 Lettine van den Brink1
Lettine van den Brink1 Karina O. Brandão1
Karina O. Brandão1 Loukia Yiangou1
Loukia Yiangou1 Albert Blanch-Asensio1
Albert Blanch-Asensio1 Mervyn P. H. Mol1
Mervyn P. H. Mol1 Christine L. Mummery1,2
Christine L. Mummery1,2 Arie O. Verkerk3
Arie O. Verkerk3 Richard P. Davis1*
Richard P. Davis1*- 1Department of Anatomy and Embryology, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Applied Stem Cell Technologies, University of Twente, Enschede, Netherlands
- 3Department of Medical Biology, Amsterdam UMC, Amsterdam, Netherlands
While rare mutations in ion channel genes are primarily responsible for inherited cardiac arrhythmias, common genetic variants are also an important contributor to the clinical heterogeneity observed among mutation carriers. The common single nucleotide polymorphism (SNP) KCNH2-K897T is associated with QT interval duration, but its influence on the disease phenotype in patients with long QT syndrome type 2 (LQT2) remains unclear. Human induced pluripotent stem cells (hiPSCs), coupled with advances in gene editing technologies, are proving an invaluable tool for modeling cardiac genetic diseases and identifying variants responsible for variability in disease expressivity. In this study, we have used isogenic hiPSC-derived cardiomyocytes (hiPSC-CMs) to establish the functional consequences of having the KCNH2-K897T SNP in cis- or trans-orientation with LQT2-causing missense variants either within the pore-loop domain (KCNH2A561T/WT) or tail region (KCNH2N996I/WT) of the potassium ion channel, human ether-a-go-go-related gene (hERG). When KCNH2-K897T was on the same allele (cis) as the primary mutation, the hERG channel in hiPSC-CMs exhibited faster activation and deactivation kinetics compared to their trans-oriented counterparts. Consistent with this, hiPSC-CMs with KCNH2-K897T in cis orientation had longer action and field potential durations. Furthermore, there was an increased occurrence of arrhythmic events upon pharmacological blocking of hERG. Collectively, these results indicate that the common polymorphism KCNH2-K897T differs in its influence on LQT2-causing KCNH2 mutations depending on whether it is present in cis or trans. This study corroborates hiPSC-CMs as a powerful platform to investigate the modifying effects of common genetic variants on inherited cardiac arrhythmias and aids in unraveling their contribution to the variable expressivity of these diseases.
Introduction
Inherited primary arrhythmia syndromes are one of the most common causes of cardiovascular disease-related death among young people (<40 years) (Isbister and Semsarian, 2019). In long QT syndrome (LQTS), types 1–3 (LQT1–3) are the most predominant (Gnecchi et al., 2021). These are caused by primary mutations in the ion channel genes KCNQ1, KCNH2, and SCN5A, respectively, and result in a delayed ventricular repolarization, thereby predisposing patients to malignant arrhythmia and risk of sudden cardiac death. While the growing body of identified causative genes has increased our understanding of the disease (Adler et al., 2020), variable expressivity and incomplete penetrance still complicate the risk stratification and clinical management of patients with LQTS (Giudicessi and Ackerman, 2013). This heterogeneity is due to both environmental and genetic factors (Bezzina et al., 2015). It is widely recognized that besides the location and type of the primary mutation, the presence of additional common genetic variants can alter the disease expressivity (Napolitano et al., 2018; Schwartz et al., 2018).
While genome-wide association studies (GWASs) and individual genome sequencing have identified a number of potential genetic modifiers of LQTS (Lahrouchi et al., 2020; Walsh et al., 2020), establishing the contribution of these modifiers (or non-Mendelian variants) to the disease phenotype is challenging, since their effects are smaller than that of a disease-causing mutation (van den Brink et al., 2020b). Human induced pluripotent stem cells (hiPSCs), which are generated by reprogramming somatic cells from individuals (Takahashi et al., 2007), offer an approach to address this issue. These cells can be differentiated in vitro into cardiomyocytes (hiPSC-CMs) and are capable of replicating aspects of genetic disease phenotypes observed in the patients (Brandão et al., 2017; Garg et al., 2018). Combining this with advances in gene targeting technology, it has opened the door to validate variants as genetic modifiers of the disease phenotype and scrutinize their mechanism of action. Indeed, recent studies using these two techniques have identified novel modifying variants responsible for differences in the LQTS phenotype observed in patients with the same primary Mendelian variant (Chai et al., 2018; Lee et al., 2021).
The common single nucleotide polymorphism (SNP) rs1805123 (A → C) in KCNH2 has a minor allele frequency (MAF) of ∼24% in populations of European descent (Bezzina et al., 2003) and leads to the substitution of lysine for threonine at residue 897 (K897T) in the potassium ion channel human ether-a-go-go-related gene (hERG), which is responsible for conducting the rapid component of the delayed rectifier potassium current (IKr). While KCNH2-K897T has been identified as an intragenic modifier in some patients with LQT2, reports on its phenotypic impact and its effect on QT interval duration are discordant. Cohort- and family-based studies have classified KCNH2-K897T either as a protective or exacerbating modifier of the LQT2 phenotype (Crotti et al., 2005; Zhang et al., 2008; Nof et al., 2010; Jenewein et al., 2018). Similarly, in vitro studies using either heterologous expression systems or hiPSC-CMs have reported discrepant biophysical effects of K897T on hERG channel activity. In some cases, K897T has been reported to impact IKr by altering the gating properties of the channel (e.g., decelerated deactivation and inactivation, negative shift in voltage dependency), reducing IKr density, or by inhibition through Akt-mediated phosphorylation (Paavonen et al., 2003; Gentile et al., 2008; Nof et al., 2010). In contrast, other studies have observed accelerated activation and deactivation kinetics and no effects on current densities (Bezzina et al., 2003; Terrenoire et al., 2013). Furthermore, if these non-Mendelian variants occur in the same gene as the primary mutation, then phenotypic effects can differ depending on whether the variant is on the same (cis) or opposite (trans) allele (Giudicessi and Ackerman, 2013; Coll et al., 2018).
Therefore to investigate whether the chromosomal phase of KCNH2-K897T could also influence the functional consequences of primary KCNH2 mutations, we took advantage of isogenic hiPSC models that were genetically edited to harbor heterozygous LQT2-causing missense variants either within the pore-loop domain (KCNH2A561T/WT) or cytoplasmic tail region (KCNH2N996I/WT) (Brandão et al., 2020). Functional comparisons revealed differences in the biophysical properties of hERG between the cis- and trans-oriented hiPSC-CM disease models. When KCNH2-K897T was present on the same allele as the primary mutation, the hiPSC-CMs exhibited faster activation and deactivation kinetics compared to their trans-linked counterparts. This was also reflected in longer action potential (AP) and field potential (FP) durations (FPDs). Finally, the LQT2 hiPSC-CMs with KCNH2-K897T in cis exhibited an increased arrhythmogenic response to the hERG channel blocker, E4031. This study demonstrates how genetically matched hiPSC-CM disease models can facilitate investigations into the functional impact of genetic modifiers including how the linkage phase can affect disease severity.
Results
Characterization of Isogenic KCNH2 Disease hiPSC Lines With K897T in cis and trans With the Risk Allele
To establish whether the biophysical effect of the KCNH2-K897T polymorphism on mutant hERG channel activity differed depending on whether the amino acid change was on the same or different subunit to a trafficking-deficient mutation, we made use of genetically-matched hiPSC lines bearing primary LQT2-causing KCNH2 mutations. Previously, we introduced missense variants either within the pore-loop domain (A561T) or cytoplasmic tail region (N996I) in heterozygosity into a control hiPSC line (Brandão et al., 2020; Figures 1A,B). Subsequent genotyping of the hiPSC lines used in this study confirmed that the K897T polymorphism was present on the edited allele (KCNH2A561T/WT cis and KCNH2N996I/WT cis) (Figures 1C-E and Supplementary Figure 1A). Further screening also identified edited hiPSC lines with the K897T polymorphism on the opposite allele to the primary KCNH2 mutations (KCNH2A561T/WT trans and KCNH2N996I/WT trans) (Figures 1D,E). These hiPSC lines were confirmed to have a normal G-band karyotype and to express stem cell markers such as SOX2 (SRY-box transcription factor 2), OCT-4 (POU class 5 homeobox 1), NANOG, and SSEA4 (stage-specific embryonic antigen-4) (Supplementary Figures 1B–E).
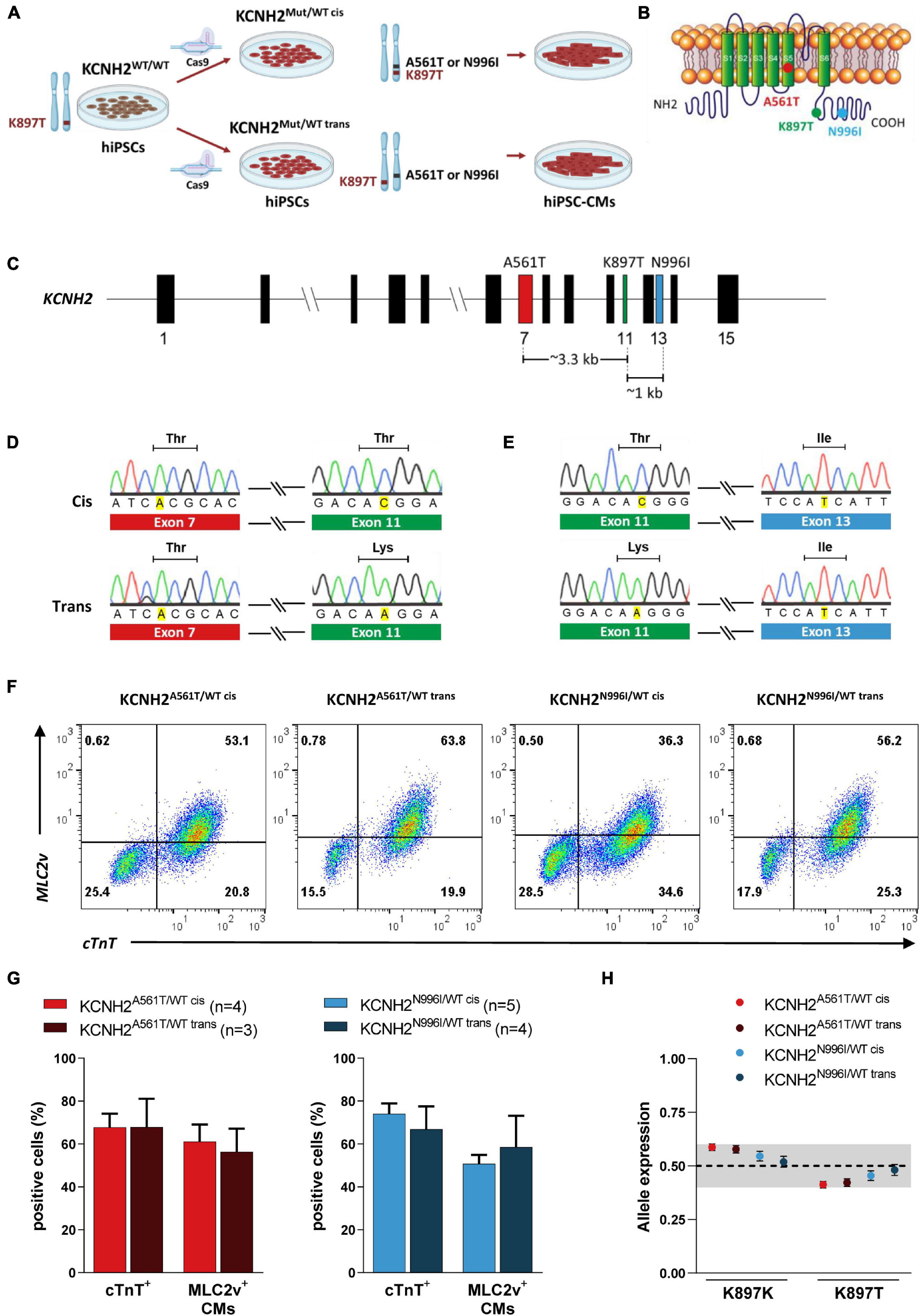
Figure 1. Genotyping of isogenic human induced pluripotent stem cell (hiPSC) lines with KCNH2 mutations and their differentiation into cardiomyocytes. (A) Schematic outlining the generation of isogenic hiPSC-CMs harboring KCNH2-K897T either in cis or trans orientation with long QT syndrome type 2 (LQT2) missense mutations, p.A561T or p.N996I. (B) Structure of KCNH2-encoded ion channel, hERG, indicating the approximate location of the primary mutations and K897T. (C) Structure of the KCNH2 genomic locus displaying the exons containing the missense mutations and the genomic distance between them. Exon numbers are indicated. (D,E) Sanger sequencing of gene-edited hiPSC lines KCNH2A561T/WT (D) and KCNH2N996I/WT (E) harboring KCNH2-K897T either in cis (top panel) or trans (bottom panel) orientation. Only the p.A561T and p.N996I mutated alleles and their corresponding K897T region of the sequences are shown. (F) Representative flow cytometry plots showing cardiac troponin T (cTnT) and myosin light chain 2v (MLC2v) expression in hiPSC-CMs at differentiation day 21 + 7. Numbers inside the plots are percentage of cells within the gated region. (G) Average percentage of hiPSC-CMs (cTnT+), and the proportion of ventricular-like (MLC2v+) cardiomyocytes within the hiPSC-CM population for the KCNH2A561T/WT (left panel) and KCNH2N996I/WT (right panel) cell lines. Values (n) indicate the number of differentiations analyzed. (H) Quantification of KCNH2 expression specifically from the K897K and K897T alleles in the differentiated hiPSC lines and displayed as fractional abundance, with the shaded area indicating the region close to 0.5 (0.4–0.6). KCNH2A561T/WT cis and KCNH2A561T/WT trans: n = 3 replicates; KCNH2N996I/WT cis and KCNH2N996I/WT trans: n = 6 replicates. Error bars are the Poisson 95% CI.
All four hiPSC lines efficiently differentiated into cardiomyocytes, with an average 67–74% of the cells (from three to five differentiations) expressing the pan-cardiomyocyte marker cardiac troponin T (cTnT) (Figures 1F,G). Similar percentages of hiPSC-CMs for each of the lines also expressed the ventricular cardiomyocyte marker, myosin light chain 2v (MLC2v; ∼50–60%) (Figures 1F,G). Additionally, hiPSC-CMs from all the four lines showed balanced allelic expression of the KCNH2 transcript (Figure 1H).
hERG Channel Kinetics in LQT2 hiPSC-CMs Is Influenced by the Linkage Phase of the K897T Polymorphism
To evaluate whether the linkage phase of the K897T polymorphism to the primary KCNH2 variants A561T and N996I resulted in differences in IKr, the current density and gating properties were determined in single hiPSC-CMs. Examples of current traces are shown in Figures 2A,B. As expected, IKr density in the disease lines was smaller compared to KCNH2WT/WT hiPSC-CMs (Supplementary Figure 2A). For example, the maximal tail current density for KCNH2WT/WT was 3.3 ± 0.2 pA/pF (n = 16), while it was 0.8 ± 0.1 pA/pF (n = 19), and 1.9 ± 0.1 pA/pF (n = 18) for KCNH2A561T/WT cis or KCNH2N996I/WT cis, respectively. Tail and steady-state IKr densities did not differ significantly between the cis and trans pairs for KCNH2A561T/WT or KCNH2N996I/WT at any voltage step (Figures 2C–F). Furthermore, voltage dependency of activation was also similar between the pairs as indicated by the overlapping normalized tail current curves (Supplementary Figures 2B,C).
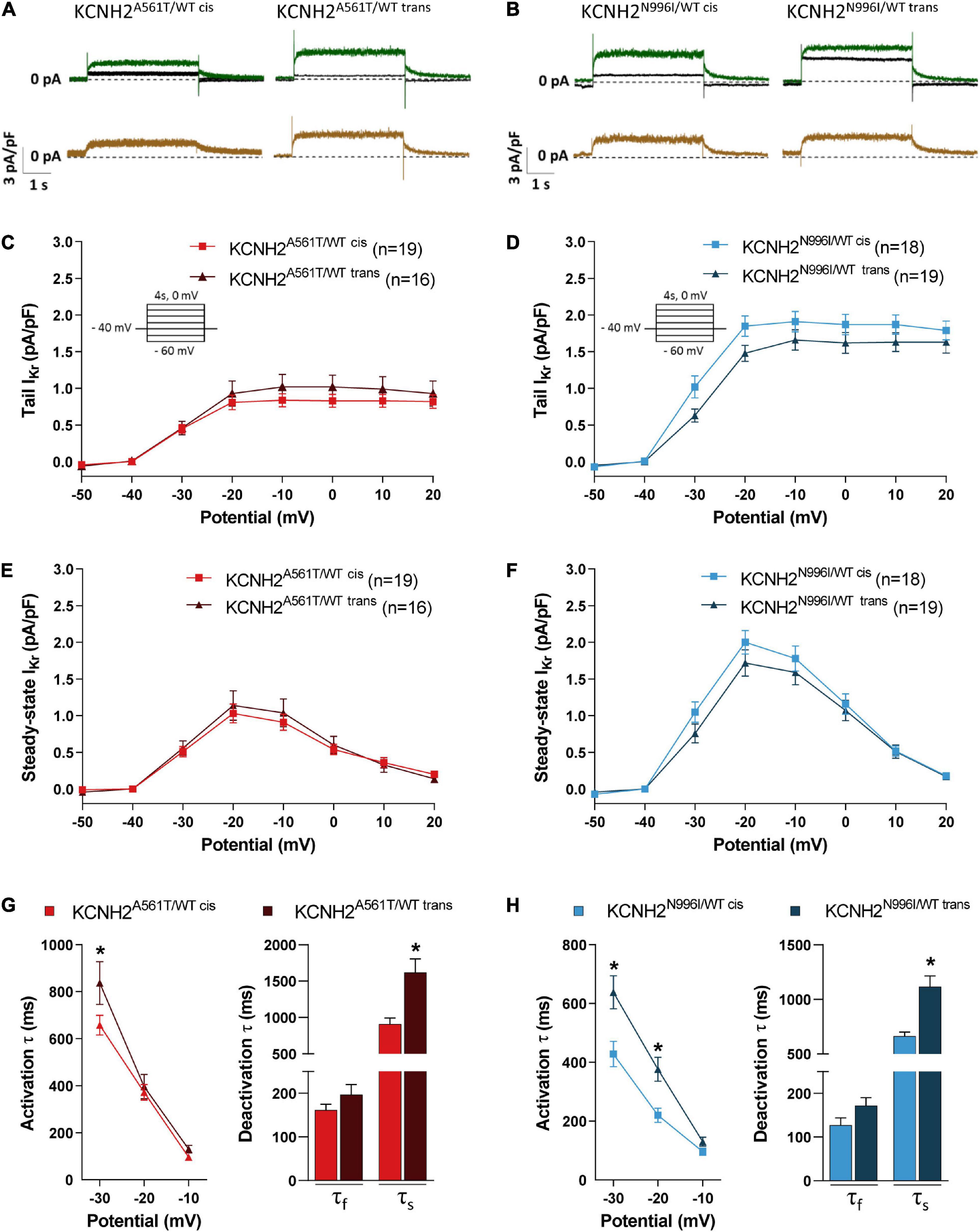
Figure 2. IKr properties in KCNH2A561T/WT and KCNH2N996I/WT hiPSC-CMs with KCNH2-K897T either in cis or trans orientation. (A,B) Representative current traces elicited upon 4 s depolarizing steps to –10 mV from a holding potential of –40 mV before (green lines) and after (black lines) application of 5 μM E4031 in isogenic sets of KCNH2A561T/WT (A) and KCNH2N996I/WT (B) hiPSC-CMs. The resulting E4031-sensitive traces are indicated in the bottom panel (brown lines). (C–F) Average current-voltage relationships for tail IKr current (C,D) and steady-state (E,F) densities in the indicated hiPSC-CMs. All the hiPSC-CMs activated upon depolarization reached a maximum steady-state current at –20 mV which decreased at more positive potentials due to the onset of inactivation. Inset: voltage protocol. Values (n) indicate the number of single hiPSC-CMs analyzed from three or four differentiations. (G,H) Average activation (left panel) and fast (τf) and slow (τs) deactivation (right panel, by stepping back from depolarizing step 0 mV to –40 mV holding potential) in KCNH2A561T/WT (G) and KCNH2N996I/WT (H) hiPSC-CMs. For KCNH2A561T/WT cis and KCNH2A561T/WT trans: activation, n = 19 and n = 16; deactivation, n = 16 and n = 10 from three differentiations; for KCNH2N996I/WT cis and KCNH2N996I/WT trans: activation n = 17 and n = 19; deactivation n = 17 and n = 19 from three or four differentiations for each cell line. *indicates statistical significance between cis-trans pairs [activation: –30 mV, KCNH2A561T/WT, p = 0.02; KCNH2N996I/WT, p < 0.001; –20 mV, KCNH2N996I/WT, p = 0.01; slow deactivation: KCNH2A561T/WT, p = 0.001; KCNH2N996I/WT, p < 0.001; the two-way ANOVA with the Sidak’s multiple comparison (activation) or the unpaired Student’s t-test (deactivation)].
However, kinetics analysis revealed significant faster channel kinetics for the hiPSC-CMs harboring K897T on the mutant allele, namely KCNH2A561T/WT cis and KCNH2N996I/WT cis (Figures 2G,H). For example, IKr activation (τ) at –30 mV was 21 and 33% faster for the KCNH2A561T/WT cis and KCNH2N996I/WT cis compared to their respective trans counterparts. The slow component (τslow) of the IKr deactivation obtained from the tail current decay also was significantly faster when K897T was in cis (Figures 2G,H). Finally, while the fast component (τfast) of deactivation was on average faster when K897T was on the same allele as the mutation, this did not reach significance for either of the KCNH2 variant pairs.
Taken together, this data indicated that while IKr current density and voltage dependency of activation in the KCNH2 variant lines were not influenced by which allele carried the K897T polymorphism, when it was on the same allele as the disease variant, an acceleration in activation and deactivation kinetics were observed. The changes in (de)activation kinetics may have a direct effect on the AP and contribute to changes in the minimum diastolic potential (mDP) in the final phase of diastole of spontaneously active hiPSC-CMs, and thereby consequently affect channel gating and permeation of other voltage-gated ion channels (Verkerk and Wilders, 2021).
Co-localization of KCNH2-K897T With the Primary Mutation Prolongs the Repolarization Period in hiPSC-CMs
Subsequently, we investigated whether the allele carrying the K897T polymorphism also altered AP parameters in the mutant lines. AP recordings were obtained from both the spontaneously beating and paced clusters of CMs (10–20 cells; Supplementary Figure 3A), with representative APs paced at 1 Hz shown in Figure 3A. As expected, the four LQT2 hiPSC-CM lines all exhibited prolonged AP durations (APDs) and, consequently, the number of cells able to follow at 4 Hz pacing was reduced compared to KCNH2WT/WT hiPSC-CMs (Supplementary Figures 3B,C). For the KCNH2-A561T lines at 1 Hz pacing, the APD at 20, 50, and 90% of repolarization (APD20, APD50, and APD90) was significantly prolonged when the K897T polymorphism was in cis compared to when it was not present on the risk allele (Figure 3B and Supplementary Table 1). Measurements from unpaced KCNH2A561T/WT hiPSC-CMs displayed similar differences in APD (Supplementary Figures 4A,B). The difference in APD90 between the KCNH2A561T/WT hiPSC-CMs was also present at slower pacing frequencies (Supplementary Figure 3B). In agreement with the shorter APD, a greater proportion of KCNH2A561T/WT trans CMs could be paced at 2 Hz or faster (Supplementary Figure 3C). While paced KCNH2N996I/WT hiPSC-CM clusters with the polymorphism in either allele showed no differences in APD or ability to follow pacing at ≤ 4 Hz (Figure 3C and Supplementary Figure 3), the APD was prolonged in spontaneously beating KCNH2N996I/WT cis hiPSC-CMs (Supplementary Figure 4C and Supplementary Table 1).
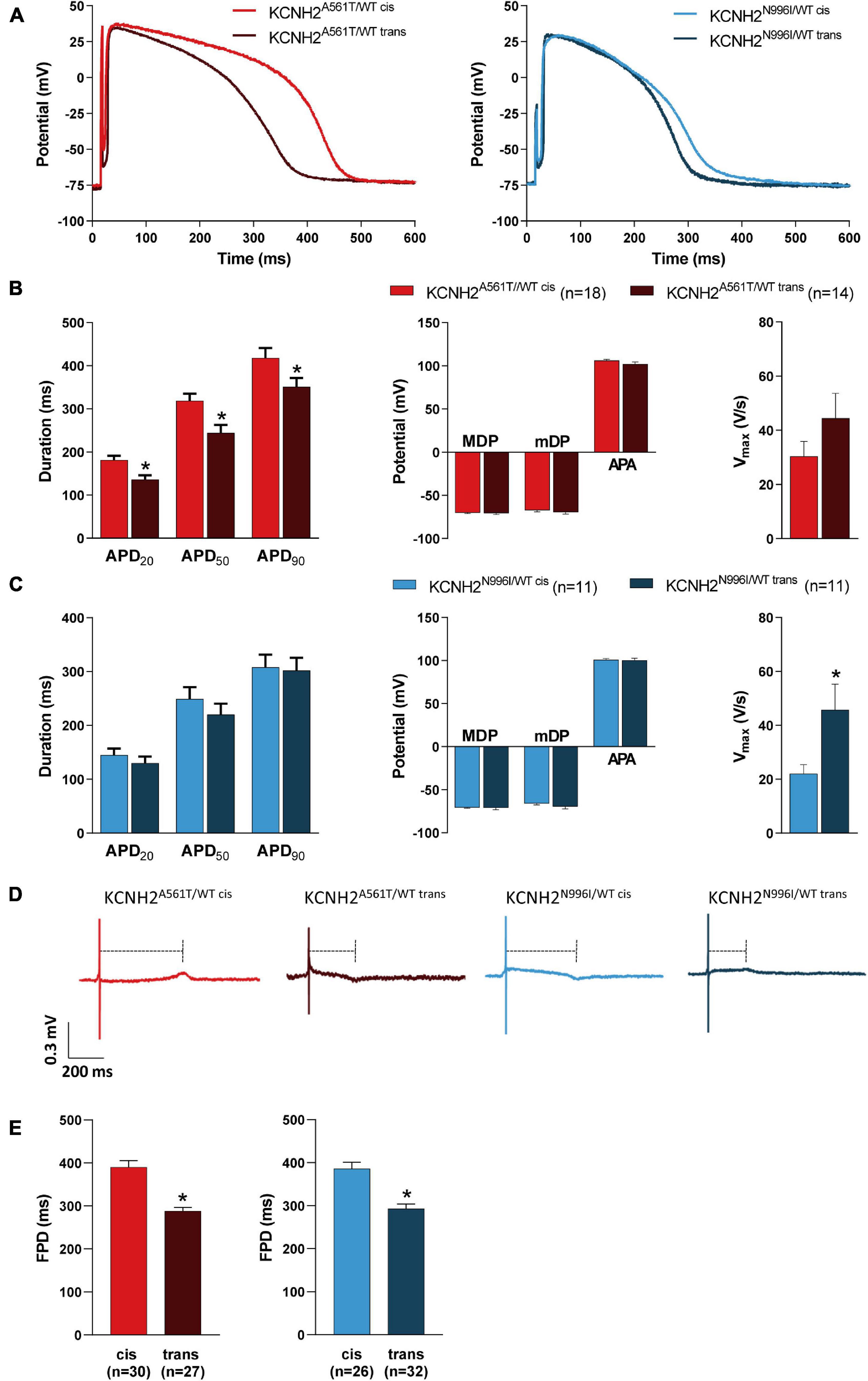
Figure 3. Electrophysiological characteristics of KCNH2A561T/WT and KCNH2N996I/WT hiPSC-CMs with KCNH2-K897T either in cis or trans orientation. (A) Representative AP traces of the pairs of KCNH2A561T/WT (left panel) and KCNH2N996I/WT (right panel) hiPSC-CMs measured at 1 Hz. (B,C) Average APD20, APD50, APD90, MDP, mDP, APA, and Vmax at 1 Hz for hiPSC-CM clusters for both KCNH2A561T/WT (B) and KCNH2N996I/WT (C) isogenic pairs. Values (n) refer to the number of individual hiPSC-CM clusters from two or three differentiations for each cell line. * indicates statistical significance [for panel (B), APD20 p = 0.003, APD50 p = 0.006, APD90 p = 0.04; for panel (C), Vmax p = 0.03; unpaired Student’s t-test]. (D) Representative MEA traces recorded at 0.8 Hz in the indicated hiPSC-CM monolayers. Dashed lines indicate the FPD. (E) Average FPD at 0.8 Hz for KCNH2A561T/WT (left panel) and KCNH2N996I/WT (right panel) isogenic pairs. Values (n) indicate the number of independent recordings analyzed from three or four differentiations. *indicates statistical significance [KCNH2A561T/WT and KCNH2N996I/WT p < 0.0001; unpaired Student’s t-test].
In agreement with the faster IKr kinetics observed in the LQT2 hiPSC-CMs where K897T was in cis with the mutation (Figures 2G,H), these lines when measured spontaneously also had a more depolarized mDP and faster cycle length compared to their trans counterparts. However the latter was only significant for the pair of KCNH2A561T/WT lines (Supplementary Figure 4 and Supplementary Table 1). It is likely that these differences contributed to the significantly lower AP upstroke velocity (Vmax) observed in the KCNH2A561T/WT cis and KCNH2N996I/WT cis hiPSC-CMs compared to their respective trans counterparts (Supplementary Figures 4B,C and Supplementary Table 1). Differences in mDP were not observed when the cells were paced because the external stimulus enabled AP initiation in a more hyperpolarized state (Figures 3A–C and Supplementary Table 1). In general, no significant differences in maximum diastolic potential (MDP) or AP amplitude (APA) were observed in either cis-trans hiPSC-CM pairs for the two KCNH2 mutations (Figures 3B,C, Supplementary Figures 4B,C, and Supplementary Table 1), though APA was smaller in spontaneous beating KCNH2A561T/WT trans compared to KCNH2A561T/WT cis hiPSC-CMs (Supplementary Figure 4B and Supplementary Table 1).
We also investigated using multi-electrode arrays (MEAs) the impact of the allele orientation of the K897T polymorphism on the KCNH2 mutations A561T and N996I in FP recordings of hiPSC-CM monolayers. To optically pace the hiPSC-CMs, the cells were transfected with Channelrhodopsin-2 (ChR2) messenger RNA (mRNA) (Supplementary Figure 5A). This did not affect the intrinsic beat rate or FPD of the hiPSC-CMs (Supplementary Figures 5B,C). Representative traces of hiPSC-CM monolayers paced at 0.8 Hz are shown in Figure 3D. All the four KCNH2 mutant lines exhibited a prolongation at multiple pacing frequencies (0.5–2.0 Hz) compared to KCNH2WT/WT hiPSC-CMs (Supplementary Figure 5D). Consistent with the AP measurements, FPD was prolonged when the K897T polymorphism was in cis (Figures 3D,E). The difference in FPD between the cis and trans pairs of hiPSC-CMs was also apparent at a range of pacing frequencies (0.5–1.5 Hz), with the longer FPD of the K897T cis-oriented hiPSC-CMs also meaning fewer monolayers than their trans counterparts could be paced at ≥1.5 Hz (Supplementary Figures 5D,E).
Collectively, these findings indicate that the differences in IKr kinetics are reflected in either the AP or FP characteristics of the hiPSC-CMs, with the lines in which KCNH2-K897T was in cis with the primary mutation having more prolonged APDs and FPDs compared to their trans counterparts.
Long QT Syndrome Type 2 hiPSC-CMs With KCNH2-K897T in cis Exhibit an Increased Arrhythmogenic Response
Finally, we investigated whether the K897T polymorphism when oriented in cis with primary KCNH2 mutations also altered the response of the hiPSC-CMs to the hERG channel blocker, E4031. Representative FP traces of hiPSC-CMs optically paced at 0.8 Hz and measured with increasing concentrations of E4031 (0–1,000 nM) are shown in Figure 4A. Because up to half of the recordings for some lines could not be paced at ≥ 30 nM E4031 due to the occurrence of arrhythmic events (Supplementary Figure 6), changes in FPD were analyzed only up to this concentration. Addition of 3–30 nM E4031 resulted in FPD prolongation when compared to baseline values and corrected for vehicle (ΔΔFPD) for all the four disease lines (Figure 4B). The linkage phase of the K897T polymorphism did not influence the change in ΔΔFPD observed in response to the cumulative addition of E4031 for the KCNH2A561T/WT lines or for the KCNH2N996I/WT hiPSC-CMs at 30 nM E4031. Although there appeared to be a difference in response between the KCNH2N996I/WT cis and KCNH2N996I/WT trans hiPSC-CMs at lower concentrations of E4031, this was not significant and was most likely due to an acute prolongation in FPD in some KCNH2N996I/WT trans hiPSC-CMs following the initial addition of the vehicle.
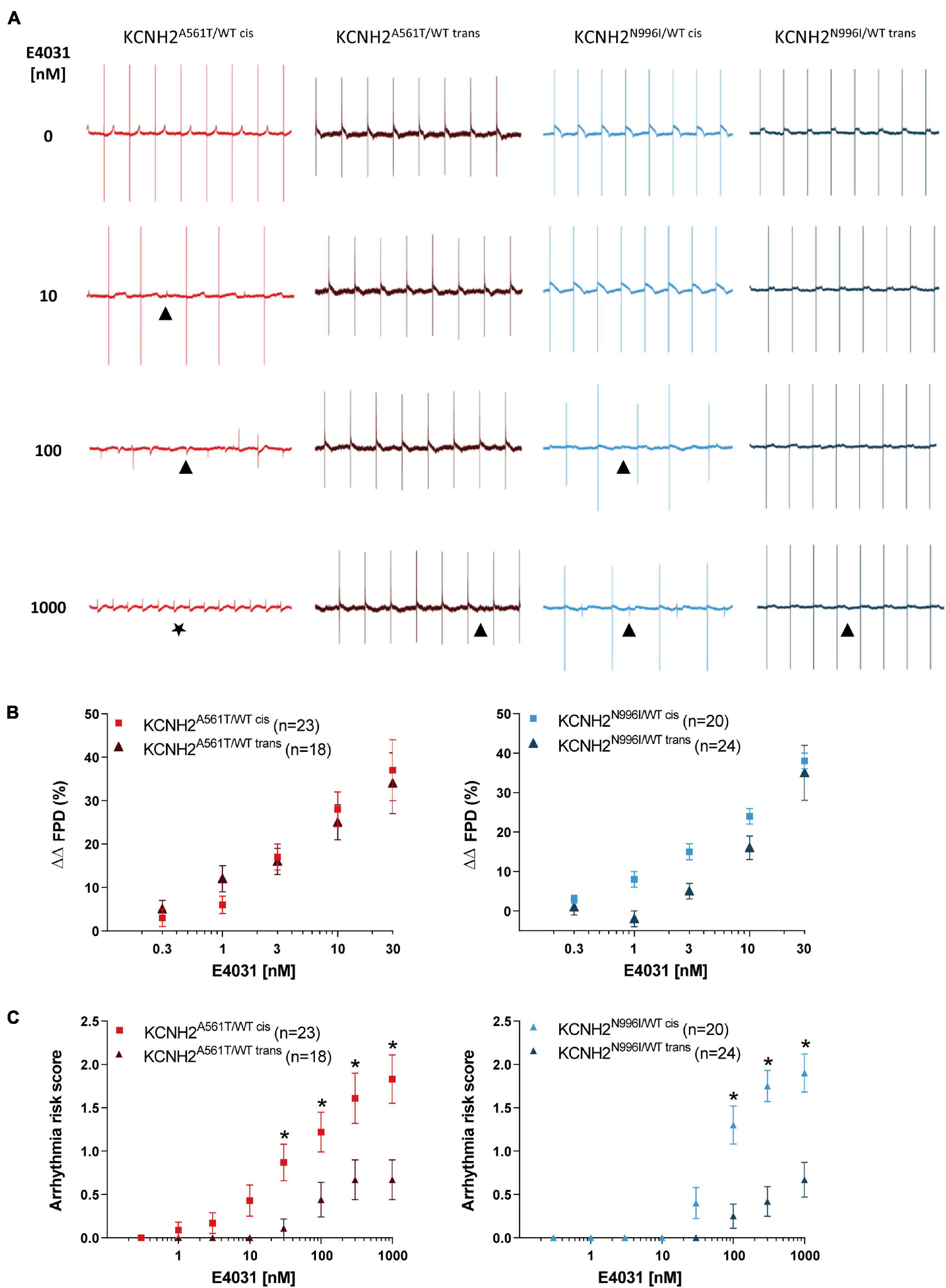
Figure 4. Effect of E4031 on FPD and arrhythmogenesis in KCNH2A561T/WT and KCNH2N996I/WT hiPSC-CMs. (A) Representative MEA traces recorded at 0.8 Hz in the indicated hiPSC-CM monolayers before and after cumulative addition of E4031 up to 1,000 nM. Symbols indicate examples of the different arrhythmia-like events observed: (▲) abnormal repolarization; (★) fibrillation. (B) Percent change in FPD relative to baseline and corrected for vehicle (ΔΔFPD) upon cumulative addition of E4031 in the KCNH2A561T/WT (left panel) and KCNH2N996I/WT (right panel) pairs of hiPSC-CMs. (C) Estimated arrhythmic risk score for the KCNH2A561T/WT (left panel) and KCNH2N996I/WT (right panel) pairs of hiPSC-CMs with cumulative addition of E4031. * indicates statistical significance between cis-trans pairs [KCNH2A561T/WT p < 0.02 and KCNH2N996I/WT p < 0.001; two-way ANOVA with Sidak’s multiple comparison test]. For panels (B,C), values (n) indicate the number of E4031-treated wells analyzed from three or four differentiations.
The E4031-induced arrhythmic events in the hiPSC-CM monolayers were classified in weighted categories [no arrhythmia; abnormal repolarization (early after depolarization (EAD)-like); fibrillation; quiescence] and a previously developed scoring system (Brandão et al., 2020) used to calculate the arrhythmogenic risk for each line. Arrhythmic events were detected in the KCNH2A561T/WT cis hiPSC-CMs from 1 nM E4031, while for the KCNH2A561T/WT trans hiPSC-CMs, these only started to occur at higher concentrations (≥30 nM; Supplementary Figure 6B). At the highest concentration of E4031 analyzed (1,000 nM), 6 out of 18 (33%) monolayers of KCNH2A561T/WT trans hiPSC-CMs exhibited abnormal repolarizations, while ∼70% of KCNH2A561T/WT cis hiPSC-CM recordings displayed arrhythmia-like events including ∼40% of recordings showing fibrillation or quiescence. This resulted in the KCNH2A561T/WT cis hiPSC-CMs having a significantly increased arrhythmia risk score compared to KCNH2A561T/WT trans hiPSC-CMs from 30 nM E4031 (Figure 4C). Similar findings were observed also with the KCNH2N996I/WT lines, with 1,000 nM E4031 causing a larger percentage of arrhythmia-like events in hiPSC-CMs in which KCNH2-K897T was on the same allele as N996I (85 vs. 33%, cis and trans respectively) (Supplementary Figure 6B). Also, KCNH2N996I/WT cis hiPSC-CMs were more susceptible to E4031-induced arrhythmic events from 100 nM (Figure 4C). Overall, this data indicates that while the chromosomal phase of the K897T polymorphism with primary KCNH2 mutations does not influence changes in E4031-induced FPD, there was an increased susceptibility to E4031-induced arrhythmic events when the SNP was in cis.
Discussion
Variability in disease phenotype is a common feature of inherited arrhythmic disorders such as LQTS and can even be present between siblings carrying the same primary disease-causing mutation (Giudicessi and Ackerman, 2013). A contributing factor to this variable disease expressivity is common genetic variants such as SNPs, either in the same LQTS-susceptibility gene or in other genes that modulate cardiac ion channel function (Giudicessi and Ackerman, 2013; Schwartz et al., 2018). Furthermore, when both the modifying SNP(s) and primary mutation are located in the same gene, then the linkage phase can also influence the disease phenotype (Amin et al., 2012). Establishing the contribution of common genetic variants to the overall disease phenotype remains a significant challenge due to small effect sizes, presence of additional polymorphisms, and/or genetic background of the patient (Napolitano et al., 2018; van den Brink et al., 2020b). However, recent studies combining hiPSC models of LQTS with gene editing have demonstrated that this approach can facilitate in determining the contribution common variants in modifying genes have on the disease phenotype and establish their mechanism of action (Chai et al., 2018; Lee et al., 2021).
In addition to these family-specific approaches, factors capable of influencing disease severity may also be identified through GWAS. The common SNP rs1805123, which leads to KCNH2-K897T, has been associated with QTc interval variability in population-based studies, with the general consensus being that it leads to a shortening of the QT interval under resting conditions (Pietilä et al., 2002; Bezzina et al., 2003; Gouas et al., 2005; Pfeufer et al., 2005; Newton-Cheh et al., 2007; Marjamaa et al., 2009). However, in patients with LQT2, results are discordant with the variant linked to both the prolonging and shortening the QTc interval (Crotti et al., 2005; Zhang et al., 2008; Nof et al., 2010; Jenewein et al., 2018). In this study, we investigated whether KCNH2-K897T can modify the electrophysiological phenotypes and susceptibility to drug-induced arrhythmias observed in two hiPSC models of KCNH2 mutations known to cause LQT2. Explicitly, we have examined whether the chromosomal phase of this common polymorphism influenced the disease phenotype by using genetically-matched pairs of hiPSC-CMs in which K897T was either in cis or trans with the KCNH2 primary mutations, A561T or N996I. Use of isogenic pairs was critical to exclude the influence of any other genetic-modifying variants possibly present in the hiPSC line. The hiPSC-CMs with KCNH2-K897T on the mutant allele (cis) presented longer APDs or FPDs and augmented susceptibility to arrhythmic events in response to E4031 compared to their trans counterpart.
Consistent with previous findings (Brandão et al., 2020), IKr density was smaller in KCNH2A561T/WT hiPSC-CMs compared to KCNH2N996I/WT hiPSC-CMs (Figures 2C,D). However, the chromosomal phase of KCNH2-K897T did not alter the IKr density further in either of these disease models, suggesting that the polymorphism does not influence intracellular trafficking of the hERG channels. A previous study of hiPSC-CMs from LQT3 mutation carriers also did not detect differences in IKr tail current density, regardless of whether KCNH2-K897T was present in heterozygosity or homozygosity (Terrenoire et al., 2013).
However, we did observe that the chromosomal phase of KCNH2-K987T impacted the time dependence of activation and deactivation. The accelerated activation kinetics in the cis-configured variant lines could contribute to a gain-of-function mechanism as it may cause a larger potassium current outflow during the depolarization phase. However, countering this, the accelerated deactivation time constant could contribute to a loss-of-function, as it decreases the potassium outflow during the repolarization phase resulting in prolongation. Given that the role of IKr is most prominent in the repolarization phase of a cardiac AP, changes in the deactivation time constant will dominate any activation time constant consequences. This will then lead to a net loss-of-function of the channel if KCNH2-K897T is present on the same allele as the primary mutation and a prolongation of the AP.
Indeed, patch clamp or MEA recordings demonstrated prolonged repolarization durations for the lines with KCNH2-K897T in cis orientation compared to their trans counterparts. As we expected any electrophysiological differences to be small, AP measurements were performed in clusters of hiPSC-CMs to reduce the variability caused by the larger seal-leak current that occurs per cell with single cells (Krogh-Madsen et al., 2005; Sheng et al., 2012). We and others have previously shown that performing electrophysiological measurements in such multicellular format enables differences in disease phenotype to be detected in hiPSC-CM models of LQT2 that were not uncovered in single cardiomyocytes (Brandão et al., 2020; Shah et al., 2020).
The accelerated slow component of deactivation in the LQT2 hiPSC-CMs harboring KCNH2-K897T on the mutant allele will also decrease the role of IKr in maintaining the diastolic potential in spontaneously active hiPSC-CMs, especially late during diastole (Doss et al., 2012). This was reflected in the spontaneously beating cis-oriented hiPSC-CMs exhibiting a less negative mDP immediately prior to the initiation of the AP. As a consequence of the more positive mDP, the maximal upstroke velocity was lower in these hiPSC-CMs, likely due to enhanced inactivation of Na+ channels (Verkerk and Wilders, 2021). The cells with KCNH2-K897T in cis configuration also exhibited a prolonged APD that was most prominent during spontaneous activity. As this prolongation was already apparent at APD20 and so outside the voltage range of IKr deactivation, the more pronounced mDP depolarization and the consequent effects on other voltage-gated ion channels may also contribute to the observed APD prolongation.
We also evaluated whether the chromosomal phase of K897T relative to the primary KCNH2 mutations altered the frequency that drug-induced arrhythmogenic events occurred in the hiPSC-CM disease models. The blocking of hERG by E4031 revealed comparable FPD prolonging effects regardless of whether the polymorphism was in cis or trans configuration. However, a significant proportion of the LQT2 hiPSC-CMs in which KCNH2-K897T was also present on the risk allele could not be paced when E4031 was > 100 nM, while most of the trans-configured cells followed pacing at all the concentrations of E4031. Classifying and scoring the types of arrhythmic events detected indicated that the polymorphism in cis with the primary mutation resulted in a higher arrhythmogenic risk when E4031 was ≥ 30 nM. These differences in arrhythmogenic susceptibility might arise from the altered IKr channel kinetics as faster deactivation properties have been proposed to aggravate the LQTS phenotype following exposure to hERG channel blockers (Romero et al., 2015).
There have been conflicting reports of the impact of the KCNH2-K897T polymorphism on the disease phenotype in patients with LQT2 with identified primary KCNH2 mutations. Several studies found LQT2 family members to only be symptomatic when they co-inherited the polymorphism (Crotti et al., 2005; Nof et al., 2010), while others observed less severe symptoms (Zhang et al., 2008; Jenewein et al., 2018). These discrepancies could be related to the type and location of the primary mutation, the presence of additional QT interval-modifying variants, or whether the disease-causing mutant is in cis or trans linkage with K897T. By using gene editing to introduce primary mutations into control hiPSC lines that are genetically defined, the variable effects that these genetic background differences introduce can be avoided. Furthermore, these models also enable the investigation of the potential differential impact that the linkage phase of the polymorphism might have with the primary KCNH2 mutation.
Although only one clone for each genotype was included in this study, we previously examined two independently derived clones for each of the KCNH2 mutant lines harboring KCNH2-K897T in cis and observed similar phenotypes in both the clonal lines (Brandão et al., 2020). In this study, we have investigated the effect of the linkage phase of K897T polymorphism for both the primary KCNH2 mutations, with similar differences observed for both when KCNH2-K897T was on the same allele as the mutation and, thus, indicating that these are unlikely to be due to variants that have arisen spontaneously in culture or from nuclease-induced off-target effects. Sequencing of KCNH2 in the control hiPSC line confirmed that no other amino acid-altering variants were present. Furthermore, KCNH2 was equally expressed from both the alleles in the hiPSC-CMs from all the lines examined, indicating that non-coding variants were not contributing to the observed differences.
Follow-up studies in which the K897T variant is corrected in the cell lines would be valuable to determine whether this common polymorphism plays a protecting or exacerbating role in QTc prolongation and the consequential effect on disease severity in patients with LQT2. Likewise, additional molecular-based investigation into whether the polymorphism influences intracellular trafficking of either wild type or mutant hERG channels, for example, by epitope tags into KCNH2 (Kanner et al., 2018), could provide further insight into the pathogenesis of LQT2. Finally, while all the hiPSC-CMs compared were culture age-matched and appeared to be of a comparable level of maturity based on MLC2v expression and similar MDP, the hiPSC-CMs are relatively immature with respect to ion channel properties compared to adult cardiomyocytes (Veerman et al., 2015).
In conclusion, we have demonstrated in hiPSC-CMs that the chromosomal phase of the KCNH2-K897T polymorphism with LQT2-associated mutations influences the biophysical properties of IKr, prolonging the repolarization phase, and increasing drug-induced arrhythmogenicity when cis-configured. This study aids in advancing hiPSC-CMs as a model to gain insight into the genetic causes of variable disease expressivity in patients with congenital LQTS and help to refine risk stratification and precision medicine approaches for these diseases.
Materials and Methods
Linkage Determination of the K897T Polymorphism in the LQT2 hiPSC Lines
The KCNH2A561T/WT and KCNH2N996I/WT hiPSC lines were generated as previously described (Brandão et al., 2020). Sequencing of parental hiPSC KCNH2WT/WT line [LUMC0020iCTRL; (Zhang et al., 2014)] determined the line was heterozygous for a substitution of lysine to threonine at position 897 of KCNH2 (K897T). Chromosomal phasing of the KCNH2-K897T polymorphism with the introduced LQT2 mutation was carried out on the resulting hiPSC clones (Supplementary Figure 1A). For KCNH2A561T/WT, this was performed using the One-Step PrimeScript RT-PCR (Takara Bio Incorporation, Kusatsu, Japan) on RNA isolated from hiPSC-CMs as described by the manufacturer; while for KCNH2N996I/WT, genomic DNA was isolated from the hiPSCs for PCR amplification (Supplementary Figure 1A). The resulting amplified products were cloned into pMiniT 2.0 [New England Biolabs (NEB), Ipswich, MA, United States] and genotyped by Sanger sequencing for both the primary mutation and the K897T polymorphism. The primers used are given in Supplementary Table 2. The previously characterized KCNH2A561T/WT and KCNH2N996I/WT hiPSC lines [LUMC0020iHERG-03 and LUMC0020iHERG-01, respectively (Brandão et al., 2020)] were confirmed to have the KCNH2-K897T SNP in cis configuration with the primary mutations. Additional targeted clones, LUMC0020iHERG-06 (KCNH2A561T/WT trans) and LUMC0020iHERG-09 (KCNH2N996I/WT trans), were identified as harboring KCNH2-K897T in trans configuration with the primary mutations.
Culture and Differentiation of hiPSCs
The hiPSCs were maintained in Essential 8 medium (Thermo Fisher Scientific, Waltham, MA, United States) on vitronectin (Thermo Fisher Scientific)-coated plates as previously described (Brandão et al., 2020). The hiPSCs were differentiated into cardiomyocytes using the Pluricyte Cardiomyocyte Differentiation kit (Ncardia, Leiden, Netherlands) according to the protocol of the manufacturer. After 20–21 days of differentiation, the hiPSC-CMs were dissociated and cryopreserved as previously described (van den Brink et al., 2020a; Campostrini et al., 2021). For replating, frozen hiPSC-CMs were thawed as described (Brandão et al., 2020) and replated in Medium C (Ncardia) with RevitaCell Supplement (1:100 dilution, Thermo Fisher Scientific). Media refreshments were performed 24 h thereafter and subsequently every 2–3 days. The hiPSC-CMs were evaluated 7–12 days after thawing, with the hiPSC-CMs age-matched in each assay (summarized in Supplementary Table 3).
Karyotyping
The hiPSC lines were karyotyped by G-banding. Chromosome spreads of 20 metaphases per cell line were performed and analyzed by the Laboratory for Diagnostic Genome Analysis (Leiden University Medical Center).
Flow Cytometric Analysis
Flow cytometry analysis of hiPSCs and hiPSC-CMs was performed as previously described (Brandão et al., 2020). The hiPSCs were incubated with the conjugated antibodies OCT-4-BV421 (1:25, Cat# 565644, BD Biosciences, Franklin Lakes, NJ, United States), SOX2-A488 (1:200, Cat# 53-9811-80, eBioscience, Waltham, MA, United States), or SSEA4-PE-vio770 (1:25, Cat# 130-105-081, Miltenyi Biotec, Bergisch Gladbach, Germany). 7–8 days post-thawing, hiPSC-CMs were fixed, permeabilized, and incubated with cTnT-Vioblue (1:11, Cat# 130-109-814, Miltenyi Biotec), and MLC2v-PE (1:11, Cat# 130-106-183, Miltenyi Biotec). A MacsQuant VYB flow cytometer (Miltenyi Biotec) was used for data acquisition and FlowJo software (FlowJo, Ashland, OR, United States) for data analysis.
Immunofluorescence Analysis
The hiPSCs were plated in 24-well cell culture plates at 0.5 × 105/cm2 and fixed 2 days later using 4% paraformaldehyde for 15 min. Fixed cells were permeabilized with phosphate buffer saline (PBS)/0.1% Triton X-100 (Sigma-Aldrich, Saint Louis, MO, United States) and blocked with 1% bovine serum albumin (Sigma-Aldrich)/0.05% Tween (Merck, Kenilworth, NJ, United States) in PBS. Cells were first incubated with NANOG (1:200, Cat# AF1997, R&D Systems, Minneapolis, MN, United States) and SSEA4 (1:200, Cat# SC59368, Santa Cruz Biotechnology, Santa Cruz, CA, United States) or with conjugated SOX2-A488 (1:200, Cat#53-9811-80, eBioscience). The primary antibodies to NANOG and SSEA4 were detected with AlexaFluor 555- (1:500, Cat#A21432, Thermo Fisher Scientific) and Alexa Fluor 488- (1:500, Cat#A-21202, Thermo Fisher Scientific) conjugated antibodies, respectively. The antibodies were diluted in permeabilization medium (medium B; Thermo Fisher Scientific) and incubated overnight at 4°C or 1.5 h at room temperature (RT) for the primary and secondary antibodies, respectively. Nuclei were labeled with 4′6-Diamidino-2-Phenylindole (DAPI) (0.3 μM). Images were captured using an EVOS FL Auto 2 Cell Imaging System (Thermo Fisher Scientific).
ddPCR
RNA was extracted from hiPSC-CMs 7–8 days post-thawing using the NucleoSpin® RNA Kit (Macherey-Nagel, Hoerdt, France) and treated with the DNA-free™ Kit (Thermo Fisher Scientific). 1 μg of RNA was transcribed into complementary DNA (cDNA) using the iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, United States). Droplet digital PCR (ddPCR) was performed using a thermocycler, the Q200 AutoDG and QX200™ Droplet Digital PCR System (Bio-Rad). Assays comprising of premixtures of a forward and reverse primer (18 μM each) with a fluorescein (FAM) or hexachlorofluorescein (HEX) conjugated hydrolysis probe (5 μM) were designed based on predefined criteria (Bell et al., 2018). Sequences for the primers and probes are given in Supplementary Table 2. The FAM-conjugated probe was designed to specifically bind the KCNH2 wild-type sequence (K897K), while the HEX-conjugated probe recognized the KCNH2 sequence harboring K897T. To improve target specificity, three or four locked nucleic acids were included per probe. Reactions (22 μl) were prepared with 2x Supermix for probes with no deoxyuridine triphosphate (Bio-Rad), 900 nM of each primer, 250 nM of each probe, and 3 μl cDNA digested with 4 U MseI (NEB). Droplet generation, PCR amplification, and analysis were all performed according to the instructions of the manufacturer.
Allelic balance was determined with the QuantaSoft software (Bio-Rad) using the “Poisson Fractional Abundance Max” and “Poisson Fractional Abundance Min” values, which define the 95% CI around the estimate of allelic balance recorded under “Fractional Abundance.” Samples were classified as having a balanced allelic expression, if the values for each allele were within 0.5 ± 0.1 (Kamitaki et al., 2018).
Patch-Clamp Electrophysiology
10 mm glass coverslips were coated with 1:100 Matrigel in DMEM-F12 (Thermo Fisher Scientific) for 2 h at 37°C before the thawed cells were seeded at 1.0–1.5 × 104/cm2. Patch-clamp measurements were performed 7–12 days later using an Axopatch 200B Amplifier (Molecular Devices, San Jose, CA, United States) and digitized with a Digidata 1440A (Molecular Devices). Data acquisition, voltage control, and analysis were performed using pClamp10.7 software (Axon Instruments, Melbourne, Australia). Potentials were corrected for the calculated liquid junction potential (Barry and Lynch, 1991). AP and IKr sampling rates were 40 and 20 kHz, respectively. IKr traces were filtered with a low-pass Gaussian filter at 0.9 Hz. Glass capillary patch pipettes (World Precision Instruments, Sarasota, FL, United States) pulled from a Sutter P97 micropipette puller (resistance 1.8–2.5 MΩ) were used and series resistance was compensated for 80%. Cell membrane capacitance (pF) was estimated by dividing the decay time constant of the capacitive transient in response to 5 mV hyperpolarizing steps from −40 mV by series resistance. All the measurements were performed at 36.0 ± 0.5°C.
Voltage Clamp Measurements
IKr was measured in spontaneously contracting cells (7–10 days post-thawing) using ruptured patch clamp methodology in Tyrode’s solution containing (in mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, 5.5 glucose, and 5.0 HEPES; pH 7.4 (NaOH). Pipettes (resistance ∼2 MΩ) were filled with (in mM): 125 K-gluconate, 20 KCl, 5 K2-ATP, 10 HEPES, and 10 EGTA; pH 7.2 (KOH). IKr was activated by 4 s hyper- and depolarizing pulses from a holding potential of –40 mV, at a cycle length of 10 s, and in the presence of 5 μM nifedipine (Sigma-Aldrich). IKr was measured as a 5 μM E4031-sensitive current (Tocris, Bioscience, Bristol, England) by subtracting current traces before and after application of E4031. Current densities were analyzed by calculating delta pA at the end of the depolarizing voltage step (steady state) or upon stepping back to the holding potential (tail) and dividing by pF. Speed of IKr activation and deactivation was analyzed using monoexponential and biexponential fittings, respectively (Clampfit 10.7).
Current Clamp Measurements
APs were recorded using amphotericin-perforated patch-clamp methodology in spontaneously contracting two-dimensional clusters of hiPSC-CMs (10–20 cells), 7–12 days after replating, in Tyrode’s solution and using a pipette solution containing (in mM): 125 K-gluconate, 20 KCl, 5.0 NaCl, 0.44 amphotericin-B, and 10 HEPES (pH 7.2; KOH). APs were evoked at 0.2–4.0 Hz by 3 ms and 1.2–1.3x threshold current pulses through the patch pipette. The AP parameters (Vmax, mDP, MDP, APA, APD20, APD50, and APD90) were analyzed from at least 10 consecutive APs and averaged.
Multi-Electrode Array Electrophysiology
For MEA experiments, 96-well Lumos MEA plates (M768-tMEA-96OPT, Axion Biosystems, Atlanta, GA, United States) were coated with human fibronectin (40 μg/ml, Alfa Aesar, Haverhill, MA, United States) for 1 h at 37°C and thawed hiPSC-CMs seeded onto the eight recording electrodes at a density of 4.4 × 105/cm2. All the experiments were performed at 37°C in a 5% carbon dioxide (CO2) controlled environment, with the hiPSC-CMs (11–12 days post-thawing) equilibrated inside the device (Maestro Pro Multiwell MEA platform; Axion Biosystems) for 10 min prior to recording. FP traces were recorded at a sampling frequency of 12.5 kHz and digital low- and high-pass filters of 2,000 Hz Kaiser window and 0.1 Hz infinite impulse response (IIR), respectively.
In vitro Transcription
The coding sequence for ChR2 fused to enhanced yellow fluorescent protein (eYFP) was PCR amplified from the plasmid pLenti-EF1a-hChR2 (H134R)-EYFP-WPRE (#20942, Addgene, Watertown, MA, United States) using primers in which the 5′ primer incorporated the T7 polymerase promoter sequence (Supplementary Table 2). Following purification, the PCR product was digested with NdeI (NEB) to generate a 5′ overhang at the 3′ end of template. The digested product was gel extracted (Wizard SV Gel and PCR Clean-Up System; Promega, Madison, WI, United States) according to the instructions of the manufacturer and subsequently concentrated (>160 ng/μl) by ethanol precipitation. In vitro transcription (IVT) was performed using the INCOGNITO T7 ARCA 5mC- and Ψ-RNA Transcription Kit (Cellscript, Madison, WI, United States) following the instructions of the manufacturer, with the resulting mRNA precipitated overnight using LiCl. To the purified mRNA, a 5′ cap nucleotide and a 3′ poly(A) tail were sequentially added using the ScriptCap Cap 1 Capping System and A-Plus Poly(A) Polymerase Tailing Kit, respectively (Cellscript). The resulting capped and tailed mRNA was precipitated again using LiCl and quantified. The mRNA integrity was confirmed by electrophoresis using the Reliant RNA Precast Gel (Lonza, Basel, Switzerland).
Optical Pacing of hiPSC-CMs
At day 7 post-seeding, the hiPSC-CMs were transfected with in vitro transcribed ChR2-eYFP mRNA. The medium was refreshed 1 h prior to mRNA transfection (40–60 ng/well). The Lipofectamine™ Stem Transfection Reagent (Thermo Fisher Scientific) was first diluted (1:14) in Opti-MEM™ Reduced Serum Medium (Cat# 51985-026, Thermo Fisher Scientific). The ChR2 mRNA was then added to the lipofectamine mixture and incubated at RT for ∼10 min to form a liposome-mRNA complex before adding to the cells (15 μl per well). Untransfected controls were transfected with lipofection mix that did not contain the ChR2-eYFP mRNA. Media was refreshed ∼18 h post-transfection and then 3–4 days later before recording. 1 μM all-trans retinal (50 μl/well) (Sigma-Aldrich) was added to all the wells at least 1 h before baseline recordings at spontaneous beat rates. Spontaneous recordings were obtained from transfected and untransfected controls. At baseline and following E4031 addition, the transfected hiPSC-CMs were optically paced at frequencies ranging from 0.5 to 3.0 Hz using the Lumos™ Optical Stimulation System (Axion Biosystems). Settings for optical stimulation with 475 nm wavelength light are shown in Supplementary Table 4.
E4031 Treatment
E4031-induced effects on the hiPSC-CMs were evaluated by sequentially increasing the concentration (300 pM to 1 μM). 10 μm E4031 (Tocris Bioscience) dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) was serially diluted in Medium C (Ncardia). Each concentration was accomplished by the stepwise removal of 7 μl medium and addition of 7 μl diluted E4031 (3.5% replacement of the total volume in each well). The hiPSC-CMs were paced at 0.8 Hz for the entire duration, with the response to E4031 recorded for 3 min after an incubation period of 10 min. The maximum amount of DMSO present in the culture medium was 0.01%, with addition of the vehicle not significantly altering the FPD or inducing arrhythmic events in any of the lines.
Field potential duration and beat period were quantified over the whole recording time using the Cardiac Analysis Tool (Axion Biosystems). Signal-to-noise ratio was high and did not require filtering prior to analysis. No frequency correction was applied on the unpaced recordings, since this overestimated the rate-dependency of the FPD. Drug responses were normalized to their baseline measurement and corrected for their response to DMSO (ΔΔFPD). Arrhythmia-like events were classified and scored in weighted categories [no arrhythmia (score 0), abnormal repolarization/EAD like (score 2), fibrillation (score 3), and quiescence (score 4)] based on a previously developed scoring system (Brandão et al., 2020). Fibrillation was defined as a severe reduction in the FP amplitude, resulting in an almost complete abolishment of the Na+ current driven depolarization and accompanied with the presence of uncoordinated electrical activity. Quiescence was always preceded by abnormal repolarization and fibrillation.
Statistical Data Analysis
All the data are presented as mean ± SEM. Statistical tests performed are indicated in the “Results” section or in the Figure legends. Comparisons of two KCNH2Mut/WT lines to the KCNH2WT/WT line (three groups) were performed using the one-way ANOVA followed by the Tukey’s multiple comparisons test for post hoc analysis. If one of the null hypotheses could be rejected, pairwise comparisons were conducted using the Student’s t-test (Goeman and Solari, 2011). Differences were considered statistically significant at p < 0.05. Analyses were conducted with GraphPad Prism 8 software (GraphPad, San Diego, CA, United States).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
LB designed and conducted experiments, performed analysis, and wrote the manuscript. KB, LY, AB-A, and MM designed and conducted experiments. CM and AV provided scientific support and edited the manuscript. RD funded, conceived and supervised the project, wrote and edited the manuscript. All authors approved the submission of manuscript.
Funding
This study was supported by STEMCARDIORISK, a Starting Grant from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation program (H2020 European Research Council; grant agreement #638030), a VIDI fellowship from the Netherlands Organization for Scientific Research (Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO); ILLUMINATE; #91715303), and the Netherlands Organ-on-Chip Initiative, an NWO Gravitation project (#024.003.001) funded by the Ministry of Education, Culture and Science of the government of the Netherlands.
Conflict of Interest
CM is a cofounder of Pluriomics B. V. (now Ncardia B. V.).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Dr. Milena Bellin for providing the control (LUMC0020iCTRL) hiPSC line and the Laboratory for Diagnostic Genome Analysis (Leiden University Medical Center) for karyotyping the hiPSC lines. Panels in some figures were created using BioRender.com.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.755642/full#supplementary-material
References
Adler, A., Novelli, V., Amin, A. S., Abiusi, E., Care, M., Nannenberg, E. A., et al. (2020). An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141, 418–428. doi: 10.1161/CIRCULATIONAHA.119.043132
Amin, A. S., Giudicessi, J. R., Tijsen, A. J., Spanjaart, A. M., Reckman, Y. J., Klemens, C. A., et al. (2012). Variants in the 3’ untranslated region of the KCNQ1-encoded K v7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur. Heart J. 33, 714–723. doi: 10.1093/eurheartj/ehr473
Barry, P. H., and Lynch, J. W. (1991). Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 121, 101–117. doi: 10.1007/BF01870526
Bell, A. D., Usher, C. L., and McCarroll, S. A. (2018). Analyzing copy number variation with droplet digital PCR. Methods Mol. Biol. 1768, 143–160. doi: 10.1007/978-1-4939-7778-9_9
Bezzina, C. R., Lahrouchi, N., and Priori, S. G. (2015). Genetics of sudden cardiac death. Circ. Res. 116, 1919–1936. doi: 10.1161/CIRCRESAHA.116.304030
Bezzina, C. R., Verkerk, A. O., Busjahn, A., Jeron, A., Erdmann, J., Koopmann, T. T., et al. (2003). A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc. Res. 59, 27–36. doi: 10.1016/S0008-6363(03)00342-0
Brandão, K. O., Tabel, V. A., Atsma, D. E., Mummery, C. L., and Davis, R. P. (2017). Human pluripotent stem cell models of cardiac disease: from mechanisms to therapies. Dis. Model. Mech. 10, 1039–1059. doi: 10.1242/dmm.030320
Brandão, K. O., van den Brink, L., Miller, D. C., Grandela, C., van Meer, B. J., Mol, M. P. H., et al. (2020). Isogenic sets of hiPSC-CMs harboring distinct KCNH2 mutations differ functionally and in susceptibility to drug-Induced arrhythmias. Stem Cell Reports 15, 1127–1139. doi: 10.1016/j.stemcr.2020.10.005
Campostrini, G., Meraviglia, V., Giacomelli, E., van Helden, R. W. J., Yiangou, L., Davis, R. P., et al. (2021). Generation, functional analysis and applications of isogenic three-dimensional self-aggregating cardiac microtissues from human pluripotent stem cells. Nat. Protoc. 16, 2213–2256. doi: 10.1038/s41596-021-00497-2
Chai, S., Wan, X., Ramirez-Navarro, A., Tesar, P. J., Kaufman, E. S., Ficker, E., et al. (2018). Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity. J. Clin. Invest. 128, 1043–1056. doi: 10.1172/JCI94996
Coll, M., Pérez-Serra, A., Mates, J., del Olmo, B., Puigmulé, M., Fernandez-Falgueras, A., et al. (2018). Incomplete penetrance and variable expressivity: Hallmarks in channelopathies associated with sudden cardiac death. Biology 7:3. doi: 10.3390/biology7010003
Crotti, L., Lundquist, A. L., Insolia, R., Pedrazzini, M., Ferrandi, C., De Ferrari, G. M., et al. (2005). KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation 112, 1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071
Doss, M. X., Di Diego, J. M., Goodrow, R. J., Wu, Y., Cordeiro, J. M., Nesterenko, V. V., et al. (2012). Maximum diastolic potential of human induced pluripotent stem cell-derived cardiomyocytes depends critically on IKr. PLoS One 7:e40288. doi: 10.1371/journal.pone.0040288
Garg, P., Garg, V., Shrestha, R., Sanguinetti, M. C., Kamp, T. J., and Wu, J. C. (2018). Human induced pluripotent Stem cell–derived cardiomyocytes as models for cardiac channelopathies. Circ. Res. 123, 224–243. doi: 10.1161/CIRCRESAHA.118.311209
Gentile, S., Martin, N., Scappini, E., Williams, J., Erxleben, C., and Armstrong, D. L. (2008). The human ERG1 channel polymorphism, K897T, creates a phosphorylation site that inhibits channel activity. Proc. Natl. Acad. Sci. U.S.A. 105, 14704–14708. doi: 10.1073/pnas.0802250105
Giudicessi, J. R., and Ackerman, M. J. (2013). Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl. Res. 161, 1–14. doi: 10.1016/j.trsl.2012.08.005
Gnecchi, M., Sala, L., and Schwartz, P. J. (2021). Precision medicine and cardiac channelopathies: when dreams meet reality. Eur. Heart J. 42, 1661–1675. doi: 10.1093/eurheartj/ehab007
Goeman, J. J., and Solari, A. (2011). Multiple testing for exploratory research. Stat. Sci. 26, 584–597. doi: 10.1214/11-STS356
Gouas, L., Nicaud, V., Berthet, M., Forhan, A., Tiret, L., Balkau, B., et al. (2005). Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur. J. Hum. Genet. 13, 1213–1222. doi: 10.1038/sj.ejhg.5201489
Isbister, J., and Semsarian, C. (2019). Cardiovascular genomics and sudden cardiac death in the young. Aust. J. Gen. Pract. 48, 90–95. doi: 10.31128/AJGP-09-18-4715
Jenewein, T., Neumann, T., Erkapic, D., Kuniss, M., Verhoff, M. A., Thiel, G., et al. (2018). Influence of genetic modifiers on sudden cardiac death cases. Int. J. Legal Med. 132, 379–385. doi: 10.1007/s00414-017-1739-7
Kamitaki, N., Usher, C. L., and McCarroll, S. A. (2018). Using droplet digital PCR to analyze allele-specific RNA expression. Methods Mol. Biol. 1768, 401–422. doi: 10.1007/978-1-4939-7778-9_23
Kanner, S. A., Jain, A., and Colecraft, H. M. (2018). Development of a high-throughput flow cytometry assay to monitor defective trafficking and rescue of long QT2 mutant hERG channels. Front. Physiol. 9:397. doi: 10.3389/fphys.2018.00397
Krogh-Madsen, T., Schaffer, P., Skriver, A. D., Taylor, L. K., Pelzmann, B., Koidl, B., et al. (2005). An ionic model for rhythmic activity in small clusters of embryonic chick ventricular cells. Am. J. Physiol. Hear. Circ. Physiol. 289, H398–H413. doi: 10.1152/ajpheart.00683.2004
Lahrouchi, N., Tadros, R., Crotti, L., Mizusawa, Y., Postema, P. G., Beekman, L., et al. (2020). Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation 142, 324–338. doi: 10.1161/CIRCULATIONAHA.120.045956
Lee, Y. K., Sala, L., Mura, M., Rocchetti, M., Pedrazzini, M., Ran, X., et al. (2021). MTMR4 SNVs modulate ion channel degradation and clinical severity in congenital long QT syndrome: Insights in the mechanism of action of protective modifier genes. Cardiovasc. Res. 117, 767–779. doi: 10.1093/cvr/cvaa019
Marjamaa, A., Newton-Cheh, C., Porthan, K., Reunanen, A., Lahermo, P., Väänänen, H., et al. (2009). Common candidate gene variants are associated with QT interval duration in the general population. J. Intern. Med. 265, 448–458. doi: 10.1111/j.1365-2796.2008.02026.x
Napolitano, C., Mazzanti, A., and Priori, S. G. (2018). Genetic risk stratification in cardiac arrhythmias. Curr. Opin. Cardiol. 33, 298–303. doi: 10.1097/HCO.0000000000000506
Newton-Cheh, C., Guo, C. Y., Larson, M. G., Musone, S. L., Surti, A., Camargo, A. L., et al. (2007). Common genetic variation in KCNH2 is associated with QT interval duration: The Framingham heart study. Circulation 116, 1128–1136. doi: 10.1161/CIRCULATIONAHA.107.710780
Nof, E., Cordeiro, J. M., Pérez, G. J., Scornik, F. S., Calloe, K., Love, B., et al. (2010). A common single nucleotide polymorphism can exacerbate long-QT type 2 syndrome leading to sudden infant death. Circ. Cardiovasc. Genet. 3, 199–206. doi: 10.1161/CIRCGENETICS.109.898569
Paavonen, K. J., Chapman, H., Laitinen, P. J., Fodstad, H., Piippo, K., Swan, H., et al. (2003). Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG). Cardiovasc. Res. 59, 603–611. doi: 10.1016/S0008-6363(03)00458-9
Pfeufer, A., Jalilzadeh, S., Perz, S., Mueller, J. C., Hinterseer, M., Illig, T., et al. (2005). Common variants in myocardial ion channel genes modify the QT interval in the general population: Results from the KORA study. Circ. Res. 96, 693–701. doi: 10.1161/01.RES.0000161077.53751.e6
Pietilä, E., Fodstad, H., Niskasaari, E., Laitinen, P. J., Swan, H., Savolainen, M., et al. (2002). Association between HERG K897T polymorphism and QT interval in middle-aged finnish women. J. Am. Coll. Cardiol. 40, 511–514. doi: 10.1016/S0735-1097(02)01979-4
Romero, L., Trenor, B., Yang, P. C., Saiz, J., and Clancy, C. E. (2015). In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J. Mol. Cell. Cardiol. 87, 271–282. doi: 10.1016/j.yjmcc.2015.08.015
Schwartz, P. J., Crotti, L., and George, A. L. (2018). Modifier genes for sudden cardiac death. Eur. Heart J. 39, 3925–3931. doi: 10.1093/eurheartj/ehy502
Shah, D., Prajapati, C., Penttinen, K., Cherian, R. M., Koivumäki, J. T., Alexanova, A., et al. (2020). hiPSC-derived cardiomyocyte model of LQT2 syndrome derived from asymptomatic and symptomatic mutation carriers reproduces clinical differences in aggregates but not in single cells. Cells 9, 1153. doi: 10.3390/cells9051153
Sheng, X., Reppel, M., Nguemo, F., Mohammad, F. I., Kuzmenkin, A., Hescheler, J., et al. (2012). Human pluripotent stem cell-derived cardiomyocytes: response to TTX and lidocain reveals strong cell to cell variability. PLoS One 7:e45963. doi: 10.1371/journal.pone.0045963
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Terrenoire, C., Wang, K., Chan Tung, K. W., Chung, W. K., Pass, R. H., Lu, J. T., et al. (2013). Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J. Gen. Physiol. 141, 61–72. doi: 10.1085/jgp.201210899
van den Brink, L., Brandão, K. O., Yiangou, L., Mol, M. P. H., Grandela, C., Mummery, C. L., et al. (2020a). Cryopreservation of human pluripotent stem cell-derived cardiomyocytes is not detrimental to their molecular and functional properties. Stem Cell Res. 43:101698. doi: 10.1016/j.scr.2019.101698
van den Brink, L., Grandela, C., Mummery, C. L., and Davis, R. P. (2020b). Inherited cardiac diseases, pluripotent stem cells, and genome editing combined—the past, present, and future. Stem Cells 38, 174–186. doi: 10.1002/stem.3110
Veerman, C. C., Kosmidis, G., Mummery, C. L., Casini, S., Verkerk, A. O., and Bellin, M. (2015). Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. 24, 1035–1052. doi: 10.1089/scd.2014.0533
Verkerk, A. O., and Wilders, R. (2021). Dynamic clamp in electrophysiological studies on stem cell-derived cardiomyocytes—why and how? J. Cardiovasc. Pharmacol. 77, 267–279. doi: 10.1097/fjc.0000000000000955
Walsh, R., Tadros, R., and Bezzina, C. R. (2020). When genetic burden reaches threshold. Eur. Heart J. 41, 3849–3855. doi: 10.1093/eurheartj/ehaa269
Zhang, M., D’Aniello, C., Verkerk, A. O., Wrobel, E., Frank, S., Ward-van Oostwaard, D., et al. (2014). Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc. Natl. Acad. Sci. U.S.A. 111, E5383–E5392. doi: 10.1073/pnas.1419553111
Keywords: long QT syndrome type 2, disease modeling, human pluripotent stem cell-derived cardiomyocytes, isogenic, genetic modifier, cardiac electrophysiology, arrhythmia, hERG
Citation: van den Brink L, Brandão KO, Yiangou L, Blanch-Asensio A, Mol MPH, Mummery CL, Verkerk AO and Davis RP (2021) The Linkage Phase of the Polymorphism KCNH2-K897T Influences the Electrophysiological Phenotype in hiPSC Models of LQT2. Front. Physiol. 12:755642. doi: 10.3389/fphys.2021.755642
Received: 09 August 2021; Accepted: 10 November 2021;
Published: 16 December 2021.
Edited by:
Ofer Binah, Technion Israel Institute of Technology, IsraelReviewed by:
Daniel Sinnecker, Technical University of Munich, GermanyDanielle Pretorius, University of Alabama at Birmingham, United States
Mari Pekkanen-Mattila, Tampere University, Finland
Copyright © 2021 van den Brink, Brandão, Yiangou, Blanch-Asensio, Mol, Mummery, Verkerk and Davis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard P. Davis, ci5wLmRhdmlzQGx1bWMubmw=