 Yan Lai1,2
Yan Lai1,2 Yongbo Huang1,2*
Yongbo Huang1,2*- 1State Key Laboratory of Respiratory Diseases, Guangzhou Institute of Respiratory Health, First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 2Department of Critical Care Medicine, First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
Mechanical ventilation is a supportive therapy for patients with acute respiratory distress syndrome (ARDS). However, it also inevitably produces or aggravates the original lung injury with pathophysiological changes of pulmonary edema caused by increased permeability of alveolar capillaries which composed of microvascular endothelium, alveolar epithelium, and basement membrane. Vascular endothelium forms a semi-selective barrier to regulate body fluid balance. Mechanical ventilation in critically ill patients produces a mechanical force on lung vascular endothelium when the endothelial barrier was destructed. This review aims to provide a comprehensive overview of molecular and signaling mechanisms underlying the endothelial barrier permeability in ventilator-induced lung jury (VILI).
Introduction
Acute respiratory distress syndrome (ARDS) is a condition in which lungs sustain an acute, diffuse, and inflammatory injury (Force et al., 2012; Fan et al., 2018). In an international, multicenter, prospective cohort study of patients admitted to ICUs, 10.4% of patients fulfilled ARDS criteria, and 23.4% require mechanical ventilation (Bellani et al., 2016). Mechanical ventilation is a supportive therapy for patients with ARDS; a lower tidal volume ventilation decreased mortality from 39.8 to 31% and increased the number of days without ventilator use (Brower et al., 2000). However, mechanical ventilation inevitably damages or aggravates the original lung injury characterized by inflammatory-cell infiltrates, formation of hyaline membranes, increased vascular permeability, and pulmonary edema (Slutsky and Ranieri, 2013). Increased permeability of alveolar capillaries enables fluid to leak into the interstitium and alveoli, resulting in clinical respiratory failure (Beasley, 2010; Murray, 2011). Exploring the mechanisms of endothelial layer responding to mechanical force is vital to develop effective endothelial-targeted treatments among patients who require mechanical ventilation. The primary purposes of this review are to (1) illustrate the increase of pulmonary microvascular endothelial permeability induced by mechanical ventilation, (2) summarize the destruction of endothelial barrier function caused by mechanical force, and 3) describe the molecular mechanisms involved in endothelial barrier disfunction.
Increased of Microvascular Permeability Caused by Mechanical Ventilation
Under physiological conditions, the alveolar capillaries act as the first defense line, restricting fluid and macromolecules from entering the interstitium from blood (Parthasarathi, 2018). Fluid leaks across the vascular endothelium and accumulates in the interstitial space and alveoli, causing cardiogenic and non-cardiogenic edema (Murray, 2011). In 1974, Webb and Tierney first used a rat model and proved that high tidal volume ventilation (HTV) can cause fulminant non-cardiogenic edema (Webb and Tierney, 1974), consistently associated with alterations in pulmonary vascular permeability barrier (Dreyfuss and Saumon, 1993). Egan and Parker found that capillary filtration coefficient was immediately increased when airway pressures exceeded 30cm H2O threshold, and lung lymph protein clearances and minimal lymph or plasma ratios of total protein were significantly higher in high peak airway pressures group than in low peak airway pressures group, indicating a significant increase in microvascular permeability (Egan, 1982; Parker et al., 1990). Later, Katira et al. reproduced the data reported by Webb and Tierney that pulmonary edema is due to a large increase in microvascular permeability after mechanical ventilation (Katira et al., 2017). In critically ill patients with mechanical ventilation, decreased plasma albumin and transferrin levels parallel increased pulmonary vascular permeability, regardless of the underlying disease or fluid status (Aman et al., 2011).
Dysfunction of Pulmonary Microvascular Endothelial Barrier
The destruction of lung gas-blood barrier function is characterized by significant changes in endothelial cell (EC) function (Birukova et al., 2006). ECs are located on the inner side of blood vessels, forming a selective semi-permeable barrier regulating body fluid balance and migration of white blood cells (Mehta and Malik, 2006). Mechanical ventilation induced excessive stretch of alveoli that transmits pathological mechanical stress to alveolar epithelium and pulmonary endothelium. In vitro studies through cyclic stretch (CS) epithelial cells indicate that 25% alveolar epithelial cell basal surface area increase is equated to exposure to 5% CS, reflects the physiological levels of mechanical stress in alveolar epithelium, and 40–50% alveolar epithelial cell basal surface area increase is equated to exposure to 18% CS, reflects high tidal volume mechanical stress in alveolar epithelium (Tschumperlin and Margulies, 1999; Wirtz and Dobbs, 2000). There are no studies report the association between capillary endothelium basal surface and ventilation, but some scholars consider that lung inflation transmits stress to capillary endothelium in a similar extent. Dreyfuss et al. found that an ultrastructural injury of ECs after mechanical ventilation occurs when the thin part of an EC is detached from the basement membrane and floats in the capillary lumen (Dreyfuss et al., 1985; Dreyfuss and Saumon, 1998). Structural and mechanical studies indicate that loss of ECs integrity induces endothelial permeability increase (Wang et al., 2012a). The endothelial permeability is mainly controlled by paracellular and transcellular permeability.
Paracellular Permeability
Interactions between cells are essential for maintaining a tight endothelial barrier. Inter-endothelial junctions including adhesion junctions (AJs), gap junctions (GJs), tight junctions (TJs), and adhesion molecules (AMs) jointly maintain paracellular permeability balance and control the passage of plasma proteins, solutes, and fluid across the endothelial barrier (Komarova and Malik, 2010). Mechanical stress associated with VILI significantly impacts the expression of EC junctions, which leads to injurious remodeling and ECs barrier disruption.
Adhesion Junctions
Adhesion junctions (AJs) are composed of vascular endothelial (VE)-cadherin complexes with catenins. VE-cadherin is an exclusive endothelium signature and the main component of endothelial AJs, comprising five extracellular cadherin domains, a transmembrane domain, and a highly conserved cytoplasmic tail, mediating intercellular adhesions of ECs through cis- and trans-dimerization (Rho et al., 2017). The cytoplasmic tail of VE-cadherin is combined with p120-catenin, β-catenin and plakoglobin. The latter two are connected to the actin cytoskeleton through the bridging effect of α-catenin (Dejana et al., 2008). VE-cadherin mediated the mechanical force transduction signals that increased integrin-dependent cell contractility and disrupted cell–cell and cell–matrix adhesions (Andresen Eguiluz et al., 2017). The expression of VE-cadherin is closely related to intercellular adhesions and ECs barrier function (Dejana and Vestweber, 2013). In tightly connected cells, phosphorylated VE-cadherin is at a low expression level, indicating a negative correlation between the integrity of AJs and phosphorylated VE-cadherin (Chiu et al., 2004). HTV combined with hyperoxia further activated Src to phosphorylate VE-cadherin at Y685 and Y658, leading to internalization and ubiquitination of VE-cadherin (Li et al., 2015; Soni et al., 2017; Zhong et al., 2019). The transmembrane domain of VE-cadherin mediates an essential adapter function by binding directly to the transmembrane domain of vascular endothelial growth factor receptor 2 (VEGFR2), thereby assembling the endothelial mechanosensory complex (Coon et al., 2015). Eighteen percent CS induced the time-dependent upregulation of VEGFR2 and dissociating VEGFR2 from VE-cadherin at cell junctions, which result in VEGFR2 activation, Src-dependent phosphorylation of VE-cadherin at Tyr658 and internalization. However, the disruption of AJs integrity was attenuated with VEGFR2 knockdown (Gawlak et al., 2016; Tian et al., 2016). The stability of AJs maintained by Yes-associated protein, and deletion of Yes-associated protein in ECs exaggerated VE-cadherin phosphorylation, downregulation of vascular endothelial protein tyrosine phosphatase, and dissociation of VE-cadherin and catenins following mechanical ventilation (Su et al., 2021). Deubiquitinating enzymeubiquitin carboxyl terminal hydrolase 1 is a protective factor in VILI of increased lung vascular permeability, as inhibition of ubiquitin carboxyl terminal hydrolase 1 was associated with decreased expression levels of VE-cadherin (Mitra et al., 2021).
Gap Junctions
Gap junctions (GJs) are mainly composed of connexins (Cx); 4–6 connexins assembled in the plasma membrane to form a connexon, allowing ions and small signaling molecules to diffuse freely between neighboring cells (Yeager and Harris, 2007; Wang et al., 2017a). In ECs, GJs play an important role in regulating the permeability of the endothelial monolayer. Cx37, Cx40, and Cx43 are abundantly expressed in different lung ECs and involved in endothelial permeability (Nagasawa et al., 2006; Hautefort et al., 2019). Cx37 functions as a break of ECs proliferation and overexpression of Cx37 facilitates ECs apoptosis, indicating that Cx37 plays a crucial role in angiogenesis and vascular repair (Seul et al., 2004; Pohl, 2020). However, there are no reports on the specific role of Cx37 in endothelial repair after mechanical ventilation or mechanical stretch. Cx40 induces Rho associated coiled-coil containing protein kinase 1 (ROCK1) upregulation and subsequent phosphorylation of myosin phosphatase target subunit and myosin light chain 20 (MLC20). Inhibition of Cx40 suppresses cells proliferation; however, genetic deficiency of Cx40 significantly attenuated inflammation, lung edema, and histological evidence of lung damage (Yin et al., 2019). Cx43 regulates lung endothelial barrier function by increasing phosphorylation of MLC and ROCK1 expression, affecting cell contraction and stress fiber formation (Zhang et al., 2015). Immunofluorescence and immunoblot analyses revealed that both Cx43 expression and micro-vessel permeability increased above baseline within a few hours after endotoxin instillation but declined progressively over the next few days, and the knockdown of vascular Cx43 by Cx43 shRNA increased VE-cadherin expression, suggesting that the reduction in Cx43 levels may modulate VE-cadherin levels in lung micro-vessels. In contrast to Cx43, the expression of micro-vessel VE-cadherin exhibited an inverse trend by initially declining below baseline and then returning to baseline at a longer duration (Kandasamy et al., 2015). Eighteen percent CS causes a significant increase of Cx43 at both protein and mRNA levels, and the GJs inhibitor, carbenoxolone, attenuates the permeability response to thrombin and inhibits MLC phosphorylation, Cx43 siRNA transfection significantly increased cell viability, inhibited cells apoptosis and caspase-3 activity (Cowan et al., 1998; O'Donnell 3rd et al., 2014). Cx37 or Cx40 seems to be less mechanosensitive, but Cx43 is mechanosensitive and plays an essential role in CS induced EC barrier regulation and possibly be a novel therapeutic target (Salameh and Dhein, 2013).
Tight Junctions
Tight junctions (TJs) are intramembrane multiprotein complexes composed of transmembrane proteins, including occludins, claudins, junction adhesion molecules, and cytoplasmic scaffold protein zonula occludins (ZO) that create a paracellular barrier in epithelial and endothelial cells and protect epithelial and endothelial cell from the external environment (Forster, 2008; Hartsock and Nelson, 2008; Cummins, 2012). Similar to AJs, the TJs is affected by VILI associated stress.
Claudins
The claudins family contains at least 24 members whose expression is tissue-specific; ECs specifically express claudin-5 (Forster, 2008). Studies established claudin-5 as being key for TJs formation in ECs. Claudin-5 gene knockout mice are selectively permeable to small molecules (Morita et al., 1999). At the transcriptional level, claudin-5 is regulated by the transcription factor Sox-18, which plays a key role in promoting blood vessel development and maintaining stability of endothelial barrier function (Fontijn et al., 2008). The shear stress up-regulates Sox-18 expression, and in turn, increases claudin-5 expression, maintains the stability of endothelial barrier function and prevents cardiogenic pulmonary edema (Gross et al., 2014). In contrast, LPS plus mechanical ventilation dramatically reduced claudin-5 expression in lung tissues, further destroyed TJs structure, and increased endothelial permeability (Li et al., 2020). Oxycodone is a synthetic opioid receptor agonist that decreased pulmonary microvascular permeability both in vivo and in vitro by upregulating the expression of claudin-5 (Li et al., 2021). The regulation of claudin-5 in the vascular endothelium can greatly impact TJs formation and endothelial barrier function (Wang et al., 2017a; Kakogiannos et al., 2020). However, our knowledge regarding the role of claudin-5 in mechanical force induced lung ECs barrier dysfunction has not been fully studied.
Occludin
Occludin is tetrameric protein that express in lung epithelial cells and ECs, and the N-terminus corresponds to several properties of the TJs barrier (Furuse et al., 1993; Zhang and Yang, 2016). The phosphorylation states of occludin affect its localization: non-phosphorylated occludin is localized at both the basolateral membrane and in cytoplasmic vesicles, and phosphorylated occludin is localized at TJs. The regulation of barrier function based on occludin in endothelial cells was correlated with its phosphorylation and expression level (Zhang and Yang, 2016). Mechanical ventilation activated PKC signaling pathway and decreased occludin expression in a HTV group than a LTV group. Pretreatment with a PKC inhibitor increased occludin expression and could reduce or delay pulmonary edema (Liu et al., 2014). Adaptive support ventilation is a closed-loop ventilation, which upregulated expression level of occludin and exhibited less lung injury and greater alveolar fluid clearance compared with the volume-control ventilation group (Dai et al., 2019b). Mechanical ventilation activated c-Src by phosphorylation and decreased occludin expression, while c-Src inhibitor upregulated occluding expression, which ameliorates barrier function (Zhao et al., 2014).
Adhesion Molecules
Adhesion molecules (AMs) include integrins and immunoglobulin superfamily cell adhesion molecules (IgCAMs), allowing the endothelium to form attachments with surrounding proteins and cells to modify endothelial barrier function and vascular permeability (Sarelius and Glading, 2015).
Integrins
Integrins are heterodimer transmembrane proteins that consist of two subunits, containing extracellular, transmembrane, and cytoplasmic domains, 18 α subunits and 8 β subunits that can assemble into 24 different receptors, which has different binding properties and different tissue distribution (Campbell and Humphries, 2011). Integrins are known to mediate vascular stability by mediating ECs adhesion to vascular basement membrane (Yamamoto et al., 2015). In ECs, integrin β1 dependent ECs basement membrane adhesions contribute to acute endothelial permeability responses, heterozygous deletion of integrin β1 protected the mice from LPS-induced vascular leakage (Hakanpaa et al., 2018). Integrins serve as bidirectional hubs transmitting signals between cells and their environment and respond to extracellular matrix, which mediates the formation of mechanosensitive structures (Kechagia et al., 2019; Sun et al., 2019a). Integrins can be activated by mechanical force and involved in vascular permeability regulation (Sarelius and Glading, 2015; Gingras and Ginsberg, 2020). Integrins β5 is specifically expressed in the pulmonary endothelium and involved in VILI. Integrin αvβ5 contributes to stress fiber formation and increases endothelial permeability by activating downstream of RhoA. Function-blocking antibody against integrin αvβ5 prevented lung vascular permeability development in VILI mice, and the same effects were also found in integrin αvβ5 gene knockout mice (Su et al., 2007). Cecal ligation and puncture (CLP) led to small but significant increases in integrin β5. However, HTV combined with CLP group increased integrin β5 expression 2–3 folds more than CLP group, and anti-integrin β5 antibodies partially inhibited the two-hit phenotype of increases in alveolar-capillary permeability, histopathologic scoring, and indices of pulmonary inflammation in mice (Ding et al., 2018). Moderate tidal volume mechanical ventilation further increased poly(I:C), an analog of natural double-strand RNA virus induced integrin β3 expression in the lung, inhibited integrin ανβ3, reduced inflammation and attenuated VILI in rats (Wang et al., 2012b; Jin et al., 2016). Mechanical ventilation or CS induces the phosphorylation of integrins at different positions (Kelly et al., 2019). Integrins activated within 1min after exposing to 15% CS, as indicated by increased phosphorylation of the T788/789 site of the integrin cytoplasmic tail (Thodeti et al., 2009). Tyrosine phosphorylation of integrin β4 at Y1440, Y1526, Y1640, or Y1422 in response to 18% CS was evident within 30min and peaked at 2h, and integrin β4 mutant mice demonstrated decreased lung vascular permeability and inflammation when subjected to mechanical ventilation (Chen et al., 2015). For other integrins, integrin β8 has a non-classical cytoplasmic domain and plays a role in angiogenesis integrin β6 is principally expressed in epithelium and play an important role in septic mice by regulating lung inflammatory and alveolar capillary permeability, integrins β2 and β7 are principally expressed in leukocytes and involve in inflammation (Tabata et al., 2008; Giusti et al., 2013; Chen et al., 2015; Ding et al., 2015, 2018; Ou et al., 2021). The involvement of integrins in mechanical force induced endothelial permeability is limited and requires further experiments.
IgCAMs
Platelet-endothelial cell adhesion molecule 1 (PECAM-1), a member of IgCAMs family, is located at ECs intercellular junctions, where it functions as a mechanosensor and maintenance of ECs junctional integrity (Privratsky and Newman, 2014; Liao et al., 2018). As an efficient signaling molecule, PECAM-1 has diverse vascular biology roles, including angiogenesis, platelet function, thrombosis, mechanosensing of fluid shear stress, and regulating leukocyte migration (Woodfin et al., 2007). Five percent CS alone did not cause any significant changes in cell area and cell aspect ratio compared to untreated control cells, but cell area increased significantly in response to concurrent shear stress and CS (Meza et al., 2019). Upon CS, the cytoplasmic domain of PECAM-1 is unfolded and exposes a phosphorylation site, allowing the rapid PECAM-1 phosphorylation, transforming a mechanical signal into a biochemical signal (Snyder et al., 2017). Five percent CS caused 42% increase in PECAM-1 phosphorylation (Meza et al., 2019). In a rat VILI model, mechanical ventilation decreased PECAM-1 expression in the lung, released soluble sPECAM1 into the blood circulation, and caused damage to the pulmonary permeability barrier as showed by pulmonary edema, leukocyte infiltration, and severe hypoxemia (Villar et al., 2014).
Transcellular Permeability
Transcellular permeability via transcellular vesicle trafficking selectively transports macromolecules such as albumin and albumin-bound ligands, insulin, lipids, and hormones from vessel lumen to interstitial space (Komarova and Malik, 2010). Caveolae, flask-shaped non-coated structures, compose 20% of cell volume in ECs and mediate transcytosis to regulate endothelial transcellular permeability (Komarova and Malik, 2010). Caveolae contain a membrane protein known as caveolin (Cav), including caveolin-1, −2, and−3. ECs express high levels of Cav-1 involved in endothelial inflammation, adhesion, and phagocytosis (Shihata et al., 2016).
Cav-1 is a structural protein of caveolae with N and C-terminal cytoplasmic tail with function domains: tyrosine phosphorylation site and oligomerization domain can regulate the endothelial transcellular permeability (Sun et al., 2011; Boscher and Nabi, 2012). To explore the role of Cav-1 in regulating lung vascular permeability, Miyawaki-Shimizu et al. used small interfering RNA to knock down Cav-1 expression in mouse lung endothelia in vivo. Caveolae disappearance in lung vessel endothelia is concomitant with increase lung vascular permeability to albumin (Miyawaki-Shimizu et al., 2006). The function of Cav-1 is regulated by Src-mediated phosphorylation of Cav-1 at Tyr14 initiating plasmalemmal vesicle fission and transendothelial vesicular transport (Sun et al., 2009; Boscher and Nabi, 2012). Src was phosphorylated and accompanied by synchronous phosphorylation of Cav-1 at Tyr14 after 30min of mechanical ventilation with a tidal volume of 21ml/kg on mice (Maniatis et al., 2012). Cav-1−/− mice show less susceptibility to pulmonary endothelial hyperpermeability induced by HTV, but rescue of Cav-1 expression restored the hyperpermeability response to HTV, indicating that caveolin-1 expression is required for mechanical force-induced endothelial hyperpermeability (Maniatis et al., 2012; Zhong et al., 2015). In contrast, Hoetzel found that ventilation with 12ml/kg for 8h increased Cav-1 expression levels, and Cav-1 deficiency aggravated lung injury characterized by protein leakage, edema formation, and macrophage infiltration, suggesting that Cav-1−/− mice were more susceptible to pulmonary endothelial hyperpermeability induced by mechanical ventilation (Hoetzel et al., 2009). The different results may depend on mechanical ventilation parameters. Cav-1 expression in transcellular permeability is controversial, and more research is needed to explore the role of Cav-1 in mechanical force-induced endothelial hyperpermeability.
Molecular Mechanisms in Endothelial Permeability
Endothelial barrier dysfunction is triggered by endothelial sense of mechanical stimuli, which are transformed into biological stimuli and trigger downstream signaling pathways, resulting in cell cytoskeleton remodeling and gene expression changes (Figure 1).
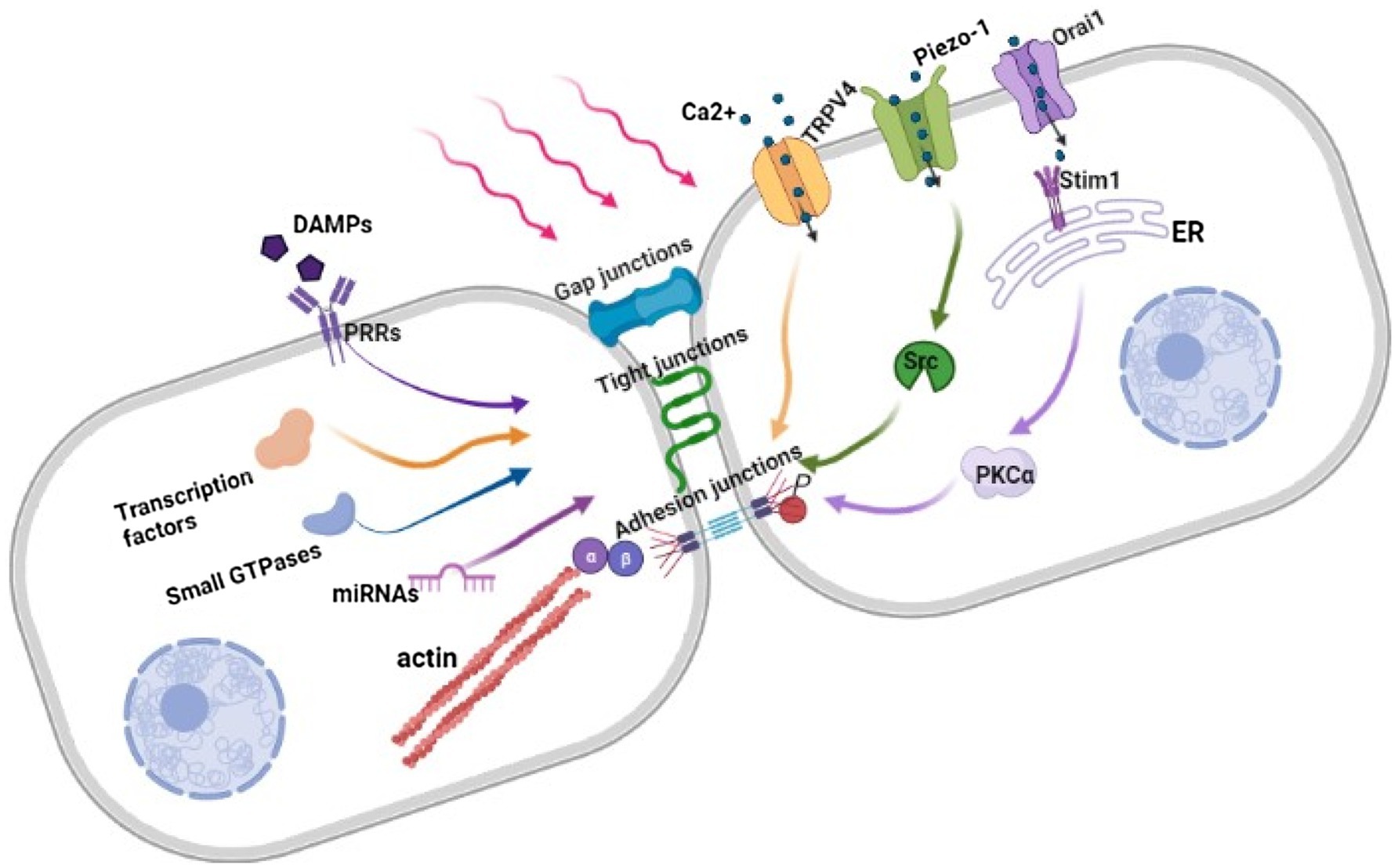
Figure 1. Regulation of Endothelial barrier in response to mechanical force. Under mechanical stimuli, the combination of DAMPs and PRRs, the influx of Ca2+ through opened mechanosensitive ion channels and changes in gene expression destruct intercellular structures further leads to cytoskeleton contracting and widening intercellular space. β, β-catenin; α, α-catenin; HMGB1, High-mobility group box 1; DAMPs, Damage-associated molecular patterns; PRRs, Pattern recognition receptors; TRPV4, Transient receptor potential vanilloid 4; Stim1, Stromal-interacting molecule 1; ER, Endoplasmic reticulum; PKCα, Protein kinase C alpha; Src, Proto-oncogene tyrosine-protein kinase.
Cation Channels
Stretch-activated cation channels act as mechanoreceptors and mechanical sensors to mediate mechanical, electrical, and mechanochemical signals (Nilius and Honore, 2012). Stretching activates cation channels by (1) direct tension on the lipid bilayer, (2) transduction of stretch via a tethering mechanism from cytoskeletal or extracellular matrix structures, or (3) through releasing or binding of another molecule that in turn activates a channel (Schwingshackl, 2016). The increased pulmonary vascular permeability which caused by excessive lung hyperinflation is associated with activation of cation channels in the vascular endothelium (Lansman et al., 1987; Parker et al., 1998). Previous studies have shown that Ca2+ influx or release of stored Ca2+ from endoplasmic reticulum activates calmodulin and MLCK. Phosphorylation of MLC increases cytoskeletal tension, intercellular space, and vascular permeability (Wysolmerski and Lagunoff, 1991; Garcia and Schaphorst, 1995; Tiruppathi et al., 2006; Dudek et al., 2010; Parker et al., 2013). Currently, Ca2+ channels including TRPs, Stim1/Orai1, and Piezo1 are widely studied.
Transient Receptor Potential Channels
Transient receptor potential channels (TRPs) are superfamily of ion channels, based on amino acid homologies, the mammalian TRP channel superfamily can be divided into six subfamilies, include TRPV (vanilloid), TRPC (classical or canonical), TRPM (melastatin), TRPP (polycystin), TRPA (ankyrin), and TRPML (mucolipin). TRPs are permeable for Ca2+ and play important role in endothelial permeability (Di and Malik, 2010). TRPV4 is abundantly expressed in the lung endothelium and attracts much attention for its influence on mechanical ventilation induced lung endothelial hyperpermeability (Simmons et al., 2019). TRPV4 activation causes Ca2+ influx, leading to an increase in the filtration coefficient of lung, but inhibition or knockout of TRPV4 improves pulmonary vascular leakage caused by HTV (Kunert-Keil et al., 2006; Hamanaka et al., 2007; Li et al., 2019; Yu et al., 2020). The protective effect of genetic deficiency or pharmacologic inhibition of TRPV4 may occur due to phosphorylation effect at its serine 824 residues by serum glucocorticoid-regulated kinase (Michalick et al., 2017). Adipose-derived stem cell exosome directly suppresses TRPV4 channel, improves ventilator-induced pulmonary microvascular hyperpermeability as evidenced by lower lung injury score, wet/dry (W/D) weight ratio, and protein concentration in bronchoalveolar lavage fluid (BAL), and increases the expression level of VE-cadherin and β-catenin. Similar to in vivo effect, Adipose-derived stem cell exosome decreased intracellular Ca2+ concentration and restored endothelial barrier in vitro (Yu et al., 2020). For other TRPs, TRPC6 functions as store-operated channels, activation of TRPCs in lung endothelium results in Ca2+ influx and subsequent vascular barrier dysfunction and endothelial contraction (Uhlig et al., 2014). TRPC6 was found to be critically involved in lung vascular leakage after stimulation with platelet-activating factor, TRPCs inhibitor or know out TRPC6 obviously attenuated vascular filtration coefficient, and lung edema formation (Samapati et al., 2012). TRPMs are a group of oxidant-activated cation channels, of which TRPM2 is highly expressed in ECs. TRPM2 channels regulate endothelial barrier integrity by facilitating Ca2+ influx to ECs, and deletion of TRPM2 in ECs significantly increases the survival rate of endotoxin mice (Mittal et al., 2017). TRPA1 is also known to be activated by oxidative stress in ECs (Simon and Liedtke, 2008). TRPML family has been identified as endolysosomal nonselective cation channels that mediate intra-endosomal calcium release in the endocytic pathway (Villalta and Townsley, 2013). Although TRPC and TRPM families involve in endothelial permeability, we have not found evidence that these channels regulate mechanical ventilation induced lung endothelial permeability (Villalta and Townsley, 2013; Smani et al., 2018; Genova et al., 2020).
Stim1/Orai1
Stromal-interacting molecule 1 (Stim1) is located in the endoplasmic reticulum and serves as an endoplasmic reticulum Ca2+ sensor. Orai1 is the prototypical calcium release-activated Ca2+ channel; activated Stim proteins translocate into endoplasmic reticulum plasma membrane junctions and combine directly with Orai1 channels to generate Ca2+ entry signals (McNally et al., 2013; Prakriya, 2013). HTV or 18% CS significantly upregulates the expression of Stim1 and Orai1 in pulmonary microvascular ECs, causing an increase in intracellular Ca2+ concentration, which activates PKCα to induce increased endothelial permeability. The application of Orai1 inhibitor prevents Ca2+ influx and reverses the high permeability of ECs (Song et al., 2018). Stim1-induced Ca2+ signaling activates Pyk2 to induce tyrosine phosphorylation of vascular endothelial protein tyrosine phosphatase, which activates Src and subsequently phosphorylates VE-cadherin to increase the endothelial permeability (Soni et al., 2017).
Piezo-1
Piezo proteins, including Piezo-1 and Piezo-2, are evolutionarily conserved and can respond to various forms of mechanical stimulation including poking, stretching, and shear stress (Geng et al., 2017). Piezo-1, a homotrimeric membrane spanning protein complex that serve as a force sensor, which then responds to mechanical force by regulating the gating of Ca2+ (Ge et al., 2015; Zhao et al., 2018). Piezo-1 was first discovered in neuroblastoma cells in mice, and subsequent studies have found that it is also expressed in ECs (Ranade et al., 2014; Cahalan et al., 2015; Ilkan et al., 2017; Liang et al., 2019). Endothelium specifical knock out Piezo-1 result in embryonic lethality, identifying this channel as a major determinant of vascular development (Li et al., 2014). Piezo-1 is a mechanosensitive Ca2+ channel, which can be activated by mechanical force, causing rapid Ca2+ influx (Coste et al., 2010). Piezo-1 signaled lung vascular hyperpermeability by promoting internalization and degradation of VE-cadherin, p120-catenin, and β-catenin in ARDS rats undergoing HVT ventilation and LPS-treated ECs exposed to 20% CS (Jiang et al., 2021). But human lung endothelial monolayers depleted of Piezo-1 and exposed to CS show increased permeability (Zhong et al., 2019). Calpain, a calcium-dependent cysteine protease, was identified as a downstream target of Piezo-1 to maintain the endothelial barrier’s stability in response to CS by cleaving Src kinase, which phosphorylates VE-cadherin at Y685 and Y658 (Zhong et al., 2019). Piezo-1 is one of the important mechanosensitive ion channels that participate in endothelial function, more research are need to explore its role in mechanical ventilation induced endothelial permeability.
Damage-Associated Molecular Patterns
Intracellular molecules released after cell death or subsequent immune cell activation and matrix degradation products are termed as alarmins or damage-associated molecular patterns (DAMPs), which activate pattern recognition receptors to induce multiple signal cascades (Bianchi, 2007). DAMPs include hyaluronan, heat shock proteins, high-mobility box group-1 (HMGB-1), S100 proteins, adenosine 5′-triphosphate, and uric acid. In particular, HMGB-1 has been studied extensively in ECs (Bianchi, 2007; Kuipers et al., 2011). Basic and clinical studies have shown that the expression of DAMPs in BAL of VILI patients was increased, and mechanical ventilation upregulated the expression of pattern recognition receptors in lung (Kuipers et al., 2011). HMGB-1 gene expression is markedly responsive to mechanical ventilation or mechanical stress. HTV of 30ml/kg increased HMGB-1 expression in BAL by five times compared with the control group. HMGB-1 expression was also significantly increased when exposed to excessive and equibiaxial CS in vitro (Ogawa et al., 2006; Wolfson et al., 2014; Dong et al., 2015; Feng et al., 2019). Increasing HMGB-1 concentrations induces mitochondrial oxidative damage, resulting in a significant decrease of VE-cadherin and ZO-1 protein that affects intercellular junctions’ integrity and increases endothelial permeability (Dong et al., 2015; Feng et al., 2019) Reducing HMGB-1 release attenuated lung W/D weight ratio and protein concentration in BAL (Wang et al., 2017b; Sun et al., 2019b). For other DAMPs, hyaluronan is a ubiquitous glycosaminoglycan of the extracellular matrix that presents in the endothelial glycocalyx covering the apical surface of ECs. As a key actor of endothelial glycocalyx, hyaluronan plays a central role in endothelial mechanosensing and barrier permeability (Dogne and Flamion, 2020). Different molecular weight of hyaluronan produce distinct biological effects, high-molecular-weight hyaluronan was proved to be an effective treatment strategy for HTV induced lung injury, but low-molecular-weight hyaluronan increased the production of IL-8, which contribute to the development of lung injury (Mascarenhas et al., 2004; Bai et al., 2005). The heat shock proteins (HSPs) are molecular chaperones abundantly present in the cytosol, high peak airway pressure ventilation significantly increased HSP70 expression in lung tissue (Kira et al., 2006). The same as hyaluronan and HSPs, S100 proteins, adenosine 5′-triphosphate, and uric acid also show increased levels during HTV, but the role of these DAMPs in mechanical ventilation induced lung endothelial permeability has not been studied extensively (Kuipers et al., 2011).
Endothelium Mechanosensitive microRNAs
microRNA (miRNA) is a small non-coding RNA with a length of ~22nt involved in the post-transcriptional regulation of gene expression by binding to 3′UTR region of its mRNA in a non-completely complementary manner (Bartel, 2004; He and Hannon, 2004). miRNAs have been found to be critical modulators of endothelial homeostasis and link to vascular diseases (Fernandez-Hernando and Suarez, 2018). miRNAs in ECs are mechanosensitive that response to mechanical stretch stimulation and exhibited obvious differential expression (Kumar et al., 2014; Zheng et al., 2015). After 1h of mechanical ventilation with a tidal volume of 40ml/kg, the expression of 50 miRNAs increased more than 2-fold, and the expression of 15 miRNAs decreased by more than half (Vaporidi et al., 2012). Among the increased miRNAs, miR-21 promoted the occurrence and development of VILI by targeting the vascular barrier protection factors bone morphogenetic protein receptor type 2 and phosphatase and tensin homolog. The administration of antagomiR-21 significantly ameliorated the indices of high-permeability pulmonary edema caused by mechanical ventilation (Vaporidi et al., 2012). miR-127 has been revealed to be highly expressed in embryos and has been implicated in lung development, placenta formation, and cellular apoptosis (Bhaskaran et al., 2009). In a mature lung, the upregulation of miR-127 expression may contribute to VILI development by increasing pulmonary permeability through NF-κB and p38 MAPK-associated signaling pathways (Li et al., 2017). In contrast to miR-127, miR-135a protects ECs from CS-induced barrier dysfunction, and the protective effects of miR-135a may be related to its inhibition of PH domain leucine-rich repeat protein phosphatase 2 and subsequent activation of PI3K/Akt signaling pathway (Yan et al., 2018). The non-muscle myosin light chain kinase isoform plays a crucial role in agonist-induced pulmonary ECs barrier regulation, and bioinformatic analysis revealed that miR-374 could potentially regulate MYLK gene expression by binding to 3′UTR region (Adyshev et al., 2013). Eighteen percent CS significantly decreases miR-374 expression levels, and functional overexpression of miR-374 attenuates lung MLC subunit activation, which induces the destruction of vascular barrier function (Adyshev et al., 2013). In the miRNA expression profile, ECs specifically express miR-126, which plays a vital role in angiogenesis, endothelial function, and vascular repair (Wang et al., 2008; Chistiakov et al., 2016). In LPS-induced endotoxic mice, miR-126a-3p overexpression decreased blood vessels’ permeability and improved the survival rate of endotoxic mice, but there is no research report on mechanical force effects on miR-126 expression in ECs (Chu et al., 2016). Although the analysis of the role of miRNAs in mechanical force induced endothelial permeability remains in its infancy, the data obtained so far suggest that specifically targeting individual miRNA is a potentially promising therapeutic approach for increased endothelium permeability.
Transcription Factors
Transcription factors are key cellular components that control gene expression by binding to DNA and other proteins, and their activities determine cells’ function and how they respond to the environment (Spitz and Furlong, 2012). Transcriptional regulation is involved in biomechanical stimuli and controls diverse biological processes (Vaquerizas et al., 2009). Eighteen percent CS significantly increases STAT5 binding to the promoter of nicotinamide phosphoribosyl-transferase (NAMPT), which can exacerbate VILI-mediated inflammation injury in vivo (Sun et al., 2014). STAT3 and Nrf2 are critical to 18% CS-induced HMGB-1 expression, which induce increased endothelial permeability in lung ECs (Wolfson et al., 2014; Dong et al., 2015). In pulmonary microvascular ECs, 18% CS reduced the expression of KLF2 by 50% compared with 5% CS, similar to in vitro experiments. After 8h of mechanical ventilation, KLF2 expression in the mouse lungs was decreased (Huang et al., 2017). KLF2 confers barrier-protection via increasing the expression level of occludin and inhibiting the phosphorylation of MLC, which augments intercellular gaps leading to increased endothelial leakage (Lin et al., 2010). KLF2 also acts as a novel activator of small GTPase Ras-related C3 botulinum toxin substrate 1 by transcriptionally controlling Rap guanine nucleotide exchange factor 3 to maintain vascular integrity (Huang et al., 2017).
Small GTPases
Small GTPases act as a molecular switch regulated by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (RhoGDI). The most characterized members, including Rho, Rac, and Cdc42, play a key role in regulating ECs permeability induced by mechanical force (Birukov, 2009). The activation of Rac, Rap1, and Rho induced by CS is amplitude-dependent (Birukov et al., 2003; Birukova et al., 2006). ECs pre- or post-conditioning at low CS magnitude induced Rap1 activation, which promotes the resealing of cell junctions disrupted by high CS magnitude. The protective effect was abolished by pharmacological or molecular inhibition of Rap1 activity (Ke et al., 2019). As a Rap1-binding protein, Krev interaction trapped-1 knockdown exacerbated Rho activation and ECs barrier disruption induced by 18% CS (Meliton et al., 2015). Five percent CS causes Rac activation and promotes the recovery of endothelium monolayer integrity after thrombin stimulation, but 18% CS increased thrombin-induced Rho activity (Birukova et al., 2006). HTV increased RhoA/Rock protein expression (Dai et al., 2019a). The activation of RhoA induces the phosphorylation of MLC and contraction of ECs, resulting in gap formation and increased endothelial permeability (Liu et al., 2013). Rock inhibitor could attenuate CS-induced lung endothelial hyperpermeability and the phosphorylation of myosin phosphatase target subunit 1 in vivo and in vitro (Wang et al., 2019). 1-palmitoyl-2-arachidonoyl-snglycero-3-phosphatidyl choline showed a protective role of stretch-induced barrier-disruptive via triggering Rac activation (Birukova et al., 2011).
Conclusion
Inappropriate mechanical ventilation is confirmed to disrupt pulmonary endothelial barrier. In this review, we discussed the endothelial barrier dysfunction caused by mechanical force. As discussed above, the maintenance of endothelial barrier depends on the stability of various intercellular structures, the open and close of mechanosensitive ion channels and changes in gene expression. The mechanisms involved in hyperpermeability of endothelial are not fully revealed, and most studies focused on individual component. We suppose that a more holistic understanding of underlying signaling mechanisms in mechanical force that induce endothelial barrier dysfunction could lead to the discovery of new therapeutic targets during VILI.
Author Contributions
YL wrote the manuscript. YH obtained funding and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81870069), the Natural Science Foundation of Guangdong Province, China (2017A030313781), and the Science and Technology Program of Guangzhou, China (202102010366).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adyshev, D. M., Moldobaeva, N., Mapes, B., Elangovan, V., and Garcia, J. G. (2013). MicroRNA regulation of nonmuscle myosin light chain kinase expression in human lung endothelium. Am. J. Respir. Cell Mol. Biol. 49, 58–66. doi: 10.1165/rcmb.2012-0397OC
Aman, J., van der Heijden, M., van Lingen, A., Girbes, A. R. J., Amerongen, G. P. V., van Hinsbergh, V. W. M., et al. (2011). Plasma protein levels are markers of pulmonary vascular permeability and degree of lung injury in critically ill patients with or at risk for acute lung injury/acute respiratory distress syndrome. Crit. Care Med. 39, 89–97. doi: 10.1097/CCM.0b013e3181feb46a
Andresen Eguiluz, R. C., Kaylan, K. B., Underhill, G. H., and Leckband, D. E. (2017). Substrate stiffness and VE-cadherin mechano-transduction coordinate to regulate endothelial monolayer integrity. Biomaterials 140, 45–57. doi: 10.1016/j.biomaterials.2017.06.010
Bai, K. J., Spicer, A. P., Mascarenhas, M. M., Yu, L., Ochoa, C. D., Garg, H. G., et al. (2005). The role of hyaluronan synthase 3 in ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 172, 92–98. doi: 10.1164/rccm.200405-652OC
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Beasley, M. B. (2010). The pathologist's approach to acute lung injury. Arch. Pathol. Lab. Med. 134, 719–727. doi: 10.5858/134.5.719
Bellani, G., Laffey, J. G., Pham, T., Fan, E., Brochard, L., Esteban, A., et al. (2016). Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315, 788–800. doi: 10.1001/jama.2016.0291
Bhaskaran, M., Wang, Y., Zhang, H., Weng, T., Baviskar, P., Guo, Y., et al. (2009). MicroRNA-127 modulates fetal lung development. Physiol. Genomics 37, 268–278. doi: 10.1152/physiolgenomics.90268.2008
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5. doi: 10.1189/jlb.0306164
Birukov, K. G. (2009). Small GTPases in mechanosensitive regulation of endothelial barrier. Microvasc. Res. 77, 46–52. doi: 10.1016/j.mvr.2008.09.006
Birukov, K. G., Jacobson, J. R., Flores, A. A., Ye, S. Q., Birukova, A. A., Verin, A. D., et al. (2003). Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L785–L797. doi: 10.1152/ajplung.00336.2002
Birukova, A. A., Chatchavalvanich, S., Rios, A., Kawkitinarong, K., Garcia, J. G., and Birukov, K. G. (2006). Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am. J. Pathol. 168, 1749–1761. doi: 10.2353/ajpath.2006.050431
Birukova, A. A., Zebda, N., Cokic, I., Fu, P., Wu, T., Dubrovskyi, O., et al. (2011). p190RhoGAP mediates protective effects of oxidized phospholipids in the models of ventilator-induced lung injury. Exp. Cell Res. 317, 859–872. doi: 10.1016/j.yexcr.2010.11.011
Boscher, C., and Nabi, I. R. (2012). Caveolin-1: role in cell signaling. Adv. Exp. Med. Biol. 729, 29–50. doi: 10.1007/978-1-4614-1222-9_3
Brower, R. G., Matthay, M. A., Morris, A., Schoenfeld, D., Thompson, B. T., and Wheeler, A. (2000). Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 342, 1301–1308. doi: 10.1056/NEJM200005043421801
Cahalan, S. M., Lukacs, V., Ranade, S. S., Chien, S., Bandell, M., and Patapoutian, A. (2015). Piezo1 links mechanical forces to red blood cell volume. elife 4:e07370. doi: 10.7554/eLife.07370
Campbell, I. D., and Humphries, M. J. (2011). Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 3:a004994. doi: 10.1101/cshperspect.a004994
Chen, W. G., Epshtein, Y., Ni, X. Q., Dull, R. O., Cress, A. E., Garcia, J. G. N., et al. (2015). Role of integrin beta 4 in lung endothelial cell inflammatory responses to mechanical stress. Sci. Rep. 5:16529. doi: 10.1038/srep16529
Chistiakov, D. A., Orekhov, A. N., and Bobryshev, Y. V. (2016). The role of miR-126 in embryonic angiogenesis, adult vascular homeostasis, and vascular repair and its alterations in atherosclerotic disease. J. Mol. Cell. Cardiol. 97, 47–55. doi: 10.1016/j.yjmcc.2016.05.007
Chiu, Y. J., Kusano, K., Thomas, T. N., and Fujiwara, K. (2004). Endothelial cell-cell adhesion and mechanosignal transduction. Endothelium 11, 59–73. doi: 10.1080/10623320490432489
Chu, M., Qin, S., Wu, R., Zhou, X., Tang, X., Zhang, S., et al. (2016). Role of MiR-126a-3p in endothelial injury in endotoxic mice. Crit. Care Med. 44, e639–e650. doi: 10.1097/CCM.0000000000001629
Coon, B. G., Baeyens, N., Han, J., Budatha, M., Ross, T. D., Fang, J. S., et al. (2015). Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 208, 975–986. doi: 10.1083/jcb.201408103
Coste, B., Mathur, J., Schmidt, M., Earley, T. J., Ranade, S., Petrus, M. J., et al. (2010). Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60. doi: 10.1126/science.1193270
Cowan, D. B., Lye, S. J., and Langille, B. L. (1998). Regulation of vascular connexin43 gene expression by mechanical loads. Circ. Res. 82, 786–793. doi: 10.1161/01.RES.82.7.786
Cummins, P. M. (2012). Occludin: one protein, many forms. Mol. Cell. Biol. 32, 242–250. doi: 10.1128/MCB.06029-11
Dai, H., Zhang, S., Du, X., Zhang, W., Jing, R., Wang, X., et al. (2019a). RhoA inhibitor suppresses the production of microvesicles and rescues high ventilation induced lung injury. Int. Immunopharmacol. 72, 74–81. doi: 10.1016/j.intimp.2019.03.059
Dai, Y. L., Wu, C. P., Yang, G. G., Chang, H., Peng, C. K., and Huang, K. L. (2019b). Adaptive support ventilation attenuates ventilator induced lung injury: human and animal study. Int. J. Mol. Sci. 20:5848. doi: 10.3390/ijms20235848
Dejana, E., Orsenigo, F., and Lampugnani, M. G. (2008). The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121, 2115–2122. doi: 10.1242/jcs.017897
Dejana, E., and Vestweber, D. (2013). The role of VE-cadherin in vascular morphogenesis and permeability control. Prog. Mol. Biol. Transl. Sci. 116, 119–144. doi: 10.1016/b978-0-12-394311-8.00006-6
Di, A., and Malik, A. B. (2010). TRP channels and the control of vascular function. Curr. Opin. Pharmacol. 10, 127–132. doi: 10.1016/j.coph.2009.11.010
Ding, X., Wang, X., Zhao, X., Jin, S., Tong, Y., Ren, H., et al. (2015). RGD peptides protects against acute lung injury in septic mice through Wisp1-integrin beta6 pathway inhibition. Shock 43, 352–360. doi: 10.1097/SHK.0000000000000313
Ding, X. B., Tong, Y., Jin, S. Q., Chen, Z. X., Li, T. L., Billiar, T. R., et al. (2018). Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1-alpha v beta 5 integrin pathway in TLR4-mediated inflammation and injury. Crit. Care 22:302. doi: 10.1186/s13054-018-2237-0
Dogne, S., and Flamion, B. (2020). Endothelial glycocalyx impairment in disease: focus on hyaluronan shedding. Am. J. Pathol. 190, 768–780. doi: 10.1016/j.ajpath.2019.11.016
Dong, W. W., Liu, Y. J., Lv, Z., Mao, Y. F., Wang, Y. W., Zhu, X. Y., et al. (2015). Lung endothelial barrier protection by resveratrol involves inhibition of HMGB1 release and HMGB1-induced mitochondrial oxidative damage via an Nrf2-dependent mechanism. Free Radic. Biol. Med. 88, 404–416. doi: 10.1016/j.freeradbiomed.2015.05.004
Dreyfuss, D., Basset, G., Soler, P., and Saumon, G. (1985). Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am. Rev. Respir. Dis. 132, 880–884. doi: 10.1164/arrd.1985.132.4.880
Dreyfuss, D., and Saumon, G. (1993). Role of tidal volume, FRC, and end-inspiratory volume in the development of pulmonary edema following mechanical ventilation. Am. Rev. Respir. Dis. 148, 1194–1203. doi: 10.1164/ajrccm/148.5.1194
Dreyfuss, D., and Saumon, G. (1998). Ventilator-induced lung injury: lessons from experimental studies. Am. J. Respir. Crit. Care Med. 157, 294–323. doi: 10.1164/ajrccm.157.1.9604014
Dudek, S. M., Chiang, E. T., Camp, S. M., Guo, Y., Zhao, J., Brown, M. E., et al. (2010). Abl tyrosine kinase phosphorylates nonmuscle myosin light chain kinase to regulate endothelial barrier function. Mol. Biol. Cell 21, 4042–4056. doi: 10.1091/mbc.E09-10-0876
Egan, E. A. (1982). Lung inflation, lung solute permeability, and alveolar edema. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 53, 121–125. doi: 10.1152/jappl.1982.53.1.121
Fan, E., Brodie, D., and Slutsky, A. S. (2018). Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA 319, 698–710. doi: 10.1001/jama.2017.21907
Feng, Z., Wang, J. W., Wang, Y., Dong, W. W., and Xu, Z. F. (2019). Propofol protects lung endothelial barrier function by suppression of high-mobility group box 1 (HMGB1) release and mitochondrial oxidative damage catalyzed by HMGB1. Med. Sci. Monit. 25, 3199–3211. doi: 10.12659/MSM.915417
Fernandez-Hernando, C., and Suarez, Y. (2018). MicroRNAs in endothelial cell homeostasis and vascular disease. Curr. Opin. Hematol. 25, 227–236. doi: 10.1097/MOH.0000000000000424
Fontijn, R. D., Volger, O. L., Fledderus, J. O., Reijerkerk, A., de Vries, H. E., and Horrevoets, A. J. (2008). SOX-18 controls endothelial-specific claudin-5 gene expression and barrier function. Am. J. Physiol. Heart Circ. Physiol. 294, H891–H900. doi: 10.1152/ajpheart.01248.2007
Force, A. D. T., Ranieri, V. M., Rubenfeld, G. D., Thompson, B. T., Ferguson, N. D., Caldwell, E., et al. (2012). Acute respiratory distress syndrome: the Berlin definition. JAMA 307, 2526–2533. doi: 10.1001/jama.2012.5669
Forster, C. (2008). Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 130, 55–70. doi: 10.1007/s00418-008-0424-9
Furuse, M., Hirase, T., Itoh, M., Nagafuchi, A., Yonemura, S., Tsukita, S., et al. (1993). Occludin – a novel integral membrane-protein localizing at tight junctions. J. Cell Biol. 123, 1777–1788. doi: 10.1083/jcb.123.6.1777
Garcia, J. G., and Schaphorst, K. L. (1995). Regulation of endothelial cell gap formation and paracellular permeability. J. Investig. Med. 43, 117–126.
Gawlak, G., Son, S., Tian, Y., O'Donnell, J. J. 3rd, Birukov, K. G., and Birukova, A. A. (2016). Chronic high-magnitude cyclic stretch stimulates EC inflammatory response via VEGF receptor 2-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 310, L1062–L1070. doi: 10.1152/ajplung.00317.2015
Ge, J., Li, W., Zhao, Q., Li, N., Chen, M., Zhi, P., et al. (2015). Architecture of the mammalian mechanosensitive Piezo1 channel. Nature 527, 64–69. doi: 10.1038/nature15247
Geng, J., Zhao, Q., Zhang, T., and Xiao, B. (2017). In touch with the mechanosensitive piezo channels: structure, ion permeation, and mechanotransduction. Curr. Top. Membr. 79, 159–195. doi: 10.1016/bs.ctm.2016.11.006
Genova, T., Gaglioti, D., and Munaron, L. (2020). Regulation of vessel permeability by TRP channels. Front. Physiol. 11:421. doi: 10.3389/fphys.2020.00421
Gingras, A. R., and Ginsberg, M. H. (2020). Signal transduction: physical deformation of the membrane activates integrins. Curr. Biol. 30, R397–R400. doi: 10.1016/j.cub.2020.02.068
Giusti, B., Margheri, F., Rossi, L., Lapini, I., Magi, A., Serrati, S., et al. (2013). Desmoglein-2-integrin beta-8 interaction regulates actin assembly in endothelial cells: deregulation in systemic sclerosis. PLoS One 8:e68117. doi: 10.1371/annotation/b41766f2-c23d-455e-8d6e-e4bce5ae1d80
Gross, C. M., Aggarwal, S., Kumar, S., Tian, J., Kasa, A., Bogatcheva, N., et al. (2014). Sox18 preserves the pulmonary endothelial barrier under conditions of increased shear stress. J. Cell. Physiol. 229, 1802–1816. doi: 10.1002/jcp.24633
Hakanpaa, L., Kiss, E. A., Jacquemet, G., Miinalainen, I., Lerche, M., Guzman, C., et al. (2018). Targeting beta1-integrin inhibits vascular leakage in endotoxemia. Proc. Natl. Acad. Sci. U. S. A. 115, E6467–E6476. doi: 10.1073/pnas.1722317115
Hamanaka, K., Jian, M. Y., Weber, D. S., Alvarez, D. F., Townsley, M. I., Al-Mehdi, A. B., et al. (2007). TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L923–L932. doi: 10.1152/ajplung.00221.2007
Hartsock, A., and Nelson, W. J. (2008). Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669. doi: 10.1016/j.bbamem.2007.07.012
Hautefort, A., Pfenniger, A., and Kwak, B. R. (2019). Endothelial connexins in vascular function. Vasc. Biol. 1, H117–H124. doi: 10.1530/VB-19-0015
He, L., and Hannon, G. J. (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. doi: 10.1038/nrg1379
Hoetzel, A., Schmidt, R., Vallbracht, S., Goebel, U., Dolinay, T., Kim, H. P., et al. (2009). Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit. Care Med. 37, 1708–1715. doi: 10.1097/CCM.0b013e31819efa31
Huang, R. T., Wu, D., Meliton, A., Oh, M. J., Krause, M., Lloyd, J. A., et al. (2017). Experimental lung injury reduces kruppel-like factor 2 to increase endothelial permeability via regulation of RAPGEF3-Rac1 signaling. Am. J. Respir. Crit. Care Med. 195, 639–651. doi: 10.1164/rccm.201604-0668OC
Ilkan, Z., Wright, J. R., Goodall, A. H., Gibbins, J. M., Jones, C. I., and Mahaut-Smith, M. P. (2017). Evidence for shear-mediated Ca2+ entry through mechanosensitive cation channels in human platelets and a megakaryocytic cell line. J. Biol. Chem. 292, 9204–9217. doi: 10.1074/jbc.M116.766196
Jiang, L., Zhang, Y., Lu, D., Huang, T., Yan, K., Yang, W., et al. (2021). Mechanosensitive Piezo1 channel activation promotes ventilator-induced lung injury via disruption of endothelial junctions in ARDS rats. Biochem. Biophys. Res. Commun. 556, 79–86. doi: 10.1016/j.bbrc.2021.03.163
Jin, S., Chen, Z., Ding, X., Zhao, X., Jiang, X., Tong, Y., et al. (2016). Mechanical ventilation augments poly(I:C)induced lung injury via A WISP1-integrin beta3 dependent pathway in mice. Mol. Med. 22, 54–63. doi: 10.2119/molmed.2015.00233
Kakogiannos, N., Ferrari, L., Giampietro, C., Scalise, A. A., Maderna, C., Rava, M., et al. (2020). JAM-A acts via C/EBP-alpha to promote claudin-5 expression and enhance endothelial barrier function. Circ. Res. 127, 1056–1073. doi: 10.1161/CIRCRESAHA.120.316742
Kandasamy, K., Escue, R., Manna, J., Adebiyi, A., and Parthasarathi, K. (2015). Changes in endothelial connexin 43 expression inversely correlate with microvessel permeability and VE-cadherin expression in endotoxin-challenged lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L584–L592. doi: 10.1152/ajplung.00211.2014
Katira, B. H., Giesinger, R. E., Engelberts, D., Zabini, D., Kornecki, A., Otulakowski, G., et al. (2017). Adverse heart-lung interactions in ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 196, 1411–1421. doi: 10.1164/rccm.201611-2268OC
Ke, Y., Karki, P., Zhang, C., Li, Y., Nguyen, T., Birukov, K. G., et al. (2019). Mechanosensitive Rap1 activation promotes barrier function of lung vascular endothelium under cyclic stretch. Mol. Biol. Cell 30, 959–974. doi: 10.1091/mbc.E18-07-0422
Kechagia, J. Z., Ivaska, J., and Roca-Cusachs, P. (2019). Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473. doi: 10.1038/s41580-019-0134-2
Kelly, G. T., Faraj, R., Zhang, Y., Maltepe, E., Fineman, J. R., Black, S. M., et al. (2019). Pulmonary endothelial mechanical sensing and signaling, a story of focal adhesions and integrins in ventilator induced lung injury. Front. Physiol. 10:511. doi: 10.3389/fphys.2019.00511
Kira, S., Mori, M., Takatani, J., Uchino, T., Yasuda, N., Miyakawa, H., et al. (2006). Effects of high peak airway pressure on the expression of heat shock protein 70 in rat lungs: a preliminary study. Acta Anaesthesiol. Scand. 50, 469–474. doi: 10.1111/j.1399-6576.2005.00942.x
Komarova, Y., and Malik, A. B. (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 72, 463–493. doi: 10.1146/annurev-physiol-021909-135833
Kuipers, M. T., van der Poll, T., Schultz, M. J., and Wieland, C. W. (2011). Bench-to-bedside review: damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit. Care 15, 235. doi: 10.1186/cc10437
Kumar, S., Kim, C. W., Simmons, R. D., and Jo, H. (2014). Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 34, 2206–2216. doi: 10.1161/ATVBAHA.114.303425
Kunert-Keil, C., Bisping, F., Kruger, J., and Brinkmeier, H. (2006). Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genomics 7:159. doi: 10.1186/1471-2164-7-159
Lansman, J. B., Hallam, T. J., and Rink, T. J. (1987). Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers? Nature 325, 811–813. doi: 10.1038/325811a0
Li, J., Hou, B., Tumova, S., Muraki, K., Bruns, A., Ludlow, M. J., et al. (2014). Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282. doi: 10.1038/nature13701
Li, J., Wen, A. M., Potla, R., Benshirim, E., Seebarran, A., Benz, M. A., et al. (2019). AAV-mediated gene therapy targeting TRPV4 mechanotransduction for inhibition of pulmonary vascular leakage. APL Bioeng. 3:046103. doi: 10.1063/1.5122967
Li, L. F., Lee, C. S., Liu, Y. Y., Chang, C. H., Lin, C. W., Chiu, L. C., et al. (2015). Activation of Src-dependent Smad3 signaling mediates the neutrophilic inflammation and oxidative stress in hyperoxia-augmented ventilator-induced lung injury. Respir. Res. 16, 112. doi: 10.1186/s12931-015-0275-6
Li, Q., Ge, Y. L., Li, M., Fang, X. Z., Yuan, Y. P., Liang, L., et al. (2017). miR-127 contributes to ventilator-induced lung injury. Mol. Med. Rep. 16, 4119–4126. doi: 10.3892/mmr.2017.7109
Li, X., Li, R., Fang, Q., Jamal, M., Wang, C., Wang, Y., et al. (2020). Oxycodone attenuates vascular leak and lung inflammation in a clinically relevant two-hit rat model of acute lung injury. Cytokine 138:155346. doi: 10.1016/j.cyto.2020.155346
Li, X., Li, R., Fang, Q., Jamal, M., Wang, C., Wang, Y., et al. (2021). Oxycodone attenuates vascular leak and lung inflammation in a clinically relevant two-hit rat model of acute lung injury. Cytokine 138:155346. doi: 10.1016/j.cyto.2020.155346
Liang, G. P., Xu, J., Cao, L. L., Zeng, Y. H., Chen, B. X., Yang, J., et al. (2019). Piezo1 induced apoptosis of type II pneumocytes during ARDS. Respir. Res. 20:118. doi: 10.1186/s12931-019-1083-1
Liao, D., Mei, H., Hu, Y., Newman, D. K., and Newman, P. J. (2018). CRISPR-mediated deletion of the PECAM-1 cytoplasmic domain increases receptor lateral mobility and strengthens endothelial cell junctional integrity. Life Sci. 193, 186–193. doi: 10.1016/j.lfs.2017.11.002
Lin, Z., Natesan, V., Shi, H., Dong, F., Kawanami, D., Mahabeleshwar, G. H., et al. (2010). Kruppel-like factor 2 regulates endothelial barrier function. Arterioscler. Thromb. Vasc. Biol. 30, 1952–1959. doi: 10.1161/ATVBAHA.110.211474
Liu, K., Mao, Y. F., Zheng, J., Peng, Z. Y., Liu, W. W., Liu, Y., et al. (2013). SC5b-9-induced pulmonary microvascular endothelial hyperpermeability participates in ventilator-induced lung injury. Cell Biochem. Biophys. 67, 1421–1431. doi: 10.1007/s12013-013-9675-8
Liu, M., Gu, C., and Wang, Y. (2014). Upregulation of the tight junction protein occludin: effects on ventilation-induced lung injury and mechanisms of action. BMC Pulm. Med. 14:94. doi: 10.1186/1471-2466-14-94
Maniatis, N. A., Kardara, M., Hecimovich, D., Letsiou, E., Castellon, M., Roussos, C., et al. (2012). Role of caveolin-1 expression in the pathogenesis of pulmonary edema in ventilator-induced lung injury. Pulm. Circ. 2, 452–460. doi: 10.4103/2045-8932.105033
Mascarenhas, M. M., Day, R. M., Ochoa, C. D., Choi, W. I., Yu, L., Ouyang, B., et al. (2004). Low molecular weight hyaluronan from stretched lung enhances interleukin-8 expression. Am. J. Respir. Cell Mol. Biol. 30, 51–60. doi: 10.1165/rcmb.2002-0167OC
McNally, B. A., Somasundaram, A., Jairaman, A., Yamashita, M., and Prakriya, M. (2013). The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J. Physiol. 591, 2833–2850. doi: 10.1113/jphysiol.2012.250456
Mehta, D., and Malik, A. B. (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367. doi: 10.1152/physrev.00012.2005
Meliton, A., Meng, F., Tian, Y., Shah, A. A., Birukova, A. A., and Birukov, K. G. (2015). Role of Krev interaction Trapped-1 in prostacyclin-induced protection against lung vascular permeability induced by excessive mechanical forces and thrombin receptor activating peptide 6. Am. J. Respir. Cell Mol. Biol. 53, 834–843. doi: 10.1165/rcmb.2014-0376OC
Meza, D., Musmacker, B., Steadman, E., Stransky, T., Rubenstein, D. A., and Yin, W. (2019). Endothelial cell biomechanical responses are dependent on both fluid shear stress and tensile strain. Cell. Mol. Bioeng. 12, 311–325. doi: 10.1007/s12195-019-00585-0
Michalick, L., Erfinanda, L., Weichelt, U., van der Giet, M., Liedtke, W., and Kuebler, W. M. (2017). Transient receptor potential vanilloid 4 and serum glucocorticoid-regulated kinase 1 are critical mediators of lung injury in overventilated mice in vivo. Anesthesiology 126, 300–311. doi: 10.1097/ALN.0000000000001443
Mitra, S., Epshtein, Y., Sammani, S., Quijada, H., Chen, W., Bandela, M., et al. (2021). UCHL1, a deubiquitinating enzyme, regulates lung endothelial cell permeability in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 320, L497–L507. doi: 10.1152/ajplung.00492.2020
Mittal, M., Nepal, S., Tsukasaki, Y., Hecquet, C. M., Soni, D., Rehman, J., et al. (2017). Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ. Res. 121, 1081–1091. doi: 10.1161/CIRCRESAHA.117.311747
Miyawaki-Shimizu, K., Predescu, D., Shimizu, J., Broman, M., Predescu, S., and Malik, A. B. (2006). siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am. J. Phys. Lung Cell. Mol. Phys. 290, L405–L413. doi: 10.1152/ajplung.00292.2005
Morita, K., Sasaki, H., Furuse, M., and Tsukita, S. (1999). Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 147, 185–194. doi: 10.1083/jcb.147.1.185
Murray, J. F. (2011). Pulmonary edema: pathophysiology and diagnosis. Int. J. Tuberc. Lung Dis. 15, 155–160.
Nagasawa, K., Chiba, H., Fujita, H., Kojima, T., Saito, T., Endo, T., et al. (2006). Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J. Cell. Physiol. 208, 123–132. doi: 10.1002/jcp.20647
Nilius, B., and Honore, E. (2012). Sensing pressure with ion channels. Trends Neurosci. 35, 477–486. doi: 10.1016/j.tins.2012.04.002
O'Donnell, J. J. 3rd, Birukova, A. A., Beyer, E. C., and Birukov, K. G. (2014). Gap junction protein connexin43 exacerbates lung vascular permeability. PLoS One 9:e100931. doi: 10.1371/journal.pone.0100931
Ogawa, E. N., Ishizaka, A., Tasaka, S., Koh, H., Ueno, H., Amaya, F., et al. (2006). Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 174, 400–407. doi: 10.1164/rccm.200605-699OC
Ou, Z. W., Dolmatova, E., Lassegue, B., and Griendling, K. K. (2021). Beta 1-and beta 2-integrins: central players in regulating vascular permeability and leukocyte recruitment during acute inflammation. Am. J. Phys. Heart Circ. Phys. 320, H734–H739. doi: 10.1152/ajpheart.00518.2020
Parker, J. C., Hashizumi, M., Kelly, S. V., Francis, M., Mouner, M., Meyer, A. L., et al. (2013). TRPV4 calcium entry and surface expression attenuated by inhibition of myosin light chain kinase in rat pulmonary microvascular endothelial cells. Physiol. Rep. 1:e00121. doi: 10.1002/phy2.121
Parker, J. C., Hernandez, L. A., Longenecker, G. L., Peevy, K., and Johnson, W. (1990). Lung edema caused by high peak inspiratory pressures in dogs. Role of increased microvascular filtration pressure and permeability. Am. Rev. Respir. Dis. 142, 321–328. doi: 10.1164/ajrccm/142.2.321
Parker, J. C., Ivey, C. L., and Tucker, J. A. (1998). Gadolinium prevents high airway pressure-induced permeability increases in isolated rat lungs. J. Appl. Physiol. 84, 1113–1118. doi: 10.1152/jappl.1998.84.4.1113
Parthasarathi, K. (2018). The pulmonary vascular barrier: insights into structure, function, and regulatory mechanisms. Adv. Anat. Embryol. Cell Biol. 228, 41–61. doi: 10.1007/978-3-319-68483-3_3
Pohl, U. (2020). Connexins: key players in the control of vascular plasticity and function. Physiol. Rev. 100, 525–572. doi: 10.1152/physrev.00010.2019
Prakriya, M. (2013). Store-operated Orai channels: structure and function. Curr. Top. Membr. 71, 1–32. doi: 10.1016/B978-0-12-407870-3.00001-9
Privratsky, J. R., and Newman, P. J. (2014). PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res. 355, 607–619. doi: 10.1007/s00441-013-1779-3
Ranade, S. S., Qiu, Z. Z., Woo, S. H., Hur, S. S., Murthy, S. E., Cahalan, S. M., et al. (2014). Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. U. S. A. 111, 10347–10352. doi: 10.1073/pnas.1409233111
Rho, S. S., Ando, K., and Fukuhara, S. (2017). Dynamic regulation of vascular permeability by vascular endothelial cadherin-mediated endothelial cell-cell junctions. J. Nippon Med. Sch. 84, 148–159. doi: 10.1272/jnms.84.148
Salameh, A., and Dhein, S. (2013). Effects of mechanical forces and stretch on intercellular gap junction coupling. Biochim. Biophys. Acta 1828, 147–156. doi: 10.1016/j.bbamem.2011.12.030
Samapati, R., Yang, Y., Yin, J., Stoerger, C., Arenz, C., Dietrich, A., et al. (2012). Lung endothelial Ca2+ and permeability response to platelet-activating factor is mediated by acid sphingomyelinase and transient receptor potential classical 6. Am. J. Respir. Crit. Care Med. 185, 160–170. doi: 10.1164/rccm.201104-0717OC
Sarelius, I. H., and Glading, A. J. (2015). Control of vascular permeability by adhesion molecules. Tissue Barriers 3:e985954. doi: 10.4161/21688370.2014.985954
Schwingshackl, A. (2016). The role of stretch-activated ion channels in acute respiratory distress syndrome: finally a new target? Am. J. Physiol. Lung Cell. Mol. Physiol. 311, L639–L652. doi: 10.1152/ajplung.00458.2015
Seul, K. H., Kang, K. Y., Lee, K. S., Kim, S. H., and Beyer, E. C. (2004). Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem. Biophys. Res. Commun. 319, 1144–1151. doi: 10.1016/j.bbrc.2004.05.097
Shihata, W. A., Michell, D. L., Andrews, K. L., and Chin-Dusting, J. P. (2016). Caveolae: a role in endothelial inflammation and mechanotransduction? Front. Physiol. 7:628. doi: 10.3389/fphys.2016.00628
Simmons, S., Erfinanda, L., Bartz, C., and Kuebler, W. M. (2019). Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 597, 997–1021. doi: 10.1113/JP276245
Simon, S. A., and Liedtke, W. (2008). How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. J. Clin. Investig. 118, 2383–2386. doi: 10.1172/JCI36111
Slutsky, A. S., and Ranieri, V. M. (2013). Ventilator-induced lung injury. N. Engl. J. Med. 369, 2126–2136. doi: 10.1056/NEJMra1208707
Smani, T., Gomez, L. J., Regodon, S., Woodard, G. E., Siegfried, G., Khatib, A. M., et al. (2018). TRP channels in angiogenesis and other endothelial functions. Front. Physiol. 9:1731. doi: 10.3389/fphys.2018.01731
Snyder, J. L., McBeath, E., Thomas, T. N., Chiu, Y. J., Clark, R. L., and Fujiwara, K. (2017). Mechanotransduction properties of the cytoplasmic tail of PECAM-1. Biol. Cell. 109, 312–321. doi: 10.1111/boc.201600079
Song, X., Liu, Y., Dong, L., and Wang, Y. (2018). Stromal-interacting molecule 1 (Stim1)/Orai1 modulates endothelial permeability in ventilator-induced lung injury. Med. Sci. Monit. 24, 9413–9423. doi: 10.12659/MSM.911268
Soni, D., Regmi, S. C., Wang, D. M., DebRoy, A., Zhao, Y. Y., Vogel, S. M., et al. (2017). Pyk2 phosphorylation of VE-PTP downstream of STIM1-induced Ca(2+) entry regulates disassembly of adherens junctions. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L1003–L1017. doi: 10.1152/ajplung.00008.2017
Spitz, F., and Furlong, E. E. (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. doi: 10.1038/nrg3207
Su, G., Hodnett, M., Wu, N., Atakilit, A., Kosinski, C., Godzich, M., et al. (2007). Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am. J. Respir. Cell Mol. Biol. 36, 377–386. doi: 10.1165/rcmb.2006-0238OC
Su, K., Wang, J., Lv, Y., Tian, M., Zhao, Y. Y., Minshall, R. D., et al. (2021). YAP expression in endothelial cells prevents ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 320, L568–L582. doi: 10.1152/ajplung.00472.2020
Sun, X. G., Elangovan, V. R., Mapes, B., Camp, S. M., Sammani, S., Saadat, L., et al. (2014). The NAMPT promoter is regulated by mechanical stress, signal transducer and activator of transcription 5, and acute respiratory distress syndrome-associated genetic variants. Am. J. Respir. Cell Mol. Biol. 51, 660–667. doi: 10.1165/rcmb.2014-0117OC
Sun, Y., Hu, G. C., Zhang, X. M., and Minshall, R. D. (2009). Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 105, 676–U669. doi: 10.1161/CIRCRESAHA.109.201673
Sun, Y., Minshall, R. D., and Hu, G. (2011). Role of caveolin-1 in the regulation of pulmonary endothelial permeability. Methods Mol. Biol. 763, 303–317. doi: 10.1007/978-1-61779-191-8_21
Sun, Z., Costell, M., and Fassler, R. (2019a). Integrin activation by Talin, kindlin and mechanical forces. Nat. Cell Biol. 21, 25–31. doi: 10.1038/s41556-018-0234-9
Sun, Z., Wang, F., Yang, Y., Wang, J., Sun, S., Xia, H., et al. (2019b). Resolvin D1 attenuates ventilator-induced lung injury by reducing HMGB1 release in a HO-1-dependent pathway. Int. Immunopharmacol. 75:105825. doi: 10.1016/j.intimp.2019.105825
Tabata, T., Kawakatsu, H., Maidji, E., Sakai, T., Sakai, K., Fang-Hoover, J., et al. (2008). Induction of an epithelial integrin alphavbeta6 in human cytomegalovirus-infected endothelial cells leads to activation of transforming growth factor-beta1 and increased collagen production. Am. J. Pathol. 172, 1127–1140. doi: 10.2353/ajpath.2008.070448
Thodeti, C. K., Matthews, B., Ravi, A., Mammoto, A., Ghosh, K., Bracha, A. L., et al. (2009). TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ. Res. 104, 1123–U1278. doi: 10.1161/CIRCRESAHA.108.192930
Tian, Y., Gawlak, G., O'Donnell, J. J. 3rd, Birukova, A. A., and Birukov, K. G. (2016). Activation of vascular endothelial growth factor (VEGF) receptor 2 mediates endothelial permeability caused by cyclic stretch. J. Biol. Chem. 291, 10032–10045. doi: 10.1074/jbc.M115.690487
Tiruppathi, C., Ahmmed, G. U., Vogel, S. M., and Malik, A. B. (2006). Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13, 693–708. doi: 10.1080/10739680600930347
Tschumperlin, D. J., and Margulies, S. S. (1999). Alveolar epithelial surface area-volume relationship in isolated rat lungs. J. Appl. Physiol. 86, 2026–2033. doi: 10.1152/jappl.1999.86.6.2026
Uhlig, S., Yang, Y., Waade, J., Wittenberg, C., Babendreyer, A., and Kuebler, W. M. (2014). Differential regulation of lung endothelial permeability in vitro and in situ. Cell. Physiol. Biochem. 34, 1–19. doi: 10.1159/000362980
Vaporidi, K., Vergadi, E., Kaniaris, E., Hatziapostolou, M., Lagoudaki, E., Georgopoulos, D., et al. (2012). Pulmonary microRNA profiling in a mouse model of ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, L199–L207. doi: 10.1152/ajplung.00370.2011
Vaquerizas, J. M., Kummerfeld, S. K., Teichmann, S. A., and Luscombe, N. M. (2009). A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252–263. doi: 10.1038/nrg2538
Villalta, P. C., and Townsley, M. I. (2013). Transient receptor potential channels and regulation of lung endothelial permeability. Pulm. Circ. 3, 802–815. doi: 10.1086/674765
Villar, J., Muros, M., Cabrera-Benitez, N. E., Valladares, F., Lopez-Hernandez, M., Flores, C., et al. (2014). Soluble platelet-endothelial cell adhesion molecule-1, a biomarker of ventilator-induced lung injury. Crit. Care 18, R41. doi: 10.1186/cc13754
Wang, B., Caluch, A., Fodil, R., Fereol, S., Zadigue, P., Pelle, G., et al. (2012a). Force control of endothelium permeability in mechanically stressed pulmonary micro-vascular endothelial cells. Biomed. Mater. Eng. 22, 163–170. doi: 10.3233/BME-2012-0703
Wang, B., Wan, J. Y., Zhang, L., and Min, S. (2012b). Synthetic RGDS peptide attenuates mechanical ventilation-induced lung injury in rats. Exp. Lung Res. 38, 204–210. doi: 10.3109/01902148.2012.664835
Wang, S., Aurora, A. B., Johnson, B. A., Qi, X., McAnally, J., Hill, J. A., et al. (2008). The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15, 261–271. doi: 10.1016/j.devcel.2008.07.002
Wang, T., Gross, C., Desai, A. A., Zemskov, E., Wu, X., Garcia, A. N., et al. (2017a). Endothelial cell signaling and ventilator-induced lung injury: molecular mechanisms, genomic analyses, and therapeutic targets. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L452–L476. doi: 10.1152/ajplung.00231.2016
Wang, X., Zhang, R. L. Z., Tong, Y., Ding, X. B., Jin, S. Q., Zhao, X., et al. (2017b). High-mobility group box 1 protein is involved in the protective effect of Saquinavir on ventilation-induced lung injury in mice. Acta Biochim. Biophys. Sin. 49, 907–915. doi: 10.1093/abbs/gmx085
Wang, Y., Gao, T. T., Xu, D. F., Zhu, X. Y., Dong, W. W., Lv, Z., et al. (2019). Upregulation of sphingosine kinase 1 contributes to ventilator-associated lung injury in a two-hit model. Int. J. Mol. Med. 44, 2077–2090. doi: 10.3892/ijmm.2019.4379
Webb, H. H., and Tierney, D. F. (1974). Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am. Rev. Respir. Dis. 110, 556–565. doi: 10.1164/arrd.1974.110.5.556
Wirtz, H. R., and Dobbs, L. G. (2000). The effects of mechanical forces on lung functions. Respir. Physiol. 119, 1–17. doi: 10.1016/S0034-5687(99)00092-4
Wolfson, R. K., Mapes, B., and Garcia, J. G. N. (2014). Excessive mechanical stress increases HMGB1 expression in human lung microvascular endothelial cells via STAT3. Microvasc. Res. 92, 50–55. doi: 10.1016/j.mvr.2013.12.005
Woodfin, A., Voisin, M. B., and Nourshargh, S. (2007). PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 27, 2514–2523. doi: 10.1161/ATVBAHA.107.151456
Wysolmerski, R. B., and Lagunoff, D. (1991). Regulation of permeabilized endothelial cell retraction by myosin phosphorylation. Am. J. Phys. 261, C32–C40. doi: 10.1152/ajpcell.1991.261.1.C32
Yamamoto, H., Ehling, M., Kato, K., Kanai, K., van Lessen, M., Frye, M., et al. (2015). Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat. Commun. 6:6429. doi: 10.1038/ncomms7429
Yan, X., Li, W., Yang, L., Dong, W., Chen, W., Mao, Y., et al. (2018). MiR-135a protects vascular endothelial cells against ventilator-induced lung injury by inhibiting PHLPP2 to activate PI3K/Akt pathway. Cell. Physiol. Biochem. 48, 1245–1258. doi: 10.1159/000492010
Yeager, M., and Harris, A. L. (2007). Gap junction channel structure in the early 21st century: facts and fantasies. Curr. Opin. Cell Biol. 19, 521–528. doi: 10.1016/j.ceb.2007.09.001
Yin, J., Lv, L., Zhai, P., Long, T., Zhou, Q., Pan, H., et al. (2019). Connexin 40 regulates lung endothelial permeability in acute lung injury via the ROCK1-MYPT1-MLC20 pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 316, L35–l44. doi: 10.1152/ajplung.00012.2018
Yu, Q., Wang, D., Wen, X., Tang, X., Qi, D., He, J., et al. (2020). Adipose-derived exosomes protect the pulmonary endothelial barrier in ventilator-induced lung injury by inhibiting the TRPV4/Ca(2+) signaling pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 318, L723–L741. doi: 10.1152/ajplung.00255.2019
Zhang, J., Yang, G. M., Zhu, Y., Peng, X. Y., Li, T., and Liu, L. M. (2015). Role of connexin 43 in vascular hyperpermeability and relationship to Rock1-MLC20 pathway in septic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1323–L1332. doi: 10.1152/ajplung.00016.2015
Zhang, Y., and Yang, W. X. (2016). Tight junction between endothelial cells: the interaction between nanoparticles and blood vessels. Beilstein J. Nanotechnol. 7, 675–684. doi: 10.3762/bjnano.7.60
Zhao, Q., Zhou, H., Chi, S., Wang, Y., Wang, J., Geng, J., et al. (2018). Structure and mechanogating mechanism of the Piezo1 channel. Nature 554, 487–492. doi: 10.1038/nature25743
Zhao, T., Liu, M., Gu, C., Wang, X., and Wang, Y. (2014). Activation of c-Src tyrosine kinase mediated the degradation of occludin in ventilator-induced lung injury. Respir. Res. 15, 158. doi: 10.1186/s12931-014-0158-2
Zheng, J., Zhang, K., Wang, Y., Cao, J., Zhang, F., Zhou, Q., et al. (2015). Identification of a microRNA signature in endothelial cells with mechanical stretch stimulation. Mol. Med. Rep. 12, 3525–3530. doi: 10.3892/mmr.2015.3835
Zhong, M., Wu, W., Kang, H., Hong, Z., Xiong, S., Gao, X., et al. (2019). Alveolar stretch activation of endothelial Piezo1 protects adherens junctions and lung vascular barrier. Am. J. Respir. Cell Mol. Biol. 62, 168–177. doi: 10.1165/rcmb.2019-0024OC
Keywords: mechanical ventilation, pulmonary vascular endothelium, lung injury, permeability, mechanisms
Citation: Lai Y and Huang Y (2021) Mechanisms of Mechanical Force Induced Pulmonary Vascular Endothelial Hyperpermeability. Front. Physiol. 12:714064. doi: 10.3389/fphys.2021.714064
Edited by:
Zhanqi Zhao, Furtwangen University, GermanyReviewed by:
Pritesh Jain, University of California, San Diego, United StatesXun Zhu, Sun Yat-sen University, China
Copyright © 2021 Lai and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongbo Huang, eW9uZ2JvMjA0NkAxNjMuY29t