 Tyler Ralph-Epps1†
Tyler Ralph-Epps1† Miriam L. Greenberg
Miriam L. Greenberg
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 28 October 2021
Sec. Lipid and Fatty Acid Research
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.768411
This article is part of the Research TopicThe Multifaceted Roles of Lipids in Physiological and Pathophysiological StatesView all 12 articles
Saccharomyces cerevisiae, commonly known as baker’s yeast, is one of the most comprehensively studied model organisms in science. Yeast has been used to study a wide variety of human diseases, and the yeast model system has proved to be an especially amenable tool for the study of lipids and lipid-related pathophysiologies, a topic that has gained considerable attention in recent years. This review focuses on how yeast has contributed to our understanding of the mitochondrial phospholipid cardiolipin (CL) and its role in Barth syndrome (BTHS), a genetic disorder characterized by partial or complete loss of function of the CL remodeling enzyme tafazzin. Defective tafazzin causes perturbation of CL metabolism, resulting in many downstream cellular consequences and clinical pathologies that are discussed herein. The influence of yeast research in the lipid-related pathophysiologies of Alzheimer’s and Parkinson’s diseases is also summarized.
Saccharomyces cerevisiae is a powerful model system used to study biological processes and human diseases. In addition to investigating the pathophysiology of diseases, yeast is also used as a model for developing and testing potential treatments. An excellent example of the power of the yeast model is the use of yeast cardiolipin (CL) synthesis mutants to understand the metabolic abnormalities in Barth syndrome (BTHS), a rare genetic disorder caused by mutations in the CL-remodeling enzyme tafazzin (Bione et al., 1996; Vreken et al., 2000). CL is a unique phospholipid localized primarily in the inner mitochondrial membrane (IMM). Yeast CL mutants have been pivotal in elucidating the role of this lipid in bioenergetics (Paradies et al., 2014; Ren et al., 2014; Raja et al., 2017a), mitochondrial metabolism (Houtkooper and Vaz, 2008; Raja et al., 2019), and programmed cell death (Manon, 2004; Eisenberg and Buttner, 2014; Lou et al., 2018b) among other cellular functions. This review aims to demonstrate how the yeast model has led the way for BTHS studies and contributed to recent advances in our understanding of other human diseases.
In 1983, physician Peter Barth reported the first description of the disorder that bears his name, describing it as “an X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle, and neutrophil leukocytes” (Barth et al., 1983). It was not until 1996 that the cause of BTHS was linked to mutations in the TAZ gene (Bione et al., 1996). Vreken et al. (2000) discovered that TAZ mutations lead to a profound defect in CL remodeling. During 2003–2004, the first research model for studying BTHS – yeast taz1Δ – was constructed (Vaz et al., 2003; Gu et al., 2004; Ma et al., 2004).
Since the emergence of the yeast taz1Δ model, numerous other BTHS model systems have been developed in different organisms through targeted disruption of the tafazzin gene. In 2006, the first whole-animal models were generated in fruit flies (Xu et al., 2006) and zebrafish (Khuchua et al., 2006). Subsequent efforts in 2011–2012 led to the development of the first mouse models of BTHS, facilitating tissue-specific studies of tafazzin deficiency in organs such as the heart and skeletal muscle (Acehan et al., 2011; Soustek et al., 2011; Phoon et al., 2012). More recently, BTHS cell models have been developed in immortalized mammalian cell lines, including mouse C2C12 myoblasts (Lou et al., 2018a) and human HEK293 kidney cells, which provide the added experimental benefit of having isogenic control cells.
As an X-linked recessive disorder, BTHS is predominantly diagnosed in male patients. It has been suggested that females carrying single-allele TAZ mutations exhibit a skewed pattern of X chromosome inactivation, resulting in a normal clinical presentation (Orstavik et al., 1998). BTHS is exceptionally rare, with only 151 living patients identified worldwide in 2012 (Clarke et al., 2013). Approximately 10 new BTHS cases are diagnosed each year in the United States, which translates to a prevalence of 1:300,000–400,000 live births (Clarke et al., 2013). However, it is likely that BTHS is underdiagnosed due to premature infant mortality and misdiagnosis of children presenting with cardiomyopathies.
BTHS results from mutations in the Tafazzin (TAZ) gene (originally referred to as G4.5). TAZ is a mitochondrial transacylase that re-acylates monolysocardiolipin (MLCL) by adding predominantly unsaturated acyl chains (Barth et al., 2004; Schlame and Xu, 2020). It is the primary enzyme that conducts this function, and as such, TAZ mutations result in elevated MLCL levels, decreased overall CL, and increased saturated fatty acid content in CL (Vreken et al., 2000; Schlame et al., 2003; Gu et al., 2004; Valianpour et al., 2005).
TAZ is located on chromosome Xq28 and contains 11 exons (Bolhuis et al., 1991; Bione et al., 1996). Although multiple mRNA splice variants exist, the only detectable form of TAZ protein in human fibroblasts lacks exon 5 (Lu et al., 2016). However, BTHS-associated mutations have been identified in all TAZ exons, including exon 5, suggesting that full-length TAZ protein is also physiologically relevant in vivo (Cantlay et al., 1999; Gonzalez, 2005). To date, more than 180 pathogenic TAZ gene mutations have been identified, ranging from single nucleotide polymorphisms to whole-gene deletion (Singh et al., 2009). A major enigma in BTHS research is the apparent discrepancy between specific TAZ mutations and the clinical phenotypes they result in. For example, individuals sharing an identical mutation can have contrasting clinical presentations that range from severe heart failure and hypotonia to being nearly asymptomatic (Ronvelia et al., 2012).
BTHS and its clinical manifestations have been previously discussed in other reviews (Raja et al., 2017b; Ghosh et al., 2019; Taylor et al., 2021; Zegallai and Hatch, 2021). Cardiomyopathy is the major clinical manifestation of BTHS, and all identified patients have developed cardiomyopathy at some point in their lives. Dilated cardiomyopathy is the most common form in BTHS patients and is often associated with hypertrophy, left-ventricular noncompaction, arrhythmia, conduction defects, and endocardial fibroelastosis (a heart disorder in children characterized by diffuse thickening of the endocardium; Brady et al., 2006; Raja et al., 2017b). Hypertrophic and dilated phases can be recurring over a patient’s lifetime (Ferreira et al., 2014). Cardiomyopathy increases the risk of arrhythmia, conduction defects, and congestive heart failure and may lead to sudden cardiac death (Spencer et al., 2006; Yen et al., 2008). In addition, cardiomyopathy in BTHS can be diagnosed late or misdiagnosed, leading to cardiac failure (Spencer et al., 2006; Mangat et al., 2007).
Fortunately, BTHS patient outcomes have improved significantly in the past two decades. Patients born after the year 2000 have a 5-year survival rate of 70% compared to 22% for those born before 2000 (Rigaud et al., 2013). This change suggests that early identification and management of heart dysfunction can significantly improve survival rates for BTHS patients. Although the molecular mechanisms are not known, studies have suggested that several factors, including mitochondrial dysfunction, defective mitochondrial protein import, autophagy, lipid storage myopathy, reduced glucose oxidation, and deficient muscle development, can all contribute to the onset of cardiomyopathy in BTHS (Shen et al., 2015; Greenwell et al., 2021).
Skeletal myopathy is widely observed in BTHS, and patients often present with a combination of muscle weakness and wasting, delayed gross motor development, pre-pubescent growth delay, and/or hypotonia (Spencer et al., 2006, 2011; Bittel et al., 2018). In BTHS, this condition is usually non-progressive and mainly affects proximal skeletal muscle (Clarke et al., 2013; Ferreira et al., 2014; Mazar et al., 2019) Growth delay usually regresses over time and is often followed by accelerated growth during mid and late puberty (Reynolds et al., 2015). Similar to cardiomyopathy, the mechanism underlying development of skeletal myopathy in BTHS is not well understood.
In a self-assessment, BTHS patients identified exercise intolerance as the clinical feature that most negatively impacts their daily life. The reduced capacity for physical activity in BTHS is thought to result from both cardiac impairment (i.e., overall endurance) and diminished skeletal muscle oxygen utilization (Spencer et al., 2011). Not only does exercise intolerance pose a physical limitation on the ability of patients to independently perform everyday tasks, but it also serves as a psychosocial barrier that likely contributes to reports of lower quality of life ratings from BTHS patients relative to their peers (Storch et al., 2009; Mazar et al., 2019).
The severity of neutropenia in BTHS ranges from mild (benign, transient neutropenia) to severe (congenital neutropenia affecting multiple organs; Folsi et al., 2014). Bacterial or viral infections resulting from neutropenia can cause significant complications throughout a patient’s life, including the risk of death by sepsis in the most extreme cases. According to the first report by Barth et al. (1983) three out of his seven patients died prematurely due to septicemia. The molecular mechanisms underlying the development of neutropenia in BTHS are still largely unknown.
While the above pathologies constitute the core clinical features of BTHS, a range of other clinical manifestations have been reported. Some patients develop facial dysmorphism characterized by a tall, broad forehead, round face, full cheeks, and large ears (Ferreira et al., 2014). Additionally, dysmorphism of the feet resulting in talipes equinovarus (clubfoot) has been reported at a higher incidence in BTHS patients (Ades et al., 1993; Spencer et al., 2011). Cognitive and neurological phenotypes have also been described. For example, a study of 15 adolescent BTHS patients found reduced abilities in mathematics and visual spatial tasks (Mazzocco et al., 2007), and a recent study also identified deficiencies in balance and motion reaction time in a group of 33 BTHS patients relative to age-matched controls (Hornby et al., 2019). Other phenotypes include a strong gag reflex (Reynolds et al., 2012), sideways curvature of the spine (Roberts et al., 2012), and increased male fetal loss, stillbirth, and neonatal death (Marziliano et al., 2007; Steward et al., 2010).
In order to comprehend how the yeast model has furthered our understanding of BTHS pathophysiology, it is necessary to understand the details of the CL biosynthetic pathway, many of which were first discovered in yeast. CL is a dimeric phospholipid in which two phosphatidyl groups are connected by a central glycerol molecule (Lecocq and Ballou, 1964). It is synthesized and localized predominantly in the inner leaflet of the IMM (Hostetler and van den Bosch, 1972; Krebs et al., 1979; Schlame and Haldar, 1993; Gebert et al., 2009; Joshi et al., 2009; Schlame and Ren, 2009; Sparagna and Lesnefsky, 2009; Osman et al., 2011).
CL biosynthesis is a four-step process that utilizes phosphatidic acid (PA) as substrate (Figure 1). PA is synthesized in both the endoplasmic reticulum (ER) and the outer leaflet of the outer mitochondrial membrane (OMM; Chakraborty et al., 1999). In both cases, the translocation of PA from the OMM to the inner leaflet of the IMM via the yeast Ups1/Mdm35 lipid transport complex (PRELID1/TRIAP1 in humans) is essential for CL synthesis (Potting et al., 2010; Tamura et al., 2010; Connerth et al., 2012). In the first step of the biosynthetic pathway, PA in the IMM is converted into cytidine diphosphate diacylglycerol (CDP-DAG) by Tam41 in yeast (TAM41 in humans; Blunsom et al., 2018). Next, the yeast mitochondrial enzyme Pgs1 or its human homolog, PGS1, converts CDP-DAG into phosphatidylglycerol phosphate (PGP) by transferring a phosphatidyl group from CDP-DAG onto glycerol-3-phosphate (G3P; Chang et al., 1998a). PGP is then dephosphorylated to phosphatidylglycerol (PG) by the yeast enzyme Gep4 (PTPMT1 in humans; Osman et al., 2010; Zhang et al., 2011). Finally, a phosphatidyl group from CDP-DAG is transferred to PG by the yeast enzyme Crd1 (hCLS1 in humans) to synthesize nascent CL (Schlame and Greenberg, 1997; Chang et al., 1998b).
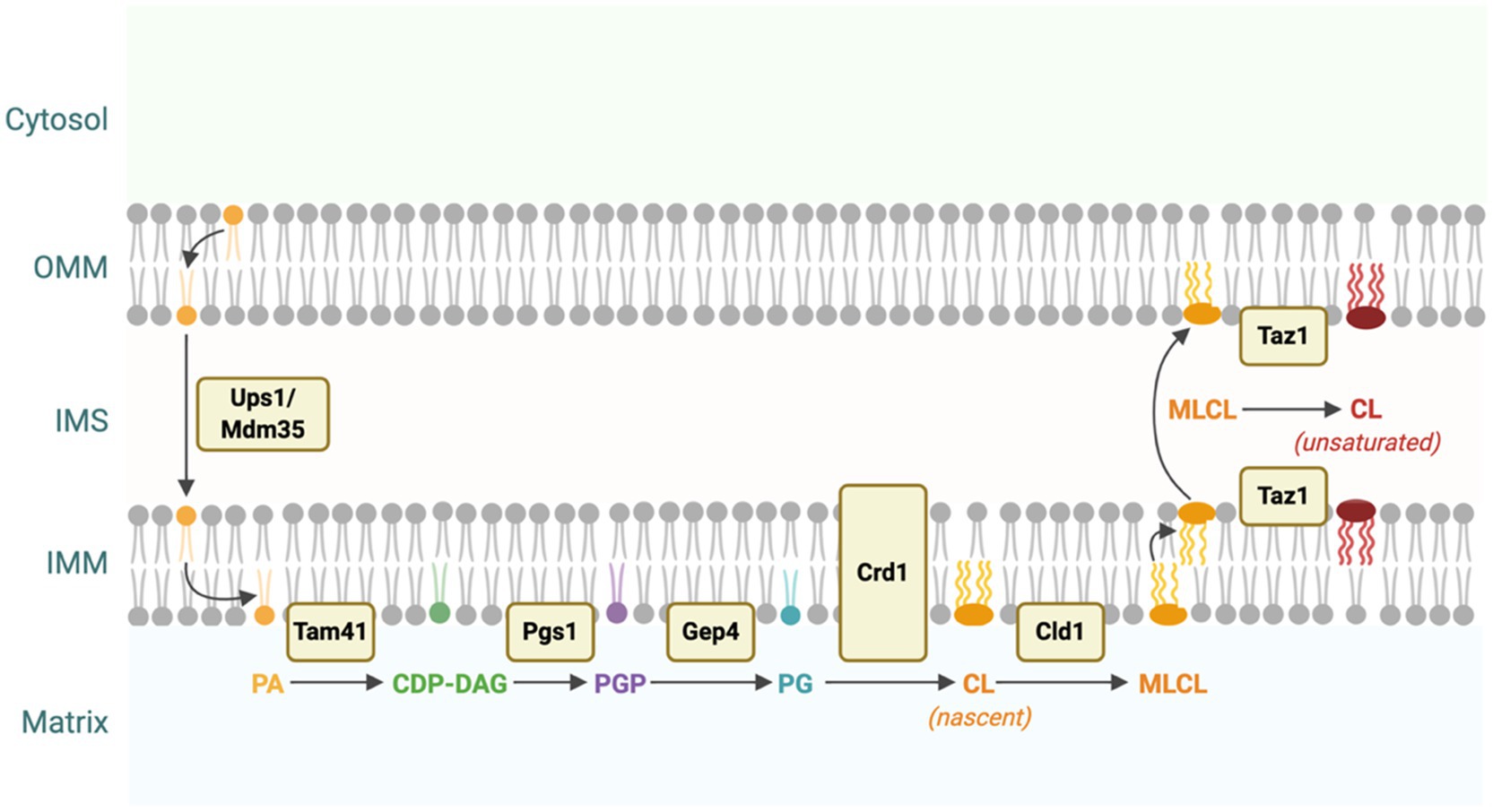
Figure 1. Cardiolipin (CL) biosynthesis in yeast. CL is synthesized from phosphatidic acid (PA) through a four-step process. PA is translocated from the outer mitochondrial membrane (OMM) to the IMM via the Ups1/Mdm35 protein complex (Potting et al., 2010; Tamura et al., 2010; Connerth et al., 2012). PA is converted into cytidine diphosphate diacylglycerol (CDP-DAG), phosphatidylglycerol phosphate (PGP), and then phosphatidylglycerol (PG) by Tam41, Pgs1, and Gep4, respectively (Chang et al., 1998a; Osman et al., 2010; Blunsom et al., 2018). Finally, PG is converted to nascent CL by the enzymatic activity of Crd1. Nascent CL, which contains predominantly saturated acyl chains, is remodeled through several cycles of deacylation and reacylation catalyzed by Cld1 and Taz1 to form primarily unsaturated CL (Xu et al., 2006; Beranek et al., 2009). This figure was created with BioRender.com.
Newly synthesized CL undergoes a unique remodeling process in which saturated acyl chains are replaced with unsaturated acyl chains through several cycles of deacylation and reacylation. The yeast enzyme Cld1 is responsible for the first step in CL remodeling by deacylating CL to form MLCL (Beranek et al., 2009). In the second step, MLCL is reacylated by tafazzin (Taz1 in yeast or TAZ in humans) to form primarily unsaturated CL (Figure 1; Xu et al., 2006).
In mammals, multiple enzymes are capable of deacylating CL, though none of them are direct homologs of the yeast enzyme Cld1. These include iPLA2γ, iPLA2β, cPLA2, and sPLA2 (Buckland et al., 1998; Mancuso et al., 2007; Dennis et al., 2011; Hsu et al., 2013). In addition to TAZ, MLCLAT1 and ALCAT1 can also catalyze the reacylation of MLCL in mammalian cells, while there are no direct homologs of these two enzymes in yeast (Ma et al., 1999; Cao et al., 2004; Li et al., 2010; Mejia et al., 2018). As a result of diminished tafazzin function, BTHS is characterized by an increase in MLCL levels, along with a decrease in unsaturated and total CL (Jiang et al., 1997; Xu et al., 2006; Lou et al., 2018a; Wang et al., 2020). The fact that these phenotypes arise despite the presence of MLCLAT1 and ALCAT1 suggests that TAZ is the primary enzyme responsible for reacylation of MLCL under physiological conditions (Saric et al., 2015).
Yeast is an excellent model for studying genetic disorders. In 1996, S. cerevisiae was the first eukaryote for which a full genome sequence was assembled (Goffeau et al., 1996; Giaever and Nislow, 2014). Subsequently, researchers generated a deletion collection containing representative mutant strains for each nonessential gene in the yeast genome (~4,800 genes; Giaever and Nislow, 2014). Using these tools, homologs of over 23% of all human genes have been identified and studied in yeast (Kachroo et al., 2015). One of the most striking discoveries from these endeavors was that many genes encoding lipid pathways are conserved from yeast to humans, thus making yeast a useful model to interrogate human diseases of lipid metabolism. The ability of yeast to grow both as haploid or diploid cells allows for the construction of double mutants by crossing single mutants, inducing sporulation, and screening the resulting haploids. In addition to being genetically tractable, yeast cells are nonpathogenic, have a short generation time, and are easy and inexpensive to culture in the laboratory.
The CL biosynthetic pathway is conserved from yeast to humans, and yeast mutants have been constructed for each step of this pathway. Among these, the crd1Δ mutant (Jiang et al., 1997; Tuller et al., 1998; Chang et al., 1998b), which lacks CL synthase and cannot synthesize CL, and the taz1Δ mutant (Vaz et al., 2003; Gu et al., 2004; Ma et al., 2004), lacking tafazzin, have been pivotal in elucidating the cellular roles of CL and understanding BTHS pathology. Yeast mutants have also been generated to recapitulate and test the functional significance of human TAZ mutations identified in BTHS patients (Claypool et al., 2006, 2011). The following sections detail pioneering discoveries relevant to BTHS pathophysiology that were first made using yeast mutants.
CL comprises 15–20% of the phospholipid content in the IMM where it plays a pivotal role in energy metabolism (Pennington et al., 2019). The mitochondrial III2IV2 supercomplex forms the terminal part of the electron transport chain and is essential for maintaining mitochondrial membrane potential and ATP synthesis (Schagger and Pfeiffer, 2000). In crd1Δ yeast, the III2IV2 supercomplex is less stable than in wildtype cells, suggesting that CL is critical for mitochondrial homeostasis and bioenergetics (Pfeiffer et al., 2003; Mileykovskaya et al., 2005; Zhang et al., 2005; Claypool et al., 2008; Bottinger et al., 2012; Bazan et al., 2013; Peyta et al., 2016; Petit et al., 2020). A deficiency in CL remodeling is also associated with reduced bioenergetics (Brandner et al., 2005; Li et al., 2007). Using taz1Δ yeast, Brandner et al. (2005) demonstrated that the absence of tafazzin results in increased dissociation of the III2IV2 supercomplex, causing the release of a complex IV monomer (Brandner et al., 2005; Claypool et al., 2008).
The initial yeast studies implicating CL and CL remodeling in bioenergetics were subsequently corroborated using mammalian BTHS models. Gonzalvez et al. (2013) and McKenzie et al. (2006) reported decreased respiratory supercomplex formation and stability in lymphoblast cells isolated from two BTHS patients relative to non-BTHS control cells. More recently, Petit et al. (2020) showed that shRNA-mediated Taz knockdown in HeLa cells results in decreased ATP synthase activity and overall ATP level, with a concomitant decrease in maximal respiratory capacity and an increase in basal oxygen consumption. Similarly, Dudek et al. (2013, 2016) showed reduced respiratory complex formation in cardiac tissue isolated from the BTHS mouse model and increased basal oxygen consumption coupled with decreased maximal respiratory capacity in induced pluripotent stem cells (iPSCs) derived from BTHS patients. Ma et al. (2004) reported a decrease in basal respiration in taz1∆ yeast, a study corroborated by Lou et al. (2018a) in C2C12 TAZ KO (Ma et al., 2004). The increased basal respiration observed in iPSC-derived cardiomyocytes could be due to increased F1F0 ATP synthase oxygen consumption and proton leak. The authors further showed that the aberrant respiration in iPSCs could be attributed to reduced respiratory complex stability, reiterating what was previously concluded from yeast. These findings underscore the power of the yeast model for interrogating the relationship between CL deficiency and the bioenergetic defects that characterize BTHS.
Iron–sulfur clusters (ISCs) are molecular assemblages of iron and sulfur atoms that act as co-factors in many cellular processes, including electron transfer within the electron transport chain and enzymatic conversion of substrate within the TCA cycle (Paul et al., 2017; Braymer et al., 2021). ISCs can exist in many configurations based on the number of iron and sulfur atoms involved (e.g., 2Fe-2S, 3Fe-4S, and 4Fe-4S). ISC biogenesis occurs in three steps within mitochondria (Lill and Freibert, 2020). The first step involves donation of sulfur from the NFS1-ISD11-ACP1 subcomplex and transfer of imported iron to the ISCU2 scaffold protein by frataxin (FXN). This forms an initial 2Fe-2S cluster. In the second step, chaperone proteins (HSC20, HSPA9, and GRPE1) bind to the 2Fe-2S cluster and transfer it first to the monothiol glutaredoxin GLRX5 and subsequently to mitochondrial ISC recipient proteins. The third step involves conversion of 2Fe-2S clusters into 4Fe-4S clusters and their delivery to recipient apoproteins (Lill and Freibert, 2020; Maio et al., 2020).
The first indication of a relationship between CL and ISC homeostasis was described in yeast. Using crd1Δ yeast, Patil et al. (2013) demonstrated that CL deficiency results in elevated mitochondrial iron and increased sensitivity to ROS and exogenously supplied iron sulfate. These phenotypes are associated with defective ISC biogenesis, and the authors subsequently showed that crd1Δ cells exhibit decreased enzymatic activity of the ISC-requiring enzymes ubiquinol-cytochrome c oxidoreductase, succinate dehydrogenase, aconitase, isopropylmalate isomerase, and sulfite reductase. Additionally, deletion of the ISC biosynthetic gene ISU1 in the crd1Δ background resulted in a synthetically sick phenotype, further supporting a role for CL in iron homeostasis (Patil et al., 2013).
These initial findings from yeast were subsequently validated in mammalian cells using the TAZ-KO C2C12 BTHS cell model. Similar to what was observed in yeast, Li et al. (2020) demonstrated that the activities of the ISC-requiring enzymes aconitase, NADH dehydrogenase, succinate dehydrogenase, and ubiquinol-cytochrome c reductase were all decreased by ~50% in TAZ-KO cells while their respective protein levels remained unchanged. TAZ-KO cells also showed increased mitochondrial iron content and elevated sensitivity to ROS and iron supplementation, mirroring the findings from yeast. This study went on to further corroborate the role of CL in ISC biogenesis by showing that the mature form of the ISC biosynthetic protein FXN is reduced in TAZ-KO cells (Li et al., 2020).
Raja et al. (2017a) provided the first evidence of a link between CL and energy metabolism by showing that crd1Δ exhibits decreased synthesis of acetyl-CoA, and that deletion of CRD1 is synthetically lethal in pyruvate dehydrogenase (PDH) mutants. Acetyl-CoA is a primary substrate utilized by the tricarboxylic acid (TCA) cycle to fuel intermediary metabolism, and under respiratory conditions it is synthesized predominantly in mitochondria through the enzymatic conversion of pyruvate by PDH (Guest et al., 1989; Raja et al., 2017a). Synthetic lethality between crd1Δ and PDH complex mutants suggests that CL plays a role in promoting acetyl-CoA synthesis and TCA cycle function. Interestingly, PDH complex mRNA and protein levels are increased in crd1Δ, but net activity of PDH is not altered (Raja et al., 2017a). This suggests that CL is required for optimal PDH function, and that in the absence of CL, upregulation of PDH cannot compensate for diminished acetyl-CoA synthesis.
Building on the findings in yeast, Li et al. (2019) found that TAZ-KO mouse C2C12 cells also show reduced carbon flux to acetyl-CoA, and this is linked to a reduction in PDH activity. PDH is regulated through phosphorylation, and the authors showed that both the inhibitory phosphorylation and enzymatic activity in mitochondrial extracts are rescued by the addition of exogenous CL. Although production of acetyl-CoA through the activity of PDH serves as an important input for the TCA cycle, the cycle itself is comprised of many steps that are each catalyzed by distinct enzymes, some of which require Fe-S cofactors for optimal activity. Li et al. (2019, 2020) identified a second way in which CL deficiency negatively impacts TCA cycle function by showing that activity of the Fe-S-requiring TCA cycle enzymes succinate dehydrogenase and aconitase are also reduced in TAZ-KO cells.
The mitochondrial calcium uniporter (MCU) is a holo-complex protein consisting of pore-forming subunit MCU, transmembrane subunit EMRE, and regulatory subunits MICU1, MICU2, and MCUb (Baughman et al., 2011; De Stefani et al., 2011; Sancak et al., 2013). MCU transports calcium from the cytosol into the mitochondria where it serves as a signal for regulating ATP synthesis and, when in excess, activating the apoptotic pathway (Carraro et al., 2020). MCU is localized in the IMM where CL is enriched, suggesting that its function could be influenced by CL (Ghosh et al., 2020).
The first evidence for CL playing a role in MCU function came from a recent study in yeast, where Ghosh et al. (2020) reported that MCU levels are decreased by 50% in CL-deficient crd1Δ cells. In this study, MCU was heterologously expressed under a strong promoter. Therefore, the authors argue that CL influences MCU stability but not expression, which is supported by the finding that the MCU turnover rate is substantially higher in crd1Δ compared to wildtype cells. As a functional consequence, mitochondrial uptake of calcium is also decreased in crd1Δ mutants. The authors further validated these yeast findings in both the TAZ-KO C2C12 mouse model and in BTHS patient-derived lymphoblasts and heart tissue. In a similar study, Ghosh et al. (2021) showed that only the MICU1 component of the mitochondrial calcium uniporter complex is decreased in crd1Δ cells. In this study, human MICU1 and Mitochondrial Calcium Uniporter Regulator 1 (MCUR1) proteins were expressed in both wildtype and crd1Δ yeast cells. The authors showed that MICU1 levels were 50% lower in crd1Δ cells compared to the wildtype, while MCUR1 levels were unaltered (Ghosh et al., 2021). This work serves as yet another example of how yeast research has led to pioneering discoveries regarding CL function and its relationship to BTHS.
The yeast model has proven to be indispensable not only in the study of CL-related pathologies, but in other lipid-related pathologies as well. In addition, the genetic tractability, rapid doubling time, and low cost often make the use of yeast preferable to other models. When modeling human diseases in yeast there are two general approaches. The first method is referred to as an orthologous approach, in which a yeast ortholog of a human gene is modified to have the same mutation as in the human disease (e.g., the crd1Δ and taz1Δ strains used to study BTHS). Using this approach researchers can identify interactions between other genes, proteins, and molecules that may interact with the aberrant disease pathway. However, this is not always possible, as the discrepancy in genome size between yeast and humans means that not all human genes have a yeast homolog. The second method, referred to as humanization, involves expressing a human disease gene in yeast. Although some tissue-wide aspects of human diseases are difficult to model in the unicellular yeast system, cellular phenotypes can often be recapitulated and studied in yeast cells. The following sections detail ways in which yeast have been used to study the lipid-related pathophysiology of two prevalent human neurodegenerative diseases, Alzheimer’s and Parkinson’s.
Alzheimer’s disease (AD) is the most common neurodegenerative disease in the world and is characterized by the extracellular accumulation of amyloid-β (Ab) peptide in senile plaques and the intracellular accumulation of neurofibrillary tangles (Masters et al., 1985; Alzheimer et al., 1995; Scheltens et al., 2016). These aggregations lead to neurodegeneration, with the main clinical manifestation being severe memory impairment and memory loss that often results in chronic dementia (Masliah et al., 1989; Bancher et al., 1997; Scheltens et al., 2016). While the molecular phenotypes of AD have been well cataloged, the exact pathophysiology of the disease is not well understood.
In order to gain insight into the pathophysiology of AD, Nair et al. (2014) conducted a genome-wide screen to identify cellular processes that influence Ab aggregation in yeast. In this study, the authors compared mutants from the yeast deletion collection expressing a GFP-tagged Ab construct (Ab-GFP) with wild-type cells to identify genes that influence the size or localization of Ab aggregations. Out of ~4,600 mutants tested, the screen identified 110 relevant genes corresponding predominantly to four major cellular processes, including phospholipid metabolism, gene expression, chromatin remodeling, and mitochondrial function. This study has been foundational in guiding subsequent AD research efforts.
In particular, dysregulation of lipid metabolism has been increasingly recognized as a contributing factor to AD pathophysiology, and the yeast model has been indispensable for exploring this link (Kao et al., 2020). One example of the link between lipid metabolism and AD pathology involves a neurotoxic species of phosphatidylcholine, referred to as PC(O-16:0/2:0), that is elevated in human AD tissue (Ryan et al., 2009). Kennedy et al. (2016) used yeast to better understand the role of this lipid species in AD pathology by combining gene expression profiling with a genome-wide chemogenomic screen. They found that elevated PC(O-16:0/2:0) causes an accumulation of ceramide that ultimately results in increased reactive oxygen species (ROS) production and mitochondrial dysfunction, cellular phenotypes that are commonly seen in AD patient cells and AD cell models (Agrawal and Jha, 2020).
Another example of the link between lipid metabolism and AD relates to tau protein phosphorylation. Tau hyperphosphorylation has been implicated as a major part of AD neurodegeneration, but the mechanism of hyperphosphorylation is not well understood (Simic et al., 2016). Using yeast, Randez-Gil et al. (2020) identified a potential mechanism wherein dysregulation of inositol phosphate signaling leads to defective sphingolipid production and a resultant increase in tau protein hyperphosphorylation. Although sphingolipid metabolism had been previously linked to neurodegenerative diseases (Alaamery et al., 2021), this was one of the first studies to suggest a role for SLs in tau hyperphosphorylation.
The previous studies directly link lipid metabolism to AD, but other aspects of lipid homeostasis have also been shown to contribute to AD pathology. Apolipoprotein E (APOE) is a lipid transporter that transports lipids between cells and tissues (Huang and Mahley, 2014). APOE is polyallelic, and the E4 allele (APOE4) is a known risk factor for AD (Farrer et al., 1997). It is well characterized that APOE4 plays a role in AD, but the mechanism by which it contributes to AD is largely unknown. Using genome-wide screens and lipidomic analysis in yeast, Sienski et al. (2021) determined that APOE4 is responsible for altered fatty acid (FA) metabolism which is commonly seen in AD cells. They found that when human APOE4 is expressed in yeast there is an increase in the degree of unsaturation in FAs accompanied by the accumulation of lipid droplets. Furthermore, they also demonstrated that choline supplementation ameliorates the aberrant FA metabolism (Sienski et al., 2021). When choline was supplemented, synthesis of the membrane phospholipid phosphatidylcholine was stimulated. This abolished APOE4 lipid-related defects and suggests an important role for phosphatidylcholine in the pathophysiology of AD. This study demonstrated not only a novel role of APOE4 in AD, but also identified a key modulator of this process, choline homeostasis.
Yeast has proved to be a powerful tool for studying lipid-related pathology in AD. By combining the genetic tractability of yeast with high-throughput screening techniques, researchers have uncovered novel mechanisms into the influence of lipid homeostasis on Ab aggregation, ROS production, tau hyperphosphorylation, and FA metabolism.
Parkinson’s disease (PD) is the second most common neurodegenerative disease world-wide. PD is characterized by degeneration of nigrostriatal dopaminergic neurons in the substantia nigra pars compacta region of the brain and the presence of intraneuronal α-synuclein (aS) inclusions known as Lewy bodies (Antony et al., 2013; Leao et al., 2015; Sveinbjornsdottir, 2016). Degradation of dopaminergic neurons in this region of the brain decreases dopamine release in the striatum, altering motor control in afflicted individuals (Antony et al., 2013; Leao et al., 2015; Sveinbjornsdottir, 2016). Common motor symptoms include bradykinesia, rigidity, tremors, and postural instability, and these presentations are often accompanied by depression, anosmia, dementia, and sleep disorders (Moustafa et al., 2016; Pfeiffer, 2016). The molecular hallmarks of PD are well characterized, but just as in AD, the exact cause of PD is not known.
One of the major molecular hallmarks of PD is the aggregation of aS, a protein commonly expressed in neurons and enriched in presynaptic terminals (Maroteaux et al., 1988; Taguchi et al., 2016). It is well known that aS aggregation has a central role in PD pathogenesis, but the mechanism by which aS aggregates form is not known. Wang et al. (2014) used the yeast model to test a potential mechanism for aS aggregation and found that phosphatidylethanolamine (PE) deficiency in yeast cells causes ER stress, vesicle defects, aS aggregation, and cells death. Subsequently, the authors demonstrated that the effects of PE deficiency can be mitigated by ethanolamine supplementation (Wang et al., 2014). This study highlights the importance of lipid homeostasis on aS aggregation.
A study by Fanning et al. (2019) sought to further interrogate the relationship between aS aggregation, lipid homeostasis, and cellular toxicity. The authors performed lipidomic profiling in yeast displaying aS aggregation to monitor changes in specific lipid classes. They determined that dysfunctional lipid homeostasis, induced by aS aggregation, leads to cytotoxicity due to the accumulation of oleic acid (OA) and diglycerides (DG) in lipid droplets of aS-expressing yeast (Fanning et al., 2019). Of particular interest from a therapeutic standpoint, they found that either preventing the conversion of triglycerides (TG) to DG or inhibiting stearoyl-CoA desaturase (SCD; the rate-limiting enzyme in the production of OA) ameliorates aS aggregation and its associated cytotoxicity.
Yeast is not only an excellent model for identifying novel pathophysiological mechanisms but also for probing and testing chemical and genetic modifiers of a disease (Griffioen et al., 2006; Williams et al., 2007; Su et al., 2010). For example, Soste et al. (2019) conducted a screen for genetic modifiers of aS aggregation and identified 33 genes that modulate aS aggregation and cytotoxicity. One of these modifiers, Pah1, is an enzyme that converts PA to DG in yeast. In accordance with the findings of Fanning et al. (2019), Soste et al. (2019) found that decreasing DG levels through the inhibition of Pah1 ameliorates aS aggregation and cytotoxicity. This suggests a key role for DG and lipid droplet homeostasis in aS aggregation and cytotoxicity in PD.
Collectively, these studies demonstrate the power of the yeast system for investigating the lipid-related pathophysiology of two prominent neurodegenerative disorders. The unicellular nature of yeast makes it particularly straightforward to track biochemical changes in molecules such as lipids.
This review outlines some of the major contributions the yeast model has made to our understanding of the lipid-related pathologies observed in BTHS, AD, and PD. Lipid homeostasis has been shown to play key roles in other diseases, including cancer (Snaebjornsson et al., 2020), heart disease (Poss et al., 2020; Richardson et al., 2020), diabetes mellitus (Katsiki et al., 2017; Bjornstad and Eckel, 2018), and other chronic conditions (Leuti et al., 2020). The multifaceted roles of lipids in human disease are only beginning to be uncovered, and as technologies such as mass spectrometry continue to evolve, the yeast model system promises to continue facilitating progress in this rapidly growing field (Zullig and Kofeler, 2021).
TR-E, CO, MS, LV, and MG all contributed to the conception, design, and writing of the manuscript. AL contributed to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
The Greenberg lab gratefully acknowledges support from grants R01 HL 117880, R01 GM 134715, and RO1 GM125082 from the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acehan, D., Vaz, F., Houtkooper, R. H., James, J., Moore, V., Tokunaga, C., et al. (2011). Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 286, 899–908. doi: 10.1074/jbc.M110.171439
Ades, L. C., Gedeon, A. K., Wilson, M. J., Latham, M., Partington, M. W., Mulley, J. C., et al. (1993). Barth syndrome: clinical features and confirmation of gene localisation to distal Xq28. Am. J. Med. Genet. 45, 327–334. doi: 10.1002/ajmg.1320450309
Agrawal, I., and Jha, S. (2020). Mitochondrial dysfunction and Alzheimer’s disease: role of microglia. Front. Aging Neurosci. 12:252. doi: 10.3389/fnagi.2020.00252
Alaamery, M., Albesher, N., Aljawini, N., Alsuwailm, M., Massadeh, S., Wheeler, M. A., et al. (2021). Role of sphingolipid metabolism in neurodegeneration. J. Neurochem. 158, 25–35. doi: 10.1111/jnc.15044
Alzheimer, A., Stelzmann, R. A., Schnitzlein, H. N., and Murtagh, F. R. (1995). An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde.” Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Antony, P. M., Diederich, N. J., Kruger, R., and Balling, R. (2013). The hallmarks of Parkinson’s disease. FEBS J. 280, 5981–5993. doi: 10.1111/febs.12335
Bancher, C., Egensperger, R., Kosel, S., Jellinger, K., and Graeber, M. B. (1997). Low prevalence of apolipoprotein E epsilon 4 allele in the neurofibrillary tangle predominant form of senile dementia. Acta Neuropathol. 94, 403–409. doi: 10.1007/s004010050726
Barth, P. G., Scholte, H. R., Berden, J. A., Van der Klei-Van Moorsel, J. M., Luyt-Houwen, I. E., Van’t Veer-Korthof, E. T., et al. (1983). An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355.
Barth, P. G., Valianpour, F., Bowen, V. M., Lam, J., Duran, M., Vaz, F. M., et al. (2004). X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am. J. Med. Genet. A 126A, 349–354. doi: 10.1002/ajmg.a.20660
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bazan, S., Mileykovskaya, E., Mallampalli, V. K., Heacock, P., Sparagna, G. C., and Dowhan, W. (2013). Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J. Biol. Chem. 288, 401–411. doi: 10.1074/jbc.M112.425876
Beranek, A., Rechberger, G., Knauer, H., Wolinski, H., Kohlwein, S. D., and Leber, R. (2009). Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578. doi: 10.1074/jbc.M805511200
Bione, S., D’Adamo, P., Maestrini, E., Gedeon, A. K., Bolhuis, P. A., and Toniolo, D. (1996). A novel X-linked gene, G4.5. Is responsible for Barth syndrome. Nat. Genet. 12, 385–389. doi: 10.1038/ng0496-385
Bittel, A. J., Bohnert, K. L., Reeds, D. N., Peterson, L. R., de Las Fuentes, L., Corti, M., et al. (2018). Reduced muscle strength in Barth syndrome may be improved by resistance exercise training: a pilot study. JIMD Rep. 41, 63–72. doi: 10.1007/8904_2018_102
Bjornstad, P., and Eckel, R. H. (2018). Pathogenesis of lipid disorders in insulin resistance: a brief review. Curr. Diabetes Rep. 18:127. doi: 10.1007/s11892-018-1101-6
Blunsom, N. J., Gomez-Espinosa, E., Ashlin, T. G., and Cockcroft, S. (2018). Mitochondrial CDP-diacylglycerol synthase activity is due to the peripheral protein, TAMM41 and not due to the integral membrane protein, CDP-diacylglycerol synthase 1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 284–298. doi: 10.1016/j.bbalip.2017.12.005
Bolhuis, P. A., Hensels, G. W., Hulsebos, T. J., Baas, F., and Barth, P. G. (1991). Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28. Am. J. Hum. Genet. 48, 481–485.
Bottinger, L., Horvath, S. E., Kleinschroth, T., Hunte, C., Daum, G., Pfanner, N., et al. (2012). Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 423, 677–686. doi: 10.1016/j.jmb.2012.09.001
Brady, A. N., Shehata, B. M., and Fernhoff, P. M. (2006). X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat. Diagn. 26, 462–465. doi: 10.1002/pd.1438
Brandner, K., Mick, D. U., Frazier, A. E., Taylor, R. D., Meisinger, C., and Rehling, P. (2005). Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214. doi: 10.1091/mbc.e05-03-0256
Braymer, J. J., Freibert, S. A., Rakwalska-Bange, M., and Lill, R. (2021). Mechanistic concepts of iron-sulfur protein biogenesis in biology. Biochim. Biophys. Acta Mol. Cell Res. 1868:118863. doi: 10.1016/j.bbamcr.2020.118863
Buckland, A. G., Kinkaid, A. R., and Wilton, D. C. (1998). Cardiolipin hydrolysis by human phospholipases A2. The multiple enzymatic activities of human cytosolic phospholipase A2. Biochim. Biophys. Acta 1390, 65–72. doi: 10.1016/s0005-2760(97)00170-7
Cantlay, A. M., Shokrollahi, K., Allen, J. T., Lunt, P. W., Newbury-Ecob, R. A., and Steward, C. G. (1999). Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J. Pediatr. 135, 311–315. doi: 10.1016/S0022-3476(99)70126-5
Cao, J., Liu, Y., Lockwood, J., Burn, P., and Shi, Y. (2004). A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734. doi: 10.1074/jbc.M402930200
Carraro, M., Carrer, A., Urbani, A., and Bernardi, P. (2020). Molecular nature and regulation of the mitochondrial permeability transition pore(s), drug target(s) in cardioprotection. J. Mol. Cell. Cardiol. 144, 76–86. doi: 10.1016/j.yjmcc.2020.05.014
Chakraborty, T. R., Vancura, A., Balija, V. S., and Haldar, D. (1999). Phosphatidic acid synthesis in mitochondria. Topography of formation and transmembrane migration. J. Biol. Chem. 274, 29786–29790. doi: 10.1074/jbc.274.42.29786
Chang, S. C., Heacock, P. N., Clancey, C. J., and Dowhan, W. (1998a). The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J. Biol. Chem. 273, 9829–9836. doi: 10.1074/jbc.273.16.9829
Chang, S. C., Heacock, P. N., Mileykovskaya, E., Voelker, D. R., and Dowhan, W. (1998b). Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J. Biol. Chem. 273, 14933–14941. doi: 10.1074/jbc.273.24.14933
Clarke, S. L., Bowron, A., Gonzalez, I. L., Groves, S. J., Newbury-Ecob, R., Clayton, N., et al. (2013). Barth syndrome. Orphanet J. Rare Dis. 8:23. doi: 10.1186/1750-1172-8-23
Claypool, S. M., Boontheung, P., McCaffery, J. M., Loo, J. A., and Koehler, C. M. (2008). The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol. Biol. Cell 19, 5143–5155. doi: 10.1091/mbc.e08-09-0896
Claypool, S. M., McCaffery, J. M., and Koehler, C. M. (2006). Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 174, 379–390. doi: 10.1083/jcb.200605043
Claypool, S. M., Whited, K., Srijumnong, S., Han, X., and Koehler, C. M. (2011). Barth syndrome mutations that cause tafazzin complex lability. J. Cell Biol. 192, 447–462. doi: 10.1083/jcb.201008177
Connerth, M., Tatsuta, T., Haag, M., Klecker, T., Westermann, B., and Langer, T. (2012). Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818. doi: 10.1126/science.1225625
Dennis, E. A., Cao, J., Hsu, Y. H., Magrioti, V., and Kokotos, G. (2011). Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 111, 6130–6185. doi: 10.1021/cr200085w
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Dudek, J., Cheng, I. F., Balleininger, M., Vaz, F. M., Streckfuss-Bomeke, K., Hubscher, D., et al. (2013). Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 11, 806–819. doi: 10.1016/j.scr.2013.05.005
Dudek, J., Cheng, I. F., Chowdhury, A., Wozny, K., Balleininger, M., Reinhold, R., et al. (2016). Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 8, 139–154. doi: 10.15252/emmm.201505644
Eisenberg, T., and Buttner, S. (2014). Lipids and cell death in yeast. FEMS Yeast Res. 14, 179–197. doi: 10.1111/1567-1364.12105
Fanning, S., Haque, A., Imberdis, T., Baru, V., Barrasa, M. I., Nuber, S., et al. (2019). Lipidomic analysis of alpha-synuclein neurotoxicity identifies stearoyl CoA desaturase as a target for Parkinson treatment. Mol. Cell 73, 1001.e1008–1014.e1008. doi: 10.1016/j.molcel.2018.11.028
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Ferreira, C., Pierre, G., Thompson, R., and Vernon, H. (2014). “Barth syndrome,” in GeneReviews. eds. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, and G. Mirzaa, et al. (Seattle, WA)
Folsi, V., Miglietti, N., Lombardi, A., Boccacci, S., Utyatnikova, T., Donati, C., et al. (2014). Cardiomyopathy in a male patient with neutropenia and growth delay. Ital. J. Pediatr. 40:45. doi: 10.1186/1824-7288-40-45
Gebert, N., Joshi, A. S., Kutik, S., Becker, T., McKenzie, M., Guan, X. L., et al. (2009). Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139. doi: 10.1016/j.cub.2009.10.074
Ghosh, S., Basu Ball, W., Madaris, T. R., Srikantan, S., Madesh, M., Mootha, V. K., et al. (2020). An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. U. S. A. 117, 16383–16390. doi: 10.1073/pnas.2000640117
Ghosh, S., Iadarola, D. M., Ball, W. B., and Gohil, V. M. (2019). Mitochondrial dysfunctions in Barth syndrome. IUBMB Life 71, 791–801. doi: 10.1002/iub.2018
Ghosh, S., Zulkifli, M., Joshi, A., Venkatesan, M., Cristel, A., Vishnu, N., et al. (2021). MCU-complex-mediated mitochondrial calcium signaling is impaired in Barth syndrome. Hum. Mol. Genet. doi: 10.1093/hmg/ddab254 [Epub ahead of print].
Giaever, G., and Nislow, C. (2014). The yeast deletion collection: a decade of functional genomics. Genetics 197, 451–465. doi: 10.1534/genetics.114.161620
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., et al. (1996). Life with 6000 genes. Science 274, 563–547. doi: 10.1126/science.274.5287.546
Gonzalez, I. L. (2005). Barth syndrome: TAZ gene mutations, mRNAs, and evolution. Am. J. Med. Genet. A 134, 409–414. doi: 10.1002/ajmg.a.30661
Gonzalvez, F., D’Aurelio, M., Boutant, M., Moustapha, A., Puech, J. P., Landes, T., et al. (2013). Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta 1832, 1194–1206. doi: 10.1016/j.bbadis.2013.03.005
Greenwell, A. A., Gopal, K., Altamimi, T. R., Saed, C. T., Wang, F., Tabatabaei Dakhili, S. A., et al. (2021). Barth syndrome-related cardiomyopathy is associated with a reduction in myocardial glucose oxidation. Am. J. Physiol. Heart Circ. Physiol. 320, H2255–H2269. doi: 10.1152/ajpheart.00873.2020
Griffioen, G., Duhamel, H., Van Damme, N., Pellens, K., Zabrocki, P., Pannecouque, C., et al. (2006). A yeast-based model of alpha-synucleinopathy identifies compounds with therapeutic potential. Biochim. Biophys. Acta 1762, 312–318. doi: 10.1016/j.bbadis.2005.11.009
Gu, Z., Valianpour, F., Chen, S., Vaz, F. M., Hakkaart, G. A., Wanders, R. J., et al. (2004). Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51, 149–158. doi: 10.1046/j.1365-2958.2003.03802.x
Guest, J. R., Angier, S. J., and Russell, G. C. (1989). Structure, expression, and protein engineering of the pyruvate dehydrogenase complex of Escherichia coli. Ann. N. Y. Acad. Sci. 573, 76–99. doi: 10.1111/j.1749-6632.1989.tb14988.x
Hornby, B., McClellan, R., Buckley, L., Carson, K., Gooding, T., and Vernon, H. J. (2019). Functional exercise capacity, strength, balance and motion reaction time in Barth syndrome. Orphanet J. Rare Dis. 14:37. doi: 10.1186/s13023-019-1006-8
Hostetler, K. Y., and van den Bosch, H. (1972). Subcellular and submitochondrial localization of the biosynthesis of cardiolipin and related phospholipids in rat liver. Biochim. Biophys. Acta 260, 380–386. doi: 10.1016/0005-2760(72)90052-5
Houtkooper, R. H., and Vaz, F. M. (2008). Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci. 65, 2493–2506. doi: 10.1007/s00018-008-8030-5
Huang, Y., and Mahley, R. W. (2014). Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 72, 3–12. doi: 10.1016/j.nbd.2014.08.025
Hsu, Y. H., Dumlao, D. S., Cao, J., and Dennis, E. A. (2013). Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS One 8:e59267. doi: 10.1371/annotation/47607b18-ed69-4a08-8619-60c39bd83a13
Jiang, F., Rizavi, H. S., and Greenberg, M. L. (1997). Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol. Microbiol. 26, 481–491. doi: 10.1046/j.1365-2958.1997.5841950.x
Joshi, A. S., Zhou, J., Gohil, V. M., Chen, S., and Greenberg, M. L. (2009). Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218. doi: 10.1016/j.bbamcr.2008.07.024
Kachroo, A. H., Laurent, J. M., Yellman, C. M., Meyer, A. G., Wilke, C. O., and Marcotte, E. M. (2015). Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348, 921–925. doi: 10.1126/science.aaa0769
Kao, Y. C., Ho, P. C., Tu, Y. K., Jou, I. M., and Tsai, K. J. (2020). Lipids and Alzheimer’s disease. Int. J. Mol. Sci. 21:1505. doi: 10.3390/ijms21041505
Katsiki, N., Mantzoros, C., and Mikhailidis, D. P. (2017). Adiponectin, lipids and atherosclerosis. Curr. Opin. Lipidol. 28, 347–354. doi: 10.1097/MOL.0000000000000431
Kennedy, M. A., Moffat, T. C., Gable, K., Ganesan, S., Niewola-Staszkowska, K., Johnston, A., et al. (2016). A signaling lipid associated with Alzheimer’s disease promotes mitochondrial dysfunction. Sci. Rep. 6:19332. doi: 10.1038/srep19332
Khuchua, Z., Yue, Z., Batts, L., and Strauss, A. W. (2006). A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ. Res. 99, 201–208. doi: 10.1161/01.RES.0000233378.95325.ce
Krebs, J. J., Hauser, H., and Carafoli, E. (1979). Asymmetric distribution of phospholipids in the inner membrane of beef heart mitochondria. J. Biol. Chem. 254, 5308–5316. doi: 10.1016/S0021-9258(18)50596-2
Leao, A. H., Sarmento-Silva, A. J., Santos, J. R., Ribeiro, A. M., and Silva, R. H. (2015). Molecular, neurochemical, and behavioral hallmarks of reserpine as a model for Parkinson’s disease: new perspectives to a long-standing model. Brain Pathol. 25, 377–390. doi: 10.1111/bpa.12253
Lecocq, J., and Ballou, C. E. (1964). On the structure of cardiolipin. Biochemistry 3, 976–980. doi: 10.1021/bi00895a023
Leuti, A., Fazio, D., Fava, M., Piccoli, A., Oddi, S., and Maccarrone, M. (2020). Bioactive lipids, inflammation and chronic diseases. Adv. Drug Deliv. Rev. 159, 133–169. doi: 10.1016/j.addr.2020.06.028
Li, G., Chen, S., Thompson, M. N., and Greenberg, M. L. (2007). New insights into the regulation of cardiolipin biosynthesis in yeast: implications for Barth syndrome. Biochim. Biophys. Acta 1771, 432–441. doi: 10.1016/j.bbalip.2006.06.007
Li, Y., Lou, W., Grevel, A., Bottinger, L., Liang, Z., Ji, J., et al. (2020). Cardiolipin-deficient cells have decreased levels of the iron-sulfur biogenesis protein frataxin. J. Biol. Chem. 295, 11928–11937. doi: 10.1074/jbc.RA120.013960
Li, Y., Lou, W., Raja, V., Denis, S., Yu, W., Schmidtke, M. W., et al. (2019). Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J. Biol. Chem. 294, 11568–11578. doi: 10.1074/jbc.RA119.009037
Li, J., Romestaing, C., Han, X., Li, Y., Hao, X., Wu, Y., et al. (2010). Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 12, 154–165. doi: 10.1016/j.cmet.2010.07.003
Lill, R., and Freibert, S. A. (2020). Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 89, 471–499. doi: 10.1146/annurev-biochem-013118-111540
Lou, W., Reynolds, C. A., Li, Y., Liu, J., Huttemann, M., Schlame, M., et al. (2018a). Loss of tafazzin results in decreased myoblast differentiation in C2C12 cells: a myoblast model of Barth syndrome and cardiolipin deficiency. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 857–865. doi: 10.1016/j.bbalip.2018.04.015
Lou, W., Ting, H. C., Reynolds, C. A., Tyurina, Y. Y., Tyurin, V. A., Li, Y., et al. (2018b). Genetic re-engineering of polyunsaturated phospholipid profile of Saccharomyces cerevisiae identifies a novel role for Cld1 in mitigating the effects of cardiolipin peroxidation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 1354–1368. doi: 10.1016/j.bbalip.2018.06.016
Lu, Y. W., Galbraith, L., Herndon, J. D., Lu, Y. L., Pras-Raves, M., Vervaart, M., et al. (2016). Defining functional classes of Barth syndrome mutation in humans. Hum. Mol. Genet. 25, 1754–1770. doi: 10.1093/hmg/ddw046
Ma, B. J., Taylor, W. A., Dolinsky, V. W., and Hatch, G. M. (1999). Acylation of monolysocardiolipin in rat heart. J. Lipid Res. 40, 1837–1845. doi: 10.1016/S0022-2275(20)34900-2
Ma, L., Vaz, F. M., Gu, Z., Wanders, R. J., and Greenberg, M. L. (2004). The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J. Biol. Chem. 279, 44394–44399. doi: 10.1074/jbc.M405479200
Maio, N., Jain, A., and Rouault, T. A. (2020). Mammalian iron-sulfur cluster biogenesis: recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 55, 34–44. doi: 10.1016/j.cbpa.2019.11.014
Mancuso, D. J., Sims, H. F., Han, X., Jenkins, C. M., Guan, S. P., Yang, K., et al. (2007). Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 282, 34611–34622. doi: 10.1074/jbc.M707795200
Mangat, J., Lunnon-Wood, T., Rees, P., Elliott, M., and Burch, M. (2007). Successful cardiac transplantation in Barth syndrome: single-Centre experience of four patients. Pediatr. Transplant. 11, 327–331. doi: 10.1111/j.1399-3046.2006.00629.x
Manon, S. (2004). Utilization of yeast to investigate the role of lipid oxidation in cell death. Antioxid. Redox Signal. 6, 259–267. doi: 10.1089/152308604322899323
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988
Marziliano, N., Mannarino, S., Nespoli, L., Diegoli, M., Pasotti, M., Malattia, C., et al. (2007). Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am. J. Med. Genet. A 143A, 907–915. doi: 10.1002/ajmg.a.31653
Masliah, E., Terry, R. D., DeTeresa, R. M., and Hansen, L. A. (1989). Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. 103, 234–239. doi: 10.1016/0304-3940(89)90582-X
Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 82, 4245–4249. doi: 10.1073/pnas.82.12.4245
Mazar, I., Stokes, J., Ollis, S., Love, E., Espensen, A., Barth, P. G., et al. (2019). Understanding the life experience of Barth syndrome from the perspective of adults: a qualitative one-on-one interview study. Orphanet J. Rare Dis. 14:243. doi: 10.1186/s13023-019-1200-8
Mazzocco, M. M., Henry, A. E., and Kelly, R. I. (2007). Barth syndrome is associated with a cognitive phenotype. J. Dev. Behav. Pediatr. 28, 22–30. doi: 10.1097/01.DBP.0000257519.79803.90
Mejia, E. M., Zegallai, H., Bouchard, E. D., Banerji, V., Ravandi, A., and Hatch, G. M. (2018). Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J. Biol. Chem. 293, 7564–7577. doi: 10.1074/jbc.RA117.001024
McKenzie, M., Lazarou, M., Thorburn, D. R., and Ryan, M. T. (2006). Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469. doi: 10.1016/j.jmb.2006.06.057
Mileykovskaya, E., Zhang, M., and Dowhan, W. (2005). Cardiolipin in energy transducing membranes. Biochemistry 70, 154–158. doi: 10.1007/s10541-005-0095-2
Moustafa, A. A., Chakravarthy, S., Phillips, J. R., Gupta, A., Keri, S., Polner, B., et al. (2016). Motor symptoms in Parkinson’s disease: a unified framework. Neurosci. Biobehav. Rev. 68, 727–740. doi: 10.1016/j.neubiorev.2016.07.010
Nair, S., Traini, M., Dawes, I. W., and Perrone, G. G. (2014). Genome-wide analysis of Saccharomyces cerevisiae identifies cellular processes affecting intracellular aggregation of Alzheimer’s amyloid-beta42: importance of lipid homeostasis. Mol. Biol. Cell 25, 2235–2249. doi: 10.1091/mbc.E13-04-0216
Orstavik, K. H., Orstavik, R. E., Naumova, A. K., D’Adamo, P., Gedeon, A., Bolhuis, P. A., et al. (1998). X chromosome inactivation in carriers of Barth syndrome. Am. J. Hum. Genet. 63, 1457–1463. doi: 10.1086/302095
Osman, C., Haag, M., Wieland, F. T., Brugger, B., and Langer, T. (2010). A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 29, 1976–1987. doi: 10.1038/emboj.2010.98
Osman, C., Voelker, D. R., and Langer, T. (2011). Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7–16. doi: 10.1083/jcb.201006159
Paradies, G., Paradies, V., De Benedictis, V., Ruggiero, F. M., and Petrosillo, G. (2014). Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 1837, 408–417. doi: 10.1016/j.bbabio.2013.10.006
Patil, V. A., Fox, J. L., Gohil, V. M., Winge, D. R., and Greenberg, M. L. (2013). Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 288, 1696–1705. doi: 10.1074/jbc.M112.428938
Paul, B. T., Manz, D. H., Torti, F. M., and Torti, S. V. (2017). Mitochondria and iron: current questions. Expert. Rev. Hematol. 10, 65–79. doi: 10.1080/17474086.2016.1268047
Pennington, E. R., Funai, K., Brown, D. A., and Shaikh, S. R. (2019). The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 1039–1052. doi: 10.1016/j.bbalip.2019.03.012
Petit, P. X., Ardilla-Osorio, H., Penalvia, L., and Rainey, N. E. (2020). Tafazzin mutation affecting cardiolipin leads to increased mitochondrial superoxide anions and mitophagy inhibition in Barth syndrome. Cell 9:2333. doi: 10.3390/cells9102333
Peyta, L., Jarnouen, K., Pinault, M., Guimaraes, C., Pais de Barros, J. P., Chevalier, S., et al. (2016). Reduced cardiolipin content decreases respiratory chain capacities and increases ATP synthesis yield in the human HepaRG cells. Biochim. Biophys. Acta 1857, 443–453. doi: 10.1016/j.bbabio.2016.01.002
Pfeiffer, R. F. (2016). Non-motor symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 22(Suppl 1), S119–S122. doi: 10.1016/j.parkreldis.2015.09.004
Pfeiffer, K., Gohil, V., Stuart, R. A., Hunte, C., Brandt, U., Greenberg, M. L., et al. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880. doi: 10.1074/jbc.M308366200
Phoon, C. K., Acehan, D., Schlame, M., Stokes, D. L., Edelman-Novemsky, I., Yu, D., et al. (2012). Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 1:e000455. doi: 10.1161/JAHA.111.000455
Poss, A. M., Holland, W. L., and Summers, S. A. (2020). Risky lipids: refining the ceramide score that measures cardiovascular health. Eur. Heart J. 41, 381–382. doi: 10.1093/eurheartj/ehz525
Potting, C., Wilmes, C., Engmann, T., Osman, C., and Langer, T. (2010). Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 29, 2888–2898. doi: 10.1038/emboj.2010.169
Raja, V., Joshi, A. S., Li, G., Maddipati, K. R., and Greenberg, M. L. (2017a). Loss of cardiolipin leads to perturbation of acetyl-CoA synthesis. J. Biol. Chem. 292, 1092–1102. doi: 10.1074/jbc.M116.753624
Raja, V., Reynolds, C. A., and Greenberg, M. L. (2017b). Barth syndrome: a life-threatening disorder caused by abnormal cardiolipin remodeling. J. Rare Dis. Res. Treat. 2, 58–62. doi: 10.29245/2572-9411/2017/2.1087
Raja, V., Salsaa, M., Joshi, A. S., Li, Y., van Roermund, C. W. T., Saadat, N., et al. (2019). Cardiolipin-deficient cells depend on anaplerotic pathways to ameliorate defective TCA cycle function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 654–661. doi: 10.1016/j.bbalip.2019.02.001
Randez-Gil, F., Bojunga, L., Estruch, F., Winderickx, J., Del Poeta, M., and Prieto, J. A. (2020). Sphingolipids and inositol phosphates regulate the tau protein phosphorylation status in humanized yeast. Front. Cell Dev. Biol. 8:592159. doi: 10.3389/fcell.2020.592159
Ren, M., Phoon, C. K., and Schlame, M. (2014). Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 55, 1–16. doi: 10.1016/j.plipres.2014.04.001
Reynolds, S., Kreider, C. M., and Bendixen, R. (2012). A mixed-methods investigation of sensory response patterns in Barth syndrome: a clinical phenotype? Am. J. Med. Genet. A 158A, 1647–1653. doi: 10.1002/ajmg.a.35413
Reynolds, S., Kreider, C. M., Meeley, L. E., and Bendixen, R. M. (2015). Taste perception and sensory sensitivity: relationship to feeding problems in boys with Barth syndrome. J. Rare Dis. 3, 1–9.
Richardson, T. G., Sanderson, E., Palmer, T. M., Ala-Korpela, M., Ference, B. A., Davey Smith, G., et al. (2020). Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 17:e1003062. doi: 10.1371/journal.pmed.1003062
Rigaud, C., Lebre, A. S., Touraine, R., Beaupain, B., Ottolenghi, C., Chabli, A., et al. (2013). Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J. Rare Dis. 8:70. doi: 10.1186/1750-1172-8-70
Roberts, A. E., Nixon, C., Steward, C. G., Gauvreau, K., Maisenbacher, M., Fletcher, M., et al. (2012). The Barth syndrome registry: distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. A 158A, 2726–2732. doi: 10.1002/ajmg.a.35609
Ronvelia, D., Greenwood, J., Platt, J., Hakim, S., and Zaragoza, M. V. (2012). Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol. Genet. Metab. 107, 428–432. doi: 10.1016/j.ymgme.2012.09.013
Ryan, S. D., Whitehead, S. N., Swayne, L. A., Moffat, T. C., Hou, W., Ethier, M., et al. (2009). Amyloid-beta42 signals tau hyperphosphorylation and compromises neuronal viability by disrupting alkylacylglycerophosphocholine metabolism. Proc. Natl. Acad. Sci. U. S. A. 106, 20936–20941. doi: 10.1073/pnas.0905654106
Sancak, Y., Markhard, A. L., Kitami, T., Kovacs-Bogdan, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. doi: 10.1126/science.1242993
Saric, A., Andreau, K., Armand, A. S., Moller, I. M., and Petit, P. X. (2015). Barth syndrome: from mitochondrial dysfunctions associated with aberrant production of reactive oxygen species to pluripotent stem cell studies. Front. Genet. 6:359. doi: 10.3389/fgene.2015.00359
Schagger, H., and Pfeiffer, K. (2000). Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783. doi: 10.1093/emboj/19.8.1777
Scheltens, P., Blennow, K., Breteler, M. M., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Schlame, M., and Greenberg, M. L. (1997). Cardiolipin synthase from yeast. Biochim. Biophys. Acta 1348, 201–206. doi: 10.1016/s0005-2760(97)00117-3
Schlame, M., and Haldar, D. (1993). Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J. Biol. Chem. 268, 74–79. doi: 10.1016/S0021-9258(18)54116-8
Schlame, M., Kelley, R. I., Feigenbaum, A., Towbin, J. A., Heerdt, P. M., Schieble, T., et al. (2003). Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999. doi: 10.1016/j.jacc.2003.06.015
Schlame, M., and Ren, M. (2009). The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 1788, 2080–2083. doi: 10.1016/j.bbamem.2009.04.019
Schlame, M., and Xu, Y. (2020). The function of tafazzin, a mitochondrial phospholipid-lysophospholipid acyltransferase. J. Mol. Biol. 432, 5043–5051. doi: 10.1016/j.jmb.2020.03.026
Shen, Z., Ye, C., McCain, K., and Greenberg, M. L. (2015). The role of cardiolipin in cardiovascular health. Biomed. Res. Int. 2015:891707. doi: 10.1155/2015/891707
Sienski, G., Narayan, P., Bonner, J. M., Kory, N., Boland, S., Arczewska, A. A., et al. (2021). APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 13:eaaz4564. doi: 10.1126/scitranslmed.aaz4564
Simic, G., Babic Leko, M., Wray, S., Harrington, C., Delalle, I., Jovanov-Milosevic, N., et al. (2016). Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomol. Ther. 6:6. doi: 10.3390/biom6010006
Singh, H. R., Yang, Z., Siddiqui, S., Pena, L. S., Westerfield, B. H., Fan, Y., et al. (2009). A novel Alu-mediated Xq28 microdeletion ablates TAZ and partially deletes DNL1L in a patient with Barth syndrome. Am. J. Med. Genet. A 149A, 1082–1085. doi: 10.1002/ajmg.a.32822
Snaebjornsson, M. T., Janaki-Raman, S., and Schulze, A. (2020). Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 31, 62–76. doi: 10.1016/j.cmet.2019.11.010
Soste, M., Charmpi, K., Lampert, F., Gerez, J. A., van Oostrum, M., Malinovska, L., et al. (2019). Proteomics-based monitoring of pathway activity reveals that blocking diacylglycerol biosynthesis rescues from alpha-synuclein toxicity. Cell Syst. 9, 309.e308–320.e308. doi: 10.1016/j.cels.2019.07.010
Soustek, M. S., Falk, D. J., Mah, C. S., Toth, M. J., Schlame, M., Lewin, A. S., et al. (2011). Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 22, 865–871. doi: 10.1089/hum.2010.199
Sparagna, G. C., and Lesnefsky, E. J. (2009). Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 53, 290–301. doi: 10.1097/FJC.0b013e31819b5461
Spencer, C. T., Bryant, R. M., Day, J., Gonzalez, I. L., Colan, S. D., Thompson, W. R., et al. (2006). Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118, e337–e346. doi: 10.1542/peds.2005-2667
Spencer, C. T., Byrne, B. J., Bryant, R. M., Margossian, R., Maisenbacher, M., Breitenger, P., et al. (2011). Impaired cardiac reserve and severely diminished skeletal muscle O(2) utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Heart Circ. Physiol. 301, H2122–H2129. doi: 10.1152/ajpheart.00479.2010
Steward, C. G., Newbury-Ecob, R. A., Hastings, R., Smithson, S. F., Tsai-Goodman, B., Quarrell, O. W., et al. (2010). Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenat. Diagn. 30, 970–976. doi: 10.1002/pd.2599
Storch, E. A., Keeley, M., Merlo, L. J., St Amant, J. B., Jacob, M., Storch, J. F., et al. (2009). Psychosocial functioning in youth with Barth syndrome. Child. Health Care 38, 137–156. doi: 10.1080/02739610902813344
Su, L. J., Auluck, P. K., Outeiro, T. F., Yeger-Lotem, E., Kritzer, J. A., Tardiff, D. F., et al. (2010). Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis. Model. Mech. 3, 194–208. doi: 10.1242/dmm.004267
Sveinbjornsdottir, S. (2016). The clinical symptoms of Parkinson’s disease. J. Neurochem. 139(Suppl. 1), 318–324. doi: 10.1111/jnc.13691
Taguchi, K., Watanabe, Y., Tsujimura, A., and Tanaka, M. (2016). Brain region-dependent differential expression of alpha-synuclein. J. Comp. Neurol. 524, 1236–1258. doi: 10.1002/cne.23901
Tamura, Y., Iijima, M., and Sesaki, H. (2010). Mdm35p imports ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J. 29, 2875–2887. doi: 10.1038/emboj.2010.149
Taylor, C., Rao, E. S., Pierre, G., Chronopoulou, E., Hornby, B., Heyman, A., et al. (2021). Clinical presentation and natural history of Barth syndrome: an overview. J. Inherit. Metab. Dis. doi: 10.1002/jimd.12422 [Epub ahead of print].
Tuller, G., Hrastnik, C., Achleitner, G., Schiefthaler, U., Klein, F., and Daum, G. (1998). YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae. FEBS Lett. 421, 15–18. doi: 10.1016/S0014-5793(97)01525-1
Valianpour, F., Mitsakos, V., Schlemmer, D., Towbin, J. A., Taylor, J. M., Ekert, P. G., et al. (2005). Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 46, 1182–1195. doi: 10.1194/jlr.M500056-JLR200
Vaz, F. M., Houtkooper, R. H., Valianpour, F., Barth, P. G., and Wanders, R. J. (2003). Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J. Biol. Chem. 278, 43089–43094. doi: 10.1074/jbc.M305956200
Vreken, P., Valianpour, F., Nijtmans, L. G., Grivell, L. A., Plecko, B., Wanders, R. J., et al. (2000). Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382. doi: 10.1006/bbrc.2000.3952
Wang, S., Li, Y., Xu, Y., Ma, Q., Lin, Z., Schlame, M., et al. (2020). AAV gene therapy prevents and reverses heart failure in a murine knockout model of Barth syndrome. Circ. Res. 126, 1024–1039. doi: 10.1161/CIRCRESAHA.119.315956
Wang, S., Zhang, S., Liou, L. C., Ren, Q., Zhang, Z., Caldwell, G. A., et al. (2014). Phosphatidylethanolamine deficiency disrupts alpha-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc. Natl. Acad. Sci. U. S. A. 111, E3976–E3985. doi: 10.1073/pnas.1411694111
Williams, R. B., Gutekunst, W. R., Joyner, P. M., Duan, W., Li, Q., Ross, C. A., et al. (2007). Bioactivity profiling with parallel mass spectrometry reveals an assemblage of green tea metabolites affording protection against human huntingtin and alpha-synuclein toxicity. J. Agric. Food Chem. 55, 9450–9456. doi: 10.1021/jf072241x
Xu, Y., Condell, M., Plesken, H., Edelman-Novemsky, I., Ma, J., Ren, M., et al. (2006). A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U. S. A. 103, 11584–11588. doi: 10.1073/pnas.0603242103
Yen, T. Y., Hwu, W. L., Chien, Y. H., Wu, M. H., Lin, M. T., Tsao, L. Y., et al. (2008). Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur. J. Pediatr. 167, 941–944. doi: 10.1007/s00431-007-0592-y
Zegallai, H. M., and Hatch, G. M. (2021). Barth syndrome: cardiolipin, cellular pathophysiology, management, and novel therapeutic targets. Mol. Cell. Biochem. 476, 1605–1629. doi: 10.1007/s11010-020-04021-0
Zhang, J., Guan, Z., Murphy, A. N., Wiley, S. E., Perkins, G. A., Worby, C. A., et al. (2011). Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13, 690–700. doi: 10.1016/j.cmet.2011.04.007
Zhang, M., Mileykovskaya, E., and Dowhan, W. (2005). Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 280, 29403–29408. doi: 10.1074/jbc.M504955200
Keywords: Saccharomyces cerevisiae, lipids, cardiolipin, Barth syndrome, pathophysiology, tafazzin
Citation: Ralph-Epps T, Onu CJ, Vo L, Schmidtke MW, Le A and Greenberg ML (2021) Studying Lipid-Related Pathophysiology Using the Yeast Model. Front. Physiol. 12:768411. doi: 10.3389/fphys.2021.768411
Edited by:
Roberto Angelini, Swansea University Medical School, United KingdomReviewed by:
Vishal M. Gohil, Texas A&M University, United StatesCopyright © 2021 Ralph-Epps, Onu, Vo, Schmidtke, Le and Greenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam L. Greenberg, bWdyZWVuYmVyZ0B3YXluZS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.