 Jean-Pierre Benitah
Jean-Pierre Benitah Romain Perrier
Romain Perrier Jean-Jacques Mercadier
Jean-Jacques Mercadier Laetitia Pereira
Laetitia Pereira Ana M. Gómez
Ana M. Gómez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 08 October 2021
Sec. Cardiac Electrophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.734210
This article is part of the Research TopicCalcium and Heart Failure: From Bench to BedsideView all 11 articles
Heart Failure (HF) is defined as the inability of the heart to efficiently pump out enough blood to maintain the body's needs, first at exercise and then also at rest. Alterations in Ca2+ handling contributes to the diminished contraction and relaxation of the failing heart. While most Ca2+ handling protein expression and/or function has been shown to be altered in many models of experimental HF, in this review, we focus in the sarcoplasmic reticulum (SR) Ca2+ release channel, the type 2 ryanodine receptor (RyR2). Various modifications of this channel inducing alterations in its function have been reported. The first was the fact that RyR2 is less responsive to activation by Ca2+ entry through the L-Type calcium channel, which is the functional result of an ultrastructural remodeling of the ventricular cardiomyocyte, with fewer and disorganized transverse (T) tubules. HF is associated with an elevated sympathetic tone and in an oxidant environment. In this line, enhanced RyR2 phosphorylation and oxidation have been shown in human and experimental HF. After several controversies, it is now generally accepted that phosphorylation of RyR2 at the Calmodulin Kinase II site (S2814) is involved in both the depressed contractile function and the enhanced arrhythmic susceptibility of the failing heart. Diminished expression of the FK506 binding protein, FKBP12.6, may also contribute. While these alterations have been mostly studied in the left ventricle of HF with reduced ejection fraction, recent studies are looking at HF with preserved ejection fraction. Moreover, alterations in the RyR2 in HF may also contribute to supraventricular defects associated with HF such as sinus node dysfunction and atrial fibrillation.
Heart failure (HF) is one of the major causes of death worldwide. It is characterized by the failure of the cardiac pump to maintain a sufficient blood flow to oxygenize and carry nutrients to the whole body. According to left ventricular systolic function, HF has been divided into two major groups: HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF). HFrEF generally occurs after cardiac injury (myocardial infarction) or under chronic stress (hypertension), leading to the alteration of contractile function of the heart. It is now well established that alteration of cardiomyocyte Ca2+ homeostasis plays a critical role in the development of the pathology, leading to cardiac remodeling, failure of the cardiac pump, and cardiac arrhythmias.
Ca2+ plays a key role in cardiomyocyte contraction. In each heartbeat, the membrane depolarization during an action potential (AP) activates L-type Ca2+ channels (LTCC), which are located in the sarcolemma and are more concentrated at the transverse (T) tubules (Figure 1). The local increase in [Ca2+]i in the dyads that follows activates the Ca2+ release channels, the ryanodine receptors (RyR2) located in the neighborhood, on the junctional sarcoplasmic reticulum (SR), resulting in the coordinated and global increase in the cell [Ca2+]i, by the Ca2+-induced Ca2+-release (CICR) mechanism, activating contractile myofibrils. Relaxation happens when the cytosolic [Ca2+]i decreases, mainly by Ca2+ re-uptake into the SR through the SarcoEndoplasmic Reticulum Ca2+ ATPase (SERCA) and extrusion out of the cell through the Na+/Ca2+ exchanger (NCX). Other systems, such as the plasmalemmal Ca2+ pump and the mitochondrial Ca2+ uniporter, play a minor role in cytosolic Ca2+ removal. The RyR2 are not so sensitive to Ca2+ so their activation probability depends on their proximity to the LTCCs, which determines the local Ca2+ concentration (Stern, 1992).
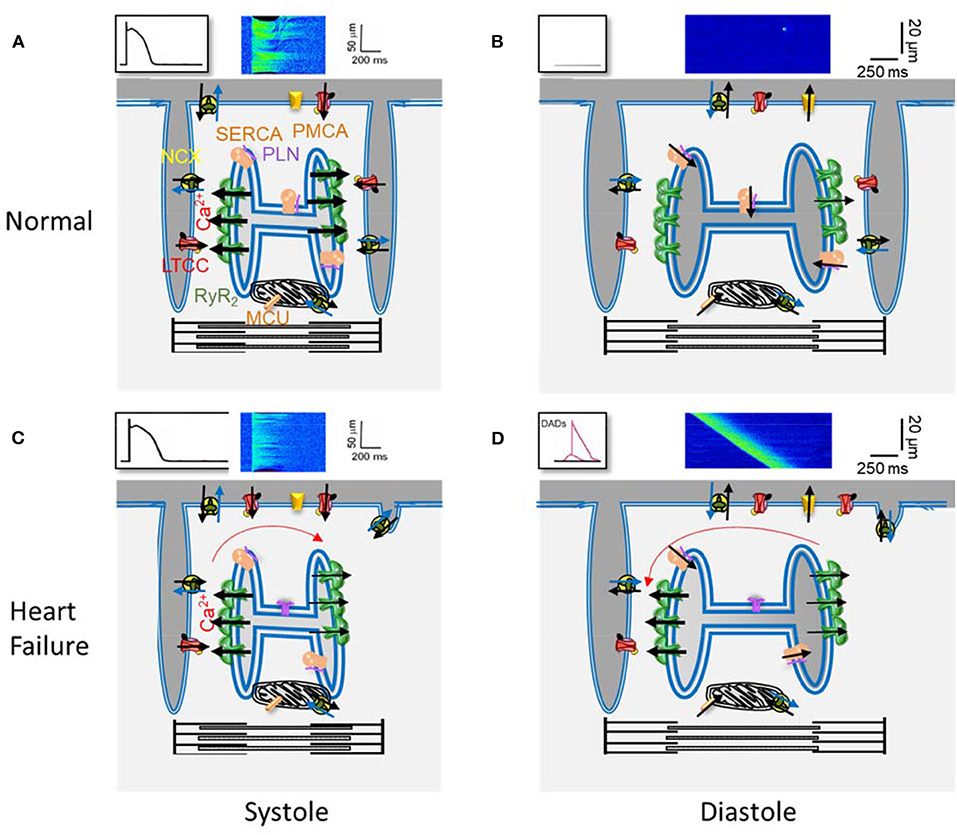
Figure 1. Main elements of Ca2+ handling in excitation-contraction coupling in a ventricular cardiomyocyte. (A) Scheme of a portion of a ventricular cardiomyocyte showing two T-tubules, with L type Ca2+ channels (LTCC) and the Na+/Ca2+ exchanger (NCX), and the junctional sarcoplasmic reticulum with Ryanodine Receptors (RyR2). Plasmalema and SarcoEndoplasmic Ca2+ ATPases (PMCA and SERCA) are also shown, with phospholamban (PLN) which slows SERCA activity, as well as the mitochondrial Ca2+ uniporter (MCU). The black arrows indicate Ca2+ movements during the action potential (drawing on top left), producing the [Ca2+]i transient (confocal line scan image on top). (B) The same elements during rest. The LTCC are inactive, and the RyR2 closed, with small leak. (C) and (D) The same as in (A) and (B) but in heart failure: the density of T-tubules is decreased as well as SERCA expression; the RyR2 are more active.
During diastole, some Ca2+ leaks outside the SR through the RyR2s, which have very low open probability at low cytosolic [Ca2+]i, but are also sensitive to luminal Ca2+, what is high during diastole. The opening of a RyR2 cluster can be viewed visualized with a confocal microscope and adequate fluorescence dyes, as rapid (~10 ms), brief (~30 ms), and local (~1.5 μm) elevations in cytosolic Ca2+, named Ca2+ sparks (Cheng and Lederer, 2008). Ca2+ sparks analyses provide insight into RyR2 function in situ, while the incorporation of RyR2 into lipid bilayers provides direct information on the channel function in a non-cellular context. During excitation-contraction coupling (ECC), Ca2+ release units (CRU) formed by a cluster of RyR2s are activated by the LTCCs located in the T tubules in front of them, which open during the AP. The spatial and temporal summation of these coordinated Ca2+ sparks results in the whole [Ca2+]i transient. The efficacy of the LTCCs to activate CRUs has been referenced as CICR gain, and it has been studied in HF combining patch-clamp to measure L-type Ca2+ current (ICa) and confocal microscopy to visualize changes in ≪Ca2+i.
In HF, there are alterations in contraction and relaxation, which may be related to alterations in cardiomyocyte Ca2+ cycling. Alterations have been reported in several cardiomyocyte Ca2+ cycling elements, such as SERCA, ratio phosphlamban (PLN)/SERCA, and single LTCCs, during both experimental and human HF. In this review, we will focus on alterations in the RyR2 function on left ventricular HF, although alterations in Ca2+ handling have also been found in right ventricular failure (Hautefort et al., 2019).
The first study of Ca2+ sparks in HF showed fewer Ca2+ sparks for a given amount of Ca2+ entry through LTCCs (ICa). This decrease in CICR gain was observed in experimental HF, due to chronic hypertension (Gomez et al., 1997) and after myocardial infarction (Gomez et al., 2001), and was reminiscent of pioneer work by Beuckelmann in human ventricular myocytes from explanted failing hearts, where they found a decrease in the global [Ca2+]i transient but normal ICa density (Beuckelmann et al., 1992). One explanation of this decrease in CICR gain was that there could be an ultrastructural disarrangement, which physically increased the distance between CRU and LTCC. In fact, while ICa density is generally maintained in HF, higher activity of at the single channel level has been reported (Schroder et al., 1998). This finding suggests fewer LTCC in the T-tubules participating in CICR or a decrease in the T-tubules density, as observed in HF (Wagner et al., 2012; Guo et al., 2013; Jones et al., 2018). But this “uncoupling” of the ECC was also suggested in larger mammals, in which the T-tubular network is less developed. Following myocardial infarction, cardiomyocytes from pigs with HF had relatively more uncoupled RyR2s, which could underlie the decreased gain of CICR (Dries et al., 2018).
Besides this decrease in CICR gain, alterations in the RyR2 itself have been reported in different models of HF.
During HF progression, the sympathetic nervous system and renin-angiotensin-aldosterone system are activated to compensate for the reduced contractile function of the heart and thus maintain a sufficient blood flow (Lymperopoulos et al., 2013). In this early stage of cardiac remodeling, ECC is enhanced, with an increased [Ca2+]i transient amplitude and contraction, together with a faster relaxation (Ohkusa et al., 1997). However, chronic activation of both systems has deleterious effects on cardiomyocytes and is responsible for the decompensation of cardiac function. This decrease in cardiac contraction is associated with an alteration of ECC (Gomez et al., 1997, 2001), the [Ca2+]i transient amplitude is decreased and its duration is increased. Besides, or on top of the ultrastructural remodeling mentioned above, the reduction in [Ca2+]i transient amplitude can be explained by a reduction of Ca2+ stored in the sarcoplasmic reticulum (Piacentino et al., 2003; Lehnart et al., 2009). While the first analyses explained this decrease in SR Ca2+ load by a lower SERCA expression and function and/or a higher expression of its natural inhibitor PLN, or a decrease in its phosphorylation (Kranias and Hajjar, 2012), some authors attributed it to RyR2 hyperactivity, which enhances diastolic SR Ca2+ leak. Indeed, an increase of the spontaneous Ca2+ leak through RyR2 has been observed in HF (Fischer et al., 2014; Ho et al., 2014; Grimm et al., 2015; Fu et al., 2016; Uchinoumi et al., 2016; Walweel et al., 2017; Dries et al., 2018). This SR Ca2+ leak, together with the reduced SERCA activity, contributes to a decrease in SR Ca2+ stores and an increase of the propensity of Ca2+ waves (Cheng et al., 1996; Venetucci et al., 2008; Curran et al., 2010; Dries et al., 2018). This contrasted with a significant decrease in the Ca2+ spark frequency reported in isolated cardiomyocytes from patients with terminal HF compared with non-failing individuals (Lindner et al., 2002). As spontaneous Ca2+ sparks depend on the amount of SR Ca2+ stored and [ATP], which are both reduced in cardiomyocytes from failing hearts, these can mask the increased activity of RyR2, whereas this higher RyR2 activity can still favor Ca2+ waves once the Ca2+ spark is produced, as the neighboring RyR2 will be more sensitive (Ruiz-Hurtado et al., 2015). Extrusion of Ca2+ constituting the Ca2+ waves during diastole via the NCX can generate delays after depolarization and trigger new action potentials that can propagate and induce arrhythmias (Pogwizd et al., 2001; Venetucci et al., 2008; Belevych et al., 2011). All these modifications of Ca2+ release have not been associated with alterations of RyR2 expression during HF (Hasenfuss and Pieske, 2002), but with post-translational modifications such as phosphorylation and oxidation (Houser, 2014). We should also keep in mind that while post-translational alterations in RyR2 tends to increase its activity, other alterations in Ca2+-handling proteins (such as SERCA reduction) and metabolic changes (that may curse with lower ATP levels) tend to reduce its activity (Ruiz-Hurtado et al., 2015).
In 2000, it was shown that hyperphosphorylation of RyR2 by PKA is responsible for the leaky RyR2 in failing hearts in humans. The authors claimed that PKA-mediated phosphorylation dissociates the FK506 binding protein (FKBP12.6) from the RyR2 leading to an increase in the channel open probability (Marx et al., 2000). FKBP12.6 is known to stabilize the RyR2 in a closed state and avoid aberrant Ca2+ leak (Brillantes et al., 1994; Gellen et al., 2008), but its removal may not affect RyR2 open probability (Xiao et al., 2007). In the early 90s, serine 2808 (S2808) was identified as the unique phosphorylation site of RyR2 (Witcher et al., 1991) and appeared to be a prominent target for PKA. In following publications, Marks' group highlighted the role of S2808 phosphorylation in the destabilization of the complex RyR2-FKBP12.6. They developed RyR2-S2808A knock-in mice, which prevent phosphorylation at this site. They showed that these mice are protected against RyR2-FKBP12.6 dissociation and leaky RyR2 in response to catecholamine stimulation (Shan et al., 2010a,b). Moreover, RyR2-S2808A knock-in mice seem to have a better cardiac function after myocardial infarction, which has been related again to the stabilization of the RyR2-FKBP12.6 complex in the absence of PKA phosphorylation (Wehrens et al., 2006). They also confirmed this major role of S2808 using RyR2-S2808D knock-in mice, which mimicked hyperphosphorylation at that site and had a reduced association of FKBP12.6 to RyR2 (Shan et al., 2010a,b). However, other groups have not been able to confirm this major role of S2808 phosphorylation in the progression of HF (Jiang et al., 2002). Valdivia and Houser's groups also developed RyR2-S2808A knock-in mice. In these mice, response to β-adrenergic stimulation on [Ca2+]i transient and ECC gain (MacDonnell et al., 2008) and progression of HF after myocardial infarction (Zhang et al., 2012) or aortic banding (Benkusky et al., 2007) were not affected no matter the genetic background of the mice (Alvarado et al., 2017). Moreover, it has also been shown that FKBP12.6 binding to RyR2 is not modified by acute β-adrenergic stimulation on wild-type mice or FKBP12.6 overexpressing mice in the heart (Gellen et al., 2008). Recently, evidences that S2808 is not the only target for RyR2 phosphorylation by PKA have been put forward; S2030 seems to be a potent target of PKA (Potenza et al., 2019) and can be involved in the progression of HF (Xiao et al., 2006; Benkusky et al., 2007). Taken together, these studies are in contradiction with Marks' group publications, and the controversy continues through the years (Bers, 2012; Alvarado and Valdivia, 2020; Dridi et al., 2020a,b). However, independently on RyR2 phosphorylation status, FKBP12.6 has been more consistently found decreased in HF (Ono et al., 2000; Gomez et al., 2009), which can by itself promote diastolic Ca2+ leak.
The Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of S2814 (2815) has also been highlighted for its role in the increase in SR Ca2+ leak through RyR2 in HF. CaMKII is activated by catecholamine and by oxidation, which is high during HF (Erickson et al., 2008). It has been shown that in failing rabbit hearts, CaMKII phosphorylation of RyR2 decreases SR load and increases Ca2+ leak by increasing RyR2 open probability (Ai et al., 2005). In these rabbits, inhibition of CaMKII, but not of PKA, reduced SR Ca2+ leak (Ai et al., 2005). In human heart failure due to ischemic or dilated cardiomyopathy, CaMKII expression has been shown to increase, contributing to augmented SR Ca2+ leak (Fischer et al., 2014). Rats subjected to myocardial infarction treated with exendin-4, which reduced CaMKII activity, showed a decreased SR Ca2+ leak (Chen et al., 2020). This major effect of CaMKII in the development of HF has been confirmed in mice lacking CaMKII. After transverse aortic constriction, these mice were protected against HF progression together with a reduced SR Ca2+ leak and RyR2 phosphorylation at S2815 (Ling et al., 2009). Similarly to CaMKII Knock out mice, RyR2-S2814A knock-in mice were protected against abnormal SR Ca2+ leak and HF after transverse aortic constriction (TAC) (van Oort et al., 2010) but surprisingly not after myocardial infarction (MI) (Respress et al., 2012). To explain how CaMKII increases SR Ca2+ leak, Uchinoumi et al. hypothesize that phosphorylation of S2814 reduces CaM affinity for the RyR2, leading to a RyR2 conformational change and leakiness of the channel (Uchinoumi et al., 2016). Supporting this hypothesis, Dantrolene, which restores RyR2 CaM affinity, suppressed SR Ca2+ leak in RyR2 S2814D knock-in mice (Uchinoumi et al., 2016).
Among the 90 cysteines present in each of the four subunits of RyR2, 21 are in a free thiol state and accessible for redox modifications during oxidative stress (Xu et al., 1998). Oxidative stress is associated with the development of several pathologies, and excessive production of reactive oxygen species has deleterious effects on protein function and cell viability. To counter these deleterious effects, cells have their own specific antioxidants machinery, including superoxide dismutase, catalase, and glutathione peroxidase, and non-specific antioxidants reduced glutathione, which is ubiquitous and the most important antioxidant system in cardiac cells (Nikolaienko et al., 2018).
During HF ROS production is chronically increased due to the uncoupling of the mitochondrial electron transport chain, the increase in energy demand, the switch from fatty acids to glucose as an energy substrate in cardiomyocytes (Mak and Newton, 2001; Ventura-Clapier et al., 2004), upregulation of nitric oxide synthase (NOS), xanthine oxidase and NADPH oxidase (NOX), and decreased reduced glutathione (Zima and Mazurek, 2016; Nikolaienko et al., 2018). ROS are also known to activate SR Ca2+ leak (Terentyev et al., 2008) by increasing spontaneous Ca2+ sparks frequency (Yan et al., 2008; Prosser et al., 2011) and Ca2+ waves (Bovo et al., 2012). Indeed, part of the beneficial effect of carvedilol on HF has been attributed to its antioxidant action on RyR2 (Mochizuki et al., 2007). ROS oxidation of RyR2 during HF induces the dissociation of FKBP12.6 leading to the aberrant Ca2+ release (Shan et al., 2010a), although other groups showed that RyR2 oxidation induces CaM dissociation without changing FKBP12.6 binding (Ono et al., 2010; Oda et al., 2015). Increased NOX activity induced by tachycardia led to an increase in RyR2 S-glutathionylation associated with an increased SR Ca2+ leak (Sanchez et al., 2005), which has been suggested to be cardio protective during preconditioning with exercise. However, the role of NOX during HF is not well-documented. The increased activity of NOX during end-stage HF (Heymes et al., 2003) can participate in the increase in diastolic SR Ca2+ leak and SR Ca2+ depletion. Even if hypernitrosylation of RyR2 is supposed to increase Ca2+ leak, decreased S-nitrosylation has been associated with increased SR Ca2+ leak during HF (Gonzalez et al., 2007; 2010). In fact, it has been proposed that hyponitrosylation during HF favors the oxidation of RyR2 by ROS, leading to this aberrant Ca2+ release. In these studies, inhibition of xanthine oxidase decreased ROS production restoring S-nitrosylation and cardiac function (Gonzalez et al., 2010). Evidence of the cardio protective effect of RyR2 S-nitrosylation has been also highlighted in a mouse model where NOS1 overexpression prevents cardiac dysfunction and delays HF in response to pressure overload (Loyer et al., 2008). On a canine model of HF, it has been suggested that the disulfide oxidation is the predominant form of redox-sensitive modulation of RyR2 compared to S-nitrosylation and S-glutathionylation to influence the RyR2-mediated leak. On a rabbit model of HF, increased SR Ca2+ leak (Mazurek et al., 2014) and Ca2+ waves (Bovo et al., 2015) could be attributed to an increase in RyR2 intersubunit disulfide cross-linking (Mazurek et al., 2014; Bovo et al., 2015, 2018).
By accessing data from different laboratories, we can gather that the RyR2 itself may be more active in HF due to several post-translational modifications such as oxidation, enhanced phosphorylation at the CaMKII site, as well as a depressed FKBP12.6 expression/binding to RyR2.
Figure 1 summarizes the key elements in Ca2+ handling in ventricular cardiomyocytes, as well as a representation of some of the reported findings in those elements, in different models of HF, which contribute to depressed contraction and pro-arrhythmogenic DADs production. The failing cell has a disruption in the T-tubular network, and a decreased SERCA expression (or augmented ratio PLN/SERCA), which together with more active RyR2 due to post-translational modifications, resulting in a drop of Ca2+ stored in the SR. As a result, and despite normal ICa density and prolonged AP, the triggered [Ca2+]i transient is of lower amplitude, contributing to diminished cell contraction. During diastole, the higher RyR2 activity favors the probability of producing Ca2+ waves, generating an inward current through the NCX, when extruding Ca2+, which produces DADs. When DADs reach the threshold, a triggered AP is generated, which may initiate arrhythmias.
HF with preserved ejection fraction (HFpEF) has been identified as a subtype of HF in addition to HFrEF. HFpEF is more frequently found in older patients with comorbidities such as hypertension, obesity, diabetes, chronic kidney diseases (Redfield et al., 2003; Bhatia et al., 2006; Owan et al., 2006). HFpEF patients present normal left ventricular systolic function with impaired left ventricular relaxation and filling. Although HFpEF represents approximately 50% of HF patients (Dunlay et al., 2017), little is known about their specific mechanisms, in comparison to HFrEF, notably regarding RyR2 and Ca2+ release. A lack of knowledge is probably related to the difficulty in generating accurate experimental models or the diversity of associated comorbidities. As described previously, in HFrEF, cardiac systolic and diastolic changes are linked to Ca2+ mishandling. In HFpEF, Ca2+ alterations are not as clearly defined and seem to differ depending on the associated comorbidity. In a rat model in HFpEF with chronic pressure overload, a significant increase in diastolic SR Ca2+ release through RyR2s leading, as in HFrEF, to higher diastolic [Ca2+] and irregular [Ca2+]i transients during pacing has been observed (Rouhana et al., 2019). This SR Ca2+ leak has been associated with an increase in PLN/SERCA ratio responsible for a delayed [Ca2+]i transient decay and diastolic dysfunction, also seen in HFpEF with chronic kidney disease (Primessnig et al., 2016) and in human HF with EF > 45% (Hohendanner et al., 2013; Ljubojevic et al., 2014). Unlike HFrEF, a SR Ca2+ leak in HFpEF depends on PKA-dependent phosphorylation of RyR2 at S2808 (Durland, 2021) rather than CaMKII-dependent phosphorylation, alteration of FKBP12.6/RyR2 ratio, or RyR2s S-nitrosylation (Adeniran et al., 2015; Frisk et al., 2021). However, phosphorylation of RyR2 at S2808 was not significantly changed in Dahl salt-sensitive rats, with HFpEF, presenting both hypertension and insulin-resistance (Kilfoil et al., 2020). This discrepancy could be explained by insulin-resistance comorbidity not present in Durland's model (Durland, 2021). Although in HFrEF an increase in SR Ca2+ leak leads to SR Ca2+ depletion and lower [Ca2+]i transient, in HFpEF, diastolic SR Ca2+ release does not induce SR Ca2+ load depletion or defective ECC. In fact, in HFpEF models, systolic Ca2+ release by the RyR2 is increased, probably as a compensatory mechanism to overcome the increased left ventricular stiffness associated with HFpEF (Selby et al., 2011; Adeniran et al., 2015; Durland, 2021). This enhanced SR Ca2+ release could be attributed to a more effective RyR2s recruitment by Ca2+ during the Ca2+ influx through the LTCC (Kilfoil et al., 2020) as well as an absence of T-tubules alteration (Durland, 2021). Indeed, in a Dahl-sensitive rat model of HFpEF (Kilfoil et al., 2020), a higher couplon recruitment improved ECC by increasing Ca2+ release synchronicity and lowering Ca2+ release latency. This should allow the heart to maintain an effective contraction besides higher ventricular wall stiffness. Moreover, T-tubule disorganization might also affect Ca2+ release effectiveness. However, the integrity of T-tubule structure in HFpEF depends on the comorbidities. Indeed, while T-tubule density is unchanged in Dahl salt-sensitive rats, in obese and diabetic Zucker rats or diabetic HFpEF patients, their density is increased in the ischemic model (post-MI with preserved EF but reduced E/A) (Frisk et al., 2021). This etiology-dependence is also found in proteins regulating SR Ca2+ release such as SERCA and NCX. In diabetic HFpEF, NCX expression and activity of SERCA decrease, which is not the case in ischemic or hypertensive disease models (Frisk et al., 2021). In type 1 diabetes, with subclinical diastolic dysfunction and normal systolic function, diastolic SR Ca2+ release and amplitude (measured as Ca2+ sparks frequency) decrease and is associated with a drop of SR Ca2+ load and [Ca2+]i transient without affecting systolic function (Lagadic-Gossmann et al., 1996; Lacombe et al., 2007; Hamouda et al., 2015). In these diabetic models, SR Ca2+ uptake by the SERCA pump appears as the main responsible for the decrease in SR Ca2+ release with, in some models, an alteration of the SERCA2/PLN ratio (Miranda-Silva et al., 2020). It is deduced from the literature that the alterations in the RyR2s and Ca2+ release diverge between HFpEF depending on their diabetic or non-diabetic etiology, with a clear implication of SERCA alteration in the Ca2+ homeostasis underlying the diastolic function in diabetes.
Interestingly, in myocardial strips from patients with hypertrophy associated with hypertension and coronary artery disease, but normal ejection fraction (LVEF ≥ 50%), diastolic dysfunction is associated with an increase in SR Ca2+ load at high pacing rates (Selby et al., 2011). However, the impact of cellular changes in Ca2+ handling does not always transduce into in vivo alterations. Indeed, in isolated myocardial strips from animal models of hypertrophy induced by aortic banding, even though cellular Ca2+ extrusion was increased due to higher NCX and SERCA activities, in vivo relaxation remained slower (Roe et al., 2017). Similarly, exacerbated cellular Ca2+ handling has been described in a rat strain with cardiac hypertrophy despite the lack of hypertension, where the increased Ca2+ influx through the LTCCs seems to underlie the increased [Ca2+]i transient amplitude and myocyte shortening. In this model, an increase in RyR2 phosphorylation by CaMKII leads to higher susceptibly to in vivo arrhythmia and spontaneous SR Ca2+ release although RyR2 expression was decreased (Curl et al., 2018).
In addition to its role in governing cardiac contraction, the RyR2-mediated Ca2+ release of the pacemaker cardiomyocytes of the heart, located at the sinus node (SAN), contributes to heart automaticity. This observation was initiated in the late 80s by the finding that ryanodine slows down the final phase of diastolic depolarization, resulting in a significant increase in cellular pacemaker cycle length in cats (Rubenstein and Lipsius, 1989). Since then, and after intense debates, it is now of general agreement that spontaneous cyclical local Ca2+ releases named late Ca2+ release (LCR), are responsible for a net depolarizing current mediated by the NCX. This process drives pacemaker depolarization, which is referred to as the “Ca2+ clock,” jointly with the “membrane or voltage clock” mediated by cyclic activation and deactivation of voltage-sensitive membrane ion channels [see for review (Carmeliet, 2019; Kohajda et al., 2020)]. This has been especially emphasized by the fact that RyR2 mutations associated with catecholaminergic polymorphic ventricular tachycardia (CPVT) induced SAN dysfunction (Neco et al., 2012; Wang et al., 2017).
Heart rate (HR) is an independent risk factor of all-cause mortality, cardiovascular mortality, and hospitalization for HF (Fox et al., 2007; Verrier and Tan, 2009). It has been long recognized that HF is associated with dysfunction of the SAN. Whereas, HR is commonly increased due to an excess of sympathetic activity and parasympathetic withdrawal, the intrinsic sinus rhythm in absence of autonomic nerve activity is depressed in HF. In the late 40s, a slower HR in isolated failing hearts was shown (Wollenberger, 1947; Wiggers, 1949). In animal models, and more importantly in humans with HF, the intrinsic HR is decreased with a reduction in SAN reserve and abnormal propagation of APs from the SAN, together with a caudal shift of the leading pacemaker site and fibrosis (Jose and Taylor, 1969; Jose and Collison, 1970; Vatner et al., 1974; Opthof et al., 2000; Verkerk et al., 2003; Sanders et al., 2004; Zicha et al., 2005; Packer et al., 2009; Swaminathan et al., 2011; Yanni et al., 2011). It has been suggested that this anatomic, structural, and functional SAN remodeling in HF might be an adaptive, protective response to improve cardiac oxygen supply to demand ratio (Mulder and Thuillez, 2006) and to prevent triggered arrhythmias enhanced by rapid HR (Opthof et al., 2000). However, reduced SAN automaticity, which favors the induction of early after-depolarization-triggered arrhythmias (Nuss et al., 1999), might translate into bradyarrhythmias or tachycardia-bradycardia syndrome (Mangrum and DiMarco, 2000). Indeed, bradyarrhythmias account for up to half of the deaths in HF (Luu et al., 1989; Stevenson et al., 1993; Uretsky and Sheahan, 1997; Faggiano et al., 2001; Packer et al., 2009; Bloch Thomsen et al., 2010; Gang et al., 2010; Glukhov et al., 2013; Lou et al., 2014). Moreover, in association with autonomic dysfunction (Colucci et al., 1989; Bristow et al., 1990; Samejima et al., 2003; Messias et al., 2016), slowing of the intrinsic HR by SAN remodeling might limit HR modulation to exercise (Sanders et al., 2004), the chronotropic incompetence observed during the HF process (Weber et al., 1982; Higginbotham et al., 1983; Brubaker et al., 2006; Brubaker and Kitzman, 2011; Benes et al., 2013).
To date, analysis of the Ca2+ clock in SAN dysfunction in HF has received little attention. In a canine model of HF induced by rapid pacing and using optical mapping, SAN bradycardia is associated with suppression of LCR together with the unresponsiveness of Ca2+ clock to isoproterenol and caffeine stimulation (Shinohara et al., 2010). In a similar rabbit HF model, alterations of SAN electro-pharmacological responses have been related to lower expression of RyR2, as well as inhibition of SERCA reuptake due to altered phosphorylation of PLN (Chang et al., 2017). By contrast, RyR2 expression, along with other Ca2+ handling proteins, is increased in the SAN but not in atrial tissue in a HF rat model (Yanni et al., 2011). The authors suggested that conjoint upregulation of RyR3 and calsequestrin 2 might however inhibit Ca2+ release resulting in slow automaticity. More recently, the same group (Yanni et al., 2020) reported that pressure overload-induced HF in mice did not change SAN mRNA for various components of the “Ca2+ clock,” including RyR2, SERCA, calsequestrin, and NCX. This is consistent with a study in isolated SAN cells from rabbit HF model of pressure and volume overload, which concluded that SAN Ca2+ cycling properties are conserved despite bradycardic effects (Verkerk et al., 2015). However, the latest study did not take into account the presence of hierarchical pacemaker clustering within the SAN that might be modified in HF (Sanders et al., 2004; Lang and Glukhov, 2021). Preliminary data from our group, using TAC-induced HF in the mouse indicate impairment of “Ca2+ clock,” characterized by slower spontaneous [Ca2+]i transients, as well as less frequent and smaller Ca2+ sparks, supported by a mechanism including depression in the CaMKII signaling pathway (Xue et al., 2020). Taking into account that only a few (and somewhat controversial) studies have evaluated SAN Ca2+ clock function in HF to date, further studies are still clearly needed.
Atrial fibrillation (AF) is a frequent complication of HF, and there is a reciprocal relationship between them. HF favors the development of AF (see below), and AF, because of its hemodynamic deleterious consequences, aggravates and may even generate HF. AF is defined as a rapid and disorganized electrical and mechanical activity of the atria, resulting in the loss of the synchronous contraction of atrial myocytes and myocardium normally occurring at end-diastole. The rapid anarchic electrical activity of the atria (around 300/min) is transmitted to the ventricle through the atrio-ventricular (AV) node, which plays the role of a filter, resulting in an irregular and nevertheless often rapid ventricular contraction rate. This has two deleterious consequences: first, the rapid irregular ventricular rate, together with the loss of atrial contraction, is responsible for a deterioration of ventricular filling, itself resulting in decreased cardiac output; second, the loss of atrial contraction favors blood stasis in the atria and especially the auricles, with the risk of thrombus formation and its migration into a peripheral artery.
As with all cardiac arrhythmias, AF necessitates the coexistence of a substrate (functional or anatomic reentry circuits, and tissue remodeling) and a trigger (increased automaticity or triggered activities), both modulated by alterations of the autonomous nervous system tone (increase in the sympathetic tone in the case of HF) or other humoral alterations such as hypokalemia, hypoxia, or acidosis (Coumel, 1993, 1996). It is important to note that HF can not only participate in but even create these three conditions.
Even if, sometimes, it may appear somewhat artificial, one may distinguish two radically different forms of AF, at least for the sake of clarity. The pathophysiology of HF-induced AF is somewhat different and much less documented than that of the “loan AF” occurring in a presumed normal heart. In the latter, AF originates from the wall of the pulmonary veins (Haissaguerre et al., 1989), starts as rare episodes of short duration (paroxysmal AF) that become more and more frequent and of longer duration with time to then finally become persistent, i.e., unable to end spontaneously to return to sinus rhythm. In this pathophysiological scheme, the recurrence of AF episodes is responsible, with time, for a progressive cellular electrical, Ca2+ signaling, and tissue remodeling that favors new AF episodes and prevents the return to sinus rhythm: AF begets AF. A detailed description of this remodeling can be found in excellent recent reviews (Landstrom et al., 2017; Denham et al., 2018).
In HF-induced AF, the pathophysiological process starts from the increased ventricular filling pressure that creates a chronic mechanical overload of the atria, associated with various degrees of neurohumoral activation leading to the atrial remodeling creating a favorable ground (both the substrate and the trigger) for the occurrence of AF. Interestingly, HF generally develops an atrial pro-arrhythmic substrate before AF arises, that may therefore immediately present as persistent (Sisti et al., 2014).
Alterations in Ca2+ signaling are instrumental to the two pathophysiological mechanisms, showing several similarities, but also subtle differences. They are involved both in the triggering of AF and in the progression of the atrial substrate that facilitates AF. These alterations are impacted by the atrial nature of cardiomyocytes. Indeed, atrial myocytes have a much less developed T-tubular network than ventricular myocytes with less junctional SR, and RyR2 clusters disconnected from T-tubules (orphan clusters). This favors a more progressive rise and delayed peak of the [Ca2+]i transient as compared to ventricular myocytes (Hatem et al., 1997; Caldwell et al., 2014). This also offers a greater opportunity for the development and propagation of Ca2+ waves. Thanks to significant technological advances in cell imaging, ECC of mice and human atrial myocytes has been recently clarified. Besides sparse T-tubule invaginations, which is associated with slow Ca2+ propagation, a voluminous axial tubular system develops extensive junctions with the SR comprising highly phosphorylated RyR2 clusters responsible, in mouse atrial myocytes, for a Ca2+ release approximately two times faster at the center of the cell than at its border, in agreement with the fast contractile activation of atrial myocytes (Brandenburg et al., 2016).
In contrast to the significant number of works exploring the role of Ca2+ mishandling in paroxysmal/persistent AF (Landstrom et al., 2017; Denham et al., 2018), far less has been published regarding the effects of HF on atrial Ca2+ handling. Studies have been carried out on various types of human atrial samples and two types of experimental models have been developed. The first uses long-lasting rapid ventricular pacing that finally results in increased atrial pressures and atrial dilation associated with various patterns of ventricular remodeling (Yeh et al., 2008; Dibb et al., 2009). More consistent with the actual HF pathophysiology, the second (e.g., MI in rat or rabbit, TAC in the mouse), aims to obtain left ventricular failure with only secondary impact on the left atrium (Boixel et al., 2001; Kettlewell et al., 2013; Brandenburg et al., 2016).
ICa is generally found to be decreased in atrial myocytes of HF models as well as in dilated atria or other pathological conditions associated with increased susceptibility to AF (Dinanian et al., 2008). In the rat MI model, the decrease in ICa density was related to a decrease in basal cAMP-dependent regulation of the current (Boixel et al., 2001). ICa density and the resulting [Ca2+]i transient amplitude are also decreased under β-adrenergic stimulation in atrial myocytes isolated from rabbits with MI and increased susceptibility to AF (Kettlewell et al., 2013). In the tachypacing HF model in dogs, [Ca2+]i transient amplitude and SR Ca2+ load were increased, associated with an increased diastolic Ca2+ concentration (Yeh et al., 2008). This was associated with decreased RyR2 and calsequestrin protein expression and increased CaMKII-dependent PLN phosphorylation whereas RyR2 phosphorylation was unchanged. In the mouse TAC model, left atrial hypertrophy is associated with marked proliferation of axial tubules and an increase in phosphorylated RyR2 at S2808, but not S2814, thereby accelerating SR Ca2+ release through non-junctional RyR2 cluster sites, despite decreases in RyR2 cluster density and RyR2 protein expression (Brandenburg et al., 2016). In a rabbit model of combined pressure and volume overload, diastolic Ca2+ concentration was also increased with [Ca2+]i transient of larger amplitude due to enhanced IP3 receptor-induced Ca2+ release originating from central non-junctional SR, associated with increased frequency of spontaneous Ca2+ waves, increased activity of NCX, and Ca2+ wave-triggered action potentials (Hohendanner et al., 2015). Interestingly, reduced RyR2 expression associated with increased sensitivity to ryanodine occurs in the atrioventricular node and participate in the slowing of AV conduction observed with aging (Saeed et al., 2018). This process, probably enhanced in failing hearts, would increase the role of the filter assigned to the AV node and therefore protect the failing ventricles from the deleterious consequences of high beating rates.
As seen in paroxysmal/persistent AF, HF-induced AF is also associated with increased diastolic Ca2+ leak despite no change or even decreased RyR2 expression. However, in contrast to the former, in which RyR2 phosphorylation is increased at both the PKA and CaMKII sites, at the stage of persistent AF, RyR2 phosphorylation does not appear to change in HF-induced AF. Therefore, the increased leak is more likely due to the increase in SR Ca2+ content, generally observed in models of HF in big mammals (Yeh et al., 2008; Clarke et al., 2015; Aistrup et al., 2017). The increased diastolic Ca2+ leak is often associated with increased NCX activity that favors the occurrence of DADs.
Recently, a striated muscle preferentially expressed protein kinase (SPEG) has been shown to play a role in the pathophysiology of paroxysmal/persistent AF (Campbell et al., 2020). Unlike PKA and CaMKII that increase RyR2 activity, SPEG phosphorylation at S2367 reduces RyR2-mediated SR Ca2+ release. SPEG protein levels, and RyR2 S2367 phosphorylation are decreased in atrial biopsies from patients with paroxysmal AF and transgenic RyR2-S2367A mice, in which the site cannot be phosphorylated, exhibited an increased susceptibility to pacing-induced AF. Whether such a pathophysiological mechanism exists in HF-induced AF remains to be established.
At last, it should be noted that HF also affects the pulmonary veins by increasing their electrical activity, thereby favoring the incidence of the trigger of paroxysmal/persistent AF in HF-induced AF (Lin et al., 2016).
In summary, the pathophysiology of HF-induced AF is complex and shares many features with paroxysmal/persistent AF, at least regarding alterations in Ca2+ signaling. More than in other fields of experimental cardiology, we often observed conflicting results due to differences in the species, experimental models, and disease stage studied. HF-induced AF pathophysiology may differ in humans according to the HF type considered. Indeed, HFpEF, in addition to the hemodynamic overload of the left atrium, may comprise specific pathophysiological mechanisms operating from the disease onset such as those of diabetes, inflammation, oxidation, etc., which may play a direct role simultaneously on the ventricles and atria through a “common atrial and ventricular myopathy” (Packer et al., 2020). Unequivocal information on the pathophysiology of HF-induced AF will be obtained from well-characterized standardized experimental models targeting left ventricular failure with secondary progressive atrial remodeling studied at the various stages of the pathological process.
In summary, alterations in RyR2 posttranslational modifications, location due to T-tubule remodeling, expression, and binding to accessory proteins have been all found in HF with variable conclusions depending on the experimental models or human etiology. What is clear is that RyR2 alteration contributes to the pathology of HF by participating in the depressed [Ca2+]i transient, which is important for depression of cell contraction and in favoring arrhythmogenic Ca2+ waves during diastole.
All authors contributed to the conception of the manuscript, wrote, edited the manuscript, contributed to the article, and approved the submitted version.
This work was funded by Inserm, University Paris-Saclay, and grants from ANR (ANR-15-CE14–0005 and ANR-19-CE14-0031-01) and NIH (2R01HL055438-22).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adeniran, I., MacIver, D. H., Hancox, J. C., and Zhang, H. (2015). Abnormal calcium homeostasis in heart failure with preserved ejection fraction is related to both reduced contractile function and incomplete relaxation: an electromechanically detailed biophysical modeling study. Front. Physiol.6:78. doi: 10.3389/fphys.2015.00078
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Aistrup, G. L., Arora, R., Grubb, S., Yoo, S., Toren, B., Kumar, M., et al. (2017). Triggered intracellular calcium waves in dog and human left atrial myocytes from normal and failing hearts. Cardiovasc. Res. 113, 1688-1699. doi: 10.1093/cvr/cvx167
Alvarado, F. J., Chen, X., and Valdivia, H. H. (2017). Ablation of the cardiac ryanodine receptor phospho-site Ser2808 does not alter the adrenergic response or the progression to heart failure in mice. Elimination of the genetic background as critical variable. J. Mol. Cell Cardiol. 103, 40-47. doi: 10.1016/j.yjmcc.2017.01.001
Alvarado, F. J., and Valdivia, H. H. (2020). Mechanisms of ryanodine receptor 2 dysfunction in heart failure. Nat. Rev. Cardiol. 17:748. doi: 10.1038/s41569-020-00443-x
Belevych, A. E., Terentyev, D., Terentyeva, R., Nishijima, Y., Sridhar, A., Hamlin, R. L., et al. (2011). The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc. Res. 90, 493-502. doi: 10.1093/cvr/cvr025
Benes, J., Kotrc, M., Borlaug, B. A., Lefflerova, K., Jarolim, P., Bendlova, B., et al. (2013). Resting heart rate and heart rate reserve in advanced heart failure have distinct pathophysiologic correlates and prognostic impact: a prospective pilot study. JACC Heart Fail. 1, 259-266. doi: 10.1016/j.jchf.2013.03.008
Benkusky, N. A., Weber, C. S., Scherman, J. A., Farrell, E. F., Hacker, T. A., John, M. C., et al. (2007). Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ. Res. 101, 819-829. doi: 10.1161/CIRCRESAHA.107.153007
Bers, D. M. (2012). Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ. Res. 110, 796-799. doi: 10.1161/CIRCRESAHA.112.265579
Beuckelmann, D. J., Nabauer, M., and Erdmann, E. (1992). Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 85, 1046-1055. doi: 10.1161/01.CIR.85.3.1046
Bhatia, R. S., Tu, J. V., Lee, D. S., Austin, P. C., Fang, J., Haouzi, A., et al. (2006). Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl. J. Med. 355, 260-269. doi: 10.1056/NEJMoa051530
Bloch Thomsen, P. E., Jons, C., Raatikainen, M. J., Moerch Joergensen, R., Hartikainen, J., Virtanen, V., et al. (2010). Long-term recording of cardiac arrhythmias with an implantable cardiac monitor in patients with reduced ejection fraction after acute myocardial infarction: the cardiac Arrhythmias and risk stratification after acute myocardial infarction (CARISMA) study. Circulation. 122, 1258-1264. doi: 10.1161/CIRCULATIONAHA.109.902148
Boixel, C., Gonzalez, W., Louedec, L., and Hatem, S. N. (2001). Mechanisms of L-type Ca(2+) current downregulation in rat atrial myocytes during heart failure. Circ. Res. 89, 607-613. doi: 10.1161/hh1901.096702
Bovo, E., Lipsius, S. L., and Zima, A. V. (2012). Reactive oxygen species contribute to the development of arrhythmogenic Ca(2)(+) waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 590, 3291-3304. doi: 10.1113/jphysiol.2012.230748
Bovo, E., Mazurek, S. R., Fill, M., and Zima, A. V. (2015). Cytosolic Ca(2)(+) buffering determines the intra-SR Ca(2)(+) concentration at which cardiac Ca(2)(+) sparks terminate. Cell Calcium. 58, 246-253. doi: 10.1016/j.ceca.2015.06.002
Bovo, E., Mazurek, S. R., and Zima, A. V. (2018). The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca(2+) transient amplitude in rabbit failing myocytes. Pflugers Arch. 470, 959-968. doi: 10.1007/s00424-018-2122-3
Brandenburg, S., Kohl, T., Williams, G. S., Gusev, K., Wagner, E., Rog-Zielinska, E. A., et al. (2016). Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Invest. 126, 3999-4015. doi: 10.1172/JCI88241
Brillantes, A. B., Ondrias, K., Scott, A., Kobrinsky, E., Ondriasova, E., Moschella, M. C., et al. (1994). Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77, 513–523. doi: 10.1016/0092-8674(94)90214-3
Bristow, M. R., Hershberger, R. E., Port, J. D., Gilbert, E. M., Sandoval, A., Rasmussen, R., et al. (1990). Beta-adrenergic pathways in non-failing and failing human ventricular myocardium. Circulation 82, 12–25.
Brubaker, P. H., Joo, K. C., Stewart, K. P., Fray, B., Moore, B., and Kitzman, D. W. (2006). Chronotropic incompetence and its contribution to exercise intolerance in older heart failure patients. J. Cardiopulm. Rehabil. 26, 86-89. doi: 10.1097/00008483-200603000-00007
Brubaker, P. H., and Kitzman, D. W. (2011). Chronotropic incompetence: causes, consequences, and management. Circulation 123, 1010-1020. doi: 10.1161/CIRCULATIONAHA.110.940577
Caldwell, J. L., Smith, C. E., Taylor, R. F., Kitmitto, A., Eisner, D. A., Dibb, K. M., et al. (2014). Dependence of cardiac transverse tubules on the BAR domain protein amphiphysin II (BIN-1). Circ. Res. 115, 986-996. doi: 10.1161/CIRCRESAHA.116.303448
Campbell, H. M., Quick, A. P., Abu-Taha, I., Chiang, D. Y., Kramm, C. F., Word, T. A., et al. (2020). Loss of SPEG inhibitory phosphorylation of ryanodine receptor type-2 promotes atrial fibrillation. Circulation 142, 1159-1172. doi: 10.1161/CIRCULATIONAHA.120.045791
Carmeliet, E. (2019). Pacemaking in cardiac tissue. From IK2 to a coupled-clock system. Physiol. Rep. 7:e13862. doi: 10.14814/phy2.13862
Chang, S. L., Chuang, H. L., Chen, Y. C., Kao, Y. H., Lin, Y. K., Yeh, Y. H., et al. (2017). Heart failure modulates electropharmacological characteristics of sinoatrial nodes. Exp. Ther. Med. 13, 771-779. doi: 10.3892/etm.2016.4015
Chen, J., Xu, S., Zhou, W., Wu, L., Wang, L., and Li, W. (2020). Exendin-4 reduces ventricular arrhythmia activity and calcium sparks-mediated sarcoplasmic reticulum Ca leak in rats with heart failure. Int. Heart J. 61, 145-152. doi: 10.1536/ihj.19-327
Cheng, H., Lederer, M. R., Lederer, W. J., and Cannell, M. B. (1996). Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 270, 148–159. doi: 10.1152/ajpcell.1996.270.1.C148
Cheng, H., and Lederer, W. J. (2008). Calcium sparks. Physiol. Rev. 88, 1491-1545. doi: 10.1152/physrev.00030.2007
Clarke, J. D., Caldwell, J. L., Horn, M. A., Bode, E. F., Richards, M. A., Hall, M. C., et al. (2015). Perturbed atrial calcium handling in an ovine model of heart failure: potential roles for reductions in the L-type calcium current. J. Mol. Cell Cardiol. 79, 169-179. doi: 10.1016/j.yjmcc.2014.11.017
Colucci, W. S., Ribeiro, J. P., Rocco, M. B., Quigg, R. J., Creager, M. A., Marsh, J. D., et al. (1989). Impaired chronotropic response to exercise in patients with congestive heart failure. Role of postsynaptic beta-adrenergic desensitization. Circulation 80, 314-323. doi: 10.1161/01.CIR.80.2.314
Coumel, P. (1993). Cardiac arrhythmias and the autonomic nervous system. J. Cardiovasc. Electrophysiol. 4, 338-355. doi: 10.1111/j.1540-8167.1993.tb01235.x
Coumel, P. (1996). Autonomic influences in atrial tachyarrhythmias. J. Cardiovasc. Electrophysiol. 7, 999-1007. doi: 10.1111/j.1540-8167.1996.tb00474.x
Curl, C. L., Danes, V. R., Bell, J. R., Raaijmakers, A. J. A., Ip, W. T. K., Chandramouli, C., et al. (2018). Cardiomyocyte functional etiology in heart failure with preserved ejection fraction is distinctive-a new preclinical model. J. Am. Heart Assoc. 7:e007451. doi: 10.1161/JAHA.117.007451
Curran, J., Brown, K. H., Santiago, D. J., Pogwizd, S., Bers, D. M., and Shannon, T. R. (2010). Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J. Mol. Cell Cardiol. 49, 25-32. doi: 10.1016/j.yjmcc.2010.03.013
Denham, N. C., Pearman, C. M., Caldwell, J. L., Madders, G. W. P., Eisner, D. A., Trafford, A. W., et al. (2018). Calcium in the pathophysiology of atrial fibrillation and heart failure. Front. Physiol. 9:1380. doi: 10.3389/fphys.2018.01380
Dibb, K. M., Clarke, J. D., Horn, M. A., Richards, M. A., Graham, H. K., Eisner, D. A., et al. (2009). Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2, 482-489. doi: 10.1161/CIRCHEARTFAILURE.109.852228
Dinanian, S., Boixel, C., Juin, C., Hulot, J. S., Coulombe, A., Rucker-Martin, C., et al. (2008). Downregulation of the calcium current in human right atrial myocytes from patients in sinus rhythm but with a high risk of atrial fibrillation. Eur. Heart J. 29, 1190-1197. doi: 10.1093/eurheartj/ehn140
Dridi, H., Kushnir, A., Zalk, R., Yuan, Q., Melville, Z., and Marks, A. R. (2020a). Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 17, 732-747. doi: 10.1038/s41569-020-0394-8
Dridi, H., Kushnir, A., Zalk, R., Yuan, Q., Melville, Z., and Marks, A. R. (2020b). Reply to “Mechanisms of ryanodine receptor 2 dysfunction in heart failure.” Nat. Rev. Cardiol. 17, 749-750. doi: 10.1038/s41569-020-00444-w
Dries, E., Santiago, D. J., Gilbert, G., Lenaerts, I., Vandenberk, B., Nagaraju, C. K., et al. (2018). Hyperactive ryanodine receptors in human heart failure and ischaemic cardiomyopathy reside outside of couplons. Cardiovasc. Res. 114, 1512-1524. doi: 10.1093/cvr/cvy088
Dunlay, S. M., Roger, V. L., and Redfield, M. M. (2017). Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 14, 591-602. doi: 10.1038/nrcardio.2017.65
Durland, L. (2021). Distinguishing HF with reduced and preserved ejection fraction at the level of individual cardiomyocytes: implications for therapeutic development. J. Physiol. 599, 1027-1029. doi: 10.1113/JP280739
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462-474. doi: 10.1016/j.cell.2008.02.048
Faggiano, P., d'Aloia, A., Gualeni, A., Gardini, A., and Giordano, A. (2001). Mechanisms and immediate outcome of in-hospital cardiac arrest in patients with advanced heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am. J. Cardiol. 87, 655-657. doi: 10.1016/S0002-9149(00)01450-8
Fischer, T. H., Eiringhaus, J., Dybkova, N., Forster, A., Herting, J., Kleinwachter, A., et al. (2014). Ca(2+)/calmodulin-dependent protein kinase II equally induces sarcoplasmic reticulum Ca(2+) leak in human ischaemic and dilated cardiomyopathy. Eur. J. Heart Fail. 16, 1292-1300. doi: 10.1002/ejhf.163
Fox, K., Borer, J. S., Camm, A. J., Danchin, N., Ferrari, R., Lopez Sendon, J. L., et al. (2007). Resting heart rate in cardiovascular disease. J. Am. Coll. Cardiol. 50, 823-830. doi: 10.1016/j.jacc.2007.04.079
Frisk, M., Le C Shen, X., Roe, A. T., Hou, Y., Manfra, O., Silva, G. J. J., et al. (2021). Etiology-dependent impairment of diastolic cardiomyocyte calcium homeostasis in heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. 77, 405-419. doi: 10.1016/j.jacc.2020.11.044
Fu, Y., Shaw, S. A., Naami, R., Vuong, C. L., Basheer, W. A., Guo, X., et al. (2016). Isoproterenol promotes rapid ryanodine receptor movement to bridging integrator 1 (BIN1)-organized dyads. Circulation 133, 388-397. doi: 10.1161/CIRCULATIONAHA.115.018535
Gang, U. J., Jons, C., Jorgensen, R. M., Abildstrom, S. Z., Haarbo, J., Messier, M. D., et al. (2010). Heart rhythm at the time of death documented by an implantable loop recorder. Europace 12, 254-260. doi: 10.1093/europace/eup383
Gellen, B., Fernandez-Velasco, M., Briec, F., Vinet, L., LeQuang, K., Rouet-Benzineb, P., et al. (2008). Conditional FKBP12.6 overexpression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation-contraction coupling. Circulation 117, 1778-1786. doi: 10.1161/CIRCULATIONAHA.107.731893
Glukhov, A. V., Hage, L. T., Hansen, B. J., Pedraza-Toscano, A., Vargas-Pinto, P., Hamlin, R. L., et al. (2013). Sinoatrial node reentry in a canine chronic left ventricular infarct model: role of intranodal fibrosis and heterogeneity of refractoriness. Circ. Arrhythm. Electrophysiol. 6, 984-994. doi: 10.1161/CIRCEP.113.000404
Gomez, A. M., Guatimosim, S., Dilly, K. W., Vassort, G., and Lederer, W. J. (2001). Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation 104, 688-693. doi: 10.1161/hc3201.092285
Gomez, A. M., Rueda, A., Sainte-Marie, Y., Pereira, L., Zissimopoulos, S., Zhu, X., et al. (2009). Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation 119, 2179-2187. doi: 10.1161/CIRCULATIONAHA.108.805804
Gomez, A. M., Valdivia, H. H., Cheng, H., Lederer, M. R., Santana, L. F., Cannell, M. B., et al. (1997). Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276, 800-806. doi: 10.1126/science.276.5313.800
Gonzalez, D. R., Beigi, F., Treuer, A. V., and Hare, J. M. (2007). Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 20612–20617. doi: 10.1073/pnas.0706796104
Gonzalez, D. R., Treuer, A. V., Castellanos, J., Dulce, R. A., and Hare, J. M. (2010). Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxidase activity contributes to calcium leak in heart failure. J. Biol. Chem. 285, 28938–28945. doi: 10.1074/jbc.M110.154948
Grimm, M., Ling, H., Willeford, A., Pereira, L., Gray, C. B., Erickson, J. R., et al. (2015). CaMKIIdelta mediates beta-adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic beta-adrenergic stimulation. J. Mol. Cell Cardiol. 85, 282-291. doi: 10.1016/j.yjmcc.2015.06.007
Guo, A., Zhang, C., Wei, S., Chen, B., and Song, L. S. (2013). Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc. Res. 98, 204-215. doi: 10.1093/cvr/cvt020
Haissaguerre, M., Warin, J. F., Lemetayer, P., Saoudi, N., Guillem, J. P., and Blanchot, P. (1989). Closed-chest ablation of retrograde conduction in patients with atrioventricular nodal reentrant tachycardia. N. Engl. J. Med. 320, 426-433. doi: 10.1056/NEJM198902163200704
Hamouda, N. N., Sydorenko, V., Qureshi, M. A., Alkaabi, J. M., Oz, M., and Howarth, F. C. (2015). Dapagliflozin reduces the amplitude of shortening and Ca(2+) transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol. Cell Biochem. 400, 7-68. doi: 10.1007/s11010-014-2262-5
Hasenfuss, G., and Pieske, B. (2002). Calcium cycling in congestive heart failure. J. Mol. Cell Cardiol. 34, 951-969. doi: 10.1006/jmcc.2002.2037
Hatem, S. N., Benardeau, A., Rucker-Martin, C., Marty, I., de Chamisso, P., Villaz, M., et al. (1997). Different compartments of sarcoplasmic reticulum participate in the excitation-contraction coupling process in human atrial myocytes. Circ. Res. 80, 345-353. doi: 10.1161/01.RES.80.3.345
Hautefort, A., Mendes-Ferreira, P., Sabourin, J., Manaud, G., Bertero, T., Rucker-Martin, C., et al. (2019). Bmpr2 mutant rats develop pulmonary and cardiac characteristics of pulmonary arterial hypertension. Circulation 139, 932-948. doi: 10.1161/CIRCULATIONAHA.118.033744
Heymes, C., Bendall, J. K., Ratajczak, P., Cave, A. C., Samuel, J. L., Hasenfuss, G., et al. (2003). Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll Cardiol. 41, 2164-2171. doi: 10.1016/S0735-1097(03)00471-6
Higginbotham, M. B., Morris, K. G., Conn, E. H., Coleman, R. E., and Cobb, F. R. (1983). Determinants of variable exercise performance among patients with severe left ventricular dysfunction. Am. J. Cardiol. 51, 52-60. doi: 10.1016/S0002-9149(83)80010-1
Ho, H. T., Liu, B., Snyder, J. S., Lou, Q., Brundage, E. A., Velez-Cortes, F., et al. (2014). Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc. Res. 101, 165-174. doi: 10.1093/cvr/cvt233
Hohendanner, F., Ljubojevic, S., MacQuaide, N., Sacherer, M., Sedej, S., Biesmans, L., et al. (2013). Intracellular dyssynchrony of diastolic cytosolic [Ca(2)(+)] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ. Res. 113, 527-538. doi: 10.1161/CIRCRESAHA.113.300895
Hohendanner, F., Walther, S., Maxwell, J. T., Kettlewell, S., Awad, S., Smith, G. L., et al. (2015). Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. J. Physiol. 593, 1459-1477. doi: 10.1113/jphysiol.2014.283226
Houser, S. R. (2014). Role of RyR2 phosphorylation in heart failure and arrhythmias: protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ. Res. 114, 1320-1327. doi: 10.1161/CIRCRESAHA.114.300569
Jiang, M. T., Lokuta, A. J., Farrell, E. F., Wolff, M. R., Haworth, R. A., and Valdivia, H. H. (2002). Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ. Res. 91, 1015–1022. doi: 10.1161/01.RES.0000043663.08689.05
Jones, P. P., MacQuaide, N., and Louch, W. E. (2018). Dyadic Plasticity in Cardiomyocytes. Front. Physiol. 9:1773. doi: 10.3389/fphys.2018.01773
Jose, A. D., and Collison, D. (1970). The normal range and determinants of the intrinsic heart rate in man. Cardiovasc. Res. 4, 160-167. doi: 10.1093/cvr/4.2.160
Jose, A. D., and Taylor, R. R. (1969). Autonomic blockade by propranolol and atropine to study intrinsic myocardial function in man. J. Clin. Invest. 48, 2019-2031. doi: 10.1172/JCI106167
Kettlewell, S., Burton, F. L., Smith, G. L., and Workman, A. J. (2013). Chronic myocardial infarction promotes atrial action potential alternans, afterdepolarizations, and fibrillation. Cardiovasc. Res. 99, 215-224. doi: 10.1093/cvr/cvt087
Kilfoil, P. J., Lotteau, S., Zhang, R., Yue, X., Aynaszyan, S., Solymani, R. E., et al. (2020). Distinct features of calcium handling and beta-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J. Physiol. 598, 5091-5108. doi: 10.1113/JP280425
Kohajda, Z., Loewe, A., Toth, N., Varro, A., and Nagy, N. (2020). The cardiac pacemaker story-fundamental role of the Na(+)/Ca(2+) exchanger in spontaneous automaticity. Front. Pharmacol. 11:516. doi: 10.3389/fphar.2020.00516
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646-1660. doi: 10.1161/CIRCRESAHA.111.259754
Lacombe, V. A., Viatchenko-Karpinski, S., Terentyev, D., Sridhar, A., Emani, S., Bonagura, J. D., et al. (2007). Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, 1787-1797. doi: 10.1152/ajpregu.00059.2007
Lagadic-Gossmann, D., Buckler, K. J., Le Prigent, K., and Feuvray, D. (1996). Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am. J. Physiol. 270, 1529–1537. doi: 10.1152/ajpheart.1996.270.5.H1529
Landstrom, A. P., Dobrev, D., and Wehrens, X. H. T. (2017). Calcium signaling and cardiac arrhythmias. Circ. Res. 120, 1969-1993. doi: 10.1161/CIRCRESAHA.117.310083
Lang, D., and Glukhov, A. V. (2021). Cellular and molecular mechanisms of functional hierarchy of pacemaker clusters in the sinoatrial node: new insights into sick sinus syndrome. J. Cardiovasc. Dev. Dis. 8:43. doi: 10.3390/jcdd8040043
Lehnart, S. E., Maier, L. S., and Hasenfuss, G. (2009). Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail. Rev. 14, 213-224. doi: 10.1007/s10741-009-9146-x
Lin, Y. K., Chen, Y. C., Chen, Y. A., Yeh, Y. H., Chen, S. A., and Chen, Y. J. (2016). B-type natriuretic peptide modulates pulmonary vein arrhythmogenesis: a novel potential contributor to the genesis of atrial tachyarrhythmia in heart failure. J. Cardiovasc. Electrophysiol. 27, 1462-1471. doi: 10.1111/jce.13093
Lindner, M., Brandt, M. C., Sauer, H., Hescheler, J., Böhle, T., and Beuckelmann, D. J. (2002). Calcium sparks in human ventricular cardiomyocytes from patients with terminal heart failure. Cell Calcium. 31, 175-182. doi: 10.1054/ceca.2002.0272
Ling, H., Zhang, T., Pereira, L., Means, C. K., Cheng, H., Gu, Y., et al. (2009). Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Invest. 119, 1230-1240. doi: 10.1172/JCI38022
Ljubojevic, S., Radulovic, S., Leitinger, G., Sedej, S., Sacherer, M., Holzer, M., et al. (2014). Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation 130, 244-255. doi: 10.1161/CIRCULATIONAHA.114.008927
Lou, Q., Hansen, B. J., Fedorenko, O., Csepe, T. A., Kalyanasundaram, A., Li, N., et al. (2014). Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation 130, 315-324. doi: 10.1161/CIRCULATIONAHA.113.007086
Loyer, X., Gomez, A. M., Milliez, P., Fernandez-Velasco, M., Vangheluwe, P., Vinet, L., et al. (2008). Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation 117, 3187–3198. doi: 10.1161/CIRCULATIONAHA.107.741702
Luu, M., Stevenson, W. G., Stevenson, L. W., Baron, K., and Walden, J. (1989). Diverse mechanisms of unexpected cardiac arrest in advanced heart failure. Circulation 80, 1675-1680. doi: 10.1161/01.CIR.80.6.1675
Lymperopoulos, A., Rengo, G., and Koch, W. J. (2013). Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ. Res. 113, 739-753. doi: 10.1161/CIRCRESAHA.113.300308
MacDonnell, S. M., Garcia-Rivas, G., Scherman, J. A., Kubo, H., Chen, X., Valdivia, H., et al. (2008). Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ. Res. 102, 65-72. doi: 10.1161/CIRCRESAHA.108.174722
Mak, S., and Newton, G. E. (2001). The oxidative stress hypothesis of congestive heart failure: radical thoughts. Chest 120, 2035-2046. doi: 10.1378/chest.120.6.2035
Mangrum, J. M., and DiMarco, J. P. (2000). The evaluation and management of bradycardia. N. Engl. J. Med. 342, 703-709. doi: 10.1056/NEJM200003093421006
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
Mazurek, S. R., Bovo, E., and Zima, A. V. (2014). Regulation of sarcoplasmic reticulum Ca(2+) release by cytosolic glutathione in rabbit ventricular myocytes. Free Radic. Biol. Med. 68, 159-167. doi: 10.1016/j.freeradbiomed.2013.12.003
Messias, L. R., Messias, A. C., de Miranda, S. M., Wiefels, C. C., Ferreira, A. G., Santos, L. M., et al. (2016). Abnormal adrenergic activation is the major determinant of reduced functional capacity in heart failure with preserved ejection fraction. Int. J. Cardiol. 203, 900-902. doi: 10.1016/j.ijcard.2015.10.224
Miranda-Silva, D., Wust, R. C. I., Conceicao, G., Goncalves-Rodrigues, P., Goncalves, N., Goncalves, A., et al. (2020). Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol (Oxf). 228:e13378. doi: 10.1111/apha.13378
Mochizuki, M., Yano, M., Oda, T., Tateishi, H., Kobayashi, S., Yamamoto, T., et al. (2007). Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J. Am. Coll. Cardiol. 49, 1722-1732. doi: 10.1016/j.jacc.2007.01.064
Mulder, P., and Thuillez, C. (2006). Heart rate slowing for myocardial dysfunction/heart failure. Adv. Cardiol. 43, 97-105. doi: 10.1159/000095431
Neco, P., Torrente, A. G., Mesirca, P., Zorio, E., Liu, N., Priori, S. G., et al. (2012). Paradoxical effect of increased diastolic Ca(2+) release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 126, 392-401. doi: 10.1161/CIRCULATIONAHA.111.075382
Nikolaienko, R., Bovo, E., and Zima, A. V. (2018). Redox dependent modifications of ryanodine receptor: basic mechanisms and implications in heart diseases. Front. Physiol. 9:1775. doi: 10.3389/fphys.2018.01775
Nuss, H. B., Kaab, S., Kass, D. A., Tomaselli, G. F., and Marbán, E. (1999). Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am. J. Physiol. 277, 80–91. doi: 10.1152/ajpheart.1999.277.1.H80
Oda, T., Yang, Y., Uchinoumi, H., Thomas, D. D., Chen-Izu, Y., Kato, T., et al. (2015). Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell Cardiol. 85, 240-248. doi: 10.1016/j.yjmcc.2015.06.009
Ohkusa, T., Hisamatsu, Y., Yano, M., Kobayashi, S., Tatsuno, H., Saiki, Y., et al. (1997). Altered cardiac mechanism and sarcoplasmic reticulum function in pressure overload-induced cardiac hypertrophy in rats. J. Mol. Cell Cardiol. 29, 45-54. doi: 10.1006/jmcc.1996.0250
Ono, K., Yano, M., Ohkusa, T., Kohno, M., Hisaoka, T., Tanigawa, T., et al. (2000). Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal Ca(2+) release in heart failure. Cardiovasc. Res. 48, 323-331. doi: 10.1016/S0008-6363(00)00191-7
Ono, M., Yano, M., Hino, A., Suetomi, T., Xu, X., Susa, T., et al. (2010). Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca(2+) release in heart failure. Cardiovasc. Res. 87, 609-617. doi: 10.1093/cvr/cvq108
Opthof, T., Coronel, R., Rademaker, H. M., Vermeulen, J. T., Wilms-Schopman, F. J., and Janse, M. J. (2000). Changes in sinus node function in a rabbit model of heart failure with ventricular arrhythmias and sudden death. Circulation 101, 2975-2980. doi: 10.1161/01.CIR.101.25.2975
Owan, T. E., Hodge, D. O., Herges, R. M., Jacobsen, S. J., Roger, V. L., and Redfield, M. M. (2006). Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 355, 251-259. doi: 10.1056/NEJMoa052256
Packer, D. L., Prutkin, J. M., Hellkamp, A. S., Mitchell, L. B., Bernstein, R. C., Wood, F., et al. (2009). Impact of implantable cardioverter-defibrillator, amiodarone, and placebo on the mode of death in stable patients with heart failure: analysis from the sudden cardiac death in heart failure trial. Circulation 120, 2170-2176. doi: 10.1161/CIRCULATIONAHA.109.853689
Packer, M., Lam, C. S. P., Lund, L. H., and Redfield, M. M. (2020). Interdependence of atrial fibrillation and heart failure with a preserved ejection fraction reflects a common underlying atrial and ventricular myopathy. Circulation 141, 4-6. doi: 10.1161/CIRCULATIONAHA.119.042996
Piacentino, V. 3rd, Weber, C. R., Chen, X., Weisser-Thomas, J., Margulies, K. B., Bers, D. M., and Houser, S. R. (2003). Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92, 651–658. doi: 10.1161/01.RES.0000062469.83985.9B
Pogwizd, S. M., Schlotthauer, K., Li, L., Yuan, W., and Bers, D. M. (2001). Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 88, 1159-1167. doi: 10.1161/hh1101.091193
Potenza, D. M., Janicek, R., Fernandez-Tenorio, M., Camors, E., Ramos-Mondragon, R., Valdivia, H. H., et al. (2019). Phosphorylation of the ryanodine receptor 2 at serine 2030 is required for a complete beta-adrenergic response. J. Gen. Physiol. 151, 131-145. doi: 10.1085/jgp.201812155
Primessnig, U., Schonleitner, P., Holl, A., Pfeiffer, S., Bracic, T., Rau, T., et al. (2016). Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 18, 987-997. doi: 10.1002/ejhf.524
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440-1445. doi: 10.1126/science.1202768
Redfield, M. M., Jacobsen, S. J., Burnett, J. C. Jr., Mahoney, D. W., Bailey, K. R., and Rodeheffer, R. J. (2003). Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA 289, 194-202. doi: 10.1001/jama.289.2.194
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474-1483. doi: 10.1161/CIRCRESAHA.112.268094
Roe, A. T., Aronsen, J. M., Skardal, K., Hamdani, N., Linke, W. A., Danielsen, H. E., et al. (2017). Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc. Res. 113, 1161-1172. doi: 10.1093/cvr/cvx087
Rouhana, S., Farah, C., Roy, J., Finan, A., Rodrigues de Araujo, G., Bideaux, P., et al. (2019). Early calcium handling imbalance in pressure overload-induced heart failure with nearly normal left ventricular ejection fraction. Biochim. Biophys. Acta. Mol. Basis Dis. 1865, 230-242. doi: 10.1016/j.bbadis.2018.08.005
Rubenstein, D. S., and Lipsius, S. L. (1989). Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ. Res. 64, 648-657. doi: 10.1161/01.RES.64.4.648
Ruiz-Hurtado, G., Li, L., Fernandez-Velasco, M., Rueda, A., Lefebvre, F., Wang, Y., et al. (2015). Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. J. Gen. Physiol. 146, 295-306. doi: 10.1085/jgp.201511366
Saeed, Y., Temple, I. P., Borbas, Z., Atkinson, A., Yanni, J., Maczewski, M., et al. (2018). Structural and functional remodeling of the atrioventricular node with aging in rats: the role of hyperpolarization-activated cyclic nucleotide-gated and ryanodine 2 channels. Heart Rhythm 15, 752-760. doi: 10.1016/j.hrthm.2017.12.027
Samejima, H., Omiya, K., Uno, M., Inoue, K., Tamura, M., Itoh, K., et al. (2003). Relationship between impaired chronotropic response, cardiac output during exercise, and exercise tolerance in patients with chronic heart failure. Jpn. Heart J. 44, 515-525. doi: 10.1536/jhj.44.515
Sanchez, G., Pedrozo, Z., Domenech, R. J., Hidalgo, C., and Donoso, P. (2005). Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J. Mol. Cell Cardiol. 39, 982-991. doi: 10.1016/j.yjmcc.2005.08.010
Sanders, P., Kistler, P. M., Morton, J. B., Spence, S. J., and Kalman, J. M. (2004). Remodeling of sinus node function in patients with congestive heart failure: reduction in sinus node reserve. Circulation 110, 897-903. doi: 10.1161/01.CIR.0000139336.69955.AB
Schroder, F., Handrock, R., Beuckelmann, D. J., Hirt, S., Hullin, R., Priebe, L., et al. (1998). Increased availability and open probability of single L-type calcium channels from failing compared with non-failing human ventricle. Circulation 98, 969–976. doi: 10.1161/01.CIR.98.10.969
Selby, D. E., Palmer, B. M., LeWinter, M. M., and Meyer, M. (2011). Tachycardia-induced diastolic dysfunction and resting tone in myocardium from patients with a normal ejection fraction. J. Am. Coll. Cardiol. 58, 147-154. doi: 10.1016/j.jacc.2010.10.069
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010a). Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Invest. 120, 4375-4387. doi: 10.1172/JCI37649
Shan, J., Kushnir, A., Betzenhauser, M. J., Reiken, S., Li, J., Lehnart, S. E., et al. (2010b). Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Invest. 120, 4388-4398. doi: 10.1172/JCI32726
Shinohara, T., Park, H. W., Han, S., Shen, M. J., Maruyama, M., Kim, D., et al. (2010). Ca2+ clock malfunction in a canine model of pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 299, 1805-1811. doi: 10.1152/ajpheart.00723.2010
Sisti, A. D. E., Leclercq, J. F., Halimi, F., Fiorello, P., Bertrand, C., and Attuel, P. (2014). Evaluation of time course and predicting factors of progression of paroxysmal or persistent atrial fibrillation to permanent atrial fibrillation. Pacing Clin. Electrophysiol. 37, 345-355. doi: 10.1111/pace.12264
Stern, M. D. (1992). Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63, 497-517. doi: 10.1016/S0006-3495(92)81615-6
Stevenson, W. G., Stevenson, L. W., Middlekauff, H. R., and Saxon, L. A. (1993). Sudden death prevention in patients with advanced ventricular dysfunction. Circulation 88, 2953-2961. doi: 10.1161/01.CIR.88.6.2953
Swaminathan, P. D., Purohit, A., Soni, S., Voigt, N., Singh, M. V., Glukhov, A. V., et al. (2011). Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Invest. 121, 3277-3288. doi: 10.1172/JCI57833
Terentyev, D., Gyorke, I., Belevych, A. E., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2008). Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466-1472. doi: 10.1161/CIRCRESAHA.108.184457
Uchinoumi, H., Yang, Y., Oda, T., Li, N., Alsina, K. M., Puglisi, J. L., et al. (2016). CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J. Mol. Cell Cardiol. 98, 62-72. doi: 10.1016/j.yjmcc.2016.06.007
Uretsky, B. F., and Sheahan, R. G. (1997). Primary prevention of sudden cardiac death in heart failure: will the solution be shocking? J. Am. Coll. Cardiol. 30, 1589-1597. doi: 10.1016/S0735-1097(97)00361-6
van Oort, R. J., McCauley, M. D., Dixit, S. S., Pereira, L., Yang, Y., Respress, J. L., et al. (2010). Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 122, 2669-2679. doi: 10.1161/CIRCULATIONAHA.110.982298
Vatner, S. F., Higgins, C. B., and Braunwald, E. (1974). Sympathetic and parasympathetic components of reflex tachycardia induced by hypotension in conscious dogs with and without heart failure. Cardiovasc. Res. 8, 153-161. doi: 10.1093/cvr/8.2.153
Venetucci, L. A., Trafford, A. W., O'Neill, S. C., and Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292. doi: 10.1093/cvr/cvm009
Ventura-Clapier, R., Garnier, A., and Veksler, V. (2004). Energy metabolism in heart failure. J. Physiol. 555, 1-13. doi: 10.1113/jphysiol.2003.055095
Verkerk, A. O., van Borren, M. M., van Ginneken, A. C., and Wilders, R. (2015). Ca(2+) cycling properties are conserved despite bradycardic effects of heart failure in sinoatrial node cells. Front. Physiol. 6:18. doi: 10.3389/fphys.2015.00018
Verkerk, A. O., Wilders, R., Coronel, R., Ravesloot, J. H., and Verheijck, E. E. (2003). Ionic remodeling of sinoatrial node cells by heart failure. Circulation 108, 760-766. doi: 10.1161/01.CIR.0000083719.51661.B9
Verrier, R. L., and Tan, A. (2009). Heart rate, autonomic markers, and cardiac mortality. Heart Rhythm 6, 68-75. doi: 10.1016/j.hrthm.2009.07.017
Wagner, E., Lauterbach, M. A., Kohl, T., Westphal, V., Williams, G. S., Steinbrecher, J. H., et al. (2012). Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 111, 402-414. doi: 10.1161/CIRCRESAHA.112.274530
Walweel, K., Molenaar, P., Imtiaz, M. S., Denniss, A., Dos Remedios, C., van Helden, D. F., et al. (2017). Ryanodine receptor modification and regulation by intracellular Ca(2+) and Mg(2+) in healthy and failing human hearts. J. Mol. Cell Cardiol. 104, 53-62. doi: 10.1016/j.yjmcc.2017.01.016
Wang, Y. Y., Mesirca, P., Marques-Sule, E., Zahradnikova, A. Jr., Villejoubert, O., D'Ocon, P., et al. (2017). catecholaminergic polymorphic ventricular tachycardia mutation induces bradycardia by disturbing the coupled clock pacemaker mechanism. JCI Insight 2:e91872. doi: 10.1172/jci.insight.91872
Weber, K. T., Kinasewitz, G. T., Janicki, J. S., and Fishman, A. P. (1982). Oxygen utilization and ventilation during exercise in patients with chronic cardiac failure. Circulation 65, 1213-1223. doi: 10.1161/01.CIR.65.6.1213
Wehrens, X. H., Lehnart, S. E., Reiken, S., Vest, J. A., Wronska, A., and Marks, A. R. (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U.S.A. 103, 511-518. doi: 10.1073/pnas.0510113103
Witcher, D. R., Kovacs, R. J., Schulman, H., Cefali, D. C., and Jones, L. R. (1991). Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266, 11144-11152. doi: 10.1016/S0021-9258(18)99140-4
Wollenberger, A. (1947). On the energy-rich phosphate supply of the failing heart. Am. J. Physiol. 150, 733-745. doi: 10.1152/ajplegacy.1947.150.4.733
Xiao, B., Zhong, G., Obayashi, M., Yang, D., Chen, K., Walsh, M. P., et al. (2006). Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem. J. 396, 7-16. doi: 10.1042/BJ20060116
Xiao, J., Tian, X., Jones, P. P., Bolstad, J., Kong, H., Wang, R., et al. (2007). Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem. 282, 34828–34838. doi: 10.1074/jbc.M707423200
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234-237. doi: 10.1126/science.279.5348.234
Xue, J., Arbel Ganon, L., Gomez, S., Benitah, J. P., and Gomez, A. M. (2020). SAN function is altered in a mice model of heart failure. J. Mol. Cell Cardiol. 140:149. doi: 10.1016/j.yjmcc.2019.11.018
Yan, Y., Liu, J., Wei, C., Li, K., Xie, W., Wang, Y., et al. (2008). Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 77, 432-441. doi: 10.1093/cvr/cvm047
Yanni, J., D'Souza, A., Wang, Y., Li, N., Hansen, B. J., Zakharkin, S. O., et al. (2020). Silencing miR-370-3p rescues funny current and sinus node function in heart failure. Sci. Rep. 10:11279. doi: 10.1038/s41598-020-67790-0
Yanni, J., Tellez, J. O., Maczewski, M., Mackiewicz, U., Beresewicz, A., Billeter, R., et al. (2011). Changes in ion channel gene expression underlying heart failure-induced sinoatrial node dysfunction. Circ. Heart Fail. 4, 496-508. doi: 10.1161/CIRCHEARTFAILURE.110.957647
Yeh, Y. H., Wakili, R., Qi, X. Y., Chartier, D., Boknik, P., Kaab, S., et al. (2008). Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol. 1, 93-102. doi: 10.1161/CIRCEP.107.754788
Zhang, H., Makarewich, C. A., Kubo, H., Wang, W., Duran, J. M., Li, Y., et al. (2012). Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ. Res. 110, 831-840. doi: 10.1161/CIRCRESAHA.111.255158
Zicha, S., Fernandez-Velasco, M., Lonardo, G., L'Heureux N., and Nattel, S. (2005). Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc. Res. 66, 472-481. doi: 10.1016/j.cardiores.2005.02.011
Keywords: ryanodine receptor, heart failure, calcium, excitation contraction coupling, sinus node, atrial fibrillation
Citation: Benitah J-P, Perrier R, Mercadier J-J, Pereira L and Gómez AM (2021) RyR2 and Calcium Release in Heart Failure. Front. Physiol. 12:734210. doi: 10.3389/fphys.2021.734210
Received: 30 June 2021; Accepted: 30 August 2021;
Published: 08 October 2021.
Edited by:
Daniel M. Johnson, The Open University, United KingdomReviewed by:
Bjorn C. Knollmann, Vanderbilt University, United StatesCopyright © 2021 Benitah, Perrier, Mercadier, Pereira and Gómez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana M. Gómez, YW5hLW1hcmlhLmdvbWV6QGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.