease: a controlled pilot trial. Clin. Park Relat. Disord. 1, 41–47. doi: 10.1016/j.prdoa.2019.07.006
Lauritzen, K. H., Morland, C., Puchades, M., Holm-Hansen, S., Hagelin, E. M., Lauritzen, F., et al. (2014). Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 24, 2784–2795. doi: 10.1093/cercor/bht136
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Leino, R. L., Gerhart, D. Z., Duelli, R., Enerson, B. E., and Drewes, L. R. (2001). Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 38, 519–527. doi: 10.1016/s0197-0186(00)00102-9
Lennox, R., Porter, D. W., Flatt, P. R., Holscher, C., Irwin, N., and Gault, V. A. (2014). Comparison of the independent and combined effects of sub-chronic therapy with metformin and a stable GLP-1 receptor agonist on cognitive function, hippocampal synaptic plasticity and metabolic control in high-fat fed mice. Neuropharmacology 86, 22–30. doi: 10.1016/j.neuropharm.2014.06.026
Lerchundi, R., Fernandez-Moncada, I., Contreras-Baeza, Y., Sotelo-Hitschfeld, T., Machler, P., Wyss, M. T., et al. (2015). NH4(+) triggers the release of astrocytic lactate via mitochondrial pyruvate shunting. Proc. Natl. Acad. Sci. U.S.A. 112, 11090–11095. doi: 10.1073/pnas.1508259112
Lester-Coll, N., Rivera, E. J., Soscia, S. J., Doiron, K., Wands, J. R., and De La Monte, S. M. (2006). Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis. 9, 13–33. doi: 10.3233/jad-2006-9102
Lev-Vachnish, Y., Cadury, S., Rotter-Maskowitz, A., Feldman, N., Roichman, A., Illouz, T., et al. (2019). L-lactate promotes adult hippocampal neurogenesis. Front Neurosci. 13:403. doi: 10.3389/fnins.2019.00403
Li, F., Sami, A., Noristani, H. N., Slattery, K., Qiu, J., Groves, T., et al. (2020). Glial metabolic rewiring promotes axon regeneration and functional recovery in the central nervous system. Cell Metab. 32, 767–785.e7. doi: 10.1016/j.cmet.2020.08.015
Li, Y., Duffy, K. B., Ottinger, M. A., Ray, B., Bailey, J. A., Holloway, H. W., et al. (2010). GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimers Dis. 19, 1205–1219. doi: 10.3233/JAD-2010-1314
Li, Y., Perry, T., Kindy, M. S., Harvey, B. K., Tweedie, D., Holloway, H. W., et al. (2009). GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 106, 1285–1290. doi: 10.1073/pnas.0806720106
Liang, W. S., Reiman, E. M., Valla, J., Dunckley, T., Beach, T. G., Grover, A., et al. (2008). Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4441–4446. doi: 10.1073/pnas.0709259105
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lin, A. L., Jahrling, J. B., Zhang, W., Derosa, N., Bakshi, V., Romero, P., et al. (2017). Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 37, 217–226. doi: 10.1177/0271678X15621575
Liu, L., Mackenzie, K. R., Putluri, N., Maletic-Savatic, M., and Bellen, H. J. (2017). The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 26:e716. doi: 10.1016/j.cmet.2017.08.024
Liu, W., Jalewa, J., Sharma, M., Li, G., Li, L., and Holscher, C. (2015). Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 303, 42–50. doi: 10.1016/j.neuroscience.2015.06.054
Lourenco, M. V., Clarke, J. R., Frozza, R. L., Bomfim, T. R., Forny-Germano, L., Batista, A. F., et al. (2013). TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. doi: 10.1016/j.cmet.2013.11.002
Luchsinger, J. A., Perez, T., Chang, H., Mehta, P., Steffener, J., Pradabhan, G., et al. (2016). Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J. Alzheimers Dis. 51, 501–514. doi: 10.3233/JAD-150493
Machler, P., Wyss, M. T., Elsayed, M., Stobart, J., Gutierrez, R., Von Faber-Castell, A., et al. (2016). In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 23, 94–102. doi: 10.1016/j.cmet.2015.10.010
MacVicar, B. A., and Newman, E. A. (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7:a020388. doi: 10.1101/cshperspect.a020388
Magistretti, P. J., and Allaman, I. (2015). A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. doi: 10.1016/j.neuron.2015.03.035
Magistretti, P. J., and Allaman, I. (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 19, 235–249. doi: 10.1038/nrn.2018.19
Magistretti, P. J., and Chatton, J. Y. (2005). Relationship between L-glutamate-regulated intracellular Na+ dynamics and ATP hydrolysis in astrocytes. J. Neural Transm. (Vienna) 112, 77–85. doi: 10.1007/s00702-004-0171-6
Magistretti, P. J., and Pellerin, L. (1996). Cellular bases of brain energy metabolism and their relevance to functional brain imaging: evidence for a prominent role of astrocytes. Cereb. Cortex 6, 50–61. doi: 10.1093/cercor/6.1.50
Mahley, R. W., and Rall, S. C. Jr. (2000). Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537. doi: 10.1146/annurev.genom.1.1.507
Mahley, R. W., Weisgraber, K. H., and Huang, Y. (2006). Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5644–5651. doi: 10.1073/pnas.0600549103
Manninen, T., Saudargiene, A., and Linne, M. L. (2020). Astrocyte-mediated spike-timing-dependent long-term depression modulates synaptic properties in the developing cortex. PLoS Comput. Biol. 16:e1008360. doi: 10.1371/journal.pcbi.1008360
Marcus, D. L., and Freedman, M. L. (1997). Decreased brain glucose metabolism in microvessels from patients with Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 248–253. doi: 10.1111/j.1749-6632.1997.tb48476.x
Margineanu, M. B., Mahmood, H., Fiumelli, H., and Magistretti, P. J. (2018). L-lactate regulates the expression of synaptic plasticity and neuroprotection genes in cortical neurons: a transcriptome analysis. Front Mol Neurosci 11:375. doi: 10.3389/fnmol.2018.00375
Masliah, E., Mallory, M., Hansen, L., Deteresa, R., and Terry, R. D. (1993). Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 43, 192–197. doi: 10.1212/wnl.43.1_part_1.192
Mathiisen, T. M., Lehre, K. P., Danbolt, N. C., and Ottersen, O. P. (2010). The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58, 1094–1103. doi: 10.1002/glia.20990
Mattay, V. S., Fera, F., Tessitore, A., Hariri, A. R., Berman, K. F., Das, S., et al. (2006). Neurophysiological correlates of age-related changes in working memory capacity. Neurosci. Lett. 392, 32–37. doi: 10.1016/j.neulet.2005.09.025
McClean, P. L., Jalewa, J., and Holscher, C. (2015). Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav. Brain Res. 293, 96–106. doi: 10.1016/j.bbr.2015.07.024
McClean, P. L., Parthsarathy, V., Faivre, E., and Holscher, C. (2011). The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 31, 6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011
McKenna, M. C. (2007). The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J. Neurosci. Res. 85, 3347–3358. doi: 10.1002/jnr.21444
McNeilly, A. D., Williamson, R., Balfour, D. J., Stewart, C. A., and Sutherland, C. (2012). A high-fat-diet-induced cognitive deficit in rats that is not prevented by improving insulin sensitivity with metformin. Diabetologia 55, 3061–3070. doi: 10.1007/s00125-012-2686-y
Merlini, M., Meyer, E. P., Ulmann-Schuler, A., and Nitsch, R. M. (2011). Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 122, 293–311. doi: 10.1007/s00401-011-0834-y
Minoshima, S., Giordani, B., Berent, S., Frey, K. A., Foster, N. L., and Kuhl, D. E. (1997). Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 42, 85–94. doi: 10.1002/ana.410420114
Mongeon, R., Venkatachalam, V., and Yellen, G. (2016). Cytosolic NADH-NAD(+) redox visualized in brain slices by two-photon fluorescence lifetime biosensor imaging. Antioxid. Redox Signal. 25, 553–563. doi: 10.1089/ars.2015.6593
Mooradian, A. D., Chung, H. C., and Shah, G. N. (1997). GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 18, 469–474. doi: 10.1016/s0197-4580(97)00111-5
Moore, R. Y., and Bloom, F. E. (1979). Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu. Rev. Neurosci. 2, 113–168. doi: 10.1146/annurev.ne.02.030179.000553
Morland, C., Lauritzen, K. H., Puchades, M., Holm-Hansen, S., Andersson, K., Gjedde, A., et al. (2015). The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: expression and action in brain. J. Neurosci. Res. 93, 1045–1055. doi: 10.1002/jnr.23593
Muddapu, V. R., Dharshini, S. A. P., Chakravarthy, V. S., and Gromiha, M. M. (2020). Neurodegenerative diseases - is metabolic deficiency the root cause? Front. Neurosci. 14:213. doi: 10.3389/fnins.2020.00213
Muraleedharan, R., Gawali, M. V., Tiwari, D., Sukumaran, A., Oatman, N., Anderson, J., et al. (2020). AMPK-regulated astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell Rep. 32:108092. doi: 10.1016/j.celrep.2020.108092
Nagele, R. G., Wegiel, J., Venkataraman, V., Imaki, H., Wang, K. C., and Wegiel, J. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674. doi: 10.1016/j.neurobiolaging.2004.01.007
Nehlig, A. (2004). Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fatty Acids 70, 265–275. doi: 10.1016/j.plefa.2003.07.006
Newman, L. A., Korol, D. L., and Gold, P. E. (2011). Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427. doi: 10.1371/journal.pone.0028427
Novak, P., Pimentel Maldonado, D. A., and Novak, V. (2019). Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: a double-blinded placebo-controlled pilot study. PLoS One 14:e0214364. doi: 10.1371/journal.pone.0214364
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009
Oksanen, M., Petersen, A. J., Naumenko, N., Puttonen, K., Lehtonen, S., Gubert Olive, M., et al. (2017). PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 9, 1885–1897. doi: 10.1016/j.stemcr.2017.10.016
Ong, Q. R., Chan, E. S., Lim, M. L., Cole, G. M., and Wong, B. S. (2014). Reduced phosphorylation of brain insulin receptor substrate and Akt proteins in apolipoprotein-E4 targeted replacement mice. Sci. Rep. 4:3754. doi: 10.1038/srep03754
Owen, O. E., Morgan, A. P., Kemp, H. G., Sullivan, J. M., Herrera, M. G., and Cahill, G. F. Jr. (1967). Brain metabolism during fasting. J. Clin. Invest. 46, 1589–1595. doi: 10.1172/JCI105650
Palmer, A. M. (1999). The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J. Neural Transm. (Vienna) 106, 317–328. doi: 10.1007/s007020050161
Paoli, A., Bianco, A., Damiani, E., and Bosco, G. (2014). Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed. Res. Int. 2014:474296. doi: 10.1155/2014/474296
Parikh, H., Carlsson, E., Chutkow, W. A., Johansson, L. E., Storgaard, H., Poulsen, P., et al. (2007). TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 4:e158. doi: 10.1371/journal.pmed.0040158
Patching, S. G. (2017). Glucose transporters at the blood-brain barrier: function, regulation and gateways for drug delivery. Mol. Neurobiol. 54, 1046–1077. doi: 10.1007/s12035-015-9672-6
Pawlosky, R. J., Kemper, M. F., Kashiwaya, Y., King, M. T., Mattson, M. P., and Veech, R. L. (2017). Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 141, 195–207.
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Pellerin, L., and Magistretti, P. J. (2012). Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 32, 1152–1166. doi: 10.1038/jcbfm.2011.149
Perry, T., and Greig, N. H. (2005). Enhancing central nervous system endogenous GLP-1 receptor pathways for intervention in Alzheimer’s disease. Curr. Alzheimer Res. 2, 377–385. doi: 10.2174/1567205054367892
Perry, T., Holloway, H. W., Weerasuriya, A., Mouton, P. R., Duffy, K., Mattison, J. A., et al. (2007). Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp. Neurol. 203, 293–301. doi: 10.1016/j.expneurol.2006.09.028
Peterson, A. C., and Li, C. R. (2018). Noradrenergic dysfunction in Alzheimer’s and Parkinson’s diseases-an overview of imaging studies. Front. Aging Neurosci. 10:127. doi: 10.3389/fnagi.2018.00127
Pintana, H., Apaijai, N., Pratchayasakul, W., Chattipakorn, N., and Chattipakorn, S. C. (2012). Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats. Life Sci. 91, 409–414. doi: 10.1016/j.lfs.2012.08.017
Porras, O. H., Ruminot, I., Loaiza, A., and Barros, L. F. (2008). Na(+)-Ca(2+) cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia 56, 59–68. doi: 10.1002/glia.20589
Prapong, T., Buss, J., Hsu, W. H., Heine, P., West Greenlee, H., and Uemura, E. (2002). Amyloid beta-peptide decreases neuronal glucose uptake despite causing increase in GLUT3 mRNA transcription and GLUT3 translocation to the plasma membrane. Exp. Neurol. 174, 253–258. doi: 10.1006/exnr.2001.7861
Puchalska, P., and Crawford, P. A. (2017). Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 25, 262–284. doi: 10.1016/j.cmet.2016.12.022
Qi, L., Ke, L., Liu, X., Liao, L., Ke, S., Liu, X., et al. (2016). Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3beta pathway in an amyloid beta protein induced Alzheimer disease mouse model. Eur. J. Pharmacol. 783, 23–32. doi: 10.1016/j.ejphar.2016.04.052
Raber, J., Huang, Y., and Ashford, J. W. (2004). ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 25, 641–650. doi: 10.1016/j.neurobiolaging.2003.12.023
Rachmany, L., Tweedie, D., Li, Y., Rubovitch, V., Holloway, H. W., Miller, J., et al. (2013). Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age (Dordr) 35, 1621–1636. doi: 10.1007/s11357-012-9464-0
Raichle, M. E., and Mintun, M. A. (2006). Brain work and brain imaging. Annu. Rev. Neurosci. 29, 449–476. doi: 10.1146/annurev.neuro.29.051605.112819
Rajkowska, G., and Stockmeier, C. A. (2013). Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr. Drug Targets 14, 1225–1236. doi: 10.2174/13894501113149990156
Rasmussen, P., Wyss, M. T., and Lundby, C. (2011). Cerebral glucose and lactate consumption during cerebral activation by physical activity in humans. FASEB J. 25, 2865–2873. doi: 10.1096/fj.11-183822
Reger, M. A., Watson, G. S., Green, P. S., Baker, L. D., Cholerton, B., Fishel, M. A., et al. (2008a). Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis. 13, 323–331. doi: 10.3233/jad-2008-13309
Reger, M. A., Watson, G. S., Green, P. S., Wilkinson, C. W., Baker, L. D., Cholerton, B., et al. (2008b). Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 70, 440–448.
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. U.S.A. 101, 284–289. doi: 10.1073/pnas.2635903100
Reiner, D. J., Mietlicki-Baase, E. G., Mcgrath, L. E., Zimmer, D. J., Bence, K. K., Sousa, G. L., et al. (2016). Astrocytes regulate GLP-1 receptor-mediated effects on energy balance. J. Neurosci. 36, 3531–3540.
Resnick, S. M., Pham, D. L., Kraut, M. A., Zonderman, A. B., and Davatzikos, C. (2003). Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J. Neurosci. 23, 3295–3301. doi: 10.1523/jneurosci.23-08-03295.2003
Rivera, E. J., Goldin, A., Fulmer, N., Tavares, R., Wands, J. R., and De La Monte, S. M. (2005). Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J. Alzheimers Dis. 8, 247–268.
Riveros, N., Fiedler, J., Lagos, N., Munoz, C., and Orrego, F. (1986). Glutamate in rat brain cortex synaptic vesicles: influence of the vesicle isolation procedure. Brain Res. 386, 405–408.
Robinson, M. M., Lowe, V. J., and Nair, K. S. (2018). Increased brain glucose uptake after 12 weeks of aerobic high-intensity interval training in young and older adults. J. Clin. Endocrinol. Metab. 103, 221–227. doi: 10.1210/jc.2017-01571
Rodriguez, J. J., Olabarria, M., Chvatal, A., and Verkhratsky, A. (2009). Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16, 378–385.
Roosterman, D., and Cottrell, G. S. (2020). Astrocytes and neurons communicate via a monocarboxylic acid shuttle. AIMS Neurosci. 7, 94–106. doi: 10.3934/Neuroscience.2020007
Ros, J., Pecinska, N., Alessandri, B., Landolt, H., and Fillenz, M. (2001). Lactate reduces glutamate-induced neurotoxicity in rat cortex. J. Neurosci. Res. 66, 790–794. doi: 10.1002/jnr.10043
Rosenbloom, M., Barclay, T. R., Kashyap, B., Hage, L., O’keefe, L. R., Svitak, A., et al. (2021). A phase ii, single-center, randomized, double-blind, placebo-controlled study of the safety and therapeutic efficacy of intranasal glulisine in amnestic mild cognitive impairment and probable mild Alzheimer’s disease. Drugs Aging 38, 407–415. doi: 10.1007/s40266-021-00845-7
Rotermund, C., Machetanz, G., and Fitzgerald, J. C. (2018). The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. (Lausanne) 9:400. doi: 10.3389/fendo.2018.00400
Roumes, H., Jolle, C., Blanc, J., Benkhaled, I., Chatain, C. P., Massot, P., et al. (2021). Lactate transporters in the rat barrel cortex sustain whisker-dependent BOLD fMRI signal and behavioral performance. Proc. Natl. Acad. Sci. U.S.A. 118:e2112466118. doi: 10.1073/pnas.2112466118
Rub, U., Del Tredici, K., Schultz, C., Thal, D. R., Braak, E., and Braak, H. (2001). The autonomic higher order processing nuclei of the lower brain stem are among the early targets of the Alzheimer’s disease-related cytoskeletal pathology. Acta Neuropathol. 101, 555–564. doi: 10.1007/s004010000320
Saab, A. S., Tzvetavona, I. D., Trevisiol, A., Baltan, S., Dibaj, P., Kusch, K., et al. (2016). Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91, 119–132. doi: 10.1016/j.neuron.2016.05.016
Sada, N., Lee, S., Katsu, T., Otsuki, T., and Inoue, T. (2015). Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367. doi: 10.1126/science.aaa1299
Salat, D. H., Buckner, R. L., Snyder, A. Z., Greve, D. N., Desikan, R. S., Busa, E., et al. (2004). Thinning of the cerebral cortex in aging. Cereb. Cortex 14, 721–730.
Samaras, K., Makkar, S., Crawford, J. D., Kochan, N. A., Wen, W., Draper, B., et al. (2020). Metformin use is associated with slowed cognitive decline and reduced incident dementia in older adults with type 2 diabetes: the sydney memory and ageing study. Diabetes Care 43, 2691–2701. doi: 10.2337/dc20-0892
Schurr, A., Payne, R. S., Miller, J. J., and Rigor, B. M. (1997). Glia are the main source of lactate utilized by neurons for recovery of function posthypoxia. Brain Res. 774, 221–224. doi: 10.1016/s0006-8993(97)81708-8
Schurr, A., Payne, R. S., Miller, J. J., Tseng, M. T., and Rigor, B. M. (2001). Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 895, 268–272.
Simpson, I. A., Chundu, K. R., Davies-Hill, T., Honer, W. G., and Davies, P. (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 35, 546–551.
Sluggett, J. K., Koponen, M., Bell, J. S., Taipale, H., Tanskanen, A., Tiihonen, J., et al. (2020). Metformin and risk of Alzheimer’s disease among community-dwelling people with diabetes: a national case-control study. J. Clin. Endocrinol. Metab. 105:dgz234. doi: 10.1210/clinem/dgz234
Smith, D., Pernet, A., Hallett, W. A., Bingham, E., Marsden, P. K., and Amiel, S. A. (2003). Lactate: a preferred fuel for human brain metabolism in vivo. J. Cereb. Blood Flow Metab. 23, 658–664. doi: 10.1097/01.WCB.0000063991.19746.11
Sowell, E. R., Peterson, B. S., Thompson, P. M., Welcome, S. E., Henkenius, A. L., and Toga, A. W. (2003). Mapping cortical change across the human life span. Nat. Neurosci. 6, 309–315. doi: 10.1038/nn1008
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimers Dis. 7, 63–80. doi: 10.3233/jad-2005-7107
Sullivan, P. G., Rippy, N. A., Dorenbos, K., Concepcion, R. C., Agarwal, A. K., and Rho, J. M. (2004). The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 55, 576–580. doi: 10.1002/ana.20062
Supplie, L. M., Duking, T., Campbell, G., Diaz, F., Moraes, C. T., Gotz, M., et al. (2017). Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci. 37, 4231–4242. doi: 10.1523/JNEUROSCI.0756-16.2017
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823. doi: 10.1016/j.cell.2011.02.018
Talbot, K., Wang, H. Y., Kazi, H., Han, L. Y., Bakshi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. doi: 10.1172/JCI59903
Tang, B. L. (2020). Glucose, glycolysis, and neurodegenerative diseases. J. Cell. Physiol. 235, 7653–7662. doi: 10.1002/jcp.29682
Taouis, M., and Torres-Aleman, I. (2019). Editorial: Insulin and The Brain. Front Endocrinol (Lausanne) 10:299.
Thomas, S. C., Alhasawi, A., Appanna, V. P., Auger, C., and Appanna, V. D. (2015). Brain metabolism and Alzheimer’s disease: the prospect of a metabolite-based therapy. J. Nutr. Health Aging 19, 58–63. doi: 10.1007/s12603-014-0511-7
Tieu, K., Perier, C., Caspersen, C., Teismann, P., Wu, D. C., Yan, S. D., et al. (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 112, 892–901. doi: 10.1172/JCI18797
Tomi, M., Zhao, Y., Thamotharan, S., Shin, B. C., and Devaskar, S. U. (2013). Early life nutrient restriction impairs blood-brain metabolic profile and neurobehavior predisposing to Alzheimer’s disease with aging. Brain Res. 1495, 61–75. doi: 10.1016/j.brainres.2012.11.050
Uemura, E., and Greenlee, H. W. (2001). Amyloid beta-peptide inhibits neuronal glucose uptake by preventing exocytosis. Exp. Neurol. 170, 270–276. doi: 10.1006/exnr.2001.7719
Vadini, F., Simeone, P. G., Boccatonda, A., Guagnano, M. T., Liani, R., Tripaldi, R., et al. (2020). Liraglutide improves memory in obese patients with prediabetes or early type 2 diabetes: a randomized, controlled study. Int. J. Obes. (Lond.). 44, 1254–1263. doi: 10.1038/s41366-020-0535-5
Van der Auwera, I., Wera, S., Van Leuven, F., and Henderson, S. T. (2005). A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. (Lond.). 2:28. doi: 10.1186/1743-7075-2-28
van Gijsel-Bonnello, M., Baranger, K., Benech, P., Rivera, S., Khrestchatisky, M., De Reggi, M., et al. (2017). Metabolic changes and inflammation in cultured astrocytes from the 5xFAD mouse model of Alzheimer’s disease: alleviation by pantethine. PLoS One 12:e0175369. doi: 10.1371/journal.pone.0175369
Vandoorne, T., De Bock, K., and Van Den Bosch, L. (2018). Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathol. 135, 489–509. doi: 10.1007/s00401-018-1835-x
Vardjan, N., Chowdhury, H. H., Horvat, A., Velebit, J., Malnar, M., Muhic, M., et al. (2018). Enhancement of astroglial aerobic glycolysis by extracellular lactate-mediated increase in cAMP. Front. Mol. Neurosci. 11:148. doi: 10.3389/fnmol.2018.00148
Veech, R. L. (2004). The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids 70, 309–319. doi: 10.1016/j.plefa.2003.09.007
Wang, J., Gallagher, D., Devito, L. M., Cancino, G. I., Tsui, D., He, L., et al. (2012). Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 11, 23–35. doi: 10.1016/j.stem.2012.03.016
Watson, K. T., Wroolie, T. E., Tong, G., Foland-Ross, L. C., Frangou, S., Singh, M., et al. (2019). Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav. Brain Res. 356, 271–278.
Weise, C. M., Chen, K., Chen, Y., Kuang, X., Savage, C. R., Reiman, E. M., et al. (2018). Left lateralized cerebral glucose metabolism declines in amyloid-beta positive persons with mild cognitive impairment. Neuroimage Clin. 20, 286–296. doi: 10.1016/j.nicl.2018.07.016
Westhaus, A., Blumrich, E. M., and Dringen, R. (2017). The antidiabetic drug metformin stimulates glycolytic lactate production in cultured primary rat astrocytes. Neurochem. Res. 42, 294–305. doi: 10.1007/s11064-015-1733-8
Williams, H. C., Farmer, B. C., Piron, M. A., Walsh, A. E., Bruntz, R. C., Gentry, M. S., et al. (2020). APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol. Dis. 136:104742. doi: 10.1016/j.nbd.2020.104742
Wilson, R. S., Nag, S., Boyle, P. A., Hizel, L. P., Yu, L., Buchman, A. S., et al. (2013). Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology 80, 1202–1208. doi: 10.1212/WNL.0b013e3182897103
Wong-Riley, M. T. (1989). Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci. 12, 94–101.
Woo, B. K., Harwood, D. G., Melrose, R. J., Mandelkern, M. A., Campa, O. M., Walston, A., et al. (2010). Executive deficits and regional brain metabolism in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 25, 1150–1158.
Wu, L., Zhang, X., and Zhao, L. (2018). Human ApoE Isoforms differentially modulate brain glucose and ketone body metabolism: implications for Alzheimer’s disease risk reduction and early intervention. J. Neurosci. 38, 6665–6681. doi: 10.1523/JNEUROSCI.2262-17.2018
Xu, P. T., Schmechel, D., Rothrock-Christian, T., Burkhart, D. S., Qiu, H. L., Popko, B., et al. (1996). Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol. Dis. 3, 229–245. doi: 10.1006/nbdi.1996.0023
Xu, Q., Bernardo, A., Walker, D., Kanegawa, T., Mahley, R. W., and Huang, Y. (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006
Xu, Q., Zhang, Y., Zhang, X., Liu, L., Zhou, B., Mo, R., et al. (2020). Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer’s disease patients with APOE4(-/-): a double-blind, randomized, placebo-controlled crossover trial. Clin. Nutr. 39, 2092–2105. doi: 10.1016/j.clnu.2019.10.017
Yan, X., Hu, Y., Wang, B., Wang, S., and Zhang, X. (2020). Metabolic dysregulation contributes to the progression of Alzheimer’s disease. Front. Neurosci. 14:530219. doi: 10.3389/fnins.2020.530219
Yang, J., Ruchti, E., Petit, J. M., Jourdain, P., Grenningloh, G., Allaman, I., et al. (2014). Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. U.S.A. 111, 12228–12233. doi: 10.1073/pnas.1322912111
Yang, Y., Bender, A. R., and Raz, N. (2015). Age related differences in reaction time components and diffusion properties of normal-appearing white matter in healthy adults. Neuropsychologia 66, 246–258. doi: 10.1016/j.neuropsychologia.2014.11.020
Yao, J., and Brinton, R. D. (2011). Targeting mitochondrial bioenergetics for Alzheimer’s prevention and treatment. Curr. Pharm. Des. 17, 3474–3479. doi: 10.2174/138161211798072517
Yildirim Simsir, I., Soyaltin, U. E., and Cetinkalp, S. (2018). Glucagon like peptide-1 (GLP-1) likes Alzheimer’s disease. Diabetes Metab. Syndr. 12, 469–475. doi: 10.1016/j.dsx.2018.03.002
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 100, 108–122. doi: 10.1016/j.freeradbiomed.2016.04.200
Yu, Y., Herman, P., Rothman, D. L., Agarwal, D., and Hyder, F. (2018). Evaluating the gray and white matter energy budgets of human brain function. J. Cereb. Blood Flow Metab. 38, 1339–1353. doi: 10.1177/0271678X17708691
Zhang, J., Cao, Q., Li, S., Lu, X., Zhao, Y., Guan, J. S., et al. (2013). 3-Hydroxybutyrate methyl ester as a potential drug against Alzheimer’s disease via mitochondria protection mechanism. Biomaterials 34, 7552–7562. doi: 10.1016/j.biomaterials.2013.06.043
Zhao, N., Liu, C. C., Van Ingelgom, A. J., Martens, Y. A., Linares, C., Knight, J. A., et al. (2017). Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron 96, 115–129.e5. doi: 10.1016/j.neuron.2017.09.003
Zhao, W., Varghese, M., Vempati, P., Dzhun, A., Cheng, A., Wang, J., et al. (2012). Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS One 7:e49191. doi: 10.1371/journal.pone.0049191
Zhao, W. Q., and Townsend, M. (2009). Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta 1792, 482–496. doi: 10.1016/j.bbadis.2008.10.014
Zheng, J., Xie, Y., Ren, L., Qi, L., Wu, L., Pan, X., et al. (2021). GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 47:101180. doi: 10.1016/j.molmet.2021.101180
Zhou, J., Liu, T., Guo, H., Cui, H., Li, P., Feng, D., et al. (2018). Lactate potentiates angiogenesis and neurogenesis in experimental intracerebral hemorrhage. Exp. Mol. Med. 50, 1–12. doi: 10.1038/s12276-018-0113-2
Keywords: astrocytes, lactate, glucose, brain, energy, metabolism, new therapeutic approach, GliaPharm
Citation: Beard E, Lengacher S, Dias S, Magistretti PJ and Finsterwald C (2022) Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 12:825816. doi: 10.3389/fphys.2021.825816
Received: 30 November 2021; Accepted: 20 December 2021;
Published: 11 January 2022.
Edited by:
Lin-Hua Jiang, University of Leeds, United KingdomReviewed by:
Rosa Maria Moresco, University of Milano-Bicocca, ItalyCopyright © 2022 Beard, Lengacher, Dias, Magistretti and Finsterwald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Finsterwald, Y2ZpbnN0ZXJ3YWxkQGdsaWFwaGFybS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.