 Encan Li
Encan Li Vera Loen
Vera Loen Willem B. van Ham
Willem B. van Ham Willy Kool1
Willy Kool1 Marcel A. G. van der Heyden
Marcel A. G. van der Heyden Hiroki Takanari
Hiroki Takanari
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 25 January 2022
Sec. Membrane Physiology and Membrane Biophysics
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.812572
Alteration of the inward rectifier current IK1, carried by KIR2.1 channels, affects action potential duration, impacts resting membrane stability and associates with cardiac arrhythmias. Congenital and acquired KIR2.1 malfunction frequently associates with aberrant ion channel trafficking. Cellular processes underlying trafficking are intertwined with cytoskeletal function. The extent to which the cytoskeleton is involved in KIR2.1 trafficking processes is unknown. We aimed to quantify the dependence of KIR2.1 trafficking on cytoskeleton function. GFP or photoconvertible Dendra2 tagged KIR2.1 constructs were transfected in HEK293 or HeLa cells. Photoconversion of the Dendra2 probe at the plasma membrane and subsequent live imaging of trafficking processes was performed by confocal laser-scanning microscopy. Time constant of green fluorescent recovery (τg,s) represented recruitment of new KIR2.1 at the plasma membrane. Red fluorescent decay (τr,s) represented internalization of photoconverted KIR2.1. Patch clamp electrophysiology was used to quantify IKIR2.1. Biochemical methods were used for cytoskeleton isolation and detection of KIR2.1-cytoskeleton interactions. Cytochalasin B (20 μM), Nocodazole (30 μM) and Dyngo-4a (10 nM) were used to modify the cytoskeleton. Chloroquine (10 μM, 24 h) was used to impair KIR2.1 breakdown. Cytochalasin B and Nocodazole, inhibitors of actin and tubulin filament formation respectively, strongly inhibited the recovery of green fluorescence at the plasma membrane suggestive for inhibition of KIR2.1 forward trafficking [τg,s 13 ± 2 vs. 131 ± 31* and 160 ± 40* min, for control, Cytochalasin B and Nocodazole, respectively (*p < 0.05 vs. control)]. Dyngo-4a, an inhibitor of dynamin motor proteins, strongly slowed the rate of photoconverted channel internalization, whereas Nocodazole and Cytochalasin B had less effect [τr,s 20 ± 2 vs. 87 ± 14*, 60 ± 16 and 64 ± 20 min (*p < 0.05 vs. control)]. Cytochalasin B treatment (20 μM, 24 h) inhibited IKIR2.1. Chloroquine treatment (10 μM, 24 h) induced intracellular aggregation of KIR2.1 channels and enhanced interaction with the actin/intermediate filament system (103 ± 90 fold; p < 0.05 vs. control). Functional actin and tubulin cytoskeleton systems are essential for forward trafficking of KIR2.1 channels, whereas initial backward trafficking relies on a functional dynamin system. Chronic disturbance of the actin system inhibits KIR2.1 currents. Internalized KIR2.1 channels become recruited to the cytoskeleton, presumably in lysosomes.
Inward rectifier potassium channels from the KIR2.x protein family are present in many different excitable cell types. Ventricular cardiomyocytes strongly express KIR2.1 and KIR2.2 channel proteins (De Boer et al., 2010). Homo- or heterotetrameric KIR2.x assemblies form the functional channels that underlie the inward rectifier current (IK1) (Hibino et al., 2010). IK1 contributes to the final repolarization phase of the cardiac action potential and stabilizes the resting membrane potential of these cells (Van der Heyden and Jespersen, 2016). IK1 loss-of-function, either genetic or pharmacological, can result in cardiac action potential prolongation and ectopic activity in both model systems and patients (Biliczki et al., 2002; Tristani-Firouzi et al., 2002; Takanari et al., 2013; Myles et al., 2015; Reilly et al., 2020). In contrast, IK1 gain-of-function associates with action potential shortening and reentry-based arrhythmia mechanisms, as for example atrial fibrillation (Xia et al., 2005; Kharche et al., 2008; Hattori et al., 2012; Deo et al., 2013).
KIR2.1 proteins possess a short N-terminal and long C-terminal intracellular domain, two transmembrane domains, and two very short extracellular domains divided by the transmembrane pore domain. The intracellular domains contain several specific highly conserved sequences for intracellular transport, i.e., endoplasmic reticulum and Golgi export sequences, and sequences for subcellular anchoring and protein-protein interaction, such as the C-terminal PDZ domain (Stockklausner et al., 2001; Sampson et al., 2003; Stockklausner and Klocker, 2003; Leonoudakis et al., 2004a,b; Hofherr et al., 2005; Ma et al., 2011; Houtman et al., 2014; Li et al., 2016). Proper expression of the necessary amount of KIR2.1 proteins at the plasma membrane is of crucial importance for normal channel function in the context of an action potential (Willis et al., 2015). The cytoskeleton has an important role in processes that transport KIR2.1, and other cardiac ion channels, to and from the plasma membrane, known as forward and backward trafficking, and in anchoring KIR2.1 at the plasma membrane (Vatta and Faulkner, 2006; Steele and Fedida, 2014). Importantly, many loss-of-function mutations in the KIR2.1 gene associated with clinical phenotypes result in aberrant trafficking (Zangerl-Plessl et al., 2019).
The cytoskeleton consists of four types of filaments (Mostowy and Cossart, 2012). Actin filaments are 7 nm diameter, double stranded, helical assemblies of α, β or γ-type ± 42 kDa globular actin monomers. Tubulin filaments are 23 nm diameter hollow cylinders formed by α/β-tubulin (±50 kDa) dimers. A number of different intermediate filaments (11 nm diameter) proteins exist that may display cell type specific expression, such as vimentin in mesenchymal and keratin proteins in epithelial cells. Finally, septin proteins (30–66 kDa) form complexes that can organize as filaments which can further assemble as bundles and even circular structures. Actin and tubulin filaments are mainly involved in trafficking processes of ion channels (Steele and Fedida, 2014). Specific motor proteins interact with ion channels and transport their cargo along the filament systems. For example, KIR2.1 internalization depends on functional dynamin motor proteins, as their inhibition by dynasore or overexpression of the dynactin p50, increases expression at the plasma membrane (Loewen et al., 2009; Varkevisser et al., 2013). Pharmacological disruption of the actin and tubulin filament systems can be achieved experimentally by the use of Cytochalasin B and Nocodazole, respectively (De Brabander et al., 1976; MacLean-Fletcher and Pollard, 1980). Remarkably, in contrast to several other potassium channels whose expression at the plasma membrane increases upon Nocodazole mediated disruption of the tubulin system, KIR2.1 expression was found to decrease (Loewen et al., 2009). The authors suggest that substantial tubulin-dependent forward trafficking might explain this phenomenon. Currently, however, quantitative data on the roles of the actin and tubulin on KIR2.1 forward and backward trafficking are not available.
We demonstrated previously that the antimalarial drug Chloroquine (CQ) increased KIR2.1 expression and IK1. CQ also induced intracellular accumulation of KIR2.1-GFP and led to the presence of an N-terminal cleavage product (Jansen et al., 2008; Varkevisser et al., 2013). The latter two phenomena most likely result from direct or indirect alkalization of the lysosome by CQ affecting its function (Ohkuma and Poole, 1978; Ponsford et al., 2021). In kidney cells, CQ has also been shown to affect the cytoskeleton (Kang et al., 2020). To which extent this affects interaction of KIR2.1 with the cytoskeleton is unknown.
In the current work we made use of fluorescently C-terminally tagged KIR2.1 proteins (GFP and Dendra2), which are amendable for fluorescent microscopy and western blot analysis, and do not interfere with electrophysiological properties (De Boer et al., 2006b; Korte et al., 2017). The Dendra2 tag can be irreversibly photoconverted from green to red fluorescence by short UV illumination (Chudakov et al., 2007). In real-time microscopy this allows visualization of trafficking effects of specific pools of the KIR2.1-Dendra2 protein. By use of this technology, we determined alterations in KIR2.1 forward and backward trafficking kinetics in response to cytoskeleton-modifying compounds. Furthermore, we assessed the interaction of KIR2.1 with the cytoskeleton and the effects of Chloroquine treatment on these.
HeLa cells were cultured in low glucose Dulbeco’s Modified Eagle Medium supplemented with 10% fetal calf serum and 1% penicillin/streptomycin, all purchased from Wako (Osaka, Japan). Human Embryonal Kidney 293 cells stably expressing KIR2.1 channel tagged with GFP [HEK-KWGF (De Boer et al., 2006b)] and Chinese Hamster Ovary cells stably expressing KIR2.1 channel tagged with Dendra2 [CHO-Dendra2 (Korte et al., 2017)] were cultured in the same medium, but purchased from Lonza (Walkersville, MD, United States). In time course experiments, cells were seeded and harvested on identical days.
We purchased Dyngo-4a and Nocodazole from Abcam (Cambridge, United Kingdom). Cytochalasin B was purchased from Sigma Aldrich Japan (Kawasaki, Japan). The chemicals were dissolved in DMSO to provide a stock solution of 10 mM. All compounds were aliquoted and stored at −20°C until use. Cells were exposed to the drugs by adding stock solutions to the cell medium at the following concentrations: Dyngo-4a, 10 nM; Nocodazole, 30 μM; Cytochalasin B, 20 μM, Chloroquine, 10 μM.
Cell lysates were prepared in buffer D [20 mM HEPES, 125 mM NaCl, 10% glycerol, 1 mM EGTA, 1 mM dithiothreitol, 1 mM EDTA, and 1% Triton X-100 (pH 7.6)] supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10 μg⋅mL–1 aprotinin. Lysates were clarified by centrifugation at 14,000 rpm for 10 min at 4°C. Protein concentrations were established by BCA Protein Assay (Thermo Fisher Scientific, Breda, Netherlands). Samples were added to loading buffer and 25 μg of proteins were separated by 7.5% SDS–PAGE and blotted on a nitrocellulose membrane (Bio-Rad Laboratories, Veenendaal, Netherlands). Ponceau S staining was used for subsequent quantification. Blots were blocked with 5% Protifar dissolved in Tris-buffered saline/Tween 20 [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.05% Tween-20 (v/v)] for 1 h at room temperature. Blots were incubated with Dendra2 (Origene, Rockville, MD, United States) primary antibody and peroxidase-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, United States). Standard ECL procedure was used for final detection (Amersham Bioscience, Buckinghamshire, United Kingdom). Quantification was done by Image Lab software version 6.1 (Bio-Rad Laboratories, Veenendaal, Netherlands).
HeLa cells were cultured on 10% poly-L-lysine-coated small glass-bottom dishes (Iwaki, Shizuoka, Japan). Cells were transfected with the KIR2.1-Dendra2 construct using Lipofectamine 2000 (Thermo Fisher Scientific, Yokohama, Japan). Twenty-four hours after transfection, the medium of the transfected dishes was replaced by fresh supplemented DMEM. At least 1 h after changing the medium, dishes were treated with various drugs when not used for baseline measurements. In case of baseline measurements, the DMEM of the dishes was changed just before imaging to DMEM without phenol red supplemented with 10% fetal calf serum, 1% penicillin/streptomycin and 25 mM HEPES buffer (Dojin, Kyoto, Japan). In case of measurements of treated cells, dishes were treated 24 h prior to imaging. For treatment the DMEM of the dishes was changed to DMEM without phenol red supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, 25 mM HEPES buffer and either 10 nM Dyngo-4a, 30 μM Nocodazole or 20 μM Cytochalasin B. Imaging was carried out using a Nikon A1R confocal microscope (Nikon, Tokyo, Japan) with a 60x oil immersion objective. The cells were maintained using a stage-top incubator providing a 37°C, 5% CO2, humid environment (TOKAI HIT, Fujinomiya, Japan). Photoconversion of KIR2.1-Dendra2 was carried out on a small region of interest (ROI) by irradiation of a blue laser (405 nm) at 6% power for 1 s. To prevent substantial photobleaching, the specific recommendations of Chudakov et al. (2007) with respect to Dendra2 were applied. Green signal was excited by a 488-nm laser with 1.5% power and emitted light was obtained through a FITC band-path filter (535/45 nm). Red signal was excited by a 562-nm laser with 3% power and fluorescence was recorded through a TRITC band-path filter (605/55 nm). In total, following photoconversion 26 images were taken for each channel. Data were processed using NIS Elements software (Nikon, Tokyo, Japan). Fluorescence data for the red and green signal in each (partially) converted cell was divided by its maximum value to make all cells intercomparable. Values for green and red fluorescence intensity were plotted against time (s) and were fitted by the following double exponential equation.
Then the value τ1 and τ2 for each data series was estimated as time constant for fast and slow component, respectively, to indicate the speed of increase in green fluorescence or decay in red fluorescence. Double exponential fitting was performed using Origin version 8 (Microcal software, Northampton, MA, United States).
HEK-KWGF cells were grown on 0.1% gelatin (Bio-Rad, Veenendaal, Netherlands) coated Ø 12-mm cover slips in a 24-well plate. Cells were randomly divided into two groups; the treatment groups were treated with 20 μM Cytochalasin B for 24 h, while the control group did not undergo any treatment. IKIR2.1 from single cells was recorded in whole-cell voltage clamp mode using an Axopatch 200B amplifier (Axon Instruments, Union City, CA, United States) controlled by Clamp10.4 software (Molecular Devices, LLC., San Jose, CA, United States) at room temperature. Voltage clamp measurements were performed by applying 1s test pulses ranging between −120 and +30 mV, in 10 mV increments. Signals were low-pass filtered at 2 kHz and sampled at 4 kHz from a holding potential of −40 mV, with series resistance compensation of at least 70%. Liquid junction potential (LJP) was determined with the built in “Junction Potential Calculator” application of pCLAMP. Using the current solutions, LJP was 13.8 mV. Steady-state current at the end of the pulse was normalized to cell capacitance and plotted versus test potential (corrected for LJP). Patch pipettes were made with a Sutter P-2000 puller (HEKA Elektronik, Lambrecht, Germany) and had resistances of 2–3 MΩ. Extracellular solution for whole cell IKIR2.1 measurements contained (in mmol/L): NaCl 140, KCl 5.4, CaCl2 1, MgCl2 1, glucose 6, NaHCO3 17.5, HEPES 15, pH 7.4/NaOH. Pipette solution contained potassium gluconate 125, KCl 10, HEPES 5, EGTA 5, MgCl2 2, CaCl2 0.6, Na2ATP 4, pH 7.20/KOH.
Fractionation was performed essentially as described earlier (Van Bergen en Henegouwen et al., 1992). In short, cells were cultured on two 100 mm Ø petri dishes per treatment, until 90% confluency was reached. Then, cells were washed twice with cold PBS. One petri dish was lysed with buffer D, scraped and transferred into a precooled tube. Lysate was spun at 14,000 rpm at 4°C for 5 min (supernatant = whole cell fraction, WC). The second petri dish was lysed with cytoskeleton buffer (10 mM PIPES, 250 mM sucrose, 3 mM MgCl2, 150 mM KCl and 1 mM EGTA) with 1% (v/v) Triton X-100 (CSK-100), scraped and transferred into a precooled tube. Lysate was spun at 14,000 rpm at 4°C for 10 min (supernatant = membrane and cytosol fraction, CM). The pellet was resuspended in CSK-100 and spun at 14,000 rpm at 4°C for 10 min. Supernatant was removed and the pellet was resuspended in RIPA lysis buffer (20 mM Tris, 150 mM NaCl, 10 mM Na2HPO4, 1% (v/v) Triton X-100, 1% (w/v) Na-deoxycholaat, 0.1% (w/v) SDS, 1 mM EDTA and 50 mM NaF) supplemented with 200 mM PMSF and 10 μg/ml aprotinin and spun down at 14,000 rpm at 4°C for 10 min (supernatant = cytoskeleton fraction, CSK).
HEK-KWGF cells were cultured on 15 mm Ø 0.1% gelatin coated coverslips. Cells were washed with PBS++ (PBS supplemented with 1 mM MgCl2 and 1 mM CaCl2) and either stayed in PBS++ or were incubated two times 5 min with cold CSK-100 buffer at room temperature to isolate the cytoskeleton. Cells were then fixed with ice-cold methanol at −20°C for 20 min. Cells were quenched with 50 mM glycine in PBS two times for 10 min, and blocked with NET-gel (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.2% (w/v) gelatin, 0.05% Igepal and 0.02% NaN3) two times for 15 min. Coverslips were incubated overnight with primary antibody (β-actin, Sigma-Aldrich cat. no. A5444) in NET-gel at room temperature. KIR2.1-GFP was imaged using the GFP fluorophore. Secondary antibody (Jackson Laboratories) incubation was done in NET-gel for 120 min at room temperature. Images were made with a Nikon eclipse 80i light microscope with a 60x oil immersion lens and NIS elements software (Nikon Europe B.V., Amsterdam, Netherlands).
Data are expressed as mean ± SD. Differences between group averages were tested using a one-way ANOVA with a post hoc test (Tukey’s HSD), or unpaired T-test and considered significant when the p-value was less than 0.05. Statistical analysis was performed using either GraphPad Prism version 9 (GraphPad software, San Diego, CA, United States) or Origin version 8 (Microcal software, Northampton, MA, United States).
Since interpretation of local subcellular signals obtained by fluorescent microscopy might be hindered by rapid changes in overall expression levels of the fluorescent protein, we first determined expression levels of KIR2.1-Dendra2 in response to Dyngo-4a, Nocodazole and Cytochalasin B for up to 6 h by western blot analysis. In transiently transfected HeLa cells (Figure 1A) and stable CHO-KIR2.1-Dendra2 cells (Figure 1B), Dyngo-4a, Nocodazole or Cytochalasin B did not significantly alter KIR2.1-Dendra2 expression levels for up to 6 h (Figures 1C,D). Therefore, within the intended fluorescent imaging time frame of 1 h (next section), no effects of treatment on overall KIR2.1-Dendra2 expression levels were expected.
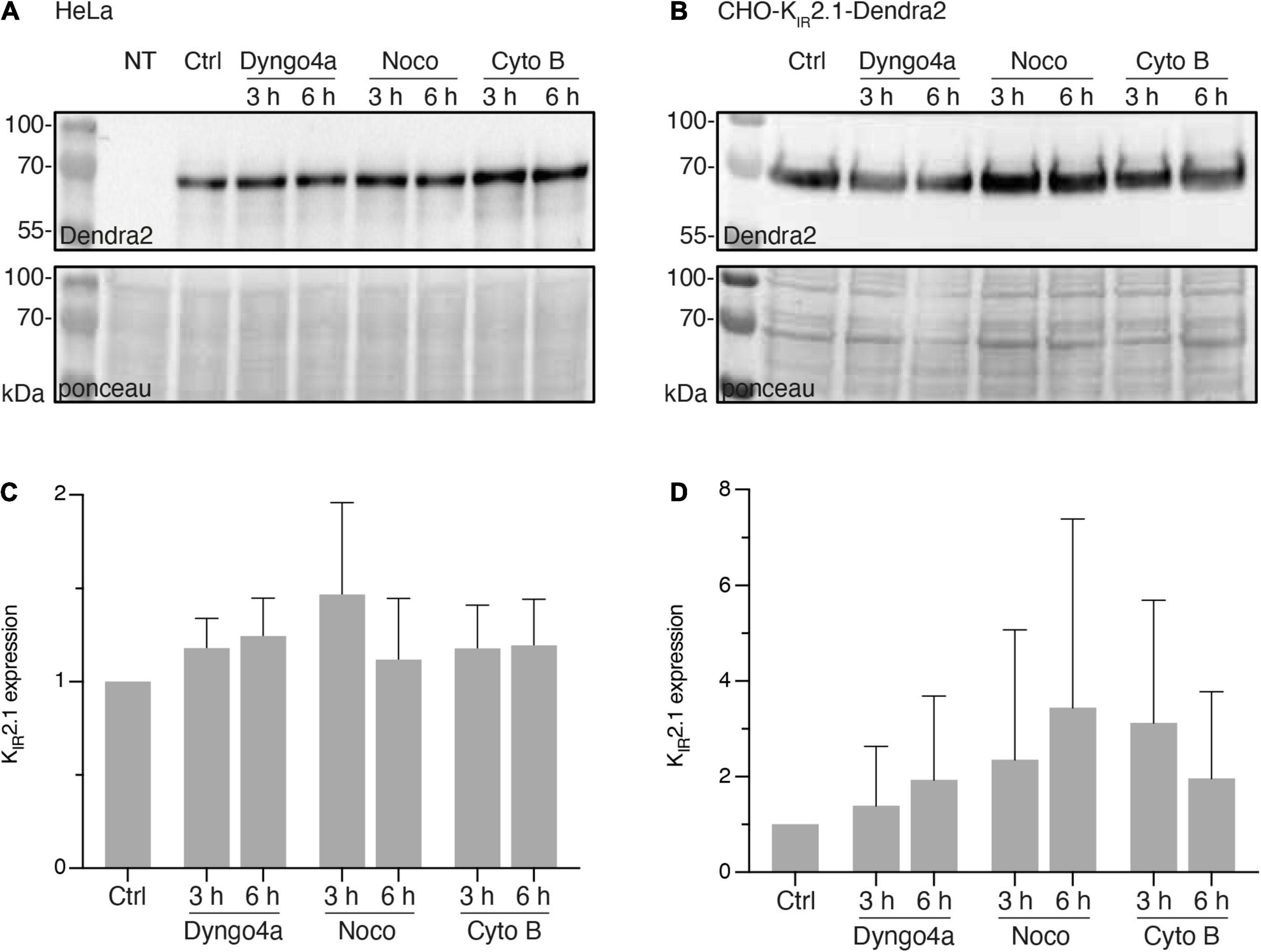
Figure 1. Nocodazole, Cytochalasin B or Dyngo-4a treatment up to 6 h do not affect KIR2.1-Dendra2 protein expression level. (A) Western blot analysis of KIR2.1-Dendra2 expression in HeLa Cells treated for different time period with Dyngo-4a (10 nM), Nocodazole (30 μM) or Cytochalasin B (20 μM) (n = 5). Non-transfected cells (NT) were used as negative control. (B) Western blot analysis of KIR2.1-Dendra2 expression in CHO cells (n = 6). (C) Summarized results of KIR2.1-Dendra2 expression in control and HeLa cells treated for 3 h or 6 h (n = 5). Data are presented as the fold of control. (D) Summarized results of (B). Data are showed as mean ± SD, control protein level was designed as 100% after correction. Ponceau staining was used as a loading control.
Next, HeLa cells were transiently transfected with KIR2.1-Dendra2 to assess forward and backward trafficking of KIR2.1 channel. Upon proper plasma membrane expression, local laser-assisted Dendra2 green-to-red conversion was performed using a confocal microscope. Recovery of the green signal (planar movement and forward trafficking) and decay of the red signal (planar movement and backward trafficking) were assessed for up to 60 min. Figure 2 shows representative time-lapse images of HeLa cells transfected with KIR2.1-Dendra before and after photoconversion. White circles indicate the region irradiated by the blue laser for photoconversion of Dendra2. Green fluorescence decreased and red fluorescence increased at the region of photoconversion just after blue laser irradiation, followed by time-dependent recovery of green fluorescence and reduction of red fluorescence. Although the shape and the size of the cells were affected by each chemical treatment, the trend of fluorescent changes was the same in all groups. Among them, the decay of red fluorescence appeared to be slower in the cells treated with Dyngo-4a and the recovery of green fluorescence was slower in the cells treated with Nocodazole and Cytochalasin B. Summarized data of green and red fluorescent intensity also revealed that Nocodazole and Cytochalasin B slowed down the recovery of the green signal (blue and green triangles, respectively, in Figure 3A), while Dyngo-4a prolonged the decay of the red signal (red circles in Figure 3B). The maximum recovery rate of green fluorescence at 1 h after photoconversion seemed to be reduced by Nocodazole and Cytochalasin B, especially the treatment with Cytochalasin B resulted in significant decrease in the maximum recovery rate by approximately 40% (inset Figure 3A). Recovery and decay of each fluorescence by time could be best described with a double exponential fitting resulting in two time constants, fast (τ1) and slow (τ2) component, respectively. In non-treated cells, green and red signals displayed a fast time constant (1.4 ± 1.3 min n = 14 and 2.8 ± 2.3 min n = 12, respectively) presumed as being planar movements. No significant changes in the fast components were observed with Dyngo-4a, Nocodazole or Cytochalasin B treatment (Figures 3C,D). In non-treated cells, τ2 for green recovery and red decay were approximately 8–10 times higher (13.1 ± 7.7 min, n = 14 and 20.2 ± 6.7 min, n = 12, respectively) than the fast component, and presumably reflect forward and backward trafficking of KIR2.1-Dendra2 channels, respectively. Slow recovery of green signal, forward trafficking, was strongly affected by Nocodazole (160.0 ± 177 min, P < 0.05, n = 20, 12-fold) and Cytochalasin B (131.5 ± 119 min, P < 0.05 n = 15, 10-fold) (Figure 3E). Dyngo-4a significantly increased τ2 for the red decay, i.e., slowing backward trafficking, (86.7 ± 56 min, P < 0.05, n = 15) (Figure 3F). Apparent increases in τ2 for red decay by Nocodazole (60.1 ± 72.4 min, n = 20) and Cytochalasin B (64.3 ± 81.2 min, n = 17) did not reach statistical significance. As impaired forward trafficking might result in intracellular KIR2.1-Dendra2 accumulation, we assessed vesicular accumulation (<10 μm structures) and massive accumulation (intracellular structures >10 μm). Indeed, disruption of the cytoskeleton by Nocodazole or Cytochalasin B increased the ratio of cells displaying intracellular accumulations (Supplementary Figure 1).

Figure 2. Time-lapse confocal microscopic images of HeLa cells transfected with KIR2.1-Dendra2 before and after photoconversion. The cells without any treatment (A) and the cells treated with Dyngo-4a (B), Nocodazole (C), Cytochalasin B (D) were imaged. The green fluorescence shows the localization of KIR2.1 channel before photoconversion and the red fluorescence shows the localization of KIR2.1 channel after photoconversion. White circle in each figure indicates the region where a blue laser was irradiated for photoconversion of Dendra2. Scale bar in each images indicate 10 μm.
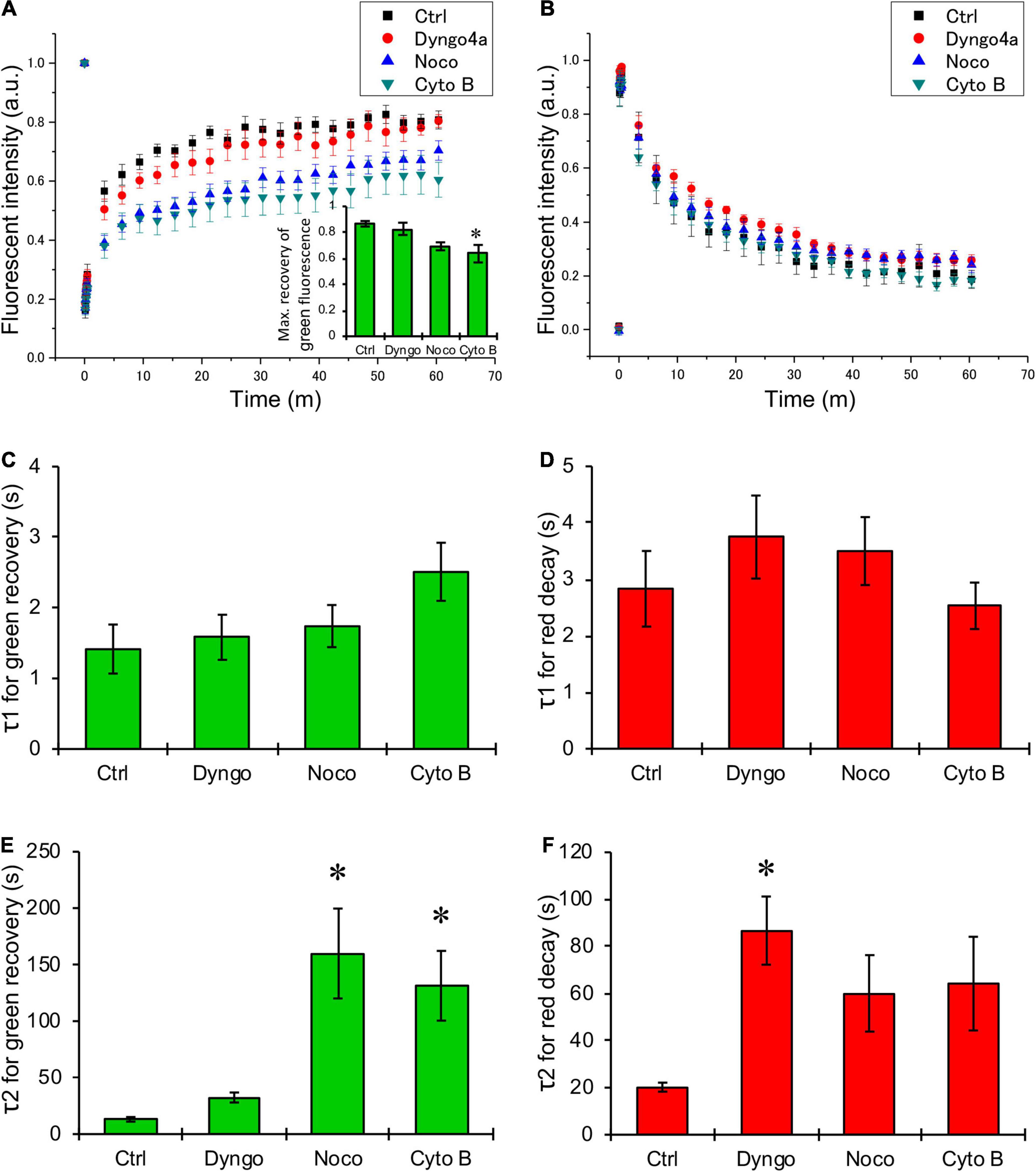
Figure 3. Quantification of changes in green and red fluorescence of KIR2.1-Dendra2. (A,B) Summarized data of time-dependent recovery in green fluorescence (A) and decay of red fluorescence (B). Black boxes are for control (n = 14), and red circles, blue triangles, green triangles are for the data from the cells treated with Dyngo-4a (n = 15), Nocodazole (n = 20), Cytochalasin B (n = 17), respectively. (C,D) Summarized data of τ1 (C) and τ2 (D), fast and slow component of time constant for the recovery of green fluorescence after photoconversion, respectively. (E,F) Summarized data of τ1 (E) and τ2 (F), fast and slow component of time constant for the decay of red fluorescence after photoconversion, respectively. (A, inset) The maximum recovery rate of green fluorescence after photoconversion. *: p < 0.05 vs. control (one-way ANOVA).
Previous and the above results demonstrate that Cytochalasin B and Nocodazole (Loewen et al., 2009) strongly inhibit KIR2.1 forward trafficking over backward trafficking, which would result in decreased IKIR2.1. Therefore, IKIR2.1 current generated by KIR2.1 channels in HEK-KWGF cells was determined following 24 h application of Cytochalasin B by whole-cell patch clamp. It was found that both the inward (from −120 to −90 mV) and outward (−70 to + 30 mV) IKIR2.1 components were significantly decreased when compared with non-treated HEK-KWGF cells (Figure 4). Similar results were found previously for Nocodazole (Loewen et al., 2009).
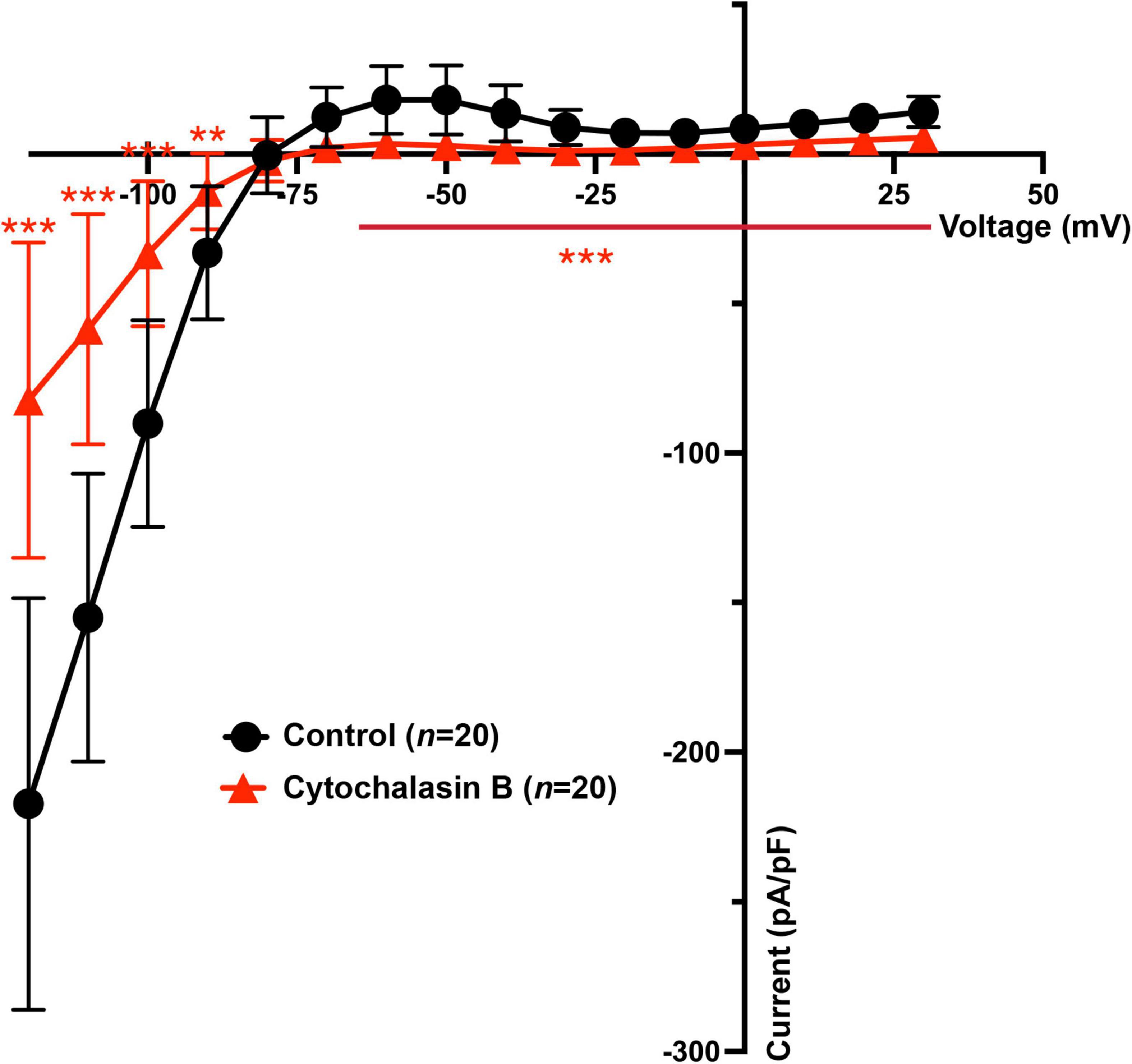
Figure 4. Cytochalasin B treatment decreases both inward and outward IKIR2.1 in HEK-KWGF cells. Current-voltage relationship of mean IKIR2.1 current values ± SD. In the treatment group, cells were treated with Cytochalasin B for 24 h with a final concentration of 20 μM. The control group did not undergo any treatment. n = 20 cells for each group. **p < 0.01, ***p < 0.001.
We previously observed that CQ treatment resulted in increased KIR2.1 expression levels in HEK293 cells (Jansen et al., 2008; Varkevisser et al., 2013). In view of the recent work of Kang et al. (2020) that concluded that CQ affects cytoskeleton stability in podocytes, we assessed a potential effect of CQ on KIR2.1 interaction with the cytoskeleton. Therefore, HEK-KWGF cells were treated with 10 μM CQ for 24 h. Subsequently, whole cell (WC), cytoplasm + membrane (CM) and cytoskeleton (CSK) fractions were prepared and analyzed by western blot (Figure 5A). CQ treatment appeared to increase KIR2.1-GFP expression levels in all fractions, however, the strongest increase was found in the cytoskeleton fraction (2.6 ± 0.9 fold, 2.2 ± 0.5 and 103 ± 90 fold for WC, CM and CSK fractions, respectively) (Figure 5B). The fractionation procedure, at 4°C, resulted in isolation of cytoskeletons consisting mainly of actin and intermediate filaments, but no tubulin (Figure 5A). Upon CQ, a KIR2.1-GFP degradation product that results from N-terminal cleavage in the KIR2.1 protein (Varkevisser et al., 2013), became apparent in all three fractions (Figure 5A, marked with *). This cleaved product associated with the CSK as well, even reaching higher cleaved/full length ratios than in WC and CM fractions, which is indicative for a certain amount of enrichment (Figure 5C).
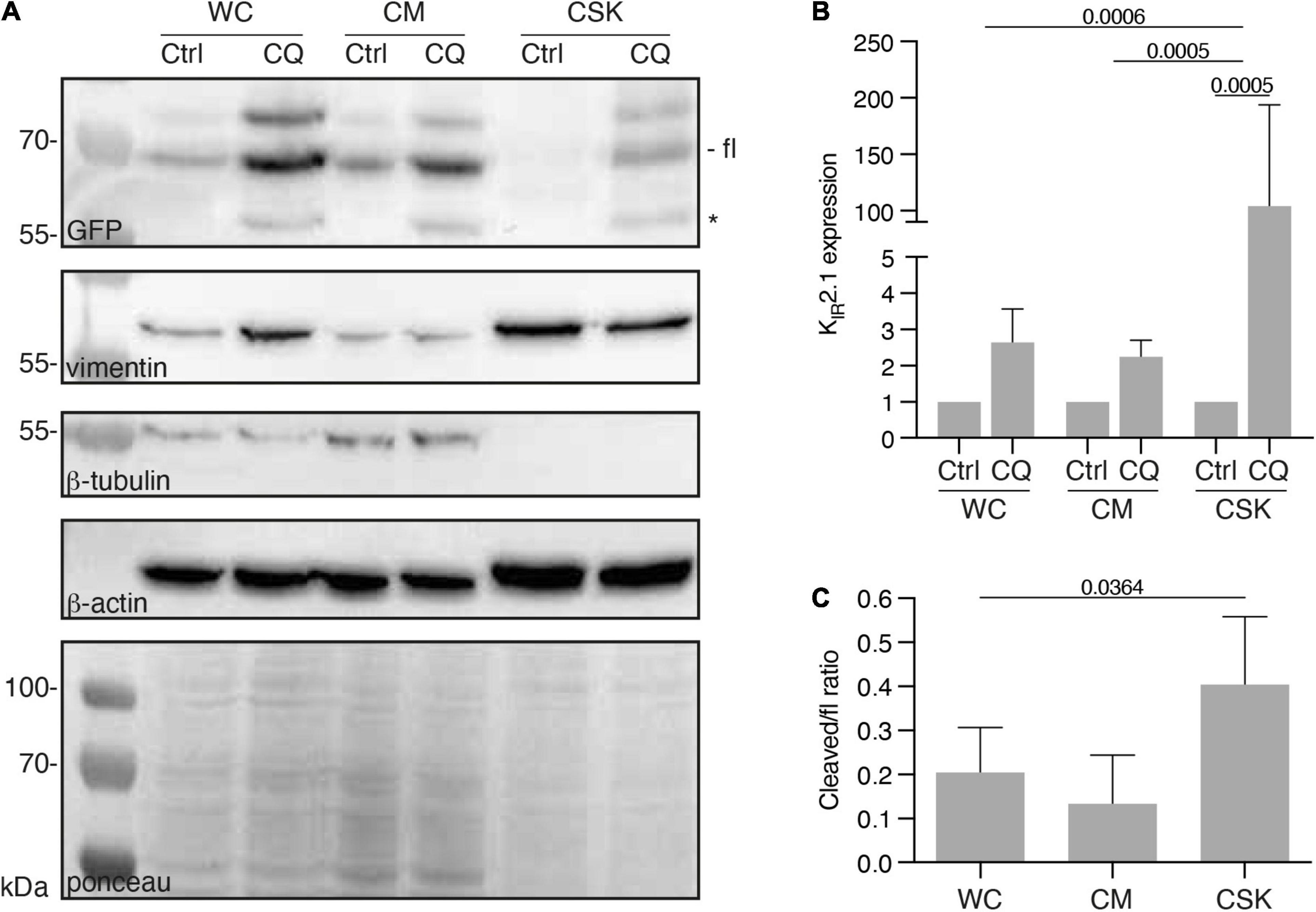
Figure 5. Presence of KIR2.1-GFP in the cytoskeleton containing cell fraction, induced by Chloroquine treatment. (A) Western blot analysis of whole cell lysates (WC), cytosol and membrane fractions (CM) and cytoskeleton fractions (CSK) of non-treated (NT) and 10 μM Chloroquine (CQ) (24 h) treated HEK-KWGF cells. All three cytoskeleton components were included as well as a ponceau staining, which was used as loading control. Cleaved KIR2.1-GFP, indicated with an asterisk, is only present after CQ treatment. Fl, full-length KIR2.1-GFP. (B) Quantified results of the CQ induced induction of KIR2.1-GFP expression (n = 6). One-way ANOVA was used to calculate significance. (C) Quantified results of cleaved/fl KIR2.1-GFP ratios in all cell fractions (n = 6). One-way ANOVA was used to calculate significance.
Since the KIR2.1-GFP degradation product is not found at the plasma membrane (Varkevisser et al., 2013), our findings suggest interaction of non-plasma membrane associated KIR2.1-GFP with the cytoskeleton following CQ treatment. Fluorescent microscopy on whole cells and isolated cytoskeletons (Figure 6) demonstrates intracellular accumulation of KIR2.1-GFP upon CQ treatment (Figures 6A,B) as shown before (Jansen et al., 2008; Varkevisser et al., 2013). Indeed, these intracellular KIR2.1-GFP loaded structures are strongly associated with isolated cytoskeleton (Figure 6D). Close inspection demonstrates that KIR2.1-GFP signals in at least some of the intracellular vesicles are surrounded by and/or in close vicinity of actin, while all vesicles are in subcellular areas with dense actin signals (Figure 6D, inset).
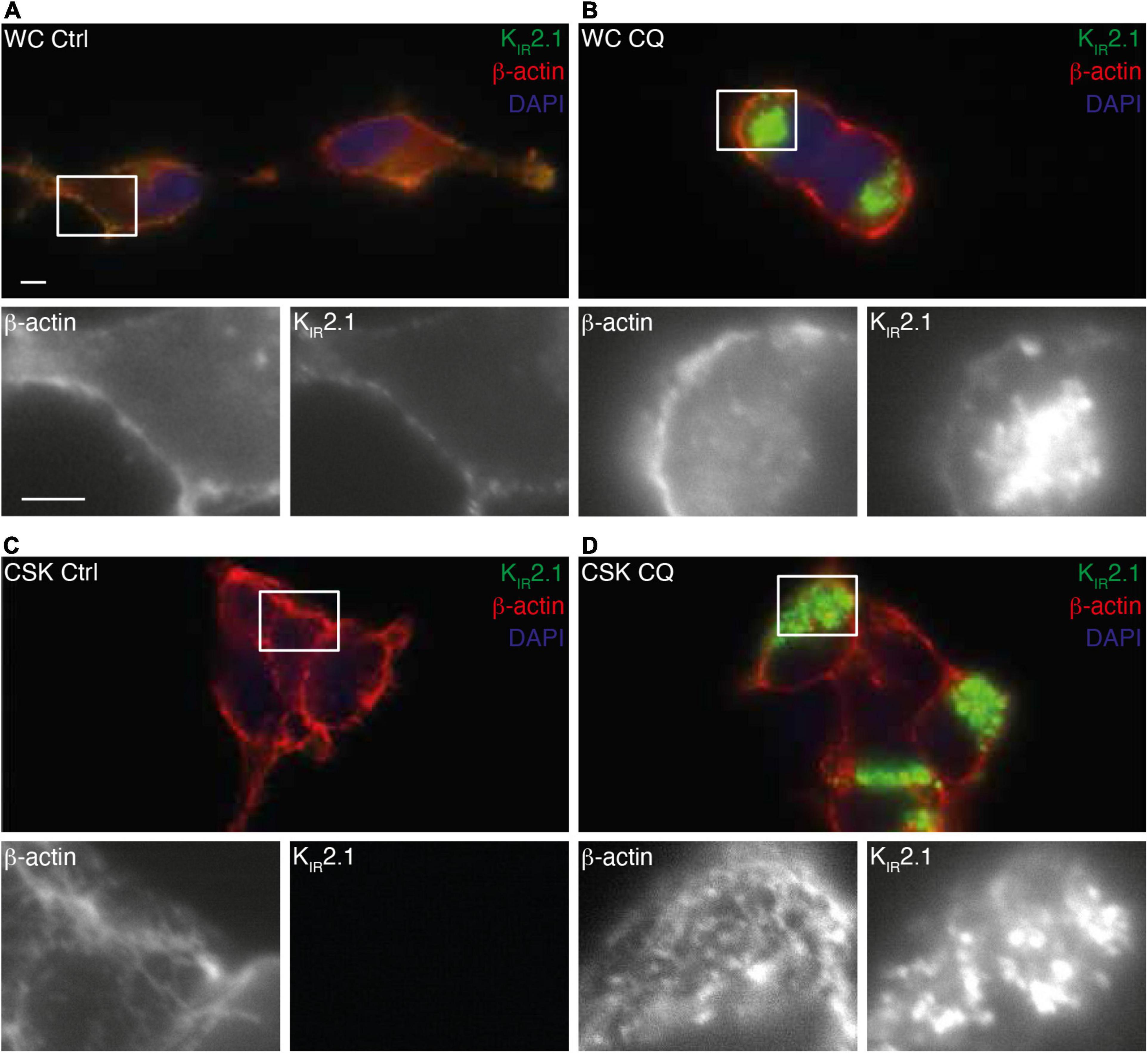
Figure 6. Interaction of KIR2.1-GFP with the cell cytoskeleton following Chloroquine treatment. Co-staining of KIR2.1-GFP (green) and β-actin in non-treated whole cell (WC) (A,B) or cytoskeleton isolated (CSK) (C,D) HEK-KWGF cells. Cells were either non-treated (Ctrl) (A,C) or treated with 10 μM CQ for 24 h (B,D). Single staining patterns of the boxed areas are shown below each picture. Scale bar represents 5 μm.
Using a transient transfection system of the photoconvertible fluorescent protein Dendra2, we were able to optically quantify the differences in the dynamics of “old” and “new” proteins on the plasma membrane based on their red and green fluorescence intensity, respectively. Furthermore, we showed how disruption of the cytoskeleton slowed down forward and backward trafficking of the KIR2.1 channel protein. The intensity of green and red fluorescence was fitted by a double exponential function to separate the fast (τ1) and slow (τ2) component of fluorescent changes by time. The fast component of green recovery and red decay did not change significantly by the treatment with either drug, suggesting that τ1 for the fluorescence changes was due to the planar movement of the Dendra2-tagged proteins along the plasma membrane and therefore out of the ROI for fluorescent measurement. In control cells, we observed that the slow time constants for forward trafficking and backward trafficking were in the same order of magnitude (13.1 vs. 20.2 min, respectively), as could be expected under conditions of homeostasis at 48 h after transfection. Cytoskeletal modification affected the slow component of forward and backward trafficking of KIR2.1 channels. The τ2 for green fluorescence recovery prolonged significantly through Nocodazole and Cytochalasin B, indicating that the forward trafficking of KIR2.1 channels strongly depends on the tubulin and actin network. Although the differences were not statistically significant, the τ2 for red fluorescent decay in the cells treated with Nocodazole and Cytochalasin B was also prolonged compared to control cells. Loewen et al. (2009) speculated that KIR2.1 forward trafficking was particularly sensitive for Nocodazole-mediated tubulin disruption in contrast to some other cardiac potassium channels. Indeed, our time constant for forward trafficking was more strongly affected by Nocodazole compared to backward trafficking (approximately 12 vs. 3-fold), consistent with a net decrease in KIR2.1 ion channels at the plasma membrane. Furthermore, disruption of the actin skeleton also affected forward trafficking more potently than backward trafficking (approximately 10 vs. 3-fold). Consequently, we show that 24 h Cytochalasin B treatment decreased functional KIR2.1 channels at the plasma membrane as was evident from a decrease in IKIR2.1. However, we cannot exclude that channel activation, rather than expression, also contributes to a decrease in IKIR2.1. For example, Cytochalasin D has been shown to affect enzymes in the phospatidyl-4,5-biphosphate (PIP2) metabolic pathways (Vasilev et al., 2021). PIP2 is an important factor for KIR2.1 activity (Huang et al., 1998). Inhibition of dynamin motor proteins, as performed here through treatment with Dyngo-4a, was more potent on backward than on forward trafficking (approx. 4.3 vs. 2.5-fold). This latter finding is consistent with earlier work from Loewen et al. (2009) and Varkevisser et al. (2013) who used other molecular and pharmacological tools for dynamin inhibition, resulting in an increased ectopic KIR2.1 current in HEK293 cells.
The cytoskeleton contributes not only to the transport of membrane proteins but also to many other cellular biological processes including maintenance of cell shape, cell migration, cell division, endocytosis, and cell polarization (e.g., Pollard and Cooper, 2009; Kopf et al., 2020). Therefore, when using pharmacological approaches for cytoskeletal modulation as in our study, it should be considered that various cell biological processes are also affected by the drug mediated cytoskeleton modifications, as this altered cell biology may have indirect effects on ion channel trafficking too.
Obviously, KIR2.1 trafficking depends on many other interacting proteins, of which a number are known already like clathrin and AP1 (e.g., Tong et al., 2001; Ma et al., 2011). Furthermore, channel complexes, like KIR2.1 and Nav1.5 may actively cooperate to regulate their trafficking (Ponce-Balbuena et al., 2018). It can be assumed that the cytoskeleton may also have a role in steering these interacting proteins in the processes of KIR2.1 trafficking.
In earlier studies we demonstrated that (1) CQ induces dose- and time-dependent increased expression levels of non-tagged or tagged KIR2.1 in four different cell lines, which was accompanied with intracellular accumulation of full-length and KIR2.1 degradation products in lysosomes, (2) that CQ could counteract drug-induced defects in KIR2.1 expression, and (3) that CQ induced increased KIR2.1 dependent inward rectifier current (Jansen et al., 2008; Nalos et al., 2011; Varkevisser et al., 2013; Ji et al., 2017; Korte et al., 2017). However, a link to the cytoskeleton was not made earlier, which led us to perform cell fractionation assays and KIR2.1 imaging on isolated cytoskeletons. Our cell fractionation method was based on established protocols in which 0.5% non-ionic detergent Triton X-100 was combined with tubulin depolarization at low temperature. This concentration of Triton X-100 extracts all non-cytoskeleton bound proteins and phospholipids from the cytoskeleton (Van Bergen en Henegouwen et al., 1989), whereas extraction at low temperature (4°C) (Van Bergen en Henegouwen et al., 1992) yields a cytoskeleton fraction nearly devoid of tubulin, as shown here also in Figure 5A. Therefore, KIR2.1 interaction with the cytoskeleton shown here most likely represents KIR2.1 interaction with the actin and/or intermediate filament system. Given the proven interaction of KIR2.1 with actin binding proteins (Sampson et al., 2003), we may speculate that KIR2.1-actin interaction is dominant in the isolated CSK fraction. Besides KIR2.1 trafficking being dependent on a functional actin network, as shown here, it was demonstrated recently that membrane-localized functional KIR2.1 channels modulate actin filament dynamics (Wu et al., 2020). These authors suggest a role for KIR2.1 channels on cellular morphology, migration and adhesion, via the actin filament system.
The interaction of lysosomal localized KIR2.1 channel proteins with the actin/intermediate filament system must be considered in view of the crucial role of these cytoskeletal systems in late-endosome and lysosome organelle transport within the cell (Styers et al., 2004; Bonifacino and Neefjes, 2017), and therefore it may not be surprising that the late-endosome/lysosome cargo also interacts with these cytoskeletal elements, especially when the cargo itself is an actin binding protein such as KIR2.1. Since tubulin filaments depolarize at low temperatures as used in our fractionation and immunofluorescent microscopy assays (Van Bergen en Henegouwen et al., 1992), we cannot rule out that late-endosomal/lysosomal localized KIR2.1 channels also associate with tubulin filaments in intact cells.
In the failing heart, cardiomyocytes have less KIR2.1 expression compared to healthy counterparts (Borlak and Thum, 2003), whereas KIR2.1 expression is increased in cardiac fibroblasts in profibrillatory atrial tissue (Qi et al., 2015). Although changes in KIR2.1 channels and IK1 in failing hearts have been explained by changes in mRNA and channel proteins so far, this study emphasizes the need to consider the membrane transport of channels via the cytoskeleton too. It has been reported that cytoskeletal remodeling occurs in the failing hearts. In particular, tubulin, a component of microtubules, has been found to display an increased expression and excessive polymerization in pressure-loaded animal models (Stones et al., 2012) and human failing heart muscle in dilated cardiomyopathy (Heling et al., 2000). Recent studies have demonstrated that detyrosination, which inhibits excessive polymerization of tubulin, might improve contractile and diastolic function of cardiomyocytes in the failing myocardium (Chen et al., 2018). Microtubule-associated proteins (MAPs), such as tau, MAP4, and EB-1, are essential for the transport of membrane proteins, and the balance between microtubule polymerization and MAP binding determines membrane transport. The KIR2.1 protein interacts with actin through filamin-A, an actin-binding protein, but how KIR2.1 might interact with tubulin filaments is not yet known. Further studies are needed to understand how microtubule remodeling in the failing heart affects membrane trafficking of channel proteins, and live imaging using the Dendra2 protein used in this study might be useful.
Our work comes with several limitations. We studied the role of the cytoskeleton in KIR2.1-Dendra2 trafficking in human cell lines, rather than in adult ventricular human cardiomyocytes, for several practical and scientific reasons. Adult human ventricular cardiomyocytes are scarcely available for basic research. Furthermore, our approach requires cells to become transfected with KIR2.1-Dendra2 and treated with drugs for considerable timeframes, which would require culturing of the isolated cells. However, this would result in dedifferentiation of the rod-shaped cells (e.g., Bugaisky and Zak, 1989; De Boer et al., 2006a), most likely affecting also the cytoskeleton structure, and thus would still not mimic human adult ventricular cardiomyocytes completely. We considered human (induced pluripotent) stem cell derived cardiomyocytes, but this approach would suffer from their immature character (e.g., absence of an anisotropic phenotype, absence of T-tubuli, spontaneous active action potential formation, depolarized membrane potential, immature calcium handling). This limitation could be largely overcome by generating KIR2.1-Dendra2 transgenic animal models and isolating the adult ventricular cardiomyocytes. Of particular interest would be crossings of such a transgenic model with animal models of cardiac disease, or interventions resulting in for instance in heart failure, like ischemia/reperfusion or chronic AV block. Therefore, our technologies and findings may be an important first step toward better understanding of the cytoskeleton’s role in KIR2.1 trafficking in the healthy and diseased heart.
TIRF microscopy is currently the best tool to separate fluorescent information from the plasma membrane from intracellular fluorescence. However, the combination of the available TIRF setup with the technical challenging and time-restricted processes of photoconversion and dual-channel live imaging prevented the use of this technology. We decided to perform imaging by better established and validated confocal laser-scanning microscopy instead. By using the flat HeLa cells, rather than more rounded HEK293 cells for imaging, we were able to photoconvert only the proteins on the cell membrane as far as possible.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
EL, VL, WH, WK, and HT performed research. MH, EL, VL, WH, WK, and HT analysed the results. MH and HT designed the study and wrote the manuscript. All authors reviewed and approved the final manuscript.
EL was supported by the Chinese Scholarship Council. VL was supported by the Van Wijck-Stam Caspersfonds. HT was supported by JSPS KAKENHI grant numbers JP16H05238 and JP19H04038, Yokoyama Foundation for Clinical Pharmacology, and SENSHIN Medical Research Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Kosaku Ota and Kazuki Horikawa in Tokushima Bioimaging Station for their technical instructions on microscopic operation.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.812572/full#supplementary-material
Biliczki, P., Virág, L., Iost, N., Papp, J. G., and Varró, A. (2002). Interaction of different potassium channels in cardiac repolarization in dog ventricular preparations: role of repolarization reserve. Br. J. Pharmacol. 137, 361–368. doi: 10.1038/sj.bjp.0704881
Bonifacino, J. S., and Neefjes, J. (2017). Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 47, 1–8. doi: 10.1016/j.ceb.2017.01.008
Borlak, J., and Thum, T. (2003). Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. 17, 1592–1608. doi: 10.1096/fj.02-0889com
Bugaisky, L. B., and Zak, R. (1989). Differentiation of adult rat cardiac myocytes in cell culture. Circ. Res. 64, 493–500. doi: 10.1161/01.res.64.3.493
Chen, C. Y., Caporizzo, M. A., Bedi, K., Vite, A., Bogush, A. I., Robison, P., et al. (2018). Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat. Med. 24, 1225–1233. doi: 10.1038/s41591-018-0046-2
Chudakov, D. M., Lukyanov, S., and Lukyanov, K. A. (2007). Using photoactivatable fluorescent protein Dendra2 to track protein movement. Biotechniques 42:553. doi: 10.2144/000112470
De Boer, T. P., Houtman, M. J., Compier, M., and van der Heyden, M. A. (2010). The mammalian K(IR)2.x inward rectifier ion channel family: expression pattern and pathophysiology. Acta Physiol (Oxf). 199, 243–256. doi: 10.1111/j.1748-1716.2010.02108.x
De Boer, T. P., van Veen, T. A., Houtman, M. J., Jansen, J. A., van Amersfoorth, S. C., Doevendans, P. A., et al. (2006b). Inhibition of cardiomyocyte automaticity by electrotonic application of inward rectifier current from Kir2.1 expressing cells. Med. Biol. Eng. Comput. 44, 537–542. doi: 10.1007/s11517-006-0059-8
De Boer, T. P., van der Heyden, M. A., Rook, M. B., Wilders, R., Broekstra, R., Kok, B., et al. (2006a). Pro-arrhythmogenic potential of immature cardiomyocytes is triggered by low coupling and cluster size. Cardiovasc. Res. 71, 704–714. doi: 10.1016/j.cardiores.2006.06.001
De Brabander, M. J., Van de Veire, R. M., Aerts, F. E., Borgers, M., and Janssen, P. A. (1976). The effects of methyl (5-(2-thienylcarbonyl)-1H-benzimidazol-2-yl) carbamate, (R 17934; NSC 238159), a new synthetic antitumoral drug interfering with microtubules, on mammalian cells cultured in vitro. Cancer Res. 36, 905–916.
Deo, M., Ruan, Y., Pandit, S. V., Shah, K., Berenfeld, O., Blaufox, A., et al. (2013). KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc. Natl. Acad. Sci. U. S. A. 110, 4291–4296. doi: 10.1073/pnas.1218154110
Hattori, T., Makiyama, T., Akao, M., Ehara, E., Ohno, S., Iguchi, M., et al. (2012). A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc. Res. 93, 666–673. doi: 10.1093/cvr/cvr329
Heling, A., Zimmermann, R., Kostin, S., Maeno, Y., Hein, S., Devaux, B., et al. (2000). Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ. Res. 86, 846–853. doi: 10.1161/01.RES.86.8.846
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Hofherr, A., Fakler, B., and Klöcker, N. (2005). Selective Golgi export of Kir2.1 controls the stoichiometry of functional Kir2.x channel heteromers. J. Cell Sci. 118, 1935–1943. doi: 10.1242/jcs.02322
Houtman, M. J., Korte, S. M., Ji, Y., Kok, B., Vos, M. A., Stary-Weinzinger, A., et al. (2014). Insights in KIR2.1 channel structure and function by an evolutionary approach; cloning and functional characterization of the first reptilian inward rectifier channel KIR2.1, derived from the California kingsnake (Lampropeltis getula californiae). Biochem. Biophys. Res. Commun. 452, 992–997. doi: 10.1016/j.bbrc.2014.09.031
Huang, C. L., Feng, S., and Hilgemann, D. W. (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391, 803–806. doi: 10.1038/35882
Jansen, J. A., de Boer, T. P., Wolswinkel, R., van Veen, T. A., Vos, M. A., van Rijen, H. V., et al. (2008). Lysosome mediated Kir2.1 breakdown directly influences inward rectifier current density. Biochem. Biophys. Res. Commun. 367, 687–692. doi: 10.1016/j.bbrc.2007.12.168
Ji, Y., Veldhuis, M. G., Zandvoort, J., Romunde, F. L., Houtman, M. J. C., Duran, K., et al. (2017). PA-6 inhibits inward rectifier currents carried by V93I and D172N gain-of-function KIR2.1 channels, but increases channel protein expression. J. Biomed. Sci. 24:44. doi: 10.1186/s12929-017-0352-x
Kang, Y., Law, H. K., Zhang, Y., Wu, Y., Zhu, G. H., Saleem, M. A., et al. (2020). TMT-based proteomic analysis reveals the effects of chloroquine on human podocytes. Am. J. Transl. Res. 12, 4290–4301.
Kharche, S., Garratt, C. J., Boyett, M. R., Inada, S., Holden, A. V., Hancox, J. C., et al. (2008). Atrial proarrhythmia due to increased inward rectifier current (I(K1)) arising from KCNJ2 mutation–a simulation study. Prog. Biophys. Mol. Biol. 98, 186–197. doi: 10.1016/j.pbiomolbio.2008.10.010
Kopf, A., Renkawitz, J., Hauschild, R., Girkontaite, I., Tedford, K., Merrin, J., et al. (2020). Microtubules control cellular shape and coherence in amoeboid migrating cells. J. Cell Biol. 219:e201907154. doi: 10.1083/jcb.201907154
Korte, S. M., Houtman, M. J. C., Meeuwsen, J. A. L., Kercher, I., Zandvoort, J., Vos, M. A., et al. (2017). “KIR2.1-Dendra2: a new tool to study drug-induced ion channel trafficking defects,” in Adaptation Biology and Medicine, eds Y. Kawai, A. R. Hargens, and P. K. Singal (New Delhi: Narosa), 361–372.
Leonoudakis, D., Conti, L. R., Anderson, S., Radeke, C. M., McGuire, L. M., Adams, M. E., et al. (2004a). Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J. Biol. Chem. 279, 22331–22346. doi: 10.1074/jbc.M400285200
Leonoudakis, D., Conti, L. R., Radeke, C. M., McGuire, L. M., and Vandenberg, C. A. (2004b). A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J. Biol. Chem. 279, 19051–19063. doi: 10.1074/jbc.M400284200
Li, X., Ortega, B., Kim, B., and Welling, P. A. A. (2016). Common signal patch drives AP-1 protein-dependent golgi export of inwardly rectifying potassium channels. J. Biol. Chem. 291, 14963–14972. doi: 10.1074/jbc.M116.729822
Loewen, M. E., Wang, Z., Eldstrom, J., Dehghani Zadeh, A., Khurana, A., Steele, D. F., et al. (2009). Shared requirement for dynein function and intact microtubule cytoskeleton for normal surface expression of cardiac potassium channels. Am. J. Physiol. Heart Circ. Physiol. 296, H71–H83. doi: 10.1152/ajpheart.00260.2008
Ma, D., Taneja, T. K., Hagen, B. M., Kim, B. Y., Ortega, B., Lederer, W. J., et al. (2011). Golgi export of the Kir2.1 channel is driven by a trafficking signal located within its tertiary structure. Cell 145, 1102–1115. doi: 10.1016/j.cell.2011.06.007
MacLean-Fletcher, S., and Pollard, T. D. (1980). Mechanism of action of cytochalasin B on actin. Cell 20, 329–341. doi: 10.1016/0092-8674(80)90619-4
Mostowy, S., and Cossart, P. (2012). Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 13, 183–194. doi: 10.1038/nrm3284
Myles, R. C., Wang, L., Bers, D. M., and Ripplinger, C. M. (2015). Decreased inward rectifying K+ current and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J. Physiol. 593, 1479–1493. doi: 10.1113/jphysiol.2014.279638
Nalos, L., de Boer, T. P., Houtman, M. J., Rook, M. B., Vos, M. A., and van der Heyden, M. A. G. (2011). Inhibition of lysosomal degradation rescues pentamidine-mediated decreases of K(IR)2.1 ion channel expression but not that of K(v)11.1. Eur. J. Pharmacol. 652, 96–103. doi: 10.1016/j.ejphar.2010.10.093
Ohkuma, S., and Poole, B. (1978). Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. U. S. A. 75, 3327–3331. doi: 10.1073/pnas.75.7.3327
Pollard, T. A., and Cooper, J. A. (2009). Actin, a central player in cell shape and movement. Science 326, 1208–1212. doi: 10.1126/science.1175862
Ponce-Balbuena, D., Guerrero-Serna, G., Valdivia, C. R., Caballero, R., Diez-Guerra, F. J., Jiménez-Vázquez, E. N., et al. (2018). Cardiac Kir2.1 and Na(V)1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 122, 1501–1516. doi: 10.1161/CIRCRESAHA.117.311872
Ponsford, A. H., Ryan, T. A., Raimondi, A., Cocucci, E., Wycislo, S. A., Fröhlich, F., et al. (2021). Live imaging of intra-lysosome pH in cell lines and primary neuronal culture using a novel genetically encoded biosensor. Autophagy 17, 1500–1518. doi: 10.1080/15548627.2020.1771858
Qi, X. Y., Huang, H., Ordog, B., Luo, X., Naud, P., Sun, Y., et al. (2015). Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial remodeling. Circ. Res. 116, 836–845. doi: 10.1161/CIRCRESAHA.116.305326
Reilly, L., Alvarado, F. J., Lang, D., Abozeid, S., Van Ert, H., Spellman, C., et al. (2020). Genetic loss of IK1 causes adrenergic-induced phase 3 early afterdepolariz ations and polymorphic and bidirectional ventricular tachycardia. Circ. Arrhythm Electrophysiol. 13:e008638. doi: 10.1161/CIRCEP.120.008638
Sampson, L. J., Leyland, M. L., and Dart, C. (2003). Direct interaction between the actin-binding protein filamin-A and the inwardly rectifying potassium channel, Kir2.1. J Biol Chem. 278, 41988–41997. doi: 10.1074/jbc.M307479200
Steele, D. F., and Fedida, D. (2014). Cytoskeletal roles in cardiac ion channel expression. Biochim. Biophys. Acta. 1838, 665–673. doi: 10.1016/j.bbamem.2013.05.001
Stockklausner, C., and Klocker, N. (2003). Surface expression of inward rectifier potassium channels is controlled by selective Golgi export. J. Biol. Chem. 278, 17000–17005. doi: 10.1074/jbc.M212243200
Stockklausner, C., Ludwig, J., Ruppersberg, J. P., and Klöcker, N. (2001). A sequence motif responsible for ER export and surface expression of Kir2.0 inward rectifier K(+) channels. FEBS Lett. 493, 129–133. doi: 10.1016/s0014-5793(01)02286-4
Stones, R., Benoist, D., Peckham, M., and White, E. (2012). Microtubule proliferation in right ventricular myocytes of rats with monocrotaline-induced pulmonary hypertension. J. Mol. Cell Cardiol. 56, 91–96. doi: 10.1016/j.yjmcc.2012.12.010
Styers, M. L., Salazar, G., Love, R., Peden, A. A., Kowalczyk, A. P., and Faundez, V. (2004). The endo-lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol. Biol. Cell 15, 5369–5382. doi: 10.1091/mbc.e04-03-0272
Takanari, H., Nalos, L., Stary-Weinzinger, A., de Git, K. C., Varkevisser, R., Linder, T., et al. (2013). Efficient and specific cardiac IK1 inhibition by a new pentamidine analogue. Cardiovasc. Res. 99, 203–14. doi: 10.1093/cvr/cvt103
Tong, Y., Brandt, G. S., Li, M., Shapovalov, G., Slimko, E., Karschin, A., et al. (2001). Tyrosine decaging leads to substantial membrane trafficking during modulation of an inward rectifier potassium channel. J. Gen. Physiol. 117, 103–118. doi: 10.1085/jgp.117.2.103
Tristani-Firouzi, M., Jensen, J. L., Donaldson, M. R., Sansone, V., Meola, G., Hahn, A., et al. (2002). Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J. Clin. Invest. 110, 381–388. doi: 10.1172/JCI15183
Van Bergen en Henegouwen, P. M., Defize, L. H., de Kroon, J., van Damme, H., Verkleij, A. J., and Boonstra, J. (1989). Ligand-induced association of epidermal growth factor receptor to the cytoskeleton of A431 cells. J. Cell Biochem. 39, 455–465. doi: 10.1002/jcb.240390411
Van Bergen en Henegouwen, P. M., den Hartigh, J. C., Romeyn, P., Verkleij, A. J., and Boonstra, J. (1992). The epidermal growth factor receptor is associated with actin filaments. Exp. Cell Res. 199, 90–97. doi: 10.1016/0014-4827(92)90465-k
Van der Heyden, M. A., and Jespersen, T. (2016). Pharmacological exploration of the resting membrane potential reserve: impact on atrial fibrillation. Eur. J. Pharmacol. 771, 56–64. doi: 10.1016/j.ejphar.2015.11.026
Varkevisser, R., Houtman, M. J., Waasdorp, M., Man, J. C., Heukers, R., Takanari, H., et al. (2013). Inhibiting the clathrin-mediated endocytosis pathway rescues K(IR)2.1 downregulation by pentamidine. Pflugers Arch. 465, 247–259. doi: 10.1007/s00424-012-1189-5
Vasilev, F., Ezhova, Y., and Chun, J. T. (2021). Signaling enzymes and ion channels being modulated by the actin cytoskeleton at the plasma membrane. Int. J. Mol. Sci. 22:10366. doi: 10.3390/ijms221910366
Vatta, M., and Faulkner, G. (2006). Cytoskeletal basis of ion channel function in cardiac muscle. Future Cardiol. 2, 467–476. doi: 10.2217/14796678.2.4.467
Willis, B. C., Ponce-Balbuena, D., and Jalife, J. (2015). Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 308, H1463–H1473. doi: 10.1152/ajpheart.00176.2015
Wu, L., Wang, Q., Gu, J., Zhang, H., and Gu, Y. (2020). Modulation of actin filament dynamics by inward rectifying of potassium channel Kir2.1. Int. J. Mol. Sci. 21:7479. doi: 10.3390/ijms21207479
Xia, M., Jin, Q., Bendahhou, S., He, Y., Larroque, M. M., Chen, Y., et al. (2005). A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 332, 1012–9. doi: 10.1016/j.bbrc.2005.05.054
Keywords: ion channel, trafficking, cytoskeleton, Chloroquine, Nocodazole, Cytochalasin, Dendra2, patch clamp
Citation: Li E, Loen V, van Ham WB, Kool W, van der Heyden MAG and Takanari H (2022) Quantitative Analysis of the Cytoskeleton’s Role in Inward Rectifier KIR2.1 Forward and Backward Trafficking. Front. Physiol. 12:812572. doi: 10.3389/fphys.2021.812572
Received: 11 November 2021; Accepted: 23 December 2021;
Published: 25 January 2022.
Edited by:
Henrique Girao, University of Coimbra, PortugalReviewed by:
Michael Tamkun, Colorado State University, United StatesCopyright © 2022 Li, Loen, van Ham, Kool, van der Heyden and Takanari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcel A. G. van der Heyden, bS5hLmcudmFuZGVyaGV5ZGVuQHVtY3V0cmVjaHQubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.