
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 21 October 2021
Sec. Respiratory Physiology and Pathophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.720196
This article is part of the Research Topic Respiratory Barrier Versus Exogenous and Endogenous Noxious Stimuli: at the Crossroad Between Integrity and Dysfunction View all 9 articles
 Hussein Traboulsi1,2,3
Hussein Traboulsi1,2,3 Angela Rico de Souza1,2
Angela Rico de Souza1,2 Benoit Allard1
Benoit Allard1 Zahraa Haidar1,2,3
Zahraa Haidar1,2,3 Mark Sorin1,2,3
Mark Sorin1,2,3 Vanessa Moarbes1,2,3
Vanessa Moarbes1,2,3 Elizabeth D. Fixman1,2,3
Elizabeth D. Fixman1,2,3 James G. Martin1,2,3
James G. Martin1,2,3 David H. Eidelman1,2,3
David H. Eidelman1,2,3 Carolyn J. Baglole1,2,3,4,5*
Carolyn J. Baglole1,2,3,4,5*The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that regulates the metabolism of xenobiotics. There is growing evidence that the AhR is implicated in physiological processes such proliferation, differentiation, and immune responses. Recently, a role of the AhR in regulating allergic asthma has been suggested, but whether the AhR also regulates other type of asthma, particularly occupational/irritant-induced asthma, remains unknown. Using AhR-deficient (Ahr−/−) mice, we compared the function of the AhR in the response to ovalbumin (OVA; allergic asthma) vs. chlorine (Cl2; irritant-induced asthma) exposure. Lung inflammation and airway hyperresponsiveness were assessed 24h after exposure to Cl2 or OVA challenge in Ahr−/− and heterozygous (Ahr+/−) mice. After OVA challenge, absence of AhR was associated with significantly enhanced eosinophilia and lymphocyte influx into the airways of Ahr−/− mice. There were also increased levels of interleukin-4 (IL-4) and IL-5 in the airways. However, OVA-induced airway hyperresponsiveness was not affected. In the irritant-induced asthma model caused by exposure to Cl2, the AhR did not regulate the inflammatory response. However, absence of AhR reduced Cl2-induced airway hyperresponsiveness. Collectively, these results support a differential role for the AhR in regulating asthma outcomes in response to diverse etiological agents.
Exposure of the lungs to environmental toxicants such as pesticides, solvents, and air pollutants may lead to acute and chronic pulmonary inflammation that is associated with the development of asthma (Wong et al., 2016). With the increased prevalence in the second half of the 20th century, it is estimated that over 300 million people world-wide have asthma (Braman, 2006; Asher et al., 2020), making it a major health burden (Boonpiyathad et al., 2019). Asthma is a heterogeneous chronic disease of the airways characterized by inflammation, airway hyperresponsiveness and narrowing that result from airway smooth muscle (ASM) contraction and airway remodeling (James et al., 2009; Asher et al., 2020). In susceptible individuals, these pathological characteristics cause recurrent episodes of wheezing, breathlessness, chest tightness, and coughing (Maslan and Mims, 2014).
Asthma is classified as allergic asthma or non-allergic asthma, and this is based on the age of onset (e.g., adult-onset asthma or childhood asthma). Also it can clinically be classified by the patient history, symptoms and the predominant type of leukocytes in the sputum (e.g., eosinophilic, neutrophilic, or paucigranulocytic; Simpson et al., 2006; Barnes, 2018; Papi et al., 2018). Eosinophilic asthma is one of the most common subtypes of asthma diagnosed in children and adults and can be either allergic or non-allergic. The allergic form is an adaptive T helper 2-driven disease characterized by elevated levels of interleukin (IL)-5, interleukin-4 (IL-4), and IL-13, associated with enhanced levels of circulating and lung eosinophils, elevated serum IgE, mucus hypersecretion and airway hyperresponsiveness (Holgate and Polosa, 2008; Chiba et al., 2009; Pelaia et al., 2015). In the non-allergic form, innate lymphoid cells (ILC2) produce IL-5 to recruit eosinophils into the airway (Jonckheere et al., 2019). Neutrophilic-asthma (non-eosinophilic) is triggered by Th1 and Th17 lymphocytes with the release of cytokines (e.g., IFN-γ and IL-17) which favor the development of a cellular immune response, activation of macrophages, and release of neutrophil chemokines (Papi et al., 2018). Environmental stimuli such as chlorine (Cl2) can trigger Th1 and Th17-mediated airway inflammation that is implicated in the development of severe neutrophilic asthma (Fisk et al., 2010; Pelaia et al., 2015). Chlorine and Cl2 derivatives are present in disinfecting agents that are widely used by cleaning personnel and are linked to the development of occupational asthma (De Genaro et al., 2018). Chronic low dose exposure to chlorine also occurs through frequentation of chlorinated swimming pools (Ferrari et al., 2011). Thus, exposure to environmental toxicants not only contributes to the increasing prevalence of asthma, but these exposures can also affect disease outcomes.
The molecular and cellular mechanisms involved in the pathogenesis of the asthmatic phenotype particularly allergic vs. non-allergic asthma are not fully understood. Recently, it has been shown that the aryl hydrocarbon receptor (AhR) may be involved in suppressing the development of allergic asthma (Jeong et al., 2012; Chang et al., 2020). The AhR is a ligand-activated transcription factor that belongs to the basic helix loop helix (bHLH)/PER-ARNT-SIM (PAS) family and is highly expressed in the lung. Historically the AhR is known for its ability to mediate the deleterious effects of the environmental toxicant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD; dioxin). In the absence of ligand, the AhR remains in the cytoplasm. After ligand binding, it translocates to the nucleus and forms a heterodimer with the AhR nuclear transporter (ARNT). This complex binds to DNA sequences termed the dioxin response element (DRE), initiating the transcription of genes that comprise the AhR gene battery such as cytochrome P450 (CYP) enzymes (Guerrina et al., 2018). Although historically, the AhR has been largely associated with xenobiotic metabolism leading to toxicity, we have shown that the AhR suppresses the development of chronic obstructive pulmonary disease (COPD; Guerrina et al., 2021), an obstructive lung disease caused predominantly by cigarette smoke. Mechanistically, the AhR also suppresses neutrophil recruitment to the lungs in response to cigarette smoke (Thatcher et al., 2007; De Souza et al., 2014; Rico De Souza et al., 2021). While a role of the AhR in controlling asthma related-outcomes has emerged (Xu et al., 2015; Thatcher et al., 2016; Chang et al., 2020; Poulain-Godefroy et al., 2020), these studies utilized mouse models of eosinophilic allergic asthma. However, a role for the AhR in suppressing asthma caused by other environmental triggers, particularly those that are associated with neutrophilic asthma, remains unknown.
Therefore, we sought to understand whether the AhR can control the development of the asthmatic phenotype using two different triggers: ovalbumin (OVA) and Cl2. Ovalbumin induces an eosinophilic asthma phenotype and thus is a model of allergen-induced asthma. For the second model, we used acute Cl2 exposure as a model of neutrophilic asthma. In these two models, we set out to study the extent of airway and parenchymal inflammation as well as airway hyperresponsiveness using AhR knock out (Ahr−/−) mice. Herein, we demonstrated an important role of the AhR in decreasing pulmonary inflammation in the OVA mouse model, but not in the Cl2 mouse model. These data highlight the differential role that AhR may play in controlling asthma phenotypes.
All chemicals were purchased from Sigma (St. Louis, MO, United States) unless otherwise indicated. 6-Formylindoleo [3,2-b] carbazole (FICZ) was from Tocris Bioscience (Minneapolis, MN, United States).
Mice heterozygotes for AhR (Ahr+/−) and knockout (Ahr−/−) mice (strain B6.129-Ahrtm1Bra) were bred and maintained in the Research Institute of the McGill University Health Centre (RI MUHC) as previously described (Rico De Souza et al., 2021). This strain carries a targeted deletion of exon 2 of the Ahr gene and was backcrossed for 12 generations onto C57BL/6. As Ahr+/+ or Ahr+/− mice do not exhibit any phenotypic difference in the ability to be activated by AhR ligands (Thatcher et al., 2007; De Souza et al., 2014). The Ahr+/− mice were used as littermate controls in this study. Mice were maintained on an ad libitum diet with free access to food and water and subjected to a 12-h light cycle. Male and female mice were used in experiments unless otherwise indicated. All animal procedures were approved by the McGill University Animal Care Committee (2,010–5,933), were carried out in accordance with the guidelines of the Canadian Council on Animal Care and followed the ARRIVE guidelines for the design, analysis, and reporting of research with animals (Kilkenny et al., 2010).
Chlorine exposure was performed as previously described (Allard et al., 2019). Briefly, 8–12-week-old mice were exposed to Cl2 for 5min using a nose-only exposure device at a concentration of 100ppm. Chlorine was mixed with room air using a standardized calibrator (VICIMetronics, Dynacalibrator, Model230-28A). The AhR ligand FICZ was dissolved in DMSO and administered intraperitoneally (i.p.; 1μg per mouse). A single injection of FICZ or DMSO was given to mice on day 0 and 1h before the Cl2 exposure. Mice were sacrificed 24h after the Cl2 exposure.
Mice were sensitized by i.p injection of 1mg/ml chicken OVA solution mixed with Imject alum adjuvant (1:4 dilution in PBS; Thermofisher Scientific). The control group received 200μl PBS mixed with of Imject alum alone. After 14days, sensitized mice were challenged on days 14, 15, and 16 by intranasal administration of 10μg of OVA diluted in 30μl of PBS. The PBS group received 30μl of PBS. Animals were sacrificed on day 18.
Mice were sedated with an i.p injection of xylazine (8mg/kg) and anaesthetized with i.p. injection of sodium pentobarbital (30mg/kg). Next, the mouse was tracheostomized using at 18-gauge cannula and connected to the flexiVent. Muscle paralysis was induced with rocuronium pentabromide (2mg/kg). Finally, the mouse was mechanically ventilated using the following settings [tidal volume of 10ml/kg, maximum inflation pressure of 30cm H20, a positive end expiratory pressure (PEEP) of 3cm H20 and a frequency of 150/min]. Following a standardized deep inflation, two lung inflations to a transrespiratory pressure of 25cm H2O were performed and baseline measurements were recorded in six replicates. Respiratory mechanics were estimated using a single compartment model and commercial software (Scireq). These included a low-frequency range of oscillations (1–20.5Hz) used to calculate Newtonian resistance (resistancen), an estimate of central airways resistance, and a single sinusoidal waveform (2.5Hz) used to calculate total respiratory system elastance and resistance. These procedures were also performed directly after inhalation of increasing concentrations of aerosolized methacholine (5–50mg/ml; Ano et al., 2017).
Lungs were excised and PBS (0.5ml) was injected twice to lavage the lungs. The bronchoalveolar lavage (BAL) was centrifuged at 3,000rpm for 5min and the supernatant was separated from the cells. The cells were resuspended in PBS, counted and cytospin slides (CytoSpin, Thermofisher Scientific) were stained with HEMA 3 STAT PACK (Fisher Scientific).
Interleukin-4, IL-5, and IL-13 were quantified in BAL fluid collected as described above using a cytokine multiplex analysis (Milliplex MAP, Millipore) according to the manufacturer’s instructions and were read on a Luminex 100 System.
Lungs were collected, minced, and digested using collagenase IV at a concentration of 150units per ml at 37° C for 1h in RPMI medium containing 10% FBS. Single cell populations were then obtained by gently rubbing lung tissue over nylon mesh with 70μm pores. The nylon mesh was washed twice with medium and the lung homogenates were centrifuged at 1,500rpm for 5min at 4° C. Red blood cells were lysed by adding 2ml of ACK lysing buffer (Thermofisher Scientific) for 2min at room temperature. Cells were resuspended in 0.5ml of medium. Bronchoalveolar lavage cells were centrifuged and re-suspended. Lysis of red blood cells in BAL fluid was performed, when necessary, using ACK lysing buffer. Concentrations of all live cell suspensions were determined by trypan blue exclusion. Aliquots of 105 for the BAL cells or 106 for the lung cells were seeded in round-bottom 96-well plates. Fc receptors were then blocked with FC block anti-CD16/32 antibody (BD Biosciences) for 20min at 4°C. Cells were then labeled with viability dye (eFluor 780), anti-CD45 (Indo-1 violet) anti-Ly6G (Alexa fluor 700), anti-CD11c (APC), anti-SiglecF (PE), anti-CD3 (FITC), anti-CD4 (Pacific blue), and CD8 (PerCP.Cy5.5; BD Biosciences). Cells were analyzed immediately by flow cytometry. Cells were acquired using a BD FACSCanto flow cytometer (BD Biosciences) and data were analyzed by Flowjo software. Fluorescence minus one (FMO) was used to set the gates for the flow cytometry. Eosinophils were determined by first excluding the neutrophils (Ly6Ghi). Eosinophils were identified as Ly6Glow CD11c−/low Siglec-Fmed/high as previously described (Abdala Valencia et al., 2016).
Results are reported as means±SEM. Statistical differences between group-mean values were determined using Prism 6 (GraphPad software) by two-way ANOVA followed by the Tukey’s multiple comparisons test. A value of p<0.05 was considered statistically significant.
As OVA is a well-characterized allergic asthma model (Gueders et al., 2009; Pareek et al., 2019), we utilized this model to test the importance of AhR expression on the suppression of this asthma phenotype. Here, evaluation of BAL cells in mice sensitized and challenged with OVA revealed that OVA significantly increased the number of total cells compared with the PBS group in both Ahr−/− and Ahr+/− mice (Figures 1A,B). Total BAL cells were significantly higher in OVA-exposed Ahr−/− mice compared with OVA-exposed Ahr+/− mice (Figure 1B). While there was no difference in macrophage numbers (Figure 1C), there were significantly more eosinophils and lymphocytes in the airways of mice sensitized and challenged with OVA in both Ahr−/− and Ahr+/− mice compared with PBS control mice (Figures 1D,E). Reflecting the increase in total cell numbers in Ahr−/− mice, there were also significantly more eosinophils and lymphocytes in the Ahr−/− mice compared with Ahr+/− mice; the percentages of eosinophils and lymphocytes were also significantly higher (Figure 2). Neutrophils were not detected. Thus, these data recapitulate that the AhR suppresses eosinophilic airway inflammation in an allergic model.
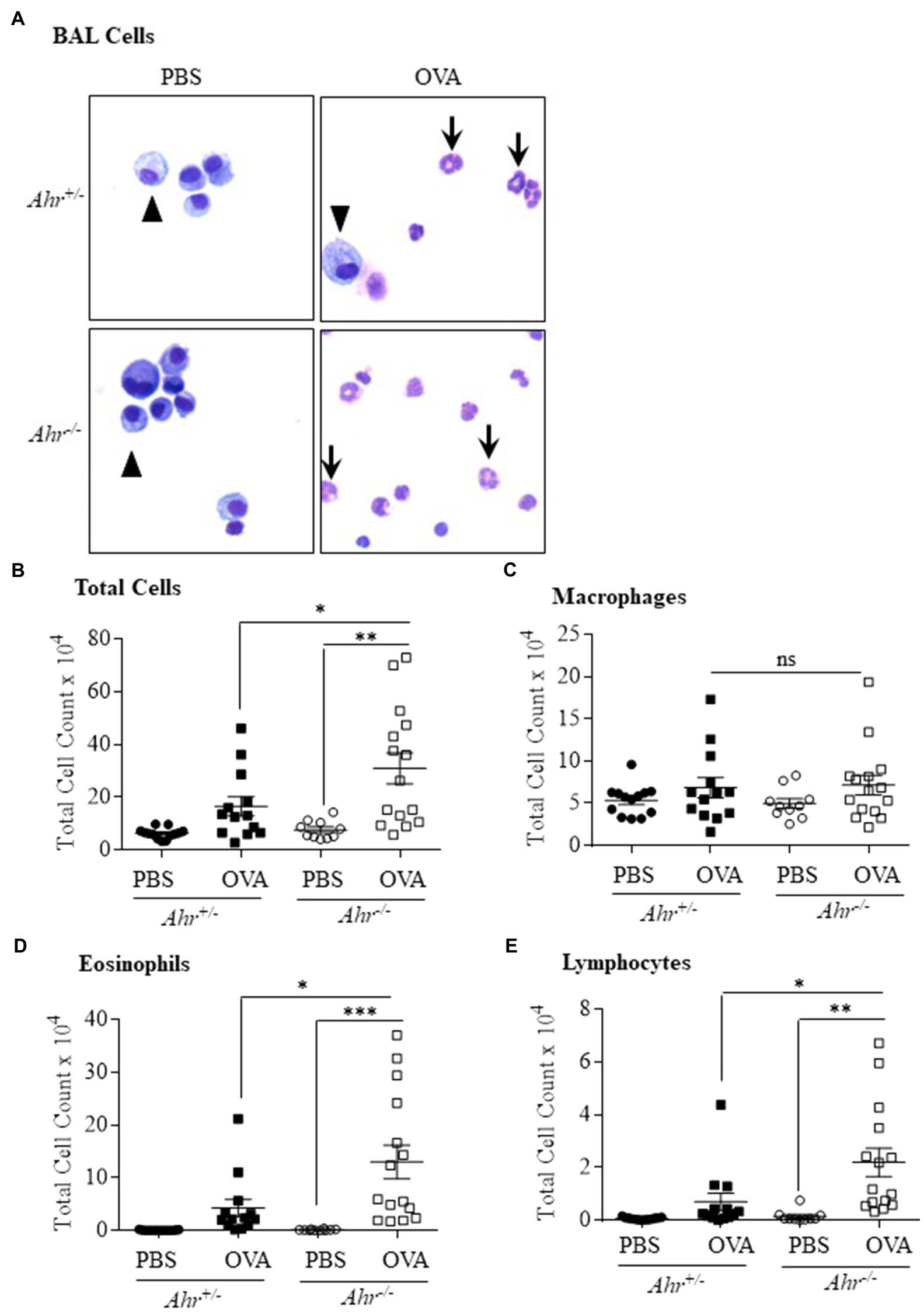
Figure 1. Aryl hydrocarbon receptor (AhR) reduces ovalbumin (OVA)-induced airway inflammation. (A) Bronchoalveolar lavage (BAL) cells – presence of macrophages (arrowheads) in the BAL as the predominant cell type in PBS-exposed mice. There were more eosinophils (arrows) in the OVA-exposed Ahr−/− as well as Ahr+/− mice. (B) Total Cells – there was a significant increase in total cells in Ahr−/− mice exposed to OVA (**p =0.001 OVA compared with PBS; *p =0.0451 OVA-exposed Ahr−/− mice vs. OVA-exposed Ahr+/− mice). (C) Macrophages – there were no significant differences in macrophages numbers between the Ahr−/− and Ahr+/− exposed to OVA. (D) Eosinophils – there was a significant increase in eosinophils in OVA-exposed Ahr−/− mice compared with both PBS control (***p =0.0005) as well as OVA-exposed Ahr+/− mice (*p =0.0148). (E) Lymphocytes – the number of lymphocytes in OVA-exposed Ahr−/− mice was significantly higher than in OVA-exposed Ahr+/− mice compared with PBS control mice (**p =0.0016) as well as OVA-exposed Ahr+/− mice (*p =0.018). Results are expressed as the mean±SEM; values for individual mice from two independent experiments are shown.
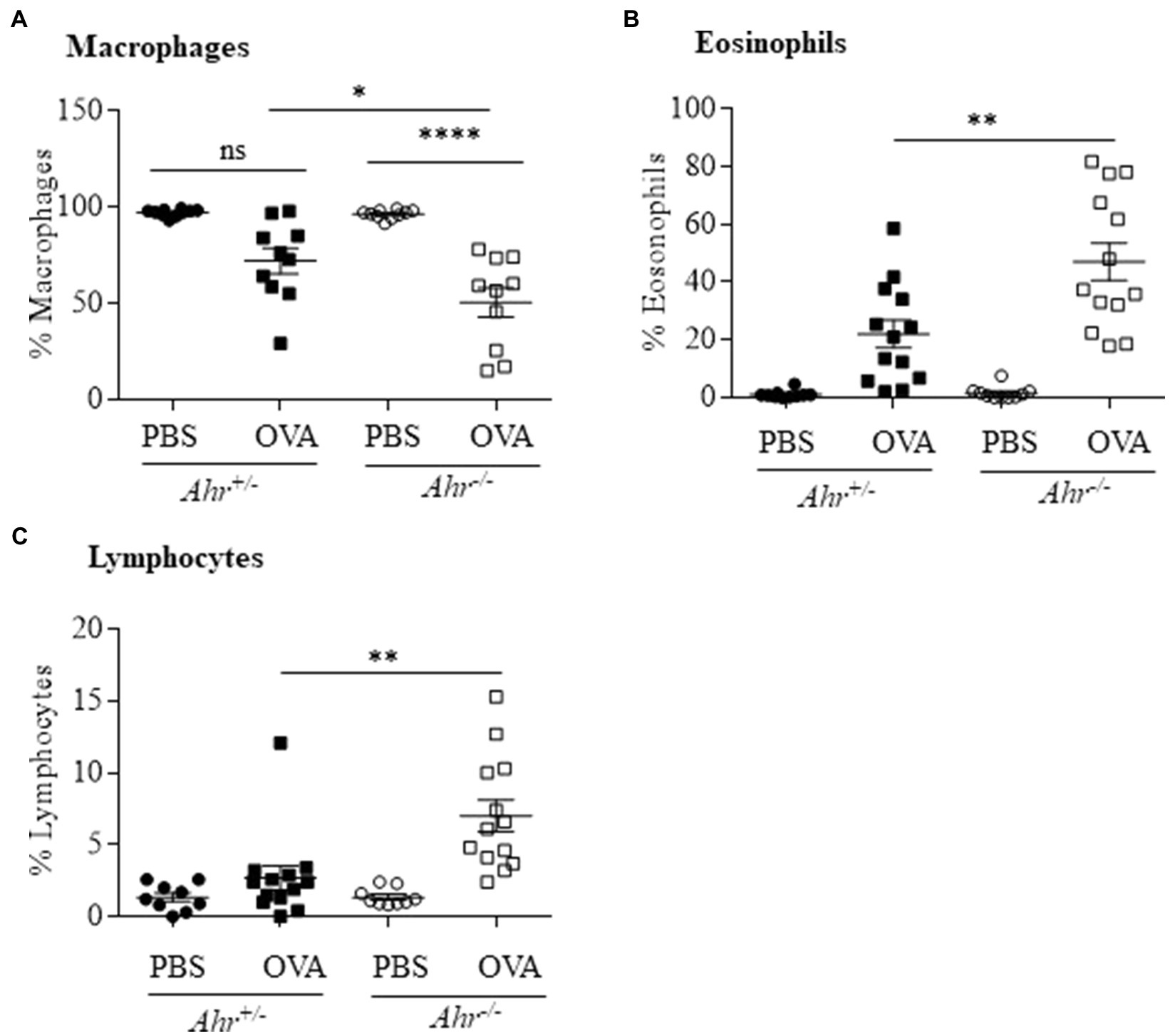
Figure 2. Percentage of immune cells in OVA-induced airway inflammation. (A) Macrophages – there was a significant difference in the percentage of macrophages between OVA-challenged Ahr−/− and Ahr+/− mice (*p =0.0232; ****p =0.0001 between PBS and OVA-challenged Ahr−/− mice). (B) Eosinophils – there was a significantly higher percentage of eosinophils in OVA-exposed Ahr−/− mice compared with OVA-exposed Ahr+/− mice (**p =0.0011). (C) Lymphocytes – there was a significantly higher percentage of lymphocytes in OVA-exposed Ahr−/− mice compared with OVA-exposed Ahr+/− mice (**p =0.0016). Results are expressed as the mean±SEM from two independent experiments.
Our finding that the AhR reduces allergen-induced eosinophil influx into the airways led us to speculate whether this suppression also occurred in the lung parenchyma. To more comprehensively profile the eosinophil phenotype, lung cells from OVA-challenged mice were isolated 48h post challenge, and mature (SiglecFint CD11c−) and activated (SiglecFhi CD11clo) eosinophils were identified by flow cytometry. The gating strategy used to quantify mature vs. activated eosinophils is presented in Figure 3A (Abdala Valencia et al., 2016). There was a significant increase in total eosinophils only in the lung tissue of the OVA-exposed Ahr−/− mice compared with PBS controls (Figure 3B) but no change in total eosinophils was found in OVA-exposed Ahr+/− mice. There was also a significant increase in both mature (Figure 3C) and activated (Figure 3D) eosinophils in OVA-exposed Ahr−/− mice compared with PBS-exposed Ahr−/− mice. Overall, these new data suggest that Ahr−/− mice challenged with OVA recruit more eosinophils into the lung, which subsequently upregulate CD11c, after which they migrate into the airways. This enhanced response does not occur in Ahr+/− mice.
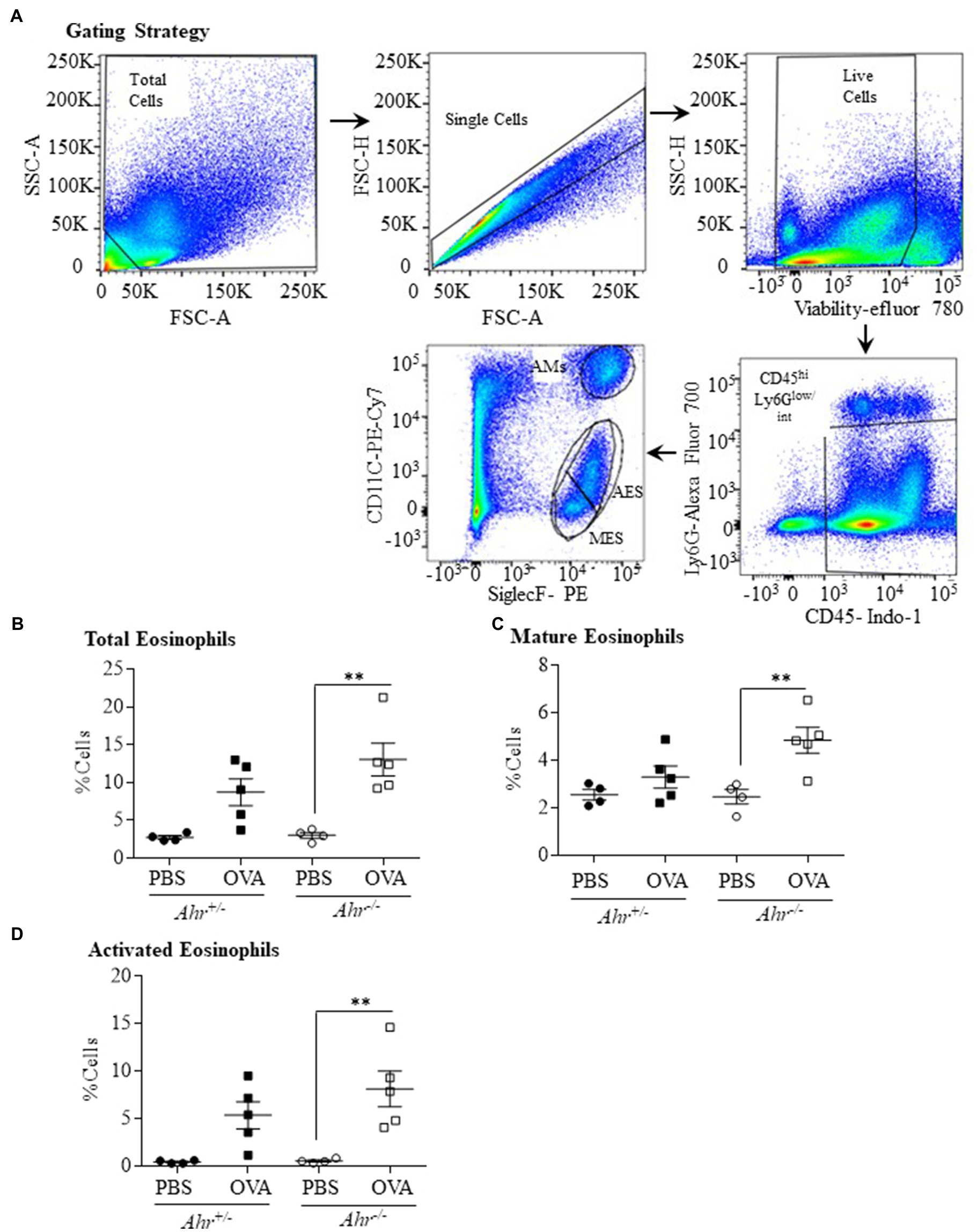
Figure 3. Aryl hydrocarbon receptor expression decreases the percentage of eosinophils in the lung parenchyma after exposure to OVA. (A) Gating Strategy – the gating strategy utilized for flow cytometry to quantify mature vs. activated eosinophils in lungs tissue is shown. The percentage of total (B), mature (C), and activated (D) eosinophils in lung tissue was significantly increased in Ahr−/− mice exposed to OVA compared with PBS control mice (**p =0.0028; 0.0088, and 0.0065, respectively). Results are expressed as the mean±SEM; values for individual mice are shown.
Because, we observed that the AhR reduces eosinophil recruitment into the lungs, we sought to determine whether the AhR regulates the secretion of these Th2 cytokines in OVA-challenged mice. Using a multiplex assay to quantify levels of IL-4, IL-5, IL-13 in the BAL fluid, we found that there was a significant increase in IL-4 (Figure 4A) and IL-5 (Figure 4B) only in Ahr−/− mice after OVA challenge. IL-4 was also significantly higher in OVA-exposed Ahr−/− mice compared with the Ahr+/− mice (Figure 4A). Interestingly, there was no significant change in IL-13 in any of the groups (Figure 4C).
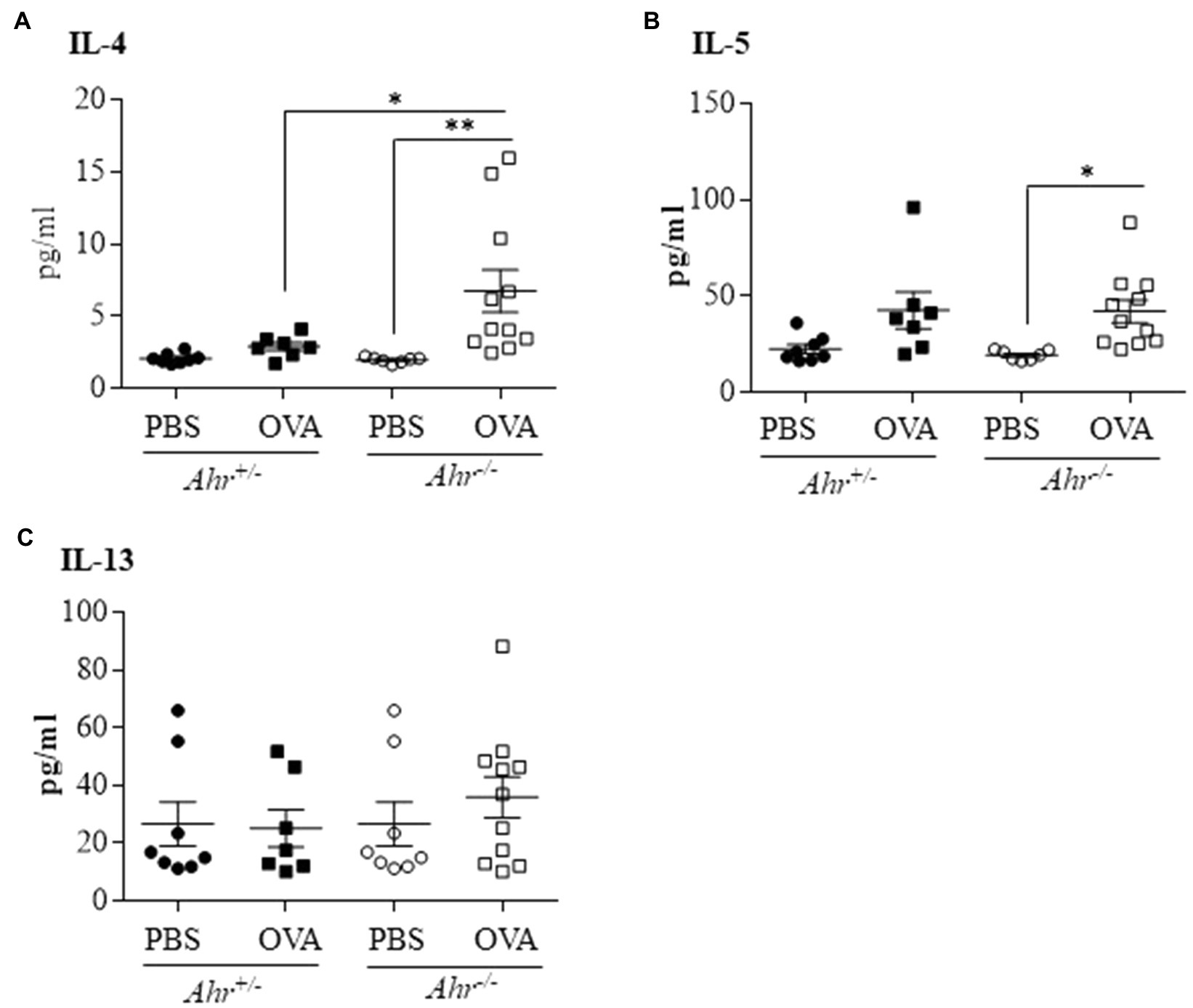
Figure 4. Ahr−/− mice exhibit increased levels of IL-4 and IL-5 following OVA challenge. Protein levels of interleukin-4 (IL-4), IL-5, and IL-13 were measured in the BAL fluid by multiplex assay. (A) IL-4 – there was a significant increase of IL-4 in the Ahr−/− mice exposed to OVA compared with the Ahr−/− mice exposed to PBS (**p =0.0095) and to the Ahr+/− mice exposed to OVA (*p =0.0466). (B) IL-5 – there was a significant increase of IL-5 only in the Ahr−/− mice exposed to OVA (*p =0.0396). (C) IL-13 – IL-13 was not significantly increased between any of the groups. Results are expressed as the mean±SEM; values for individual mice are shown.
Next, we investigated whether the AhR regulates airway hyperresponsiveness in OVA challenged mice using a flexiVent to measure airway resistance upon exposure with increasing concentrations of aerosolized methacholine. Consistent with the lack of change in levels of IL-13, there was no significant difference in resistance and elastance between OVA-exposed Ahr−/− and Ahr+/− mice (Figures 5A,B). Thus, although the AhR controls immune cell infiltration to the lungs in the OVA asthma model, the AhR exerts minimal influence on airway function.
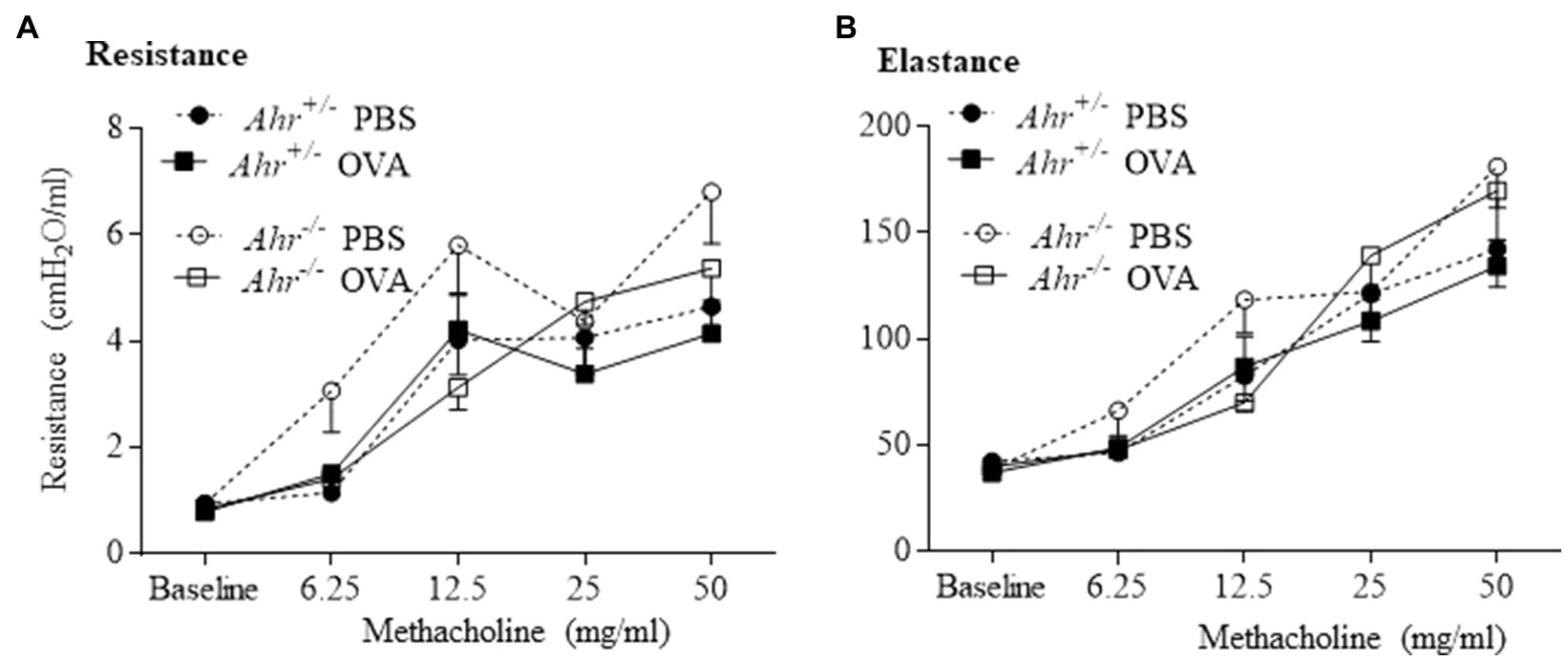
Figure 5. Aryl hydrocarbon receptor does not influence lung function in the allergic asthma model. Lung mechanics were evaluated by flexiVent. There was no significant difference between OVA-immunized Ahr−/− and Ahr+/− mice in any of the parameters evaluated including resistance (A) and elastance (B). Results are expressed as the mean±SEM.
We next utilized a model of irritant-induced asthma that provokes a neutrophilic response in the lungs and airways to evaluate whether the AhR can also suppress neutrophilia in response to diverse etiologic agents. For these experiments, we utilized Cl2 as a representative trigger of the irritant-induced asthma phenotype. Here, airway inflammation was observed in both Ahr+/− and Ahr−/− mice after Cl2 exposure, where there was a significant increase in the number of total cells in the BAL in Ahr+/− and Ahr−/− mice exposed to Cl2 compared with air-only controls (Figure 6A). In addition, the level of inflammatory cell infiltration was also significantly increased in mice exposed to Cl2 (Figure 6B). Chlorine also caused a significant increase in the number of epithelial cells in the BAL of Ahr+/− mice; there was a trend toward an increase in Ahr−/− mice although this did not reach statistical significance (Figure 6C). Recruitment of inflammatory cells to the lungs in response to Cl2 was also significantly increased compared with air-exposed mice and was dominated by macrophages (Figure 6D) and neutrophils (Figure 6E). There was also a significant increase in the number of eosinophils with Cl2 exposure only in Ahr−/− mice (Figure 6F). However, there was no significant difference in any of these cell types between Cl2-exposed Ahr+/− and Ahr−/− mice.
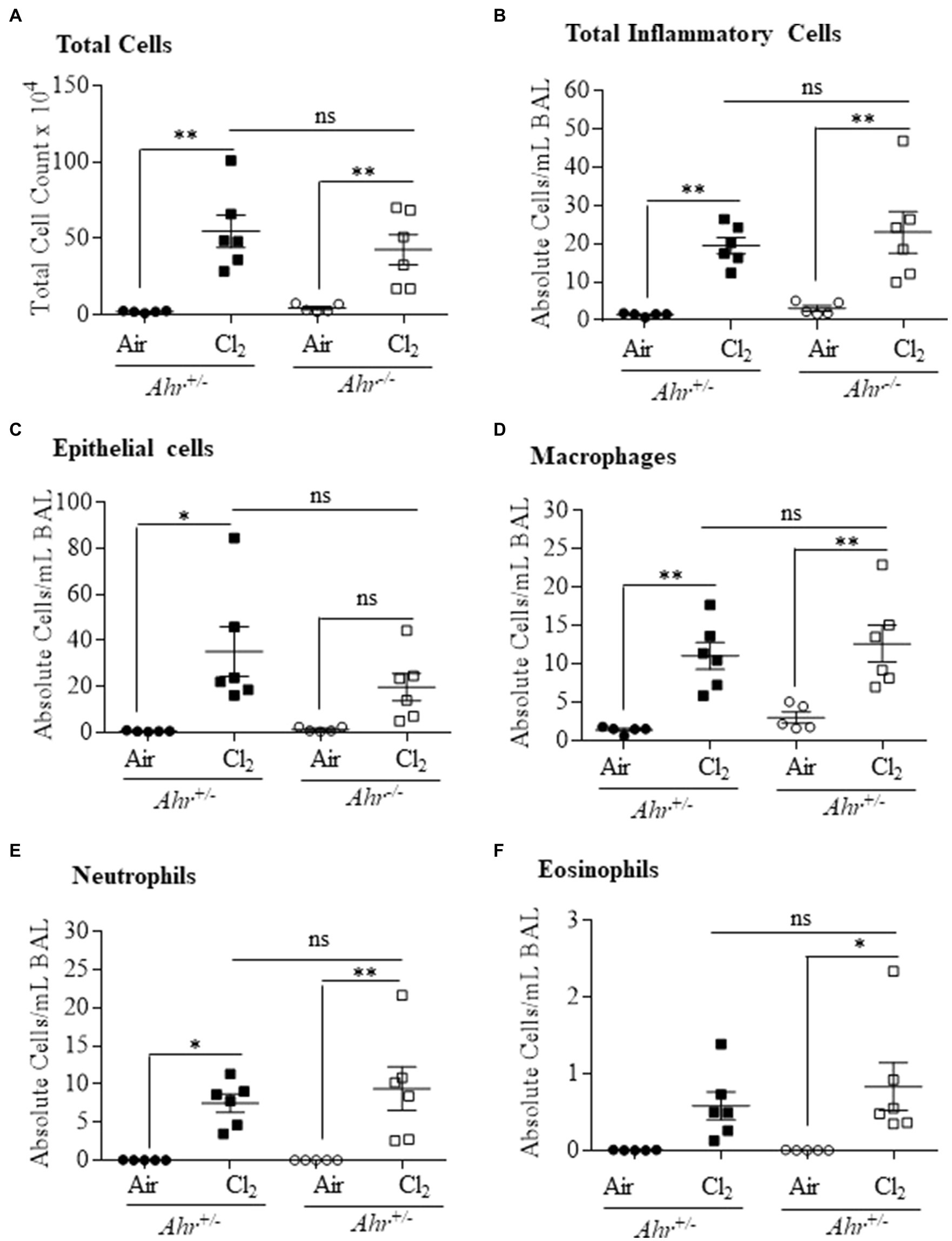
Figure 6. Aryl hydrocarbon receptor does not control chlorine (Cl2)-induced airway inflammation. Total and differential cell counts of the BAL of Ahr+/− and Ahr−/− was evaluated 24h after exposure to Cl2. There was a significant increase in total cells (A), total inflammatory cells (*p =0.0023; B), epithelial cells (*p =0.06001; C), macrophages **p =0.0039 (D) and neutrophils (*p =0.0297; **p =0.0055; E) and eosinophils (*p =0.0402; F) in mice exposed to Cl2 compared with the control mice. There was no significant difference (ns) between the Ahr+/− and Ahr−/− mice. Results are expressed as the mean±SEM; values for individual female mice are shown.
Next, we evaluated airway hyperresponsiveness after exposure to Cl2. These data revealed the general regulation of lung function by the AhR in this model. First, respiratory resistance, which reflects airway hyperresponsiveness to inhaled aerosolized methacholine, was greater in Cl2 exposed mice (Figure 7A). In the absence of AhR, resistance was significantly lower compared with Ahr+/− mice (Figure 7A). We also evaluated other parameters of respiratory mechanics such as elastance (Figure 7B). These data suggest that the AhR actually promotes airway hyperresponsiveness after Cl2 exposure.
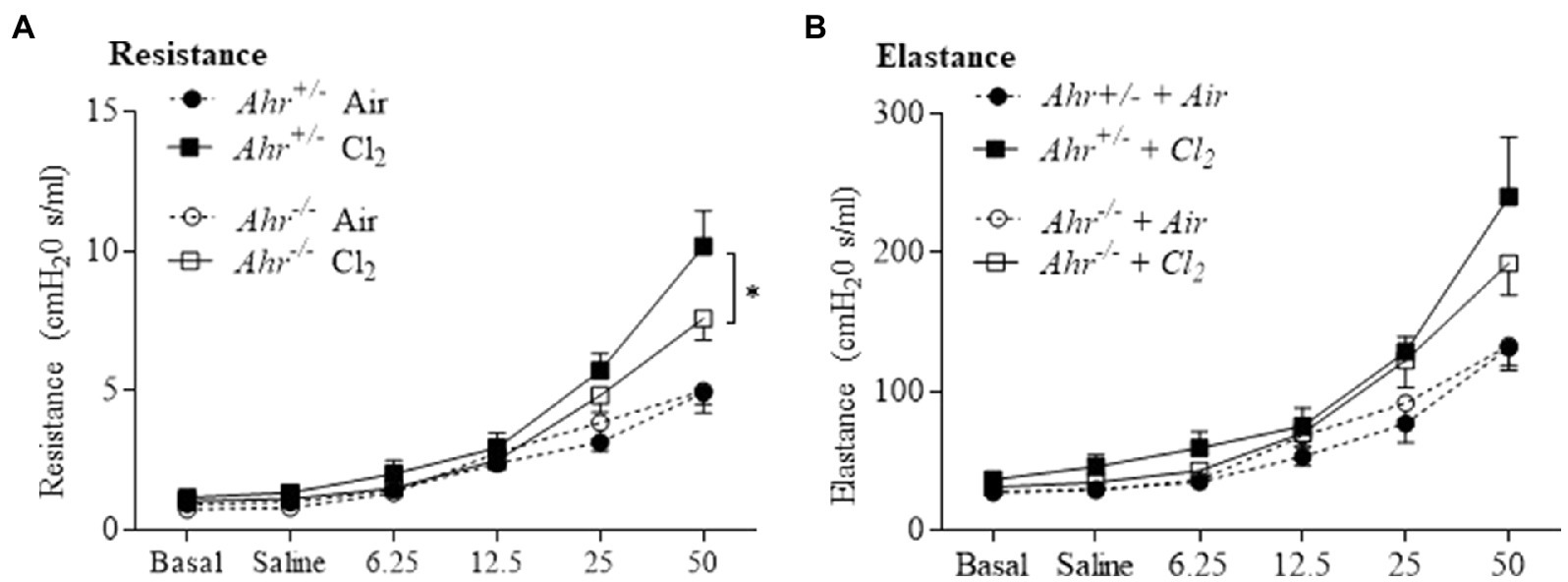
Figure 7. Aryl hydrocarbon receptor increases airway hyperresponsiveness after Cl2 exposure. Lung mechanics parameters included assessment of resistance (A) and elastance (B) by flexiVent in response to methacholine in Ahr+/− and Ahr−/− at 24h after exposure to air or Cl2. Cl2 increased resistance compared with the control. Respiratory resistance was significantly lower in Ahr−/− mice compared with Ahr+/− mice at the highest dose of methacholine (*p =0.0358). Results are expressed as the mean±SEM.
6-Formylindoleo [3,2-b] carbazole is an endogenous AhR ligand and derivative of tryptophan that is produced in the skin after ultraviolet exposure (Wei et al., 1998; Wincent et al., 2009). Our data show that FICZ attenuates acute lung neutrophilia caused by cigarette smoke (Rico De Souza et al., 2021). Therefore, we next tested whether activation of the AhR by FICZ would protect against lung inflammation in response to Cl2. Using Ahr+/− mice, these data show that there was a significant increase in the total BAL cells from Cl2 exposure (Figures 8A,B). There was no increase in macrophages (Figure 8C). There was a significant increase in both neutrophils (Figure 8D) and epithelial cells (Figure 8E) in response to Cl2. However, FICZ did not significantly change the levels of cells in the BAL in response to Cl2, suggesting that FICZ does not alter the inflammatory response to Cl2.
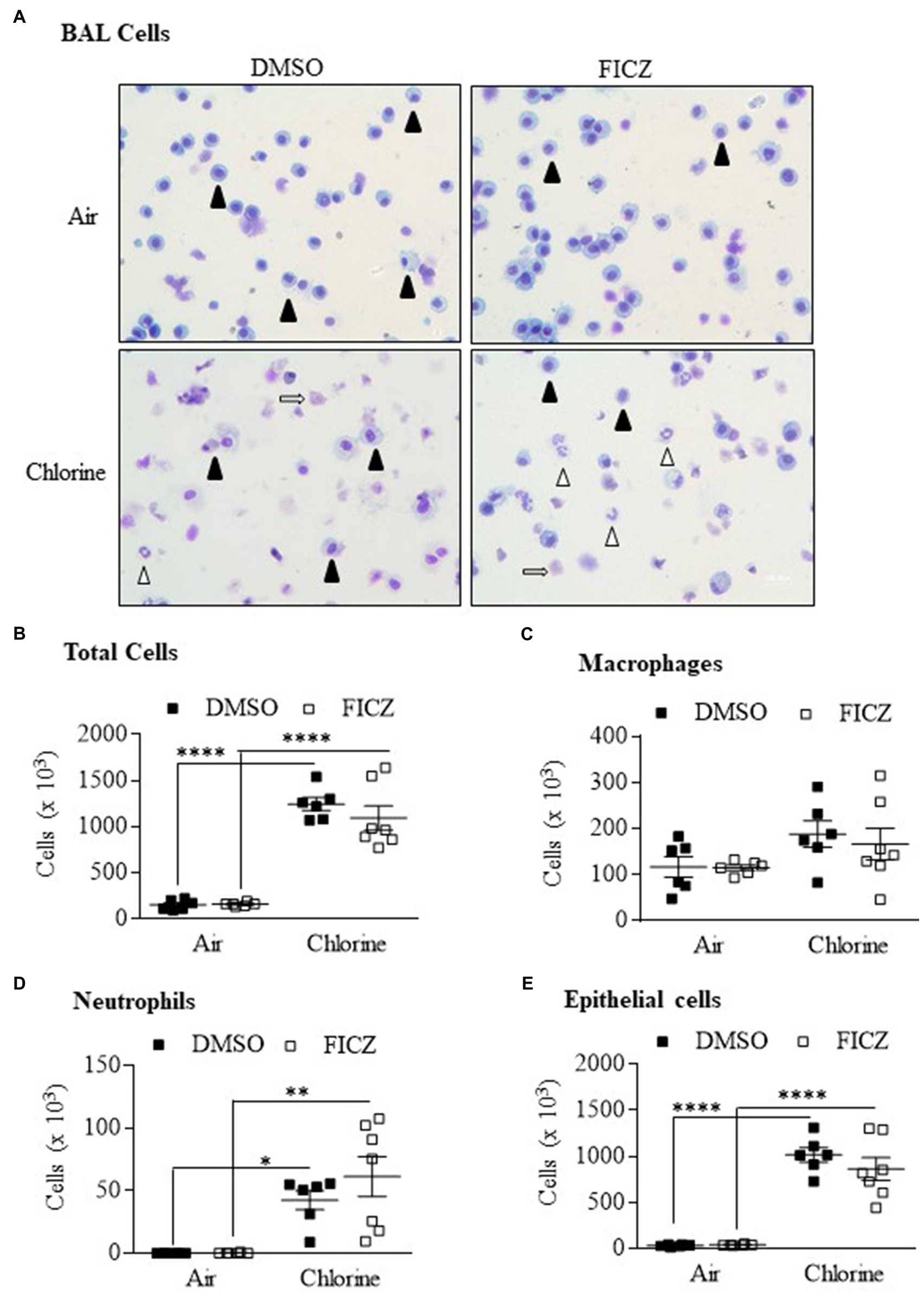
Figure 8. 6-Formylindoleo [3,2-b] carbazole (FICZ) does not attenuate Cl2-induced airway inflammation. (A) BAL cells – there was an increase in neutrophils (open arrowheads) and epithelial cells (open arrows) 24h after exposure to Cl2. Macrophages are indicated as closed arrowheads. (B) Total Cells – there was a significant increase in total cells in mice exposed to Cl2 (****p =0.0001). FICZ had no effect on the total number of cells. (C) Macrophages – FICZ did not change macrophages in response to Cl2. (D) Neutrophils – there was a significant increase in neutrophils in response to Cl2 (*p =0.0313 and **p =0.001 in DMSO and FICZ treated mice, respectively). (E) Epithelial cells – there was a significant increase in BAL epithelial cells in mice treated with DMSO or FICZ and exposed to Cl2 (****p =0.0001). There was no significant difference between FICZ and DMSO-treated mice exposed to Cl2. Results are expressed as the mean±SEM; values for individual male mice are shown.
Asthma is a complex disease triggered by environmental factors that lead to dysregulation of the immune system. Herein, we report the novel findings that there is differential regulation of the asthma phenotype between OVA and Cl2. Our novel results show that expression of the AhR had minimal effect on Cl2-induced neutrophilia, but attenuated OVA-induced pulmonary inflammation that is typified by eosinophilia. This latter finding is consistent with previous publications showing that AhR-deficient mice developed allergic asthma in preclinical OVA and cockroach allergen models (Xu et al., 2015; Thatcher et al., 2016; Chang et al., 2020). These confirmatory data strongly support the general regulation of the allergic asthma phenotype by the AhR. However, one of the unresolved questions in AhR biology is why there can be similar responses in ligand-treated mice and AhR deficient mice. In the context of asthma, AhR activation by TCDD or benzo[a]pyrene (B[a]P) can be pro-inflammatory and induce MUC5AC expression, leading to mucus hypersecretion, airway remodeling, dysregulation of antigen-presenting cells, and exacerbation of asthma (Wong et al., 2018; Poulain-Godefroy et al., 2020). Yet, our data herein confirm that AhR expression also protects against the development of allergic asthma. Although, we did not measure AhR levels, we posit that this discrepancy may be related to changes in AhR expression after its activation. Because a key event in AhR-mediated signaling is prolonged AhR degradation following ligand binding (Prokipcak and Okey, 1991; Pollenz, 2007), it could be that decreased AhR expression due to persistent activation by TCDD or B[a]P is sufficient to mimic an Ahr−/− phenotype, thereby yielding similar results. This is supported by the observation that both Ahr−/− mice and TCDD-exposed mice have impaired hippocampal neurogenesis (Latchney et al., 2013). This may also be why both Ahr−/− cells as well as cells exposed to AhR ligands exhibit decreased proliferation (Elizondo et al., 2000; Faust et al., 2013; Hecht et al., 2014). Thus, overall AhR expression levels in the lungs may be an important determinant of its ability to mitigate damage associated with environmental exposures, and further highlight the importance of the AhR in the maintenance of lung health in response to diverse environmental exposures (Guerrina et al., 2018).
As previous work has shown that Ahr−/− mice have enhanced airway inflammation and hyperresponsiveness in response to OVA (Chang et al., 2020), our results are comparable to previous publications in the allergic asthma models in that there is suppression of airway inflammation by the AhR, including reduced infiltration of eosinophils and lymphocytes (Chang et al., 2020). There are, however, differences in the response between these studies and ours. For example, Chang et al. (2020) reported that there was an increase in macrophages and neutrophils in the OVA model which was not observed in our study. We also did not see an increase in IL-13 as reported by others (Chang et al., 2020). Finally, our results also differ from that of Chang et al. (2020) in that Ahr−/− mice in their study exhibited an enhancement of airway hyperresponsiveness after OVA immunization. These variations in results could be due to differences in the protocol in OVA administration and/or duration between studies. However, despite these differences, our results further reaffirm the importance of the AhR in suppressing inflammation associated with the allergic asthma phenotype.
Although, the AhR mitigates allergic asthma, whether the AhR affects the pathogenesis of other types of asthma, such as irritant-induced asthma, was not known. Surprisingly, the AhR did not regulate inflammation, including neutrophilia in response to Cl2. Based on the lack of difference in immune cell infiltration due to AhR expression, we did not measure cytokine production in this model. However, Cl2 is a gas that causes oxidative stress and airway dysfunction following inhalation, the effects of which can be ameliorated by administration of antioxidants (Ano et al., 2017). It is known that AhR-deficient mice are more susceptible to hyperoxic lung injury due to decreased expression of antioxidant enzymes such as cytochrome P4501A, NAD(P)H quinone reductase-1 (NQO1) and microsomal glutathione S-transferase (GST; Zhang et al., 2015). Despite not having an effect on the inflammatory response, the AhR did aggravate Cl2-induced airway hyperresponsiveness. Persistent airway hyperresponsiveness is caused by inflammatory and structural changes in the airways (Gabehart et al., 2013). Our data suggest that changes in the inflammatory response are unlikely to be how the AhR aggravates the airway reactivity after Cl2 exposure. Other possibilities to explain these results include that the AhR may have direct effects in tissues that influence airway hyperresponsiveness such as the epithelium or smooth muscle.
As Th2-mediated responses that underlie airway eosinophilia and airway hyperresponsiveness have been linked to IL-4, IL-5, and IL-13, we measured the levels of these cytokines in the BAL of OVA-exposed mice. Of these, IL-4 and IL-5 were significantly increased in Ahr−/− mice, a finding that may explain the increased lung eosinophil influx (Kips et al., 2001; Maes et al., 2012). These results also supported an important role for the AhR in suppressing lung inflammation and are consistent with previous studies demonstrating an anti-inflammatory role for the AhR in asthma models, including studies that also utilized cockroach allergen challenge (Xu et al., 2015; Thatcher et al., 2016). Our results are therefore consistent with the hypothesis that the AhR serves as an important negative regulator of inflammation in the lungs. However, one of the limitations of our study is the focus on an acute model of chlorine and OVA exposure, as this does not allow for the development of structural changes and airway remodeling, pathological features of asthma that contribute to the clinical manifestations of the disease. Other limitations of the OVA model include that the pattern and distribution of lung inflammation in the lower airway of mice differs from humans due to differences in lung branching (Kim et al., 2019). It also needs to be noted that there are differences between these asthma models in terms of the number and duration of the exposure (e.g., three nasal OVA challenges vs. a single chlorine) that may influence interpretation of these data. The adaptation of mice to repeated chlorine exposures prevents the application of identical exposure protocols (Allard et al., 2019). However, despite these limitations, these exposure regimes allowed us to compare the role of the AhR using two models of exposure that induce different asthma phenotypes.
Thus, we show that AhR differentially affects the development asthma-like disease, with the majority of AhR-dependent effects involving the suppression of inflammation associated with the allergic phenotype. In conjunction with our previous work establishing the AhR attenuates tobacco smoke-induced inflammation (Rogers et al., 2017; Rico De Souza et al., 2021), these findings position the AhR as a homeostatic modulator of pulmonary inflammation in response to diverse etiologic agents. A better understanding of the connection between the AhR and its role in pulmonary inflammation may aid the development of therapeutic agents to combat specific inflammatory lung diseases.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All animal procedures were approved by the McGill University Animal Care Committee and were carried out in accordance with the Canadian Council on Animal Care (Protocol Number: 5933).
HT, MS, AR, and BA: data curation and/or analysis. CB: funding acquisition. HT, AR, BA, VM, and JM: methodology. HT and CB: project administration. CB and EF: supervision. HT, CB, DE, EF, VM, ZH, and JM: intellectual contributions. HT, ZH, CB, DE, JM, and EF: manuscript writing, review, and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the Canada Foundation for Innovation (CFI), the Canadian Institutes for Health Research Project Grants (168836 and 162273), and the Natural Sciences and Engineering Research Council of Canada (NSERC). CB was supported by a salary award from the Fonds de recherche du Quebec-Sante (FRQ-S). HT was supported by a Réseau de recherche en santé réspiratoire du Québec (RSR) Scholarship and a Meakins-Christie Laboratories Collaborative Research Award.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdala Valencia, H., Loffredo, L. F., Misharin, A. V., and Berdnikovs, S. (2016). Phenotypic plasticity and targeting of Siglec-F(high) CD11c(low) eosinophils to the airway in a murine model of asthma. Allergy 71, 267–271. doi: 10.1111/all.12776
Allard, B., Panariti, A., Pernet, E., Downey, J., Ano, S., Dembele, M., et al. (2019). Tolerogenic signaling of alveolar macrophages induces lung adaptation to oxidative injury. J. Allergy Clin. Immunol. 144, 945–961.e949. doi: 10.1016/j.jaci.2019.07.015
Ano, S., Panariti, A., Allard, B., O'sullivan, M., Mcgovern, T. K., Hamamoto, Y., et al. (2017). Inflammation and airway hyperresponsiveness after chlorine exposure are prolonged by Nrf2 deficiency in mice. Free Radic. Biol. Med. 102, 1–15. doi: 10.1016/j.freeradbiomed.2016.11.017
Asher, M. I., Garcia-Marcos, L., Pearce, N. E., and Strachan, D. P. (2020). Trends in worldwide asthma prevalence. Eur. Respir. J. 56:2002094. doi: 10.1183/13993003.02094-2020
Barnes, P. J. (2018). Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 18, 454–466. doi: 10.1038/s41577-018-0006-6
Boonpiyathad, T., Sozener, Z. C., Satitsuksanoa, P., and Akdis, C. A. (2019). Immunologic mechanisms in asthma. Semin. Immunol. 46:101333. doi: 10.1016/j.smim.2019.101333
Braman, S. S. (2006). The global burden of asthma. Chest 130, 4S–12S. doi: 10.1378/chest.130.1_suppl.4S
Chang, Y. D., Li, C. H., Tsai, C. H., Cheng, Y. W., Kang, J. J., and Lee, C. C. (2020). Aryl hydrocarbon receptor deficiency enhanced airway inflammation and remodeling in a murine chronic asthma model. FASEB J. 34, 15300–15313. doi: 10.1096/fj.202001529R
Chiba, Y., Nakazawa, S., Todoroki, M., Shinozaki, K., Sakai, H., and Misawa, M. (2009). Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am. J. Respir. Cell Mol. Biol. 40, 159–167. doi: 10.1165/rcmb.2008-0162OC
De Genaro, I. S., De Almeida, F. M., Hizume-Kunzler, D. C., Moriya, H. T., Silva, R. A., Cruz, J. C. G., et al. (2018). Low dose of chlorine exposure exacerbates nasal and pulmonary allergic inflammation in mice. Sci. Rep. 8:12636. doi: 10.1038/s41598-018-36667-8
De Souza, A. R., Zago, M., Eidelman, D. H., Hamid, Q., and Baglole, C. J. (2014). Aryl hydrocarbon receptor (AhR) attenuation of subchronic cigarette smoke-induced pulmonary neutrophilia is associated with retention of nuclear RelB and suppression of intercellular adhesion molecule-1 (ICAM-1). Toxicol. Sci. 140, 204–223. doi: 10.1093/toxsci/kfu068
Elizondo, G., Fernandez-Salguero, P., Sheikh, M. S., Kim, G. Y., Fornace, A. J., Lee, K. S., et al. (2000). Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol. 57, 1056–1063.
Faust, D., Kletting, S., Ueberham, E., and Dietrich, C. (2013). Aryl hydrocarbon receptor-dependent cell cycle arrest in isolated mouse oval cells. Toxicol. Lett. 223, 73–80. doi: 10.1016/j.toxlet.2013.08.022
Ferrari, M., Schenk, K., Mantovani, W., Papadopoulou, C., Posenato, C., Ferrari, P., et al. (2011). Attendance at chlorinated indoor pools and risk of asthma in adult recreational swimmers. J. Sci. Med. Sport 14, 184–189. doi: 10.1016/j.jsams.2010.12.006
Fisk, M. Z., Steigerwald, M. D., Smoliga, J. M., and Rundell, K. W. (2010). Asthma in swimmers: a review of the current literature. Phys. Sportsmed. 38, 28–34. doi: 10.3810/psm.2010.12.1822
Gabehart, K. E., Royce, S. G., Maselli, D. J., Miyasato, S. K., Davis, E. C., Tang, M. L., et al. (2013). Airway hyperresponsiveness is associated with airway remodeling but not inflammation in aging Cav1−/− mice. Respir. Res. 14:110. doi: 10.1186/1465-9921-14-110
Gueders, M. M., Paulissen, G., Crahay, C., Quesada-Calvo, F., Hacha, J., Van Hove, C., et al. (2009). Mouse models of asthma: a comparison between C57BL/6 and BALB/c strains regarding bronchial responsiveness, inflammation, and cytokine production. Inflamm. Res. 58, 845–854. doi: 10.1007/s00011-009-0054-2
Guerrina, N., Traboulsi, H., Eidelman, D. H., and Baglole, C. J. (2018). The aryl hydrocarbon receptor and the maintenance of lung health. Int. J. Mol. Sci. 19:3882. doi: 10.3390/ijms19123882
Guerrina, N., Traboulsi, H., Rico De Souza, A., Bosse, Y., Thatcher, T. H., Robichaud, A., et al. (2021). Aryl hydrocarbon receptor deficiency causes the development of chronic obstructive pulmonary disease through the integration of multiple pathogenic mechanisms. FASEB J. 35:e21376. doi: 10.1096/fj.202002350R
Hecht, E., Zago, M., Sarill, M., Rico De Souza, A., Gomez, A., Matthews, J., et al. (2014). Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation. Toxicol. Appl. Pharmacol. 280, 511–525. doi: 10.1016/j.taap.2014.08.023
Holgate, S. T., and Polosa, R. (2008). Treatment strategies for allergy and asthma. Nat. Rev. Immunol. 8, 218–230. doi: 10.1038/nri2262
James, A. L., Bai, T. R., Mauad, T., Abramson, M. J., Dolhnikoff, M., Mckay, K. O., et al. (2009). Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. Eur. Respir. J. 34, 1040–1045. doi: 10.1183/09031936.00181608
Jeong, K. T., Hwang, S. J., Oh, G. S., and Park, J. H. (2012). FICZ, a tryptophan photoproduct, suppresses pulmonary eosinophilia and Th2-type cytokine production in a mouse model of ovalbumin-induced allergic asthma. Int. Immunopharmacol. 13, 377–385. doi: 10.1016/j.intimp.2012.04.014
Jonckheere, A. C., Bullens, D. M. A., and Seys, S. F. (2019). Innate lymphoid cells in asthma: pathophysiological insights from murine models to human asthma phenotypes. Curr. Opin. Allergy Clin. Immunol. 19, 53–60. doi: 10.1097/ACI.0000000000000497
Kilkenny, C., Browne, W., Cuthill, I. C., Emerson, M., and Altman, D. G., Group, N.C.R.R.G.W (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 160, 1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x
Kim, D. I., Song, M. K., and Lee, K. (2019). Comparison of asthma phenotypes in OVA-induced mice challenged via inhaled and intranasal routes. BMC Pulm. Med. 19:241. doi: 10.1186/s12890-019-1001-9
Kips, J. C., Tournoy, K. G., and Pauwels, R. A. (2001). New anti-asthma therapies: suppression of the effect of interleukin (IL)-4 and IL-5. Eur. Respir. J. 17, 499–506. doi: 10.1183/09031936.01.17304990
Latchney, S. E., Hein, A. M., O'banion, M. K., Dicicco-Bloom, E., and Opanashuk, L. A. (2013). Deletion or activation of the aryl hydrocarbon receptor alters adult hippocampal neurogenesis and contextual fear memory. J. Neurochem. 125, 430–445. doi: 10.1111/jnc.12130
Maes, T., Joos, G. F., and Brusselle, G. G. (2012). Targeting interleukin-4 in asthma: lost in translation? Am. J. Respir. Cell Mol. Biol. 47, 261–270. doi: 10.1165/rcmb.2012-0080TR
Maslan, J., and Mims, J. W. (2014). What is asthma? Pathophysiology, demographics, and health care costs. Otolaryngol. Clin. N. Am. 47, 13–22. doi: 10.1016/j.otc.2013.09.010
Papi, A., Brightling, C., Pedersen, S. E., and Reddel, H. K. (2018). Asthma. Lancet 391, 783–800. doi: 10.1016/S0140-6736(17)33311-1
Pareek, S., Traboulsi, H., Allard, B., Rico De Souza, A., Eidelman, D. H., and Baglole, C. J. (2019). Pulmonary neutrophilia caused by absence of the NF-kappaB member RelB is dampened by exposure to cigarette smoke. Mol. Immunol. 114, 395–409. doi: 10.1016/j.molimm.2019.08.011
Pelaia, G., Vatrella, A., Busceti, M. T., Gallelli, L., Calabrese, C., Terracciano, R., et al. (2015). Cellular mechanisms underlying eosinophilic and neutrophilic airway inflammation in asthma. Mediat. Inflamm. 2015:879783. doi: 10.1155/2015/879783
Pollenz, R. S. (2007). Specific blockage of ligand-induced degradation of the Ah receptor by proteasome but not calpain inhibitors in cell culture lines from different species. Biochem. Pharmacol. 74, 131–143. doi: 10.1016/j.bcp.2007.03.021
Poulain-Godefroy, O., Boute, M., Carrard, J., Alvarez-Simon, D., Tsicopoulos, A., and De Nadai, P. (2020). The aryl hydrocarbon receptor in asthma: friend or foe? Int. J. Mol. Sci. 21:8797. doi: 10.3390/ijms21228797
Prokipcak, R. D., and Okey, A. B. (1991). Downregulation of the Ah receptor in mouse hepatoma cells treated in culture with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Can. J. Physiol. Pharmacol. 69, 1204–1210. doi: 10.1139/y91-176
Rico De Souza, A., Traboulsi, H., Wang, X., Fritz, J. H., Eidelman, D. H., and Baglole, C. J. (2021). The aryl hydrocarbon receptor attenuates acute cigarette smoke-induced airway neutrophilia independent of the dioxin response element. Front. Immunol. 12:630427. doi: 10.3389/fimmu.2021.630427
Rogers, S., De Souza, A. R., Zago, M., Iu, M., Guerrina, N., Gomez, A., et al. (2017). Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure. Sci. Rep. 7:40539. doi: 10.1038/srep40539
Simpson, J. L., Scott, R., Boyle, M. J., and Gibson, P. G. (2006). Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology 11, 54–61. doi: 10.1111/j.1440-1843.2006.00784.x
Thatcher, T. H., Maggirwar, S. B., Baglole, C. J., Lakatos, H. F., Gasiewicz, T. A., Phipps, R. P., et al. (2007). Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am. J. Pathol. 170, 855–864. doi: 10.2353/ajpath.2007.060391
Thatcher, T. H., Williams, M. A., Pollock, S. J., Mccarthy, C. E., Lacy, S. H., Phipps, R. P., et al. (2016). Endogenous ligands of the aryl hydrocarbon receptor regulate lung dendritic cell function. Immunology 147, 41–54. doi: 10.1111/imm.12540
Wei, Y. D., Helleberg, H., Rannug, U., and Rannug, A. (1998). Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem. Biol. Interact. 110, 39–55. doi: 10.1016/S0009-2797(97)00111-7
Wincent, E., Amini, N., Luecke, S., Glatt, H., Bergman, J., Crescenzi, C., et al. (2009). The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 284, 2690–2696. doi: 10.1074/jbc.M808321200
Wong, T. H., Lee, C. L., Su, H. H., Wu, C. C., Wang, C. C., Sheu, C. C., et al. (2018). A prominent air pollutant, Indeno[1,2,3-cd]pyrene, enhances allergic lung inflammation via aryl hydrocarbon receptor. Sci. Rep. 8:5198. doi: 10.1038/s41598-018-23542-9
Wong, J., Magun, B. E., and Wood, L. J. (2016). Lung inflammation caused by inhaled toxicants: a review. Int. J. Chronic Obstr. Pulm. Dis. 11, 1391–1401. doi: 10.2147/COPD.S106009
Xu, T., Zhou, Y., Qiu, L., Do, D. C., Zhao, Y., Cui, Z., et al. (2015). Aryl hydrocarbon receptor protects lungs from cockroach allergen-induced inflammation by modulating mesenchymal stem cells. J. Immunol. 195, 5539–5550. doi: 10.4049/jimmunol.1501198
Zhang, S., Patel, A., Chu, C., Jiang, W., Wang, L., Welty, S. E., et al. (2015). Aryl hydrocarbon receptor is necessary to protect fetal human pulmonary microvascular endothelial cells against hyperoxic injury: mechanistic roles of antioxidant enzymes and RelB. Toxicol. Appl. Pharmacol. 286, 92–101. doi: 10.1016/j.taap.2015.03.023
Keywords: aryl hydrocarbon receptor, allergic asthma, chlorine, inflammation, lungs, neutrophils, occupational asthma
Citation: Traboulsi H, de Souza AR, Allard B, Haidar Z, Sorin M, Moarbes V, Fixman ED, Martin JG, Eidelman DH and Baglole CJ (2021) Differential Regulation of the Asthmatic Phenotype by the Aryl Hydrocarbon Receptor. Front. Physiol. 12:720196. doi: 10.3389/fphys.2021.720196
Edited by:
Rosalinda Sorrentino, University of Salerno, ItalyReviewed by:
Jill Johnson, Aston University, United KingdomCopyright © 2021 Traboulsi, de Souza, Allard, Haidar, Sorin, Moarbes, Fixman, Martin, Eidelman and Baglole. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn J. Baglole, Y2Fyb2x5bi5iYWdsb2xlQG1jZ2lsbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.