
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 28 May 2021
Sec. Cardiac Electrophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.661413
This article is part of the Research TopicInherited Arrhythmias of the Cardiac Sodium Channel Nav1.5View all 8 articles
 Nicolas Doisne1,2†
Nicolas Doisne1,2† Marta Grauso1,2†
Marta Grauso1,2† Nathalie Mougenot1,2,3Michel Clergue1,2
Nathalie Mougenot1,2,3Michel Clergue1,2 Charlotte Souil1,2Alain Coulombe1,2Pascale Guicheney1,2
Charlotte Souil1,2Alain Coulombe1,2Pascale Guicheney1,2 Nathalie Neyroud1,2*
Nathalie Neyroud1,2*Loss-of-function mutations in the cardiac Na+ channel α-subunit Nav1.5, encoded by SCN5A, cause Brugada syndrome (BrS), a hereditary disease characterized by sudden cardiac death due to ventricular fibrillation. We previously evidenced in vitro the dominant-negative effect of the BrS Nav1.5-R104W variant, inducing retention of wild-type (WT) channels and leading to a drastic reduction of the resulting Na+ current (INa). To explore this dominant-negative effect in vivo, we created a murine model using adeno-associated viruses (AAVs).
Methods: Due to the large size of SCN5A, a dual AAV vector strategy was used combining viral DNA recombination and trans-splicing. Mice were injected with two AAV serotypes capsid 9: one packaging the cardiac specific troponin-T promoter, the 5′ half of hSCN5A cDNA, a splicing donor site and a recombinogenic sequence; and another packaging the complementary recombinogenic sequence, a splicing acceptor site, the 3′ half of hSCN5A cDNA fused to the gfp gene sequence, and the SV40 polyA signal. Eight weeks after AAV systemic injection in wild-type (WT) mice, echocardiography and ECG were recorded and mice were sacrificed. The full-length hSCN5A-gfp expression was assessed by western blot and immunohistochemistry in transduced heart tissues and the Na+ current was recorded by the patch-clamp technique in isolated adult GFP-expressing heart cells.
Results: Almost 75% of the cardiomyocytes were transduced in hearts of mice injected with hNav1.5 and ∼30% in hNav1.5-R104W overexpressing tissues. In ventricular mice cardiomyocytes expressing R104W mutant channels, the endogenous INa was significantly decreased. Moreover, overexpression of R104W channels in normal hearts led to a decrease of total Nav1.5 expression. The R104W mutant also induced a slight dilatation of mice left ventricles and a prolongation of RR interval and P-wave duration in transduced mice. Altogether, our results demonstrated an in vivo dominant-negative effect of defective R104W channels on endogenous ones.
Conclusion: Using a trans-splicing and viral DNA recombination strategy to overexpress the Na+ channel in mouse hearts allowed us to demonstrate in vivo the dominant-negative effect of a BrS variant identified in the N-terminus of Nav1.5.
Brugada syndrome (BrS) is an inherited autosomal-dominant cardiac channelopathy with incomplete penetrance, characterized by a typical electrocardiographic (ECG) pattern showing an ST-segment elevation in the right precordial leads (V1–V3) and an increased risk of sudden cardiac death due to ventricular fibrillation in structurally normal hearts (Brugada et al., 2018). Mutations in the SCN5A gene, encoding the cardiac voltage-gated sodium channel Nav1.5, have been identified in around 25% of affected individuals (Watanabe and Minamino, 2015) and commonly reveal loss-of-function properties reducing the sodium current INa either by gating abnormalities, trafficking defects, or premature stop codons leading to haploinsufficiency (Wilde and Brugada, 2011).
Nav1.5 constitutes the α-subunit of the cardiac Na+ channel complex, which includes other transmembrane subunits and intracellular partners that participate in its expression and function (Abriel et al., 2015). Unlike potassium channel genes, which encode monomers associating in tetramers to constitute the functional channel, Nav1.5 channels were thought to be structured as single entities. It was thus unexpected to report Nav1.5 mutants with a dominant-negative effect on wild-type (WT) channels, as we and others did a few years ago (Keller et al., 2005; Clatot et al., 2012; Mercier et al., 2012; Hoshi et al., 2014; Pambrun et al., 2014; Wang et al., 2020). In these studies, a decrease of INa exceeding the 50% of current density expected in case of haploinsufficiency was indeed observed when co-expressing some mutants with WT channels in a 1:1 ratio to mimic patient heterozygosity (Keller et al., 2005; Clatot et al., 2012; Hoshi et al., 2014). For example, we have reported that co-expression of the BrS R104W mutant and WT channels in HEK293 cells caused a loss of 80% of INa compared to WT channels expressed alone and demonstrated that this dominant-negative effect was due to an interaction between R104W α-subunits retained in the endoplasmic reticulum and WT channels (Clatot et al., 2012). It was then established that Nav1.5 α-subunits form dimers through an interaction site located in the domain I-II linker, and that Nav1.5 channels not only interact but also gate as dimers (Clatot et al., 2017).
Animal and cellular models have been created to simulate BrS, including transgenic mice, canine heart preparations, transgenic pork, expression of mutant SCN5A in different cellular models and, more recently, induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) (Sendfeld et al., 2019). Knowledge gained from numerous studies achieved using experimental models has contributed to our current understanding of the pathophysiological mechanisms involved in BrS. Nevertheless, each of these models has revealed inherent limitations, e.g., the lack of cardiac background in heterologous expression systems, time and cost required to generate transgenic animal models and immaturity of iPS-CM.
In this study, we aimed to develop a versatile animal model of BrS using adeno-associated Viruses (AAVs) injection into mice. During the last two decades, AAVs turned out to be useful tools in gene therapy (Kaplitt et al., 1994; Guggino and Cebotaru, 2020) for the reason that they are small non-pathogenic and non-replicative DNA viruses with tissue-specific tropism extremely efficient for targeting in vivo transgene delivery (Prasad et al., 2011). One limitation of the use of AAVs as vectors for gene delivery is their intrinsic small packaging capacity of 5 kb (Dong et al., 1996). Nevertheless, the development of a dual-vector trans-splicing approach allowed to overcome this package-capacity limit (Duan et al., 2000; Sun et al., 2000; Ghosh et al., 2008, 2011). In this approach, the cDNA of a large gene can be split into two parts at the level of an intron and separately packaged into two individual AAVs, which will recombine in host cells and will be spliced in a mature full-length mRNA (Duan et al., 2000; Sun et al., 2000; Ghosh et al., 2008, 2011).
Taking advantage of the ability of AAVs to concatemerize, confirmed in other contexts like skeletal muscle (Sondergaard et al., 2015) and retina (Trapani et al., 2014), we used the dual-AAV trans-splicing strategy to overexpress in mice hearts and characterize in vivo the Nav1.5-R104W mutant previously reported to display a strong dominant-negative effect in vitro (Clatot et al., 2012). Our results showed for the first time that this dual-AAV trans-splicing approach allows overexpression of the full human SCN5A gene in up to 75% of injected-mice heart cells. Importantly, we recorded a significantly decreased endogenous INa in cardiomyocytes overexpressing R104W mutant channels and a reduction of the total Nav1.5 expression, demonstrating in vivo the dominant-negative effect of this BrS mutation in Nav1.5 on endogenous wild type (WT) channels. The R104W mutant overexpression also induced a slight dilatation of mice left ventricles, confirming that impairment of INa may be responsible for early stages of heart failure. Altogether our results demonstrated that the use of AAVs to overexpress SCN5A mutants in vivo is a relevant approach to create a versatile and valuable animal model of BrS.
The AAV vectors pAcTnT-S and pAcTnT-eGFP were described previously (Prasad et al., 2011) and kindly provided by Dr. B. A. French (Virginia University, United States). The human Nav1.5 sequence hH1a (RefSeq accession number NM_000335.4) was subcloned from plasmid pcDNA3.1-hH1a, a gift of Dr. H. Abriel (University of Bern, Bern, Switzerland). The 75-bp donor and 58-bp acceptor consensus sequences were subcloned from the chimeric intron of the pCI mammalian expression vector (Promega, Madison, WI, United States). The 288-bp alkaline phosphatase (AP) sequence [pAG71 plasmid (Ghosh et al., 2011)] was kindly provided by Dr. D. Duan (University of Missouri, Columbia, MO, United States). The helper and packaging plasmids pXX6 and pAAV2-9 were a kind gift of Dr. S. Benkhelifa-Ziyyat (Institute of Myology, Paris, France). All plasmids were purified with the NucleoBond® EF kit (Macherey Nagel, Düren, Germany) and sequenced for unwanted mutations (GATC, Konstanz, Germany).
Residue 104 of Nav1.5 is highly conserved between species and among sodium channels (Clatot et al., 2012) and, as a general concern, human and murine cardiac sodium channel sequences share a high homology of 95%. We thus decided to overexpress the human SCN5A-gene sequence carrying the R104W BrS variant into mice.
All elements for AAV recombination and splicing were inserted at the exon 17–18 junction of Nav1.5 cDNA (hH1a isoform; RefSeq NM_000335.4) using overlap extension PCR cloning to create 5′ and 3′ Nav1.5 halves for separate cloning in AAV vectors. The hNav1.5 trans-splicing construct was generated by inserting a chimeric intron from the pCI vector in Nav1.5 cDNA at the junction between exons 17 and 18. The 133-bp chimeric intron was amplified on the pCI vector using primers hH1a.ex17-pCI.forward and hH1a.ex18-pCI.reverse (Table 1). The PCR was done with Phusion High-Fidelity DNA polymerase (Finnzymes, Waltham, MA, United States) at a hybridization temperature of 56°C and 35 cycles. After purification, the amplified pCI chimeric intron was inserted by overlap extension PCR (Bryksin and Matsumura, 2010) in the Nav1.5 cDNA. The overlap extension PCR was done at a hybridization temperature of 65°C and 25 cycles. Finally, the Nav1.5-pCI chimeric intron plasmid was obtained by transforming Escherichia coli cells after digestion of the overlap-extension PCR amplicons by DpnI.

Table 1. Primer sequences.
In a second step, a successful clone was used to insert a reverse/complement-oriented recombinogenic AP sequence inside pCI chimeric intron sequence in a manner to obtain a 75 bp donor and a 58 bp acceptor consensus sequence for intron splicing. A 288 bp AP sequence was amplified on pAG71 plasmid using primers: hH1a-iDO-AP.forward and hH1a-AP-iAC.reverse (Table 1). PCR amplification protocol was obtained with a hybridization temperature of 65°C and 25 cycles. Purified AP amplification was inserted by overlap extension PCR in the previously obtained Nav1.5-pCI chimeric intron plasmid using an insert/plasmid ratio of 1:250, a hybridization temperature of 65°C and 25 cycles. The DpnI digested overlap extension PCR amplification was used to transform E. coli cells to obtain a Nav1.5-WT plasmid containing the whole recombinogenic/splicing cassette at the exon 17–18 junction.
R104W site-directed mutagenesis was then achieved on the Nav1.5-WT recombinogenic/splicing cassette-containing plasmid using the kit QuikChange II XL (Stratagene, Santa Clara, CA, United States) with the complementary primers R104W.forward and R104W.reverse (Table 1) following the manufacturer’s instructions.
The 5′ hNav1.5 recombinogenic/splicing half part was amplified by PCR on the Nav1.5 recombinogenic/splicing cassette-containing plasmid, WT or R104W mutated, using primers 5′hH1a-iDO-AP.forward and 5′hH1a-AP-iAC.reverse (Table 1). The purified amplification product was first cloned in pCRBlunt vector using TOP10 chemically competent E. coli (Invitrogen). The 3,653-bp HindIII-SalI fragment from a positive clone was then subcloned in the HindIII-SalI digested pAcTnT-S vector, using SURE2 competent cells (Stratagene), to obtain the pAcTnT.5′Nav1.5WT or pAcTnT.5′Nav1.5R104W viral plasmids.
The 3′hNav1.5 recombinogenic/splicing half part was amplified by PCR on the Nav1.5 recombinogenic/splicing cassette-containing plasmid using primers AP-iAC-3′hH1a.forward and 3′hH1a-iAC-AP.reverse (Table 1). The purified amplification product was first cloned in pCRBlunt vector using TOP10 chemically competent E. coli. The 3172-bp XbaI-SacII digested fragment from a positive clone was then subcloned in the XbaI-SacII digested pAcTnT-eGFP vector, using SURE2 competent cells, to obtain the pA.3′Nav1.5-eGFP viral plasmid. Absence of insert recombination during bacterial amplification was verified by digestion pattern of restriction sites SmaI and MscI in AAV inverted terminal repeat (ITR) sequence.
Adeno-associated viruses were either in-house prepared or produced by the Viral Vector Core of Nantes University (France). We used the triple transfection of HEK293T cells with pXX6 and pAAV2-9 as helper plasmids respectively to obtain AAVs of serotype-9 capsids. Viral:helper plasmid molar ratio for HEK293T transfection was 1:1 and a total of 145 μg of DNA was used to transfect 500 square centimeter of sub-confluent HEK293T cells with polyethylenimine (PEI) as transfecting agent in 2%-fetal calf serum DMEM media. Cells were collected 3 days after transfection to recover AAV particles. AAVs were purified after ammonium-sulfate precipitation (∼50% saturation) and Benzonase (Sigma-Aldrich, United States) digestion of free DNA by ultracentrifugation on iodixanol gradient. Viral particles were finally concentrated using Vivaspin 100 kD columns (Sartorius, Göttingen, Germany).
To assess viral genome titer, we performed the Universal Real-Time PCR with AAV2 ITR specific primers and probe as described previously (Aurnhammer et al., 2012) but using the LightCycler 480 probes master kit (Roche, Basel, Switzerland) in a final reaction volume of 20 μl. PCR mix contained a final concentration of 0.5 μM of each primer and 0.1 μM of probe and 2 μl of template. PCR protocol consisted in one denaturation of 15 min at 95°C, 45 amplification cycles of 1 min at 95°C and 1 min at 60°C and a final cooling cycle of 10 s at 40°C. Viral template was added in four 1:10 serial dilutions from 10–2 to 10–5 of purified stock. For standard curve, we used six serial dilutions of viral vector pAcTnT-eGFP, NdeI linearized, containing from 5.8 107 to 5.8 102 copy number per μl. The viral vector pAcTnT-eGFP stock concentration was determined with Quant-iTTM PicoGreen® dsDNA reagent and kit (Molecular Probes, United States).
Three to five-days old C57Bl6/J mice were injected through one jugular vein with a maximum of 100 μl of either AAV-cTnT-eGFP or a mix of AAV-cTnT-5′hNav1.5 (WT or R104W) and AAV-3′hNav1.5-eGFP. Depending on the titer of AAV preparations, we injected between 2.8 1011 and 1.45 1012 viral particles for AAV-cTnT-eGFP and between 6.84 1011 and 2.6 1012 total viral particles for the mix of AAV-cTnT-5′hNav1.5 and AAV-3′hNav1.5-eGFP in a 1:1 molar ratio. Seven weeks after injection, ECG and echocardiography were performed. Mice were then sacrificed and the heart was excised for ex vivo and in vitro experiments.
Echocardiography was performed on lightly anesthetized 8-week-old mice under isoflurane. Non-invasive measurements of left ventricular dimensions were evaluated using echocardiography-Doppler (Vivid 7 Dimension/Vivid7 PRO; GE Medical System Co., Vélizy, France) with a probe emitting ultrasounds from 9- to 14-MHz frequency. The two-dimensionally guided Time Motion mode recording (parasternal long-axis view) of the left ventricle (LV) provided the following measurements: diastolic and systolic septal (IVSd and IVSs) and posterior wall thicknesses (LVPWd and LVPWs), internal end-diastolic diameter (LVEDD) and end-systolic diameter (LVESD), and heart rate. Each set of measurements was obtained from the same cardiac cycle. At least three sets of measurements were obtained from three different cardiac cycles. Fractional shortening was calculated by the following formula: [(LVEDD–LVESD)/LVEDD] × 100. Cardiac output was measured by Pulse Wave Doppler using the following formula: [(πd2/4) × VTI × HR], where d is the diameter of aorta, VTI, the subaortic velocity time integral and HR the heart rate.
Surface ECG measurements were also performed under isoflurane anesthesia on 8-week-old mice. Two-lead ECGs were recorded with 29-gauge subcutaneous electrodes on a computer using an analog-digital converter (iox 2.4.2.6; emka Technologies, Paris, France) for monitoring and analyzed with ecgAUTO software (emka Technologies, Paris, France). Recordings were filtered at 50 Hz, and a stable signal was reliably obtained before proceeding. ECG traces were signal averaged and analyzed for heart rate (RR interval), P wave, PR and QRS interval duration.
Total proteins were extracted from frozen pieces of hearts from mice injected with either AAV-GFP or the mix of Nav1.5 AAVs, in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% Triton and complete protease inhibitor cocktail from Roche) for 1.5 h at 4°C on a wheel. The soluble fractions from 30-min centrifugation at 13,000 × g (4°C) were then used for western blot experiments.
Total extracted proteins were separated on a 3–8% acrylamide SDS-PAGE gel and transferred to a nitrocellulose membrane. After the membrane was cut horizontally between 250 and 130 kD, and vertically on the molecular weight, it was incubated with primary antibodies followed by infrared IR-Dye secondary antibodies (LI-COR Biosciences, United States). Primary antibodies used were as follows: rabbit anti-GFP (1:2000, Torrey Pines Biolabs, United States), rabbit anti-Nav1.5 (1:200, Alomone Labs, Israel) and mouse anti-α-tubulin (1:1000, Sigma-Aldrich, United States). Proteins were detected using the Odyssey Infrared Imaging System (LI-COR Biosciences, United States). Signals were quantified using ImageJ software. Total protein signals were normalized to α-tubulin levels in hearts expressing GFP as a control of mice injection.
Indirect immunofluorescence was performed on 10 μm control (GFP) Nav1.5-WT or R104W-injected mouse ventricle cryosections fixed with paraformaldehyde for 15 min. Sections were washed twice for 5 min with phosphate buffer saline (PBS), blocked in PBS-5% bovine serum albumin (BSA) for 30 min at room temperature. Sections were then incubated overnight with primary antibodies at 4°C: the rabbit anti-GFP (1:2000, Torrey Pines Biolabs, United States) to detect exogenous hNav1.5-GFP, and mouse anti-α-actinin 2 (1:500, Sigma-Aldrich, United States). Heart sections were then washed twice with PBS and incubated 1 h with secondary antibodies: goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 594 (1:1000, Molecular Probes, Thermo Fisher Scientific, United States), and the nuclear dye DAPI (1:2000, Merck, Germany) diluted in the blocking buffer. Control experiments were performed by omitting primary antibodies.
Labeled ventricle sections were observed with a DeltaVision epifluorescent microscope (20× or 60×). Images were analyzed with DeltaVision imaging system (GE Healthcare, Seattle, WA, United States) equipped with 3D-deconvolution. For each sample, series of consecutive plans were acquired (sectioning step: 0.2 μm).
Eight- to ten-week-old mice were anesthetized and heart was quickly excised. After cannulation of the aorta, hearts were mounted to a constant pressure Langendorff system. First, hearts were rinsed for around 4 min by perfusion of a free-Ca2+ Tyrode solution (see composition below in Solutions) containing 10 mM BDM (ButaneDione Monoxime) and 20 mM Taurine and subsequently perfused with enzymatic solution containing 0.09 mM Ca2+ Tyrode with 3 mg/ml collagenase type 2 (Worthington Biochemical Corporation, Lakewood, NJ, United States) for 6–10 min. Hearts were then immersed in a 0.18 mM Ca2+ Tyrode containing 5 mg/ml BSA and ventricles were cut into small pieces and further dissociated into single cells by gentle shaking. The Tyrode Ca2+ concentration was then two times doubled every 5 min to reach 0.72 mM. Cells were kept in this 0.72 mM Ca2+ Tyrode solution and used within 5 h after isolation.
Patch-clamp recordings were carried out at room temperature (22 ± 1°C). Ionic currents were recorded by the whole-cell patch-clamp technique with the amplifier VE-2 (Alembic, Canada). Patch pipettes (Corning Kovar Sealing code 7052, WPI) had resistances of 1–1.5 MΩ. Currents were filtered at 5 kHz (23 dB, 8-pole low-pass Bessel filter) and digitized at 30 kHz (NI PCI-6251, National Instruments, Austin, TX, United States). Data were acquired and analyzed with ELPHY software (G. Sadoc, CNRS, Gif/Yvette, France). To measure peak INa amplitude and determine current–voltage (I/V curves) and activation-Vm relationships, currents were elicited by test potentials of 0.2 Hz frequency to −100 to +60 mV for 50 msec by increments of 5 or 10 mV from a holding potential of −120 mV. The steady-state inactivation-Vm protocol was established from a holding potential of −120 mV and a 2 s conditioning pre-pulse was applied in 5 or 10 mV increments between −140 and +20 mV, followed by a 50 msec test pulse to −20 mV at 0.2 Hz frequency. Data for the activation-Vm and steady-state availability-Vm relationships of INa were fitted to the Boltzmann equation: Y = 1/{1 + exp[-(Vm-V1/2)/k]}, where Vm is the membrane potential, V1/2 is the half-activation or half-availability potential, k is the slope factor and Y represents the relative conductance.
Composition of free-Ca2+ Tyrode solution was (in mM): 135 NaCl, 4 KCl, 2.5 MgCl2, 10 HEPES, 10 glucose, pH 7.4 (NaOH). Cells were bathed in an extracellular Tyrode solution containing (in mM): 135 NaCl, 4 KCl, 2.5 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, pH 7.4 (NaOH). Patch pipette medium was (in mM): 135 CsCl, 1 CaCl2, 2 MgCl2, 4 Mg-ATP, 15 EGTA, 10 HEPES, pH 7.2 (CsOH). During current recording, cells were perfused with an external solution with reduced Na+ concentration containing (in mM): 10 NaCl, 123.5 CsCl, 2 CaCl2, 2.5 MgCl2, 10 HEPES, 10 glucose, 20 Tetra Ethyl Ammonium, 3 4-AP, 3 CoCl2, pH 7.4 (CsOH).
Data are represented as mean ± SEM. Statistical significance was estimated with GraphPad Prism® software (San Diego, CA, United States) by Student t-test after population normality, checked by Shapiro–Wilk test, was assessed in each group. P < 0.05 was considered significant.
One limitation of the use of AAVs as vectors for transgene delivery is their intrinsic small packaging capacity of 5 kb (Dong et al., 1996), which is less than the full-length SCN5A cDNA size (6048 bp). To overcome this restriction, we developed in this study a dual-vector trans-splicing approach permitting to double AAVs’ package-capacity limit, as previously published (Duan et al., 2000; Sun et al., 2000; Ghosh et al., 2008, 2011). In this approach, after host infection by both AAV populations and viral genome unpackaging, the transgene cDNA is reconstituted thanks to a short highly recombinogenic sequence and its excision from the pre-mRNA through intron splicing.
To this end, we first chose a suitable region in SCN5A to insert a DNA cassette composed of the first third of the human AP sequence (Ghosh et al., 2011) with splicing donor and acceptor consensus sites from the chimeric intron of pCI vector located at its 5′ and 3′ ends (Figure 1A). Using the Human Splicing Finder tool1, we obtained the best score for the junction between exons 17 and 18 of SCN5A, which also permitted to cut the full cDNA into two portions of equivalent size and to obtain dual hybrid AAV genomes of 4.4 and 4.7 kb, respectively. To avoid appearance of cloning restriction sites, we inserted the recombinogenic/splicing cassette by overlap extension PCR cloning, a technique allowing for insertion of large sequences (Bryksin and Matsumura, 2010).

Figure 1. Design of 5′ and 3′ AAV-hNav1.5 expression vectors. (A) The recombinogenic/splicing cassette inserted at the junction between exons 17 and 18 of SCN5A cDNA contains splicing donor and acceptor sequences from the chimeric intron of pCI vector and a reverse-complementary 288 bp sequence from human placental alkaline phosphatase. (B) Schematic representation of plasmids pAcTnT.5′Nav1.5-R104W and pA.3′Nav1.5-eGFP cloned to produce two distinct trans-splicing AAV populations.
With the aim to restrain SCN5A overexpression to heart tissue, we combined the use of AAV serotype 9 for its tropism for heart tissue and the chicken cardiac troponin T (cTnT) promoter for its cardiac specificity (Prasad et al., 2011), to design viral genome plasmids. Also, in order to quantify heart-tissue viral transduction and trans-splicing efficiency and to visualize cardiomyocytes overexpressing human Nav1.5 channels during patch-clamp recordings, we fused the sequence of eGFP to the 3′-end of SCN5A.
As cloning final results, we obtained the AAV plasmids named pAcTnT.5′hH1a and pAcTnT.5′hH1a-R104W containing the hNav1.5 or hNav1.5-R104W cDNA from ATG to nucleotide 3228 (exon 2–17), splicing donor site and AP sequences, and the AAV plasmid named pA.3′hH1a-eGFP carrying the AP, splicing acceptor site and hNav1.5 cDNA from nucleotide 3229–6045 (exon 18 to 28 with deletion of the stop codon) sequences as shown on Figure 1B. We also designed pAcTnT-eGFP for control experiments.
To assess whether our strategy of dual vectors was efficient to express the full-length human Nav1.5-GFP channel in mouse cardiac tissue and to compare its efficiency to single-AAV transduction, we first made 10 μm cryosections of injected-mouse ventricles to study expression and localization of hNav1.5-GFP or GFP alone by immunohistochemistry. As shown on Figure 2, we observed a strong expression of GFP in ∼75% of heart cells in AAV-GFP injected mouse cardiac ventricles (Figures 2A,D) and an equally important expression (∼75%) of hNav1.5-WT in ventricular cardiomyocytes of mice injected with dual trans-splicing vectors AAV-cTnT-5′Nav1.5-WT and AAV-3′hNav1.5-eGFP (Figures 2B,E). A robust but lower expression of hNav1.5 channels (∼30%) was observable in mice injected with hNav1.5-R104W dual AAVs (Figures 2C,F).
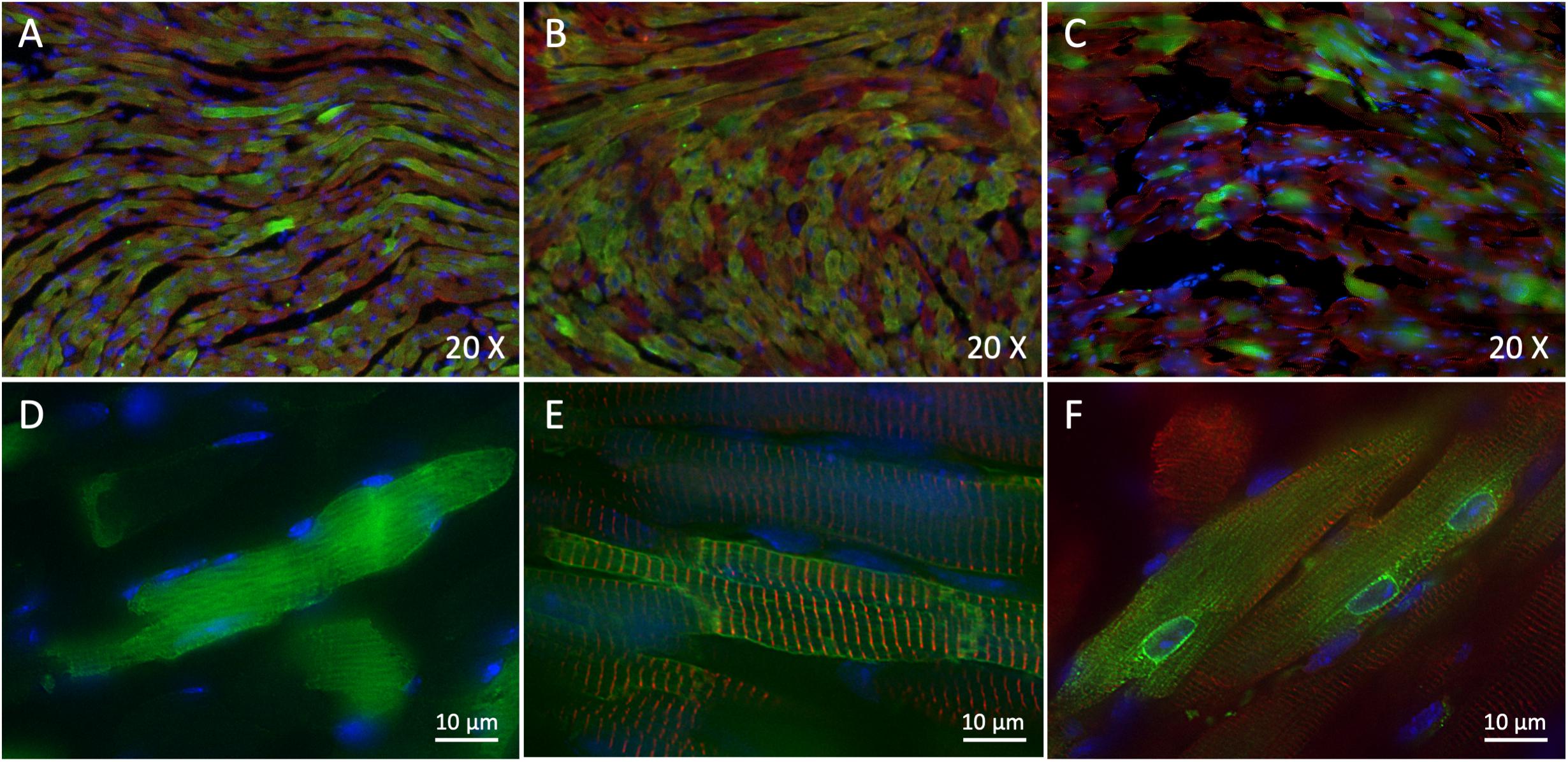
Figure 2. Trans-splicing dual AAVs efficiently transduce heart tissue. (A,D) Representative 3-dimensional deconvolution images of GFP (green) and α-actinin 2 (red) immunostaining of 10 μm GFP-injected mouse heart cryosections. (A): 20×; (D): 60×; scale bar: 10 μm. (B,E) Representative 3-dimensional deconvolution images of hNav1.5 (green) and α-actinin 2 (red) immunostaining of 10 μm hNav1.5-injected mouse heart cryosections. (B): 20×; (E): 60×; scale bar: 10 μm. (C,F) Representative 3-dimensional deconvolution images of hNav1.5-R104W (green) and α-actinin 2 (red) immunostaining of 10 μm R104W-injected mouse heart cryosections. (C): 20×; (F): 60×; scale bar: 10 μm. Exogenous hNav1.5-GFP channels were stained in green by the anti-GFP antibody, and nuclei in blue using DAPI. Note that AAV-GFP transduced approximately 75% of injected mice cardiomyocytes, as did the trans-splicing AAVs expressing hNav1.5, while the hNav1.5-R104W mutant channels were observable in around 1/3 of heart cells. The GFP protein was expressed in the whole cytoplasm of transduced cells, whereas the hNav1.5 channels were expressed at the cell surface and hNav1.5 channels carrying the R104W variant were mostly retained in the perinuclear area of cardiomyocytes.
It is noteworthy that GFP overexpressed alone was localized in the whole cytoplasm (Figure 2D), while hNav1.5-WT channels were mainly expressed at the cell surface (Figure 2E). This was not the case for hNav1.5-R104W channels retained in the perinuclear area of heart cells, suggesting a retention of the mutant sodium channels in endoplasmic reticulum (Figure 2F), as we already observed in HEK293 cells (Clatot et al., 2012).
Our first concern was to verify that AAV systemic injection in newborn mice had no consequences on heart function assessed by cardiac echocardiography. We compared echocardiography parameters in non-injected and AAV-GFP injected mice and observed no significant differences (Supplementary Table 1). We thus considered AAV-GFP-injected mice as the control group in our study. On the other hand, overexpression of the R104W mutated sodium channel significantly increased left ventricular diameter (0.32 ± 0.006 cm in WT, n = 20 vs. 0.36 ± 0.006 cm in R104W, n = 21 for diastolic diameter, P < 0.001 and 0.18 ± 0.004 cm in WT, n = 20 vs. 0.21 ± 0.006 cm in R104W, n = 21 for systolic diameter, P < 0.0001) and decreased left ventricular ejection fraction (82 ± 0.5 % in WT, n = 20 vs. 77 ± 1.2 % in R104W, n = 21, P < 0.005) and fractional shortening (45 ± 0.5 % in WT, n = 20 vs. 40 ± 1.1 %, n = 21 in R104W, P < 0.005) when compared to control mice, as shown in Figure 3A. Moreover, overexpression of hNav1.5-R104W channels significantly increased end-diastolic (EDV) and stroke volumes (SV) compared to WT (Table 2). Altogether these data suggest that overexpression of the R104W mutant sodium channel in mouse heart leads to early stages of dilated cardiomyopathy.
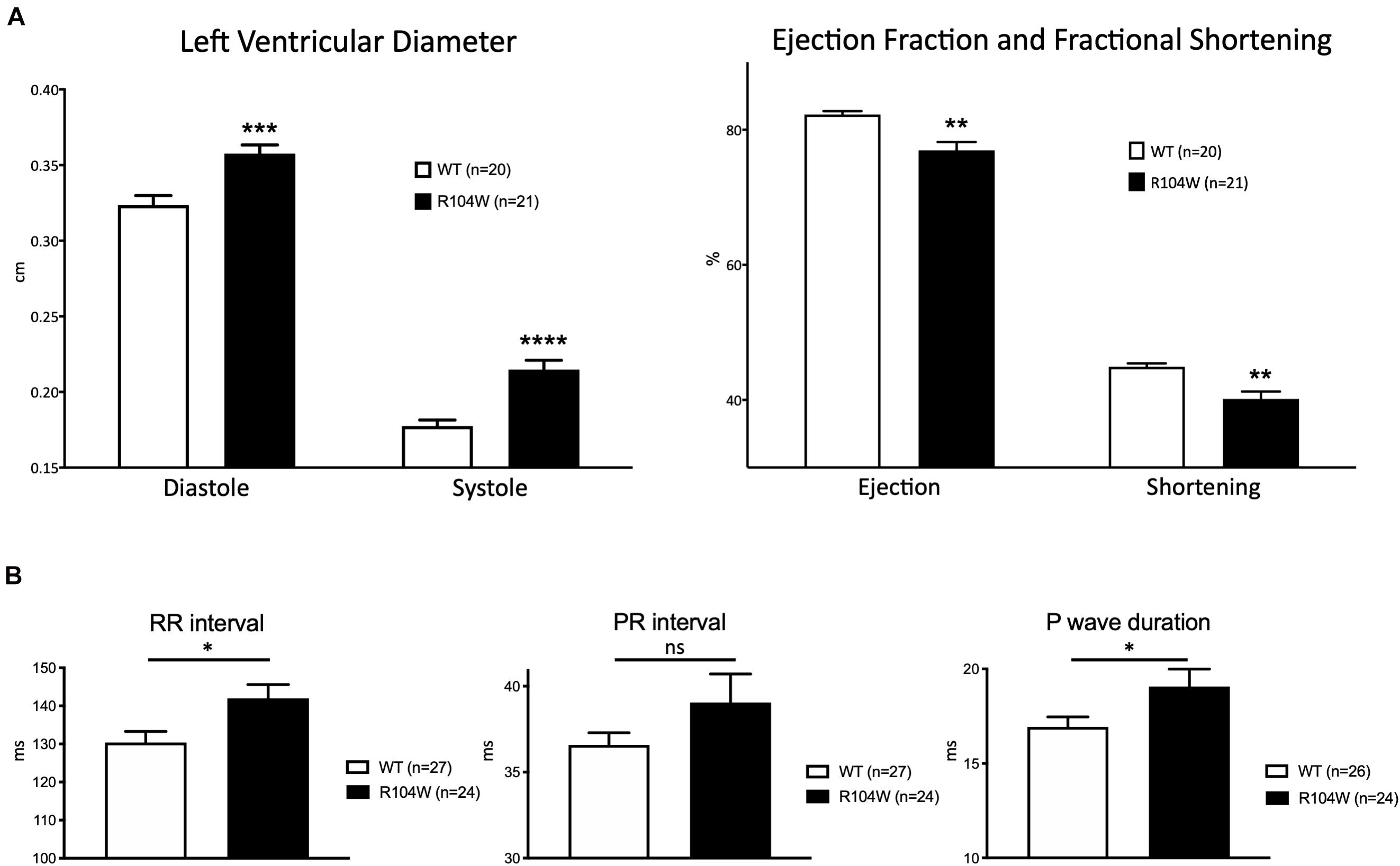
Figure 3. Cardiac functional effects of the dominant-negative mutant R104W in Nav1.5. (A) Left ventricular diameter (in cm), left ventricular ejection fraction and fractional shortening (in %) measured by echocardiography in GFP-injected (n = 20) and Nav1.5-R104W-injected mice (n = 21). Overexpression of the hNav1.5-R104W channel induced a significant left ventricular dilation combined to a significant reduction of left ventricular ejection fraction and fractional shortening. (B) RR interval, PR interval, and P wave duration (in ms) measured on ECG recordings in control (n = 27) and hNav1.5-R104W-injected mice (n = 24). Overexpression of the hNav1.5-R104W mutant channel led to a slight but significant prolongation of RR interval and P wave duration. PR interval was also slightly increased in R104W overexpressing mice when compared to controls, but not significantly. *: P < 0.05; **: P < 0.005; ***: P < 0.001; ****: P < 0.0001.
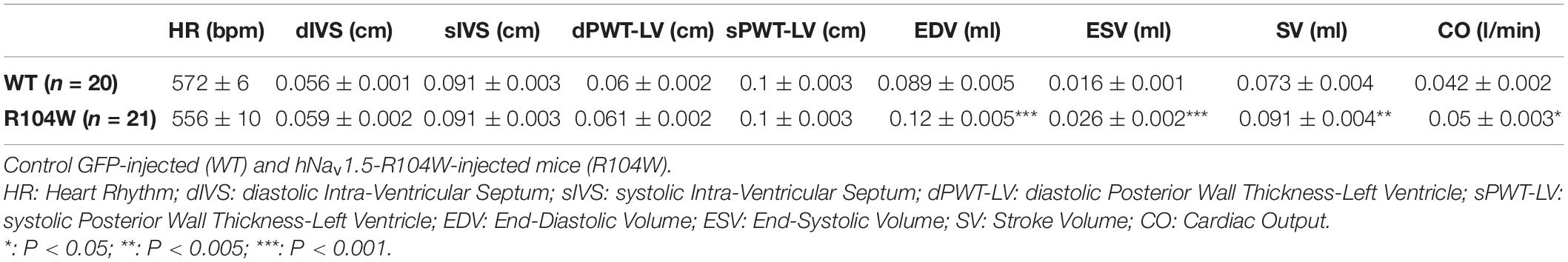
Table 2. Echocardiographic parameters.
To evaluate the effects of the dominant-negative BrS variant R104W overexpression on mice ECG parameters, we recorded surface ECG in injected mice (Figure 3B and Table 3) and observed a small but significant reduction of heart rate (RR interval: 130 ± 2.9 ms in WT, n = 27 vs. 142 ± 3.6 ms in R104W, n = 24, P < 0.05) and a significant prolongation of the P wave duration (17 ± 0.5 ms in WT, n = 26 vs. 19 ± 0.9 ms in R104W, n = 24, P < 0.05). We observed no other significant differences in ECG parameters between both groups, while PR interval slightly prolonged in R104W overexpressing mice (37 ± 0.7 ms in WT, n = 27 vs. 39 ± 1.7 ms in R104W, n = 24) (Table 3).
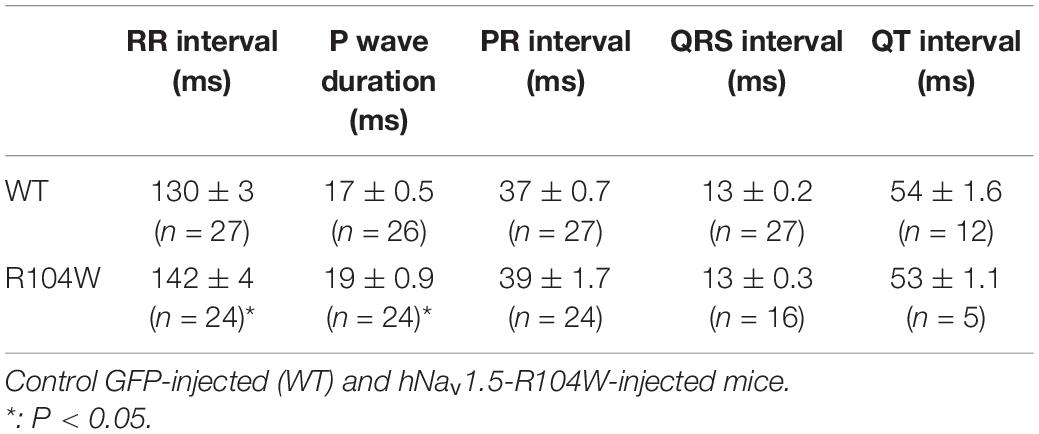
Table 3. Electrocardiographic parameters.
Since hNav1.5-R104W overexpression in mice hearts using AAV dual vectors seemed to cause a cardiac dysfunction, we then sought to record the sodium current in cardiomyocytes isolated from injected mice hearts. We recorded the global INa resulting from co-expression of endogenous WT murine channels and hNav1.5-R104W channels by the patch-clamp technique in whole cell configuration (Figure 4). In hNav1.5-R104W-expressing cardiomyocytes, the INa peak amplitude at −30 mV was significantly decreased by 15 % when compared to control cardiomyocytes (−40 ± 1.7 pA/pF in WT, n = 38 vs. −34 ± 1.6 pA/pF in R104W, n = 54, P < 0.05) as shown in Figures 4A,B, suggesting an in vivo dominant-negative effect of this BrS variant. In Figure 4C, we represented all peak INa amplitude recorded in each cell to highlight cellular variation of INa in the R104W group: from 0 (indicated by an arrow) to −55 pA/pF.
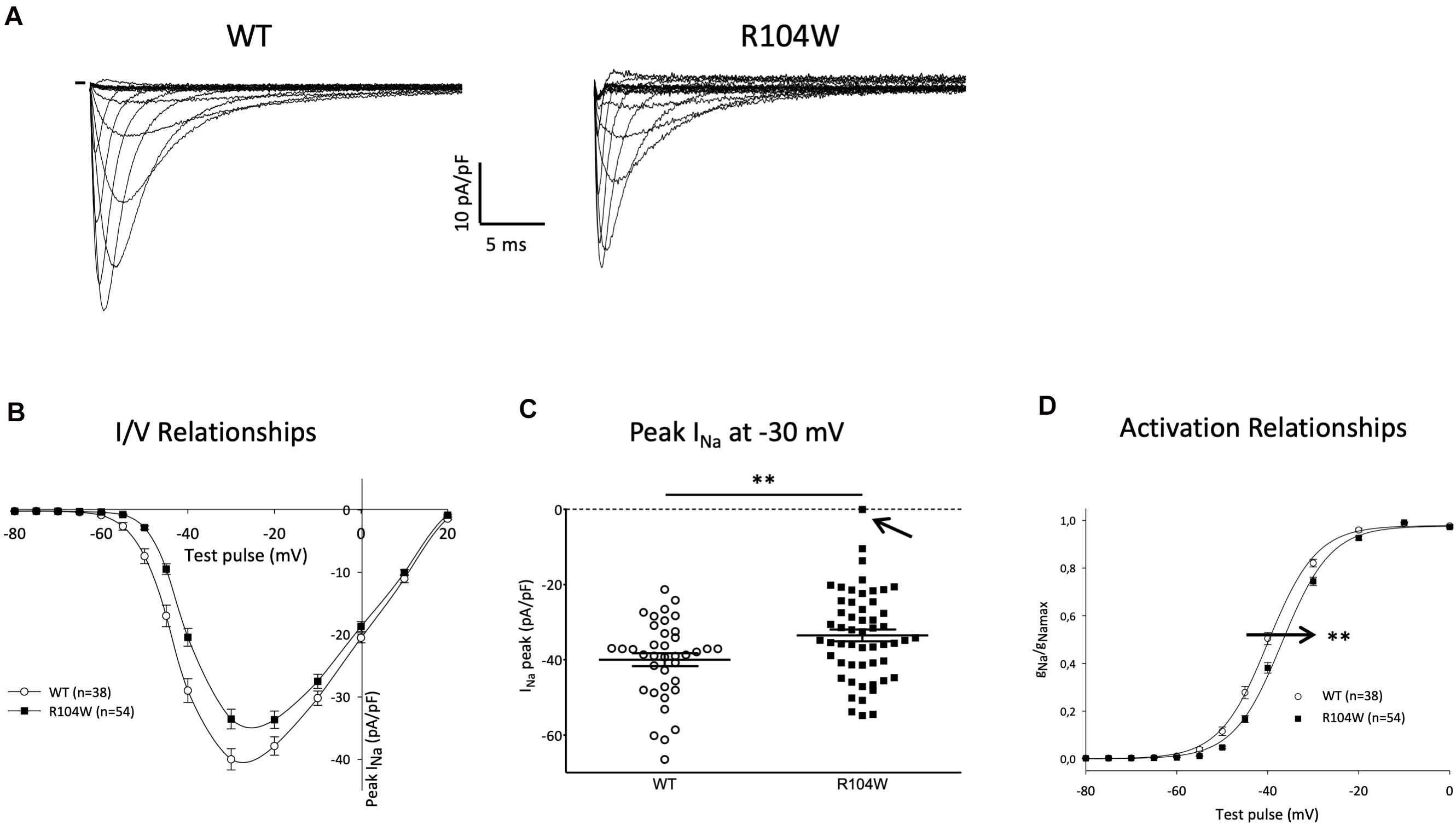
Figure 4. Overexpression of the hNav1.5-R104W mutant decreased endogenous INa. (A) Typical Na+ currents recorded in control (WT, 10 mM Na+ outside) and hNav1.5-R104W-injected mouse cardiomyocytes (R104W, 10 mM Na+ outside). (B) I/V relationships of peak Na+ current recorded in control (WT, n = 38) and hNav1.5-R104W-injected mouse cardiomyocytes (R104W, n = 54). (C) Distribution of peak Na+ current recorded at –30 mV in control (WT, n = 38) and hNav1.5-R104W-injected mouse cardiomyocytes (R104W, n = 54). Note that, in one cell indicated by an arrow, INa was null. (D) Activation/voltage relationships of peak Na+ current in control (WT, n = 38) and hNav1.5-R104W-injected mouse cardiomyocytes (R104W, n = 54). **: P < 0.005. Overexpression of the hNav1.5-R104W mutant significantly decreased INa recorded in injected mouse and shifted activation relationship to more positive potentials.
As shown on Figure 4D, activation curve of R104W overexpressing cells was significantly shifted to more positive potentials by 3 mV, compared to controls (V1/2 = −39.9 ± 0.7 mV in WT, n = 38 vs. −36.9 ± 0.6 mV in R104W, n = 54, P < 0.005), suggesting a loss-of-function of mutant channels, as it was already shown in transfected HEK293 cells (Clatot et al., 2012). No significant difference was observed in inactivation curves (V1/2 = −77 ± 1.1 mV in WT, n = 33 vs. −76.3 ± 0.7 mV in R104W, n = 42).
In order to demonstrate that our strategy of dual AAVs was efficient to modulate the endogenous sodium current, we also recorded INa in cardiomyocytes isolated from mice overexpressing hNav1.5-WT. We observed a significant and important increase (65 %) of total INa in AAV-hNav1.5-WT injected hearts (Supplementary Figure 1). Altogether our electrophysiological results suggest that the use of the Troponin T cardiac-specific promoter to drive Nav1.5 overexpression together with a dual AAV vector approach was powerful to modulate the murine INa.
To confirm the in vivo dominant-negative effect of Nav1.5-R104W BrS variant, we explored the possible explanation of this effect at the cellular level by quantifying the expression of the total Nav1.5 protein in injected-mouse hearts on western-blots (Figure 5 and Supplementary Figure 2 for raw data). We observed that the quantity of total Nav1.5 protein, revealed by a specific anti-Nav1.5 antibody, was decreased in hNav1.5-R104W-injected mouse hearts compared to controls (P < 0.05; Figure 5B), suggesting a degradation of endogenous murine channels.
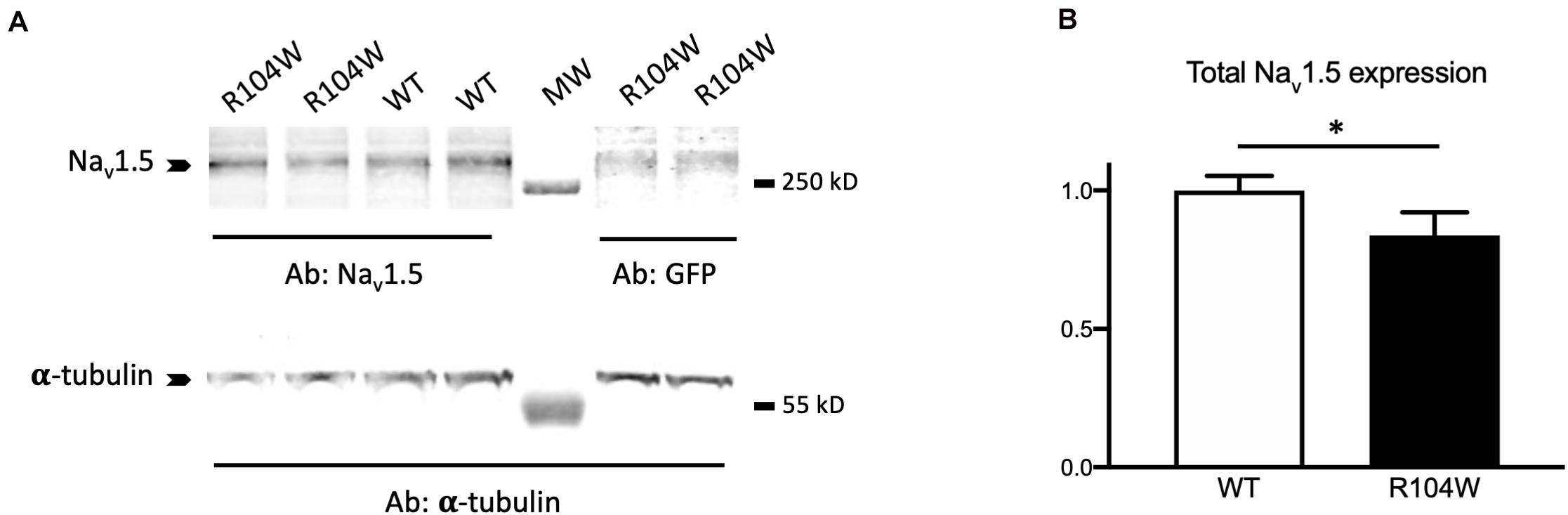
Figure 5. hNav1.5-R104W overexpression decreased the Nav1.5-protein total quantity. (A) Representative western blot of total cardiac proteins extracted from GFP (WT) or R104W injected mice. Total Nav1.5 proteins were revealed using the anti-Nav1.5 antibody and normalized to α-tubulin levels in hearts expressing GFP as a control of AAV injection and expression. (B) Total Nav1.5 protein expression was significantly decreased in R104W injected mice (n = 12), when compared to control ones (n = 10). *: P < 0.05.
Although several BrS-causing SCN5A mutations have been characterized using cellular and animal models, integrated understanding of the mechanisms linking sodium channel dysfunction to cardiac pathophysiology is still lacking. With the aim to develop a versatile and ready-for-use BrS animal model of SCN5A variant characterization, we used AAVs to generate a powerful system of overexpression of a large gene targeted to mice heart tissue. We demonstrated that in vivo expression of a human BrS SCN5A variant was responsible for a dominant-negative effect, which confirmed what was previously observed in vitro. Indeed, hNav1.5-R104W cardiac overexpression in mice decreased global INa and total Nav1.5-protein expression, prolonged PR interval and P-wave duration, and led to early stages of dilated cardiomyopathy.
Taking advantage of a technique published in 2000 (Duan et al., 2000; Sun et al., 2000), we developed dual hybrid AAV vectors carrying the Troponin T cardiac-specific promoter and the full-length SCN5A-gene sequence fused to the eGFP reporter gene, to overexpress Nav1.5 variants in their physiological cardiac background. The challenge was to overcome limited cargo capacity of AAVs and to design vectors capable of recombination and trans-splicing to reconstitute the full-length SCN5A-eGFP sequence, once in mice cardiac cells.
This strategy proof-of-principle was established by Dr. Duan’s group in 2007 in a study designed to assess mice whole-body transduction using trans-splicing AAVs (Ghosh et al., 2007, 2008). Then, dual AAV hybrid vectors have been exploited in human gene therapy in the past years, demonstrating the interest of this technique for in vivo efficient gene transfer (Koo et al., 2014; Trapani et al., 2014; Trapani, 2018; Barbon et al., 2021). To the best of our knowledge, we present here, for the first time, overexpression of a cardiac large gene in ∼75% of mice cardiomyocytes. Most importantly, our results demonstrated that dual AAVs could be used to create animal models mimicking human diseases. If the use of the cTnT promoter confirmed a robust cardiomyocyte-specific expression of the transgene (Prasad et al., 2011), we also believe that systemic injection of AAVs in the early stages of life (3–5 days after birth) helped to reduce mice immune response and to favor cell transduction efficiency (Ghosh et al., 2007; Hu et al., 2010).
A limitation of our strategy was the choice to design a protein fusion of Nav1.5 and eGFP. Indeed, the dominant-negative effect of the R104W variant implied retention and degradation of Nav1.5 channels, leading to degradation of the GFP and loss of its reporter gene function. If in GFP and hNav1.5-injected mouse cardiomyocytes, green positive cells were easy to visualize for patch-clamp recordings, native fluorescence was almost not visible in hNav1.5-R104W overexpressing cardiomyocytes, and we could not select the transduced cells to record. This was most probably responsible for the large variation of currents represented in Figure 4C, and likely for an underestimation of the in vivo dominant-negative effect of R104W on endogenous WT channels. Nevertheless, it is worth to note that one INa recorded in a hNav1.5-R104W-injected mouse cardiomyocyte (indicated by an arrow in Figure 4C) was null, even if recorded with 135 mM Na+ in the outer solution. We suspect the degradation of mutated channels to lead also to the apparent lower transduction rate observed for hNav1.5-R104W AAVs compared to hNav1.5-WT ones (Figures 2B,C). Nevertheless, we believe that the hNav1.5-R104W-transduction rate was underestimated as a result of GFP degradation, since the transduction rate of hNav1.5-WT reached 75%, with the exact same dual AAV genome, except for the R104W missense mutation. On another hand, immunostaining of the GFP fused to the channel allowed to localize mutant channels and to confirm that hNav1.5-R104W was overexpressed in injected-mice cardiomyocytes (Figures 2C,F) and in injected-mice cardiac tissues analyzed in western-blots (Figure 5A), an observation that would have not been possible using a reporter gene not fused to the channel.
A mouse model with targeted disruption of Scn5a has been established in 2002 (Papadatos et al., 2002). If homozygous knock out (KO) mouse embryos die during mid-gestation due to structural abnormalities of the heart, heterozygous mice show normal survival and several cardiac electrical defects such as decreased atrial, atrioventricular, and ventricular conduction and increased susceptibility to pacing-induced ventricular arrhythmias (Papadatos et al., 2002; van Veen et al., 2005). Despite being detectable in less than a half of mice cardiomyocytes, the R104W dominant-negative variant induced a prolongation of RR interval, P wave duration and PR interval as a consequence of the significant decrease of INa, like observed in Scn5a+/– mice (Leoni et al., 2010). However, we did not observe any prolongation of QRS intervals, compared to transgenic deficient mice. It is worth to note that we recorded a small but significant rightward shift of R104W activation curve in transduced mice cardiomyocytes, as in HEK293 cells (Clatot et al., 2012), accounting for the loss-of-function characteristics of this variant.
Two knock in (KI) models of SCN5A mutants have been developed (Sendfeld et al., 2019): one in mice, exhibiting an overlap syndrome whose conduction-defect severity was strain-dependent (Remme et al., 2006), and one in pig, showing prolonged P and QRS wave duration and prolonged PR intervals, consistent with slowed cardiac conduction (Park et al., 2015). Both KI transgenic models, like our model of overexpression using viral vectors, suggest that expressing a specific mutation led to a particular phenotype, recapitulating the complexity of BrS.
Overexpression of SCN5A in transgenic mice has been shown to shorten P wave duration and PR interval, while QRS and QT intervals remained unchanged (Zhang et al., 2007). This was consistent with the observation that mice overexpressing SCN5A exhibit accelerated atrioventricular, atrial, and ventricular conduction (Liu et al., 2015). It was therefore not surprising to record an increase of the P wave duration and the PR interval, even if not significant, in our model of overexpression of a dominant-negative Nav1.5 mutation. At this stage, we can hypothesize that action potential upstroke velocity (dV/dt) is impaired in R104W-overexpressing mice since their cardiac INa is significantly decreased, but further experiments of action potential recordings should be realized to confirm this hypothesis.
We chose to develop our dual AAVs strategy in the mouse since several other genetically modified mouse models were available to compare our results with (Derangeon et al., 2012; Sendfeld et al., 2019) and because mouse is the most utilized mammal in scientific research for its size and similarities with human. Nevertheless, further studies should be conducted to adapt this approach to bigger animals with features closer to human cardiac physiology. Moreover, our model, as others, has inherent limitations, as it is a model of overexpression using a promoter chosen to drive a robust cardiac specific expression, but which did not allow to control the transgene expression level.
It has long been assumed that cardiac structural abnormalities are undetectable in patients with loss of-function SCN5A channelopathies, in coherence with the conventional concept that Nav1.5 is only involved in maintaining cardiac electrical integrity. However, this paradigm has been challenged in the last years as loss-of-function SCN5A mutations are found in a growing number of patients with dilated cardiomyopathy (Zaklyazminskaya and Dzemeshkevich, 2016; Asatryan, 2019), by the demonstration that Nav1.5 is part of a macromolecular complex which contains cytoskeleton proteins (Rook et al., 2012) and by the observation of dilatation and impairment in ventricular contractile function in patients carrying loss-of-function BrS SCN5A variants (van Hoorn et al., 2012). As recently reviewed by Rivaud et al. (2020), an alternative concept is emerging in which Nav1.5 may also be involved in maintaining cardiac structural integrity by non-ionic mechanisms. In the light of this analysis, we can hypothesize that impairment of INa may be responsible for cardiac dilatation and early stages of heart failure.
In the strict sense of the term, a dominant-negative effect is observed when a decrease of INa exceeding the 50% of current density expected in case of haploinsufficiency is recorded while co-expressing mutants with WT channels in a 1:1 ratio to mimic patient heterozygosity. As discussed above, degradation of the reporter protein fused to the mutated channel very likely underestimated the mutant functional effects on INa. Considering this limitation, we understand the significantly reduced INa and the decrease of total Nav1.5 protein expression in R104W-injected mice as an evidence of the in vivo dominant-negative effect of R104W. At this stage, we can only speculate that WT endogenous Nav1.5 channels degradation occurred through their interaction with R104W α-subunits, as shown previously in vitro (Clatot et al., 2012). This hypothesis was further supported by the abnormal perinuclear localization of mutant channels, compared to WT-overexpressed ones (Figures 2E,F). Nevertheless, further studies should be conducted to demonstrate the interaction between hNav1.5-R104W and mNav1.5 endogenous channels as the mechanism of the in vivo dominant-negative effect of R104W mutant channels.
To summarize, our results showed for the first time that a dual-AAV trans-splicing approach allows overexpression of a large gene encoding an ion channel in up to 75% of injected-mice cardiomyocytes. Applied to overexpression of a BrS variant in mouse heart, this strategy enabled us to confirm in vivo the R104W variant dominant-negative effect previously observed in vitro. Altogether our results demonstrated that the use of AAVs to overexpress SCN5A mutants in vivo is a relevant approach to create a versatile and valuable animal model of BrS. Furthermore, the success of our approach of dual trans-splicing AAVs to overexpress SCN5A in the heart constitutes the proof-of-concept of future work aimed at developing novel treatment for malignant arrhythmias observed in SCN5A loss-of-function-related channelopathies.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Comité d’éthique en expérimentation animale Charles Darwin N°5 INSERM & Sorbonne Université.
ND and MG realized all the experiments of molecular biology and electrophysiology. ND, MG, and NN analyzed the data. NM recorded echocardiographies and ECGs. MC produced some AAV preparations. CS performed western blots. ND, MG, AC, PG, and NN contributed to manuscript writing. PG and NN funded the project. All authors contributed to the article and approved the submitted version.
This work was supported by the Fondation pour la Recherche Médicale (FRM grant DPC20111122989), Institut National de la Santé et de la Recherche Médicale (INSERM), and Sorbonne Université.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors want to acknowledge the Viral Vector Core of Nantes University (France) for AAV production, B. A. French (Virginia University, United States) for the gift of plasmids pAcTnT-S and pAcTnT-eGFP, H. Abriel (University of Bern, Bern, Switzerland) for plasmid pcDNA3.1-hH1a, D. Duan (University of Missouri, Columbia, MO, United States) for the plasmid pAG71, and S. Benkhelifa-Ziyyat (Myology Institute, Paris, France) for plasmids pXX6 and pAAV2-9. The authors also wish to thank Tiphaine Héry for her precious help in AAV titration.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.661413/full#supplementary-material
Abriel, H., Rougier, J.-S., and Jalife, J. (2015). Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ. Res. 116, 1971–1988. doi: 10.1161/CIRCRESAHA.116.305017
Asatryan, B. (2019). Cardiac sodium channel dysfunction and dilated cardiomyopathy: a contemporary reappraisal of pathophysiological concepts. J. Clin. Med. 8:1029. doi: 10.3390/jcm8071029
Aurnhammer, C., Haase, M., Muether, N., Hausl, M., Rauschhuber, C., Huber, I., et al. (2012). Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods 23, 18–28. doi: 10.1089/hgtb.2011.034
Barbon, E., Kawecki, C., Marmier, S., Sakkal, A., Collaud, F., Charles, S., et al. (2021). Development of a dual hybrid AAV vector for endothelial-targeted expression of von Willebrand factor. Gene Ther. 1–10. doi: 10.1038/s41434-020-00218-6
Brugada, J., Campuzano, O., Arbelo, E., Sarquella-Brugada, G., and Brugada, R. (2018). Present status of brugada syndrome: JACC state-of-the-art review. J. Am. Coll. Cardiol. 72, 1046–1059. doi: 10.1016/j.jacc.2018.06.037
Bryksin, A. V., and Matsumura, I. (2010). Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques 48, 463–465. doi: 10.2144/000113418
Clatot, J., Hoshi, M., Wan, X., Liu, H., Jain, A., Shinlapawittayatorn, K., et al. (2017). Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 8:2077. doi: 10.1038/s41467-017-02262-0
Clatot, J., Ziyadeh-Isleem, A., Maugenre, S., Denjoy, I., Liu, H., Dilanian, G., et al. (2012). Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 α-subunits. Cardiovasc. Res. 96, 53–63. doi: 10.1093/cvr/cvs211
Derangeon, M., Montnach, J., Baró, I., and Charpentier, F. (2012). Mouse models of SCN5A-related cardiac arrhythmias. Front. Physiol. 3:210. doi: 10.3389/fphys.2012.00210
Dong, J. Y., Fan, P. D., and Frizzell, R. A. (1996). Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 7, 2101–2112. doi: 10.1089/hum.1996.7.17-2101
Duan, D., Yue, Y., Yan, Z., and Engelhardt, J. F. (2000). A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat. Med. 6, 595–598. doi: 10.1038/75080
Ghosh, A., Yue, Y., and Duan, D. (2011). Efficient transgene reconstitution with hybrid dual AAV vectors carrying the minimized bridging sequences. Hum. Gene Ther. 22, 77–83. doi: 10.1089/hum.2010.122
Ghosh, A., Yue, Y., Lai, Y., and Duan, D. (2008). A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol. Ther. J. Am. Soc. Gene Ther. 16, 124–130. doi: 10.1038/sj.mt.6300322
Ghosh, A., Yue, Y., Long, C., Bostick, B., and Duan, D. (2007). Efficient whole-body transduction with trans-splicing adeno-associated viral vectors. Mol. Ther. J. Am. Soc. Gene Ther. 15, 750–755. doi: 10.1038/sj.mt.6300081
Guggino, W. B., and Cebotaru, L. (2020). Gene therapy for cystic fibrosis paved the way for the use of adeno-associated virus in gene therapy. Hum. Gene Ther. 31, 538–541. doi: 10.1089/hum.2020.046
Hoshi, M., Du, X. X., Shinlapawittayatorn, K., Liu, H., Chai, S., Wan, X., et al. (2014). Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ. Cardiovasc. Genet. 7, 123–131. doi: 10.1161/CIRCGENETICS.113.000292
Hu, C., Busuttil, R. W., and Lipshutz, G. S. (2010). RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J. Gene Med. 12, 766–778. doi: 10.1002/jgm.1496
Kaplitt, M. G., Leone, P., Samulski, R. J., Xiao, X., Pfaff, D. W., O’Malley, K. L., et al. (1994). Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 8, 148–154. doi: 10.1038/ng1094-148
Keller, D. I., Rougier, J.-S., Kucera, J. P., Benammar, N., Fressart, V., Guicheney, P., et al. (2005). Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc. Res. 67, 510–519. doi: 10.1016/j.cardiores.2005.03.024
Koo, T., Popplewell, L., Athanasopoulos, T., and Dickson, G. (2014). Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum. Gene Ther. 25, 98–108. doi: 10.1089/hum.2013.164
Leoni, A.-L., Gavillet, B., Rougier, J.-S., Marionneau, C., Probst, V., Le Scouarnec, S., et al. (2010). Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/-) mouse model. PLoS One 5:e9298. doi: 10.1371/journal.pone.0009298
Liu, G. X., Remme, C. A., Boukens, B. J., Belardinelli, L., and Rajamani, S. (2015). Overexpression of SCN5A in mouse heart mimics human syndrome of enhanced atrioventricular nodal conduction. Heart Rhythm 12, 1036–1045. doi: 10.1016/j.hrthm.2015.01.029
Mercier, A., Clément, R., Harnois, T., Bourmeyster, N., Faivre, J.-F., Findlay, I., et al. (2012). The β1-subunit of Na(v)1.5 cardiac sodium channel is required for a dominant negative effect through α-α interaction. PLoS One 7:e48690. doi: 10.1371/journal.pone.0048690
Pambrun, T., Mercier, A., Chatelier, A., Patri, S., Schott, J.-J., Le Scouarnec, S., et al. (2014). Myotonic dystrophy type 1 mimics and exacerbates Brugada phenotype induced by Nav1.5 sodium channel loss-of-function mutation. Heart Rhythm 11, 1393–1400. doi: 10.1016/j.hrthm.2014.04.026
Papadatos, G. A., Wallerstein, P. M. R., Head, C. E. G., Ratcliff, R., Brady, P. A., Benndorf, K., et al. (2002). Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. U.S.A. 99, 6210–6215. doi: 10.1073/pnas.082121299
Park, D. S., Cerrone, M., Morley, G., Vasquez, C., Fowler, S., Liu, N., et al. (2015). Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J. Clin. Invest. 125, 403–412. doi: 10.1172/JCI76919
Prasad, K.-M. R., Xu, Y., Yang, Z., Acton, S. T., and French, B. A. (2011). Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 18, 43–52. doi: 10.1038/gt.2010.105
Remme, C. A., Verkerk, A. O., Nuyens, D., van Ginneken, A. C. G., van Brunschot, S., Belterman, C. N. W., et al. (2006). Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114, 2584–2594. doi: 10.1161/CIRCULATIONAHA.106.653949
Rivaud, M. R., Delmar, M., and Remme, C. A. (2020). Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: ionic and non-ionic mechanisms. Cardiovasc. Res. 116, 1557–1570. doi: 10.1093/cvr/cvaa082
Rook, M. B., Evers, M. M., Vos, M. A., and Bierhuizen, M. F. A. (2012). Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc.Res. 93, 12–23. doi: 10.1093/cvr/cvr252
Sendfeld, F., Selga, E., Scornik, F. S., Pérez, G. J., Mills, N. L., and Brugada, R. (2019). Experimental models of brugada syndrome. Int. J. Mol. Sci. 20, 2123. doi: 10.3390/ijms20092123
Sondergaard, P. C., Griffin, D. A., Pozsgai, E. R., Johnson, R. W., Grose, W. E., Heller, K. N., et al. (2015). AAV.dysferlin overlap vectors restore function in dysferlinopathy animal models. Ann. Clin. Transl. Neurol. 2, 256–270. doi: 10.1002/acn3.172
Sun, L., Li, J., and Xiao, X. (2000). Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med. 6, 599–602. doi: 10.1038/75087
Trapani, I. (2018). Dual AAV vectors for stargardt disease. Methods Mol. Biol. Clifton NJ 1715, 153–175. doi: 10.1007/978-1-4939-7522-8_11
Trapani, I., Colella, P., Sommella, A., Iodice, C., Cesi, G., de Simone, S., et al. (2014). Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 6, 194–211. doi: 10.1002/emmm.201302948
van Hoorn, F., Campian, M. E., Spijkerboer, A., Blom, M. T., Planken, R. N., van Rossum, A. C., et al. (2012). SCN5A mutations in Brugada syndrome are associated with increased cardiac dimensions and reduced contractility. PloS One 7:e42037. doi: 10.1371/journal.pone.0042037
van Veen, T. A. B., Stein, M., Royer, A., Le Quang, K., Charpentier, F., Colledge, W. H., et al. (2005). Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 112, 1927–1935. doi: 10.1161/CIRCULATIONAHA.105.539072
Wang, Z., Vermij, S. H., Sottas, V., Shestak, A., Ross-Kaschitza, D., Zaklyazminskaya, E. V., et al. (2020). Calmodulin binds to the N-terminal domain of the cardiac sodium channel Nav1.5. Channels Austin Tex 14, 268–286. doi: 10.1080/19336950.2020.1805999
Watanabe, H., and Minamino, T. (2015). Genetics of Brugada syndrome. J. Hum. Genet. 61, 57–60. doi: 10.1038/jhg.2015.97
Wilde, A. A. M., and Brugada, R. (2011). Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res. 108, 884–897. doi: 10.1161/CIRCRESAHA.110.238469
Zaklyazminskaya, E., and Dzemeshkevich, S. (2016). The role of mutations in the SCN5A gene in cardiomyopathies. Biochim. Biophys. Acta 1863, 1799–1805. doi: 10.1016/j.bbamcr.2016.02.014
Keywords: Brugada syndrome, Nav1.5, SCN5A, animal model, electrophysiology, AAV
Citation: Doisne N, Grauso M, Mougenot N, Clergue M, Souil C, Coulombe A, Guicheney P and Neyroud N (2021) In vivo Dominant-Negative Effect of an SCN5A Brugada Syndrome Variant. Front. Physiol. 12:661413. doi: 10.3389/fphys.2021.661413
Received: 30 January 2021; Accepted: 21 April 2021;
Published: 28 May 2021.
Edited by:
Mohamed-Yassine Amarouch, Sidi Mohamed Ben Abdellah University, MoroccoCopyright © 2021 Doisne, Grauso, Mougenot, Clergue, Souil, Coulombe, Guicheney and Neyroud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Neyroud, bmF0aGFsaWUubmV5cm91ZEBzb3Jib25uZS11bml2ZXJzaXRlLmZy
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.