 Xiao Chun Li*
Xiao Chun Li* Ana Paula Oliveira Leite
Ana Paula Oliveira Leite Jia Long Zhuo
Jia Long Zhuo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 02 August 2021
Sec. Renal Physiology and Pathophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.702797
This article is part of the Research Topic Hypertension and Chronic Kidney Injury or Failure, Volume II View all 6 articles
Hypertension is well recognized to be the most important risk factor for cardiovascular diseases, stroke, and end-stage kidney failure. A quarter of the world’s adult populations and 46% of the US adults develop hypertension and currently require antihypertensive treatments. Only 50% of hypertensive patients are responsive to current antihypertensive drugs, whereas remaining patients may continue to develop cardiovascular, stroke, and kidney diseases. The mechanisms underlying the poorly controlled hypertension remain incompletely understood. Recently, we have focused our efforts to uncover additional renal mechanisms, pathways, and therapeutic targets of poorly controlled hypertension and target organ injury using novel animal models or innovative experimental approaches. Specifically, we studied and elucidated the important roles of intratubular, intracellular, and mitochondrial angiotensin II (Ang II) system in the development of Ang II-dependent hypertension. The objectives of this invited article are to review and discuss our recent findings that (a) circulating and intratubular Ang II is taken up by the proximal tubules via the (AT1) AT1a receptor-dependent mechanism, (b) intracellular administration of Ang II in proximal tubule cells or adenovirus-mediated overexpression of an intracellular Ang II fusion protein selectively in the mitochonria of the proximal tubules induces blood pressure responses, and (c) genetic deletion of AT1 (AT1a) receptors or the Na+/H+ exchanger 3 selectively in the proximal tubules decreases basal blood pressure and attenuates Ang II-induced hypertension. These studies provide a new perspective into the important roles of the intratubular, intracellular, and mitochondrial angiotensin II/AT1 (AT1a) receptor signaling in Ang II-dependent hypertensive kidney diseases.
Hypertension is a well-established risk factor for morbidity and mortality associated with coronary artery disease, stroke, and chronic kidney disease (Muntner et al., 2018; Whelton et al., 2018; Carey et al., 2019). According to the most recent American College of Cardiology/American Heart Association Guidelines, 46% of the United States’ adults have hypertension and will require antihypertensive treatments in their life time (Calhoun et al., 2008; Lloyd-Jones et al., 2010; Sarafidis et al., 2013; Whelton et al., 2018). Yet, only about 50% of hypertensive patients have their blood pressure adequately controlled with current antihypertensive drugs (Carey et al., 2018, 2019; Muntner et al., 2018; Whelton et al., 2018). Although distal nephron-targeting diuretics, the blockers of the renin-angiotensin-aldosterone system, calcium channel blockers, adrenergic β receptor antagonists, and renal nerve radiofrequency ablation are widely used to treat hypertension and prevent target organ damage, some hypertensive patients continue to develop cardiovascular, stroke, and renal injury (Jorde et al., 2000; Calhoun et al., 2008, 2019; Lloyd-Jones et al., 2010; Sarafidis et al., 2013). The mechanisms underlying poorly controlled hypertension and kidney injury and the reasons why it is so difficult to treat these patients still remain incompletely understood. Although circulating Ang II levels are not always elevated in most hypertensive patients, renin inhibitors (O’Brien et al., 2007; Oparil et al., 2007; Lambers Heerspink et al., 2009), ACE inhibitors (Wing et al., 2003; Lloyd-Jones et al., 2010; Muntner et al., 2018; Whelton et al., 2018; Carey et al., 2019), and AT1 receptor blockers (ARBs) significantly lower the blood pressure in a large number of hypertensive patients (Casas et al., 2005; Lloyd-Jones et al., 2010; Muntner et al., 2018; Whelton et al., 2018; Carey et al., 2019). However, clinical trials have shown that not all the RAS-targeting drugs or other classes of antihypertensive drugs afford the same degree of antihypertensive effects and cardiovascular and renal protection (Casas et al., 2005; Bomback and Toto, 2009; Lloyd-Jones et al., 2010; Muntner et al., 2018; Whelton et al., 2018; Carey et al., 2019). It is therefore imperative to continue to uncover new mechanisms and targets of hypertension and design new antihypertensive drugs to prevent and treat poorly controlled hypertension and target organ injury.
In this invited article, we review and discuss the evidence and recently published studies supporting our hypothesis that intratubular Ang II and AT1 (AT1a) receptors in the proximal tubules of the kidney are required for maintaining basal blood pressure homeostasis and for the development of Ang II-induced hypertension and renal injury, and that deletion of AT1a receptors selectively in the proximal tubules will attenuate Ang II-dependent hypertension and renal injury. As the proof of concept studies, we recently used highly innovative proximal tubule-specific, genetically modified mouse models with loss of function (knockout) or gain of function (overexpression) to test this hypothesis and determine: (a) whether intratubular Ang II and AT1a receptors or the downstream target the Na+/H+ exchanger 3 (NHE3) in the proximal tubules are required for maintaining basal blood pressure homeostasis by regulating the pressure natriuresis response (Li et al., 2018, 2019a, 2020, 2021), (b) whether Ang II and AT1a receptors in the proximal tubules are required for the development of Ang II-infused hypertension by resetting the pressure natriuresis response (Li et al., 2021), and (c) whether deletion of Ang II and AT1a receptors or NHE3 selectively in the proximal tubules attenuates Ang II-dependent hypertension and renal injury (Li et al., 2018, 2019a, 2021). The results obtained from these recent in vitro and in vivo studies likely provide new insights and perspectives into the potential roles of the intratubular, intracellular, and mitochondrial Ang II/AT1 (AT1a) receptor signaling in Ang II-dependent hypertensive and kidney diseases. It is hoped that the new knowledge may help stimulate further debates or new studies, and potentially lead to a paradigm shift in our understanding of what roles the proximal tubules and the intratubular Ang II system may play in the pathogenesis of hypertension and renal injury. This new knowledge in turn may help develop novel proximal tubule-targeting drugs to prevent and treat poorly controlled hypertension and kidney injury in humans.
The RAS is now not only recognized as a circulating or endocrine system (tissue-to-tissue) but also increasingly viewed as a functional paracrine (cell-to-cell) and intracrine (intracellular and/or nuclear) system in the proximal nephron of the kidney (De Mello and Danser, 2000; Cook et al., 2006; Zhuo et al., 2006a; Kobori et al., 2007; Kumar et al., 2008; Li et al., 2008, 2015b). Ang II is the most powerful peptide of the RAS to induce classical cardiovascular, renal, and hypertensive effects by activating AT1 (AT1a) receptors (Timmermans et al., 1993; De Gasparo et al., 2000; Touyz and Schiffrin, 2000; Carey and Siragy, 2003; Crowley et al., 2005; Higuchi et al., 2007). In vitro, sustained stimulation of the AT1 (AT1a) receptors leads to its desensitization and loss of vasoconstrictive responses to Ang II (Hein et al., 1997; Zhang et al., 1997; Ferguson, 2001). This phenomenon has led a long-held paradigm for G protein-coupled receptor (GPCR) pharmacology that repeated stimulation of GPCR by agonists will not have long-term pharmacological effects because of its receptor desensitization. However, we and others have shown that infusion of Ang II for weeks continues to induce progressive hypertension and target organ kidney injury (von Thun et al., 1994; Zou et al., 1996; Zhuo et al., 2002; Li et al., 2007; Li and Zhuo, 2008b, 2011, 2013), suggesting that this classical paradigm should be revised to include the intracellular system (Kurtz and Gardner, 1998; De Mello and Danser, 2000; Re, 2000; Kumar et al., 2008). Indeed, Ang II is rapidly internalized with AT1 (AT1a) receptors in target cells, but not all of internalized Ang II is sorted to the lysosome degradation pathway in proximal tubule cells (van Kats et al., 2001; Li et al., 2006, 2007, 2009, 2014; Li and Zhuo, 2014). Some internalized Ang II bypasses the lysosome degradation pathway and is transported to the mitochondria, endoplasmic reticulum, and nucleus, where it continues to induce signaling and long-term transcriptional responses or long-lasting genomic effects by activating mitochondrial and nuclear AT1a receptors (Kurtz and Gardner, 1998; van Kats et al., 2001; Bivona and Philips, 2003; Cottrell et al., 2009; Murphy et al., 2009). We and others have evidence that intracellular administration of Ang II induces the expression of nuclear factor-κB (Brasier et al., 2000; Ruiz-Ortega et al., 2000; Zhuo et al., 2006b, 2016;Schupp et al., 2007; Li and Zhuo, 2008a), monocyte chemoattractant protein 1 (MCP-1; Zhuo, 2004; Li and Zhuo, 2008a; Takahashi et al., 2008), TNF-α (Takahashi et al., 2008), TGF-β1, and NHE3 (Kagami et al., 1994; Wolf et al., 1999; Weigert et al., 2002), and induces production in the mitochondria and nucleus of the proximal tubule cells (Gwathmey et al., 2010a,b; Li et al., 2020). Furthermore, global or proximal tubule-specific overexpression of an intracellular ANG II fusion protein selectively in the proximal tubules of the kidney, Ad-sglt2-ECFP/Ang II (Figure 1), or in the mitochondria of the proximal tubules, Ad-sglt2-mito-ECFP/Ang II, developed antinatriuretic responses and elevated blood pressure by altering the mitochondrial functions (Li et al., 2011b, 2020, 2021; Li and Zhuo, 2013). Overall, these proof of concept studies strongly support a new paradigm of a functional proximal tubule intratubular, intracellular, and mitochondrial Ang II system in the development of hypertension and renal injury.
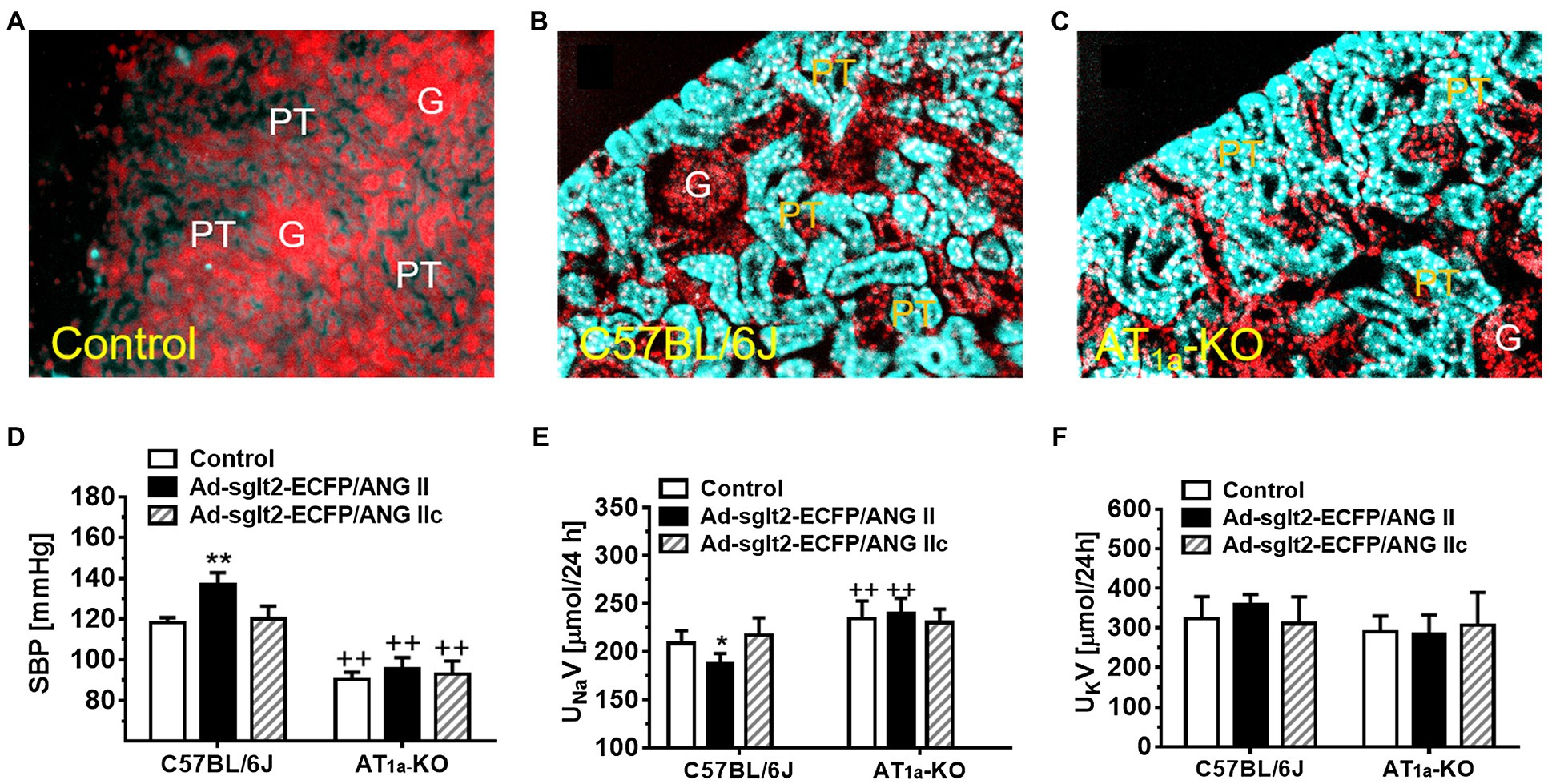
Figure 1. Overexpression of intracellular ECFP/ANG II selectively in the proximal tubule of the kidney in C57BL/6J or AT1a-KO mice and its effects on systolic blood pressure and 24 h urinary sodium (UNaV) and potassium excretion (UKV). (A) A representative control C57BL/6 J kidney showing a low level of autofluorescence in the cortex. (B) A representative control C57BL/6 J kidney showing overexpression of ECFP/ANG II selectively in the proximal tubule of the superficial cortex in blue-green color. (C) A representative AT1a-KO mouse kidney also showing overexpression of ECFP/ANG II selectively in the proximal tubule. Red represents DAPI-stained nuclei in the cortex after conversion from blue color. (D) Effect on systolic blood pressure. (E) Effect on UNaV. (F) Effect on UKV. **p < 0.01 vs. control, whereas ++p < 0.01 vs. ECFP/ANG II overexpression. N = 6–8 for each group. Reproduced with permission (Zhuo et al., 2016).
The pressure-natriuresis response is a central element of the overall feedback mechanism for long-term control of arterial pressure, in which an increase in arterial pressure will lead to a decrease in Na+ reabsorption and a natriuresis response in the kidney and restore blood pressure to normal (Roman, 1986; Cowley and Roman, 1996; Hall et al., 1996; Granger et al., 2002; Li et al., 2018, 2019a, 2021). The pressure natriuresis response is reportedly mediated by: (a) inhibition of proximal tubule Na+ transport (Moreno et al., 2001; Dos Santos et al., 2004), (b) increase in renal interstitial hydrostatic pressure (Li and Zhuo, 2013), (c) increase in renal medullary blood flow (Roman, 1986; Williams et al., 2007), (d) increase in 20-HETE production (Moreno et al., 2001; Dos Santos et al., 2004; Williams et al., 2007), (e) increase in AT2-mediated cGMP production (Siragy and Carey, 1996; Jin et al., 2001, 2004), (f) increased dopamine-induced signaling (Hussain and Lokhandwala, 1998; Banday and Lokhandwala, 2008; Wang et al., 2009), or (g) increased renal nitric oxide (Majid et al., 1993, 1998). None of these factors, however, adequately explains the pressure natriuresis response in hypertension. We reasoned that the proximal tubules are responsible for reabsorbing ~65–~70% of filtered Na+ (Wilcox et al., 1992; Wang et al., 2009; Li and Zhuo, 2013) and Ang II exerts a powerful stimulatory effect on proximal tubule Na+ reabsorption (Harris and Navar, 1985; Cogan, 1990; Wang and Chan, 1990; Li and Zhuo, 2013). We hypothesized that intratubular Ang II via activating AT1 (AT1a) receptors in the proximal tubules plays a key role in the regulation of the pressure natriuresis response and it is resetting in Ang II-dependent hypertension. Indeed, an impaired pressure natriuresis response has been reported in SHR (Roman and Cowley, 1985; Roman, 1987) and animal models of L-NAME- (Majid et al., 1993; Granger and Alexander, 2000), 2-Kidney, 1-Clip (Rostand et al., 1982), TGR (mRen-2)27- (Gross et al., 1994; Zhuo et al., 1999), and Ang II-induced hypertension (Mattson et al., 1991; Wang et al., 2000; Zhuo et al., 2002; Li et al., 2011a, 2021). Most, if not all, of these hypertension models involve the activation of intratubular Ang II and AT1 (AT1a) receptors in the proximal tubules, which stimulates proximal tubule Na+ reabsorption and induces Na+ retention. Nevertheless, the roles of intratubular AT1 (AT1a) receptors in the proximal tubules in the regulation of the pressure natriuresis response and hypertension have not been investigated using strictly proximal tubule-specific, genetically modified animal models. Although AT1 (AT1a) receptors were reportedly deleted from the proximal tubules using the phosphoenolpyruvate carboxykinase (PEPCK) promoter-driven Cre (Gurley et al., 2011) or the androgen-dependent promoter (KAP2)-driven Cre approach (Li et al., 2011a), the specificity or selectivity of PEPCK and KAP2 to drive Cre expression selectively in the proximal tubules remains uncertain. PEPCK is abundantly expressed in the epithelial cells of liver and the digestive system, whereas KAP2 is also expressed extensively in many other androgen-responsive tissues or tubular segments in the kidney (Ding et al., 1997; Li et al., 2008). Despite the lack of specificity, however, these studies were still able to demonstrate an important and lower basal blood pressure phenotype (Gurley et al., 2011; Li et al., 2011b). It may be reasonably argued that the blood pressure phenotype and its response to Ang II in these mutant mouse models may not be due only to the deletion of AT1a receptors in the proximal tubules, but also likely involve the absence of AT1a receptors in other tissues with the expression of PEPCK and KAP2.
To overcome the limitation of these technical approaches, we have recently used the Sglt2-Cre/Agtr1a-foxed recombination to delete AT1 (AT1a) receptors selectively in the proximal tubules of the kidney and to determine the specific roles of intratubular Ang II and AT1 (AT1a) receptors in basal blood pressure homeostasis and the development of hypertension induced by circulating or intracellular Ang II (Rubera et al., 2004; Li et al., 2011b, 2021; Rateri et al., 2011). The hypothesis to be tested was that intratubular Ang II and AT1a receptors in the proximal tubules are required for maintaining normal blood pressure and the development of Ang II-induced hypertension. We treated adult male wild-type, global Agtr1a−/−, and PT-Agtr1a−/− mice with osmotic minipump infusion of a high pressor dose of Ang II (1.5 mg/kg/day, i.p.), a slow pressor dose of Ang II (0.5 mg/kg/day, i.p.), or with adenovirus-mediated overexpression of an intracellular Ang II fusion protein in the proximal tubules of the kidney for 2 weeks (Figure 2; Li et al., 2021). Deletion of AT1a receptors in the proximal tubules led to a decrease in basal telemetry blood pressure by ~15 ± 3 mmHg in PT-Agtr1a−/− than wild-type mice, which was ~13 ± 3 mmHg higher than the whole-body Agtr1a−/− mice. The lower basal blood pressure phenotype was associated with an increase in basal glomerular filtration by ~23.9%, a decrease in fractional proximal tubule Na+ reabsorption, and augmented the pressure-natriuresis response and natriuretic responses to salt loading or Ang III infusion in PT-Agtr1a−/− mice (Li et al., 2021). Furthermore, deletion of AT1a receptors in the proximal tubules attenuated ~50% of Ang II-induced hypertension in PT-Agtr1a−/− mice, compared with wild-type mice, but completely blocked intracellular Ang II fusion protein-induced hypertension in PT-Agtr1a−/− mice (Li et al., 2021). Taken together, the results of this study provide new insights into the critical role of intratubular Ang II/AT1 (AT1a) pathways in the proximal tubules in normal blood pressure control and the development of Ang II-induced hypertension.
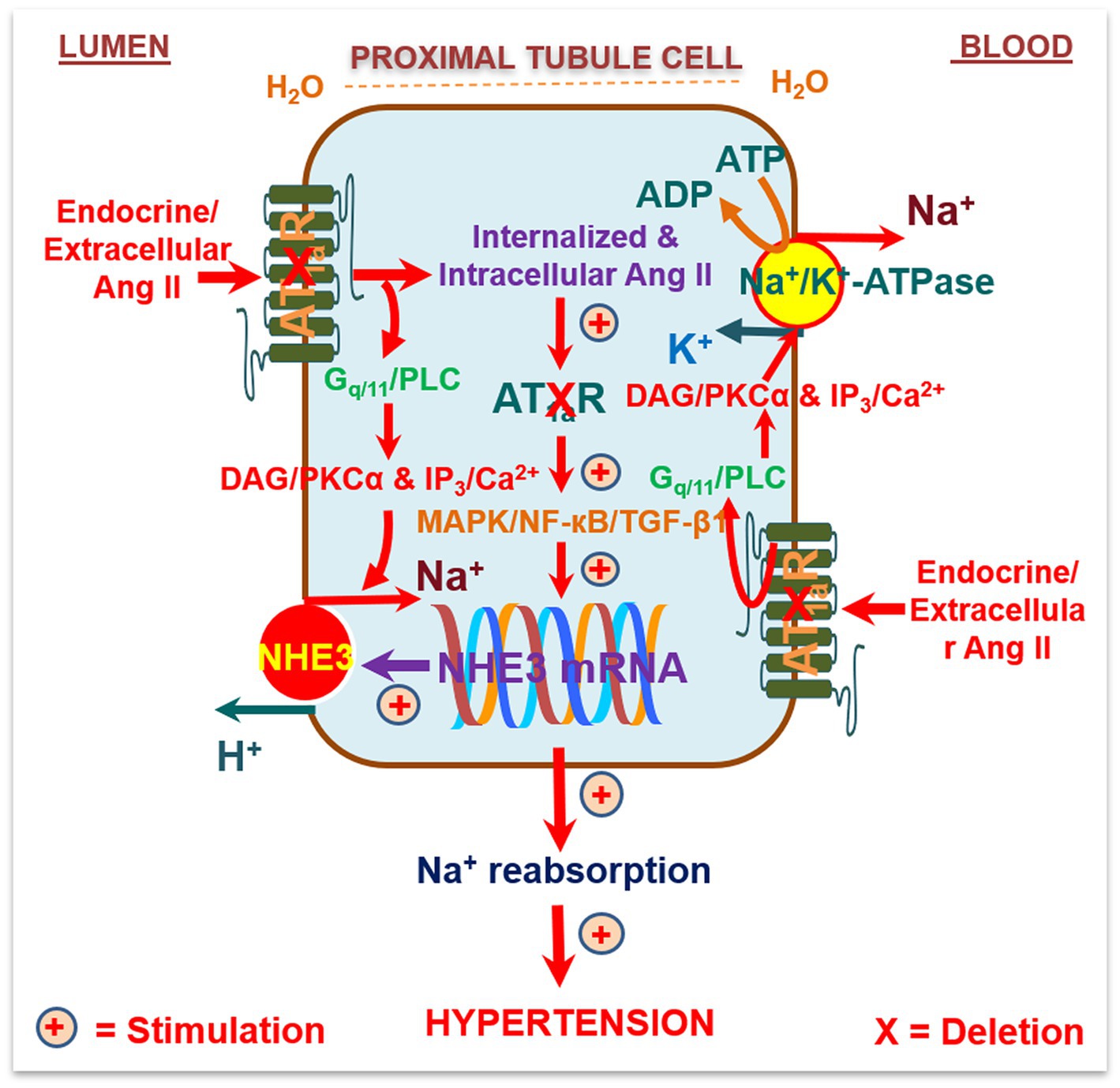
Figure 2. Schematic diagram showing the hypothesis that circulating/extracellular Ang/intratubular II binds to apical (lumen) and basolateral membrane (blood) AT1a receptors, and activates G protein-coupled Gq11/phospholipase C (PLC)/diacylglycerol(DAG)/protein kinase Cα (PKCα) signaling, and/or inositol trisphosphate (IP3)/Ca2+ signaling pathways. This activated signaling increases the activities and expression of Na+/H+ exchanger 3 (NHE3), Na+/K+-ATPase, and other Na+ cotransporters in the proximal tubules to increase Na+ reabsorption and maintain arterial blood pressure under physiological conditions and elevate it during Ang II-induced hypertension. Alternatively, circulating/extracellular Ang II is taken up by the proximal tubules via AT1a receptors and the endocytic receptor protein megalin under physiological conditions and during Ang II-induced hypertension to act as an intracellular/intracrine peptide. The latter will in turn induce long-term transcriptional effects to increase the expression of NHE3, Na+/K+-ATPase, and other Na+ cotransporters in the proximal tubules, promote proximal tubule Na+ reabsorption, and elevate blood pressure. As expected, deletion of AT1a receptors selectively in the proximal tubules of the kidney will attenuate circulating/extracellular and intratubular/intracellular Ang II-induced hypertension in PT-Agtr1a−/− mice. Reproduced with permission (Li et al., 2021).
However, it remains unknown whether deletion of AT1a receptors selectively in the proximal tubules of the kidney may alter the expression of other peptide receptors, such as AT1b, AT2, dopamine, or endothelin (ET) receptors alone, or alter other heterodimer receptors in the proximal tubules. Heterodimer Ang II AT1a and D3 dopamine receptors (Zeng et al., 2003) or heterodimer AT1a and endothelin ETB receptors have been reported previously in renal proximal tubule cells of SHR (Zeng et al., 2005). In the absence of AT1 (AT1a) receptors in the proximal tubules, these receptors may act to inhibit proximal tubule Na+ reabsorption, promote the pressure-natriuresis response, and lower the basal blood pressure or help attenuate Ang II-induced hypertension. Further studies are necessary to determine the roles or interactions between intratubular Ang II, dopamine, natriuretic peptide, or ETB receptor signaling pathways in the proximal tubules in blood pressure control and body salt and fluid balance.
The NHE3 is well recognized to be the most important Na+/H+ antiporter member in the proximal tubules of the kidney (Lorenz et al., 1999; Wang et al., 1999; Vallon et al., 2000; McDonough, 2010; Li et al., 2018, 2019a). NHE3 acts directly to extrude H+ from proximal tubule cells in exchange for luminal Na+ entry, directly contributing to ~25% of active Na+ reabsorption, and after generating a luminal Cl− gradient, to drive passive reabsorption of additional >30% of the filtered Na+ load in the proximal tubules (Aronson, 1983; Rector, 1983; Schafer et al., 1984; Li and Zhuo, 2013). We and others have shown that global knockout of the Nhe3 gene in Nhe3−/− mice decreases Na+ reabsorption in the proximal convoluted tubule by 50% and lowers basal blood pressure by about 15 mmHg (Schultheis et al., 1998; Woo et al., 2003; Noonan et al., 2005; Li et al., 2015a,b). Even with the transgenic rescue of the Nhe3 gene selectively in small intestines of the gastrointestinal tract in tgNhe3−/− mice, basal blood pressure remained significantly lower, suggesting that NHE3 in the kidney plays a critical role in maintain basal blood pressure homeostasis (Woo et al., 2003; Noonan et al., 2005; Li et al., 2015b). Indeed, Fenton et al. were instrumental in generating a new kidney-selective Nhe3−/− mouse model using the Pax8-Cre/NHE3loxlox approach to determine the role of renal tubule NHE3 in blood pressure regulation (Fenton et al., 2017). This approach appears to be superior to tgNhe3−/− mice with the transgenic rescue of the Nhe3 gene selectively in small intestines (Woo et al., 2003; Noonan et al., 2005; Li et al., 2015b). However, Pax8, paired box gene 8, is still expressed widely in the epithelial cells of the kidney tubules, endocervix, endometrium, ovary, Fallopian tube, seminal vesicle, epididymis, pancreatic islet cells, and lymphoid cells (Poleev et al., 1992; Thompson et al., 2021). Thus, this mouse Nhe3−/− model may still be considered as a panepithelial cell-specific or whole-kidney tubule-specific Nhe3−/− model. Despite this limitation, basal blood pressure was found to be 10–20 mmHg lower in Pax8-Cre/NHE3loxlox mice when fed with low or high Na+ diet. Because this basal blood pressure phenotype is largely similar to those of tgNhe3−/− mice, the results of this study are consistent with the hypothesis that NHE3 in the kidney plays a critical role in maintaining basal blood pressure homeostasis.
Recently, we employed a different and more specific approach to generate a mutant mouse model with proximal tubule-specific deletion of NHE3, PT-Nhe3−/−, to test our hypothesis on the important roles of NHE3 in the proximal tubules in basal blood pressure control and Ang II-induced hypertension (Li et al., 2018, 2019a). Specifically, PT-Nhe3−/− mice were generated using the Sglt2-Cre/Nhe3loxlox approach, whereas Ang II-induced hypertension was induced by Ang II infusion via osmotic minipump for 2 weeks (Li et al., 2018, 2019a). We demonstrated that under basal conditions, systolic blood pressure, diastolic blood pressure, and mean arterial blood pressure were significantly lower in male and female PT-Nhe3−/− than wild-type mice. The lower blood pressure phenotype was again associated with significant inhibition of proximal tubule Na+ reabsorption, resulting in significant natriuretic responses and augmented pressure-natriuresis response in PT-Nhe3−/− mice (Li et al., 2018). As expected, Ang II induced robust hypertension in wild-type mice, but the hypertensive effect of Ang II was attenuated by about 50% in male and female PT-Nhe3−/− mice (Figure 3). Furthermore, the pressure-natriuresis response was impaired in Ang II-infused wild-type mice but was augmented in male and female PT-Nhe3−/− mice infused with Ang II (Li et al., 2019a). These results were largely reproduced in wild-type mice infused with Ang II and concurrently treated with an orally absorbable NHE3 inhibitor, AVE-0657 (20 mg/kg/day for 14–28 days), which also significantly attenuated Ang II-induced hypertension in C57BL/6J mice (Li et al., 2019a). Taken together, our studies in PT-Nhe3−/− mice provide the evidence that NHE3 in the proximal tubules of the kidney plays an important physiological role in proximal tubule Na+ reabsorption and basal blood pressure homeostasis, and in the development of Ang II-induced hypertension. NHE3 in the proximal tubules of the kidney may serve as a potential therapeutical target in hypertension associated with the activation of intratubular Ang II system or with increased NHE3 expression in the proximal tubules.
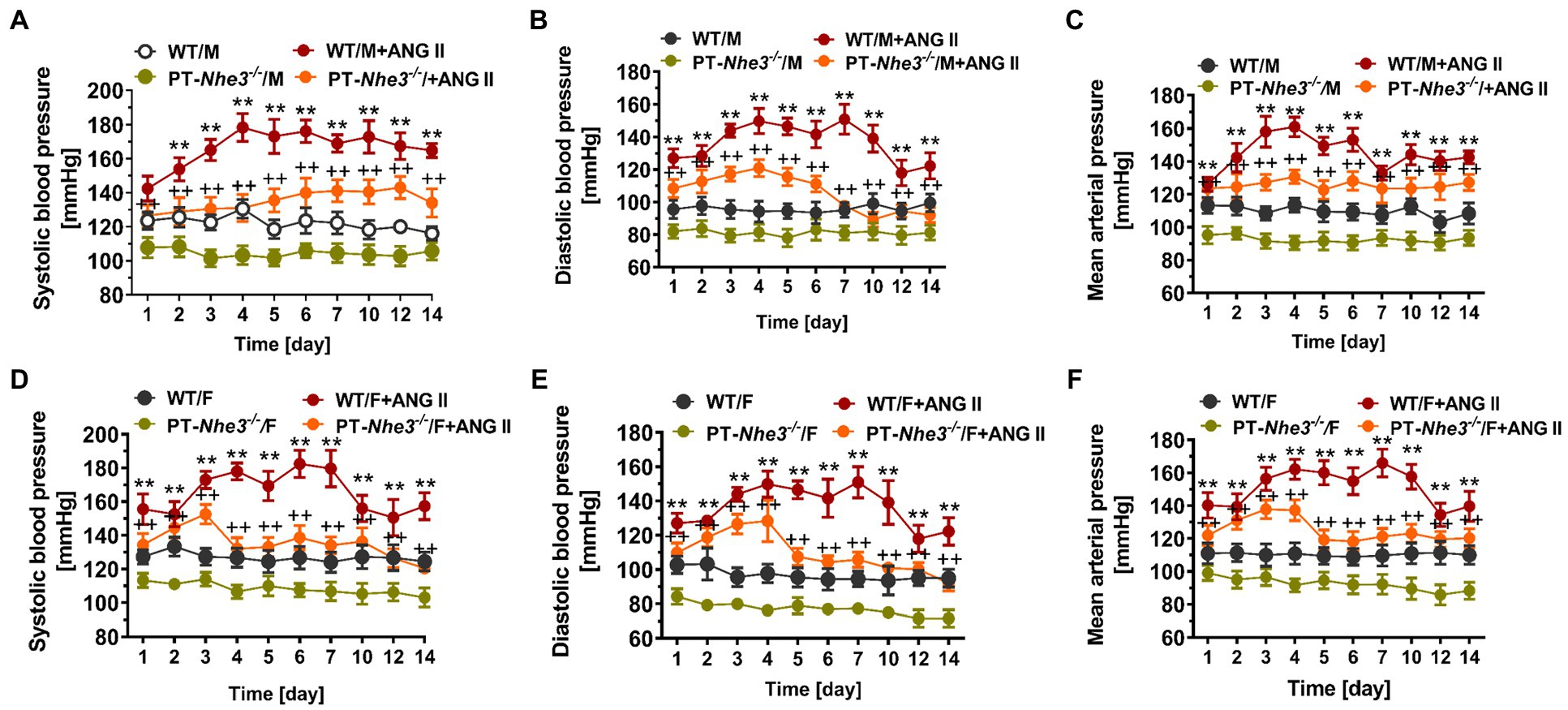
Figure 3. Systolic, diastolic, and mean arterial blood pressure responses to a pressor dose of Ang II infusion, 1.5 mg/kg/day, i.p., via osmotic minipump in conscious, adult male (M) and female (F) wild-type (WT) and PT-Nhe3−/− mice, as measured continuously for 14 days using the direct implanted telemetry technique. Please note the time-dependent increases in systolic (A,D), diastolic (B,E), and mean arterial blood pressure responses (C,F) in male and female WT mice, and significantly attenuated hypertensive responses to Ang II in male and female PT-Nhe3−/− mice. WT/M, male WT; WT/M+Ang II, male WT with Ang II infusion; PT-Nhe3−/−/M, male PT-Nhe3−/−; and PT-Nhe3−/−/+Ang II, male PT-Nhe3−/− with Ang II infusion. F represents female WT or PT-Nhe3−/− mice. **p < 0.01 vs. WT time-control group and ++p < 0.01 vs. PT-Nhe3−/− time-control group, respectively. N = 5–12 per group. Reproduced with permission (Li et al., 2019a).
In addition to hypertension, renal ischemia and reperfusion (I/R) injury is a leading factor in the pathogenesis of acute kidney diseases commonly developed due to severe hypotension, sepsis, cardiac bypass surgery, and kidney transplantation (Basile et al., 2012; Zuk and Bonventre, 2016; Smith et al., 2019). Renal I/R injury is characterized by temporary loss of blood supply to the kidney, followed by reperfusion, with subsequent activation of intratubular humoral factors, generation of reactive oxygen species (ROS), and initiation of a cascade of proinflammatory and profibrotic responses, and glomerular and tubulointerstitial injury (Basile et al., 2012; Zuk and Bonventre, 2016; Smith et al., 2019). The mechanisms underlying the development of renal I/R injury are extremely complex, involving the RAS (Johnson et al., 1992; Kontogiannis and Burns, 1998; Rodriguez-Romo et al., 2016), ROS (Paller et al., 1984; Morpurgo et al., 1996; Choi et al., 2015), NF-кB (Wan et al., 2011; Xue et al., 2014; Nishikawa et al., 2018), Toll-Like receptor 4 (TLR4; Chen et al., 2011; Trentin-Sonoda et al., 2015; Biancardi et al., 2017), sphingosine-1-phosphate 1 (S1P1) receptor (Bajwa et al., 2010; Deng et al., 2010; Park et al., 2012), and hypoxia-inducible factors (HIF-1α; Tanaka and Nangaku, 2010; Zhu et al., 2011; Luo et al., 2015). However, none of these factors adequately explains how it induces renal I/R injury, and a unified hypothesis may be therefore required. We hypothesize that during the development of renal I/R injury, intratubular and intracellular Ang II and AT1a receptors are activated in the proximal tubules, which play a key role in the pathogenesis of renal I/R by impairing mitochondrial function, and that deletion of AT1a receptors selectively in the proximal tubules attenuates renal I/R injury by blocking AT1a-mediated, intracellular Ang II-induced activation of proinflammatory cytokine and chemokine production and profibrotic responses. This hypothesis is supported by studies in which Ang II induced marked vascular, glomerular, and tubulointerstitial macrophage and monocyte infiltration, type IV collagen deposition, and tubulointerstitial fibrosis (Johnson et al., 1992; Kontogiannis and Burns, 1998; Mezzano et al., 2001; Ruiz-Ortega et al., 2002; Rodriguez-Romo et al., 2016). Ang II reportedly activates TLR4 in the proximal tubules of the kidney (Wolf et al., 2006; De Batista et al., 2014; Pushpakumar et al., 2017), S1P1 receptors (Bajwa et al., 2010; Park et al., 2012), and HIF-1α (Tanaka and Nangaku, 2010; Zhu et al., 2011; Luo et al., 2015).
Indeed, intratubular and intracellular Ang II and AT1a receptor signaling pathways in the proximal tubules are expected to be activated by induction of renal ischemia and reperfusion, which may play an important role in inducing mitochondrial dysfunction and kidney injury. Angiotensin II has been linked to mitochondrial dysfunction associated with hypertension and renal injury (de Cavanagh et al., 2007, 2011; Re and Cook, 2010). ARBs improve mitochondrial function and slow the aging process (de Cavanagh et al., 2011), whereas knockout of AT1a receptors prolongs longevity by increasing the number of mitochondria and improving mitochondrial function (Benigni et al., 2009). Whether intratubular Ang II, especially mitochondrial Ang II, via activation of mitochondrial AT1a receptors, induces mitochondrial dysfunction in renal I/R injury remains unknown. We hypothesize that intratubular and intracellular Ang II activates cell surface as well as mitochondrial AT1 (AT1a) receptors to induce activation of the Nox/NADPH or redox-sensitive signaling cascade in the mitochondria (Montezano and Touyz, 2012; Montezano et al., 2015; Li et al., 2020). Increased production by Ang II leads to uncoupling eNOS and activation of proinflammatory, profibrotic, and mitogenic responses, contributing to renal I/R injury (Montezano and Touyz, 2012; Montezano et al., 2015; Li et al., 2020). As one of the Nox/NADPH families, Nox4 is highly expressed in proximal tubule cells (Nlandu et al., 2012; Sedeek et al., 2013). Ang II inhibits the expression of mitochondrial electron transport chain and TCA cycle-modifying genes, induces mitochondrial oxidative stress, and decreases mitochondrial membrane potential (∆ψm) via mitochondrial (Kimura et al., 2005; Zhang et al., 2007). Consistent with studies, we and others have localized internalized [125I]- and FITC-labeled Ang II and AT1 receptors in endosomal, mitochondrial, and nuclear compartments in proximal tubule cells in vitro and in vivo (Zhuo et al., 2002; Li et al., 2007, 2009, 2020; Gwathmey et al., 2010a,b). Intracellular administration of Ang II to mimic internalized Ang II stimulated intracellular Ca2+ mobilization in VSMCs and proximal tubule cells (Haller et al., 1996, 1998; Zhuo et al., 2006a), whereas Ca2+ uptake in the mitochondria is closely associated with mitochondrial ATP synthesis (Jouaville et al., 1999; Duchen, 2000) and mitochondrial membrane potential ∆ψm (Hall et al., 2009, 2013). By increasing mitochondrial , decreasing NO bioavailability, and impairing mitochondrial function (de Cavanagh et al., 2007; Dikalova et al., 2010), Ang II is expected to contribute to Ang II-induced hypertension and renal I/R injury.
As a proof-of-concept study to demonstrate that the mitochondrial Ang II may directly alter mitochondrial function via activation of AT1/AT2 receptor signaling, we have recently constructed an adenoviral construct encoding a proximal tubule-specific, mitochondria-targeting intracellular Ang II fusion protein, Ad-sglt2-mito-ECFP/Ang II, for its overexpression selectively in the mitochondria of the proximal tubules (Li et al., 2020). We hypothesized that overexpression of Ad-sglt2-mito-ECFP/Ang II selectively in the mitochondria of mouse proximal tubule cells is expected to induce mitochondrial oxidative and glycolytic responses and elevates blood pressure via the Ang II/AT1a receptor//NHE3-dependent mechanisms. The expression of mito-ECFP/Ang II in the mitochondria of the proximal tubules was confirmed by the colocalization with MitoTracker Red FM or TMRM in the proximal tubules (Li et al., 2020). In vitro, mito-ECFP/Ang II markedly increased oxygen consumption rate (OCR) as an index of mitochondrial oxidative response and extracellular acidification rate (ECAR) as an index of mitochondrial glycolytic response. As the AT1 blocker losartan and a mitochondria-targeting superoxide scavenger mito-TEMPO blocked, whereas the nonselective NO inhibitor L-NAME alone increased, the mito-ECFP/Ang II-induced OCR and ECAR responses, our results suggest that mitochondrial Ang II may directly activate AT1 receptors to induce production in the mitochondria of proximal tubule cells (Li et al., 2020). In the kidney, overexpression of mito-ECFP/Ang II selectively in the mitochondria of the proximal tubules moderately increased systolic blood pressure by 12 ± 3 mmHg, and the blood pressure-elevating effect of mito-ECFP/Ang II was attenuated in PT-Agtr1a−/− and PT-Nhe3−/− mice. Interestingly, overexpression of AT2 receptors selectively in the mitochondria of the proximal tubules induced moderate natriuretic responses in PT-Agtr1a−/− and PT-Nhe3−/− mice. Taken together, these results provide new evidence for a physiological role of proximal tubule mitochondrial Ang II/AT1a/superoxide/NHE3 and Ang II/AT2/NO/NHE3 signaling pathways in maintaining blood pressure homeostasis (Figure 4; Li et al., 2020). Whether intracellular Ang II via AT1 (AT1a) or AT2 receptors in the mitochondria induces similar mitochondrial responses in other cells or tissues remains unknown. Given the important roles of Ang II in inducing mitochondrial dysfunction in hypertensive, cardiovascular, and kidney diseases, further studies using innovative, mitochondria-targeting approaches to determine the direct roles of Ang II and underlying mechanisms in the mitochondria are necessary.
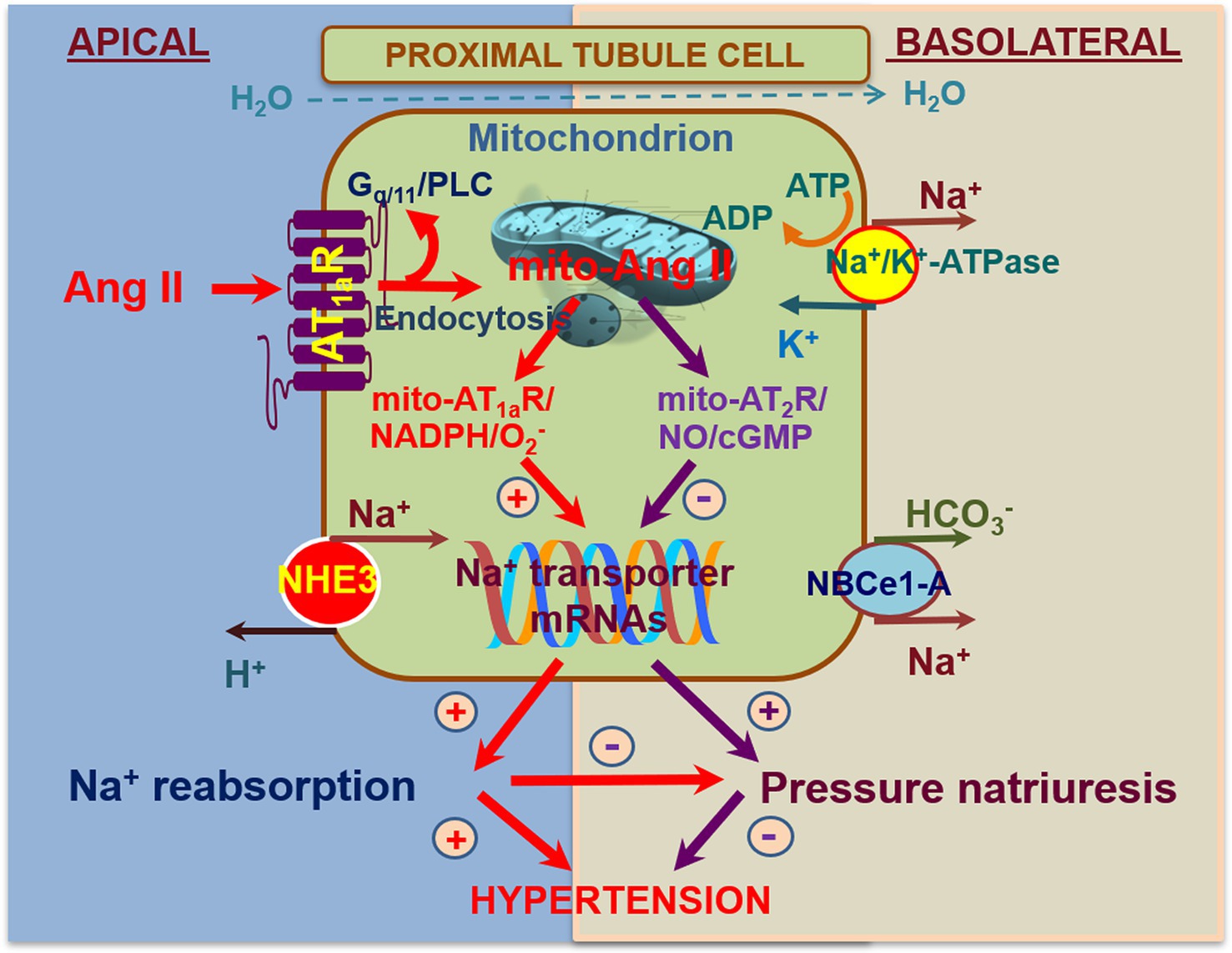
Figure 4. Schematic diagram showing the hypothesis for novel biological and physiological roles of intracellular Ang II system in the mitochondria of the proximal tubules in the regulation of proximal tubule Na+ reabsorption and blood pressure homeostasis. In addition to local onsite generation, extracellular (endocrine and paracrine) Ang II is taken up by the proximal tubule cells via the AT1 (AT1a) receptor-mediated mechanism. Some internalized Ang II/AT1 receptor complexes bypass the lysosomal degradation pathway and be transported to other intracellular organelles, including the mitochondria and the nucleus, where Ang II activates AT1 and/or AT2 receptors in the mitochondria to alter mitochondrial oxidative and glycolysis stress responses. This may in turn alter the expression or activity of NHE3 on the apical membranes or Na+/K+-ATPase on the basolateral membranes in the proximal tubules. Thus, activation of the mito-Ang II/AT1/ signaling will stimulate proximal tubule Na+ reabsorption and elevate blood pressure. Conversely, activation of the mito-Ang II/AT2/NO/cGMP signaling by overexpressing AT2 receptors selectively in the mitochondria will likely inhibit proximal tubule Na+ reabsorption, induce natriuretic response, and lower the blood pressure. Reproduced with permission (Li et al., 2020).
Sirtuin 3 (SIRT3) is a member of the sirtuin family of protein deacetylases and plays important roles in maintaining mitochondrial function in humans (Miyazaki et al., 2008; Gao et al., 2014; Kitada et al., 2014). SIRT3 is primarily localized in the mitochondria matrix, where it acts as a mitochondrial NAD+-dependent protein deacetylase to regulate mitochondrial function (Onyango et al., 2002; Kitada et al., 2014; Liu et al., 2015). The primary roles of SIRT3 in the mitochondria include anti-oxidative, anti-aging, anti-inflammation, and blood pressure-regulating effects by decreasing ROS/O2− production via activation of long chain fatty acyl-CoA dehydrogenase, succinate dehydrogenase, and NADH dehydrogenase (Onyango et al., 2002; Schwer et al., 2002; Ahn et al., 2008; Someya et al., 2010; Kitada et al., 2014; Liu et al., 2015). By contrast, Ang II acts as an important pro-oxidative, pro-growth, proinflammatory, and hypertensive peptide in part by suppressing SIRT3 expression in the mitochondria, whereas global deletion of AT1a receptors reportedly increases the expression of SIRT3 (Benigni et al., 2009). SIRT3 also appears to be protective against acute kidney injury by improving mitochondrial dynamics (Morigi et al., 2015). However, the roles of mitochondrial SIRT3 in the proximal tubules in Ang II-induced hypertension and renal injury have not been investigated using mouse model with proximal tubule-specific knockout of SIRT3.
We have recently tested the hypothesis that genetic deletion of SIRT3 selectively in the proximal tubules of the kidney aggravates Ang II-induced hypertension in proximal tubule-specific SIRT3 knockout mice, PT-SIRT3−/−. PT-SIRT3−/− mice were generated using the SGLT2-Cre/SIRT3-loxP approach (Li et al., 2019b). Ang II-dependent hypertension was induced by infusing a slow pressor dose of Ang II, 0.5 mg/kg/day, i.p., and a 2% Na+ diet for 2 weeks, and compared the hypertensive effect in adult male wild-type and PT-SIRT3−/− mice. Interestingly, basal systolic, diastolic, and mean arterial pressure were significantly lower, whereas urinary Na+ excretion was significantly higher in PT-SIRT3−/− mice than WT mice, without altering urinary K+ excretion (Li et al., 2019b). Furthermore, deletion of SIRT3 selectively in the proximal tubules of the kidney significantly augmented Ang II-induced hypertension in PT-SIRT3−/− mice (Li et al., 2019b). Further studies are ongoing to test whether genetic deletion of mitochondrial SIRT3 in the proximal tubules of the kidney aggravates Ang II-induced hypertension by impairing the pressure-natriuretic response and inducing Na+ retention in PT-SIRT3−/− mice.
In summary, preclinical animal and human clinical studies over the last few decades have firmly established the important role of the kidney in blood pressure regulation and the development of hypertension by controlling urinary Na+ excretion (Cowley and Roman, 1996; Hall et al., 1996; Carey and Siragy, 2003; Crowley et al., 2005; McDonough, 2010; Coffman, 2011; Li and Zhuo, 2013). However, the precise renal mechanisms involved and the relative contributions of renal hemodynamics and tubular transporter systems to basal blood pressure homeostasis and the development of hypertension remain incompletely understood. Indeed, although the loop of Henle- or distal tubule-targeting diuretics has been widely prescribed as a first line of antihypertensive drug in humans, some hypertensive patients still have difficulty in controlling their blood pressure and preventing target organ complications even treated with three different classes of antihypertensive drugs (Casas et al., 2005; Bomback and Toto, 2009; Lloyd-Jones et al., 2010; Muntner et al., 2018; Whelton et al., 2018; Carey et al., 2019). The mechanisms responsible for poorly controlled hypertension remain to be further studied. Based on recent studies from our and other’s laboratories, we hypothesize that the intratubular, intracellular, and mitochondrial Ang II/AT1a/NHE3 signaling pathways in the proximal tubules of the kidney may serve as new renal mechanisms and therapeutic targets at least in hypertension and kidney diseases associated with activation of the intratubular renin-angiotensin system. This hypothesis is supported by our recent studies using novel mutant mouse model with proximal tubule-specific deletion (loss of function) or overexpression (gain of function) of major components of the intratubular RAS in the kidney (Li et al., 2018, 2019a, 2020, 2021). Specifically, we have used the state-of-the-art SGLT2-Cre/LoxP approach to delete AT1a receptors (Li et al., 2021), the major Na+ transporter NHE3 (Li et al., 2018, 2019a), or a key mitochondrial protein SIRT3 selectively in the S1 and S2 segments of the proximal tubules in the kidney (Li et al., 2019b). Since proximal tubule-specific deletion of AT1a, NHE3, or SIRT3 decreases basal blood pressure, and attenuates or augments Ang II-induced hypertension, we conclude that intratubular Ang II via AT1a, NHE3, or SIRT3 in the proximal tubules plays an important role in maintaining basal blood pressure and the development of hypertension and kidney injury. We believe that these studies are highly significant and clinically relevant, and the new knowledge may lead to a paradigm shift on understanding new renal mechanisms of hypertension and kidney injury, and help develop proximal tubule-targeting drugs to treat poorly controlled hypertension and kidney diseases.
JZ and XL drafted, reviewed, and finalized the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported in part by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK123144-01, 2R01DK067299-10A1, and 2R01DK102429-03A1) and the National Heart, Lung, and Blood Institute (1R56HL130988-01) to JZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Most of authors’ recent studies were carried out in the laboratory of JZ at the University of Mississippi Medical Center in Jackson, Mississippi, and the Tulane University School of Medicine, New Orleans, Louisiana, respectively. We would like to thank Drs. Isabelle Rubera and Michell Tauc from the Laboratoire de Physiomédecine Moléculaire, LP2M, UMR-CNRS 7370, Université Côte d’Azur, Nice Cedex 2, France, for generously providing us with breeding pairs of iL1-SGLT2-Cre mouse strain, Dr. Manoocher Soleimani of the University of Cincinnati School of Medicine for providing breeding pairs of NHE3-floxed mice, Dr. Jian-Xiong Chen of the University of Mississippi Medical Center for providing breeding pairs of SIRT3-floxed mice, Dr. Julia Cook of Ochsner Clinic for providing the intracellular Ang II fusion protein construct, and Vector BioLabs of Philadelphia for constructing, amplifying, and purifying the adenoviral vectors, Ad-sglt2-ECFP/Ang II, Ad-sglt2-mito-ECFP/Ang II, and Ad-sglt2-mito-GFP/AT1a receptors for our in vitro and in vivo studies. Our previous and present technicians (Elise Miguel-Qin, Victor Gu, Fang Chen, Rui Xu, and Ruman Hassan) and postdoctoral fellows’ excellent technical support and assistance over many years are greatly appreciated (Xu Chen, Jianfeng Zhang, Xiaowen Zheng, and Chunling Zhao).
Ahn, B. H., Kim, H. S., Song, S., Lee, I. H., Liu, J., Vassilopoulos, A., et al. (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U. S. A. 105, 14447–14452. doi: 10.1073/pnas.0803790105
Aronson, P. S. (1983). Mechanisms of active H+ secretion in the proximal tubule. Am. J. Phys. 245, F647–F659.
Bajwa, A., Jo, S. K., Ye, H., Huang, L., Dondeti, K. R., Rosin, D. L., et al. (2010). Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia- reperfusion injury. J. Am. Soc. Nephrol. 21, 955–965. doi: 10.1681/ASN.2009060662
Banday, A. A., and Lokhandwala, M. F. (2008). Dopamine receptors and hypertension. Curr. Hypertens. Rep. 10, 268–275. doi: 10.1007/s11906-008-0051-9
Basile, D. P., Anderson, M. D., and Sutton, T. A. (2012). Pathophysiology of acute kidney injury. Compr. Physiol. 2, 1303–1353. doi: 10.1002/cphy.c110041
Benigni, A., Corna, D., Zoja, C., Sonzogni, A., Latini, R., Salio, M., et al. (2009). Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 119, 524–530. doi: 10.1172/JCI36703
Biancardi, V. C., Bomfim, G. F., Reis, W. L., Al-Gassimi, S., and Nunes, K. P. (2017). The interplay between angiotensin II, TLR4 and hypertension. Pharmacol. Res. 120, 88–96. doi: 10.1016/j.phrs.2017.03.017
Bivona, T. G., and Philips, M. R. (2003). Ras pathway signaling on endomembranes. Curr. Opin. Cell Biol. 15, 136–142. doi: 10.1016/S0955-0674(03)00016-4
Bomback, A. S., and Toto, R. (2009). Dual blockade of the renin-angiotensin-aldosterone system: beyond the ACE inhibitor and angiotensin-II receptor blocker combination. Am. J. Hypertens. 22, 1032–1040. doi: 10.1038/ajh.2009.138
Brasier, A. R., Jamaluddin, M., Han, Y., Patterson, C., and Runge, M. S. (2000). Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-кB (NF-кB) transcription factor. Mol. Cell. Biochem. 212, 155–169. doi: 10.1023/A:1007133710837
Calhoun, D. A., Jones, D., Textor, S., Goff, D. C., Murphy, T. P., Toto, R. D., et al. (2008). Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for high blood pressure research. Hypertension 51, 1403–1419. doi: 10.1161/HYPERTENSIONAHA.108.189141
Calhoun, D. A., Schiffrin, E. L., and Flack, J. M. (2019). Resistant hypertension: an update. Am. J. Hypertens. 32, 1–3. doi: 10.1093/ajh/hpy156
Carey, R. M., Calhoun, D. A., Bakris, G. L., Brook, R. D., Daugherty, S. L., Dennison-Himmelfarb, C. R., et al. (2018). Resistant hypertension: detection, evaluation, and management: a scientific statement from the American Heart Association. Hypertension 72, e53–e90. doi: 10.1161/HYP.0000000000000084
Carey, R. M., Sakhuja, S., Calhoun, D. A., Whelton, P. K., and Muntner, P. (2019). Prevalence of apparent treatment-resistant hypertension in the United States. Hypertension 73, 424–431. doi: 10.1161/HYPERTENSIONAHA.118.12191
Carey, R. M., and Siragy, H. M. (2003). Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr. Rev. 24, 261–271. doi: 10.1210/er.2003-0001
Casas, J. P., Chua, W., Loukogeorgakis, S., Vallance, P., Smeeth, L., Hingorani, A. D., et al. (2005). Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet 366, 2026–2033. doi: 10.1016/S0140-6736(05)67814-2
Chen, J., John, R., Richardson, J. A., Shelton, J. M., Zhou, X. J., Wang, Y., et al. (2011). Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 79, 288–299. doi: 10.1038/ki.2010.381
Choi, E. K., Jung, H., Kwak, K. H., Yeo, J., Yi, S. J., Park, C. Y., et al. (2015). Effects of allopurinol and Apocynin on renal ischemia-reperfusion injury in rats. Transplant. Proc. 47, 1633–1638. doi: 10.1016/j.transproceed.2015.06.007
Coffman, T. M. (2011). Under pressure: the search for the essential mechanisms of hypertension. Nat. Med. 17, 1402–1409. doi: 10.1038/nm.2541
Cogan, M. G. (1990). Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension 15, 451–458. doi: 10.1161/01.HYP.15.5.451
Cook, J. L., Mills, S. J., Naquin, R., Alam, J., and Re, R. N. (2006). Nuclear accumulation of the AT1 receptor in a rat vascular smooth muscle cell line: effects upon signal transduction and cellular proliferation. J. Mol. Cell. Cardiol. 40, 696–707. doi: 10.1016/j.yjmcc.2005.11.014
Cottrell, G. S., Padilla, B. E., Amadesi, S., Poole, D. P., Murphy, J. E., Hardt, M., et al. (2009). Endosomal endothelin-converting enzyme-1: a regulator of beta-arrestin-dependent ERK signaling. J. Biol. Chem. 284, 22411–22425. doi: 10.1074/jbc.M109.026674
Cowley, A. W. Jr., and Roman, R. J. (1996). The role of the kidney in hypertension. JAMA 275, 1581–1589. doi: 10.1001/jama.1996.03530440061038
Crowley, S. D., Gurley, S. B., Oliverio, M. I., Pazmino, A. K., Griffiths, R., Flannery, P. J., et al. (2005). Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J. Clin. Invest. 115, 1092–1099. doi: 10.1172/JCI23378
De Batista, P. R., Palacios, R., Martin, A., Hernanz, R., Medici, C. T., Silva, M. A., et al. (2014). Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS One 9:e104020. doi: 10.1371/journal.pone.0104020
de Cavanagh, E. M., Inserra, F., and Ferder, L. (2011). Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc. Res. 89, 31–40. doi: 10.1093/cvr/cvq285
de Cavanagh, E. M., Inserra, F., Ferder, M., and Ferder, L. (2007). From mitochondria to disease: role of the renin-angiotensin system. Am. J. Nephrol. 27, 545–553. doi: 10.1159/000107757
De Gasparo, M., Catt, K. J., Inagami, T., Wright, J. W., and Unger, T. (2000). International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472.
De Mello, W. C., and Danser, A. H. (2000). Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension 35, 1183–1188. doi: 10.1161/01.HYP.35.6.1183
Deng, A., Arndt, M. A., Satriano, J., Singh, P., Rieg, T., Thomson, S., et al. (2010). Renal protection in chronic kidney disease: hypoxia-inducible factor activation vs. angiotensin II blockade. Am. J. Physiol. Ren. Physiol. 299, F1365–F1373. doi: 10.1152/ajprenal.00153.2010
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., McCann, L., Lewis, W., et al. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. doi: 10.1161/CIRCRESAHA.109.214601
Ding, Y., Davisson, R. L., Hardy, D. O., Zhu, L. J., Merrill, D. C., Catterall, J. F., et al. (1997). The kidney androgen- regulated protein promoter confers renal proximal tubule cell-specific and highly androgen-responsive expression on the human angiotensinogen gene in transgenic mice. J. Biol. Chem. 272, 28142–28148. doi: 10.1074/jbc.272.44.28142
Dos Santos, E. A., Dahly-Vernon, A. J., Hoagland, K. M., and Roman, R. J. (2004). Inhibition of the formation of EETs and 20- HETE with 1-aminobenzotriazole attenuates pressure natriuresis. Am. J. Phys. Regul. Integr. Comp. Phys. 287, R58–R68. doi: 10.1152/ajpregu.00713.2003
Duchen, M. R. (2000). Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 28, 339–348. doi: 10.1054/ceca.2000.0170
Fenton, R. A., Poulsen, S. B., de la Mora, C. S., Soleimani, M., Dominguez Rieg, J. A., and Rieg, T. (2017). Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int. 92, 397–414. doi: 10.1016/j.kint.2017.02.001
Ferguson, S. S. (2001). Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24.
Gao, P., Xu, T. T., Lu, J., Li, L., Xu, J., Hao, D. L., et al. (2014). Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. 92, 347–357. doi: 10.1007/s00109-013-1111-4
Granger, J. P., and Alexander, B. T. (2000). Abnormal pressure-natriuresis in hypertension: role of nitric oxide. Acta Physiol. Scand. 168, 161–168. doi: 10.1046/j.1365-201x.2000.00655.x
Granger, J. P., Alexander, B. T., and Llinas, M. (2002). Mechanisms of pressure natriuresis. Curr. Hypertens. Rep. 4, 152–159. doi: 10.1007/s11906-002-0040-3
Gross, V., Roman, R. J., and Cowley, A. W. Jr. (1994). Abnormal pressure-natriuresis in transgenic renin gene rats. J. Hypertens. 12, 1029–1034.
Gurley, S. B., Riquier-Brison, A. D., Schnermann, J., Sparks, M. A., Allen, A. M., Haase, V. H., et al. (2011). AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 13, 469–475. doi: 10.1016/j.cmet.2011.03.001
Gwathmey, T. M., Pendergrass, K. D., Reid, S. D., Rose, J. C., Diz, D. I., and Chappell, M. C. (2010b). Angiotensin-(1-7)-angiotensin- converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension 55, 166–171. doi: 10.1161/HYPERTENSIONAHA.109.141622
Gwathmey, T. M., Westwood, B. M., Pirro, N. T., Tang, L., Rose, J. C., Diz, D. I., et al. (2010a). Nuclear angiotensin-(1-7) receptor is functionally coupled to the formation of nitric oxide. Am. J. Physiol. Ren. Physiol. 299, F983–F990. doi: 10.1152/ajprenal.00371.2010
Hall, A. M., Rhodes, G. J., Sandoval, R. M., Corridon, P. R., and Molitoris, B. A. (2013). In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 83, 72–83. doi: 10.1038/ki.2012.328
Hall, A. M., Unwin, R. J., Parker, N., and Duchen, M. R. (2009). Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J. Am. Soc. Nephrol. 20, 1293–1302. doi: 10.1681/ASN.2008070759
Hall, J. E., Brands, M. W., and Shek, E. W. (1996). Central role of the kidney and abnormal fluid volume control in hypertension. J. Hum. Hypertens. 10, 633–639.
Haller, H., Lindschau, C., Erdmann, B., Quass, P., and Luft, F. C. (1996). Effects of intracellular angiotensin II in vascular smooth muscle cells. Circ. Res. 79, 765–772. doi: 10.1161/01.RES.79.4.765
Haller, H., Maasch, C., Lindschau, C., Brachmann, M., Buchner, K., and Luft, F. C. (1998). Intracellular targeting and protein kinase C in vascular smooth muscle cells: specific effects of different membrane-bound receptors. Acta Physiol. Scand. 164, 599–609. doi: 10.1046/j.1365-201X.1998.00448.x
Harris, P. J., and Navar, L. G. (1985). Tubular transport responses to angiotensin II. Am. J. Physiol. Ren. Physiol. 248, F621–F630. doi: 10.1152/ajprenal.1985.248.5.F621
Hein, L., Meinel, L., Pratt, R. E., Dzau, V. J., and Kobilka, B. K. (1997). Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol. Endocrinol. 11, 1266–1277. doi: 10.1210/mend.11.9.9975
Higuchi, S., Ohtsu, H., Suzuki, H., Shirai, H., Frank, G. D., and Eguchi, S. (2007). Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin. Sci. 112, 417–428. doi: 10.1042/CS20060342
Hussain, T., and Lokhandwala, M. F. (1998). Renal dopamine receptor function in hypertension. Hypertension 32, 187–197. doi: 10.1161/01.HYP.32.2.187
Jin, X. H., McGrath, H. E., Gildea, J. J., Siragy, H. M., Felder, R. A., and Carey, R. M. (2004). Renal interstitial guanosine cyclic 3', 5'- monophosphate mediates pressure-natriuresis via protein kinase G. Hypertension 43, 1133–1139. doi: 10.1161/01.HYP.0000123574.60586.7d
Jin, X. H., Siragy, H. M., and Carey, R. M. (2001). Renal interstitial cGMP mediates natriuresis by direct tubule mechanism. Hypertension 38, 309–316. doi: 10.1161/01.HYP.38.3.309
Johnson, R. J., Alpers, C. E., Yoshimura, A., Lombardi, D., Pritzl, P., Floege, J., et al. (1992). Renal injury from angiotensin II-mediated hypertension. Hypertension 19, 464–474. doi: 10.1161/01.HYP.19.5.464
Jorde, U. P., Ennezat, P. V., Lisker, J., Suryadevara, V., Infeld, J., Cukon, S., et al. (2000). Maximally recommended doses of angiotensin-converting enzyme (ACE) inhibitors do not completely prevent ACE-mediated formation of angiotensin II in chronic heart failure. Circulation 101, 844–846. doi: 10.1161/01.CIR.101.8.844
Jouaville, L. S., Pinton, P., Bastianutto, C., Rutter, G. A., and Rizzuto, R. (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. U. S. A. 96, 13807–13812. doi: 10.1073/pnas.96.24.13807
Kagami, S., Border, W. A., Miller, D. E., and Noble, N. A. (1994). Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J. Clin. Invest. 93, 2431–2437. doi: 10.1172/JCI117251
Kimura, S., Zhang, G. X., Nishiyama, A., Shokoji, T., Yao, L., Fan, Y. Y., et al. (2005). Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 45, 438–444. doi: 10.1161/01.HYP.0000157169.27818.ae
Kitada, M., Kume, S., and Koya, D. (2014). Role of sirtuins in kidney disease. Curr. Opin. Nephrol. Hypertens. 23, 75–79. doi: 10.1097/01.mnh.0000437330.85675.ac
Kobori, H., Nangaku, M., Navar, L. G., and Nishiyama, A. (2007). The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 59, 251–287. doi: 10.1124/pr.59.3.3
Kontogiannis, J., and Burns, K. D. (1998). Role of AT1 angiotensin II receptors in renal ischemic injury. Am. J. Phys. 274, F79–F90.
Kumar, R., Singh, V. P., and Baker, K. M. (2008). The intracellular renin-angiotensin system: implications in cardiovascular remodeling. Curr. Opin. Nephrol. Hypertens. 17, 168–173. doi: 10.1097/MNH.0b013e3282f521a8
Kurtz, T. W., and Gardner, D. G. (1998). Transcription-modulating drugs: a new frontier in the treatment of essential hypertension. Hypertension 32, 380–386. doi: 10.1161/01.HYP.32.3.380
Lambers Heerspink, H. J., Perkovic, V., and De, Z. D. (2009). Renal and cardio-protective effects of direct renin inhibition: a systematic literature review. J. Hypertens. 27, 2321–2331. doi: 10.1097/HJH.0b013e3283310f92
Li, H., Weatherford, E. T., Davis, D. R., Keen, H. L., Grobe, J. L., Daugherty, A., et al. (2011a). Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am. J. Phys. Regul. Integr. Comp. Phys. 301, R1067–R1077. doi: 10.1152/ajpregu.00124.2011
Li, H., Zhou, X., Davis, D. R., Xu, D., and Sigmund, C. D. (2008). An androgen-inducible proximal tubule-specific Cre recombinase transgenic model. Am. J. Physiol. Ren. Physiol. 294, F1481–F1486. doi: 10.1152/ajprenal.00064.2008
Li, H. C., Du, Z., Barone, S., Rubera, I., McDonough, A. A., Tauc, M., et al. (2013). Proximal tubule specific knockout of the Na/H exchanger NHE3: effects on bicarbonate absorption and ammonium excretion. J. Mol. Med. 91, 951–963. doi: 10.1007/s00109-013-1015-3
Li, X. C., Carretero, O. A., Navar, L. G., and Zhuo, J. L. (2006). AT1 receptor-mediated accumulation of extracellular angiotensin II in proximal tubule cells: role of cytoskeleton microtubules and tyrosine phosphatases. Am. J. Physiol. Ren. Physiol. 291, F375–F383. doi: 10.1152/ajprenal.00405.2005
Li, X. C., Chen, X., Zheng, X., Zhao, C., Chen, J. X., and Zhuo, J. L. (2019b). Genetic deletion of sirtuin 3 (SIRT3) in the mitochondria of the proximal tubules aggravates angiotensin II-induced hypertension by impairing the pressure-natriuresis response in proximal tubule-specific SIRT3 knockout mice. Hypertension 74:A102. doi: 10.1161/hyp.74.suppl_1.102
Li, X. C., Cook, J. L., Rubera, I., Tauc, M., Zhang, F., and Zhuo, J. L. (2011b). Intrarenal transfer of an intracellular cyan fluorescent fusion of angiotensin II selectively in proximal tubules increases blood pressure in rats and mice. Am. J. Physiol. Ren. Physiol. 300, F1076–F1088. doi: 10.1152/ajprenal.00329.2010
Li, X. C., Gu, V., Miguel-Qin, E., and Zhuo, J. L. (2014). Role of caveolin 1 in AT1a receptor-mediated uptake of angiotensin II in the proximal tubule of the kidney. Am. J. Physiol. Ren. Physiol. 307, F949–F961. doi: 10.1152/ajprenal.00199.2014
Li, X. C., Hopfer, U., and Zhuo, J. L. (2009). AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through the microtubule-dependent endocytic pathway. Am. J. Physiol. Ren. Physiol. 297, F1342–F1352. doi: 10.1152/ajprenal.90734.2008
Li, X. C., Leite, A. P. O., Zheng, X., Zhao, C., Chen, X., Zhang, L., et al. (2021). Proximal tubule-specific deletion of angiotensin II type 1a receptors in the kidney attenuates circulating and intratubular angiotensin II-induced hypertension in PT-Agtr1a−/− mice. Hypertension 77, 1285–1298. doi: 10.1161/HYPERTENSIONAHA.120.16336
Li, X. C., Navar, L. G., Shao, Y., and Zhuo, J. L. (2007). Genetic deletion of AT1a receptors attenuates intracellular accumulation of angiotensin II in the kidney of AT1a receptor-deficient mice. Am. J. Physiol. Ren. Physiol. 293, F586–F593. doi: 10.1152/ajprenal.00489.2006
Li, X. C., Shull, G. E., Miguel-Qin, E., Chen, F., and Zhuo, J. L. (2015b). Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension in NHE3-deficient mice with transgenic rescue of NHE3 in small intestines. Phys. Rep. 3:e12605. doi: 10.14814/phy2.12605
Li, X. C., Shull, G. E., Miguel-Qin, E., and Zhuo, J. L. (2015a). Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiol. Genomics 47, 479–487. doi: 10.1152/physiolgenomics.00056.2015
Li, X. C., Soleimani, M., Zhu, D., Rubera, I., Tauc, M., Zheng, X., et al. (2018). Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) promotes the pressure-natriuresis response and lowers blood pressure in mice. Hypertension 72, 1328–1336. doi: 10.1161/HYPERTENSIONAHA.118.10884
Li, X. C., Zhou, X., and Zhuo, J. L. (2020). Evidence for a physiological mitochondrial angiotensin II system in the kidney proximal tubules: novel roles of mitochondrial Ang II/AT1a/O2− and Ang II/AT2/NO signaling. Hypertension 76, 121–132. doi: 10.1161/HYPERTENSIONAHA.119.13942
Li, X. C., Zhu, D., Chen, X., Zheng, X., Zhao, C., Zhang, J., et al. (2019a). Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates Ang II (angiotensin II)-induced hypertension in mice. Hypertension 74, 526–535. doi: 10.1161/HYPERTENSIONAHA.119.13094
Li, X. C., and Zhuo, J. L. (2008a). Nuclear factor-кB as a hormonal intracellular signaling molecule: focus on angiotensin II- induced cardiovascular and renal injury. Curr. Opin. Nephrol. Hypertens. 17, 37–43. doi: 10.1097/MNH.0b013e3282f2903c
Li, X. C., and Zhuo, J. L. (2008b). In vivo regulation of AT1a receptor-mediated intracellular uptake of [125I]-Val5-angiotensin II in the kidneys and adrenal glands of AT1a receptor-deficient mice. Am. J. Physiol. Ren. Physiol. 294, F293–F302. doi: 10.1152/ajprenal.00398.2007
Li, X. C., and Zhuo, J. L. (2008c). Intracellular Ang II directly induces in vitro transcription of TGF-beta1, MCP-1, and NHE- 3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am. J. Phys. Cell Phys. 294, C1034–C1045. doi: 10.1152/ajpcell.00432.2007
Li, X. C., and Zhuo, J. L. (2011). Phosphoproteomic analysis of AT1 receptor-mediated signaling responses in proximal tubules of angiotensin II-induced hypertensive rats. Kidney Int. 80, 620–632. doi: 10.1038/ki.2011.161
Li, X. C., and Zhuo, J. L. (2013). Proximal tubule-dominant transfer of AT1a receptors induces blood pressure responses to intracellular angiotensin II in AT1a receptor-deficient mice. Am. J. Phys. Regul. Integr. Comp. Phys. 304, R588–R598. doi: 10.1152/ajpregu.00338.2012
Li, X. C., and Zhuo, J. L. (2014). Mechanisms of AT1a receptor-mediated uptake of angiotensin II by proximal tubule cells: a novel role of the multiligand endocytic receptor megalin. Am. J. Physiol. Ren. Physiol. 307, F222–F233. doi: 10.1152/ajprenal.00693.2013
Liu, H., Chen, T., Li, N., Wang, S., and Bu, P. (2015). Role of SIRT3 in angiotensin II-induced human umbilical vein endothelial cells dysfunction. BMC Cardiovasc. Disord. 15:81. doi: 10.1186/s12872-015-0075-4
Lloyd-Jones, D., Adams, R. J., Brown, T. M., Carnethon, M., Dai, S., De, S. G., et al. (2010). Heart disease and stroke statistics-2010 update: a report from the American Heart Association. Circulation 121, e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667
Lorenz, J. N., Schultheis, P. J., Traynor, T., Shull, G. E., and Schnermann, J. (1999). Micropuncture analysis of single-nephron function in NHE3-deficient mice. Am. J. Phys. 277, F447–F453.
Luo, R., Zhang, W., Zhao, C., Zhang, Y., Wu, H., Jin, J., et al. (2015). Elevated endothelial hypoxia-inducible factor-1alpha contributes to glomerular injury and promotes hypertensive chronic kidney disease. Hypertension 66, 75–84. doi: 10.1161/HYPERTENSIONAHA.115.05578
Majid, D. S., Omoro, S. A., Chin, S. Y., and Navar, L. G. (1998). Intrarenal nitric oxide activity and pressure natriuresis in anesthetized dogs. Hypertension 32, 266–272. doi: 10.1161/01.HYP.32.2.266
Majid, D. S., Williams, A., and Navar, L. G. (1993). Inhibition of nitric oxide synthesis attenuates pressure-induced natriuretic responses in anesthetized dogs. Am. J. Phys. 264, F79–F87.
Mattson, D. L., Raff, H., and Roman, R. J. (1991). Influence of angiotensin II on pressure natriuresis and renal hemodynamics in volume-expanded rats. Am. J. Phys. 260, R1200–R1209.
McDonough, A. A. (2010). Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Phys. Regul. Integr. Comp. Phys. 298, R851–R861. doi: 10.1152/ajpregu.00002.2010
Mezzano, S. A., Ruiz-Ortega, M., and Egido, J. (2001). Angiotensin II and renal fibrosis. Hypertension 38, 635–638. doi: 10.1161/hy09t1.094234
Miyazaki, R., Ichiki, T., Hashimoto, T., Inanaga, K., Imayama, I., Sadoshima, J., et al. (2008). SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 28, 1263–1269. doi: 10.1161/ATVBAHA.108.166991
Montezano, A. C., and Touyz, R. M. (2012). Oxidative stress, Noxs, and hypertension: experimental evidence and clinical controversies. Ann. Med. 44(Suppl. 1), S2–S16. doi: 10.3109/07853890.2011.653393
Montezano, A. C., Tsiropoulou, S., Dulak-Lis, M., Harvey, A., Camargo, L. L., and Touyz, R. M. (2015). Redox signaling, Nox5 and vascular remodeling in hypertension. Curr. Opin. Nephrol. Hypertens. 24, 425–433. doi: 10.1097/MNH.0000000000000153
Moreno, C., Maier, K. G., Hoagland, K. M., Yu, M., and Roman, R. J. (2001). Abnormal pressure-natriuresis in hypertension: role of cytochrome P450 metabolites of arachidonic acid. Am. J. Hypertens. 14(Suppl. 1), S90–S97. doi: 10.1016/s0895-7061(01)02075-1
Morigi, M., Perico, L., Rota, C., Longaretti, L., Conti, S., Rottoli, D., et al. (2015). Sirtuin 3- dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest. 125, 715–726. doi: 10.1172/JCI77632
Morpurgo, E., Cadrobbi, R., Morpurgo, M., Rigotti, P., Schiavon, F., Schiavon, O., et al. (1996). Protective effect of superoxide dismutase and polyethylene glycol-linked superoxide dismutase against renal warm ischemia/reperfusion injury. Transplantation 62, 1221–1223. doi: 10.1097/00007890-199611150-00006
Muntner, P., Carey, R. M., Gidding, S., Jones, D. W., Taler, S. J., Wright, J. T. Jr., et al. (2018). Population impact of the 2017 ACC/AHA high blood pressure guideline. Circulation 137, 109–118. doi: 10.1161/CIRCULATIONAHA.117.032582
Murphy, J. E., Padilla, B. E., Hasdemir, B., Cottrell, G. S., and Bunnett, N. W. (2009). Endosomes: a legitimate platform for the signaling train. Proc. Natl. Acad. Sci. U. S. A. 106, 17615–17622. doi: 10.1073/pnas.0906541106
Nishikawa, H., Taniguchi, Y., Matsumoto, T., Arima, N., Masaki, M., Shimamura, Y., et al. (2018). Knockout of the interleukin-36 receptor protects against renal ischemia-reperfusion injury by reduction of proinflammatory cytokines. Kidney Int. 93, 599–614. doi: 10.1016/j.kint.2017.09.017
Nlandu, K. S., Dizin, E., Sossauer, G., Szanto, I., Martin, P. Y., Feraille, E., et al. (2012). NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol. 23, 1967–1976. doi: 10.1681/ASN.2012040373
Noonan, W. T., Woo, A. L., Nieman, M. L., Prasad, V., Schultheis, P. J., Shull, G. E., et al. (2005). Blood pressure maintenance in NHE3-deficient mice with transgenic expression of NHE3 in small intestine. Am. J. Phys. Regul. Integr. Comp. Phys. 288, R685–R691. doi: 10.1152/ajpregu.00209.2004
O’Brien, E., Barton, J., Nussberger, J., Mulcahy, D., Jensen, C., Dicker, P., et al. (2007). Aliskiren reduces blood pressure and suppresses plasma renin activity in combination with a thiazide diuretic, an angiotensin- converting enzyme inhibitor, or an angiotensin receptor blocker. Hypertension 49, 276–284. doi: 10.1161/01.HYP.0000253780.36691.4f
Onyango, P., Celic, I., McCaffery, J. M., Boeke, J. D., and Feinberg, A. P. (2002). SIRT3, a human SIR2 homologue, is an NAD- dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U. S. A. 99, 13653–13658. doi: 10.1073/pnas.222538099
Oparil, S., Yarows, S. A., Patel, S., Fang, H., Zhang, J., and Satlin, A. (2007). Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trial. Lancet 370, 221–229. doi: 10.1016/S0140-6736(07)61124-6
Paller, M. S., Hoidal, J. R., and Ferris, T. F. (1984). Oxygen free radicals in ischemic acute renal failure in the rat. J. Clin. Invest. 74, 1156–1164. doi: 10.1172/JCI111524
Park, S. W., Kim, M., Brown, K. M., D'Agati, V. D., and Lee, H. T. (2012). Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 23, 266–280. doi: 10.1681/ASN.2011050503
Poleev, A., Fickenscher, H., Mundlos, S., Winterpacht, A., Zabel, B., Fidler, A., et al. (1992). PAX8, a human paired box gene: isolation and expression in developing thyroid, kidney and Wilms’ tumors. Development 116, 611–623. doi: 10.1242/dev.116.3.611
Pushpakumar, S., Ren, L., Kundu, S., Gamon, A., Tyagi, S. C., and Sen, U. (2017). Toll-like receptor 4 deficiency reduces oxidative stress and macrophage mediated inflammation in hypertensive kidney. Sci. Rep. 7, 6349–6484. doi: 10.1038/s41598-017-06484-6
Rateri, D. L., Moorleghen, J. J., Balakrishnan, A., Owens, A. P. III, Howatt, D. A., Subramanian, V., et al. (2011). Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor−/− mice. Circ. Res. 108, 574–581. doi: 10.1161/CIRCRESAHA.110.222844
Re, R. N. (2000). On the biological actions of intracellular angiotensin. Hypertension 35, 1189–1190. doi: 10.1161/01.HYP.35.6.1189
Re, R. N., and Cook, J. L. (2010). The mitochondrial component of intracrine action. Am. J. Physiol. Heart Circ. Physiol. 299, H577–H583. doi: 10.1152/ajpheart.00421.2010
Rector, F. C. Jr. (1983). Sodium, bicarbonate, and chloride absorption by the proximal tubule. Am. J. Phys. 244, F461–F471.
Rodriguez-Romo, R., Benitez, K., Barrera-Chimal, J., Perez-Villalva, R., Gomez, A., Aguilar-Leon, D., et al. (2016). AT1 receptor antagonism before ischemia prevents the transition of acute kidney injury to chronic kidney disease. Kidney Int. 89, 363–373. doi: 10.1038/ki.2015.320
Roman, R. J. (1986). Pressure diuresis mechanism in the control of renal function and arterial pressure. Fed. Proc. 45, 2878–2884.
Roman, R. J. (1987). Altered pressure-natriuresis relationship in young spontaneously hypertensive rats. Hypertension 9, III130–III136. doi: 10.1161/01.hyp.9.6_pt_2.iii130
Roman, R. J., and Cowley, A. W. Jr. (1985). Abnormal pressure-diuresis-natriuresis response in spontaneously hypertensive rats. Am. J. Phys. 248, F199–F205.
Rostand, S. G., Lewis, D., Watkins, J. B., Huang, W. C., and Navar, L. G. (1982). Attenuated pressure natriuresis in hypertensive rats. Kidney Int. 21, 330–338. doi: 10.1038/ki.1982.26
Rubera, I., Poujeol, C., Bertin, G., Hasseine, L., Counillon, L., Poujeol, P., et al. (2004). Specific Cre/lox recombination in the mouse proximal tubule. J. Am. Soc. Nephrol. 15, 2050–2056. doi: 10.1097/01.ASN.0000133023.89251.01
Ruiz-Ortega, M., Lorenzo, O., Ruperez, M., Konig, S., Wittig, B., and Egido, J. (2000). Angiotensin II activates nuclear transcription factor kappaB through AT1 and AT2 in vascular smooth muscle cells: molecular mechanisms. Circ. Res. 86, 1266–1272. doi: 10.1161/01.RES.86.12.1266
Ruiz-Ortega, M., Ruperez, M., Lorenzo, O., Esteban, V., Blanco, J., and Mezzano, S. (2002). Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int. Suppl. 82, 12–22. doi: 10.1046/j.1523-1755.62.s82.4.x
Sarafidis, P. A., Georgianos, P., and Bakris, G. L. (2013). Resistant hypertension—its identification and epidemiology. Nat. Rev. Nephrol. 9, 51–58. doi: 10.1038/nrneph.2012.260
Schafer, J. A., Robert, F., and Pitts Memorial Lecture (1984). Mechanisms coupling the absorption of solutes and water in the proximal nephron. Kidney Int. 25, 708–716. doi: 10.1038/ki.1984.78
Schultheis, P. J., Clarke, L. L., Meneton, P., Miller, M. L., Soleimani, M., Gawenis, L. R., et al. (1998). Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 19, 282–285. doi: 10.1038/969
Schupp, N., Schmid, U., Rutkowski, P., Lakner, U., Kanase, N., Heidland, A., et al. (2007). Angiotensin II-induced genomic damage in renal cells can be prevented by angiotensin II type 1 receptor blockage or radical scavenging. Am. J. Physiol. Ren. Physiol. 292, F1427–F1434. doi: 10.1152/ajprenal.00458.2006
Schwer, B., North, B. J., Frye, R. A., Ott, M., and Verdin, E. (2002). The human silent information regulator (sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 158, 647–657. doi: 10.1083/jcb.200205057
Sedeek, M., Nasrallah, R., Touyz, R. M., and Hebert, R. L. (2013). NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J. Am. Soc. Nephrol. 24, 1512–1518. doi: 10.1681/ASN.2012111112
Siragy, H. M., and Carey, R. M. (1996). The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3', 5'- monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J. Clin. Invest. 97, 1978–1982. doi: 10.1172/JCI118630
Smith, S. F., Hosgood, S. A., and Nicholson, M. L. (2019). Ischemia-reperfusion injury in renal transplantation: 3 key signaling pathways in tubular epithelial cells. Kidney Int. 95, 50–56. doi: 10.1016/j.kint.2018.10.009
Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann, J. M., Leeuwenburgh, C., et al. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812. doi: 10.1016/j.cell.2010.10.002
Takahashi, M., Suzuki, E., Takeda, R., Oba, S., Nishimatsu, H., Kimura, K., et al. (2008). Angiotensin II and tumor necrosis factor-alpha synergistically promote monocyte chemoattractant protein-1 expression: roles of NF-кB, p38, and reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 294, H2879–H2888. doi: 10.1152/ajpheart.91406.2007
Tanaka, T., and Nangaku, M. (2010). The role of hypoxia, increased oxygen consumption, and hypoxia-inducible factor- 1 alpha in progression of chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 19, 43–50. doi: 10.1097/MNH.0b013e3283328eed
Thompson, B., Davidson, E. A., Liu, W., Nebert, D. W., Bruford, E. A., Zhao, H., et al. (2021). Overview of PAX gene family: analysis of human tissue-specific variant expression and involvement in human disease. Hum. Genet. 140, 381–400. doi: 10.1007/s00439-020-02212-9
Timmermans, P. B., Wong, P. C., Chiu, A. T., Herblin, W. F., Benfield, P., Carini, D. J., et al. (1993). Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol. Rev. 45, 205–251.
Touyz, R. M., and Schiffrin, E. L. (2000). Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 52, 639–672.
Trentin-Sonoda, M., da Silva, R. C., Kmit, F. V., Abrahao, M. V., Monnerat, C. G., Brasil, G. V., et al. (2015). Knockout of toll-Like receptors 2 and 4 prevents renal ischemia-reperfusion-induced cardiac hypertrophy in mice. PLoS One 10:e0139350. doi: 10.1371/journal.pone.0139350
Vallon, V., Schwark, J. R., Richter, K., and Hropot, M. (2000). Role of Na+/H+ exchanger NHE3 in nephron function: micropuncture studies with S3226, an inhibitor of NHE3. Am. J. Physiol. Ren. Physiol. 278, F375–F379. doi: 10.1152/ajprenal.2000.278.3.F375
van Kats, J. P., Schalekamp, M. A., Verdouw, P. D., Duncker, D. J., and Danser, A. H. (2001). Intrarenal angiotensin II: interstitial and cellular levels and site of production. Kidney Int. 60, 2311–2317. doi: 10.1046/j.1523-1755.2001.00049.x
von Thun, A. M., Vari, R. C., El Dahr, S. S., and Navar, L. G. (1994). Augmentation of intrarenal angiotensin II levels by chronic angiotensin II infusion. Am. J. Phys. 266, F120–F128.
Wan, X., Fan, L., Hu, B., Yang, J., Li, X., Chen, X., et al. (2011). Small interfering RNA targeting IKKbeta prevents renal ischemia-reperfusion injury in rats. Am. J. Physiol. Ren. Physiol. 300, F857–F863. doi: 10.1152/ajprenal.00547.2010
Wang, C. T., Chin, S. Y., and Navar, L. G. (2000). Impairment of pressure-natriuresis and renal autoregulation in Ang II-infused hypertensive rats. Am. J. Physiol. Ren. Physiol. 279, F319–F325. doi: 10.1152/ajprenal.2000.279.2.F319
Wang, T., and Chan, Y. L. (1990). Mechanism of angiotensin II action on proximal tubular transport. J. Pharmacol. Exp. Ther. 252, 689–695.
Wang, T., Yang, C. L., Abbiati, T., Schultheis, P. J., Shull, G. E., Giebisch, G., et al. (1999). Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. Am. J. Phys. 277, F298–F302.
Wang, X., Armando, I., Upadhyay, K., Pascua, A., and Jose, P. A. (2009). The regulation of proximal tubular salt transport in hypertension: an update. Curr. Opin. Nephrol. Hypertens. 18, 412–420. doi: 10.1097/MNH.0b013e32832f5775
Weigert, C., Brodbeck, K., Klopfer, K., Haring, H. U., and Schleicher, E. D. (2002). Angiotensin II induces human TGF-β1 promoter activation: similarity to hyperglycaemia. Diabetologia 45, 890–898. doi: 10.1007/s00125-002-0843-4
Whelton, P. K., Carey, R. M., Aronow, W. S., Casey, D. E. Jr., Collins, K. J., Dennison, H. C., et al. (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation 138, e426–e483. doi: 10.1161/CIR.0000000000000597
Wilcox, C. S., Baylis, C., and Wingo, C. S. (1992). “Glomerular-tubular balance and proximal regulation,” in The Kidney: Physiology and Pathophysiology. eds. D. W. Seldin and G. Giebisch (New York: Raven Press), 1807–1842.
Williams, J. M., Sarkis, A., Lopez, B., Ryan, R. P., Flasch, A. K., and Roman, R. J. (2007). Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 49, 687–694. doi: 10.1161/01.HYP.0000255753.89363.47
Wing, L. M., Reid, C. M., Ryan, P., Beilin, L. J., Brown, M. A., Jennings, G. L., et al. (2003). A comparison of outcomes with angiotensin-converting-enzyme inhibitors and diuretics for hypertension in the elderly. N. Engl. J. Med. 348, 583–592. doi: 10.1056/NEJMoa021716
Wolf, G., Bohlender, J., Bondeva, T., Roger, T., Thaiss, F., and Wenzel, U. O. (2006). Angiotensin II upregulates toll-like receptor 4 on mesangial cells. J. Am. Soc. Nephrol. 17, 1585–1593. doi: 10.1681/ASN.2005070699
Wolf, G., Ziyadeh, F. N., and Stahl, R. A. (1999). Angiotensin II stimulates expression of transforming growth factor beta receptor type II in cultured mouse proximal tubular cells. J. Mol. Med. 77, 556–564. doi: 10.1007/s001099900028
Woo, A. L., Noonan, W. T., Schultheis, P. J., Neumann, J. C., Manning, P. A., Lorenz, J. N., et al. (2003). Renal function in NHE3-deficient mice with transgenic rescue of small intestinal absorptive defect. Am. J. Physiol. Ren. Physiol. 284, F1190–F1198. doi: 10.1152/ajprenal.00418.2002
Xue, C., Liu, Y., Li, C., Li, Y., Yang, T., Xie, L., et al. (2014). Powerful protection against renal ischemia reperfusion injury by T cell-specific NF-kappaB inhibition. Transplantation 97, 391–396. doi: 10.1097/01.TP.0000438622.89310.95
Zeng, C., Asico, L. D., Wang, X., Hopfer, U., Eisner, G. M., Felder, R. A., et al. (2003). Angiotensin II regulation of AT1 and D3 dopamine receptors in renal proximal tubule cells of SHR. Hypertension 41, 724–729. doi: 10.1161/01.HYP.0000047880.78462.0E
Zeng, C., Wang, Z., Asico, L. D., Hopfer, U., Eisner, G. M., Felder, R. A., et al. (2005). Aberrant ETB receptor regulation of AT1 receptors in immortalized renal proximal tubule cells of spontaneously hypertensive rats. Kidney Int. 68, 623–631. doi: 10.1111/j.1523-1755.2005.00440.x
Zhang, G. X., Lu, X. M., Kimura, S., and Nishiyama, A. (2007). Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc. Res. 76, 204–212. doi: 10.1016/j.cardiores.2007.07.014
Zhang, J., Ferguson, S. S., Barak, L. S., Aber, M. J., Giros, B., Lefkowitz, R. J., et al. (1997). Molecular mechanisms of G protein-coupled receptor signaling: role of G protein-coupled receptor kinases and arrestins in receptor desensitization and resensitization. Recept. Channels 5, 193–199.
Zhu, Q., Wang, Z., Xia, M., Li, P. L., Van Tassell, B. W., Abbate, A., et al. (2011). Silencing of hypoxia-inducible factor-1alpha gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension 58, 657–664. doi: 10.1161/HYPERTENSIONAHA.111.177626
Zhuo, J. L. (2004). Monocyte chemoattractant protein-1: a key mediator of angiotensin II-induced target organ damage in hypertensive heart disease? J. Hypertens. 22, 451–454. doi: 10.1097/00004872-200403000-00003
Zhuo, J. L., Carretero, O. A., and Li, X. C. (2006b). Effects of AT1 receptor-mediated endocytosis of extracellular Ang II on activation of nuclear factor-kappa B in proximal tubule cells. Ann. N. Y. Acad. Sci. 1091, 336–345. doi: 10.1196/annals.1378.078
Zhuo, J. L., Imig, J. D., Hammond, T. G., Orengo, S., Benes, E., and Navar, L. G. (2002). Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT1 receptor. Hypertension 39, 116–121. doi: 10.1161/hy0102.100780
Zhuo, J. L., Kobori, H., Li, X. C., Satou, R., Katsurada, A., and Navar, L. G. (2016). Augmentation of angiotensinogen expression in the proximal tubule by intracellular angiotensin II via AT1a/MAPK/NF-кB signaling pathways. Am. J. Physiol. Ren. Physiol. 310, F1103–F1112. doi: 10.1152/ajprenal.00350.2015
Zhuo, J. L., and Li, X. C. (2013). Proximal nephron. Compr. Physiol. 3, 1079–1123. doi: 10.1002/cphy.c110061
Zhuo, J. L., Li, X. C., Garvin, J. L., Navar, L. G., and Carretero, O. A. (2006a). Intracellular angiotensin II induces cytosolic Ca2+ mobilization by stimulating intracellular AT1 receptors in proximal tubule cells. Am. J. Physiol. Ren. Physiol. 290, F1382–F1390. doi: 10.1152/ajprenal.00269.2005
Zhuo, J. L., Ohishi, M., and Mendelsohn, F. A. (1999). Roles of AT1 and AT2 receptors in the hypertensive Ren-2 gene transgenic rat kidney. Hypertension 33, 347–353. doi: 10.1161/01.HYP.33.1.347
Zou, L. X., Imig, J. D., von Thun, A. M., Hymel, A., Ono, H., and Navar, L. G. (1996). Receptor-mediated intrarenal angiotensin II augmentation in angiotensin II-infused rats. Hypertension 28, 669–677. doi: 10.1161/01.HYP.28.4.669
Keywords: angiotensin II, AT1 receptors/AT2 receptors, hypertension, kidney, proximal tubule
Citation: Li XC, Wang C-H, Leite APO and Zhuo JL (2021) Intratubular, Intracellular, and Mitochondrial Angiotensin II/AT1 (AT1a) Receptor/NHE3 Signaling Plays a Critical Role in Angiotensin II-Induced Hypertension and Kidney Injury. Front. Physiol. 12:702797. doi: 10.3389/fphys.2021.702797
Edited by:
Jennife Sullivan, Augusta University, United StatesReviewed by:
Steven Crowley, Duke University, United StatesCopyright © 2021 Li, Wang, Leite and Zhuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Long Zhuo, anpodW9AdHVsYW5lLmVkdQ==; Xiao Chun Li, eGxpNjhAdHVsYW5lLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.