 Charles A. LeDuc
Charles A. LeDuc Alicja A. Skowronski
Alicja A. Skowronski Michael Rosenbaum
Michael Rosenbaum
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 10 December 2021
Sec. Metabolic Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.789519
This article is part of the Research TopicLeptin in Physiology and DiseaseView all 5 articles
LEP is a pleiotropic gene and the actions of leptin extend well beyond simply acting as the signal of the size of adipose tissue stores originally proposed. This is a discussion of the multi-system interactions of leptin with the development of the neural systems regulating energy stores, and the subsequent maintenance of energy stores throughout the lifespan. The prenatal, perinatal, and later postnatal effects of leptin on systems regulating body energy stores and on the energy stores themselves are heavily influenced by the nutritional environment which leptin exposure occurs. This review discusses the prenatal and perinatal roles of leptin in establishing the neuronal circuitry and other systems relevant to the adiposity set-point (or “threshold”) and the role of leptin in maintaining weight homeostasis in adulthood. Therapeutic manipulation of the intrauterine environment, use of leptin sensitizing agents, and identification of specific cohorts who may be more responsive to leptin or other means of activating the leptin signaling pathway are ripe areas for future research.
In 1973, Coleman (1973) demonstrated that parabiosis of the obese (later Lepob) mice with diabetes (later Leprdb) mice and wild type mice resulted in hypophagia and starvation of the Lepob mice while not affecting the phenotype of the Leprdb mice. He postulated that “the obese mouse is able to produce sufficient satiety factor to regulate its food consumption, whereas the diabetes mouse produces satiety factor, but cannot respond to it”. Subsequently, Leibel and Hirsch (1984) and others (Welle et al., 1984) found that reduced body weight maintenance was accompanied by a decline in energy expenditure and an increase in hunger disproportionate to changes in body weight and composition that strongly resembled the metabolic state of the Lepob and Leprdb mice. These observations were consistent with the so called “lipostatic” theory of body weight maintenance in which a “signal” reflecting adipose tissue mass affected hypothalamic neural circuitry regulating energy intake and expenditure (Kennedy, 1953; Mayer, 1955; Hetherington and Ranson, 1983).
The advent of large-scale genome-wide association studies (GWAS) combined with polygenic risk scoring has facilitated the identification of aggregate genetic factors determining body weight and the underlying energy homeostatic mechanisms that regulate it. By calculating an obesity propensity score from 2.1 million SNPs, individuals can be categorized into “obesity risk” deciles (Khera et al., 2019) with an overall correlation of genetic propensity score (GPS) and body mass index (BMI) of 0.29.
Needing 2.1 million SNPs to determine an obesity propensity score indicates that there are an almost uncountable number of minute genetic contributors that act in concert to determine a person’s genetic predisposition to adiposity. Mendelian breeding strategies, which can be employed in rodents, facilitate the identification of critical genetic variants. It is not surprising that the most important genes affecting energy homeostasis, including those first postulated by Coleman, are involved in the leptin-melanocortin pathway [including Lep, Lepr, melanocortin 4 receptor (Mc4r), pro-opiomelanocortin (Pomc)] were all first discovered in rodent models of extreme obesity and only subsequently identified in humans. Other critical feeding circuit genes in the same pathway were also first identified in animal models. Npy was discovered due to peptide abundance in porcine intestine extract, its co-localization in the murine brain (Tatemoto et al., 1982), and potent stimulation of energy intake following intracerebroventricular (i.c.v.) administration to rats (Clark et al., 1984). The orexigenic and thyroid releasing hormone (TRH) suppressive effects of agouti-related peptide (Agrp) were first recognized primarily because Agrp expression increased 10-fold in Lepob mice (Manne et al., 1995).
It is now understood that LEP is a pleiotropic gene and that the actions of leptin extend well beyond simply acting as the signal of the size of adipose tissue stores originally proposed by Coleman. This is a discussion of the multi-system interactions of leptin with the development of the neural systems regulating energy stores, and the subsequent maintenance of energy stores throughout the lifespan. This review will attempt to show predominantly animal data regarding the prenatal and perinatal roles of leptin in establishing the neuronal circuitry of the adiposity set-point (or “threshold”) and the role of leptin as the signal regulating the “deployment” of those systems to maintain energy homeostasis in both humans and rodents.
The developmental environment of mice can be manipulated for prospective studies of weight regulation whereas human studies of the effects of obesogenic gene exposure are, of necessity, observational and epidemiological. Though data from rodent experiments are the most informative in elucidating the mechanistic developmental impact of leptin, there is compelling evidence indicating that both the development and regulation of body weight in humans reflects both pre- and post- natal gene × environment interactions.
The interactions between leptin and the developing neurocircuitry in rodents are dependent upon the nutritional environment in which they occur. The impact of maternal metabolic status and nutritional status of the pups in the prenatal and perinatal periods on body weight in adulthood can be directly investigated in rodents, and there have been numerous studies to this effect. Both maternal under- and over- nutrition during gestation affect these systems in a manner that favors the development of obesity (Breton, 2013). In general, offspring of dams who were malnourished during pregnancy show structural disorganization of the hypothalamic systems regulating appetite (Breton et al., 2009). Prenatal maternal undernutrition reduces the response of POMC neurons to energy status and food intake rhythm (Breton et al., 2009). Maternal overfeeding, especially with a high fat diet (see below), results in altered brain appetite regulators in the offspring (Rajia et al., 2010).
Rodent models of perinatal undernutrition include maternal caloric or protein restriction during gestation and/or lactation (Vickers et al., 2000; Yura et al., 2005; Delahaye et al., 2008; Cripps et al., 2009) and increasing litter size to lower milk availability per pup in a litter (Aubert et al., 1980; Marangon et al., 2020). Perinatal maternal undernutrition drastically reduces the postnatal surge of plasma leptin, disturbing particularly the hypothalamic wiring as well as the gene expression of the anorexigenic POMC neurons in male rat pups (Delahaye et al., 2008).
The macronutrient composition of gestational undernutrition and the perinatal environment interact in their effects on adult rodent adiposity. While the association of gestational undernutrition by caloric restriction induces only a small degree of increased weight gain in the adult offspring (Lagisz et al., 2014), the evidence is stronger that specifically restricting protein in pregnant dams results in increased body weight of the offspring which is exacerbated by HFD exposure (Ozanne et al., 2004; Lagisz et al., 2014; Juan De Solis et al., 2016). Caloric or protein restriction during the suckling period and rearing in large litters has an opposite effect in that the offspring gain less weight during the first postnatal weeks and this lower weight persists in these pups throughout lifetime (Ozanne et al., 2004; Patterson et al., 2010).
There is strong evidence that maternal HFD feeding during perinatal period as well as overnutrition of sucking pups (by decreasing litter size) increases body weight and predisposes the offspring to greater weight gain when exposed to HFD in adulthood (Ainge et al., 2011; Lagisz et al., 2015; Ribaroff et al., 2017). Some of the leptin-related consequences of overfeeding in rodents are related to the magnitude of the postnatal leptin surge (Marangon et al., 2020; Skowronski et al., 2021), leptin sensitivity in the CNS (Kirk et al., 2009), neuroanatomy of the leptin-dependent feeding circuits (Kirk et al., 2009; Vogt et al., 2014), and epigenetic changes—specifically hypermethylation of the hypothalamic POMC promoter (Plagemann et al., 2009; Marco et al., 2014).
In humans, epigenetic studies have examined the effects of the intrauterine environment, primarily in the form of factors affecting DNA methylation, histone acetylation, and expression of microRNAs, on gene expression relevant to obesity and its co-morbidities. Increased DNA methylation decreases the transcription of relevant genes and is affected by parental obesity, maternal diet (e.g., nutrition, folic acid content, and other methyl donors), gestational diabetes (see below), maternal medications (antibiotics and antipsychotics), smoking, and exposure to chemicals such as bisphenol (Inadera, 2013; Reynolds et al., 2013). Major intrauterine environmental influences on the risk of subsequent obesity in the offspring via these processes and others include maternal adiposity and gestational weight gain, under- and over- nutrition, maternal stress, and various chemicals, pharmaceuticals, etc. to which the mother and fetus may be exposed to during pregnancy are summarized in Table 1.

Table 1. Overview of intrauterine epigenetic factors relevant to subsequent adiposity.
Leptin and its signaling pathways figure prominently in these intrauterine and perinatal systems affecting the development and maintenance of energy stored as adipose tissue. Leptin is an adipocyte-derived hormone providing a long-term signal to the CNS regarding quantity of stored body adiposity, largely by binding to the long form of leptin receptor, LepRb, in POMC and AgRP/NPY) neurons. These first order neurons are primarily located in the arcuate nucleus of the hypothalamus (ARH) and are well described in mediating neuroendocrine systems related to energy homeostasis (Schwartz et al., 2000). Activation of the Jak2/STAT3 pathway by leptin signaling increases the activity of POMC neurons and the expression of Pomc and Cart (Cocaine and amphetamine-related transcript) while inhibiting the orexigenic AgRP/NPY neurons and decreasing the expression of Agrp and Npy (Hahn et al., 1998; Cowley et al., 2001). POMC is posttranslationally cleaved by proconvertases (PC1 and PC2) and other peptidases to create several smaller peptides including β-endorphin and α-melanocyte stimulating hormone (α-MSH) (Benjannet et al., 1991). α-MSH inhibits energy intake and stimulates energy expenditure via melanocortin 4 receptors (MC4R) and, to a lesser degree, melanocortin 3 receptors (MC3R) located on second order neurons (Seeley et al., 1997; Sweeney et al., 2021). AgRP is an antagonist of MC4R (Ollmann et al., 1997) and opposes the effects of POMC. NPY is an agonist of the NPY receptors which mediates additional orexigenic effects (Schwartz et al., 2000). Mutations in the POMC gene lead to severe human obesity (Krude et al., 1998) while rodent Pomc knockouts are obese and less sensitive to leptin (Challis et al., 2004). While congenital mouse AgRP knockouts have a limited metabolic phenotype (Qian et al., 2002) with normal body weight, adiposity, and food intake, conditional ablation of AgRP neurons in adulthood induces an ultimately lethal anorexia (Luquet et al., 2005).
In addition to adipose tissue, leptin is produced by the placenta (Hassink et al., 1997) and stomach (Mix et al., 1999) in humans. In embryonic mice, leptin is also produced by hair follicles, liver, heart, bone, and cartilage (with both protein and mRNA detected) (Hoggard et al., 2000). During pregnancy in humans, circulating leptin is increased by 1.5–3 fold in the second and third trimesters of pregnancy (Butte et al., 1997; Hardie et al., 1997; Highman et al., 1998; Sattar et al., 1998), followed by a significant drop in the early postpartum period, as early as 24 h after delivery (Sivan et al., 1998; Lage et al., 1999). Leptin is detected in fetal circulation as early as 19 weeks of gestation with a rapid increase in fetal leptin between weeks 33 and 41 and correlates with fetal size (Butte et al., 1997). At delivery, newborn plasma leptin correlates with gestational age and birthweight (Cetin et al., 2000) with subsequent decreases to low levels within the first few days after parturition (Schubring et al., 1999).
The changing functions of leptin with regards to neurogenesis and adipose tissue from conception to adulthood in rodents are illustrated in Figure 1. Since the fetal intrauterine environment receives a continuous transplacental supply of glucose and other nutrients necessary for growth, leptin does not regulate “appetite” in the traditional sense in utero, but such wide expression of Lep could have other functions. The complete impact of prenatal leptin on energy homeostatic and other systems is not fully understood, but there is clear evidence that it acts as a neurogenic factor in utero. Compared to wild type mice, mice that lack leptin have brains that are both smaller by weight and have reduced cortical volume (Bereiter and Jeanrenaud, 1979), have fewer cells at embryonic day 16 (E16) and E18, and fewer recently born cells at E14 and E16 in the neuroepithelium. Intracerebroventricular leptin injection of E14 Lepob embryos normalized the number of neuroepithelium cells at E16 (Udagawa et al., 2006). The neurogenic effects of leptin are still evident postnatally. Intraperitoneal administration of leptin daily for 2 to 4-week-old Lepob mice resulted in increased dry brain weight due, at least partially, to an increase in cell number as indicated by total brain DNA which increased at a greater rate than brain weight (Steppan and Swick, 1999).
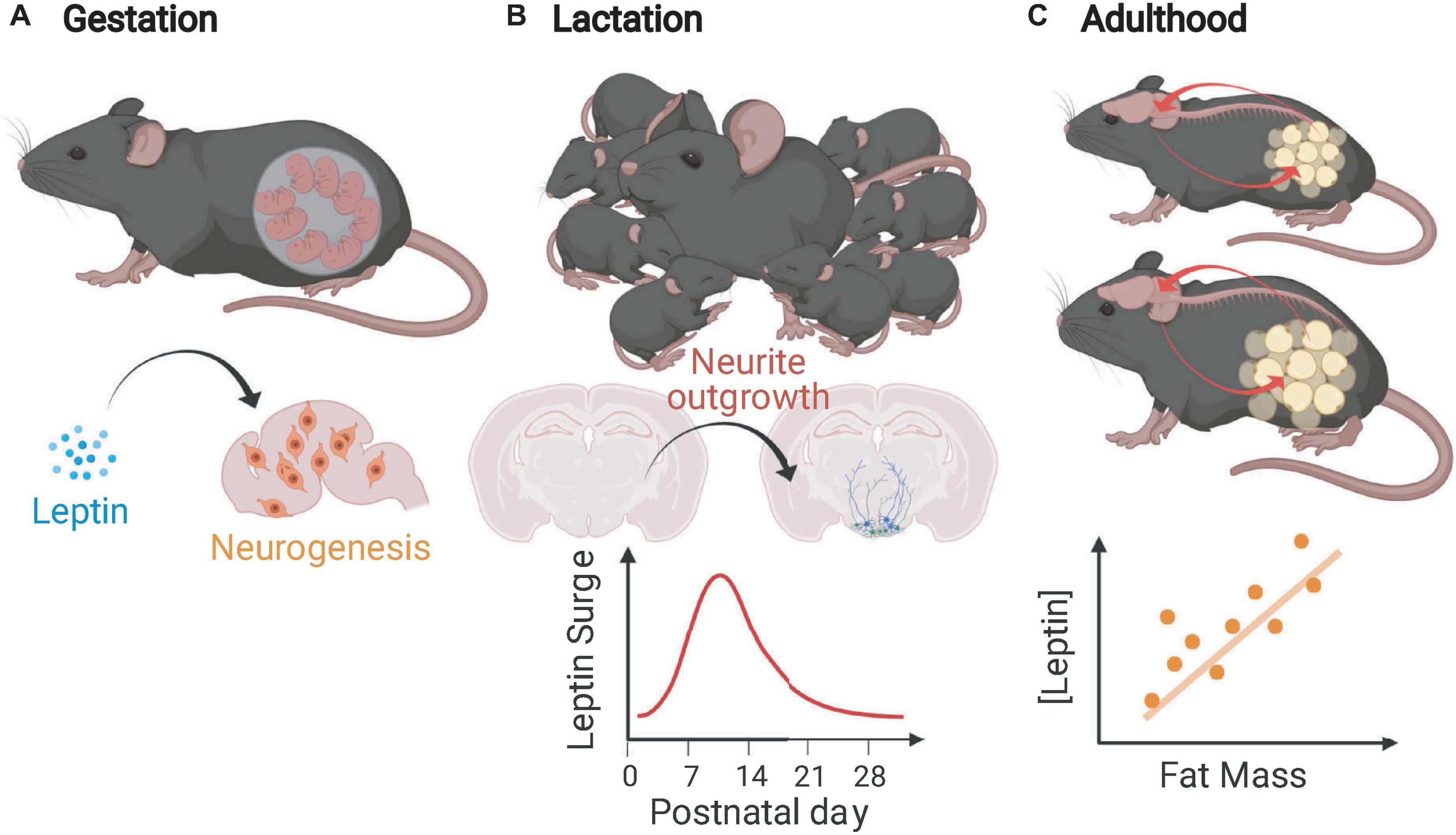
Figure 1. Leptin functions in mice during gestation, lactation, and adulthood. (A) In the gestational period, leptin mediates neurogenesis and proliferation of other brain cells (not limited to gestation). (B) During the immediate postnatal (lactational) period, mice undergo a leptin surge that is critical for the outgrowth of projections from feeding circuit essential neurons. (C) In adult mice, leptin is produced in rough proportion to stable fat mass, informs the CNS about energy stores, and protects against fat loss (Figure created with BioRender.com).
Deficiency of leptin during development impairs the formation of the feeding circuits; the density of projections originating from the ARH to other hypothalamic regions critical in energy homeostasis [including the paraventricular nucleus (PVH), the dorsomedial hypothalamic nucleus (DMH), and the lateral hypothalamic area (LHA)] is decreased (Bouret et al., 2004). Ahima et al. (1998) first demonstrated that mice undergo a postnatal leptin surge during which the circulating leptin concentration is significantly higher than predicted by fat mass and is unrelated to amount of stored energy (Bouret et al., 2004). The timing of the leptin surge is critical; it overlaps temporally with the generation of homeostatic feeding circuits in the hypothalamus and brain stem (Bouret et al., 2004; Biddinger et al., 2020). Studies in Lepob mice directly indicate that leptin acts as a neurotrophic factor during the perinatal period. Administration of exogenous leptin to Lepob mice to mimic the naturally occurring leptin surge (P4–P12) rescues the axonal density of feeding circuit neurons while supplementation of leptin in adult Lepob mice fails to restore these hypothalamic projection densities (Bouret et al., 2004). In addition to the hypothalamus, the nucleus of the solitary tract (NTS) within the brain stem is critical in energy homeostasis, especially for the integration of the viscerosensory signals. The majority of Glp-1 expressing neurons within the NTS express LepRb and project primarily to the PVH. The density of GLP-1 innervation from the NTS to the PVH is augmented in Lepob mice indicating leptin’s role in the development of this circuit (Biddinger et al., 2020).
Leptin influences astrocyte development. There is a marked increase in glia cell number between postnatal weeks 2 and 3 in rodents (Bandeira et al., 2009) coinciding with the natural leptin surge (Ahima et al., 1998). Astrocytes express the long form of the leptin receptor (LepRb) (Pan et al., 2008; Kim et al., 2014). Exogenous leptin administration between P8 and P12 increases the proliferation of astrocytes in the hypothalamus. This is a direct effect of leptin since proliferation is decreased when LepRb is conditionally removed from these cells (Rottkamp et al., 2015). Conditional deletion of LepRb in adult mouse astrocytes leads to glial morphological changes and increased synaptic inputs onto hypothalamic POMC and AgRP neurons (Kim et al., 2014). These mice also show diminished leptin-regulated feeding suppression, suggesting a direct impact of leptin on astrocyte development and function in adult mice (Kim et al., 2014) which is supported by studies demonstrating leptin-mediated neurogenesis post-stroke (Avraham et al., 2013) as well as a model of Alzheimer’s disease (Calio et al., 2021) in rodents.
In rats and mice (Marangon et al., 2020; Skowronski et al., 2021), maternal high fat diet (HFD) feeding during gestation and/or lactation, or overfeeding the pups via reduced litter size augments the postnatal leptin surge (see below) and subsequent weight in the offspring. Caloric or protein restriction of dams or underfeeding the pups by increased litter size reduces and delays the leptin surge with a reduction in weight into adulthood (Delahaye et al., 2008). Skowronski et al. (2021) demonstrated that all pups undergo a postnatal leptin surge, but in the underfed state, the surge is delayed and transitory. Excess or deficiency in leptin does not affect body weight and adiposity of pups prior to the second week of life. There is no difference in body weight or composition between Lepob mice, hyperleptinemic, and wild type mice at postnatal day 10 (Mistry et al., 1999). Unlike adult mice with mature feeding circuits, in the first 2 weeks of mouse life, leptin does not influence feeding, instead, it is critical in the proper formation of these circuits.
In addition to changes in magnitude and timing of the leptin surge, maternal diet influences the development of neurocircuitry relevant to energy intake. HFD feeding during pregnancy in rodents is associated with disruptions in the normal patterns of projections in the hypothalamic feeding circuits, including decreased AgRP immunoreactive fibers in the PVH (Kirk et al., 2009; Vogt et al., 2014) and reduced density of α-MSH projections from the ARH to PVH, DMH and LHA in 8-week old progeny (Vogt et al., 2014). These are the same projections disrupted in congenitally leptin deficient mice suggesting that effects of maternal diet on the weight of the offspring may be mediated through effects on the postnatal leptin surge which, in turn, alters the development of the feeding circuitry.
Elevated circulating leptin concentrations during the rodent suckling period leads to increased adiposity and weight gain in adult offspring when exposed to obesogenic diets (Vickers et al., 2008; Skowronski et al., 2020). In leptin transgenic TET-ON mice, oral doxycycline (DOX) causes leptin overexpression in proportion to the concentration of DOX, allowing for the transient elevation of leptin without increasing fat mass and consequently avoiding any obesity co-morbidities. These leptin transgenic mice were transiently exposed to elevated leptin during the first 3 weeks of life (to mimic the augmented leptin surge induced by postnatal overfeeding) and in adulthood were metabolically identical to control littermates which were not postnatally supplemented with excess leptin. However, when the adult mice were exposed to a HFD challenge, the postnatally hyperleptinemic mice gained more fat mass and weight than littermate controls and the difference in body weight gain was statistically significant within 3 days (Skowronski et al., 2020). Since this experiment isolated the augmented leptin surge from the other confounds of postnatally overfeeding pups, it suggests that leptin, in isolation, can reprogram the body weight set point such that mice are less sensitive to future increases in leptin. Male rat offspring given exogenous leptin IP during the first 2 weeks of life resulted in increased diet-induced weight and fat gain in adulthood (de Oliveira Cravo et al., 2002; Vickers et al., 2008). Interestingly, Sanchez et al. (2005) administered leptin orally to postnatal pups to investigate the role of leptin in breastmilk. This oral leptin was absorbed by the immature gastric epithelium of the neonate and down-regulated endogenous leptin production in the pup, suggesting leptin’s potential role in the short-term control on food intake during the lactation period. Additionally, orally-fed leptin to postnatal pups (P0–P20) had the opposite effect of IP leptin injections—the leptin-fed offspring gained less weight in adulthood and had a lower preference for fat-rich foods when exposed to HFD (Pico et al., 2007) compared to their controls suggesting that postnatal oral leptin may permanently reduce endogenous leptin production and lead to increased responsiveness to leptin in adult rats.
Humans and mice stably maintain body energy stores (fat) without conscious effort to adjust food intake or energy expenditure. Adult humans, regardless of adiposity, gain weight at an average of approximately 0.3–0.5 kg/year (Zheng et al., 2017) (∼3,000 kcal stored energy) between the ages of 18–55 years in females and 21–55 years in males, while ingesting over 800,000 kcal/year (Ford and Dietz, 2013) thereby suggesting the operation of homeostatic mechanisms for body weight regulation. Hyperphagia and hypometabolism (including decreased circulating concentrations of bioactive thyroid hormones and sympathetic nervous system tone and increased parasympathetic nervous system tone) act together to favor weight regain after successful weight reduction and oppose efforts by most individuals to sustain weight loss (Leibel and Rosenbaum, 2010; Thomas et al., 2014; Hall and Kahan, 2018). The level of body energy stores that are “defended” in a given individual depends upon homeostatic systems—the structure and function of which are, at least partially, determined by early leptin exposure. As discussed above, the degree and timing of leptin exposure reflects a number of key factors in the intrauterine environment including maternal health and nutrition.
The effects of leptin on energy homeostasis are not limited to the development of these systems. Leptin serves as a marker of adipose tissue stores and energy balance and is a major signal governing the extent to which individuals respond to attempts to lose weight and keep it off.
There are changes in energy intake and expenditure which act concordantly to oppose weight loss and the maintenance of reduced body weight. Changes related to energy intake include increased, hunger, delayed satiation, increased neuronal responses to food in the orbital frontal cortex and areas related to food reward and decreased responses in the prefrontal cortex and areas related to food restraint. Homeostatic changes in energy expenditure are due, at least in part, to changes in skeletal muscle via increased work efficiency due to increased expression of the more efficient myosin heavy chain I (MHC) and sarcoplasmic endoplasmic reticulum Ca++-dependent ATPase 2 (SERCA2). This hypometabolic state is augmented by changes in neuroendocrine function (decreased circulating concentrations of bioactive thyroid hormones and leptin) and, at least during reduced weight maintenance, increased parasympathetic and decreased sympathetic nervous tone (Larrouy et al., 2008; Leibel and Rosenbaum, 2010; Rosenbaum and Leibel, 2014; Dulloo and Schutz, 2015). The multi-system interactions that oppose the maintenance of a reduced body weight are summarized in Figure 2.
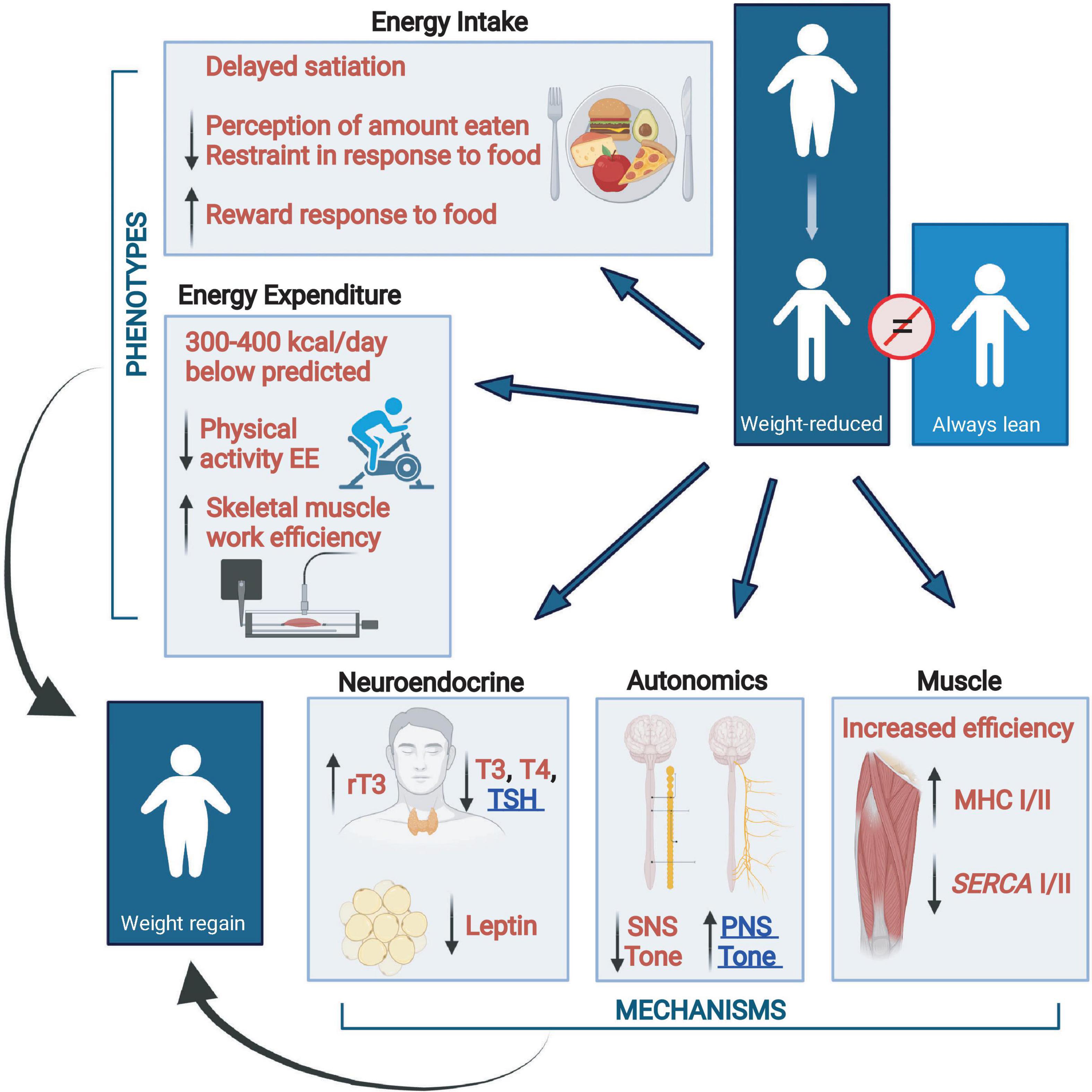
Figure 2. Changes from baseline in energy balance and homeostatic systems during maintenance of a 10% or greater reduced body weight and their responsiveness to exogenous leptin in individuals who initially had obesity or never had obesity (Rosenbaum and Leibel, 2014). Energy expenditure due to physical activity is calculated as the difference between direct measurement of 24-h energy expenditure and measurement of resting energy expenditure plus diet-induced thermogenesis. Eating behavior, including energy intake, is examined by visual analog scales during a fixed liquid formula meal, kcal of the liquid formula consumed to reach satiation, and by fMRI studies of brain responses to food. Assessments of autonomic nervous system activity were made by analyses of heart rate variability during sequential blockade of the parasympathetic and sympathetic nervous systems with atropine and esmolol, respectively, and by 24-h urine catecholamine excretion. Skeletal muscle contractile efficiency was measured by graded bicycle ergometry. Myosin heavy chain (MHC) and sarcoplasmic endoplasmic reticulum Ca++-dependent ATPase (SERCA) muscle gene expression studies were done by mRNA quantification in biopsies of vastus lateralis muscle. All phenotypes opposing sustained weight loss are responsive to leptin repletion except for PNS tone and TSH which are underlined in blue. SNS, sympathetic nervous system; PNS, parasympathetic nervous system; T3, triiodothyronine; T4, thyroxine; rT3, reverse T3; TSH, thyroid stimulating hormone; MHC, myosin heavy chain; SERCA, sarcoplasmic endoplasmic reticulum Ca++-dependent ATPase (Figure created with BioRender.com).
As noted above, body fat stores, are regulated by multiple systems that conspire to defend energy stores (fat) against energy imbalance by adjusting energy intake and output to maintain a relatively constant level of available energy over time. Leptin provides a signal to the brain regarding the quantity of fat stores as well as energy balance (weight loss in particular). The result is that the intensity of the leptin signal, and the energy homeostatic responses to changes in that intensity, are determined by both the ambient leptin concentration (Myers et al., 2008) and by the nutritional state of the organism. It is notable that leptin mediates the development of the same brain regions that ultimately influence neuroendocrine functions, autonomic efferents, and food-related behaviors (Korner et al., 1999, 2001; Korner and Aronne, 2003).
Leptin-mediated signals regulate a complex neural system that mediates what is physiologically apparent as the regulation of body weight via the integration of short- (e.g., gut-derived hormones and glucose) and longer- (e.g., leptin, insulin, and free fatty acids) term signals related to energy homeostasis (Korner et al., 1999, 2001; Schwartz et al., 2000; Korner and Aronne, 2003). Teleologically, these body weight regulatory systems should be biased toward “defending” against sustained weight loss which could threaten reproductive capacity/fertility and/or survival (Rosenbaum and Leibel, 1998). It is not surprising that the effects of leptin administration show a strong functional bias in favor of the preservation of body fat stores versus their reduction as discussed below.
Shortly after leptin was cloned from the Lepob mouse (Zhang et al., 1994), humans with the nonsense mutations in the LEP gene were reported (Montague et al., 1997). Similar to mice, humans that are leptin deficient have morbid obesity and reduced muscle mass, are hyperphagic, and hypometabolic. When leptin deficient humans (Farooqi et al., 1999) or mice (Hwa et al., 1997) are supplemented with leptin, they reverse these phenotypes. Short term administration of leptin to lean, Lepob, or to diet-induced obese mice reduces appetite, body weight, and adiposity (Campfield et al., 1995; Halaas et al., 1995; Pelleymounter et al., 1995). The treatment of leptin in these initial studies was short (Pelleymounter et al., 1995) (from two successive 5-day treatments to 28 days at the longest) but the effects were striking; Lepob mice were normalized and lean mice reduced body fat from 12.2 to 0.7% (Halaas et al., 1995).
The leptin receptor is highly expressed in the cells of the hypothalamic nuclei–the development of which is mediated by leptin (discussed above). These nuclei play prominent roles in homeostatic weight regulation (Pan and Mg Myers, 2018) as well as communicate with other telencephalic and diencephalic neurons that mediate behavioral responses to food. Many neurons outside the hypothalamus also express the receptor, though their functional roles in these cells remain unclear.
As discussed above, the interactions of leptin with the developing neurocircuitry are dependent upon the nutritional environment in which those circuits develop. Similarly, the effects of leptin on energy homeostatic systems are dependent upon the nutritional environment (fat mass and energy balance) in which leptin is administered. Most of these systems are more sensitive to leptin following weight loss (especially during maintenance of reduced weight) compared to initiation of weight loss or during dynamic weight loss; and these energy homeostatic responses are stronger to leptin depletion than excess (Leibel, 2002; see Table 1).
The discovery of leptin was initially expected to ameliorate the obesity epidemic; however, this expectation has never been met. Unlike mice, administration of exogenous leptin to humans with or without obesity has little or no effect on body weight even at grossly supraphysiological doses (Heymsfield et al., 1999). Leptin administration to individuals during caloric restriction (negative energy balance), with concomitant declines in circulating leptin concentrations, results in a small reduction in appetite but no significant changes in energy expenditure or neuroendocrine function (Hukshorn et al., 2000, 2002, 2003).
Leptin administration does not seem to induce or perpetuate weight loss in humans. Heymsfield et al. (1999) administered leptin in placebo, physiological, and supraphysiological doses to 54 lean and 73 obese subjects for 4 weeks; and to 47 obese subjects for 24 weeks. Participants with obesity were placed on diets restricting caloric intake to about 500 kcal/day below usual, but dietary compliance was not assessed and there was no significant weight loss in the placebo group. After 4 weeks, overall weight reduction in leptin-treated subjects with or without obesity was not different from placebo-treated. Participants with obesity, not those without obesity, who received the highest doses of exogenous leptin (sufficient to raise circulating leptin concentrations more than 20-fold above initial) for a period of 24 weeks showed a small significant weight loss (2.3 kg more than placebo) and a small but statistically insignificant decrease in daily energy intake. The high circulating leptin concentrations and low levels of weight loss in participants with obesity following exogenous leptin administration have been interpreted to indicate “leptin resistance” (Friedman and Halaas, 1998; Kalra, 2001; Lee et al., 2001; Scarpace and Zhang, 2007). This conclusion that leptin resistance at usual weight reflects leptin resistance only in individuals with obesity is not supported by the data since individuals without obesity were not more responsive to leptin. If anything, these data would suggest that any individual at usual weight is likely to be leptin resistant.
Similarly, Moon et al. (2011), found no significant effects of 10 mg BID of subcutaneous leptin administration to 71 weight stable participants with obesity and type 2 diabetes managed by diet alone. Mackintosh and Hirsch (2001) reported no effects of high dose (0.3 mg/kg/day) leptin administration—the highest dose used by Heymsfield et al. (1999)—on autonomic nervous system (ANS) tone in weight-stable lean subjects which is significant compared to the significant effect of leptin repletion on sympathetic tone noted following weight loss.
The lack of effects of leptin on weight loss induction or potentiation, even in supraphysiological doses, are in stark contrast to the potent effects of leptin in weight-reduced individuals who are weight stable (i.e., in energy balance) (see Figure 2). Simple leptin repletion to levels present prior to weight loss in this population “reverses” most, if not all, of the metabolic and behavioral phenotypes that oppose the reduction of energy stores. These include the increased hunger and delayed satiation as well as the hypometabolism and its components (increased muscle efficiency, decreased sympathetic nervous system tone and circulating concentration of bioactive thyroid hormones) that favor weight regain (Rosenbaum et al., 2005, 2008b,a; Kissileff et al., 2012; Hinkle et al., 2013). Taken together, these data suggest that the major function of leptin in energy homeostasis is to signal the organism that energy stores are low and/or that energy intake is inadequate to maintain weight. The individual is much more responsive to a drop in circulating concentrations of leptin below a personalized leptin threshold than to a rise in leptin above that threshold.
Circulating leptin concentrations are determined by fat mass (cell number x size) but the relationship is modified downward (decreased leptin per unit of fat mass) at reduced weight and even more so during negative energy balance during weight loss (Halle et al., 1999). The actions of some hormones, such as the effects of insulin on glucose utilization, are linear within a physiological range (Rizza et al., 1981). However, leptin actions are non-linear and most consistent with a “threshold model” wherein leptin effects are triggered when leptin levels fall below a certain individualized “set-point” and leptin response is attenuated if administered when circulating concentrations are above that point. The “threshold” for sufficiency of leptin action for any individual is determined by genetic, developmental and environmental factors that influence both the structure of relevant parts of the CNS as well as the acute responses of those cells to ambient leptin. Individuals who have been obese have higher thresholds and more leptin (fat mass) is needed to create a state of sufficiency in the CNS. Once that level is achieved, further increases in circulating leptin concentrations have little physiological effect.
When fat mass is reduced by caloric restriction, a fall in circulating leptin concentrations below the threshold “informs” specific neurons that invoke behavioral (hunger) and metabolic (reduced energy expenditure) changes that act coordinately to restore body fat (leptin). This individual molecular-cellular leptin threshold does not decrease with weight loss or with prolonged maintenance of reduced weight (Rosenbaum et al., 2008a); hence, the metabolic/behavioral response to reduced leptin does not abate. The threshold is an individualized molecular-cellular phenotype such that response to declines in leptin below the threshold are similar regardless of initial adiposity. What differs between individuals with and without obesity is the threshold. Therefore leptin signal transduction is dependent upon an individual’s “usual weight” and any ongoing or previous negative energy balance (Morabito et al., 2017).
Response to exogenous leptin is also dependent on energy stores and balance. When leptin concentrations are raised above an individual “threshold” (which is higher in individuals with obesity), the relevant neuronal tracts are less sensitive (LeDuc and Leibel, 2019; Zhao et al., 2019) and further leptin administration evokes little if any response in humans (Rosenbaum et al., 2018b). For reasons not yet understood, when leptin concentrations are reduced below the threshold and the individual is in negative energy balance (caloric restriction) the effect of leptin repletion is small (Hukshorn et al., 2000, 2002, 2003). However, in low leptin states where there is little energy imbalance, such as reduced weight maintenance, congenital leptin deficiency, or lipodystrophy, most, if not all, of the metabolic and behavioral effects of low leptin are at least partially relieved (Farooqi et al., 1999; Oral et al., 2002; McDuffie et al., 2004; Rosenbaum et al., 2005, 2008b,b; Park et al., 2007; Kissileff et al., 2012; Hinkle et al., 2013; Brown et al., 2018).
The threshold model described above presumes that the primary function of leptin is to preserve fat mass in times of perceived undernutrition in defense of preserving reproductive integrity and survival of a species. This is supported by the effects of leptin repletion in states of hypothalamic amenorrhea which can be caused by excessive exercise or decreased food intake and leads to infertility and bone loss. Short term treatment with leptin from 3 months to 36 weeks recovers menstruation and corrects the abnormalities in the gonadal, thyroid, growth hormone, and adrenal axes (Welt et al., 2004; Chou et al., 2011). This is consistent with the role of leptin acting as a starvation signal; the drop in circulating leptin concentration signals the gonadal system to decrease procreation when energy stores are scarce to prevent pregnancy that would be overly challenging to both the mother and fetus.
The critical roles played by leptin in the early development of systems regulating body weight and its subsequent actions within those symptoms has implications for the prevention and treatment of obesity. Obesity risk could theoretically be altered via modification of the development of leptin-mediated neuronal circuitry regulating body weight toward the defense of a lower body weight in those at risk. Manipulation of the intrauterine environment by diet or other means to reduce fetal overnutrition or undernutrition may reduce the propensity toward later obesity (Delahaye et al., 2008; Breton et al., 2009; Breton, 2013). Though it is unlikely that leptin will be effective as a weight loss medication – its efficacy might be increased in the setting of leptin sensitizing agent (Ravussin et al., 2009) or in individuals with disproportionately low levels of leptin (Ahima, 2008). The potent effects of leptin after weight loss indicates the need for longer term studies of the effects of leptin on the likelihood of successful reduced weight maintenance.
A critical question is whether the leptin threshold is changeable, particularly in a downward direction. While there are clearly genetic effects on human adiposity, and while the majority of these genes are expressed in the central nervous system, the precise manner(s) in which these effects are integrated is not yet clear (Loos, 2018). The threshold model is consistent with the phenomenology of the similarity of responses to weight loss among obese and non-obese individuals. In humans the phenotypes associated with increased metabolic efficiency and drive to eat do not abate with time (Shick et al., 1998; DelParigi et al., 2007; Rosenbaum et al., 2008a), suggesting that prolonged maintenance of reduced body weight can only be achieved by indefinite attention to both food intake and exercise (Wing and Hill, 2001). However, it is possible that, in addition to the effects of allelic variants, there are developmental effects on energy homeostatic circuits that can influence an individual’s apparent threshold for minimum body fat that could potentially be modified through earlier intervention in those “at-risk” for subsequent adiposity. An example of this potential to manipulate the “set-point” prenatally is the reduction in fatness, blood pressure, circulating concentrations of insulin and gene expression relevant to diabetes, autoimmune disease, and vascular disease in children who develop in a post-bariatric surgery intrauterine environment compared to their siblings who were gestated prior to surgical weight loss in the mother (Guenard et al., 2013).
Leptin is a highly pleiotropic molecule that influences the prenatal and perinatal development of the major neuronal tracts that determine the body weight that is “defended” over a lifetime. Subsequently, it provides the primary signal that determines the activity of the same regulatory systems.
Leptin, importantly, functions primarily as a signal of decreased energy stores and/or negative energy balance from the periphery to the CNS. In the state of negative energy balance, the resulting rapid decrease in circulating leptin concentration is sensed by the CNS and, in response, drives hunger, suppresses energy expenditure, and reduces reproductive competence; despite potentially underlying obesity (Boden et al., 1996). Similar effects are seen as a result of a lesser degree of relative hypoleptinemia following weight loss at which time most individuals are quite responsive to leptin repletion. This “predictive” characteristic of leptin production by adipose stores acts to increase fat mass, and thereby protect reproductive integrity in times of undernutrition, is arguably the leptin function that is most important in an evolutionary context. Going forward, leptin therapy during adulthood is likely to be a factor in the maintenance of reduced weight after successful treatment by weight loss.
CL, AS, and MR contributed equally to the preparation of this manuscript and approved the submitted version.
This research done by the investigators was supported by NIH grants R01 DK30583, R01 DK64773, R01 DK052431, P30 DK063608, P30 DK026687, UL1 TR00040, and the Russell Berrie Foundation Program in the Neurobiology of Body Weight Regulation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge the contributions of Rudolph L. Leibel, Wendy K. Chung, and the Division of Molecular Genetics at Columbia University Irving Medical Center to the studies performed by the authors. We would also like to thank the nursing, nutritional, and support staff of the Clinical Research Resources at Rockefeller University and Columbia University Irving Medical Center for their support during the performance of many of the cited clinical studies. Finally, we would like to thank all our volunteers who participated in these studies.
Ahima, R. S. (2008). Revisiting leptin’s role in obesity and weight loss. J. Clin. Invest. 118, 2380–2383. doi: 10.1172/JCI36284
Ahima, R. S., Prabakaran, D., and Flier, J. S. (1998). Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J. Clin. Invest. 101, 1020–1027. doi: 10.1172/JCI1176
Ainge, H., Thompson, C., Ozanne, S. E., and Rooney, K. B. (2011). A systematic review on animal models of maternal high fat feeding and offspring glycaemic control. Int. J. Obes. (Lond). 35, 325–335. doi: 10.1038/ijo.2010.149
Aubert, R., Suquet, J. P., and Lemonnier, D. (1980). Long-term morphological and metabolic effects of early under- and over-nutrition in mice. J. Nutr. 110, 649–661. doi: 10.1093/jn/110.4.649
Avraham, Y., Dayan, M., Lassri, V., Vorobiev, L., Davidi, N., Chernoguz, D., et al. (2013). Delayed leptin administration after stroke induces neurogenesis and angiogenesis. J. Neurosci. Res. 91, 187–195. doi: 10.1002/jnr.23147
Bandeira, F., Lent, R., and Herculano-Houzel, S. (2009). Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc. Natl. Acad. Sci. U. S. A. 106, 14108–14113. doi: 10.1073/pnas.0804650106
Barker, D. J. (1997). Maternal nutrition, fetal nutrition, and disease in later life. Nutrition 13, 807–813.
Benjannet, S., Rondeau, N., Day, R., Chretien, M., and Seidah, N. G. (1991). PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proc. Natl. Acad. Sci. U. S. A. 88, 3564–3568.
Bereiter, D. A., and Jeanrenaud, B. (1979). Altered neuroanatomical organization in the central nervous system of the genetically obese (ob/ob) mouse. Brain Res. 165, 249–260. doi: 10.1016/0006-8993(79)90557-2
Biddinger, J. E., Lazarenko, R. M., Scott, M. M., and Simerly, R. (2020). Leptin suppresses development of GLP-1 inputs to the paraventricular nucleus of the hypothalamus. Elife 9:e59857. doi: 10.7554/eLife.59857
Boden, G., Chen, X., Mozzoli, M., and Ryan, I. (1996). Effect of fasting on serum leptin in normal human subjects. J. Clin. Endocrinol. Metab. 81, 3419–3423. doi: 10.1210/jcem.81.9.8784108
Bouret, S. G., Draper, S. J., and Simerly, R. B. (2004). Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304, 108–110.
Breton, C. (2013). The hypothalamus-adipose axis is a key target of developmental programming by maternal nutritional manipulation. J. Endocrinol. 216, R19–R31. doi: 10.1530/JOE-12-0157
Breton, C., Lukaszewski, M. A., Risold, P. Y., Enache, M., Guillemot, J., Riviere, G., et al. (2009). Maternal prenatal undernutrition alters the response of POMC neurons to energy status variation in adult male rat offspring. Am. J. Physiol. Endocrinol. Metab. 296, E462–E472. doi: 10.1152/ajpendo.90740.2008
Brown, R. J., Valencia, A., Startzell, M., Cochran, E., Walter, P. J., Garraffo, H. M., et al. (2018). Metreleptin-mediated improvements in insulin sensitivity are independent of food intake in humans with lipodystrophy. J. Clin. Invest. 128, 3504–3516. doi: 10.1172/JCI95476
Butte, N. F., Hopkinson, J. M., and Nicolson, M. A. (1997). Leptin in human reproduction: serum leptin levels in pregnant and lactating women. J. Clin. Endocrinol. Metab. 82, 585–589. doi: 10.1210/jcem.82.2.3731
Calio, M. L., Mosini, A. C., Marinho, D. S., Salles, G. N., Massinhani, F. H., Ko, G. M., et al. (2021). Leptin enhances adult neurogenesis and reduces pathological features in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 148:105219. doi: 10.1016/j.nbd.2020.105219
Campfield, L. A., Smith, F. J., Guisez, Y., Devos, R., and Burn, P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546–549.
Cetin, I., Morpurgo, P. S., Radaelli, T., Taricco, E., Cortelazzi, D., Bellotti, M., et al. (2000). Fetal plasma leptin concentrations: relationship with different intrauterine growth patterns from 19 weeks to term. Pediatr. Res. 48, 646–651. doi: 10.1203/00006450-200011000-00016
Challis, B. G., Coll, A. P., Yeo, G. S., Pinnock, S. B., Dickson, S. L., Thresher, R. R., et al. (2004). Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-36). Proc. Natl. Acad. Sci. U. S. A. 101, 4695–4700.
Chou, S. H., Chamberland, J. P., Liu, X., Matarese, G., Gao, C., Stefanakis, R., et al. (2011). Leptin is an effective treatment for hypothalamic amenorrhea. Proc. Natl. Acad. Sci. U. S. A. 108, 6585–6590. doi: 10.1073/pnas.1015674108
Clark, J. T., Kalra, P. S., Crowley, W. R., and Kalra, S. P. (1984). Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 115, 427–429.
Coleman, D. L. (1973). Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 9, 294–298. doi: 10.1007/bf01221857
Cowley, M. A., Smart, J. L., Rubinstein, M., Cerdan, M. G., Diano, S., Horvath, T. L., et al. (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. doi: 10.1038/35078085
Cripps, R. L., Martin-Gronert, M. S., Archer, Z. A., Hales, C. N., Mercer, J. G., and Ozanne, S. E. (2009). Programming of hypothalamic neuropeptide gene expression in rats by maternal dietary protein content during pregnancy and lactation. Clin. Sci. (Lond). 117, 85–93. doi: 10.1042/CS20080393
de Oliveira Cravo, C., Teixeira, C. V., Passos, M. C., Dutra, S. C., de Moura, E. G., and Ramos, C. (2002). Leptin treatment during the neonatal period is associated with higher food intake and adult body weight in rats. Horm. Metab. Res. 34, 400–405. doi: 10.1055/s-2002-33473
Delahaye, F., Breton, C., Risold, P. Y., Enache, M., Dutriez-Casteloot, I., Laborie, C., et al. (2008). Maternal perinatal undernutrition drastically reduces postnatal leptin surge and affects the development of arcuate nucleus proopiomelanocortin neurons in neonatal male rat pups. Endocrinology 149, 470–475. doi: 10.1210/en.2007-1263
DelParigi, A., Chen, K., Salbe, A. D., Hill, J. O., Wing, R. R., Reiman, E. M., et al. (2007). Successful dieters have increased neural activity in cortical areas involved in the control of behavior. Int. J. Obes. (Lond). 31, 440–448. doi: 10.1038/sj.ijo.0803431
Dulloo, A. G., and Schutz, Y. (2015). Adaptive thermogenesis in resistance to obesity therapies: issues in quantifying thrifty energy expenditure phenotypes in humans. Curr. Obes. Rep. 4, 230–240. doi: 10.1007/s13679-015-0156-9
Entringer, S., Buss, C., Swanson, J. M., Cooper, D. M., Wing, D. A., Waffarn, F., et al. (2012). Fetal programming of body composition, obesity, and metabolic function: the role of intrauterine stress and stress biology. J. Nutr. Metab. 2012:632548.
Entringer, S., and Wadhwa, P. D. (2013). Developmental programming of obesity and metabolic dysfunction: role of prenatal stress and stress biology. Nestle Nutr. Inst. Workshop Ser. 74, 107–120. doi: 10.1159/000348454
Farooqi, I. S., Jebb, S. A., Langmack, G., Lawrence, E., Cheetham, C. H., Prentice, A. M., et al. (1999). Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 341, 879–884. doi: 10.1056/NEJM199909163411204
Ford, E. S., and Dietz, W. H. (2013). Trends in energy intake among adults in the United States: findings from NHANES. Am. J. Clin. Nutr. 97, 848–853.
Friedman, J. M., and Halaas, J. L. (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. doi: 10.1038/27376
Godfrey, K. M., and Barker, D. J. (2000). Fetal nutrition and adult disease. Am. J. Clin. Nutr. 71, 1344S–1352S.
Guenard, F., Deshaies, Y., Cianflone, K., Kral, J. G., Marceau, P., and Vohl, M. C. (2013). Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc. Natl. Acad. Sci. U. S. A. 110, 11439–11444. doi: 10.1073/pnas.1216959110
Hahn, T. M., Breininger, J. F., Baskin, D. G., and Schwartz, M. W. (1998). Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1, 271–272. doi: 10.1038/1082
Halaas, J. L., Gajiwala, K. S., Maffei, M., Cohen, S. L., Chait, B. T., Rabinowitz, D., et al. (1995). Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269, 543–546. doi: 10.1126/science.7624777
Hall, K. D., and Kahan, S. (2018). Maintenance of Lost Weight and Long-Term Management of Obesity. Med. Clin. North Am. 102, 183–197. doi: 10.1016/j.mcna.2017.08.012
Halle, M., Berg, A., Garers, U., Grathwohl, D., Kniser, W., and Keul, J. (1999). Concurrent reductions of serum leptin and lipids during weight loss in obese men with type II diabetes. Am. J. Physiol. 277, E277–E282. doi: 10.1152/ajpendo.1999.277.2.E277
Hardie, L., Trayhurn, P., Abramovich, D., and Fowler, P. (1997). Circulating leptin in women: a longitudinal study in the menstrual cycle and during pregnancy. Clin. Endocrinol. (Oxf). 47, 101–106. doi: 10.1046/j.1365-2265.1997.2441017.x
Hassink, S. G., de Lancey, E., Sheslow, D. V., Smith-Kirwin, S. M., O’Connor, D. M., Considine, R. V., et al. (1997). Placental leptin: an important new growth factor in intrauterine and neonatal development? Pediatrics 100:E1.
Hattersley, A. T., and Tooke, J. E. (1999). The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet 353, 1789–1792. doi: 10.1016/S0140-6736(98)07546-1
Hetherington, A. W., and Ranson, S. W. (1983). Hypothalamic Lesions and Adiposity in the Rat. Nutr. Rev. 41, 124–127. doi: 10.1111/j.1753-4887.1983.tb07169.x
Heymsfield, S. B., Greenberg, A. S., Fujioka, K., Dixon, R. M., Kushner, R., Hunt, T., et al. (1999). Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA 282, 1568–1575. doi: 10.1001/jama.282.16.1568
Highman, T. J., Friedman, J. E., Huston, L. P., Wong, W. W., and Catalano, P. M. (1998). Longitudinal changes in maternal serum leptin concentrations, body composition, and resting metabolic rate in pregnancy. Am. J. Obstet. Gynecol. 178, 1010–1015. doi: 10.1016/s0002-9378(98)70540-x
Hinkle, W., Cordell, M., Leibel, R., Rosenbaum, M., and Hirsch, J. (2013). Effects of reduced weight maintenance and leptin repletion on functional connectivity of the hypothalamus in obese humans. PLoS One 8:e59114. doi: 10.1371/journal.pone.0059114
Hoggard, N., Hunter, L., Lea, R. G., Trayhurn, P., and Mercer, J. G. (2000). Ontogeny of the expression of leptin and its receptor in the murine fetus and placenta. Br. J. Nutr. 83, 317–326. doi: 10.1017/s0007114500000398
Hukshorn, C. J., Menheere, P. P., Westerterp-Plantenga, M. S., and Saris, W. H. (2003). The effect of pegylated human recombinant leptin (PEG-OB) on neuroendocrine adaptations to semi-starvation in overweight men. Eur. J. Endocrinol. 148, 649–655. doi: 10.1530/eje.0.1480649
Hukshorn, C. J., Saris, W. H., Westerterp-Plantenga, M. S., Farid, A. R., Smith, F. J., and Campfield, L. A. (2000). Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J. Clin. Endocrinol. Metab. 85, 4003–4009. doi: 10.1210/jcem.85.11.6955
Hukshorn, C. J., van Dielen, F. M., Buurman, W. A., Westerterp-Plantenga, M. S., Campfield, L. A., and Saris, W. H. (2002). The effect of pegylated recombinant human leptin (PEG-OB) on weight loss and inflammatory status in obese subjects. Int. J. Obes. Relat. Metab. Disord. 26, 504–509. doi: 10.1038/sj.ijo.0801952
Hwa, J. J., Fawzi, A. B., Graziano, M. P., Ghibaudi, L., Williams, P., Van Heek, M., et al. (1997). Leptin increases energy expenditure and selectively promotes fat metabolism in ob/ob mice. Am. J. Physiol. 272, R1204–R1209.
Inadera, H. (2013). Developmental origins of obesity and type 2 diabetes: molecular aspects and role of chemicals. Environ. Health Prev. Med. 18, 185–197. doi: 10.1007/s12199-013-0328-8
Juan De Solis, A., Baquero, A. F., Bennett, C. M., Grove, K. L., and Zeltser, L. M. (2016). Postnatal undernutrition delays a key step in the maturation of hypothalamic feeding circuits. Mol. Metab. 5, 198–209. doi: 10.1016/j.molmet.2016.01.003
Kalra, S. P. (2001). Circumventing leptin resistance for weight control. Proc. Natl. Acad. Sci. U. S. A. 98, 4279–4281. doi: 10.1073/pnas.091101498
Kennedy, G. C. (1953). The role of depot fat in the hypothalamic control of food intake in the rat. Proc. R. Soc. Lond. B Biol. Sci. 140, 578–596. doi: 10.1098/rspb.1953.0009
Khera, A. V., Chaffin, M., Wade, K. H., Zahid, S., Brancale, J., Xia, R., et al. (2019). Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 177, 587–596.e9. doi: 10.1016/j.cell.2019.03.028
Kim, J. G., Suyama, S., Koch, M., Jin, S., Argente-Arizon, P., Argente, J., et al. (2014). Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci. 17, 908–910. doi: 10.1038/nn.3725
Kirk, S. L., Samuelsson, A. M., Argenton, M., Dhonye, H., Kalamatianos, T., Poston, L., et al. (2009). Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One 4:e5870. doi: 10.1371/journal.pone.0005870
Kissileff, H. R., Thornton, J. C., Torres, M. I., Pavlovich, K., Mayer, L. S., Kalari, V., et al. (2012). Leptin reverses declines in satiation in weight-reduced obese humans. Am. J. Clin. Nutr. 95, 309–317. doi: 10.3945/ajcn.111.012385
Korner, J., and Aronne, L. J. (2003). The emerging science of body weight regulation and its impact on obesity treatment. J. Clin. Invest. 111, 565–570. doi: 10.1172/jci17953
Korner, J., Chua, S. C. Jr., Williams, J. A., Leibel, R. L., and Wardlaw, S. L. (1999). Regulation of hypothalamic proopiomelanocortin by leptin in lean and obese rats. Neuroendocrinology 70, 377–383.
Korner, J., Savontaus, E., Chua, S. C. Jr., Leibel, R. L., and Wardlaw, S. L. (2001). Leptin regulation of Agrp and Npy mRNA in the rat hypothalamus. J. Neuroendocrinol. 13, 959–966. doi: 10.1046/j.1365-2826.2001.00716.x
Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., and Gruters, A. (1998). Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157. doi: 10.1038/509
Lage, M., Garcia-Mayor, R. V., Tome, M. A., Cordido, F., Valle-Inclan, F., Considine, R. V., et al. (1999). Serum leptin levels in women throughout pregnancy and the postpartum period and in women suffering spontaneous abortion. Clin. Endocrinol. (Oxf). 50, 211–216. doi: 10.1046/j.1365-2265.1999.00637.x
Lagisz, M., Blair, H., Kenyon, P., Uller, T., Raubenheimer, D., and Nakagawa, S. (2014). Transgenerational effects of caloric restriction on appetite: a meta-analysis. Obes. Rev. 15, 294–309. doi: 10.1111/obr.12138
Lagisz, M., Blair, H., Kenyon, P., Uller, T., Raubenheimer, D., and Nakagawa, S. (2015). Little appetite for obesity: meta-analysis of the effects of maternal obesogenic diets on offspring food intake and body mass in rodents. Int. J. Obes. (Lond). 39, 1669–1678. doi: 10.1038/ijo.2015.160
Larrouy, D., Barbe, P., Valle, C., Dejean, S., Pelloux, V., Thalamas, C., et al. (2008). Gene expression profiling of human skeletal muscle in response to stabilized weight loss. Am. J. Clin. Nutr. 88, 125–132. doi: 10.1093/ajcn/88.1.125
LeDuc, C. A., and Leibel, R. L. (2019). Auto-Regulation of leptin neurobiology. Cell Metab. 30, 614–616. doi: 10.1016/j.cmet.2019.09.006
Lee, J. H., Reed, D. R., and Price, R. A. (2001). Leptin resistance is associated with extreme obesity and aggregates in families. Int. J. Obes. Relat. Metab. Disord. 25, 1471–1473. doi: 10.1038/sj.ijo.0801736
Leibel, R. L. (2002). The role of leptin in the control of body weight. Nutr. Rev. 60, S15–9. doi: 10.1301/002966402320634788
Leibel, R. L., and Hirsch, J. (1984). Diminished energy requirements in reduced-obese patients. Metabolism 33, 164–170. doi: 10.1016/0026-0495(84)90130-6
Leibel, R. L., and Rosenbaum, M. (2010). “Metabolic responses to weight perturbation,” in Novel Insights into Adipose Cell Functions, eds Y. Christen, K. Clément, and B. M. Spiegelman (Berlin: Springer), 121–133. doi: 10.1007/978-3-642-13517-0_12
Loos, R. J. (2018). The genetics of adiposity. Curr. Opin. Genet. Dev. 50, 86–95. doi: 10.1016/j.gde.2018.02.009
Luquet, S., Perez, F. A., Hnasko, T. S., and Palmiter, R. D. (2005). NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 310, 683–685. doi: 10.1126/science.1115524
Mackintosh, R. M., and Hirsch, J. (2001). The effects of leptin administration in non-obese human subjects. Obes. Res. 9, 462–469. doi: 10.1038/oby.2001.60
Manne, J., Argeson, A. C., and Siracusa, L. D. (1995). Mechanisms for the pleiotropic effects of the agouti gene. Proc. Natl. Acad. Sci. U. S. A. 92, 4721–4724. doi: 10.1073/pnas.92.11.4721
Marangon, P. B., Mecawi, A. S., Antunes-Rodrigues, J., and Elias, L. L. K. (2020). Perinatal over- and underfeeding affect hypothalamic leptin and ghrelin neuroendocrine responses in adult rats. Physiol. Behav. 215:112793. doi: 10.1016/j.physbeh.2019.112793
Marco, A., Kisliouk, T., Tabachnik, T., Meiri, N., and Weller, A. (2014). Overweight and CpG methylation of the Pomc promoter in offspring of high-fat-diet-fed dams are not “reprogrammed” by regular chow diet in rats. FASEB J. 28, 4148–4157. doi: 10.1096/fj.14-255620
Mayer, J. (1955). Regulation of energy intake and the body weight: the glucostatic theory and the lipostatic hypothesis. Ann. N. Y. Acad. Sci. 63, 15–43. doi: 10.1111/j.1749-6632.1955.tb36543.x
McDuffie, J. R., Riggs, P. A., Calis, K. A., Freedman, R. J., Oral, E. A., DePaoli, A. M., et al. (2004). Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J. Clin. Endocrinol. Metab. 89, 4258–4263. doi: 10.1210/jc.2003-031868
Mistry, A. M., Swick, A., and Romsos, D. R. (1999). Leptin alters metabolic rates before acquisition of its anorectic effect in developing neonatal mice. Am. J. Physiol. 277, R742–R747. doi: 10.1152/ajpregu.1999.277.3.R742
Mix, H., Manns, M. P., Wagner, S., Widjaja, A., and Brabant, G. (1999). Expression of leptin and its receptor in the human stomach. Gastroenterology 117:509. doi: 10.1053/gast.1999.0029900509b
Montague, C. T., Farooqi, I. S., Whitehead, J. P., Soos, M. A., Rau, H., Wareham, N. J., et al. (1997). Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908.
Moon, H. S., Matarese, G., Brennan, A. M., Chamberland, J. P., Liu, X., Fiorenza, C. G., et al. (2011). Efficacy of metreleptin in obese patients with type 2 diabetes: cellular and molecular pathways underlying leptin tolerance. Diabetes 60, 1647–1656. doi: 10.2337/db10-1791
Moore, V. M., Cockington, R. A., Ryan, P., and Robinson, J. S. (1999). The relationship between birth weight and blood pressure amplifies from childhood to adulthood. J. Hypertens. 17, 883–888. doi: 10.1097/00004872-199917070-00003
Morabito, M. V., Ravussin, Y., Mueller, B. R., Skowronski, A. A., Watanabe, K., Foo, K. S., et al. (2017). Weight perturbation alters leptin signal transduction in a region-specific manner throughout the brain. PLoS One 12:e0168226. doi: 10.1371/journal.pone.0168226
Myers, M. G., Cowley, M. A., and Munzberg, H. (2008). Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 70, 537–556. doi: 10.1146/annurev.physiol.70.113006.100707
Oken, E., Kleinman, K. P., Belfort, M. B., Hammitt, J. K., and Gillman, M. W. (2009). Associations of gestational weight gain with short- and longer-term maternal and child health outcomes. Am. J. Epidemiol. 170, 173–180. doi: 10.1093/aje/kwp101
Ollmann, M. M., Wilson, B. D., Yang, Y. K., Kerns, J. A., Chen, Y., Gantz, I., et al. (1997). Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 278, 135–138. doi: 10.1126/science.278.5335.135
Oral, E. A., Simha, V., Ruiz, E., Andewelt, A., Premkumar, A., Snell, P., et al. (2002). Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 346, 570–578.
Ozanne, S. E., Lewis, R., Jennings, B. J., and Hales, C. N. (2004). Early programming of weight gain in mice prevents the induction of obesity by a highly palatable diet. Clin. Sci. (Lond). 106, 141–145. doi: 10.1042/CS20030278
Pan, W., Hsuchou, H., He, Y., Sakharkar, A., Cain, C., Yu, C., et al. (2008). Astrocyte leptin receptor (ObR) and leptin transport in adult-onset obese mice. Endocrinology 149, 2798–2806. doi: 10.1210/en.2007-1673
Pan, W., and Mg Myers, J. (2018). Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 19, 95–105. doi: 10.1038/nrn.2017.168
Park, J. Y., Javor, E. D., Cochran, E. K., DePaoli, A. M., and Gorden, P. (2007). Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism 56, 508–516. doi: 10.1016/j.metabol.2006.11.010
Patterson, C. M., Bouret, S. G., Park, S., Irani, B. G., Dunn-Meynell, A. A., and Levin, B. E. (2010). Large litter rearing enhances leptin sensitivity and protects selectively bred diet-induced obese rats from becoming obese. Endocrinology 151, 4270–4279. doi: 10.1210/en.2010-0401
Pelleymounter, M. A., Cullen, M. J., Baker, M. B., Hecht, R., Winters, D., Boone, T., et al. (1995). Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269, 540–543. doi: 10.1126/science.7624776
Pettitt, D. J., Aleck, K. A., Baird, H. R., Carraher, M. J., Bennett, P. H., and Knowler, W. C. (1988). Congenital susceptibility to NIDDM. Role of intrauterine environment. Diabetes 37, 622–628. doi: 10.2337/diab.37.5.622
Pettitt, D. J., Baird, H. R., Aleck, K. A., Bennett, P. H., and Knowler, W. C. (1983). Excessive obesity in offspring of Pima Indian women with diabetes during pregnancy. N. Engl. J. Med. 308, 242–245. doi: 10.1056/NEJM198302033080502
Pettitt, D. J., Knowler, W. C., Bennett, P. H., Aleck, K. A., and Baird, H. R. (1987). Obesity in offspring of diabetic Pima Indian women despite normal birth weight. Diab. Care 10, 76–80. doi: 10.2337/diacare.10.1.76
Pico, C., Oliver, P., Sanchez, J., Miralles, O., Caimari, A., Priego, T., et al. (2007). The intake of physiological doses of leptin during lactation in rats prevents obesity in later life. Int. J. Obes. (Lond). 31, 1199–1209. doi: 10.1038/sj.ijo.0803585
Plagemann, A., Harder, T., Brunn, M., Harder, A., Roepke, K., Wittrock-Staar, M., et al. (2009). Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J. Physiol. 587, 4963–4976. doi: 10.1113/jphysiol.2009.176156
Qian, S., Chen, H., Weingarth, D., Trumbauer, M. E., Novi, D. E., Guan, X., et al. (2002). Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol. Cell Biol. 22, 5027–5035. doi: 10.1128/mcb.22.14.5027-5035.2002
Rajia, S., Chen, H., and Morris, M. J. (2010). Maternal overnutrition impacts offspring adiposity and brain appetite markers-modulation by postweaning diet. J. Neuroendocrinol. 22, 905–914. doi: 10.1111/j.1365-2826.2010.02005.x
Ravelli, A. C., van der Meulen, J. H., Michels, R. P., Osmond, C., Barker, D. J., Hales, C. N., et al. (1998). Glucose tolerance in adults after prenatal exposure to famine. Lancet 351, 173–177. doi: 10.1016/s0140-6736(97)07244-9
Ravelli, G. P., Stein, Z. A., and Susser, M. W. (1976). Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 295, 349–353. doi: 10.1056/NEJM197608122950701
Ravussin, E., Smith, S. R., Mitchell, J. A., Shringarpure, R., Shan, K., Maier, H., et al. (2009). Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity (Silver Spring) 17, 1736–1743. doi: 10.1038/oby.2009.184
Reynolds, R. M., Jacobsen, G. H., and Drake, A. J. (2013). What is the evidence in humans that DNA methylation changes link events in utero and later life disease? Clin. Endocrinol. (Oxf). 78, 814–822. doi: 10.1111/cen.12164
Ribaroff, G. A., Wastnedge, E., Drake, A. J., Sharpe, R. M., and Chambers, T. J. G. (2017). Animal models of maternal high fat diet exposure and effects on metabolism in offspring: a meta-regression analysis. Obes. Rev. 18, 673–686. doi: 10.1111/obr.12524
Rizza, R. A., Mandarino, L. J., and Gerich, J. E. (1981). Dose-response characteristics for effects of insulin on production and utilization of glucose in man. Am. J. Physiol. 240, E630–E639.
Rosenbaum, M., Goldsmith, R., Bloomfield, D., Magnano, A., Weimer, L., Heymsfield, S., et al. (2005). Low-dose leptin reverses skeletal muscle, autonomic, and neuroendocrine adaptations to maintenance of reduced weight. J. Clin. Invest. 115, 3579–3586. doi: 10.1172/JCI25977
Rosenbaum, M., Goldsmith, R. L., Haddad, F., Baldwin, K. M., Smiley, R., Gallagher, D., et al. (2018a). Triiodothyronine and leptin repletion in humans similarly reverse weight-loss-induced changes in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 315, E771–E779. doi: 10.1152/ajpendo.00116.2018
Rosenbaum, M., Heaner, M., Goldsmith, R. L., Christian Schulze, P., Shukla, A., Shen, W., et al. (2018b). Resistance training reduces skeletal muscle work efficiency in weight-reduced and non-weight-reduced subjects. Obesity (Silver Spring) 26, 1576–1583. doi: 10.1002/oby.22274
Rosenbaum, M., Sy, M., Pavlovich, K., Leibel, R. L., and Hirsch, J. (2008b). Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J. Clin. Invest. 118, 2583–2591. doi: 10.1172/JCI35055
Rosenbaum, M., Hirsch, J., Gallagher, D. A., and Leibel, R. L. (2008a). Long-term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am. J. Clin. Nutr. 88, 906–912. doi: 10.1093/ajcn/88.4.906
Rosenbaum, M., and Leibel, R. L. (1998). Leptin: a molecule integrating somatic energy stores, energy expenditure and fertility. Trends Endocrinol. Metab. 9, 117–124. doi: 10.1016/s1043-2760(98)00028-9
Rosenbaum, M., and Leibel, R. L. (2014). 20 years of leptin: role of leptin in energy homeostasis in humans. J. Endocrinol. 223, T83–T96.
Rottkamp, D. M., Rudenko, I. A., Maier, M. T., Roshanbin, S., Yulyaningsih, E., Perez, L., et al. (2015). Leptin potentiates astrogenesis in the developing hypothalamus. Mol. Metab. 4, 881–889. doi: 10.1016/j.molmet.2015.08.005
Sanchez, J., Oliver, P., Miralles, O., Ceresi, E., Pico, C., and Palou, A. (2005). Leptin orally supplied to neonate rats is directly uptaken by the immature stomach and may regulate short-term feeding. Endocrinology 146, 2575–2582. doi: 10.1210/en.2005-0112
Sattar, N., Greer, I. A., Pirwani, I., Gibson, J., and Wallace, A. M. (1998). Leptin levels in pregnancy: marker for fat accumulation and mobilization? Acta Obstet. Gynecol. Scand. 77, 278–283. doi: 10.1034/j.1600-0412.1998.770304.x
Scarpace, P. J., and Zhang, Y. (2007). Elevated leptin: consequence or cause of obesity? Front. Biosci. 12, 3531–3544. doi: 10.2741/2332
Schubring, C., Siebler, T., Kratzsch, J., Englaro, P., Blum, W. F., Triep, K., et al. (1999). Leptin serum concentrations in healthy neonates within the first week of life: relation to insulin and growth hormone levels, skinfold thickness, body mass index and weight. Clin. Endocrinol. (Oxf). 51, 199–204. doi: 10.1046/j.1365-2265.1999.00761.x
Schwartz, M. W., Woods, S. C., Porte, D. Jr., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671.
Seeley, R. J., Yagaloff, K. A., Fisher, S. L., Burn, P., Thiele, T. E., van Dijk, G., et al. (1997). Melanocortin receptors in leptin effects. Nature 390, 349.
Shick, S. M., Wing, R. R., Klem, M. L., McGuire, M. T., Hill, J. O., and Seagle, H. (1998). Persons successful at long-term weight loss and maintenance continue to consume a low-energy, low-fat diet. J. Am. Diet. Assoc. 98, 408–413. doi: 10.1016/s0002-8223(98)00093-5
Sivan, E., Whittaker, P. G., Sinha, D., Homko, C. J., Lin, M., Reece, E. A., et al. (1998). Leptin in human pregnancy: the relationship with gestational hormones. Am. J. Obstet. Gynecol. 179, 1128–1132. doi: 10.1016/s0002-9378(98)70118-8
Skowronski, A. A., LeDuc, C. A., Foo, K. S., Goffer, Y., Burnett, L. C., Egli, D., et al. (2020). Physiological consequences of transient hyperleptinemia during discrete developmental periods on body weight in mice. Sci. Transl. Med. 12:eaax6629. doi: 10.1126/scitranslmed.aax6629
Skowronski, A. A., Shaulson, E. D., Leibel, R. L., and LeDuc, C. A. (2021). The postnatal leptin surge in mice is variable in both time and intensity and reflects nutritional status. Int. J. Obes. [Epub Online ahead of print]. doi: 10.1038/s41366-021-00957-5
Steppan, C. M., and Swick, A. G. (1999). A role for leptin in brain development. Biochem. Biophys. Res. Commun. 256, 600–602.
Sweeney, P., Bedenbaugh, M. N., Maldonado, J., Pan, P., Fowler, K., Williams, S. Y., et al. (2021). The melanocortin-3 receptor is a pharmacological target for the regulation of anorexia. Sci. Transl. Med. 13:eabd6434. doi: 10.1126/scitranslmed.abd6434
Tatemoto, K., Carlquist, M., and Mutt, V. (1982). Neuropeptide Y–a novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature 296, 659–660. doi: 10.1038/296659a0
Thomas, J. G., Bond, D. S., Phelan, S., Hill, J. O., and Wing, R. R. (2014). Weight-loss maintenance for 10 years in the national weight control registry. Am. J. Prev. Med. 46, 17–23.
Udagawa, J., Hashimoto, R., Suzuki, H., Hatta, T., Sotomaru, Y., Hioki, K., et al. (2006). The role of leptin in the development of the cerebral cortex in mouse embryos. Endocrinology 147, 647–658.
Vickers, M. H., Breier, B. H., Cutfield, W. S., Hofman, P. L., and Gluckman, P. D. (2000). Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol. Endocrinol. Metab. 279, E83–E87. doi: 10.1152/ajpendo.2000.279.1.E83
Vickers, M. H., Gluckman, P. D., Coveny, A. H., Hofman, P. L., Cutfield, W. S., Gertler, A., et al. (2008). The effect of neonatal leptin treatment on postnatal weight gain in male rats is dependent on maternal nutritional status during pregnancy. Endocrinology 149, 1906–1913. doi: 10.1210/en.2007-0981
Vogt, M. C., Paeger, L., Hess, S., Steculorum, S. M., Awazawa, M., Hampel, B., et al. (2014). Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 156, 495–509. doi: 10.1016/j.cell.2014.01.008
Welle, S. L., Amatruda, J. M., Forbes, G. B., and Lockwood, D. H. (1984). Resting metabolic rates of obese women after rapid weight loss. J. Clin. Endocrinol. Metab. 59, 41–44. doi: 10.1210/jcem-59-1-41
Welt, C. K., Chan, J. L., Bullen, J., Murphy, R., Smith, P., DePaoli, A. M., et al. (2004). Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 351, 987–997.
Wing, R. R., and Hill, J. O. (2001). Successful weight loss maintenance. Annu. Rev. Nutr. 21, 323–341.
Yarbrough, D. E., Barrett-Connor, E., Kritz-Silverstein, D., and Wingard, D. L. (1998). Birth weight, adult weight, and girth as predictors of the metabolic syndrome in postmenopausal women: the Rancho Bernardo Study. Diab. Care 21, 1652–1658. doi: 10.2337/diacare.21.10.1652
Yura, S., Itoh, H., Sagawa, N., Yamamoto, H., Masuzaki, H., Nakao, K., et al. (2005). Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab. 1, 371–378. doi: 10.1016/j.cmet.2005.05.005
Zhang, Y., Proenca, R., Maffei, M., Barone, M., Leopold, L., and Friedman, J. M. (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432. doi: 10.1038/372425a0
Zhao, S., Zhu, Y., Schultz, R. D., Li, N., He, Z., Zhang, Z., et al. (2019). Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab. 30, 706–719.e6. doi: 10.1016/j.cmet.2019.08.005
Keywords: leptin, adipose tissue, development, maintenance, energy balance
Citation: LeDuc CA, Skowronski AA and Rosenbaum M (2021) The Role of Leptin in the Development of Energy Homeostatic Systems and the Maintenance of Body Weight. Front. Physiol. 12:789519. doi: 10.3389/fphys.2021.789519
Received: 05 October 2021; Accepted: 17 November 2021;
Published: 10 December 2021.
Edited by:
Harbindarjeet Singh, Universiti Teknologi MARA, MalaysiaReviewed by:
Nu-Chu Liang, University of Illinois at Urbana-Champaign, United StatesCopyright © 2021 LeDuc, Skowronski and Rosenbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Rosenbaum, bXI0NzVAY3VtYy5jb2x1bWJpYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.