
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 12 October 2021
Sec. Cardiac Electrophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.744730
This article is part of the Research Topic Calcium and Heart Failure: From Bench to Bedside View all 11 articles
 Andrew G. Edwards1,2
Andrew G. Edwards1,2 Halvor Mørk1
Halvor Mørk1 Mathis K. Stokke1,3,4David B. Lipsett1Ivar Sjaastad1,3
Mathis K. Stokke1,3,4David B. Lipsett1Ivar Sjaastad1,3 Sylvain Richard5
Sylvain Richard5 Ole M. Sejersted1
Ole M. Sejersted1 William E. Louch1,3*
William E. Louch1,3*Dysfunctional sarcoplasmic reticulum Ca2+ handling is commonly observed in heart failure, and thought to contribute to arrhythmogenesis through several mechanisms. Some time ago we developed a cardiomyocyte-specific inducible SERCA2 knockout mouse, which is remarkable in the degree to which major adaptations to sarcolemmal Ca2+ entry and efflux overcome the deficit in SR reuptake to permit relatively normal contractile function. Conventionally, those adaptations would also be expected to dramatically increase arrhythmia susceptibility. However, that susceptibility has never been tested, and it is possible that the very rapid repolarization of the murine action potential (AP) allows for large changes in sarcolemmal Ca2+ transport without substantially disrupting electrophysiologic stability. We investigated this hypothesis through telemetric ECG recording in the SERCA2-KO mouse, and patch-clamp electrophysiology, Ca2+ imaging, and mathematical modeling of isolated SERCA2-KO myocytes. While the SERCA2-KO animals exhibit major (and unique) electrophysiologic adaptations at both the organ and cell levels, they remain resistant to arrhythmia. A marked increase in peak L-type calcium (ICaL) current and slowed ICaL decay elicited pronounced prolongation of initial repolarization, but faster late repolarization normalizes overall AP duration. Early afterdepolarizations were seldom observed in KO animals, and those that were observed exhibited a mechanism intermediate between murine and large mammal dynamical properties. As expected, spontaneous SR Ca2+ sparks and waves were virtually absent. Together these findings suggest that intact SR Ca2+ handling is an absolute requirement for triggered arrhythmia in the mouse, and that in its absence, dramatic changes to the major inward currents can be resisted by the substantial K+ current reserve, even at end-stage disease.
Dysfunctional sarcoplasmic reticulum (SR) Ca2+ handling is known to destabilize cardiac electrophysiology in a broad range of arrhythmogenic diseases, from rare channelopathies (Priori et al., 2001, 2002) to prevalent acquired diseases such as heart failure (Pogwizd et al., 2001; Pogwizd and Bers, 2002). However, the role of the SR as an intracellular Ca2+ store able to drive cardiac contraction is fundamental to normal function of the heart, and evolutionary processes have carefully balanced the electrical stability of cardiac excitation-contraction (EC) coupling with the requirement for a high-gain intracellular Ca2+ release system. This balance has important implications for arrhythmia mechanisms in the heart, and these can be clearly illustrated by considering differences across mammalian species.
In healthy large mammals, approximately 30% of the Ca2+ that fuels cardiac contraction is obtained from L-type Ca2+ current (ICaL)-mediated Ca2+ influx (Shannon et al., 2004; Fearnley et al., 2011), and this can increase to near 50% in chronic diseases such as heart failure (Pogwizd and Bers, 2002). Such large transmembrane Ca2+ fluxes are permitted by a prolonged action potential (AP) plateau, which itself results from a relatively delicate balance of inward and outward currents in large mammals (Weiss et al., 2010). These characteristics of EC coupling in large mammals tend to favor repolarization instabilities, and potentiation of ICaL by frequency, neurohormonal stimuli, and genetic abnormalities are well known to destabilize repolarization in humans (Piot et al., 1996; Splawski et al., 2004, 2005; Sato et al., 2009; Tran et al., 2009). Indeed, it is for exactly this reason that significant investments have been made to establish and study large animal models of human arrhythmogenic diseases thought to result from repolarization abnormalities (Brunner et al., 2008; Koren, 2009).
In contrast, the structure of murine EC coupling pushes the mouse heart toward arrhythmogenic mechanisms that rely upon unstable SR Ca2+ handling, and away from mechanisms that result directly from repolarization instabilities due to ICa,L or other surface membrane currents. In particular, the large and rapidly activating outward K+ currents in the mouse and rat permit only a very brief AP. This both limits the degree to which ICaL can contribute to contractile Ca2+ and necessitates a larger contribution from the SR Ca2+ store (∼92%) (Bers, 2001). This promotes instability in both systolic and diastolic Ca2+ handling. Indeed, the mouse has proven to be a very useful model for studying arrhythmia phenotypes resulting from aberrant spontaneous SR Ca2+ release (Lehnart et al., 2008; Kashimura et al., 2010; Bai et al., 2013), and we have also shown that unstable triggered Ca2+ release recruits unique EAD dynamics in mice (Edwards et al., 2014). In vivo studies utilizing simultaneous ECG and surface mapping (monophasic AP or optical mapping) have suggested that the dominant mechanisms of arrhythmia in these mice are focal activity (perhaps originating from DADs in the His-Purkinje network), and APD alternans driven by aberrant SR Ca2+ handling (Lehnart et al., 2006; Cerrone et al., 2007). Together, these characteristics have led us to hypothesize that destabilized Ca2+ handling is a fundamental requirement for triggered arrhythmia in the mouse, and that in the absence of intact SR Ca2+ release, even a dramatically altered balance of sarcolemmal currents is not sufficient to elicit arrhythmia in response to commonly applied neurohormonal challenge.
To interrogate this hypothesis, we have examined in vivo and cellular arrhythmogenesis in a conditional SR Ca2+ ATPase type 2 (SERCA2) knockout mouse (KO). These mice progress to contractile failure in 7–10 weeks and display remarkable adaptations to the rapid loss of cardiac SR Ca2+ reuptake (Andersson et al., 2009; Liu et al., 2011; Li et al., 2012; Swift et al., 2012; Land et al., 2013). Most prominently, the Ca2+ fluxes responsible for supporting myofilament activation shift from the SR to the sarcolemma, with several fold increases in the inward currents attributable to ICaL and forward mode Na+-Ca2+ exchange (INaCa). In combination, these adaptations constitute a genetic model of extreme loss of ICaL control in the mouse, and present a unique opportunity to directly dissect the role of sarcolemmal versus intracellular mechanisms in murine arrhythmia.
At end-stage life we found SERCA2 KO mice are remarkably resistant to arrhythmia, both at the cellular level and in intact conscious animals. This resistance exists in the face of large increases in both ICaL and Na+-Ca2+ exchange, both of which contribute to marked prolongation of the AP and are ordinarily interpreted as strongly proarrhythmic changes. In combination, these data strongly suggest that SR Ca2+ release is required for cellular arrhythmogenesis and tissue-level triggered events in the mouse.
All experiments were performed in accordance with the Norwegian Animal Welfare Act, which conforms to NIH guidelines (NIH publication No 85-23, revised 1996).
The SERCA2 knock out mouse (KO) has previously been described and studied in detail as a model of contractile failure (Andersson et al., 2009; Liu et al., 2011; Li et al., 2012; Swift et al., 2012; Land et al., 2013). Briefly, cardiac-specific SERCA2 KO excision was achieved by tamoxifen activation of Cre-recombinase (α-MHC driven MerCreMer) via a single tamoxifen injection at 8–12 weeks of age. Here we studied these animals near to their mean age of death at 7 weeks after tamoxifen injection, when they exhibit pronounced contractile dysfunction, and markedly reduced cardiac output (Andersson et al., 2009). Age-matched flox/flox mice (FF) were used as controls.
Telemetry transmitters (Physiotel ETA-F10, Data Sciences International, St. Paul, MN, United States) were used to record electrocardiograms in freely moving animals. Implantation was performed 6 weeks after tamoxifen injection, and as previously described (Manotheepan et al., 2016). Intraperitoneal Xylazine hydrochloride and ketamine hydrochloride were administered in combination with isoflurane inhalation (1–4%) during the procedure, and subcutaneous buprenorphine was given toward the end of the procedure for postoperative analgesia. The transmitters were placed subcutaneously in the dorsal thoracolumbar region, and stabilized by ligatures to the dorsal muscles. Leads were attached to the pectoral muscles by ligatures in the upper left and lower right pectoral regions. Electrocardiograms were recorded after 7 days recovery from surgery, and recordings were analyzed for heart rate as well as ECG parameters during baseline conditions, and after a subsequent intraperitoneal injection of adrenalin (Epi, 0.5mg/kg). Analysis was performed manually and in fully-blinded fashion by the same experienced technician using both proprietary software (Matlab version 2013b, The Mathworks, Natick, MA, United States), and Ponemah (Data Sciences International, St. Paul, MN, United States). Steady state ECG analyses were performed by applying conventional definitions for all intervals. Of note, because the mouse often exhibits a pronounced J-wave we have defined QRS width (duration) as the time from first negative deflection after the P-wave to the first time after the S-wave minimum at which the ascending voltage signal crosses the isoelectric line. Rate dependent QT correction was performed by the Bazzet formula:
Premature ventricular complexes (PVCs) were manually identified, and episodes of ventricular tachycardia (VT) were defined as 4 or more PVCs occurring in sequence. Runs of VT were defined as non-sustained (NSVT) if they lasted less than 20-s, and as sustained (SVT) if longer.
Cardiomyocytes were isolated from ventricles of FF and KO mice similarly to our prior studies (Ottesen et al., 2015; Manotheepan et al., 2016). Briefly, mice were isoflurane-anesthetized (98% oxygen, 2.0% isoflurane), and euthanized by cervical dislocation. Excised hearts were first rinsed in ice-cold isolation buffer containing (mM): 130 NaCl, 25 Hepes, 22 D-glucose, 5.4 KCl, 0.5 MgCl2, 0.4 NaH2PO4 (pH 7.4). Each heart was then retrograde-perfused under constant flow conditions (3 ml/min) with warmed (37°C) isolation buffer for 4 min, and then with the same buffer supplemented with 400 U/ml collagenase Type II (Worthington Biochemical Corporation, Lakewood, NJ, United States) and 0.015 mmol/L Ca2+. After 10 min of enzyme perfusion, hearts were cut down and the left ventricle was removed, diced, and triturated in collagenase-free isolation buffer including 1% BSA and 0.02 U/L deoxyribonuclease I (Worthington), again at 37°C. The resulting cell suspension was filtered (200 μm nylon mesh) and sedimented, the cell pellet was washed in isolation buffer supplemented with 1% BSA, and Ca2+ was progressively reintroduced (0.05, 0.1, 0.2, and 0.5 mmol/L). The Ca2+-tolerant cardiomyocytes were stored at room temperature until use, which occurred within 8 h of isolation.
Single rod-shaped cardiomyocytes with clear striations were patch-clamped in whole-cell configuration using Axoclamp 2A and 2B amplifiers. Borosilicate patch-pipettes had resistances of 2–3 MOhms with the corresponding internal and external solutions. We used identical solutions for current clamp AP recordings and end-pulse K+ currents in voltage clamp, where the internal solution contained (in mM: 120 K-Asp, 25 KCl, 0.5 MgCl2, 6 NaCl, 4 K2-ATP, 0.06 EGTA, 10 HEPES, and 10 D-Glucose, pH corrected to 7.2 with KOH), and external (in mM): NaCl 140, HEPES 5, KCl 5.4, MgCl2⋅6H2O 0.5, Glucose 5.5, NaH2PO4H2O 0.4, CaCl2 1. For voltage clamp recordings of the transient outward potassium current (Ito), the internal solution contained (in mM) 110 K-Asp, 20 KCl, 0.5 MgCl2, 4 K2-ATP, 5 EGTA, 5 HEPES, and 10 D-Glucose - pH 7.2 with KOH, with a Na+ and Ca2+ free external solution (mM): 140 CholineCl, 1 CdCl2, 0.5 MgCl2, 5 HEPES, 5.5 Glucose, 5.4 KCl, pH 7.2 with KOH. For ICa,L the internal solution was (mM) 130 CsCl, 0.33 MgCl2, 4 Mg-ATP, 0.06 EGTA, 10 HEPES and 20 tetraethylammonium chloride - pH 7.2 with CsOH, and the bathing solution included (mM) 135 NaCl, 20 CsCl, 1 MgCl2, 10 glucose, 10 HEPES, 1 CaCl2, and 4 4-AP (pH 7.4).
Cells were superfused with external solution in all experiments, and all current measurements were made during step-pulse protocols at 1 Hz. Following ten 50-ms conditioning pulses (also 1 Hz) to 0 mV from a holding potential of −70 mV, ICaL was elicited by a 200-ms depolarizing voltage step (Mørk et al., 2009). Test potentials ranged from −40 to +50 mV with 10-mV increments from a post-train holding potential of −40 mV (to inactivate INa). Current amplitude was measured as the difference between peak inward current and steady-state current at the end of the test pulse. Ito (peak IK) was elicited by 300-ms steps to test potentials between −40 and +60 mV in 10-mV increments from a holding potential of −70 mV (Louch et al., 2010a). Peak IK was calculated as the peak outward current less the steady-state end pulse current from these 300-ms steps. The pedestal or plateau component of IK (IKp) was calculated as the mean of the final 20-ms of 500-ms test-pulse steps between −100 and +60 mV, again at 10 mV increments from a –70 mV holding potential. All currents were normalized to capacitance, which was calculated as the integral of the transient current response to a 10-mV hyperpolarizing step (150 ms) from a holding potential of −70 mV.
Action potential waveforms were also recorded at 1 Hz in all experimental conditions, and at least 20 sequential beats were allowed to achieve steady state. In experiments assessing EAD and DAD susceptibility, Iso-containing superfusate (1 μM) was applied after 30-s of baseline recording, and maintained for at least 3 min further.
The computational models used here are modifications of those published by Li et al. (2012). Briefly, both the FF and KO models were constructed on the basis of extensive data collected in the FF and KO mice at the same age and time after SERCA gene excision as studied here. Our modifications here were made sequentially to establish the requirements for the observed AP changes accompanying SERCA-KO. Thus, we describe them in that sequence in the results section. All parameter changes and details for code access are provided in the Supplementary Material.
Steady state electrocardiographic parameters were assessed by repeated measures ANOVA (RMANOVA: Genotype × Epi) with repeated measures for Epi. ECG arrhythmia outcomes (PVC and NSVT frequency) were non-normally distributed, and interrogated by Kruskal-Wallis rank sum test after being separated into baseline and Epi recordings. Similarly, skewed data for EAD frequency were assessed via the Kruskal-Wallis test, and the Fisher exact test was used to determine differences in EAD incidence. Peak IK, IKp, ICaL, and AP durations at 20, 50, 70, and 90% repolarization were all tested by RMANOVA where step potential was repeated for current measurements, and treatment conditions (Ca2+ concentration, Nifedipine, or Isoproterenol) were repeated for AP recordings.
For all RMANOVAs, the Holm-Sidak test was used to identify pairwise effects post hoc. In the event that the sphericity assumption was not valid, RMANOVA was replaced by the Mann-Whitney-Wilcoxon test. Significant effects were defined at p < 0.05, and p-values are explicitly stated for all marginal results (0.05 ≤ p ≤ 0.1). All statistical analyses were performed either with SigmaPlot (Systat Software Inc., CA, United States), or R software (version 3.2.1, The R Foundation of Statistical Computing).
Seven weeks after gene excision, KO mice exhibit a range of electrophysiologic dysfunction as measured by telemetered ECG. The most pronounced effects are slowed ventricular activation observed as increased QRS width (p < 0.01, Figure 1A, left), and a much more prominent and positive-going T-wave than is normally measurable via standard lead II recordings in the mouse (p < 0.0001), particularly during epinephrine challenge (Figures 1B,C). Interestingly, these changes exist in the absence of significant rate-corrected QT prolongation (p = 0.07, Figure 1A, right), thus suggesting that terminal repolarization is not delayed in the KO mice. These outcomes will be discussed in more detail later, but are consistent with a markedly altered AP morphology in the KO myocytes.
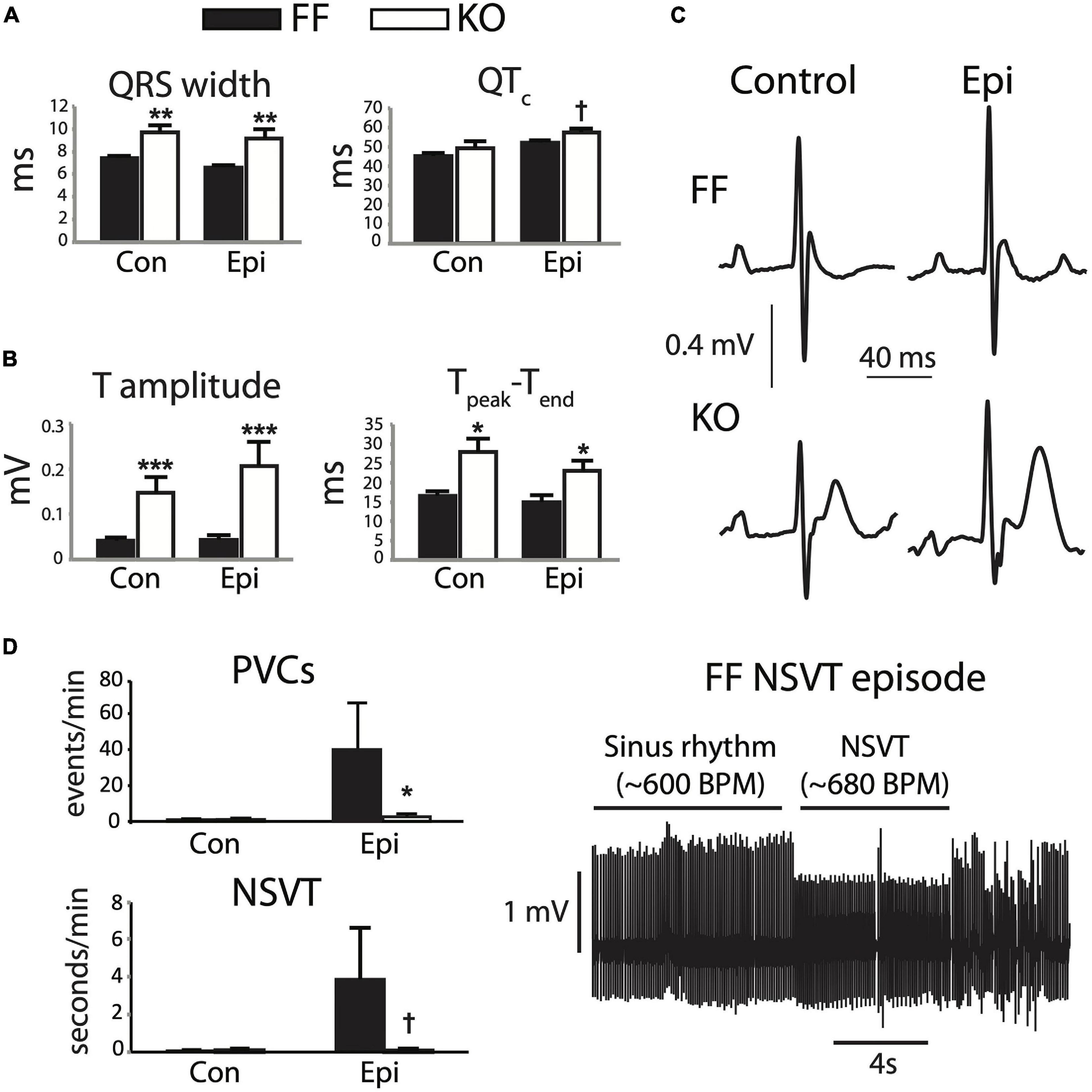
Figure 1. SERCA2-KO mice exhibit unique alterations in global cardiac electrophysiology. Telemetered ECG recordings from 8 animals in each group were first analyzed for steady state characteristics from periods exhibiting only normal sinus activation sequence (A,B). KO mice showed increased QRS width (A, left), and marked increases in T-wave amplitude and duration (B). (C) Typical sinus waveforms recorded from FF and KO animals. Ectopic activity was also analyzed (D), and KO animals surprisingly exhibited a reduced frequency of total ectopy (PVC frequency, top left), and a trend for fewer runs of non-sustained VT (bottom left), during Epi challenge. All panels: †p < 0.1 KO vs. FF, *p < 0.05 KO vs. FF, **p < 0.01 KO vs. FF, ***p < 0.001 KO vs. FF. Data are means ± SEM.
The combined influence of these electrocardiographic abnormalities did not result in a higher frequency of either isolated PVCs or runs of non-sustained (or sustained) VT (p > 0.1). In fact, during Epi challenge we observed reduced frequency of PVCs (p < 0.05) and a tendency for reduced NSVT frequency in the KO group (p = 0.09, Figure 1D). Together, these observations suggest that while the KO mice exhibit clear disturbances to global cardiac electrophysiology, they remain resilient to arrhythmia.
Figure 2 shows the effect of SERCA2 loss on the cardiomyocyte AP and underlying currents. At the level of the AP (Figure 2A), the outstanding feature is a brief early plateau in KO myocytes, which is reminiscent of the phase 2 plateau in large mammals albeit much shorter (∼20 ms). Mechanistically, this plateau indicates that the balance of currents in early repolarization is shifted inwardly, particularly in the region between +20 and −20 mV. This effect was pronounced, and can be seen as a 273% longer APD50 and 230% longer APD70 in KO myocytes (both p < 0.01). Panels B and C show that this effect is due to modest potentiation of peak ICaL (39% increase at 0 mV, p < 0.001), and dramatically slowed ICaL inactivation (50% relaxation time at 0 mV is increased by 178% in KO, p < 0.001), which together result in a 200% increase in Ca2+ influx, measured as the ICaL integral during the square-pulse voltage clamp protocol (p < 0.001). Exacerbating this gain in ICaL was a moderate reduction of end-pulse potassium current (IKp) at positive potentials (p < 0.05). While we did not attempt to experimentally dissect the different components of this current, we used a computational model to assess the ability of those components to contribute to the observed changes in AP morphology. These analyses are described in further detail below.
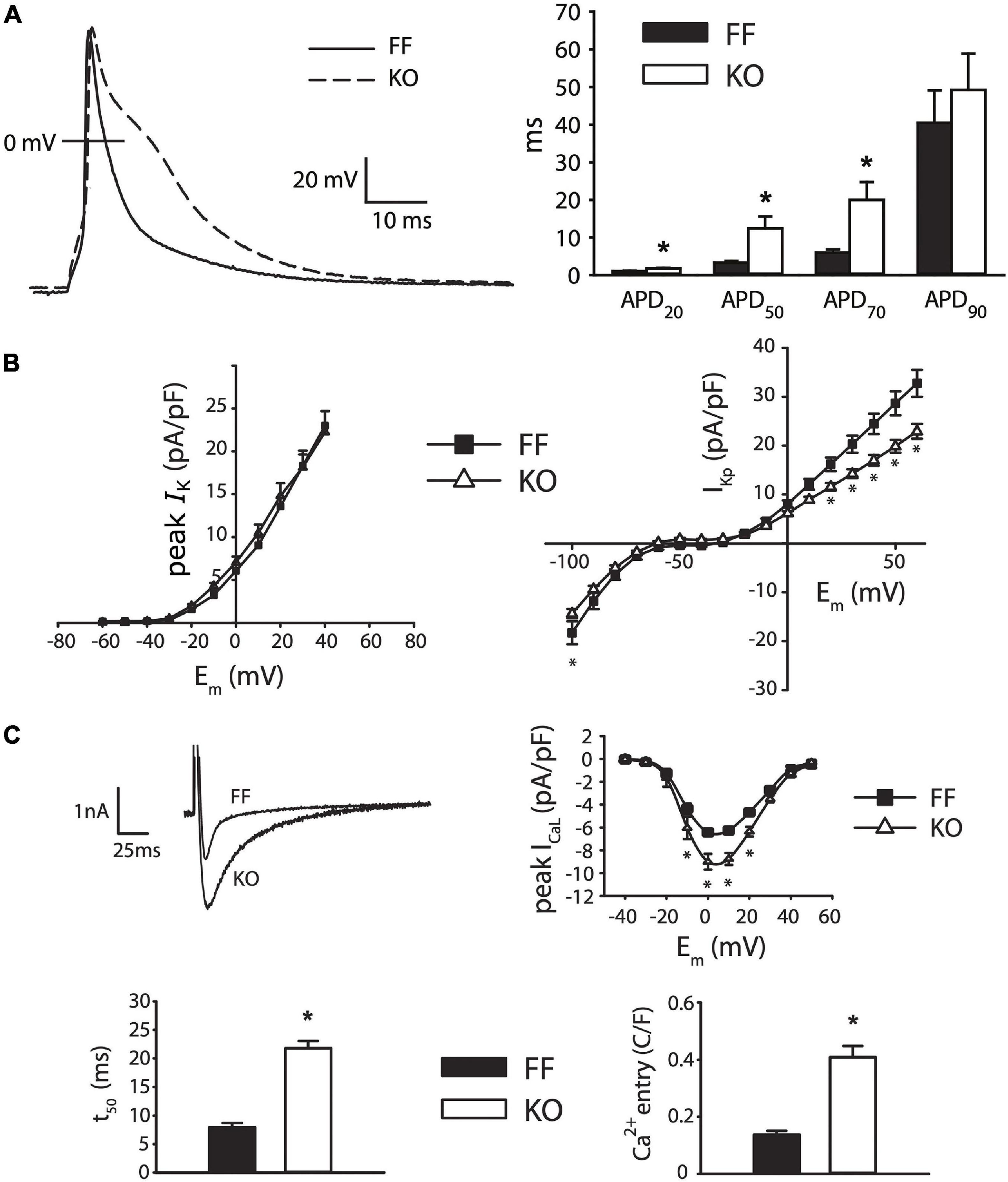
Figure 2. Myocytes from failing SERCA2-KO hearts exhibit prolonged early repolarization due to exaggerated sarcolemmal calcium flux. (A) KO myocytes (n = 10) display marked prolongation of early repolarization compared to FF (n = 11), although this difference is normalized by 90% repolarization. (B) The dominant outward current during early repolarization (Ito), is unaltered in the failing KO myocytes relative to FF (n = 10, both groups; left panel), while slowly inactivating K+ current components (measured as end-pulse, “pedestal” or “plateau,” K+ current – IKp) were slightly suppressed in KO cells (n = 17) relative to FF (n = 20). (C) However, peak ICaL is potentiated and ICaL inactivation is dramatically slowed in KO cells (n = 14) versus FF (n = 12), leading to a marked increase in calcium influx. All panels: *p < 0.05. Data are means ± SEM.
In Figure 3 we elaborate on the role of ICaL in slowing early repolarization either by acutely changing superfusate Ca2+ concentration, or applying ICaL blockade by Nifedipine (Nif, 1 μM). Figure 3A shows that the difference in APD50 and APD70 is removed when Ca2+ is excluded from the bath, and slightly exaggerated when it is increased to 1.8 mM (both p < 0.05). These alterations in extracellular [Ca2+] did not have statistically discernible effects on the kinetics of early repolarization in FF myocytes (p > 0.1). Because field-screening effects and non LCC-specific Ca fluxes2+ (such as Na+/Ca2+ exchange) will be altered by modulation of extracellular Ca2+, we also used Nifedipine to pharmacologically antagonize ICaL (Figure 3C). As for removal of extracellular Ca2+, this maneuver eliminated virtually all of the delay in early repolarization present in the KO myocytes – APD50 and APD70 were almost completely normalized (both p < 0.01 vs. control superfusion). This suggests that inward ICaL is the major contributor to the small phase 2 plateau in KO cells. Finally, to determine whether maneuvers that ordinarily modulate Ca2+-dependent ICaL inactivation (via SR Ca2+ release) are ineffective in KO mice, we also assessed the frequency-dependence of peak ICaL and ICaL inactivation (Supplementary Figure 1). In moving from 0.1 to 1 Hz FF animals exhibit slowed ICaL inactivation due to well-known Ca2+-dependent facilitation, and this property is dependent on cytosolic Ca2+ (Fauconnier et al., 2003). In KO animals this frequency-dependent modulation of ICaL inactivation is completely absent (given the already very slow inactivation in KO myocytes), further indicating the loss of SR-dependent ICaL control in KO animals.
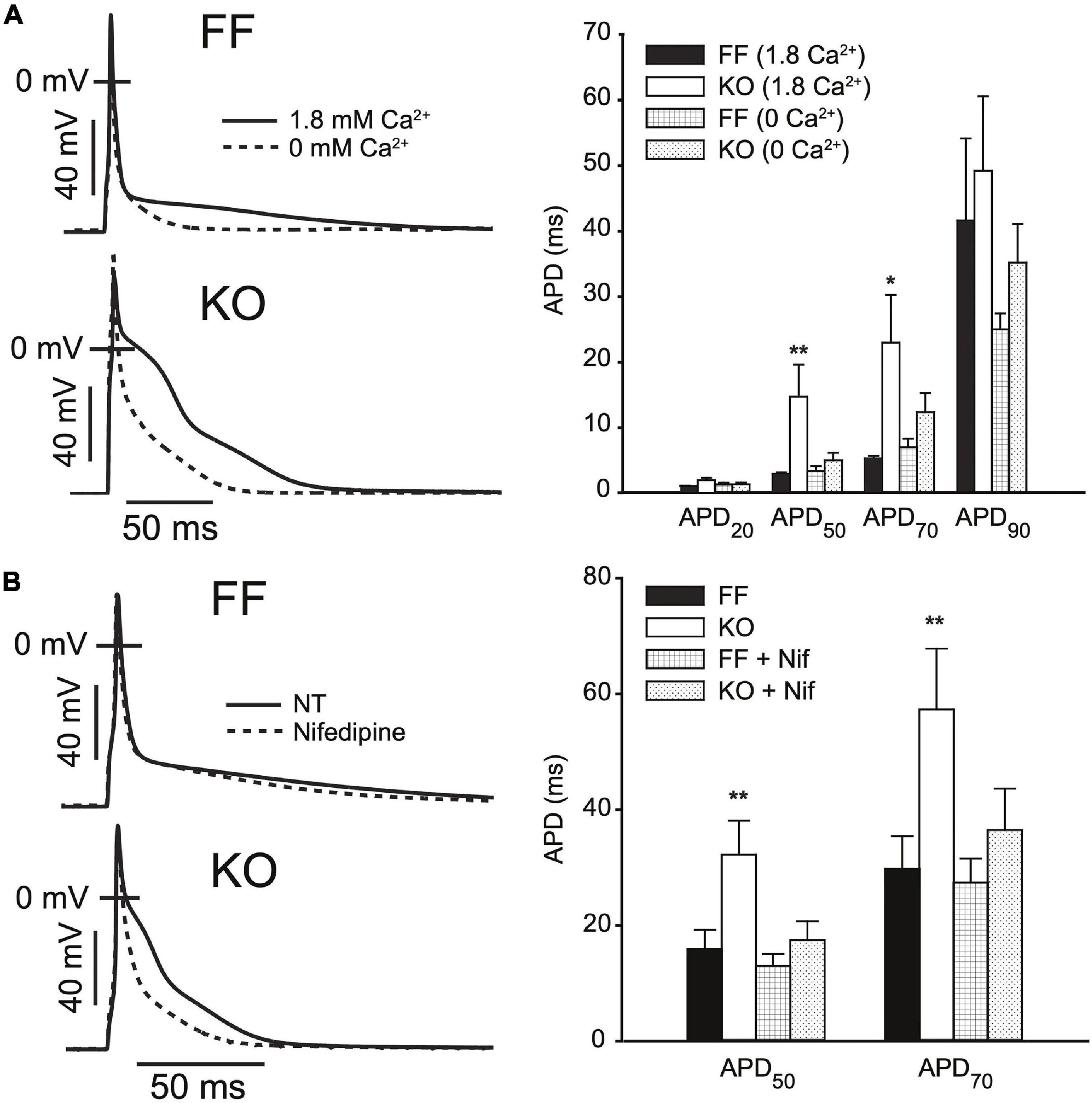
Figure 3. AP prolongation in SERCA2-KO myocytes relies upon augmented L-type calcium current. (A) Superfusing FF (n = 7) and KO (n = 8) myocytes with nominally Ca2+ free extracellular solution abolishes early AP prolongation in KO myocytes, while elevating extracellular Ca2+ to 1.8 mM exaggerates the difference in APD50 and APD70 (**p < 0.05 FF vs. KO at 1.8 mM Ca2+; and p < 0.05 KO at 1.8 mM vs. 0 mM Ca2+, *p < 0.0 KO at 1.8 mM vs. 0 mM Ca2+). (B) ICaL blockade via Nifedipine (1 μM) achieves similar normalization of APD50 and APD70 in KO myocytes (n = 7) relative to FF (n = 7). **p < 0.05 FF vs. KO in control superfusate; and p < 0.05 KO in control vs. Nifedipine. All panels: data are means ± SEM.
Importantly, this shift in balance during early repolarization is not mirrored by slowed terminal repolarization. That is, the differences present at APD50 and APD70 are lost by 90% (APD90) repolarization. The dominant inward currents modulating this late phase of repolarization in the mouse are INaCa and recovering INa (Edwards et al., 2014; Morotti et al., 2014). As described further below these inward currents compete with several components of IK, particularly the inward rectifier K+ current (IK1) to shape terminal repolarization. While we have not directly assessed the balance of these currents during late repolarization in KO myocytes, the near complete absence of triggered Ca2+ release (Andersson et al., 2009) drastically reduces the potential for inward INaCa. The slower early repolarization would also be expected to limit INa recovery and the potential for INa reactivation.
In larger mammals, the increases in ICaL peak current and 50% relaxation time would be expected to promote repolarization instabilities and EADs. To assess EAD susceptibility we challenged KO and FF myocytes with Isoproterenol (1 μM) for at least 3 min while pacing at 1 Hz in current clamp, and defined EADs as any upward deflection in the voltage signal exceeding 3 mV. Figure 4A shows that under these conditions EAD incidence and frequency were not increased in KO cells (both p > 0.1), rather EAD frequency tended to be higher (albeit non-significantly, p > 0.05) in the FF (9.3 ± 4.7%) than KO group (2.1 ± 1.8%). In FF cells, EADs exhibited signature properties identified in previous work (Sato et al., 2009; Tran et al., 2009). These features included periods with very high APD variability, indicating variable timing of terminal repolarization, but also intermittent beats that did not exhibit EADs. Furthermore, analysis of EAD take-off potentials (Figure 4B) suggests that EADs in the FF group exhibited very similar underlying dynamics to those we have described previously in the mouse, and different to those in larger mammals (Edwards et al., 2014). That is, they initiated at potentials too negative (−55 ± 7 mV) to be carried by ICaL, and occurred too soon after stimulation (34.6 ± 2.7 ms; approximately coinciding with the peak of the bulk cytosolic Ca2+ transient) to result from subcellular Ca2+ waves. Thus, it is very likely that EADs in FF myocytes were carried almost entirely by non-equilibrium reactivation of the fast component of the sodium current (INa) (Edwards et al., 2014). In contrast, the EADs present in KO myocytes initiated at more positive take-off potentials (−41.8 ± 2 mV, p < 0.001), were much larger in amplitude (KO = 26.4 ± 9 mV vs. FF = 7.8 ± 2.6 mV; p < 0.001), and lasted much longer than EADs in the FF myocytes (KO = 124 ± 24 ms vs. FF = 55 ± 27 ms; p < 0.001). While these EADs initiated later than those in FF cells, because spontaneous SR Ca2+ release in KO cells is virtually negligible (Supplementary Figure 2), it is very unlikely that discoordinated SR Ca2+ release made any contribution to these events. Even after accounting for leftward shifts in ICaL activation due to β-adrenergic activation (Bean et al., 1984; Tiaho et al., 1991; Kumari et al., 2018), the −40 mV take-off potentials are still only just approaching the lower limit of the ICaL activation range, thus the ability for ICaL to have directly contributed to initiation of these EADs is somewhat limited. However, once initiated, the large amplitude excursions of these EADs (see for example Figure 4B bottom left) suggest that ICaL reactivation is pronounced and likely to be the dominant contributor to EAD dynamics, as it is in ventricular myocytes of large mammals (Weiss et al., 2010).
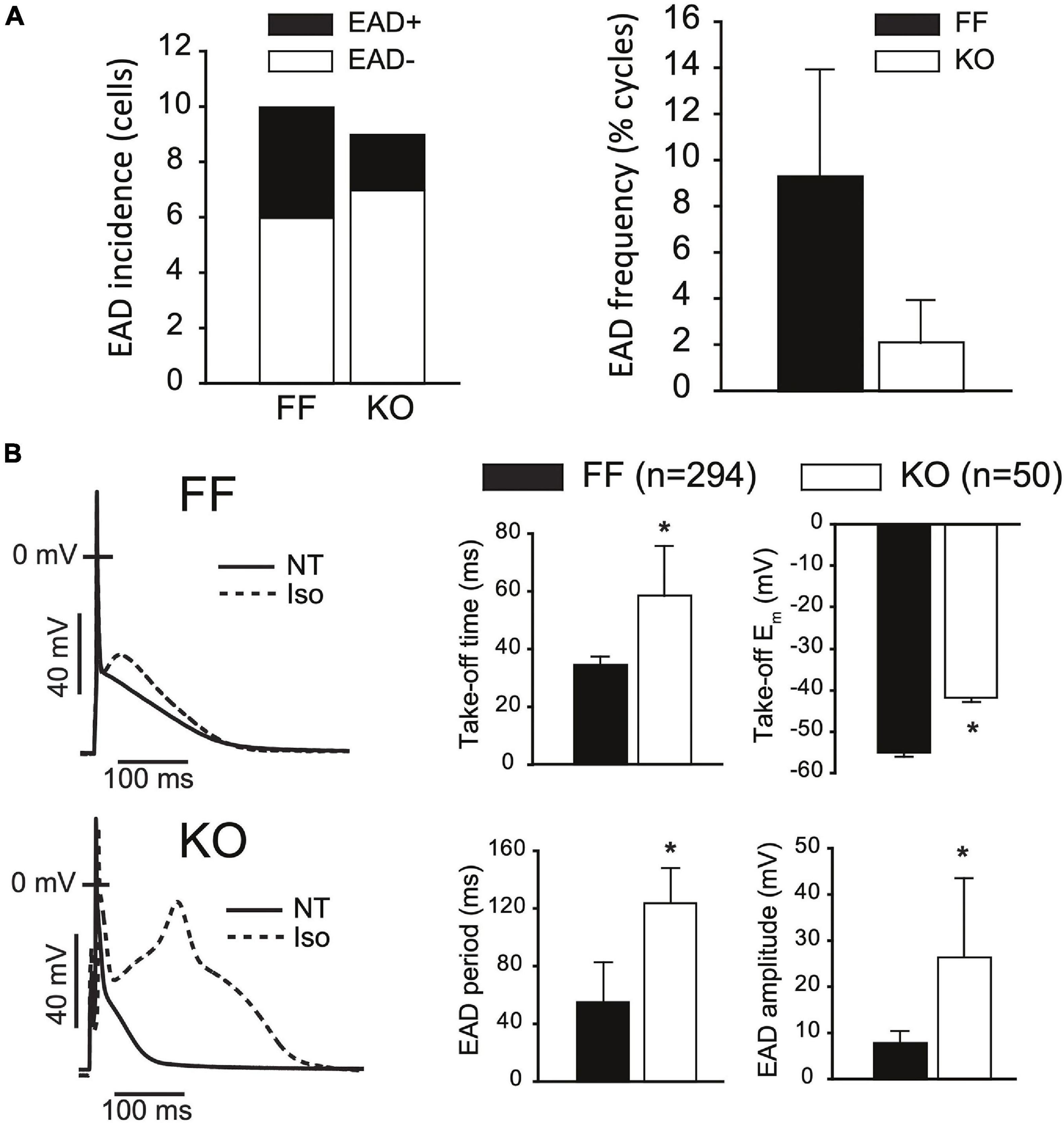
Figure 4. SERCA2-KO myocytes are not more susceptible to EADs but exhibit altered EAD dynamics. (A) KO cells (n = 9) did not exhibit a higher incidence (left panel: 2/9 KO cells vs. 4/10 FF cells, p > 0.01), or frequency (right panel: 50/3037 total KO cycles vs. 293/2593 total FF cycles, p > 0.01) of EADs relative to FF (n = 10). (B) However, KO EADs exhibit altered dynamics as indicated by increased amplitude, period and time to initiation, and reduced (more positive) take-off potentials. All panels: *p < 0.05. Data are means ± SEM.
In considering the balance of currents shaping the trajectory of repolarization in the mouse, an important quantitative aspect is how rapid early repolarization favors ICaL deactivation and limits the opportunity for Ca2+-dependent ICaL inactivation, which is prominent in larger species. Combining this with the known redundancy and different kinetic characteristics among the various K+ currents active in this early phase of repolarization, it becomes much more difficult to predict or account for how changes in ICaL regulation can contribute to modulating early repolarization. To interrogate these dynamics, we employed a published computational model of the mouse cardiomyocyte, which is specifically parameterized to incorporate the reduced SR Ca2+ handling and altered Na+ balance in failing KO myocytes [16]. This model already incorporates the increased Cav1.2 and NCX1 expression in KO cells, which leads to exaggerated peak ICaL and INaCa. However, it does not fully capture the pronounced slowing of ICaL inactivation we have observed in KO myocytes at 7 weeks of age, and incorporating this characteristic was our first alteration. Figure 5A shows the behavior of the reparameterized ICaL model in square-pulse voltage-clamp protocols identical to our experiments (all parameter changes are provided in full in Supplementary Table 1). Even for its larger peak current and much slower inactivation, this ICaL model was still overwhelmed by the fast component of the transient outward current (Ito,f) during early repolarization in both the FF and KO models, and had relatively little impact on the trajectory of early repolarization (APD20, Figure 5C).
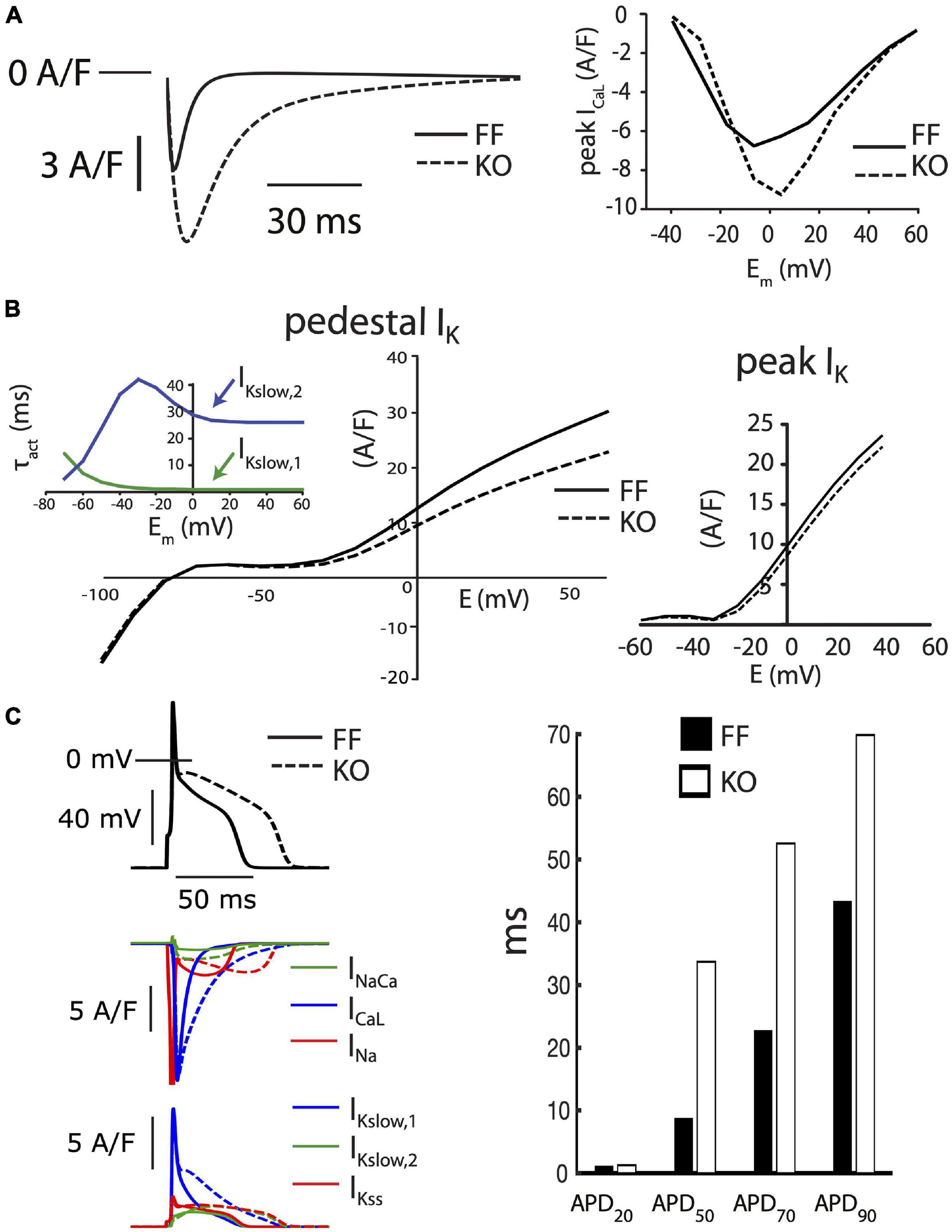
Figure 5. Baseline FF and KO computational models. (A) ICaL in the KO model was parameterized to permit the markedly slowed ICaL inactivation accompanying loss of SR calcium release (left panel) and moderately increased peak current observed in KO myocytes (right panel). (B) Conductances for the slower activating component of IKslow (Kv2.1, IKslow,2) and the steady state K+ current (IKss) were both reduced by 25% to fit end-pulse “pedestal” total IK (main left panel). Simulated currents were assessed as the sum of all end-pulse K+ currents as for the experimental measurements shown in Figure 2, where this compound current is referred to as IKp. To simultaneously fit the pedestal and peak components of the compound IK, as well as differences in ICaL and steady state APD, it was necessary to implement the slower activation kinetics of Kv2.1/IKslow,2 (inset left). Peak IK was straightforwardly fit through scaling the conductance of Ito,f in both the FF and KO models (right panel). (C) Together, these alterations resulted in slowed intermediate repolarization (APD50 and APD70) similar to that observed for KO myocytes. However, the slower early repolarization and rapid late repolarization in KO cells were much more difficult to capture with models that remained faithful to the voltage-clamp measurements. See Supplementary Table 1 for complete details of differences between these two baseline models.
As mentioned above, KO myocytes also exhibit a reduction in the measurable total IK at the end of our 500 ms test steps. The molecular identity of K+ channels contributing to this slowly inactivating current are not fully reconciled, although it is clear that the slow component of the transient outward current (carried by the Kv1.4 alpha subunit) is minimally expressed in mice, and that currents carried by Kv1.5 and Kv2.1 make important contributions (Nerbonne, 2014). Together these channels carry the slowly inactivating current (IKslow), also commonly referred to as the ultrarapidly activating delayed rectifier (IKur) K+ current. IKslow is a dominant contributor to the end-pulse K+ current in mice. It combines with the steady state K+ current (IKss, primarily carried by TASK1 and TREK1 channels (Nerbonne, 2014), and IK1 at more negative potentials, to comprise the compound end-pulse IK that we term the “pedestal” or “plateau” IKp. Due to difficulties in separating the activation kinetics of the Kv1.5 and Kv2.1 contributions to IKslow in cardiac cells, most models have assumed that this current is a single functional entity, with very rapid activation and slow inactivation. One relatively recent model separated the currents for the purpose of implementing differing phosphoregulation and inactivation kinetics (Morotti et al., 2014). To simultaneously fit intermediate AP prolongation (APD50 and APD70) and measured end pulse IK with the KO model, we also had to employ the approach of Morotti et al. (2014) and separate IKslow into Kv1.5-(IKslow,1) and Kv2.1-(IKslow,2) specific components. However, unlike Morotti et al. (2014) we implemented slower activation kinetics for IKslow,2, as shown in Figure 5B, and which can be measured for Kv2.1 in heterologous systems (Gordon et al., 2006). These slower activation kinetics of Kv2.1 slowed the onset of IKslow enough to permit ICaL modulation of intermediate repolarization, while also matching end-pulse IK and permitting stable overall repolarization, as observed in our experiments.
We employed mathematical models specific to the KO and FF myocytes (Li et al., 2012) and challenged them with a simple model implementation of β-adrenergic stimulation, which is closely analogous to our experimental challenge. This strategy involves implementing established effects of β-adrenergic regulation at ICaL (2–3 fold increased whole-cell permeability, PCaL, and 5–10 mV left-shifted steady state activation), SERCA (50–60% reduction in the Km for cytosolic Ca2+ binding), the Na+-K+ ATPase (25–30% reduction in the Km for cytosolic Na+ binding), and IKur (15–20% increase in whole cell conductance) (Edwards et al., 2014). The steady-state baseline models for the FF and KO myocytes were each simulated for 3 min at 1 Hz after applying these β-adrenergic parameter changes. Via this approach, EADs could be initiated in both the FF and KO models with identical changes to these key parameters, which remained wholly within the physiologic range. The EADs that result share several of the key features of EADs observed in our experiments during saturating Isoproterenol challenge. First the KO EADs exhibit greater ICaL reactivation, and their dynamical characteristics (oscillation period and amplitude) are increased similarly to the experimental recordings (Figure 4B). These dynamic characteristics are similar to those of ICaL-dominated EADs in large mammals (Sato et al., 2010). However, importantly, and somewhat surprisingly, even though the model EADs occurred at relatively positive potentials (approximately −38 mV) and relatively late (80 to 120 ms after the AP stimulus), they were initiated in both models by non-equilibrium reactivation of INa (Edwards et al., 2014) rather than ICaL (Figure 6C). This suggests that even for EADs that initiate 120 ms after the AP peak, recovery from inactivation of INa is sufficient to permit an arrhythmogenic current reactivation.
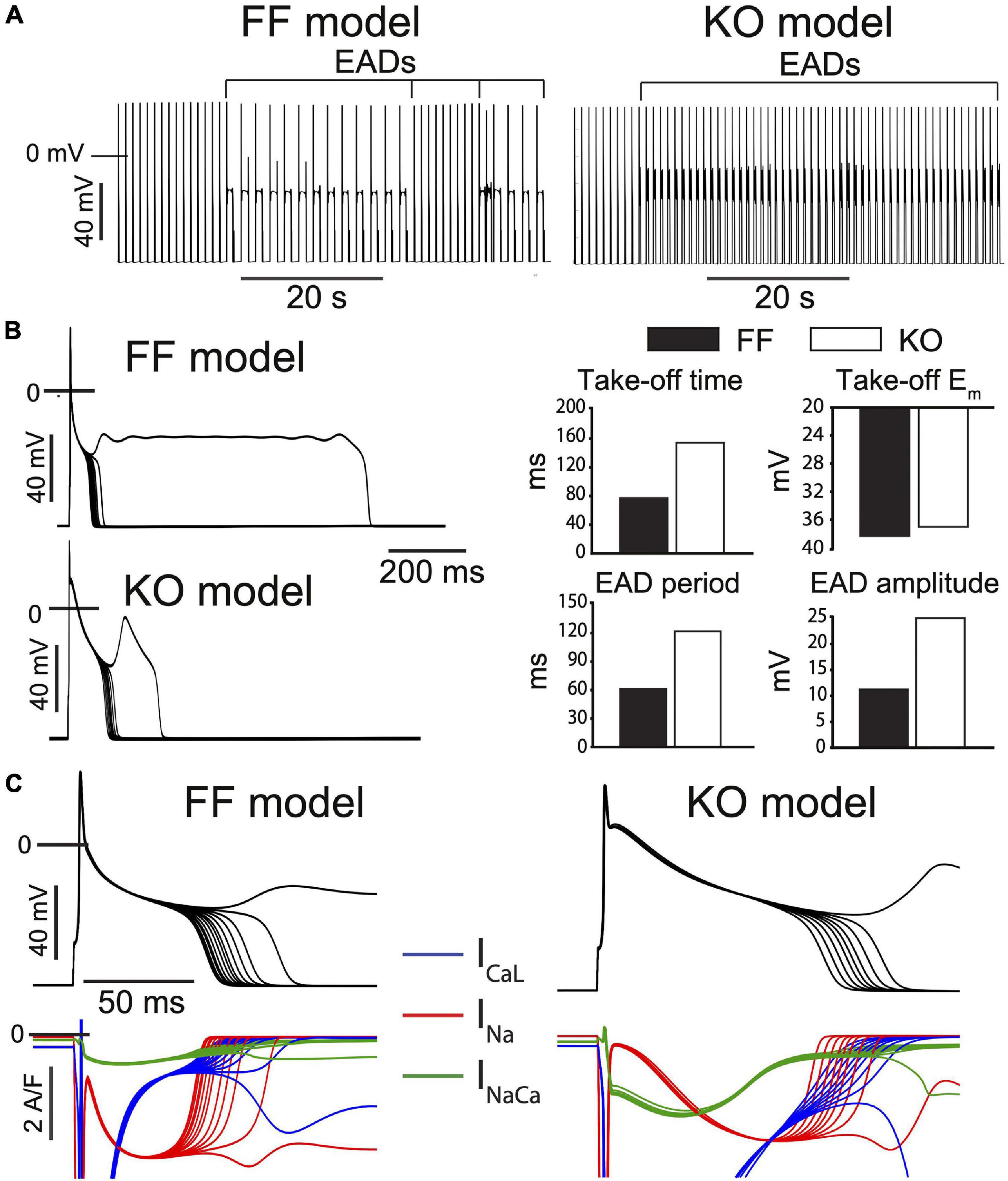
Figure 6. Progressive β-adrenergic challenge was simulated in both the FF and KO models [13] and recapitulate the major differences in EAD dynamics observed experimentally. (A) Both models transition to an unstable repolarization regime involving EADs. (B) Analysis of the EAD oscillations suggest the model dynamics are similar to those observed experimentally, where EAD amplitude, period and time of initiation are all increased in KO cells, while take-off potential is only slightly more positive. (C) Somewhat surprisingly, EAD initiation and the transition into the unstable regime is driven by non-equilibrium reactivation of INa rather than reactivation of ICaL in both models, although this dominant role for INa is slightly more clear for the FF model.
The ability for SR Ca2+ release to act as a cellular driver of cardiac arrhythmia has been appreciated for at least 30 years (Lederer and Tsien, 1976), and is widely recognized as a mechanistic contributor to all major cellular arrhythmogenic behaviors (automaticity, EADs, DADs, and APD alternans). The relative importance of SR Ca2+ fluxes in each behavior is highly dependent on the electrophysiologic context, and in this study we highlight species-specificity. By studying the electrophysiologic characteristics of murine SERCA2 knock out, we have assessed the ability for membrane versus SR mechanisms to generate arrhythmia upon the background of the characteristically large repolarizing currents in the mouse. We report four findings that are likely to hold important implications for other studies assessing electrophysiologic outcomes in mice, both in health and disease: (1) Shifting the burden of contractile calcium flux away from the SR and toward the sarcolemma elicits a pronounced, positive, and prolonged T-wave that is not normally observable in mice. While these effects are substantial, they are not sufficient to destabilize cardiac activation or repolarization. (2) The major cellular effect underlying these global changes is a very large increase in ICaL-mediated Ca2+ influx, due largely to slowed current inactivation, and which results in a pronounced delay of early repolarization creating an unusual AP plateau around 0 mV. (3) This slowing of early repolarization is difficult to capture in models due to the rapid kinetics of the large measurable peak IK, which is broadly thought to be carried by Ito,f and IKur/IKslow,1. The manner in which K+ current activation interacts with ICaL inactivation to shape early repolarization of the murine AP in this way requires further review. (4) The overall shift in the balance of membrane currents in SERCA2-KO away from a mouse-like phenotype and toward a large mammalian phenotype recruits EAD dynamics that are closer to those in large mammals. However, this shift is not sufficient to increase EAD frequency or incidence, and initiation of infrequent EADs in KO myocytes still likely relies upon INa reactivation, which rarely drives EADs in large mammalian myocytes.
The appearance of gross ECG changes in KO mice, particularly in T-wave amplitude, was remarkable and somewhat surprising given that the T-wave is often virtually absent in mice (Liu et al., 2004). However, these effects are captured by other models of pronounced induction of ICaL in mice and rats. Early rat studies of agonist dihydropiridines (Bay K 8644), which both potentiate and markedly prolong ICaL, noted similar overall changes and included additional ST segment elevation due to fusion of the QRS complex and T-wave (Abraham et al., 1987). Similarly in the mouse, β-adrenergic stimulation elicits a roughly twofold increase in T-wave amplitude (0.16 mV), and significant dispersion of repolarization as measured by T-wave decay time (Speerschneider and Thomsen, 2013). Finally here, one genetic model with very similar changes to macroscopic ICaL is Timothy syndrome (formerly type 8 long QT syndrome). While mice have been generated for major mutations known to cause this channelopathy, and brief reports suggest QT prolongation and abnormal ECG characteristics (Bett et al., 2009), we are unaware of any comprehensive description of the changes in cardiac electrophysiology present in this mouse.
Our prior work with the SERCA2-KO mouse has clearly shown that 7 weeks after genetic ablation little to no SERCA expression remains, and the SR Ca2+ store is essentially eliminated (Andersson et al., 2009; Louch et al., 2010b). To verify that spontaneous SR Ca2+ release is similarly absent in the 7-week KO mice, we also measured Ca2+ sparks and waves in isolated myocytes and observed that both were essentially abolished (Supplementary Figure 2). This is key in the context of the arrhythmogenic outcomes of interest here because it eliminates at least one major class of triggered arrhythmia resulting from spontaneous Ca2+ release and DADs. With respect to EADs, the implications of SERCA-KO are more complex. Previous studies have suggested that SR Ca2+ release is involved in murine EADs (Pott et al., 2012; Edwards et al., 2014). Specifically, we have shown that exaggerated SR Ca2+ release is the critical proximal mechanism of slowed late repolarization in mouse ventricle, and that this in turn elicits EADs through non-equilibrium reactivation of INa (Edwards et al., 2014). By this mechanism it is predictable that EADs and their arrhythmogenic potential would be inhibited in the KO mice. However, the established and very large increases in sarcolemmal Ca2+ transport in these mice create a very different electrophysiologic context. One which shifts repolarization dynamics toward those present in larger mammalian myocytes, and in principle may predispose to EADs with large mammal-like dynamics. We have shown here that this remarkable degree of plasticity is not sufficient to induce such instability.
Two important examples of genetic mouse models that exhibit very different adaptations to the SERCA-KO mouse are provided by murine knock-out and transgenic overexpression of the cardiac Na+-Ca2+ exchanger (NCX1). Henderson et al. (2004) developed a cardiac-specific NCX1 knock-out (NCX1-KO) mouse that lives to adulthood and exhibits 80-90% reduction in NCX1 expression. Through a set of (virtually opposite) adaptations to those resulting from SERCA-KO, the NCX1-KO mouse also retains remarkably viable contractile function. Unlike the SERCA-KO mice, these animals exhibit much reduced sarcolemmal Ca2+ transport but achieve sufficient contractile activation through very high gain EC coupling (Pott et al., 2005). While they exhibit clearly shortened APs (largely due to increased Ito,f) and QT intervals, they do not present an overt arrhythmia phenotype (Pott et al., 2007). In contrast, the NCX1-transgenic mouse developed by the same group exhibits pronounced slowing of terminal repolarization (prolonged APD90), EADs, and an overt susceptibility to arrhythmia (Pott et al., 2012). Taken together, these studies strongly support the remarkable ability of cardiomyocytes to autonomously reconfigure their Ca2+ handling and electrophysiologic machinery to support the Ca2+ requirements for contraction. However, they also suggest that the gene regulatory programs governing those reconfigurations are not equally apt to defend electrophysiologic stability. That is, in response to certain perturbations, the regulatory adjustments made to defend contractile Ca2+ will be arrhythmogenic. Our SERCA-KO mouse can be considered fortunate in this respect.
One immediately observable characteristic of the KO myocyte AP is marked slowing of early repolarization between +20 and −20 mV. While we did not attempt to broadly reparametrize the kinetics of the Ito,f model to fit this characteristic, it proved challenging to reconcile with our peak IK measurements, which readily overwhelmed even the large changes in peak ICaL and ICaL inactivation. While we are unaware of other studies that have systematically investigated the ability for altered ICaL inactivation kinetics to prolong this early portion of the murine AP, this characteristic has been clearly observed in previous studies. For example, acutely evacuating the SR store via caffeine both markedly prolongs early repolarization and accelerates later repolarization in a manner that is remarkably consistent with the AP morphology in KO myocytes [see Figure 3C in Edwards et al., 2014)]. Because the dynamics of interaction between ICaL and IK during this phase of the AP are critical for determining Ca2+ influx, we suggest that they deserve further investigation.
Additionally, we had limited ability to reconcile the more rapid terminal repolarization (APD70 to APD90) in KO myocytes while constraining IK1 to the measured inward end-pulse current below EK (Figure 2B, right panel). We (Edwards et al., 2014) and others (Yao et al., 1998; Pott et al., 2007, 2012; Ferreiro et al., 2012) have shown that SR Ca2+ release and resulting inward INaCa are major modulators of this phase of the murine AP. This suggests that more detailed accounting for components of IK that are active and available between −80 and −40 mV (particularly IK1), is likely necessary for properly capturing the interaction among INaCa, recovering INa and IK in determining the trajectory of terminal repolarization in the mouse.
The major shifts in sarcolemmal Ca2+ transport resulting from cardiac specific SERCA2 knock out are clearly not sufficient to destabilize myocyte repolarization or global electrophysiology in the mouse. Furthermore, those shifts only alter EAD dynamics in a relatively subtle fashion. In sum, our data suggest that, in the mouse, the destabilizing influence of extreme changes to sarcolemmal Ca2+ influx and extrusion can be readily outweighed by the stabilizing influence of an absent SR Ca2+ store and dramatic reduction in spontaneous SR Ca2+ release. Of course, this characteristic relies on the very strong overall repolarization present in the mouse, and should not be interpreted to extend to humans or indeed any larger mammals exhibiting an AP plateau.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Norwegian National Committee for Animal Welfare.
AGE collected and analyzed data, performed computational analyses, conceived and designed aspects of the study, and wrote the manuscript. HM collected and analyzed data, conceived and designed aspects of the study, and reviewed the manuscript. MKS collected and analyzed data, and reviewed the manuscript. DBL collected and analyzed data. IS conceived and designed aspects of the study, and reviewed the manuscript. SR and WEL collected and analyzed data, conceived and designed aspects of the study, reviewed the manuscript, and contributed to funding the study. OMS conceived and designed aspects of the study, reviewed the manuscript, and contributed to funding the study. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Norwegian Research Council (Grant No. 287395) and the South-Eastern Norway Regional Health Authority (Grant No. 2013075).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.744730/full#supplementary-material
Abraham, S., Amitai, G., Oz, N., and Weissman, B. A. (1987). Bay K 8644-induced changes in the ECG pattern of the rat and their inhibition by antianginal drugs. Br. J. Pharmacol. 92, 603–608. doi: 10.1111/j.1476-5381.1987.tb11362.x
Andersson, K. B., Birkeland, J. A., Finsen, A. V., Louch, W. E., Sjaastad, I., Wang, Y., et al. (2009). Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 47:187.
Bai, Y., Jones, P. P., Guo, J., Zhong, X., Clark, R. B., Zhou, Q., et al. (2013). Phospholamban knockout breaks arrhythmogenic Ca2 + ; waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ. Res. 113, 517–526. doi: 10.1161/circresaha.113.301678
Bean, B. P., Nowycky, M. C., and Tsien, R. W. (1984). β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature 307, 371–375. doi: 10.1038/307371a0
Bers, D. M. (2001). Excitation-contraction coupling and cardiac contractile force. J. Cardiova. Dis. Res. 1:45.
Bett, G. C. L., Bondarenko, V. E., and Rasmusson, R. L. (2009). Cardiac characteristics of a mouse model of timothy syndrome. Biophys. J. 96, 665a–666a.
Brunner, M., Peng, X., Liu, G. X., Ren, X. Q., Ziv, O., Choi, B. R., et al. (2008). Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J. Clin. Invest. 118, 2246–2259.
Cerrone, M., Noujaim, S. F., Tolkacheva, E. G., Talkachou, A., O’Connell, R., Berenfeld, O., et al. (2007). Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 101, 1039–1048.
Edwards, A. G., Grandi, E., Hake, J. E., Patel, S., Li, P., Miyamoto, S., et al. (2014). Nonequilibrium reactivation of Na+ current drives early afterdepolarizations in mouse ventricle. Circ. Arrhyt. Electrophys. 7:1213.
Fauconnier, J., Bedut, S., Guennec, J.-Y. L., Babuty, D., and Richard, S. (2003). Ca2 + current-mediated regulation of action potential by pacing rate in rat ventricular myocytes. Cardiovasc. Res. 57, 670–680. doi: 10.1016/s0008-6363(02)00731-9
Fearnley, C. J., Roderick, H. L., and Bootman, M. D. (2011). Calcium signaling in cardiac myocytes. CSH Perspect. Biol. 3:a004242.
Ferreiro, M., Petrosky, A. D., and Escobar, A. L. (2012). Intracellular Ca2 + release underlies the development of phase 2 in mouse ventricular action potentials. Am. J. Physiol. Heart C 302, H1160–H1172.
Gordon, E., Roepke, T. K., and Abbott, G. W. (2006). Endogenous KCNE subunits govern Kv2.1K+ channel activation kinetics in xenopus oocyte studies. Biophys. J. 90, 1223–1231. doi: 10.1529/biophysj.105.072504
Henderson, S. A., Goldhaber, J. I., So, J. M., Han, T., Motter, C., Ngo, A., et al. (2004). Functional adult myocardium in the absence of Na+-Ca2 + exchange. Circ. Res. 95, 604–611.
Kashimura, T., Briston, S. J., Trafford, A. W., Napolitano, C., Priori, S. G., Eisner, D. A., et al. (2010). In the RyR2R4496C mouse model of CPVT, β-adrenergic stimulation induces CA waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ. Res. 107, 1483–1489. doi: 10.1161/circresaha.110.227744
Koren, G. (2009). Electrical remodeling and arrhythmias in long-QT syndrome: lessons from genetic models in mice. Ann. Med. 36, 22–27. doi: 10.1080/17431380410032643
Kumari, N., Gaur, H., and Bhargava, A. (2018). Cardiac voltage gated calcium channels and their regulation by β-adrenergic signaling. Life Sci. 194, 139–149. doi: 10.1016/j.lfs.2017.12.033
Land, S., Louch, W. E., Niederer, S. A., Aronsen, J. M., Christensen, G., Sjaastad, I., et al. (2013). Beta-adrenergic stimulation maintains cardiac function in serca2 knockout mice. Biophys. J. 104, 1349–1356. doi: 10.1016/j.bpj.2013.01.042
Lederer, W. J., and Tsien, R. W. (1976). Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J. Physiol. 263, 73–100. doi: 10.1113/jphysiol.1976.sp011622
Lehnart, S. E., Mongillo, M., Bellinger, A., Lindegger, N., Chen, B. X., Hsueh, W., et al. (2008). Leaky Ca2 + release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 118, 2230–2245.
Lehnart, S. E., Terrenoire, C., Reiken, S., Wehrens, X. H., Song, L. S., Tillman, E. J., et al. (2006). Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc. Natl. Acad. Sci. U.S.A. 103:7910.
Li, L., Louch, W. E., Niederer, S. A., Aronsen, J. M., Christensen, G., Sejersted, O. M., et al. (2012). Sodium accumulation in SERCA knockout-induced heart failure. Biophys. J. 102:2048.
Liu, G., Iden, J. B., Kovithavongs, K., Gulamhusein, R., Duff, H. J., and Kavanagh, K. M. (2004). In vivo temporal and spatial distribution of depolarization and repolarization and the illusive murine T wave. J. Physiol. 555, 267–279. doi: 10.1113/jphysiol.2003.054064
Liu, X. H., Zhang, Z. Y., Andersson, K. B., Husberg, C., Enger, U. H., Ræder, M. G., et al. (2011). Cardiomyocyte-specific disruption of Serca2 in adult mice causes sarco(endo)plasmic reticulum stress and apoptosis. Cell Calcium 49, 201–207. doi: 10.1016/j.ceca.2010.09.009
Louch, W. E., Hake, J., Jølle, G. F., Mørk, H. K., Sjaastad, I., Lines, G. T., et al. (2010a). Control of Ca2 + release by action potential configuration in normal and failing murine cardiomyocytes. Biophys. J. 99, 1377–1386. doi: 10.1016/j.bpj.2010.06.055
Louch, W. E., Hougen, K., Mørk, H. K., Swift, F., Aronsen, J. M., Sjaastad, I., et al. (2010b). Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588:478.
Manotheepan, R., Danielsen, T. K., Sadredini, M., Anderson, M. E., Carlson, C. R., Lehnart, S. E., et al. (2016). Exercise training prevents ventricular tachycardia in CPVT1 due to reduced CaMKII-dependent arrhythmogenic Ca 2+ release. Cardiovasc. Res. 111, 295–306. doi: 10.1093/cvr/cvw095
Mørk, H. K., Sjaastad, I., Sejersted, O. M., and Louch, W. E. (2009). Slowing of cardiomyocyte Ca2 + release and contraction during heart failure progression in postinfarction mice. Am. J. Physiol. Heart C 296, H1069–H1079.
Morotti, S., Edwards, A. G., McCulloch, A. D., Bers, D. M., and Grandi, E. (2014). A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol. 592, 1181–1197. doi: 10.1113/jphysiol.2013.266676
Nerbonne, J. M. (2014). Mouse models of arrhythmogenic cardiovascular disease: challenges and opportunities. Curr. Opin. Pharmacol. 15, 107–114. doi: 10.1016/j.coph.2014.02.003
Ottesen, A. H., Louch, W. E., Carlson, C. R., Landsverk, O. J. B., Kurola, J., Johansen, R. F., et al. (2015). Secretoneurin is a novel prognostic cardiovascular biomarker associated with cardiomyocyte calcium handling. J. Am. Coll. Cardiol. 65:351.
Piot, C., LeMaire, S. A., Albat, B., Seguin, J., Nargeot, J., and Richard, S. (1996). High frequency–induced upregulation of human cardiac calcium currents. Circulation 93, 120–128. doi: 10.1161/01.cir.93.1.120
Pogwizd, S. M., and Bers, D. M. (2002). Calcium cycling in heart failure: the arrhythmia connection. J. Cardiovasc. Electr. 13, 88–91. doi: 10.1046/j.1540-8167.2002.00088.x
Pogwizd, S. M., Schlotthauer, K., Li, L., Yuan, W., and Bers, D. M. (2001). Arrhythmogenesis and contractile dysfunction in heart failure. Circ. Res. 88, 1159–1167. doi: 10.1161/hh1101.091193
Pott, C., Muszynski, A., Ruhe, M., Bögeholz, N., Schulte, J. S., Milberg, P., et al. (2012). Proarrhythmia in a non-failing murine model of cardiac-specific Na+/Ca 2+ exchanger overexpression: whole heart and cellular mechanisms. Basic. Res. Cardiol. 107:247.
Pott, C., Philipson, K. D., and Goldhaber, J. I. (2005). Excitation–contraction coupling in Na+–Ca2 + exchanger knockout mice. Circ. Res. 97, 1288–1295.
Pott, C., Ren, X., Tran, D. X., Yang, M. J., Henderson, S., Jordan, M. C., et al. (2007). Mechanism of shortened action potential duration in Na+-Ca2 + exchanger knockout mice. Am. J. Physiol. Cell. 292, C968–C973. doi: 10.1161/01.res.0000196563.84231.21
Priori, S. G., Napolitano, C., Memmi, M., Colombi, B., Drago, F., Gasparini, M., et al. (2002). Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106, 69–74.
Priori, S. G., Napolitano, C., Tiso, N., Memmi, M., Vignati, G., Bloise, R., et al. (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200. doi: 10.1161/01.cir.103.2.196
Sato, D., Xie, L. H., Sovari, A. A., Tran, D. X., Morita, N., Xie, F., et al. (2009). Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc. Natl. Acad. Sci. U.S.A. 106:2988.
Sato, D., Xie, L.-H., Nguyen, T. P., Weiss, J. N., and Qu, Z. (2010). Irregularly appearing early afterdepolarizations in cardiac myocytes: random fluctuations or dynamical chaos? Biophys. J. 99:773.
Shannon, T. R., Wang, F., Puglisi, J., Weber, C., and Bers, D. M. A. (2004). Mathematical treatment of integrated ca dynamics within the ventricular myocyte. Biophys J. 87, 3351–3371. doi: 10.1529/biophysj.104.047449
Speerschneider, T., and Thomsen, M. B. (2013). Physiology and analysis of the electrocardiographic T wave in mice. Acta Physiol. 209, 262–271. doi: 10.1111/apha.12172
Splawski, I., Timothy, K. W., Decher, N., Kumar, P., Sachse, F. B., Beggs, A. H., et al. (2005). Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. U.S.A. 102:8089 8096; discussion 8086-8088.
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119:31.
Swift, F., Franzini-Armstrong, C., Øyehaug, L., Enger, U. H., Andersson, K. B., Christensen, G., et al. (2012). Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc. Natl. Acad. Sci. U.S.A. 109:4001.
Tiaho, F., Nargeot, J., and Richard, S. (1991). Voltage-dependent regulation of L-type cardiac Ca channels by isoproterenol. Pflügers Arch. 419, 596–602. doi: 10.1007/bf00370301
Tran, D. X., Sato, D., Yochelis, A., Weiss, J. N., Garfinkel, A., Qu, Z., et al. (2009). Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys. Rev. Lett. 102:258103.
Weiss, J. N., Garfinkel, A., Karagueuzian, H. S., Chen, P.-S., and Qu, Z. (2010). Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 7, 1891–1899. doi: 10.1016/j.hrthm.2010.09.017
Keywords: early afterdepolarizations (EADs), delayed afterdepolarizations, species, triggered activity, repolarization
Citation: Edwards AG, Mørk H, Stokke MK, Lipsett DB, Sjaastad I, Richard S, Sejersted OM and Louch WE (2021) Sarcoplasmic Reticulum Calcium Release Is Required for Arrhythmogenesis in the Mouse. Front. Physiol. 12:744730. doi: 10.3389/fphys.2021.744730
Received: 20 July 2021; Accepted: 20 September 2021;
Published: 12 October 2021.
Edited by:
Daniel M. Johnson, The Open University, United KingdomReviewed by:
Yanmin Zhang, Xi’an Jiaotong University, ChinaCopyright © 2021 Edwards, Mørk, Stokke, Lipsett, Sjaastad, Richard, Sejersted and Louch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William E. Louch, dy5lLmxvdWNoQG1lZGlzaW4udWlvLm5v
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.