 Yu-long Bao1†Li Wang1†Hai-ting Pan1Tai-ran Zhang1Ya-hong Chen2Shan-jing Xu3Xin-li Mao3,4,5*
Yu-long Bao1†Li Wang1†Hai-ting Pan1Tai-ran Zhang1Ya-hong Chen2Shan-jing Xu3Xin-li Mao3,4,5* Shao-wei Li4,5*
Shao-wei Li4,5*- 1College of Basic Medicine, Inner Mongolia Medical University, Hohhot, China
- 2Health Management Center, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 3School of Medicine, Shaoxing University, Shaoxing, Chian
- 4Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
- 5Department of Gastroenterology, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, China
Liver fibrosis refers to the process underlying the development of chronic liver diseases, wherein liver cells are repeatedly destroyed and regenerated, which leads to an excessive deposition and abnormal distribution of the extracellular matrix such as collagen, glycoprotein and proteoglycan in the liver. Liver fibrosis thus constitutes the pathological repair response of the liver to chronic injury. Hepatic fibrosis is a key step in the progression of chronic liver disease to cirrhosis and an important factor affecting the prognosis of chronic liver disease. Further development of liver fibrosis may lead to structural disorders of the liver, nodular regeneration of hepatocytes and the formation of cirrhosis. Hepatic fibrosis is histologically reversible if treated aggressively during this period, but when fibrosis progresses to the stage of cirrhosis, reversal is very difficult, resulting in a poor prognosis. There are many causes of liver fibrosis, including liver injury caused by drugs, viral hepatitis, alcoholic liver, fatty liver and autoimmune disease. The mechanism underlying hepatic fibrosis differs among etiologies. The establishment of an appropriate animal model of liver fibrosis is not only an important basis for the in-depth study of the pathogenesis of liver fibrosis but also an important means for clinical experts to select drugs for the prevention and treatment of liver fibrosis. The present study focused on the modeling methods and fibrosis characteristics of different animal models of liver fibrosis, such as a chemical-induced liver fibrosis model, autoimmune liver fibrosis model, cholestatic liver fibrosis model, alcoholic liver fibrosis model and non-alcoholic liver fibrosis model. In addition, we also summarize the research and application prospects concerning new organoids in liver fibrosis models proposed in recent years. A suitable animal model of liver fibrosis and organoid fibrosis model that closely resemble the physiological state of the human body will provide bases for the in-depth study of the pathogenesis of liver fibrosis and the development of therapeutic drugs.
Introduction
Liver fibrosis is a pathophysiological process caused by a variety of pathogenic factors that induce the abnormal proliferation of connective tissue in the liver. The repair and healing processes of liver injury can be accompanied by the development of liver fibrosis, and if the factors underlying such injury are not addressed, the process of fibrosis continues, eventually leading to cirrhosis (Hernandez-Gea and Friedman, 2011; Parola and Pinzani, 2019). Viruses, toxins, drugs, alcohol, hereditary factors, metabolism, cholestasis and parasites, among other factors, can damage liver cells, destroying the dynamic balance between collagen fiber synthesis, deposition, degradation and absorption, thus leading to the development of liver fibrosis. Therefore, liver fibrosis is not a unique disease; instead, different factors can lead to it, and regardless of the factors causing the relevant liver injury, the fibrosis process is similar (Mehal et al., 2011; Sebastiani et al., 2014; Kamdem et al., 2018; Testino et al., 2018).
The stimulation of different factors can lead to liver cell damage, which in turn causes inflammation. A continuous inflammatory response often leads to the formation of fibrosis. This is because inflammation causes cell damage, which further enhances the release of inflammatory mediators, such as cytokines and chemokines. These mediators recruit a large number of inflammatory cells to the site of inflammation, such as lymphocytes, neutrophils, eosinophils, basophils, mast cells and macrophages. The collected inflammatory cells further activate effector cells, which promote fibrosis (Hernandez-Gea and Friedman, 2011; Koyama and Brenner, 2017; Parola and Pinzani, 2019). The activation of hepatic stellate cells (HSCs) is the core event of liver fibrosis. HSCs are activated and differentiated into myofibroblasts (MFBs), which secrete and deposit a large amount of extracellular matrix (ECM). When liver injury persists for a long time, chronic inflammatory stimulation and the continuous deposition of ECM together lead to the gradual replacement of normal liver tissue by fibrous tissue (Higashi et al., 2017; Zhangdi et al., 2019; Figure 1). As a common pathological stage of chronic liver disease, liver fibrosis is necessary for the development of liver cirrhosis and even liver cancer. Indeed, persistent liver inflammation and fibrosis are known to eventually induce cirrhosis and liver cancer (Uehara et al., 2013).
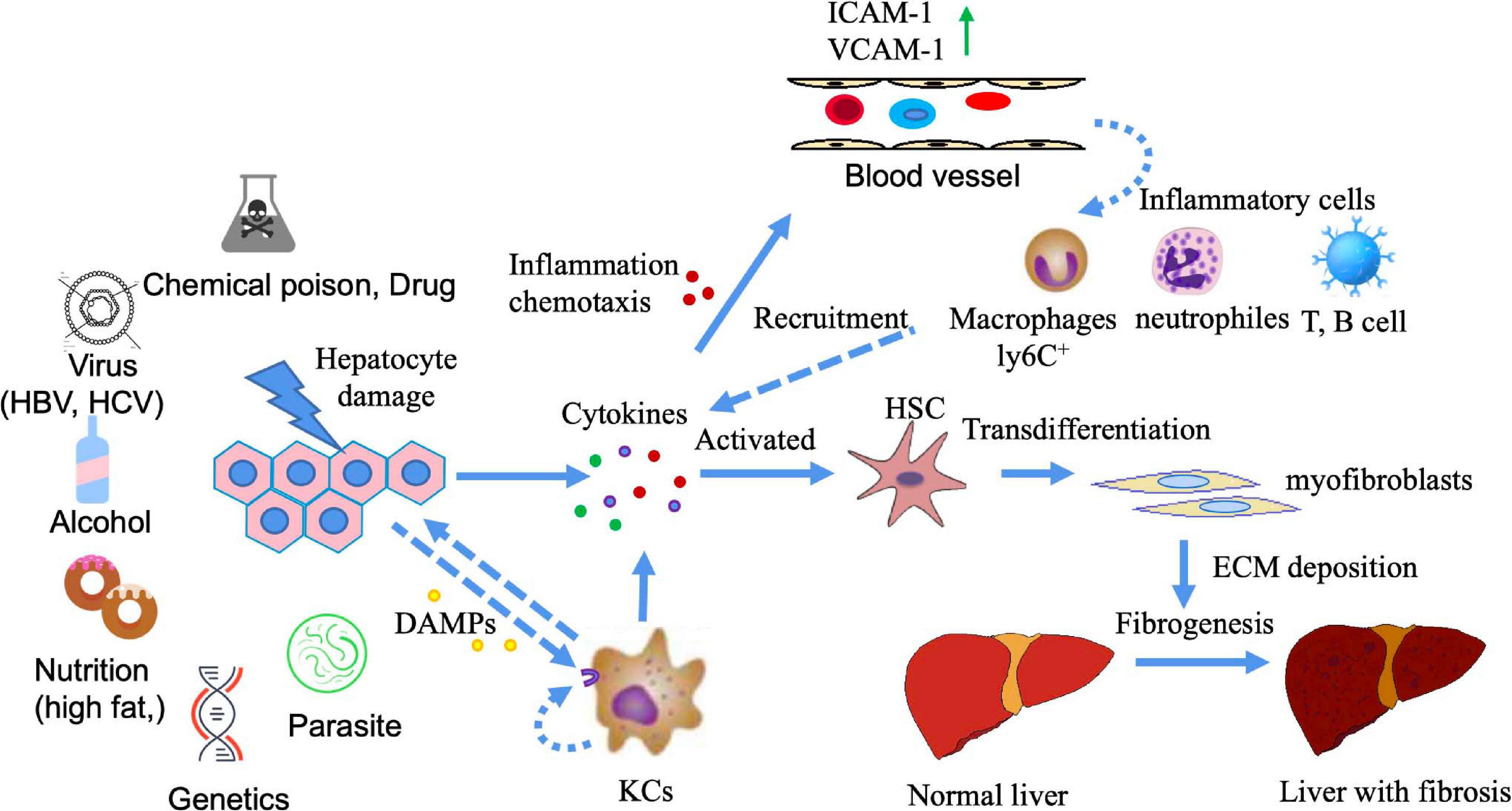
Figure 1. The mechanism of liver fibrosis. Liver damage can be caused by a variety of factors (e.g., chemical poisons, viruses, alcohol, nutrition, genetics, parasites, etc.). Injuries of liver cells release a variety of cytokines, among which damage-associated molecular patterns(DAMPs) (such as S100 family, high mobility group protein, etc.) can activate Kuffers (KCs) cells to produce inflammatory cytokines. Inflammatory cytokines can play a role through autocrine and paracrine. At this point, vascular permeability at the injured site increases, vascular endothelium simultaneously expresses high adhesion molecules, and inflammatory cells in the blood chemotaxis to the injured site under the action of chemokines, leading to the occurrence of inflammation. Multiple cytokines produced at the site of liver injury activate resting HSCs, causing HSCs to differentiate into myofibroblasts and produce ECM deposition. If the injury and inflammation are persistent or recurring, the deposition of ECM cannot be reversed, leading to liver fibrosis.
Studies in rodent models and humans have shown that liver fibrosis is reversible if the damage is ameliorated in a timely manner (Campana and Iredale, 2017). Matrix Metallopeptidase 13 (MMP13) is involved in the degradation of newly formed matrix during the recovery of liver fibrosis in rats. Although not all HSCs express MMP13, the production of MMP13 by HSCs plays a critical role in the process of fibrosis recovery (Watanabe et al., 2000). MMP9 secreted by Kupffer plays a key role in reversing hepatic fibrosis induced by thioacetamide (TAA) in mice (Feng et al., 2018). Dendritic cells can promote fibrosis regression by producing MMP9 (Jiao et al., 2012).
At present, the mechanism underlying liver fibrosis is unclear, and recent research has mostly focused on the etiology and mechanism of the disease. Although there has been some progress in the diagnosis and treatment of fibrosis, effective drugs and treatment are still lacking. The prevention, treatment and even reversal of liver fibrosis have always been the key to the successful treatment of chronic liver injury. The pathogenesis of liver fibrosis is of great clinical significance for the development of therapeutic drugs and overall improvement of therapeutic approaches. It is therefore important to study the pathogenesis of liver fibrosis and develop therapeutic drugs to construct the animal model of liver fibrosis with similar pathogeneses.
We herein report various liver fibrosis models, which are classified according to the modeling method used, to facilitate their utility as a reference for liver fibrosis researchers.
Chemical Drug-Induced Liver Fibrosis Model
Chemical liver fibrosis is induced by chemicals that can cause hepatotoxicity. The liver cells are damaged and consequently repaired, resulting in the abnormal growth of connective tissue in the liver. Models of chemical-induced liver injury are usually injected intraperitoneally, which is relatively easy to perform and results in stable development for use in studies concerning clinical liver fibrosis.
Carbon Tetrachloride (CCl4)
Carbon tetrachloride is a colorless non-polar organic compound, highly toxic, that can dissolve many substances such as fat and paint. It is a typical liver poison, but the concentration and frequency of exposure can affect its action site and toxicity. CCl4 directly damages liver cells (mainly endothelial cells and hepatic parenchyma cells in the hepatic portal vein region) by altering the permeability of lysosomes and mitochondrial membranes (Weber et al., 2003). The oxidase system in liver cells can also form highly active free radical metabolites through CYP2E1, leading to severe central lobular necrosis (Zangar et al., 2000). The damage mechanism of CCl4 is mainly oxidative damage caused by lipid peroxidation. Cytochrome P450 enzyme, especially CYP2E1, converts CCl4 into highly toxic trichloromethyl radical (⋅CCl3) and trichloromethyl peroxide (⋅CCl3O2) (Slater et al., 1985; Unsal et al., 2020). This model has been widely used to study the pathogenesis of liver fibrosis and cirrhosis.
More standardized procedures are needed for experimental liver fibrosis studies due to dramatic changes in animal welfare regulations in Europe. Scholten proposed standard operating procedure (SOPs) for the CCl4 mouse model and summarized the widely accepted experimental model for inducing liver injury leading to liver fibrosis (Scholten et al., 2015). The toxicological mechanism of liver fibrosis induced by CCl4 may be related to multiple biological processes, pathways and targets (Dong et al., 2016). After 15 weeks of CCl4 induction, multiple well-differentiated hepatocellular carcinoma (HCC) cells were found in the livers of all mice. CD133 was significantly up-regulated after CCl4 treatment, and the levels of desmin and glial fibrillary acidic protein, the representative markers of HSC, were also significantly increased. The EGF expression was significantly reduced, contrary to what has been observed in humans. In A/J mice, chronic liver injury induced by CCl4 differs from HCC induced by human cirrhosis (Fujii et al., 2010). The collagen expression was found to be significantly increased after CCl4 injury, and the number of cells expressing cytoglobin was also increased. Cytoglobin may be an early biomarker of liver fibrosis (Man et al., 2008). In addition to intraperitoneal injection, CCl4 can also be inhaled to establish a liver fibrosis model. Rats were exposed to CCl4 vapor twice a week for 30 s each time, while phenobarbital (0.3 g/L) was added to drinking water. The duration of inhalation was increased by 30 s after the first three sessions and by 1 min after every three sessions until a steady state was reached for 5 min. After 9 weeks, it can lead to liver fibrosis (Leeming et al., 2013; Marfà et al., 2016). Compared with intraperitoneal injection, inhalation route is a complex process, with great individual differences, and can cause multiple organ damage. An intraperitoneal injection can reach the liver directly from the hepatic portal vein. The animal model of liver fibrosis induced by CCl4 is relatively low-cost to develop, and the implementation method is relatively simple. Furthermore, it is a classic model and one of the earliest, most widely used and most frequently selected by researchers.
TAA
Thioacetamide is an organic compound with the molecular formula CH3CSNH2, found as a colorless or white crystal. TAA is widely used as a model for inducing experimental liver fibrosis, and can also be used to induce acute liver failure and liver tumors by controlling the dose and duration of administration. The TAA model is suitable for the study of connective tissue metabolism in fibrotic and cirrhotic models (Müller et al., 1988). TAA itself is not hepatotoxic, and its active metabolites covalently bind to proteins and lipids, causing oxidative stress leading to central lobular necrosis of the liver. Compared with CCl4, TAA resulted in more periportal inflammatory cell infiltration and more pronounced ductal hyperplasia. The intraperitoneal administration of 150 mg/kg of TAA 3 times per week for 11 weeks in rats and TAA administration in drinking water at 300 mg/L for 2-4 months in mice can successfully and repetitively cause chronic liver injury and fibrosis (Wallace et al., 2015). The continued administration of TAA (after the continuous TAA injection for more than 11 weeks) induced sustained liver fibrosis in common marmosets, and this primate-like model of liver fibrosis was thus able to be used to evaluate the therapeutic effect of liver fibrosis (Inoue et al., 2018). In Macaca fascicularis fibrosis models induced by TAA and CCl4, TAA induced significant fibrosis, but CCl4 did not. Both TAA and CCl4 increased the Child-Pugh score, but only the TAA model showed an increased retention of indocyanine green. TAA-induced M. fascicularis fibrosis was similar to Child-Pugh grade B fibrosis in humans. This model is evaluable by clinical indicators and can be used in preclinical studies (Matsuo et al., 2020). Although both CCl4 and TAA-induced liver injury and fibrosis are dependent on CYP2E1, in some cases, CYP2A5 may have a protective effect against TAA-induced liver injury and fibrosis but has no effect on the hepatotoxicity of CCl4 (Hong et al., 2016). The serum amino acid pattern in the TAA-induced chronic cirrhosis model is partially similar to the corresponding human disease (Fontana et al., 1996). The hepatic fibrosis model of rats was established by injecting TAA solution for 7 weeks. Serum and urine samples were collected weekly for a nuclear magnetic resonance metabolomics analysis to search for differential metabolites associated with TAA-induced injury. That study helped clarify the role of metabolic dynamics in the course of hepatic fibrosis disease (Wei et al., 2014). The levels of fibrogenic cytokines, such as transforming growth factor-β(TGF-β), platelet derived growth factor (PDGF) and connective tissue growth factor (CTGF), also increased in the liver tissue of all three models, but the levels of CTGF in the liver tissue and serum were the highest in the CCl4 group (Park et al., 2016). After 12-week oral administration of TAA in rats, bile duct fibrosis was induced, characterized by tubular hyperplasia surrounded by fibrous tissue (Hata et al., 2013). Both CCl4 and TAA can cause lipid oxidative damage in liver cells. The model of liver fibrosis induced by CCl4 is more suitable for studying the mechanism of spontaneous reversal of liver fibrosis. The hepatic fibrosis model induced by TAA is more suitable for the study of the mechanism of hepatic fibrosis, the screening of therapeutic drugs and the reliability evaluation of hepatic fibrosis serological markers.
DMN and Diethylnitrosamine (DEN)
The toxicity of various nitrosamines in animals and humans is well established, and trace amounts of DEN or DMN can cause severe liver injury in either the enteric or oral form. The most prominent manifestations are extensive neutrophilic infiltration, extensive central lobular hemorrhaging and necrosis, bile duct hyperplasia, fibrosis, bridging necrosis and ultimately HCC. Due to the stability of DMN- and DEN-induced liver changes, the administration of these agents to rodents has become a commonly used experimental model (Tolba et al., 2015).
Iron deposition and fat accumulation were shown to play an important role in the pathological changes of DMN-induced liver fibrosis in rats (He et al., 2007). Rats were intraperitoneally injected with DMN 3 days a week for 3 weeks. Severe central lobular congestion and hemorrhaging and necrosis were observed on day 7. On day 14, central lobular necrosis and numerous neutrophils infiltration were observed. Collagenous fibrous deposition was seen on day 21, along with severe central lobular necrosis, focal fatty changes, bile duct hyperplasia and bridging necrosis and fibrosis around the central vein. DMN-induced liver injury in rats seems to be an animal model similar to early human cirrhosis (George et al., 2001). The model shows significantly increased liver collagen fibraldehyde content due to DMN administration, and the cross-linking of liver fibrosis collagen induced by DMN is greater than that in normal liver. Furthermore, the deposition of type III collagen is more obvious than that of type I collagen in early fibrosis (George and Chandrakasan, 1996). The percentage of collagen fibrosis in rat liver fibrosis induced by DMN has been shown to be closely correlated with the serum levels of hyaluronic acid (HA), laminin (LN) and type IV collagen (Li et al., 2005). After 4 weeks of DEN treatment, 30% of zebrafish showed hyperplasia of reticular fibers. After 6 weeks, reticular and collagen fibers showed active hyperplasia, and the proliferation rate of reticular fibers increased to 80%, successfully generating a stable liver fibrosis model in zebrafish (Wang et al., 2014).
In the comparative study of dimethylnitrosamine (DMN), CCl4 and TAA rat liver fibrosis models, lipid peroxidation was highest in the CCl4 model, and the serum liver enzyme levels increased with severity. The DMN and TAA models showed significant changes in liver fibrosis. The Alpha-SAM levels significantly increased in the DMN model. In summary, while the modeling time with this method is short, its development is simple, and the fibrosis degree is stable. However, because of the toxicity of nitrosamines, researchers should ensure proper safety measures are taken.
Acetaminophen (APAP)
Acetaminophen overdose is a major cause of drug-induced acute liver failure in many developed countries. Mitochondrial oxidative stress is considered the core event of APAP-induced liver injury (Yoon et al., 2016; Yan et al., 2018). N-acetyl-p-phenylquinone imine (NAPQI), a metabolite of APAP, is hepatotoxic and can increase the mRNA expression of α-SMA, COL1A1, COL3A1 and TGF-β, inducing the phosphorylation of ERK1/2 and SMAD2/3 and nuclear translocalization of EGR-1 in hepatic stellate LX2 cells. The long-term administration of APAP can induce liver fibrosis in mice (Bai et al., 2017). When the liver is first exposed to APAP, a necrotizing inflammatory process is kicked off, followed by liver regeneration. However, the liver begins to form fibrosis after the second exposure to APAP (AlWahsh et al., 2019). Of note, a new model of cirrhosis in which rats were gavaged with corn oil daily and APAP 500 mg/day for 3 weeks resulted in the development of focal biliary cirrhosis (Tropskaya et al., 2020).
Immune Damage-Induced Liver Fibrosis Model
Immune liver injury liver fibrosis mainly refers to liver fibrosis caused by clinical autoimmune hepatitis (AIH) and virus infection, but such animal models of immune liver injury lack the sustained replication stage of human virus infection and the pathological process of sustained damage of human liver immunity.
Concanavalin A (ConA), a lectin purified from Brazilian kidney bean (Soares et al., 2011), is widely used in the mouse model of immune-mediated hepatitis. Unlike other models of liver injury, ConA-induced injury is mainly caused by the activation and recruitment of T cells to the liver (Heymann et al., 2015). Therefore, the pathogenesis of the ConA model has something in common with human immune-mediated hepatitis, such as AIH (Wang et al., 2012) and viral hepatitis. The mouse hepatitis model induced by ConA (20 mg/kg, 12 h) reflects most of the pathogenicity of human type I AIH. This provides a reliable animal model for the study of the immune pathogenesis of AIH and the rapid evaluation of new therapeutic methods (Ye et al., 2018). In acute autoimmune liver injury induced by ConA, HSCs are activated early, and the expression of TGF-β1 and TGF-β3 is unbalanced, which may be related to liver dysfunction and fibrosis development (Wang et al., 2017). Repeated injections of Con A resulted in liver fibrosis in mice (Louis et al., 2000). The model of immune fibrosis in mice was established by injecting saffra protein A (0.3 mg/body) once a week for 4 weeks. IFN-β can inhibit liver cell damage caused by repeated injections of ConA but has no effect on the development of fibrosis (Tanabe et al., 2007).
Alcohol-Induced Liver Fibrosis Model
Alcoholic liver disease (ALD) is a chronic liver disease caused by long-term heavy drinking. Fatty liver is usually present in the initial stage, which can develop into alcoholic hepatitis, alcoholic liver fibrosis and alcoholic cirrhosis. Almost all heavy drinkers develop fatty liver, but only 20-40% develop more severe ALD, and the underlying mechanism leading to disease progression is still unclear (Tsukamoto and Lu, 2001; Seitz et al., 2018). Although rodents differ from humans with regard to their alcohol metabolism (Holmes et al., 1986) and immune system, experimental animal models of ALD, especially rodent models, have been widely used in the study of human ALD (Mathews et al., 2014; Lamas-Paz et al., 2018).
After the daily administration of alcohol to rats for 16 weeks, the rates of liver steatosis, necrosis, inflammation and fibrosis were increased (Zhou et al., 2013). Chronic ethanol feeding (10 days free oral Lieber-decarli ethanol liquid diet) plus single alcoholic ethanol feeding induced liver injury, inflammation and fatty liver, simulating acute and chronic alcoholic liver injury in humans. This simple model is very useful for the study of ALD and other organs damaged by alcohol consumption (Bertola et al., 2013). Mice treated with CCl4 combined with ethanol (up to 16%) showed extremely high rates of fibrotic alcoholic fatty liver disease 7 weeks later. The pattern of steatosis, inflammation and fibrosis involved in ALD in this mouse model is similar to that in humans and is suitable as a preclinical model for drug development (Brol et al., 2019). The same CCl4 vapor exposure combined with chronic alcohol feeding resulted in extensive liver fibrosis in rats at week 5 and micronodular cirrhosis at week 10. This animal model simulates how some chronic liver damage in humans may be due to the presence of other hepatotoxins in the environment that play a role in enhancing the effects of alcohol (Hall et al., 1991). A new experimental model of porcine hepatosclerosis was established by CCl4 and ethanol. Cirrhosis was induced by the intraperitoneal injection of CCl4 twice a week for 9 weeks. Corn flour was the only food consumed during the period, and a 5% alcohol-water mixture was consumed. After 9 weeks, 83.3% of the pigs had cirrhosis, and 33.3% had died (Zhang et al., 2009). In combination with chronic alcohol administration and a non-alcoholic steatohepatitis (NASH)-induced high-fat diet, this new model enables the study of the combined effects of alcohol and a high-fat diet on liver injury, which may contribute to the development of liver fibrosis by enhancing TLR4 signaling (Gäbele et al., 2011). To cause progressive alcoholic liver injury, the animal must be given too much alcohol and maintain a persistently high blood alcohol level. Because of the rats’ natural aversion to alcohol, the method of feeding them alcoholic liquid food was greatly restricted. In addition, the experiment cycle is long, the cost is high and the success rate is low, so it has been rarely used. At present, the more commonly used method is alcohol combined with chemical poison gavage, during the control of diet to replicate the model of alcoholic liver fibrosis. The model has the advantages of simple operation, short cycle and high molding rate.
Diet Metabolism-Induced Liver Fibrosis Model
Non-alcoholic fatty liver disease (NAFLD) is a clinicopathological syndrome characterized by excessive fat deposition in hepatocytes except for alcohol and other clear liver damage factors, which is closely related to insulin resistance and genetic susceptibility of acquired metabolic stress liver injury (Cobbina and Akhlaghi, 2017). NAFLD is becoming a common chronic liver injury due to lifestyle changes. NAFLD can cause inflammation, ballooning degeneration of hepatocytes, and varying degrees of fibrosis, known as non-alcoholic steatohepatitis (NASH). Patients with advanced liver fibrosis or cirrhosis are at risk of developing complications, such as HCC and esophageal varices (Stål, 2015).
A choline-deficient high-fat (CDHF) diet induces NASH in mice. Hepatic histopathology has shown that a CDHF diet causes severe steatosis, inflammation and pericellular fibrosis (Honda et al., 2017). A modified choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) rapidly induces liver fibrosis in mice. This model will contribute to a better understanding of human NASH disease and may be useful for the development of effective treatments (Matsumoto et al., 2013). AIM–/– mice fed the D09100301 diet showed similar phenotypes to non-obese patients with NAFLD, indicating their utility as a pathophysiological model for studying obesity-induced HCC (Komatsu et al., 2019). Dietary control combined with chemical toxicants may be an effective means of reducing the modeling time of diet-induced NAFLD models. A fast food diet (FFD) combined with a trace dose of CCL4 (0.5 mL/kg body weight) for 8 weeks resulted in histological features of NAFLD, including steatosis, inflammation and fibrosis, in Wistar rat models within 8 weeks, suggesting that the model has potential utility in developing NAFLD and anti-fibrosis therapy (Chheda et al., 2014). Furthermore, using a high-fat, high-fructose and high-cholesterol diet combined with a weekly low dose of CCL4 as an accelerant shortened the cycle of a mouse NASH model of fibrosis and HCC (Tsuchida et al., 2018). The mechanism of fatty liver fibrosis is still unclear, among which oxidative stress/lipid peroxidation is an important cause of fatty liver fibrosis. Unlike alcoholic fatty liver, which leads directly to liver fibrosis, non-alcoholic fatty liver must pass through an intermediate stage of steatohepatitis before it can develop into liver fibrosis. In other words, inflammation itself is a prerequisite for fatty liver fibrosis. The diet-induced NASH mouse model is characterized by good simulation of obesity, type 2 diabetes mellitus, dyslipidemia, and metabolic syndrome, but with less liver fibrosis.
Surgery-Induced Liver Fibrosis Model
Cholestasis is an obstruction of both bile flow formation and excretion. Continuous cholestasis leads to chronic inflammation, which damages bile duct cells and liver cells, activates MFBs through a number of regulatory factors and causes the excessive deposition of ECM, leading to liver fibrosis (Li and Apte, 2015).
Surgical bile duct ligation (BDL) is one of the most widely used experimental models of cholestatic liver injury in mice and rats. The BDL model is a classic model of liver fibrosis. BDL was first achieved by the double ligation of bile ducts in rats. In brief, at 7-10 days after surgery, bile duct stenosis, increased bile duct pressure, upstream dilatation of bile ducts, increased liver volume composed of portal vein and bile duct hyperplasia are observed (Rodríguez-Garay et al., 1996). BDL protocols have been improved over time, but basically, animals are anesthetized and then undergo laparotomy. The bile duct is exposed from the abdominal cavity and ligated twice using a surgical cord. Mice and rats that undergo the procedure develop a strong fibrotic response (Kirkland et al., 2010). During the procedure, the animal can be placed on a heated plate at 37°C and permanently connected to the anesthesia system. At the time of the operation, the bile ducts are double-ligated but not dissected. This procedure induces highly repeatable morphological phenotypic changes in the liver and allows for the study of fiber formation at specific points in time (Tag et al., 2015). The standard model for cholestasis studies is total BDL (tBDL), but this model can cause severe liver damage in mice, so a new cholestasis model using partial BDL (pBDL) has been established (Heinrich et al., 2011). A mouse model of recanalization of biliary tract obstruction was previous established by performing anastomosis between the gallbladder and jejunum (G-J anastomosis), which has some value for studying the recovery from cholestasis liver fibrosis (Yoshino et al., 2021).
Transgenic Animal Liver Fibrosis Model
A chronic hepatitis C virus infection leads to liver fibrosis and cirrhosis. For a long time, chimpanzees were the only available non-human model of HCV infection (Pellicoro et al., 2014). Since the host range of HBV is relatively narrow and it only infects humans, it is very difficult to establish an animal model of HBV infection. Only chimpanzees and tupaia have previously been used for infection experiments (Walter et al., 1996; Dandri et al., 2005). The construction of humanized liver chimeric transgenic mice enables the stable regrowth of human liver cells in mice, and even the normal function and morphology of human liver, which has become an important bridge between mouse and human preclinical studies (Katoh et al., 2008). Hepatitis virus infects human TK-NOG mice and UPA-SCID mice with severe combined immunodeficiency (UPA-SCID). All human TK-NOG and UPA-SCID mice injected with hepatitis B virus infected serum developed viremia. The occurrence of HCV viremia in TK-NOG mice was significantly higher than that in UPA-SCID mice. TK-NOG mice are more beneficial for the study of hepatitis virus virology and the evaluation of antiviral drugs (Kosaka et al., 2013).
In addition to humanized mice, many transgenic mice were constructed for the study of liver fibrosis according to the different pathogenesis of liver fibrosis and the key functional genes of liver fibrosis regulation (Hayashi and Sakai, 2011). Immunodeficiency NOD induced natural killer T cell (NKT) transgenic population mediated spontaneous multi-organ chronic inflammation and fibrosis, non-obese diabetic inflammation and fibrosis (N-IF) mice. Due to fibrosis components, early onset, spontaneity, and reproducibility, this novel mouse model provides further insight into the underlying mechanisms that mediate the transformation of chronic inflammation into fibrosis (Fransén-Pettersson et al., 2016). Although the pathology of BDL is similar to chronic cholestasis in humans, the severity of surgical stress and cholestasis injury limits the application of the BDL model. MDR2 (ABCB4) is a mouse homologous gene MDR3 (ABCB4) that encodes a tubule phospholipid transporter. MDR2-/- mice, also known as ABCB4-/- mice, are another mature model of chronic cholestatic liver injury (Ikenaga et al., 2015). MDR2 knockout (MDR2 -/-) mice are a genetic model similar to patients with primary sclerosing cholangitis (Nishio et al., 2019). Transgenic mice that overexpress the transforming growth factor-β1 (TGF-β1) fusion gene [C-reactive protein (CRP)/TGF-β1] are able to control the expression level of TGF-β1. This model can be used to study the regulation of collagen synthesis, fibrinolysis and the degree of reversibility of liver fibrosis. CRP/TGF-β1 transgenic mouse model can be used as an anti-fibrofactor test model (Kanzler et al., 1999). Similarly, TGF-β1 overexpression transgenic mice were established based on the tetracycline regulation gene expression system. This model will help to analyze the role of TGF-β1 in fibrogenesis (Ueberham et al., 2003). The role of platelet-derived growth factor A (PDGF-A) in the formation of liver fibrosis in vivo can be evaluated in transgenic mice with hepatocellular specific overexpression of PDGF-A by the C-reactive protein (CRP) gene promoter (Thieringer et al., 2008). Metalloproteinase-1 tissue inhibitor (TIMP-1) is upregulated during liver fibrogenesis, but its role in liver fibrosis and carcinogenesis in mice is not necessarily direct (Thiele et al., 2017). Transgenic mice overexpressing human TIMP-1 (HTIMP-1) in the liver under the control of albumin promoter/enhancer can be used to investigate the role of TIMP-1 in promoting liver fibrosis (Yoshiji et al., 2000). Mouse models carrying human apolipoprotein E∗ 3-leiden and cholesterol ester transfer protein, fed a “Western” diet, lead to liver inflammation and fibrosis that are highly dependent on genetic background and have a large overlap of pathways between human diseases (Hui et al., 2018).
Organoid Liver Fibrosis Modes
Studying tissue and organ biology in mammals is challenging, and progress may be hampered by the availability of samples and ethical issues, especially in humans (Rossi et al., 2018). Although traditional 2D cell culture systems have many advantages, these models lack the ability to maintain in situ cellular characteristics and reflect cell-to-cell and cell-to-matrix interactions. The primary cells obtained by purification and isolation will also lose their original functions and characteristics after 2D culture in vitro. Organoids are 3D organ-like cells that are derived from embryonic or adult stem cells that are cultured in vitro and have a definite structure and function. Although these cellular structures are not human organs in the true sense, they can mimic real organs in structure and function, so they are playing an increasingly important role in scientific research. Organoids in vitro culture systems are characterized by self-renewing stem cell populations that include cells capable of differentiating into organs with similar spatial tissue functions (Artegiani and Clevers, 2018). Organoids can be used to simulate organ development and disease, and have a wide range of applications in basic research, drug development, and regenerative medicine (Lancaster and Knoblich, 2014; Huch et al., 2017; Xia et al., 2019). While mouse models and cell lines have advanced our understanding of liver biology and related diseases, they have significant drawbacks in simulating human liver tissue, particularly its complex structure and metabolic function. Currently, a variety of liver organoids have been established from induced pluripotent stem cells, embryonic stem cells, hepatoblasts and adult tissue-derived cells (Prior et al., 2019).
HepaRG (Hep) and primary human HSCs were cultured into 3D spheres in 96-well plates. The metabolic capacity of the organoid exceeds 21 days. This novel liver organ culture model is the first capable of detecting hepatocellular dependence and compound-induced HSC activation and represents an important advance in the in vitro compound assessment of drug-induced liver fibrosis (Leite et al., 2016). Induced pluripotent stem cell-hepatic stellate cells (iPSC-HSCs) are very similar to primary human HSCs at the transcriptional, cellular, and functional levels. iPSC-HSCs exhibit a static phenotype when they remain 3D spherical with HepaRG hepatocytes, but are activated in response to wound-healing mediator stimulation and hepatocytotoxicity, resulting in fibrotic responses and secretion of procollagen, and accumulation of retinol in lipid droplets, similar to their in vivo counterparts. Thus, this protocol provides a powerful in vitro system for studying stellate cell development, modeling liver fibrosis, and screening for drug toxicity (Coll et al., 2018). Activated hepatic stellate cells (aHSCs) produced by 2D culture were coated in a 3D collagen gel to form a spherical structure, which created a stiffer environment and expressed higher levels of TIMP1 and LOXL2 compared to LX-2 cells cultured in 2D culture. This model proposes a fibrosis model that can be combined with the multicellular model to more accurately reflect the impact of severe fibrosis on liver function (Brovold et al., 2020). Using organoids from intrahepatic bile ducts, APAP was used to induce organoid injury in culture medium. The injury model suggested that bile duct cell apoptosis and its fibrotic response played a role in the initiation of the fibrotic process of bile duct diseases, such as biliary atresia (BA) (Chusilp et al., 2020). Genetically susceptible NAFLD organoid systems composed of hepatocytes (HepG2) and HSCs (LX-2) can be used to clarify the molecular mechanisms underlying the accumulation of lipids that induces the early stage of fibrogenesis. In addition, these systems can be used to identify new compounds for treating NASH through high-throughput drug screening (Pingitore et al., 2019). It is difficult to select media and extracellular matrix that can co-maintain multiple cell lineages. A novel organ-like culture method was developed for co-differentiation of epithelial and mesenchymal lineages from PSCs. Using 11 different health and disease pluripotent cell lines, a repeatable method was developed to obtain multicellular human liver-like organs composed of hepatocytes, stellate cells, and Kupffer like cells that exhibit transcriptome similarity to tissue of in vivo origin. These multicellular human hepatic organs (HLOs), in combination with free fatty acid therapy, reproduce the progressive, staging nature of steatohepatitis like pathology, including steatosis, inflammation, and fibrosis, and can potentially be used for drug screening by analyzing organ hardness (Ouchi et al., 2019). Liver organoids were generated from mice with mild (NASH A), moderate (NASH B), and severe (NASH C) methionine and choline deficiency diets-induced NASH models that reproduce the characteristics of NASH disease liver tissue. The NASH liver organoid model can be used to study genetic stability and/or lipid metabolism during NAFLD/NASH transformation (Elbadawy et al., 2020).
In conclusion, organoid technology is one of the most important advances in stem cell research. Organoids are three-dimensional cell cultures that reproduce some of the key cell types and structural characteristics of the organs they represent. Organoids remove the confounding variables that might be introduced by animal models and are more complex than homogenized cell cultures. Organoid culture has a high degree of gene stability, maintaining the genotype and phenotype of the source tissue. Thus, organoids can be used to model diseases, to study the mechanisms and progression of diseases, and to predict patients’ individual responses to drug therapy (Figure 2).
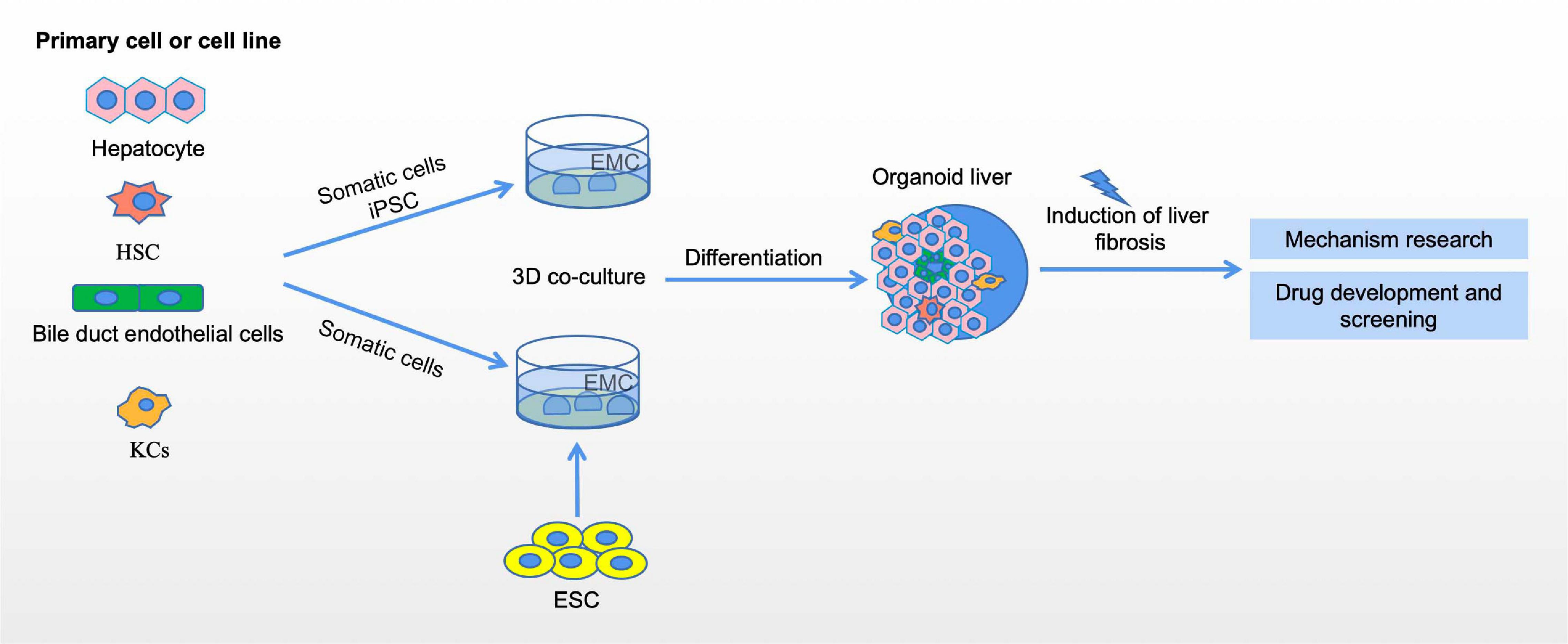
Figure 2. Organoid models of liver fibrosis. The generation of liver organoids and liver fibrosis models. liver organoid can be constructed by 3D co-culture of embryonic stem cells (ESC) and somatic hepatocytes, 3D coculture of induced pluripotent stem cells, and then through induction and differentiation of different developmental stages, and finally can be developed into liver organoids in the reactor. The establishment of liver fibrosis model using liver organoids can be used to study the mechanism of liver fibrosis and drug development and screening.
Conclusion and Perspectives
A reasonable model of liver fibrosis should resemble the characteristics and pathogenesis of human disease. It is universally recognized that “animal welfare” includes five freedoms: freedom from hunger and thirst; comfort; freedom from pain, injury and disease; freedom from fear and sadness and expression of nature. In laboratory animals, it is difficult to achieve all five freedoms at the same time. In particular, damage to these creatures’ health is often a byproduct of the natural course of research. Due to such factors as animal suffering and scientific exploration, the three R principles are widely recognized: including replacement, reduction and refinement (Lindsjö et al., 2016). This requires animal experiments to have stable experimental methods, a high mold-forming rate and good reproducibility. Thus far, researchers have successfully developed a number of hepatic fibrosis models using different experimental animals and different methods. However, due to the complexity of the pathogenesis of human liver fibrosis and differences in the genetic background between human and other animal species, there is no modeling method that can perfectly replicate the process of human liver fibrosis. Researchers can only use different models of liver fibrosis to mimic, as much as possible, the different pathologies that cause liver fibrosis in humans. Table 1 briefly compares the advantages and disadvantages of different liver fibrosis models (Table 1). Although the liver fibrosis model induced by chemical poisons is different from the pathogenesis of human liver fibrosis, it is often used to study the mechanism of liver fibrosis due to its simple operation and good reproducibility. The immune-induced liver injury fibrosis model most closely replicates the clinical situation, which is similar to the liver fibrosis caused by human AIH and virus infection. Due to the complex pathogenesis of ALD and NAFLD, and the great differences between animal genetics, metabolism and immunity and human beings, it is relatively difficult to construct a model similar to human diseases through liver fibrosis induced by alcohol and the diet metabolism. BDL can simulate liver fibrosis induced by cholestasis, requiring only a short time with good reproducibility for model construction. However, it has drawbacks of substantial operational requirements, the need for an aseptic surgical setting and high animal mortality. Humanized transgenic chimeric mice and transgenic/knockout mice are emerging modeling methods that have been established in recent decades. Humanized liver chimeric transgenic mice constitute a good animal model of hepatitis virus infection, and transgenic/knockout mice are a good animal model for studying the role of the functional genes involved in liver fibrosis. The latest organoid model makes up for the great difference between the traditional in vitro cell culture and human organs. Liver organoids are expected to be a useful new model for in vitro experiments that closely resembles the actual situation in human liver diseases. Although the techniques are becoming more advanced, liver fibrosis models are becoming more complex. Researchers continue to make various models of liver fibrosis in order to bring them closer to the true pathogenesis of human liver fibrosis (Figure 3). Each model has its advantages and disadvantages, and it remains a challenge to identify the most reasonable and stable hepatic fibrosis model.
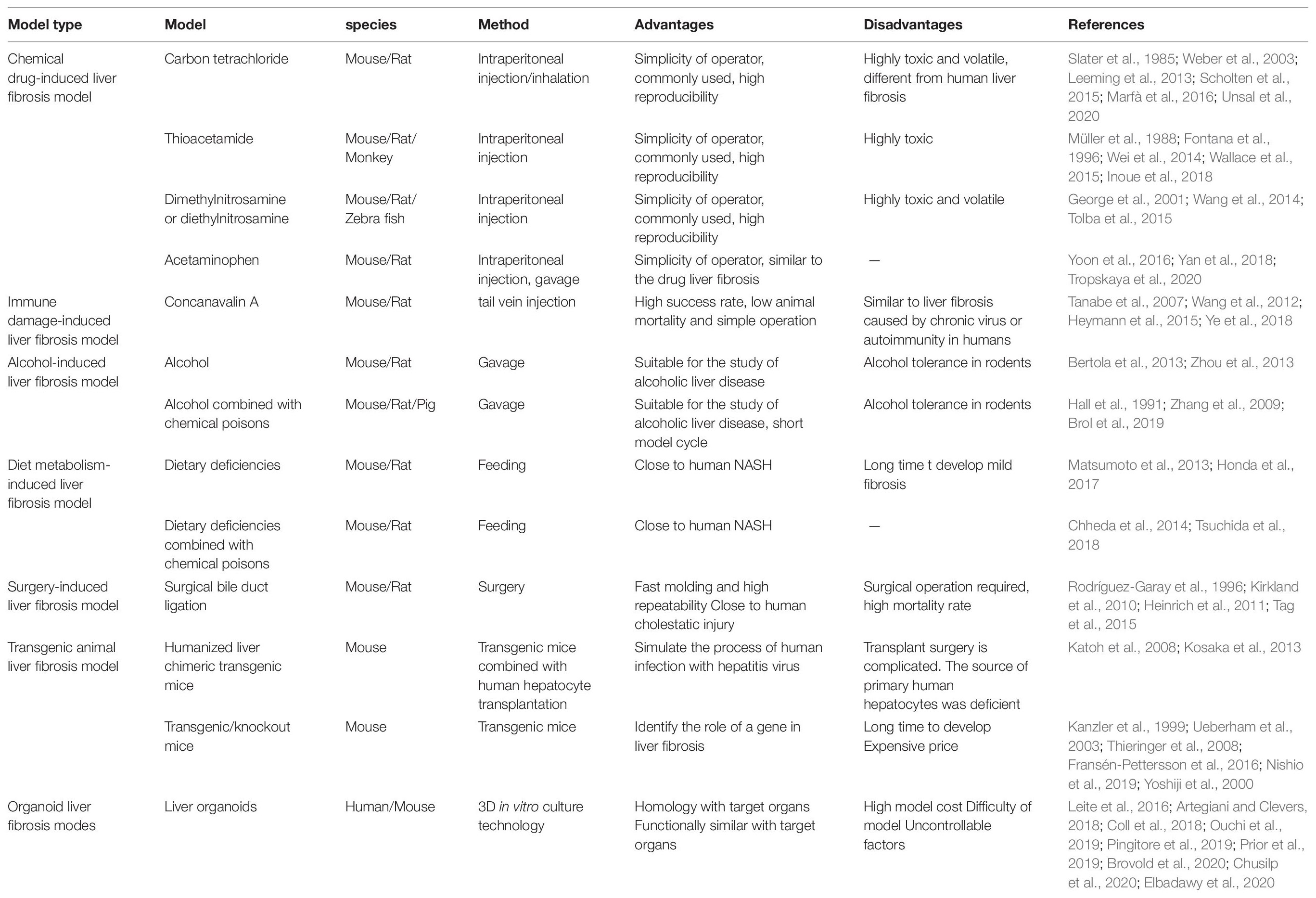
Table 1. Advantages and disadvantages of different methods in hepatic fibrosis models.
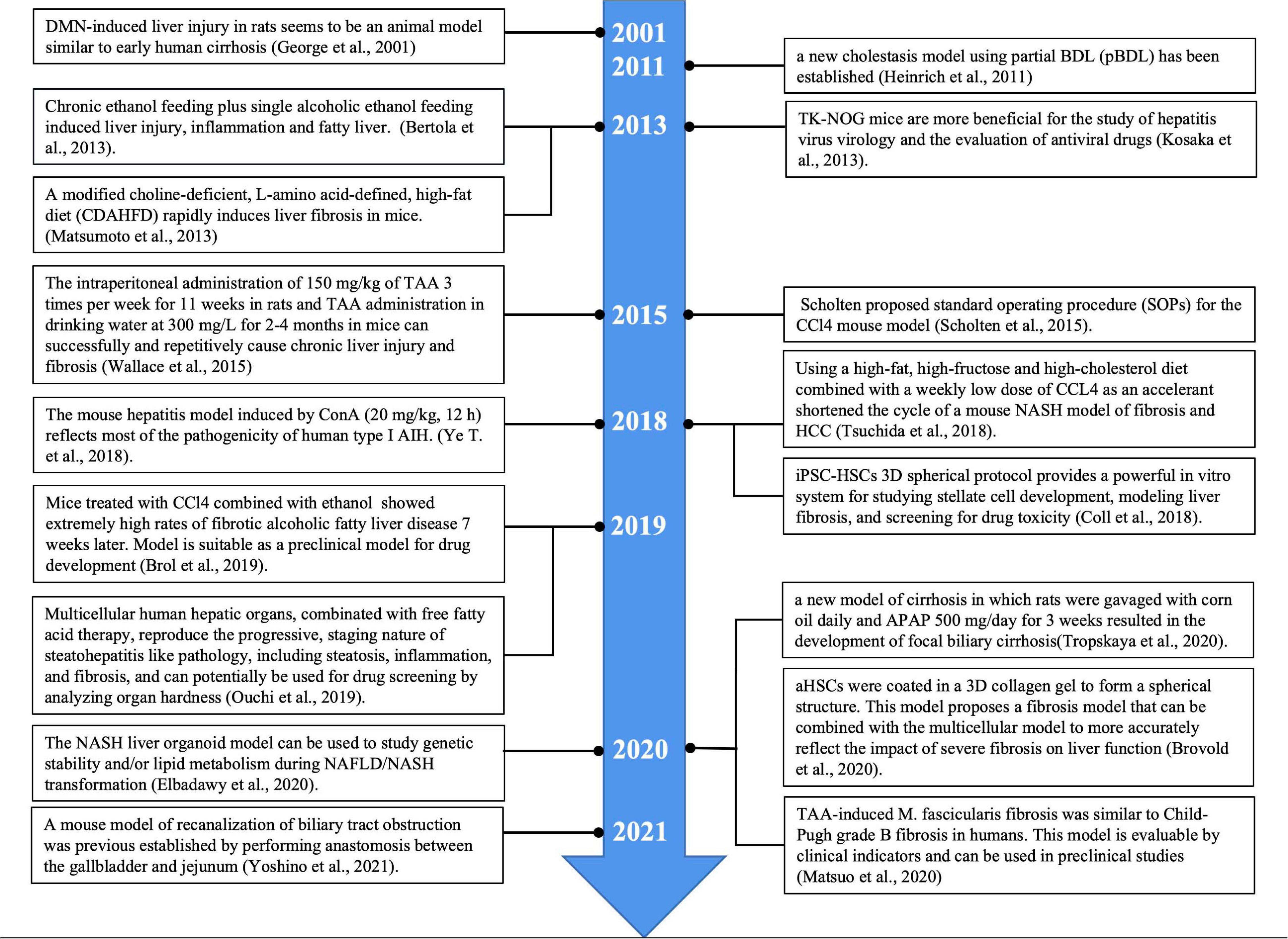
Figure 3. Timeline of animal and organoid liver fibrosis mode.
Author Contributions
All authors contributed to the writing and editing of the manuscript and contributed to the article and approved the submitted version.
Funding
This work was supported in part by the Program of Inner Mongolia Autonomous Region Tumor Biotherapy Collaborative Innovation Center, Medical Science and Technology Project of Zhejiang Province (2021PY083), Program of Taizhou Science and Technology Grant (20ywb29), Major Research Program of Taizhou Enze Medical Center Grant (19EZZDA2), Key Technology Research and Development Program of Zhejiang Province (2019C03040), and Open Fund of Key Laboratory of Minimally Invasive Techniques & Rapid Rehabilitation of Digestive System Tumor of Zhejiang Provinces (21SZDSYS01 and 21SZDSYS09).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
AlWahsh, M., Othman, A., Hamadneh, L., Telfah, A., Lambert, J., Hikmat, S., et al. (2019). Second exposure to acetaminophen overdose is associated with liver fibrosis in mice. EXCLI J. 18, 51–62.
Artegiani, B., and Clevers, H. (2018). Use and application of 3D-organoid technology. Hum. Mol. Genet. 27:ddy187. doi: 10.1093/hmg/ddy187
Bai, Q., Yan, H., Sheng, Y., Jin, Y., Shi, L., Ji, L., et al. (2017). Long-term acetaminophen treatment induced liver fibrosis in mice and the involvement of Egr-1. Toxicology 382, 47–58. doi: 10.1016/j.tox.2017.03.008
Bertola, A., Mathews, S., Ki, S. H., Wang, H., and Gao, B. (2013). Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637. doi: 10.1038/nprot.2013.032
Brol, M. J., Rösch, F., Schierwagen, R., Magdaleno, F., Uschner, F. E., Manekeller, S., et al. (2019). Combination of CCl with alcoholic and metabolic injuries mimics human liver fibrosis. Am. J. Physiol. Gastroint. Liver Physiol. 317, G182–G194. doi: 10.1152/ajpgi.00361.2018
Brovold, M., Keller, D., and Soker, S. (2020). Differential fibrotic phenotypes of hepatic stellate cells within 3D liver organoids. Biotechnol. Bioeng. 117, 2516–2526. doi: 10.1002/bit.27379
Campana, L., and Iredale, J. P. (2017). Regression of Liver Fibrosis. Semin. Liver Dis. 37:1597816. doi: 10.1055/s-0036-1597816
Chheda, T. K., Shivakumar, P., Sadasivan, S. K., Chanderasekharan, H., Moolemath, Y., Oommen, A. M., et al. (2014). Fast food diet with CCl4 micro-dose induced hepatic-fibrosis–a novel animal model. BMC Gastroenterol. 14:89. doi: 10.1186/1471-230X-14-89
Chusilp, S., Lee, C., Li, B., Lee, D., Yamoto, M., Ganji, N., et al. (2020). A novel model of injured liver ductal organoids to investigate cholangiocyte apoptosis with relevance to biliary atresia. Pediatr. Surg. Int. 36, 1471–1479. doi: 10.1007/s00383-020-04765-2
Cobbina, E., and Akhlaghi, F. (2017). Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 49, 197–211. doi: 10.1080/03602532.2017.1293683
Coll, M., Perea, L., Boon, R., Leite, S. B., Vallverdú, J., Mannaerts, I., et al. (2018). Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 23, 101.e–113.e. doi: 10.1016/j.stem.2018.05.027
Dandri, M., Volz, T. K., Lütgehetmann, M., and Petersen, J. (2005). Animal models for the study of HBV replication and its variants. J. Clin. Virol. 34(Suppl. 1), S54–S62.
Dong, S., Chen, Q.-L., Song, Y.-N., Sun, Y., Wei, B., Li, X.-Y., et al. (2016). Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 41, 561–572. doi: 10.2131/jts.41.561
Elbadawy, M., Yamanaka, M., Goto, Y., Hayashi, K., Tsunedomi, R., Hazama, S., et al. (2020). Efficacy of primary liver organoid culture from different stages of non-alcoholic steatohepatitis (NASH) mouse model. Biomaterials 237:119823. doi: 10.1016/j.biomaterials.2020.119823
Feng, M., Ding, J., Wang, M., Zhang, J., Zhu, X., and Guan, W. (2018). Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int. J. Biol. Sci. 14, 1033–1040. doi: 10.7150/ijbs.25589
Fontana, L., Moreira, E., Torres, M. I., Fernández, M. I., Ríos, A., Sánchez de Medina, F., et al. (1996). Serum amino acid changes in rats with thioacetamide-induced liver cirrhosis. Toxicology 106, 197–206. doi: 10.1016/0300-483x(95)03177-h
Fransén-Pettersson, N., Duarte, N., Nilsson, J., Lundholm, M., Mayans, S., and Larefalk, Å, et al. (2016). A New Mouse Model That Spontaneously Develops Chronic Liver Inflammation and Fibrosis. PLoS One 11:e0159850. doi: 10.1371/journal.pone.0159850
Fujii, T., Fuchs, B. C., Yamada, S., Lauwers, G. Y., Kulu, Y., Goodwin, J. M., et al. (2010). Mouse model of carbon tetrachloride induced liver fibrosis: Histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 10:79. doi: 10.1186/1471-230X-10-79
Gäbele, E., Dostert, K., Dorn, C., Patsenker, E., Stickel, F., and Hellerbrand, C. (2011). A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol Clin. Exp. Res. 35, 1361–1367. doi: 10.1111/j.1530-0277.2011.01472.x
George, J., and Chandrakasan, G. (1996). Molecular characteristics of dimethylnitrosamine induced fibrotic liver collagen. Biochim. Biophys. Acta 1292, 215–222. doi: 10.1016/0167-4838(95)00202-2
George, J., Rao, K. R., Stern, R., and Chandrakasan, G. (2001). Dimethylnitrosamine-induced liver injury in rats: the early deposition of collagen. Toxicology 156, 129–138. doi: 10.1016/s0300-483x(00)00352-8
Hall, P. D., Plummer, J. L., Ilsley, A. H., and Cousins, M. J. (1991). Hepatic fibrosis and cirrhosis after chronic administration of alcohol and “low-dose” carbon tetrachloride vapor in the rat. Hepatology 13, 815–819. doi: 10.1016/0270-9139(91)90246-r
Hata, M., Iida, H., Yamanegi, K., Yamada, N., Ohyama, H., Hirano, H., et al. (2013). Phenotypic characteristics and proliferative activity of hyperplastic ductule cells in cholangiofibrosis induced by thioacetamide in rats. Exp. Toxicol. Pathol. 65, 351–356. doi: 10.1016/j.etp.2011.11.004
Hayashi, H., and Sakai, T. (2011). Animal models for the study of liver fibrosis: new insights from knockout mouse models. Am. J. Physiol. Gastrointest Liver Physiol. 300, G729–G738. doi: 10.1152/ajpgi.00013.2011
He, J.-Y., Ge, W.-H., and Chen, Y. (2007). Iron deposition and fat accumulation in dimethylnitrosamine-induced liver fibrosis in rat. World J. Gastroenterol. 13, 2061–2065. doi: 10.3748/wjg.v13.i14.2061
Heinrich, S., Georgiev, P., Weber, A., Vergopoulos, A., Graf, R., and Clavien, P.-A. (2011). Partial bile duct ligation in mice: a novel model of acute cholestasis. Surgery 149, 445–451. doi: 10.1016/j.surg.2010.07.046
Hernandez-Gea, V., and Friedman, S. L. (2011). Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 6, 425–456. doi: 10.1146/annurev-pathol-011110-130246
Heymann, F., Hamesch, K., Weiskirchen, R., and Tacke, F. (2015). The concanavalin A model of acute hepatitis in mice. Lab. Anim. 49(1 Suppl.), 12–20. doi: 10.1177/0023677215572841
Higashi, T., Friedman, S. L., and Hoshida, Y. (2017). Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 121, 27–42. doi: 10.1016/j.addr.2017.05.007
Holmes, R. S., Duley, J. A., Algar, E. M., Mather, P. B., and Rout, U. K. (1986). Biochemical and genetic studies on enzymes of alcohol metabolism: the mouse as a model organism for human studies. Alcohol Alcohol. 21, 41–56.
Honda, T., Ishigami, M., Luo, F., Lingyun, M., Ishizu, Y., Kuzuya, T., et al. (2017). Branched-chain amino acids alleviate hepatic steatosis and liver injury in choline-deficient high-fat diet induced NASH mice. Metabolism 69, 177–187. doi: 10.1016/j.metabol.2016.12.013
Hong, F., Si, C., Gao, P., Cederbaum, A. I., Xiong, H., and Lu, Y. (2016). The role of CYP2A5 in liver injury and fibrosis: chemical-specific difference. Naunyn Schmiedebergs Arch. Pharmacol. 389, 33–43. doi: 10.1007/s00210-015-1172-8
Huch, M., Knoblich, J. A., Lutolf, M. P., and Martinez-Arias, A. (2017). The hope and the hype of organoid research. Development 144, 938–941. doi: 10.1242/dev.150201
Hui, S. T., Kurt, Z., Tuominen, I., Norheim, F., Davis, R. C., Pan, C., et al. (2018). The Genetic Architecture of Diet-Induced Hepatic Fibrosis in Mice. Hepatology 68, 2182–2196. doi: 10.1002/hep.30113
Ikenaga, N., Liu, S. B., Sverdlov, D. Y., Yoshida, S., Nasser, I., Ke, Q., et al. (2015). A new Mdr2(-/-) mouse model of sclerosing cholangitis with rapid fibrosis progression, early-onset portal hypertension, and liver cancer. Am. J. Pathol. 185, 325–334. doi: 10.1016/j.ajpath.2014.10.013
Inoue, T., Ishizaka, Y., Sasaki, E., Lu, J., Mineshige, T., Yanase, M., et al. (2018). Thioacetamide-induced hepatic fibrosis in the common marmoset. Exp. Anim. 67, 321–327. doi: 10.1538/expanim.17-0156
Jiao, J., Sastre, D., Fiel, M. I., Lee, U. E., Ghiassi-Nejad, Z., Ginhoux, F., et al. (2012). Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology 55, 244–255. doi: 10.1002/hep.24621
Kamdem, S. D., Moyou-Somo, R., Brombacher, F., and Nono, J. K. (2018). Host Regulators of Liver Fibrosis During Human Schistosomiasis. Front. Immunol. 9:2781. doi: 10.3389/fimmu.2018.02781
Kanzler, S., Lohse, A. W., Keil, A., Henninger, J., Dienes, H. P., Schirmacher, P., et al. (1999). TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am. J. Physiol. 276, G1059–G1068. doi: 10.1152/ajpgi.1999.276.4.G1059
Katoh, M., Tateno, C., Yoshizato, K., and Yokoi, T. (2008). Chimeric mice with humanized liver. Toxicology 246:12. doi: 10.1016/j.tox.2007.11.012
Kirkland, J. G., Godfrey, C. B., Garrett, R., Kakar, S., Yeh, B. M., and Corvera, C. U. (2010). Reversible surgical model of biliary inflammation and obstructive jaundice in mice. J. Surg. Res. 164, 221–227. doi: 10.1016/j.jss.2009.08.010
Komatsu, G., Nonomura, T., Sasaki, M., Ishida, Y., Arai, S., and Miyazaki, T. (2019). AIM-deficient mouse fed a high-trans fat, high-cholesterol diet: a new animal model for nonalcoholic fatty liver disease. Exp. Anim. 68, 147–158. doi: 10.1538/expanim.18-0108
Kosaka, K., Hiraga, N., Imamura, M., Yoshimi, S., Murakami, E., Nakahara, T., et al. (2013). A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem. Biophys. Res. Commun. 441, 230–235. doi: 10.1016/j.bbrc.2013.10.040
Koyama, Y., and Brenner, D. A. (2017). Liver inflammation and fibrosis. J. Clin. Invest. 127, 55–64. doi: 10.1172/JCI88881
Lamas-Paz, A., Hao, F., Nelson, L. J., Vázquez, M. T., Canals, S., Gómez Del Moral, M., et al. (2018). Alcoholic liver disease: Utility of animal models. World J. Gastroenterol. 24, 5063–5075. doi: 10.3748/wjg.v24.i45.5063
Lancaster, M. A., and Knoblich, J. A. (2014). Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345:1247125. doi: 10.1126/science.1247125
Leeming, D. J., Byrjalsen, I., Jiménez, W., Christiansen, C., and Karsdal, M. A. (2013). Protein fingerprinting of the extracellular matrix remodelling in a rat model of liver fibrosis–a serological evaluation. Liver Int. 33, 439–447. doi: 10.1111/liv.12044
Leite, S. B., Roosens, T., El Taghdouini, A., Mannaerts, I., Smout, A. J., Najimi, M., et al. (2016). Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 78:026. doi: 10.1016/j.biomaterials.2015.11.026
Li, C.-H., Piao, D.-M., Xu, W.-X., Yin, Z.-R., Jin, J.-S., and Shen, Z.-S. (2005). Morphological and serum hyaluronic acid, laminin and type IV collagen changes in dimethylnitrosamine-induced hepatic fibrosis of rats. World J. Gastroenterol. 11, 7620–7624. doi: 10.3748/wjg.v11.i48.7620
Li, T., and Apte, U. (2015). Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer. Adv. Pharmacol. 74, 263–302. doi: 10.1016/bs.apha.2015.04.003
Lindsjö, J., and Fahlman, Å, and Törnqvist, E. (2016). ANIMAL WELFARE FROM MOUSE TO MOOSE–IMPLEMENTING THE PRINCIPLES OF THE 3RS IN WILDLIFE RESEARCH. J. Wildl. Dis. 52(2 Suppl.), S65–S77. doi: 10.7589/52.2S.S65
Louis, H., Le Moine, A., Quertinmont, E., Peny, M. O., Geerts, A., Goldman, M., et al. (2000). Repeated concanavalin A challenge in mice induces an interleukin 10-producing phenotype and liver fibrosis. Hepatology 31, 381–390. doi: 10.1002/hep.510310218
Man, K.-N. M., Philipsen, S., and Tan-Un, K. C. (2008). Localization and expression pattern of cytoglobin in carbon tetrachloride-induced liver fibrosis. Toxicol. Lett. 183, 36–44. doi: 10.1016/j.toxlet.2008.09.015
Marfà, S., Morales-Ruiz, M., Oró, D., Ribera, J., Fernández-Varo, G., and Jiménez, W. (2016). Sipa1l1 is an early biomarker of liver fibrosis in CCl4-treated rats. Biol. Open 5, 858–865. doi: 10.1242/bio.018887
Mathews, S., Xu, M., Wang, H., Bertola, A., and Gao, B. (2014). Animals models of gastrointestinal and liver diseases. Animal models of alcohol-induced liver disease: pathophysiology, translational relevance, and challenges. Am. J. Physiol. Gastrointest Liver Physiol. 306, G819–G823. doi: 10.1152/ajpgi.00041.2014
Matsumoto, M., Hada, N., Sakamaki, Y., Uno, A., Shiga, T., Tanaka, C., et al. (2013). An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 94:12008. doi: 10.1111/iep.12008
Matsuo, M., Murata, S., Hasegawa, S., Hatada, Y., Ohtsuka, M., and Taniguchi, H. (2020). Novel liver fibrosis model in Macaca fascicularis induced by thioacetamide. Sci. Rep. 10:2450. doi: 10.1038/s41598-020-58739-4
Mehal, W. Z., Iredale, J., and Friedman, S. L. (2011). Scraping fibrosis: expressway to the core of fibrosis. Nat. Med. 17, 552–553. doi: 10.1038/nm0511-552
Müller, A., Machnik, F., Zimmermann, T., and Schubert, H. (1988). Thioacetamide-induced cirrhosis-like liver lesions in rats–usefulness and reliability of this animal model. Exp. Pathol. 34, 229–236. doi: 10.1016/s0232-1513(88)80155-5
Nishio, T., Hu, R., Koyama, Y., Liang, S., Rosenthal, S. B., Yamamoto, G., et al. (2019). Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J. Hepatol. 71, 573–585. doi: 10.1016/j.jhep.2019.04.012
Ouchi, R., Togo, S., Kimura, M., Shinozawa, T., Koido, M., and Koike, H. (2019). Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 30, 374.e–384.e. doi: 10.1016/j.cmet.2019.05.007
Park, H.-J., Kim, H.-G., Wang, J.-H., Choi, M.-K., Han, J.-M., Lee, J.-S., et al. (2016). Comparison of TGF-β, PDGF, and CTGF in hepatic fibrosis models using DMN, CCl4, and TAA. Drug Chem. Toxicol. 39, 111–118. doi: 10.3109/01480545.2015.1052143
Parola, M., and Pinzani, M. (2019). Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 65, 37–55. doi: 10.1016/j.mam.2018.09.002
Pellicoro, A., Ramachandran, P., Iredale, J. P., and Fallowfield, J. A. (2014). Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14, 181–194. doi: 10.1038/nri3623
Pingitore, P., Sasidharan, K., Ekstrand, M., Prill, S., Lindén, D., and Romeo, S. (2019). Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 20:20071629. doi: 10.3390/ijms20071629
Prior, N., Inacio, P., and Huch, M. (2019). Liver organoids: from basic research to therapeutic applications. Gut 68, 2228–2237. doi: 10.1136/gutjnl-2019-319256
Rodríguez-Garay, E. A., Agüero, R. M., Pisani, G., Trbojevich, R. A., Farroni, A., and Viglianco, R. A. (1996). Rat model of mild stenosis of the common bile duct. Res. Exp. Med. 196, 105–116. doi: 10.1007/s004330050017
Rossi, G., Manfrin, A., and Lutolf, M. P. (2018). Progress and potential in organoid research. Nat. Rev. Genet. 19, 671–687. doi: 10.1038/s41576-018-0051-9
Scholten, D., Trebicka, J., Liedtke, C., and Weiskirchen, R. (2015). The carbon tetrachloride model in mice. Lab. Anim. 49(1 Suppl.):23677215571192. doi: 10.1177/0023677215571192
Sebastiani, G., Gkouvatsos, K., and Pantopoulos, K. (2014). Chronic hepatitis C and liver fibrosis. World J. Gastroenterol. 20, 11033–11053. doi: 10.3748/wjg.v20.i32.11033
Seitz, H. K., Bataller, R., Cortez-Pinto, H., Gao, B., Gual, A., Lackner, C., et al. (2018). Alcoholic liver disease. Nat. Rev. Dis. Primers 4:16. doi: 10.1038/s41572-018-0014-7
Slater, T. F., Cheeseman, K. H., and Ingold, K. U. (1985). Carbon tetrachloride toxicity as a model for studying free-radical mediated liver injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 311, 633–645. doi: 10.1098/rstb.1985.0169
Soares, P. A. G., Nascimento, C. O., Porto, T. S., Correia, M. T. S., Porto, A. L. F., and Carneiro-da-Cunha, M. G. (2011). Purification of a lectin from Canavalia ensiformis using PEG-citrate aqueous two-phase system. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 879, 457–460. doi: 10.1016/j.jchromb.2010.12.030
Stål, P. (2015). Liver fibrosis in non-alcoholic fatty liver disease - diagnostic challenge with prognostic significance. World J. Gastroenterol. 21, 11077–11087. doi: 10.3748/wjg.v21.i39.11077
Tag, C. G., Sauer-Lehnen, S., Weiskirchen, S., Borkham-Kamphorst, E., Tolba, R. H., Tacke, F., et al. (2015). Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015:52438. doi: 10.3791/52438
Tanabe, J., Izawa, A., Takemi, N., Miyauchi, Y., Torii, Y., Tsuchiyama, H., et al. (2007). Interferon-beta reduces the mouse liver fibrosis induced by repeated administration of concanavalin A via the direct and indirect effects. Immunology 122, 562–570. doi: 10.1111/j.1365-2567.2007.02672.x
Testino, G., Leone, S., Fagoonee, S., and Pellicano, R. (2018). Alcoholic liver fibrosis: detection and treatment. Minerva Med. 109, 457–471. doi: 10.23736/S0026-4806.18.05844-5
Thiele, N. D., Wirth, J. W., Steins, D., Koop, A. C., Ittrich, H., Lohse, A. W., et al. (2017). TIMP-1 is upregulated, but not essential in hepatic fibrogenesis and carcinogenesis in mice. Sci. Rep. 7:714. doi: 10.1038/s41598-017-00671-1
Thieringer, F., Maass, T., Czochra, P., Klopcic, B., Conrad, I., Friebe, D., et al. (2008). Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene 423, 23–28. doi: 10.1016/j.gene.2008.05.022
Tolba, R., Kraus, T., Liedtke, C., Schwarz, M., and Weiskirchen, R. (2015). Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 49(1 Suppl.), 59–69. doi: 10.1177/0023677215570086
Tropskaya, N. S., Kislyakova, E. A., Vilkova, I. G., Kislitsyna, O. S., Gurman, Y. V., Popova, T. S., et al. (2020). Experimental Model of Cirrhosis of the Liver. Bull. Exp. Biol. Med. 169, 416–420. doi: 10.1007/s10517-020-04899-2
Tsuchida, T., Lee, Y. A., Fujiwara, N., Ybanez, M., Allen, B., Martins, S., et al. (2018). A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 69, 385–395. doi: 10.1016/j.jhep.2018.03.011
Tsukamoto, H., and Lu, S. C. (2001). Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 15, 1335–1349. doi: 10.1096/fj.00-0650rev
Ueberham, E., Löw, R., Ueberham, U., Schönig, K., Bujard, H., and Gebhardt, R. (2003). Conditional tetracycline-regulated expression of TGF-beta1 in liver of transgenic mice leads to reversible intermediary fibrosis. Hepatology 37, 1067–1078. doi: 10.1053/jhep.2003.50196
Uehara, T., Ainslie, G. R., Kutanzi, K., Pogribny, I. P., Muskhelishvili, L., Izawa, T., et al. (2013). Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol. Sci. 132, 53–63. doi: 10.1093/toxsci/kfs342
Unsal, V., Cicek, M., and Sabancilar, I. (2020). Toxicity of carbon tetrachloride, free radicals and role of antioxidants. Rev. Environ. Health 2020:48. doi: 10.1515/reveh-2020-0048
Wallace, M. C., Hamesch, K., Lunova, M., Kim, Y., Weiskirchen, R., Strnad, P., et al. (2015). Standard operating procedures in experimental liver research: thioacetamide model in mice and rats. Lab. Anim. 49(1 Suppl.), 21–29. doi: 10.1177/0023677215573040
Walter, E., Keist, R., Niederöst, B., Pult, I., and Blum, H. E. (1996). Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology 24, 1–5. doi: 10.1053/jhep.1996.v24.pm0008707245
Wang, H.-X., Liu, M., Weng, S.-Y., Li, J.-J., Xie, C., He, H.-L., et al. (2012). Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J. Gastroenterol. 18, 119–125. doi: 10.3748/wjg.v18.i2.119
Wang, K., Liu, L., Dai, W., Chen, X., Zheng, X., and Hou, J. (2014). Establishment of a hepatic fibrosis model induced by diethylnitrosamine in zebrafish. Nan Fang Yi Ke Da Xue Xue Bao 34, 777–782.
Wang, L., Tu, L., Zhang, J., Xu, K., and Qian, W. (2017). Stellate Cell Activation and Imbalanced Expression of TGF-1/TGF-3 in Acute Autoimmune Liver Lesions Induced by ConA in Mice. Biomed. Res. Int. 2017:2540540. doi: 10.1155/2017/2540540
Watanabe, T., Niioka, M., Hozawa, S., Kameyama, K., Hayashi, T., Arai, M., et al. (2000). Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J. Hepatol. 33, 224–235. doi: 10.1016/s0168-8278(00)80363-3
Weber, L. W. D., Boll, M., and Stampfl, A. (2003). Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 33, 105–136. doi: 10.1080/713611034
Wei, D.-D., Wang, J.-S., Wang, P.-R., Li, M.-H., Yang, M.-H., and Kong, L.-Y. (2014). Toxic effects of chronic low-dose exposure of thioacetamide on rats based on NMR metabolic profiling. J. Pharm. Biomed. Anal. 98, 334–338. doi: 10.1016/j.jpba.2014.05.035
Xia, X., Li, F., He, J., Aji, R., and Gao, D. (2019). Organoid technology in cancer precision medicine. Cancer Lett. 457, 20–27. doi: 10.1016/j.canlet.2019.04.039
Yan, M., Huo, Y., Yin, S., and Hu, H. (2018). Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 17, 274–283. doi: 10.1016/j.redox.2018.04.019
Ye, T., Wang, T., Yang, X., Fan, X., Wen, M., Shen, Y., et al. (2018). Comparison of Concanavalin a-Induced Murine Autoimmune Hepatitis Models. Cell Physiol. Biochem. 46, 1241–1251. doi: 10.1159/000489074
Yoon, E., Babar, A., Choudhary, M., Kutner, M., and Pyrsopoulos, N. (2016). Acetaminophen-Induced Hepatotoxicity: a Comprehensive Update. J. Clin. Transl. Hepatol. 4, 131–142. doi: 10.14218/JCTH.2015.00052
Yoshiji, H., Kuriyama, S., Miyamoto, Y., Thorgeirsson, U. P., Gomez, D. E., Kawata, M., et al. (2000). Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 32, 1248–1254. doi: 10.1053/jhep.2000.20521
Yoshino, K., Taura, K., Iwaisako, K., Masano, Y., Uemoto, Y., Kimura, Y., et al. (2021). Novel mouse model for cholestasis-induced liver fibrosis resolution by cholecystojejunostomy. J. Gastroenterol. Hepatol. 2021:15406. doi: 10.1111/jgh.15406
Zangar, R. C., Benson, J. M., Burnett, V. L., and Springer, D. L. (2000). Cytochrome P450 2E1 is the primary enzyme responsible for low-dose carbon tetrachloride metabolism in human liver microsomes. Chem. Biol. Interact. 125, 233–243. doi: 10.1016/s0009-2797(00)00149-6
Zhang, J. J., Meng, X. K., Dong, C., Qiao, J. L., Zhang, R. F., Yue, G. Q., et al. (2009). Development of a new animal model of liver cirrhosis in swine. Eur. Surg. Res. 42, 35–39. doi: 10.1159/000167855
Zhangdi, H.-J., Su, S.-B., Wang, F., Liang, Z.-Y., Yan, Y.-D., Qin, S.-Y., et al. (2019). Crosstalk network among multiple inflammatory mediators in liver fibrosis. World J. Gastroenterol. 25, 4835–4849. doi: 10.3748/wjg.v25.i33.4835
Keywords: liver, fibrosis, animal, organoid, model
Citation: Bao Y-l, Wang L, Pan H-t, Zhang T-r, Chen Y-h, Xu S-j, Mao X-l and Li S-w (2021) Animal and Organoid Models of Liver Fibrosis. Front. Physiol. 12:666138. doi: 10.3389/fphys.2021.666138
Received: 09 February 2021; Accepted: 03 May 2021;
Published: 26 May 2021.
Edited by:
Elvira Forte, Jackson Laboratory, United StatesReviewed by:
Soraia K. P. Costa, University of São Paulo, BrazilLouis Charles Penning, Utrecht University, Netherlands
Copyright © 2021 Bao, Wang, Pan, Zhang, Chen, Xu, Mao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shao-wei Li, bGlfc2hhb3dlaTgxQGhvdG1haWwuY29t; Xin-li Mao, bWFveGxAZW56ZW1lZC5jb20=
†These authors have contributed equally to this work