 Satvik Mareedu
Satvik Mareedu Emily D. Million
Emily D. Million Dongsheng Duan
Dongsheng Duan Gopal J. Babu
Gopal J. Babu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 09 April 2021
Sec. Striated Muscle Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.647010
This article is part of the Research Topic Challenges and Opportunities of Targeting Muscle for Therapy View all 10 articles
Duchenne muscular dystrophy (DMD) is an X-linked muscle-wasting disease caused by the loss of dystrophin. DMD is associated with muscle degeneration, necrosis, inflammation, fatty replacement, and fibrosis, resulting in muscle weakness, respiratory and cardiac failure, and premature death. There is no curative treatment. Investigations on disease-causing mechanisms offer an opportunity to identify new therapeutic targets to treat DMD. An abnormal elevation of the intracellular calcium () concentration in the dystrophin-deficient muscle is a major secondary event, which contributes to disease progression in DMD. Emerging studies have suggested that targeting Ca2+-handling proteins and/or mechanisms could be a promising therapeutic strategy for DMD. Here, we provide an updated overview of the mechanistic roles the sarcolemma, sarcoplasmic/endoplasmic reticulum, and mitochondria play in the abnormal and sustained elevation of levels and their involvement in DMD pathogenesis. We also discuss current approaches aimed at restoring Ca2+ homeostasis as potential therapies for DMD.
Duchenne muscular dystrophy (DMD) is X-linked and is the most common form of muscle wasting disease. It affects 1 in 3,500 to 5,000 male births (Mendell and Lloyd-Puryear, 2013). DMD is caused by mutations in the dystrophin gene, which leads to the loss of a functional dystrophin protein (Monaco et al., 1986). DMD is characterized by progressive loss of muscle mass and function due to muscle degeneration, necrosis, and fatty fibrosis, resulting in wheelchair dependence (Ervasti and Campbell, 1993; Bushby et al., 2010; Connolly et al., 2013; Aartsma-Rus et al., 2016; Duan et al., 2021). Eventually, patients die of respiratory failure and/or cardiomyopathy (Finsterer, 2006). There is currently no curative treatment for DMD. Restoring dystrophin function via gene replacement or repair therapy is attractive and promising. Although there is a favorable outcome in preclinical studies, the immunogenicity of the newly expressed dystrophin protein remains a concern. In addition, the complexity of thousands of disease-causing mutations in the dystrophin gene creates challenges for gene repair therapy. Alternatively, targeting major disease-causing mechanisms offers an opportunity to treat all DMD patients without the complications of dystrophin replacement or repair therapies.
Dystrophin is a rod-shaped cytoskeletal protein, primarily expressed in muscles. Dystrophin links the intracellular cytoskeleton network to the transmembrane components of the dystrophin–glycoprotein complex (Gao and McNally, 2015; Allen et al., 2016). Growing evidence suggests dystrophin in addition to maintaining the structural integrity of the sarcolemma plays a key role in regulating signaling pathways. This includes the nitric oxide pathway, Ca2+ entry, and the production of reactive oxygen species (ROS). In the absence of dystrophin, these pathways are damaged and contribute to muscle pathology. For details on these pathways and their clinical implications in DMD, refer to a recent review by Allen et al. (2016).
Among the various disease-causing mechanisms, changes in intracellular calcium () levels in dystrophin-deficient muscle fibers have been studied for many years. An increase in both Ca2+ influx and cytosolic Ca2+ concentration has been reported in mdx myofibers (Turner et al., 1988; Hopf et al., 1996). Although many studies have shown higher levels of Ca2+ content in the muscle fibers from mdx mice, others have reported no significant rise in Ca2+ concentration (Gillis, 1996, 1999; Gailly, 2002; Whitehead et al., 2006). Dystrophin is indispensable for maintaining the structural integrity of the striated muscle cell (Petrof et al., 1993). Thus, lack of dystrophin destabilizes sarcolemma integrity, making the sarcolemma more susceptible to contraction-induced damage. This leads to myonecrosis (necrosis of myofibers), which in turn stimulates fiber regeneration. In mdx mice, the onset of myonecrosis starts at 2 weeks of age. Myonecrosis peaks between 3 and 4 weeks of age and is significantly decreased and stabilized by 6 to 8 weeks of age. Eventually, ~80% of myofibers in adult mdx mice are regenerated fibers (Grounds et al., 2008). A sharp increase in total Ca2+ content has been shown at the peak of myonecrosis. The total Ca2+ content returns to normal level in later stages (Reeve et al., 1997). Studies from Head's laboratory suggest that regenerated fibers display branched morphology. It has been hypothesized that myofiber branching rather than dystrophin deficiency increases muscle susceptibility to damage (Head, 2010; Chan and Head, 2011). These studies further suggest that the branching of fibers resulted in altered ion channel function and excessive Ca2+ influx, which further exacerbates muscle damage. Muscle damage and degeneration are therefore suggested to play an important role in stress-induced membrane tears and dysfunction of sarcolemmal ion channels that lead to abnormal Ca2+ influx from the extracellular matrix (ECM) to the cytosol (Turner et al., 1988, 1991; Moens et al., 1993; Alderton and Steinhardt, 2000; Burr and Molkentin, 2015). In addition, there is a defect in Ca2+ cycling between the cytosol and the sarcoplasmic reticulum (SR)/endoplasmic reticulum (ER), a major internal Ca2+ store in striated muscles. These changes result in a chronic accumulation of Ca2+ in the cytoplasm. Thus, the lack of dystrophin results in muscle degeneration and improper regeneration, which may contribute to abnormal handling.
Several lines of evidence suggest that sustained elevation of cytosolic Ca2+ levels underlies muscle pathology and dysfunction in DMD. First, increased cytosolic Ca2+ levels enhance the expression and activity of calpains, the Ca2+ dependent proteases in dystrophic muscles (Spencer et al., 1995; Hussain et al., 2000; Shanmuga Sundaram et al., 2006; Voit et al., 2017). Calpain activation results in proteolytic damage to cellular proteins and the myofibrillar network (Dayton et al., 1976; MacLennan et al., 1991; Bartoli and Richard, 2005). In support of this notion, treatment with BN 82270, a membrane-permeable calpain inhibitor, improves muscle function in mdx mice (Burdi et al., 2006). Studies from our laboratory have shown normalization of cycling by improving SR Ca2+ ATPase (SERCA) activity decreases calpain activity and improves muscle function in dystrophin and utrophin double-mutant (mdx:utr−/−) mice (Voit et al., 2017). In addition to calpains, increased cytosolic Ca2+ levels also activate phospholipase A, which digests cellular membranes such as the sarcolemma (Lindahl et al., 1995). Second, the sustained elevation of concentration activates apoptotic and necrotic cell death pathways in DMD (Tidball et al., 1995; Morgan et al., 2018). Third, abnormal concentration may influence muscle differentiation and compromise muscle regeneration. In this regard, we have recently demonstrated improving cycling in dystrophic myoblasts improves myoblast fusion and differentiation (Niranjan et al., 2019). Fourth, rapid change in free Ca2+ levels in the cytoplasm is essential for proper initiation of muscle contraction and relaxation (Calderon et al., 2014). Thus, chronic accumulation of Ca2+ in the cytosol can affect muscle function in DMD. Fifth, cytoplasmic Ca2+ levels influence mitochondrial Ca2+ uptake, which in turn leads to altered metabolism and increased production of ROS. The failure to mitigate supraphysiological levels of oxygen radicals results in the loss of membrane potential and cell death (Feno et al., 2019). Collectively, abnormal cycling plays a pivotal role in DMD pathogenesis, and restoration of homeostasis may ameliorate muscle disease and cardiomyopathy in DMD. A better understanding of the molecular mechanisms underlying abnormal cycling will aid in identifying novel therapeutic targets for DMD. Below, we review Ca2+ handling in normal muscle and how changes at the sarcolemma, SR, and mitochondria cause Ca2+ dysregulation in DMD. We also highlight the current studies on improving SERCA function as a strategy to mitigate skeletal muscle disease and cardiomyopathy in DMD.
Contraction and relaxation cycles are controlled voluntarily in skeletal muscle and involuntarily in cardiac muscle and are tied with Ca2+ cycling between the SR and cytoplasm. Ca2+ cycling in normal muscle is depicted in Figure 1. In muscles, extracellular Ca2+ concentrations are ~2–4 mM, and resting cytosolic concentrations are ~50–250 nM. SR Ca2+ concentrations are ~0.4–0.5 mM (MacLennan and Kranias, 2003; Gehlert et al., 2015; Kuo and Ehrlich, 2015). In the heart, the resting diastolic Ca2+ levels are ~100 nM and during systole, cytosolic Ca2+ levels increase to ~1 μM (MacLennan and Kranias, 2003). Excitation and contraction (EC) coupling in skeletal and cardiac muscles shares many similarities. In both muscles, the action potential activates voltage-gated, L-type Ca2+ channels (Cav1.1 in skeletal muscles and Cav1.2 in cardiac muscle) in the sarcolemma, which promotes Ca2+ release from the SR via the ryanodine receptor (RyR1 in skeletal muscles and RyR2 in cardiac muscle). This process is known as Ca2+-induced Ca2+ release (CICR) (MacLennan and Kranias, 2003; Rios, 2018). Although CICR was first discovered in skeletal muscle, it does not play a major role in skeletal muscle contraction (Lamb, 2000; Endo, 2009; Rios, 2018). In cardiac muscle, the interaction between Cav1.2 and RyR2 depends on CICR, whereas in skeletal muscle, Cav1.1 and RyR1 physically interact. This interaction is independent of the Ca2+ influx via Cav1.1 (Protasi, 2002; Franzini-Armstrong, 2018). It also activates the opening of RyR1 in response to Cav1.1 voltage sensor activation, a process known as Ca2+-independent, depolarization-induced Ca2+ release (also called voltage-induced Ca2+ release) (Rios, 2018). The mechanism by which these two channels interact is not fully understood. A recent study shows junctophilins (protein localized in junctional SR) enable the Cav1.1 to efficiently couple with RyRs through protein–protein interaction. This mechanism is considered crucial for efficient EC coupling in adult skeletal muscles (Nakada et al., 2018). Studies have also shown the importance of store-operated Ca2+ entry independent of Cav1.1 in the regulation of skeletal muscle contraction (Avila-Medina et al., 2018). Store-operated Ca2+ entry machinery located in the triad of skeletal muscle contains many proteins including stromal interaction molecule 1 (STIM1), Orai, transient receptor potential canonical (TRPC) channel, and RyRs. These proteins are involved in Ca2+ store restoration during the intense work of skeletal muscles (Pan et al., 2014; Avila-Medina et al., 2018).
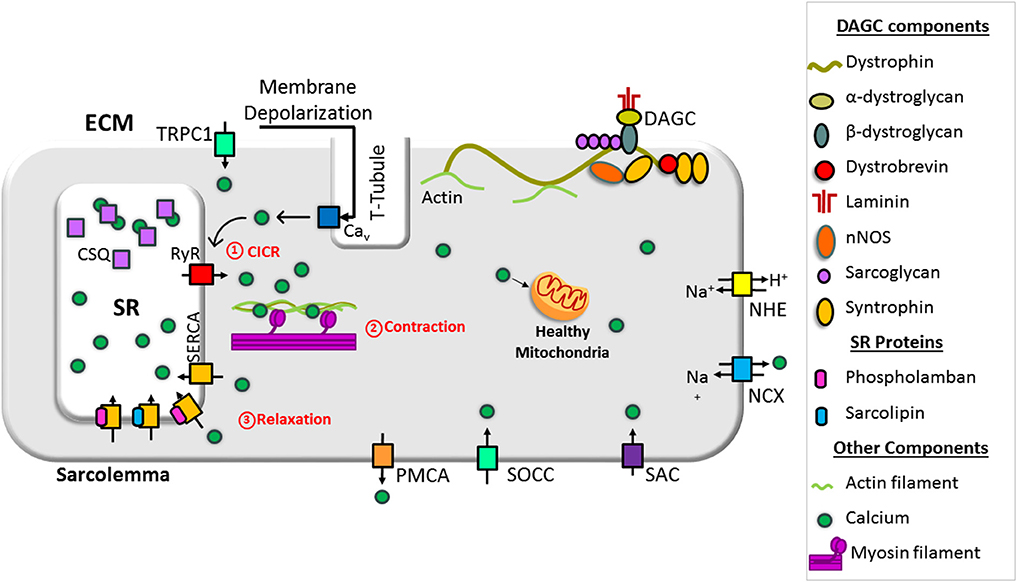
Figure 1. Schematic representation of intracellular Ca2+ cycling in a normal muscle cell. Dystrophin stabilizes muscle cells by linking the cytoskeleton (F-actin, intermediate filaments, microtubule) to the extracellular matrix via the dystrophin-associated glycoprotein complex (DAGC). Ca2+-induced Ca2+ release (CICR) occurs through activation of voltage-gated, L-type Ca2+ channels (Cav) and the release of Ca2+ from the sarcoplasmic/endoplasmic reticulum (SR) via ryanodine receptor (RyR). CICR activates muscle contraction. Resequestration of Ca2+ back into the SR by SR Ca2+ ATPase (SERCA) initiates muscle relaxation. The physiological intracellular Ca2+ level regulates mitochondrial Ca2+ content and function and maintains cellular energetics. DG, dystroglycan; NCX, sodium–calcium exchanger; NHE, sodium–proton exchanger; PMCA, plasma membrane Ca2+ ATPase; SAC, stretch-activated channels; SOCC, store-operated Ca2+ channel; TRPC1, transient receptor potential channel 1.
Both in skeletal and cardiac muscles, Ca2+ released from the SR binds to troponin C, a thin filament sarcomeric protein, and initiates muscle contraction. Muscle relaxation is initiated by Ca2+ removal from the cytoplasm. Approximately 70–90% of Ca2+ is removed by SERCA (SERCA1 and SERCA2a in skeletal muscle and SERCA2a in cardiac muscle) and resequestrated into the lumen of the SR. Most of the remaining Ca2+ is extruded out of the cell via sarcolemmal Ca2+ transport proteins. These include the sodium (Na+)–Ca2+ exchanger (NCX) and plasma membrane Ca2+ ATPase. A small amount of Ca2+ is taken up by the mitochondria via the mitochondrial uniporter (MCU). Thus, SR Ca2+ cycling is not only important for muscle contraction and relaxation cycle but also helps to maintain cytosolic Ca2+ levels. For extensive reviews on Ca2+ handling in normal skeletal muscle and heart, refer to the recent reviews (Endo, 2009; Lee, 2010; Eisner et al., 2017; Avila et al., 2019).
In dystrophin-deficient muscle, the function of sarcolemmal Ca2+ channels is altered and contributes to the abnormal elevation of cytoplasmic Ca2+ concentration. Schematic representation of abnormal Ca2+ handling via the sarcolemma in dystrophin-deficient muscle is shown in Figure 2.
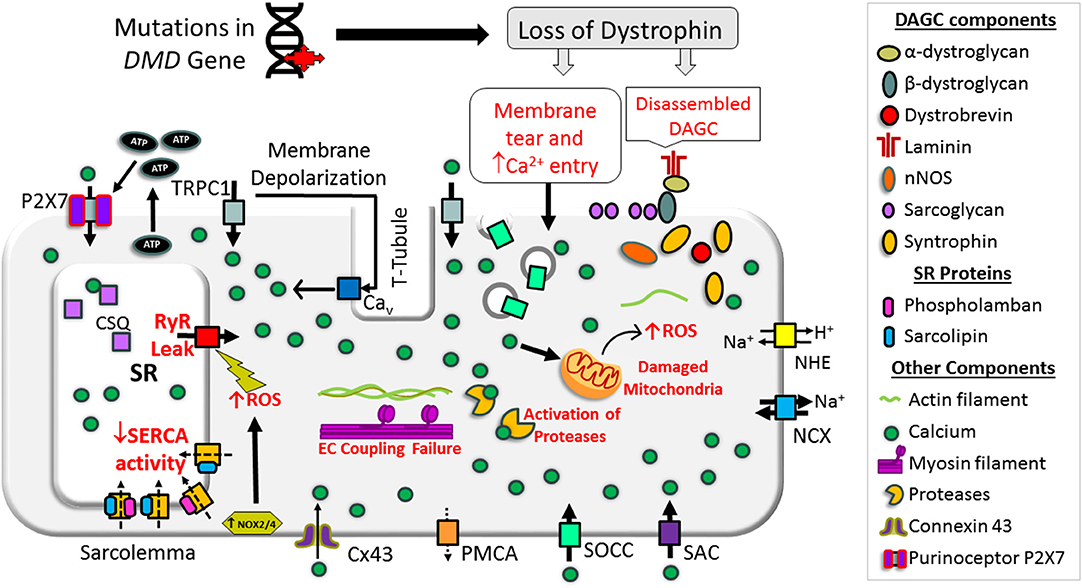
Figure 2. Schematic representation of mechanisms causing overload in a dystrophin-deficient muscle cell. Loss of dystrophin causes destabilization of dystrophin-associated glycoprotein complex (DAGC), membrane tear, and activation of store-operated calcium channels (SOCCs), resulting in abnormal Ca2+ entry. High concentrations of extracellular ATP can activate abnormal Ca2+ influx via P2X7. RyR dysfunction causes Ca2+ leak from the SR and SERCA dysfunction compromises Ca2+ resequestration. NOX activation can also increase RyR Ca2+ leak. These changes result in abnormal and chronic elevation of the cytoplasmic Ca2+ levels. Supraphysiological level Ca2+ activates Ca2+-dependent proteases and phospholipase and causes muscle necrosis and replacement of muscle by fatty and fibrotic tissues. Sustained elevation of cytoplasmic Ca2+ levels also affects mitochondrial function and increases reactive oxygen species (ROS). Collectively, these changes lead to muscle wasting and contractile dysfunction. Bold and broken arrows indicate the enhanced and decreased function of the Ca2+ channels, respectively. Cx43, connexin 43; NCX, sodium–calcium exchanger; NHE, sodium–proton exchanger; NOX, NADPH oxidases; PMCA, plasma membrane Ca2+ ATPase; P2X7, P2X purinoceptor 7; SAC, stretch-activated channels; SOCC, store-operated Ca2+ channel; TRPC1, transient receptor potential channel 1.
A primary function of dystrophin is to link the cytoskeleton with the ECM through direct or indirect interaction with the proteins in the dystrophin-associated glycoprotein complex (DAGC) and direct interaction with actin microfilaments, intermediate filaments, and microtubules (Koenig et al., 1988; Ervasti and Campbell, 1993; Petrof et al., 1993; Chakkalakal et al., 2005; Gao and McNally, 2015). The absence of dystrophin disrupts the DAGC and makes the sarcolemma more susceptible to microtears during mechanical stress (Petrof et al., 1993; Danialou et al., 2001). Increased sarcolemmal membrane permeability is an early feature of patients affected by DMD, allowing cytosolic contents such as creatine kinase to exit skeletal muscle fibers and ions such as Ca2+ to enter. On the other hand, in mdx mice, blocking NADPH oxidase 2 (NOX2), the main ROS producer, saves muscle from force loss following eccentric contraction (Olthoff et al., 2018). Similarly, mdx muscle fibers repair their sarcolemma just as efficiently as wild-type fibers following laser-induced focal damage, suggesting membrane tears of this kind are sealed promptly (Cooper and Head, 2015). Thus, it is now believed that membrane tear is not a primary pathway for Ca2+ entry in DMD (Yeung et al., 2005; Allen and Whitehead, 2011; Cooper and Head, 2015; Olthoff et al., 2018).
Calcium signaling plays an important role in sarcolemma repair (Andrews et al., 2014; Cooper and Head, 2015). Increased cytosolic Ca2+ levels trigger Ca2+-dependent repair mechanisms in which exocytic vesicles patch sarcolemmal tears. This may result in increased activity of Ca2+ leak channels because an antagonist of these channels reduced the higher levels of hydrolysis in dystrophic myotubes to nearly normal levels (Alderton and Steinhardt, 2000). Recent studies have demonstrated the involvement of transient receptor potential mucolipin 1 (TRPML1), a lysosomal Ca2+ release channel needed for lysosomal exocytosis in membrane repair in DMD (Cheng et al., 2014; Yu et al., 2020). In mice, genetic ablation of TRPML1 resulted in DMD-like phenotype with an impairment in the membrane sealing in skeletal muscles (Cheng et al., 2014). On the other hand, transgenic overexpression or pharmacological activation of TRPML1 in vivo facilitates sarcolemma repair and alleviates dystrophic phenotypes in both skeletal and cardiac muscles of mdx mice (Yu et al., 2020). These studies further show that TRPML1 activation increased lysosomal biogenesis by activating the transcription factor EB and thereby facilitated sarcolemma repair to reduce muscle damage in mdx mice (Yu et al., 2020). Therefore, targeting lysosomal Ca2+ channels may represent a promising approach to treat DMD and related muscle diseases.
Store-operated calcium channels (SOCCs) are Ca2+ channels residing at the sarcolemma that open in response to decreased SR Ca2+ concentrations. The activity of SOCCs is enhanced in dystrophic muscle cells, which further contributes to the abnormal elevation of levels in dystrophic muscles [40]. Two proteins have been identified as necessary players involved in SOCE: STIM, an ER-located Ca2+ sensor, and Orai, a highly Ca2+ selective ion channel protein in the plasma membrane (Derler et al., 2016). In dystrophin-deficient myoblasts and muscle fibers, the expression of SOCC proteins is increased (Edwards et al., 2010; Andrews et al., 2014). Interestingly, SOCCs can also be activated through a Ca2+-independent pathway. Specifically, Ca2+-independent phospholipase A2 (iPLA2) acts as an intracellular messenger and triggers Ca2+ entry through SOCCs (Smani et al., 2003). An elevation in iPLA2 levels observed in dystrophic muscles (Smani et al., 2003, 2004; Boittin et al., 2010) supports the activation of this pathway.
TRPCs are a family of plasma membrane cation channels opened by Ca2+ store depletion and/or membrane stretch. In dystrophin-deficient muscles, evidence shows Ca2+ enters via eccentric contraction-activated stretch channels (Yeung et al., 2005). Several lines of evidence suggest the opening of TRPCs could be the main contributor of Ca2+ entry through the plasma membrane. To start, overexpression of TRPC1, TRPC3, and TRPC6 has been observed in mdx muscle (Vandebrouck et al., 2007; Gervasio et al., 2008; Boittin et al., 2010; Matsumura et al., 2011; Miyatake et al., 2016). The most widely studied of these is TRPC1. In the mdx muscle, TRPC1 forms a Ca2+ influx pathway with tyrosine–protein kinase Src and caveolin-3 (Gervasio et al., 2008). TRPC1 also interacts with dystrophin and α1-syntrophin, a member of the DAGC. In one model for TRPC1 regulation, the DAGC serves as a scaffold for signaling molecules involved in the regulation of channels formed by TRPC1 and other TRPC isoforms (Sabourin et al., 2009). In the absence of dystrophin, this regulation is lost, and SOCE is increased (Vandebrouck et al., 2007). Matsumura et al. (2011) showed that the expression levels of TRPC1 correlated with the severity of muscle disease in mdx mice.
In addition to canonical TRPCs, the transient receptor potential vanilloid type 2 (TRPV2) channel may also play a role in increased Ca2+ levels. Typically, TRPV2 is localized on intracellular organelles. In dystrophin-deficient muscle, however, it translocates to the plasma membrane (Iwata et al., 2003, 2009).
In dystrophic muscle, intracellular Na+ levels are increased via the opening of voltage-gated Na+ channels (Nav1.4) (Hirn et al., 2008), Na+/H+ exchangers (NHE) type I (Iwata et al., 2007; Burr et al., 2014), and gadolinium sensitive-stretch channels (Yeung et al., 2003). In healthy muscle, the NCX removes excess Ca2+ from the cytosol in exchange for Na+. However, in the presence of excessive cytosolic Na+ (as in dystrophic muscle), NCX functions in a reverse mode to remove Na+ from the cell. In doing so, NCX moves Ca2+ into the cell. Interestingly, it has been shown that increased SR Ca2+ release in dystrophic muscle can also trigger the NCX to work in a reverse mode and thereby increase concentrations (Deval et al., 2002).
The activity of the cardiac L-type Ca2+ channel, Cav1.2, determines Ca2+ entry in the plateau phase (phase 2) of the action potential in cardiac myocytes. In mdx cardiac myocytes, Cav1.2 activation is significantly increased (Koenig et al., 2014). This leads to enhanced Ca2+ influx via Cav1.2 during the action potential. It is worth mentioning that enhanced Cav1.2 activities may disturb cardiac electrophysiology and thereby cause arrhythmias in DMD.
The role of Cav1.1 activity in abnormal Ca2+ entry in dystrophic skeletal muscle is not clear. In delta-sarcoglycan (a component of DAGC)–deficient dystrophic hamsters, the administration of diltiazem, the L-type Ca2+ antagonist, reduces muscle Ca2+ content (Bhattacharya et al., 1982). This suggests a possible pathological role played by L-type Ca2+ channels in dystrophic muscle. In contrast, Friedrich et al. found that Cav1.1 activity is significantly reduced in the fast-twitch muscles of mdx mice (Friedrich et al., 2004). These studies further suggest dystrophin, and the DAGC may regulate the interaction between L-type Ca2+ channels and RyR, which is necessary for EC coupling (Friedrich et al., 2004, 2008).
Dystrophic muscle damage triggers the release and elevation of extracellular ATP, which activates specific ionotropic purinoreceptors, P2X7, on immune cells and subsequently contributes to chronic inflammatory and immune responses (Gorecki, 2019). In addition, high concentrations of extracellular ATP have been shown to activate abnormal Ca2+ influx into dystrophic muscle cells (Young et al., 2018). This increase in is shown to be associated with increased and prolonged activation of P2X7 purinoceptors that increase the sarcolemma permeability in dystrophic muscle cells (Young et al., 2012). Degrading extracellular ATP by apyrase reduced the levels in mdx fibers (Altamirano et al., 2013). Genetic and pharmacological targeting of P2X7 reduced inflammation and increased dystrophic muscle repair (Gorecki, 2019). Thus, targeting purinergic receptors can ameliorate the abnormal Ca2+ entry in dystrophic muscles.
Connexins (Cx) are gap junction proteins, which are important for many physiological processes including coordinated depolarization of muscle, and ion movement between muscle cells (Cea et al., 2012). Cx function as gap junction channels and hemichannels, which mediate intercellular and transmembrane signaling, respectively. Hemichannels can act as conduits for Na+ and Ca2+ entry (Cea et al., 2012). In dystrophic muscles, Cx39, Cx43, and Cx45 are found to form functional hemichannels, which are absent in normal muscle fibers (Cea et al., 2016). The mdx mice deficient for Cx43/Cx45 expression in skeletal muscle show reduced basal level and necrotic phenotype (Cea et al., 2016). Cx43 levels are significantly increased and are mislocalized in the lateral sides of cardiomyocytes in mouse models of DMD (Gonzalez et al., 2018). Altered localization of Cx43 predisposed the DMD mice to cardiac arrhythmias (Gonzalez et al., 2018). A recent study shows that hypophosphorylation of Cx43 serine triplet triggers redistribution of Cx43 to the lateral sides of cardiomyocytes and contributes to the dystrophic cardiomyopathy in mdx mice (Himelman et al., 2020). The expression of phosphorylation mimic Cx43 in mdx cardiomyocytes shows improved signaling, a reduction of NOX2/ROS production and prevention of arrhythmias (Himelman et al., 2020). Taken together, these studies indicate that Cx overexpression and lateralization contribute to abnormal homeostasis in dystrophin-deficient cardiac and skeletal muscles.
The SR is the major internal Ca2+ store in striated muscles and plays a pivotal role in the regulation of EC coupling by maintaining cytoplasmic Ca2+ concentrations during the muscle contraction and relaxation cycle (Santulli et al., 2017). Numerous studies suggest SR Ca2+ cycling is compromised in dystrophic muscle (Collet et al., 1999; Woods et al., 2004; Williams and Allen, 2007a; DiFranco et al., 2008; Capote et al., 2010) (Figure 2). Below, we review the mechanisms underlying SR Ca2+ cycling defects in dystrophin-deficient cardiac and skeletal muscles.
Ca2+ release from the SR occurs via RyR, a macromolecular complex. There are three RyR isoforms reported in mammals. RyR1 is primarily expressed in skeletal muscles, RyR2 is expressed predominantly in the heart, and RyR3 is found in the brain and skeletal muscles (Conti et al., 1996; Lanner et al., 2010).
The role of the SR in cytosolic Ca2+ rise has been studied using chemically skinned muscle fibers that have not been mechanically stressed (Divet and Huchet-Cadiou, 2002). Studies on the skinned fibers isolated from extensor digitorum longus (EDL) and soleus muscles show that the SR Ca2+ release following exposure to caffeine is significantly slower in mdx mice. These studies suggest a more pronounced SR Ca2+ leak in EDL and soleus muscle fibers from mdx mice. Consistent with these findings, Robin et al. have shown increased passive SR Ca2+ leak in isolated intact myofibers prepared from FDB muscles of mdx5cv mice, an alternative dystrophin-deficient mouse model (Robin et al., 2012). On the contrary, Plant and Lynch have shown that the SR Ca2+ leak was unaltered in skinned mdx fibers (Plant and Lynch, 2003). However, peak caffeine-induced Ca2+ release was decreased in mdx fibers. In addition, all the above studies show no change in SR Ca2+ uptake in skinned fibers from mdx mice. The structural and functional defects in both cardiac and skeletal muscle RyRs have been reported in the mdx mouse model. Recent studies have shown that the SR Ca2+ release mechanism is impaired in both cardiac and skeletal muscles in DMD (Bellinger et al., 2009; Fauconnier et al., 2010). In skeletal muscle, it is believed that progressive S-nitrosylation of RyR1 and depletion of calstabin 1, a critical regulatory subunit of RyR macromolecular complex, are responsible for RyR1 Ca2+ leak (Bellinger et al., 2009). Similarly, S-nitrosylation and calstabin 2 depletion cause RyR2 Ca2+ leak and contribute to sudden cardiac arrhythmias in mdx mice (Fauconnier et al., 2010). In addition, RyR2 phosphorylation and oxidation have been linked to RyR2-mediated Ca2+ leak in the mdx heart. These studies further show that genetic inhibition of RyR2 phosphorylation at S2808 or S2814 can reduce RyR2 oxidation, suggesting a potential interaction between these posttranslational pathways (Williams and Allen, 2007b; Shannon, 2009; Prosser et al., 2011; Wang et al., 2015).
SERCA plays a key role in resequestering Ca2+ into the SR lumen during muscle relaxation. SERCA1 is expressed in fast-twitch skeletal muscles. SERCA2a is predominantly expressed in the heart and also found in slow-twitch muscles. SERCA3 is predominantly expressed in non-muscle tissues (Periasamy and Kalyanasundaram, 2007). In striated muscles, SERCA activity accounts for 70–90% of cytosolic Ca2+ removal (Periasamy and Kalyanasundaram, 2007).
It is now clear that the SERCA function is impaired in dystrophic muscles. SERCA activity can be reduced through several mechanisms, including (i) down-regulation of SERCA expression, (ii) posttranslational modification of SERCA protein, and (iii) differential expression and function of SERCA regulators. In the dystrophic myocardium of mouse models and human patients, SERCA2a levels remain unchanged (Voit et al., 2017; Wasala et al., 2020). In the mdx mouse, SERCA1a expression is increased in the spared intrinsic laryngeal and toe muscles but is reduced in the EDL muscle (Dowling et al., 2003; Ferretti et al., 2009). In the fast-twitch muscles of mdx and mdx:utr−/− mice, SERCA2a expression is significantly increased, likely due to the increased number of slow-twitch fibers (Schneider et al., 2013; Voit et al., 2017). In the extensor carpi ulnaris muscles of the canine DMD model, SERCA2a levels are decreased while SERCA1 expression is unaltered (Voit et al., 2017). These findings suggest differential expression of SERCA isoforms in different dystrophic muscles. Given the different kinetic properties of SERCA1 and SERCA2a, it is likely the muscle- and species-specific changes in SERCA isoform expression represent compensatory alterations in different dystrophic muscles.
Irrespective of SERCA levels, SR Ca2+ uptake is significantly reduced in dystrophin-deficient cardiac and skeletal muscles, indicating decreased SERCA function (Schneider et al., 2013; Voit et al., 2017; Wasala et al., 2020; Mareedu et al., 2021). Oxidative posttranslational modification has been shown to play an important role in SERCA activity in non-dystrophic heart diseases (Lancel et al., 2010; Horakova et al., 2013). As dystrophic muscle undergoes oxidative stress (Kim et al., 2013), it is likely that similar posttranslational modifications can also affect SERCA function in DMD.
SERCA function is modulated by several small-molecular-weight membrane proteins including phospholamban (PLN), sarcolipin (SLN), myoregulin (MLN), and dwarf open reading frame (DWORF) (Bhupathy et al., 2007; Anderson et al., 2015; Nelson et al., 2016; Shaikh et al., 2016). PLN, SLN, and MLN are negative regulators, whereas DWORF is a positive regulator of SERCA. PLN is highly expressed in the ventricles (Babu et al., 2007). SLN is expressed in all skeletal muscle tissues in larger mammals; however, its expression is restricted to slow-twitch muscles in rodents (Babu et al., 2007). Whereas, in the hearts of both rodents and large mammals SLN is primarily expressed in atria and expressed at a low level in ventricles (Babu et al., 2007), MLN is primarily expressed in skeletal muscles (Anderson et al., 2015). DWORF is a positive regulator of SERCA and is predominantly expressed in the heart and slow-twitch muscles (Nelson et al., 2016). In addition to these peptides, recent studies suggest that small ubiquitin-like modifier type 1 (SUMO-1) also plays a critical role in cardiac SERCA dysfunction in the setting of heart failure (Kho et al., 2015).
The PLN levels are unaltered in dystrophic skeletal and cardiac muscles, whereas SLN is significantly up-regulated in the skeletal muscles and the ventricles of mouse and dog models and DMD patients (Voit et al., 2017). Little is known about the expression of MLN, DWORF, and SUMO-1 in dystrophin-deficient cardiac and skeletal muscles. It is worth pointing out that complete elimination of PLN exacerbates mdx cardiomyopathy (Law et al., 2018), whereas partial or complete elimination of SLN significantly reduces skeletal muscle disease and cardiomyopathy in mouse models of DMD (Voit et al., 2017; Mareedu et al., 2021).
The SR luminal resident proteins, including calsequestrin (CSQ), CSQ-like proteins (CLPs), histidine-rich Ca2+-binding protein (HRCBP), calreticulin, and sarcalumenin (SLM), play an important role in buffering luminal Ca2+ concentrations and regulating SR Ca2+ uptake and release (Beard et al., 2004; Arvanitis et al., 2011; Jiao et al., 2012). Among the various SR luminal proteins, CSQ is the major Ca2+-buffering protein. It is localized within the terminal cisternae of the SR in both cardiac and skeletal muscles. CSQ exists in two isoforms. CSQ1 is mainly expressed in skeletal muscles, whereas CSQ2 is expressed in cardiac and slow-twitch muscle (Murphy et al., 2009).
Increased levels of CSQ and calmodulin (CaM) have been reported in spared extraocular and intrinsic laryngeal muscles of mdx mice, whereas these proteins are at low levels in the diaphragm of mdx mice (Pertille et al., 2010). The same study also reported that CSQ level was significantly reduced in the soleus and sternomastoid muscles but unaltered in the tibialis anterior (TA) muscles, whereas CaM level decreased only in the TA muscle (Pertille et al., 2010). Collectively, the fiber-type composition of dystrophic and spared muscles in mdx mice could contribute to the altered levels of CSQ. There is a discrepancy in the protein levels of cardiac CSQ2 in the dystrophic myocardium. It has been shown that the levels of CSQ2 were drastically reduced in the heart of mdx mice (Lohan and Ohlendieck, 2004). However, we and others have shown that CSQ2 levels are unaltered in the hearts of mdx and mdx:utr−/− mice (Pertille et al., 2010; Voit et al., 2017; Mareedu et al., 2021). Surprisingly, CSQ2 expression is aberrantly increased in the fast-twitch muscles of mdx and mdx:utr−/− mice (Schneider et al., 2013; Voit et al., 2017). The exact reason for the increase of CSQ2 is not known but may likely be due to the fast- to slow-muscle fiber type switch in mouse models of DMD. The CLPs are significantly reduced in mdx skeletal muscle (Culligan et al., 2002). The expression of HRCBP is increased in the mdx heart (Lohan and Ohlendieck, 2004). The SLM expression is reduced in both skeletal and cardiac muscles in mdx mice (Lohan and Ohlendieck, 2004). Overall, changes in the SR luminal Ca2+-binding proteins support the concept of abnormal cycling in dystrophic muscles. The differences in the expression levels of these proteins could reflect the severity of the muscle pathology, which varies between disease-free extraocular muscle, slow- and fast-twitch skeletal muscles, and cardiac muscles.
Although the role of Ca2+ dysregulation has been extensively studied in dystrophin-deficient cardiac and skeletal muscle tissues, the implications of dystrophin deficiency in smooth muscles in DMD patients or animal models have not been adequately studied. Studies on the gastric and intestinal smooth muscles of mdx mice have shown impaired muscle relaxation, which has been attributed to impairment in homeostasis (Mule et al., 1999; Mule and Serio, 2001). These studies show that in dystrophic colonic muscle, increased Ca2+ influx through L-type voltage-dependent channels is responsible for the sustained mechanical tone. A recent study showed that the enhanced stretch-induced concentration in the vascular smooth muscle cells of mdx mice occurs through the activation of TRPC1, TRPC3, and TRPC6 channels (Lopez et al., 2020). Thus, focusing on the Ca2+ handling in dystrophic smooth muscle cells may have therapeutic implications.
Mitochondria not only play a vital role in muscle bioenergetics but also contribute to Ca2+ homeostasis in striated muscles. Mitochondria store Ca2+ transiently (~10 nmol/mg of mitochondrial protein) (Finkel et al., 2015). Ca2+ levels in the mitochondria are an important determinant of mitochondrial function and cell survival (Williams et al., 2013; Finkel et al., 2015). When cytoplasmic Ca2+ levels are elevated, mitochondrial Ca2+ uptake is enhanced (Robert et al., 2001) as a compensatory mechanism to normalize the cytoplasmic Ca2+ concentration. Ca2+ uptake by mitochondria mainly occurs via the mitochondrial Ca2+ uniporter (MCU), whereas Ca2+ extrusion occurs through the mitochondrial Na+-Ca2+-Li+ exchanger (mtNCLX) (Pathak and Trebak, 2018). During conditions such as mitochondrial Ca2+ overload and mitochondrial dysfunction, the mitochondrial permeability transition pore (mPTP), a large channel in the inner mitochondrial membrane, also contributes to Ca2+ extrusion (Kwong and Molkentin, 2015). Here, we discuss the interplay between abnormal cytosolic and mitochondrial Ca2+ cycling and muscle pathogenesis in DMD (Figure 3).
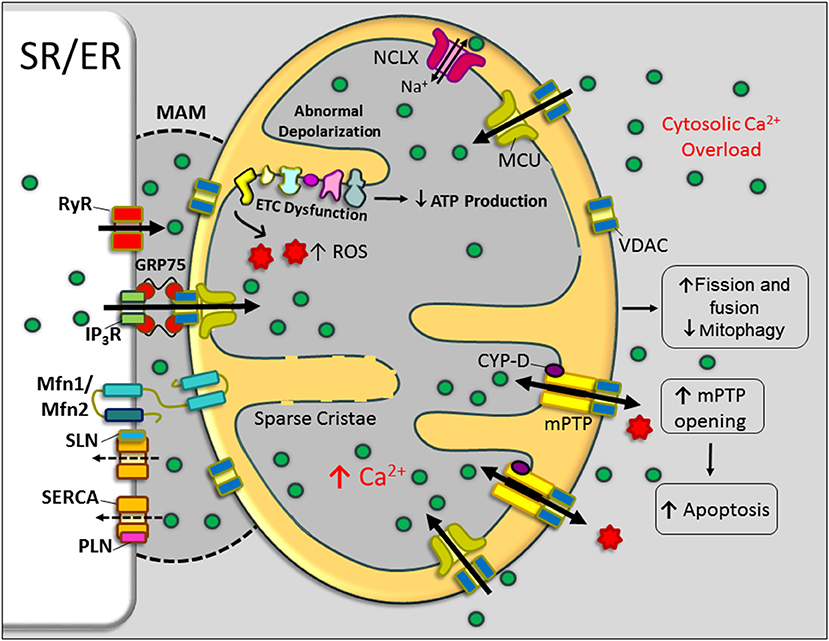
Figure 3. Mitochondrial dysfunction in DMD. Schematic representation of mitochondrial structural and functional alterations in dystrophin-deficient cardiac/skeletal muscle cells. An abnormal elevation of Ca2+ in the cytoplasm and mitochondria-associated membrane (MAM) region resulted in increased mitochondrial Ca2+ uptake and enhanced activation of mitochondrial permeability transition pore (mPTP) opening. These changes impair mitochondrial function and mitochondrial dynamics and contributing to the metabolic crisis. Currently, there is no experimental evidence for the role of the MAM region and mitochondrial Ca2+ uniporter (MCU) in the mitochondrial Ca2+ overload in DMD. Bold and broken arrows indicate the enhanced and decreased function of the Ca2+ channels, respectively. ER, endoplasmic reticulum; ETC, electron transport chain; GRP75, glucose-regulated protein 75; IP3R, inositol trisphosphate receptor; MFN, mitofusin; NCLX, Na+-Ca2+-Li+ exchanger; PLN, phospholamban; ROS, reactive oxygen species; RyR, ryanodine receptor; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase; SLN, sarcolipin; SR, sarcoplasmic reticulum; VDAC, voltage-dependent anion channel.
Mitochondrial morphology change is a salient feature of dystrophic muscle. Specifically, the mitochondrial size is markedly reduced, the electron density is decreased, and the cristae become more sparse in dystrophic muscle (Pant et al., 2015; Kang et al., 2018; Moore et al., 2020). These changes are caused by increased mitochondrial fission and fusion cycles, referred to as mitochondrial dynamics, in dystrophic skeletal muscles (Pant et al., 2015). In the mdx heart, there is substantial structural damage to mitochondria and a significant reduction in ATP production. These changes are associated with decreased mitochondrial autophagy (mitophagy) due to decreased expression of proteins involved in the PINK1/PARKIN mitophagy pathway (Kang et al., 2018). These studies indicate that increased mitochondrial dynamics and decreased mitophagy could contribute to mitochondrial dysfunction and disease pathogenesis. It is important to note that mitochondrial abnormalities are reported in the mdx heart. These changes are associated with mitochondrial Ca2+ load and are preceded by cytosolic Ca2+ overload (Jung et al., 2008; Viola et al., 2013; Kyrychenko et al., 2015b). In support of this notion, our recent studies demonstrated that normalizing the cycling improved the mitochondrial fission and fusion cycles in dystrophic myoblasts (Niranjan et al., 2019).
Several studies have shown that mitochondria are potential targets of impaired Ca2+ homeostasis in muscular dystrophy. Increased mitochondrial Ca2+ content, impaired oxidative phosphorylation, increased ROS generation, decreased ATP production, aberrant mPTP opening, and abnormal mitochondrial depolarization have all been reported in cardiac and skeletal muscles of mouse models of DMD (Kuznetsov et al., 1998; Jung et al., 2008; Rybalka et al., 2014; Kyrychenko et al., 2015b; Kang et al., 2018). Similar to animal models, decreased oxidative phosphorylation has been reported in DMD patients (Sperl et al., 1997). Thus, cytosolic Ca2+ overload appears to directly contribute to mitochondrial dysfunction. In addition, mitochondrial dysfunction resulted in decreased ATP production, loss of membrane potential, and mPTP opening, which may further aggravate cytosolic Ca2+ overload in DMD muscle. In support of this, a recent study using the Caenorhabditis elegans as a model system showed that muscle damage in DMD could be a result of mitochondrial dysfunction, leading to cytoplasmic Ca2+ overload, which in turn activates various matrix metalloproteinase–mediated collagen degradation (Sudevan et al., 2019). Mitochondrial Ca2+ levels are elevated in aged dystrophic cardiomyocytes and are associated with the excessive opening of mPTP and loss of mitochondrial membrane potential (Kyrychenko et al., 2015b). Studies from Molkentin's laboratory have suggested that altered Ca2+ handling within the dystrophic muscle cells initiates cell necrosis, in part through the triggering of mPTP opening (Millay et al., 2008). These studies further showed treatment with the cyclophilin inhibitor Debio-025, which inhibits mPTP opening reduced mitochondrial swelling and necrosis in mdx mice (Millay et al., 2008). These studies suggest that inhibition of mPTP opening could be beneficial in DMD.
Emerging studies have revealed the presence of close contact between the ER and mitochondria called mitochondria-associated membranes (MAMs) (Figure 3). MAMs are specialized subdomains of the ER/SR and outer mitochondrial membrane (Garcia-Perez et al., 2011; Patergnani et al., 2011; Csordas et al., 2018). MAMs are critical for correct communication between the ER and mitochondria, particularly, the selective transmission of physiological and pathological Ca2+ signals from the ER to mitochondria (Patergnani et al., 2011). MAMs are enriched with voltage-dependent anion channels (VDACs), outer mitochondrial membrane proteins that aid in Ca2+ entry close to the junctional SR. In addition to VDAC, glucose-regulated protein 75 (molecular chaperone present on the ER membrane), mitofusin 2 (MFN2), and MFN1 are also involved in the physical interactions between ER/SR and mitochondria (Szabadkai et al., 2006; de Brito and Scorrano, 2008). MAMs also contain proteins involved in ER-associated lipid metabolism, Ca2+-handling proteins, and mitochondrial fission and fusion proteins (Szabadkai et al., 2006; de Brito and Scorrano, 2008; Flis and Daum, 2013; Vance, 2015).
The tight tether between ER/SR and mitochondrial membranes allows Ca2+ to be rapidly transferred. This design overcomes the low apparent Ca2+ affinity (Kd ~ 15–20 M) of the MCU (Rizzuto et al., 1998; Filippin et al., 2003). Many ER/SR-associated Ca2+-handling proteins, such as SERCA, inositol 1,4,5-trisphosphate receptor type 2 (IP3R2), RyR2, SERCA2, CSQ, and PLN, have been found enriched at MAMs, supporting the close correlation between this organellar intersection and Ca2+ regulation (Simpson and Russell, 1997; Simpson et al., 1997; Garcia-Perez et al., 2008; Patergnani et al., 2011; Chen et al., 2012; Raturi and Simmen, 2013). A recent study shows that pharmacological blockade or down-regulation of IP3R expression can restore mitochondrial Ca2+ levels, mitochondrial membrane potential, mitochondrial dynamics, and mitophagy in mdx muscles (Valladares et al., 2018). Another study showed that incubation of Sol8 myotubes expressing mini-dystrophin in cyclosporine A (CsA) normalized IP3R expression levels and aided in better cell survival (Mondin et al., 2009). These studies suggest that IP3R-associated Ca2+ dysregulation in the MAM region affects mitochondrial Ca2+ content and mitochondrial function in DMD. These findings lead to the speculation that altered expression levels of RyR, IP3R, and SERCA isoforms and their regulators in the MAM region could deeply affect the signaling in this microdomain and thereby the cell metabolism and cell survival in DMD (Figure 3). A more detailed examination of MAM proteins in dystrophic muscle is needed to understand the mechanisms associated with Ca2+ mishandling in this microdomain and its contribution to DMD pathogenesis.
In summary, elevated cytoplasmic Ca2+ levels including increased Ca2+ levels in the MAM region increase mitochondrial Ca2+ load, which affects the mitochondrial structure, function, and mPTP opening. This further exacerbates cytosolic Ca2+ overload and subsequently contributes to DMD pathogenesis.
Increasing evidence suggests functional crosstalk between Ca2+ and ROS signaling systems. ROS are generated both in cytosol and mitochondria by NOX and electron complex chain, respectively (Gorlach et al., 2015). ROS produced by NOX5 has been implicated in cardiovascular diseases, renal diseases, and cancer (Touyz et al., 2019). However, studies on the role of NOX isoforms in DMD are limited. In mdx mice, NOX2 was shown to be induced in skeletal muscle, whereas NOX4 was induced in the heart (Spurney et al., 2008). Nifedipine treatment, which reduces resting concentrations, is associated with reduced expression of gp91phox/p47phox NOX2 subunits (Altamirano et al., 2013). The genetic down-regulation of NOX2 activity reduces Mn2+/Ca2+ influx across the dystrophin-deficient sarcolemma and restores autophagy and reduces muscle pathology in mdx mice (Pal et al., 2014; Loehr et al., 2016). These studies indicate the involvement of NOX2-driven oxidative stress in increased sarcolemmal Ca2+ influx in dystrophic muscles. NOX2-driven oxidative stress was also shown to be involved in dystrophic cardiomyopathy (Shirokova and Niggli, 2013). The expression of NOX2 is increased in the mdx heart along with increased superoxide production (Gonzalez et al., 2014). Furthermore, NOX2 inhibition by apocynin restored the Ca2+-handling properties, including the amplitude of Ca2+ transient and SR Ca2+ content in the dystrophic myocytes. In addition, NOX2 inhibition decreases the SR Ca2+ leak in mdx myocytes (Gonzalez et al., 2014). On the other hand, genetic inhibition of NOX2 production (by deleting the p47phox, a NOX2 regulatory subunit) failed to decrease RyR1 Ca2+ leak in mdx muscle fibers. These studies further show that NOX4 is activated in the NOX2 ROS-deficient mdx muscle and contributes to RyR1 leak (Cully and Rodney, 2020). Based on these findings, the authors proposed ROS-Ca2+ crosstalk, in which NOX4-ROS induces RyR1 Ca2+ leak and thereby increases the SR/T-tubular junctional Ca2+ concentration, which exacerbates NOX2 ROS and contributes to muscle pathology. A recent study shows the down-regulation of miR-448-3p in the mdx heart increased Ncf1 expression, which encodes p47phox (Kyrychenko et al., 2015a). These studies suggest the involvement of NOX2 in oxidative stress and enhanced Ca2+ signaling in dystrophic myocardium. In dystrophic myotubes, NOX inhibition abolished iPLA2 activity and reduced Ca2+ influx through stretch-activated and store-operated channels (Ismail et al., 2014). In addition, NOX inhibition in mdx muscle restored the force loss induced by eccentric contractions and reduced membrane damage (Ismail et al., 2014). Thus, NOX are appealing targets in developing new therapies for muscular dystrophy.
In light of the significant contribution of Ca2+ mishandling in DMD pathogenesis, strategies targeting Ca2+ handling may hold great promise to treat DMD. Preclinical studies in animal models have provided compelling evidence supporting these therapeutic modalities (Table 1).
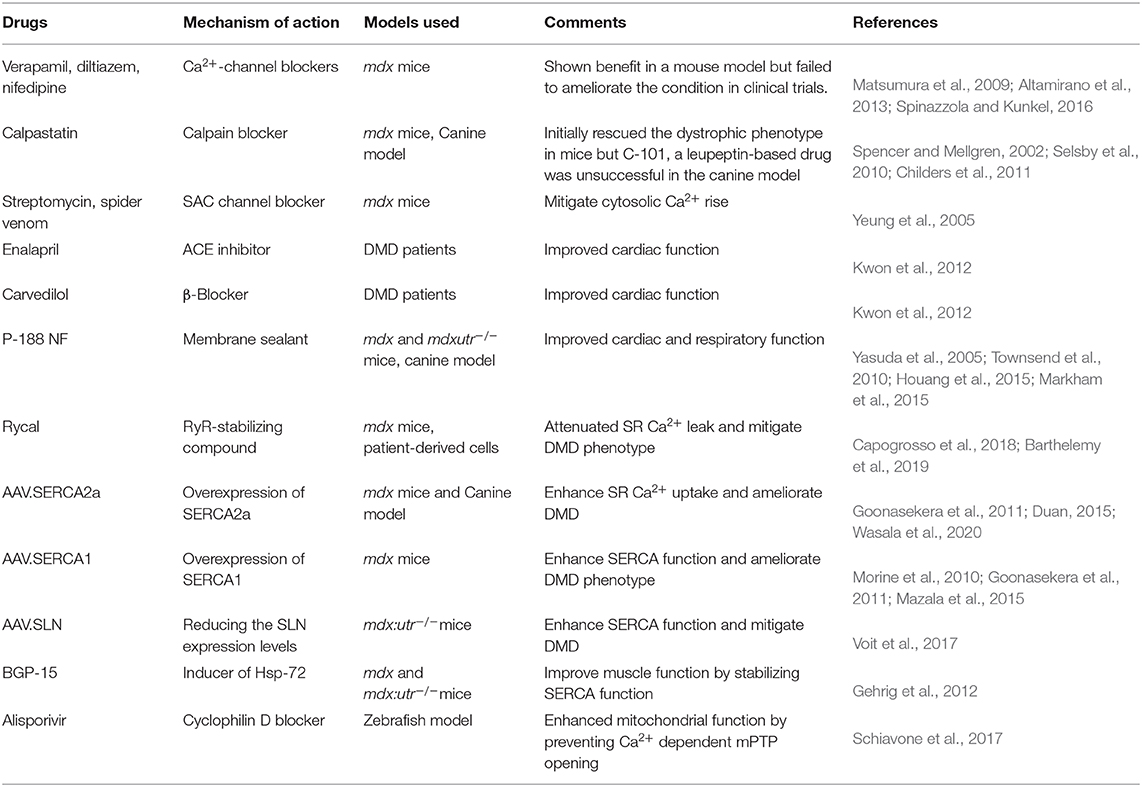
Table 1. Therapies targeting dysregulated Ca2+ directly or indirectly.
Calcium accumulation has been demonstrated in skeletal muscle biopsies from fetal human DMD (Farini et al., 2016), suggesting Ca2+ abnormalities occur at the early onset of the disease. To determine the optimal time point for Ca2+ channel blocking to prevent the pathological onset of the disease, Jorgensen et al. treated mdx mice in utero with streptomycin, a non-specific Ca2+-channel blocker that inhibits stretch-activated and mechanosensitive ion channels (Jorgensen et al., 2011). Streptomycin treatment delayed the onset of dystrophic symptoms in the limb muscles in young mdx mice but did not prevent disease progression. In mdx mice, long-term treatment with streptomycin reduced limb-muscle pathology but worsened the diaphragm and cardiac pathology (Jorgensen et al., 2011). These studies argue that blocking Ca2+ channels before disease onset is not beneficial. On the other hand, streptomycin treatment after birth reduced creatine kinase activity in the diaphragm and sternomastoid muscles of mdx mice (Matsumura et al., 2011). These studies suggest that the differential effects of streptomycin in various dystrophic muscles may be associated with the differential expression of TRPC1 (Matsumura et al., 2011). Thus, care must be taken in the long-term clinical use of non-specific Ca2+-channel blockers in DMD patients.
Membrane leaking has been suggested to contribute to cytosolic Ca2+ overload in dystrophic muscle. To address this issue, membrane sealants such as Poloxamer 188 and its derivatives have been developed to stabilize the sarcolemma. Preclinical studies in the murine and canine DMD model suggest that membrane sealants can improve cardiac function (Yasuda et al., 2005; Townsend et al., 2010; Houang et al., 2015; Markham et al., 2015). However, two studies failed to show protection in mdx mouse skeletal muscle (Quinlan et al., 2006; Terry et al., 2014). More studies are warranted to validate the therapeutic potency of P188 in preventing Ca2+ entry and ameliorating DMD.
Calcium can enter dystrophic muscle cells through the L-type Ca2+ channel. L-type Ca2+-channel blockers, such as diltiazem and verapamil, have been shown to reduce serum creatine levels and muscle necrosis in mdx mice (Matsumura et al., 2009). Similarly, nifedipine treatment restored cytosolic Ca2+ levels and improved muscle function in mouse models of DMD (Altamirano et al., 2013). Although these observations signify the therapeutic benefits of L-type Ca2+-channel blockers in animal models, several clinical trials have revealed no clinical benefit in DMD patients (Spinazzola and Kunkel, 2016). This could be due to many reasons, for example, differences in drug metabolisms and variation in drug dosing. Future studies are warranted to clarify the mechanisms underlying different outcomes in animal models and DMD patients and to develop creative strategies to improve the efficacy in human patients.
Targeting stretch-activated Ca2+ channels with streptomycin and spider venom toxin has been shown to attenuate rise and muscle damage in mdx mice (Yeung et al., 2005). Pharmacological inhibition of TRPC1 and TRPC3 channels has been shown to reduce the Ca2+ entry into dystrophic skeletal myofibers and also improve their function (Vandebrouck et al., 2002). In mdx mice, overexpression of a dominant-negative mutant of the TRPV2 ion channel protects eccentric contraction-induced damage (Zanou et al., 2009). Thus, inhibiting TRP channels is a potential therapeutic strategy to prevent Ca2+ entry.
Angiotensin II or β-receptor–mediated signaling pathways, such as G-protein–coupled receptor pathways, modulate muscle contraction–relaxation via altered handling. β2-adrenergic receptor–coupled pathway has been shown to attenuate skeletal muscle degeneration in DMD (Smith et al., 2002; Church et al., 2014; Silva et al., 2014). Treating albuterol, a β2 agonist, increased muscle strength in animal models but failed to improve muscle function in DMD patients (Skura et al., 2008). On the other hand, treating DMD patients with enalapril, an angiotensin-converting enzyme (ACE) inhibitor, or carvedilol, a β-blocker, improved the left ventricular systolic function without significant adverse effects in one patient study (Kwon et al., 2012).
SR Ca2+ leak is a significant contributor to cytosolic Ca2+ overload in DMD. Rycal, a RyR-stabilizing compound, was found to attenuate muscle disease in mdx mice (Bellinger et al., 2009; Fauconnier et al., 2010; Capogrosso et al., 2018). Rycal is suggested to improve binding of calstabin to RyR and to prevent SR Ca2+ leak, thereby restoring homeostasis (Capogrosso et al., 2018). A recent study showed that Rycal treatment bolsters antisense oligonucleotide-mediated exon skipping in patient-derived myotubes and induced pluripotent stem cell–derived diseased cardiomyocytes (Barthelemy et al., 2019). Although the therapeutic value of Rycal needs to be further validated, these studies suggest that targeting the RyR will boost treatment efficiency in DMD patients.
Enhancing cytosolic Ca2+ removal is another powerful approach to restore the homeostasis in dystrophic muscle. This can be achieved by overexpressing SERCA or by targeting its inhibitors to enhance SERCA activity. Several animal studies have shown the beneficial effects of SERCA overexpression in ameliorating muscular dystrophy in mice (Morine et al., 2010; Goonasekera et al., 2011; Shin et al., 2011; Mazala et al., 2015; Wasala et al., 2020). Transgenic overexpression of SERCA1 in skeletal muscles of mdx mice has been shown to enhance the EC coupling, improve Ca2+ removal from the cytosol, and attenuate dystrophic phenotypes such as fibrosis and increased serum creatine kinase levels (Goonasekera et al., 2011). Similarly, transgenic overexpression of SERCA1 is shown to ameliorate the structural damage and functional impairments of muscles from mdx:utr−/− mice (Mazala et al., 2015). Adeno-associated virus (AAV)-mediated overexpression of SERCA1 in the diaphragm of mdx mice increased the proportion of type IIA fibers, reduced the percentage of centrally nucleated fibers, and attenuated the loss of force production following eccentric contractions (Morine et al., 2010). In contrast to SERCA1, SERCA2a is expressed in both skeletal muscle and the heart. We evaluated AAV SERCA2a gene therapy. Delivery to 12-month-old mdx mice significantly improved cardiac electrophysiology (Shin et al., 2011). A single intravenous injection in 3-month-old mdx mice significantly improves whole-body muscle performance and ameliorates fatal dilated cardiomyopathy in mdx mice up to 21 months of age (Wasala et al., 2020). The AAV.SERCA2a vector has been extensively studied in heart failure patients (Zsebo et al., 2014; Lyon et al., 2020). Lessons learned from these studies will help facilitate the translation of AAV.SERCA2a therapy in DMD patients.
Targeting SERCA regulators such as PLN and SLN also has therapeutic implications. Reducing PLN activity is shown to improve Ca2+ handling in animal models of heart failure (Kranias and Hajjar, 2012). Surprisingly, genetic PLN knockout worsened cardiac function in mdx mice (Law et al., 2018). It is yet unclear whether the partial reduction of PLN activity is beneficial in DMD. Unlike PLN, SLN up-regulation is a common molecular signature in all dystrophin-deficient muscles. Recent studies from our laboratory and others have shown that germline ablation of SLN ameliorates severe muscular dystrophy in a mouse model of DMD (Voit et al., 2017; Tanihata et al., 2018; Mareedu et al., 2019). Most strikingly, the loss of one SLN allele extended the lifespan of mdx:utr−/− mice to 20 months. Also, our studies show that the reduction in SLN expression can improve muscle regeneration and prevent fiber-type transition in dystrophic muscles (Voit et al., 2017). Furthermore, AAV-mediated SLN reduction normalized cycling and improved fusion and differentiation of dystrophin-deficient dog myoblasts (Niranjan et al., 2019). Our recent studies have demonstrated that reducing SLN expression is sufficient to restore cardiac SERCA function and cycling and to prevent the development of cardiomyopathy in mdx mice throughout their lifespan (Mareedu et al., 2021). To translate these findings into a therapeutic strategy, we also knocked down SLN expression in 1-month-old mdx:utr−/− mice via AAV-mediated RNA interference (Voit et al., 2017). This AAV treatment markedly reduces SLN expression, attenuates muscle pathology, and improves diaphragm, skeletal muscle, and cardiac function (Voit et al., 2017). In summary, these studies suggest that targeting SLN expression or function is a promising therapeutic strategy to improve SERCA function and ameliorate skeletal muscle disease and cardiomyopathy in DMD.
The therapeutic potential of targeting DWORF has been explored in animal models of heart failure. Cardiac-specific overexpression of DWORF enhances SERCA function, Ca2+ cycling, and contractility in mice (Nelson et al., 2016). In addition, overexpression of DWORF in the heart improves Ca2+ cycling, prevents pathological remodeling, and improves cardiac function in the muscle-specific LIM protein (MLP) knockout mouse model of dilated cardiomyopathy (Makarewich et al., 2018). A recent study shows that AAV-mediated overexpression of DWORF improved cardiac function in MLP knockout mice, as well as in the myocardial infarction model of heart failure (Makarewich et al., 2020). The expression levels and function of DWORF in dystrophic cardiac and skeletal muscle are yet to be studied. Nevertheless, based on the current findings, DWORF overexpression could be a therapeutic strategy to improve SERCA function and ameliorate muscular dystrophy and cardiomyopathy in DMD.
In addition to these strategies, improving SERCA function through other mechanisms has been shown to ameliorate muscular dystrophy in mice. Transgenic overexpression of heat shock protein 72 (Hsp72) or pharmacological induction of Hsp72 improves several pathological indices in mdx and mdx:utr−/− mice, partly through the enhancement of SERCA function. These studies show that Hsp72 stabilizes the SERCA pump and increases its activity in dystrophic muscles (Gehrig et al., 2012). A recent study shows that blocking the insulin-like growth factor 2 receptor activates SERCA function and enhances the removal in muscles of mdx mice (Bella et al., 2020).
Improving mitochondrial function is another attractive therapeutic strategy for DMD. As discussed previously, Ca2+-dependent mitochondrial dysfunction could be due to the opening of mPTP (Briston et al., 2019). Thus, inhibiting cyclophilin, a key regulator of mPTP, could prevent mPTP opening and alleviate mitochondrial dysfunction in DMD. Although CsA treatment failed to improve muscle function in DMD patients (Kirschner et al., 2010), treatment with another cyclophilin inhibitor, Debio 025, which inhibits mPTP opening, has been shown to partially rescue the dystrophic phenotype in mdx mice (Reutenauer et al., 2008). Similarly, alisporivir, a derivative of CsA, has been shown to enhance the mitochondrial function in the zebrafish model of DMD (Schiavone et al., 2017). Further exploitation of these compounds and the identification of new compounds that prevent Ca2+-dependent mPTP opening provide an attractive therapeutic strategy to improve mitochondrial function and prevent muscle necrosis in DMD.
Calpain activation leads to proteolytic damage in muscle cells. Overexpression of calpastatin, an endogenous inhibitor of calpain, significantly reduced muscle necrosis in mdx mice (Spencer and Mellgren, 2002). On the contrary, the use of C101, a leupeptin-based calpain inhibitor, failed to rescue phenotypes of DMD in animal models (Selsby et al., 2010; Childers et al., 2011). There is no direct study on the PLA2 inhibition in preventing muscle damage in DMD. However, corticosteroids, in clinical practice, suppress muscle inflammation possibly via PLA2 inhibition (Hoxha, 2019).
Over the last several decades, there has been significant growth in our knowledge of the Ca2+ mishandling and Ca2+ homeostasis dysregulation in DMD. These findings have resulted in several highly promising and novel experimental therapies such as the use of the RyR stabilizer RyCal, SLN silencing, and viral vector–mediated SERCA overexpression. Yet, there are many unanswered questions and conflicting experimental results. dysregulation is not only implicated in DMD but also involved in other muscular dystrophies, such as limb-girdle muscular dystrophy and laminopathies. Further understanding of the molecular mechanisms involved in Ca2+ mishandling may shed light on the secondary disease-causing mechanisms in various forms of muscular dystrophies. This understanding will help identify new therapeutic targets for the treatment of these diseases. Based on current knowledge, future studies may be directed to the following areas for a better understanding of targeting Ca2+ dysregulation:
1) Mitochondria: Calcium is a key regulator of mitochondrial function. Mounting evidence indicates that mitochondrial dysfunction plays an important role in disease pathogenesis in DMD. Exploring the molecular mechanisms including the MAM region that link the cytoplasmic Ca2+ load and mitochondrial Ca2+ mishandling may lead to the identification of potential therapeutic targets.
2) SR Ca2+ reuptake: Based on our recent studies, enhancing SERCA2a expression or activity (through reducing SERCA inhibitors) appears to be a promising strategy to treat both cardiac and skeletal muscle defects in DMD. Therefore, exploring new strategies to improve SERCA function, developing new AAV vectors with improved tropism for both muscle and heart, and identifying small molecules that activate endogenous SERCA pumps will be useful.
3) The use of large animal models: Many Ca2+-targeted therapies were performed in mouse models of DMD. These findings need to be verified in large animal models before moving to human trials.
4) The effects of dystrophin restoration on Ca2+ handling: It is unknown whether partial dystrophin restoration alleviates Ca2+-handling defects in dystrophic muscles. This information is not available and needs to be tested. This will help to design a combination therapy of dystrophin restoration with improved Ca2+ handling.
DD is a member of the scientific advisory board for Solid Biosciences and equity holders of Solid Biosciences. The Duan laboratory has received research supports unrelated to this project from Solid Biosciences and Edgewise Therapeutics in the past 3 years. SM, EM, and GB have no conflicts of interest, financial or otherwise. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
All authors discussed, wrote, and commented on the manuscript at all stages.
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, US National Institutes of Health (NIH) grant (Grant #1R01AR069107 to GB, Grant # 1R01AR070517 to DD and GB), Jesse's Journey: The Foundation for Gene and Cell Therapy (to DD and GB), and Jackson Freel DMD Research Fund (to DD). EM was supported by the University of Missouri Life Science Fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aartsma-Rus, A., Ginjaar, I. B., and Bushby, K. (2016). The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 53, 145–151. doi: 10.1136/jmedgenet-2015-103387
Alderton, J. M., and Steinhardt, R. A. (2000). Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J. Biol. Chem. 275, 9452–9460. doi: 10.1074/jbc.275.13.9452
Allen, D. G., and Whitehead, N. P. (2011). Duchenne muscular dystrophy–what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 43, 290–294. doi: 10.1016/j.biocel.2010.11.005
Allen, D. G., Whitehead, N. P., and Froehner, S. C. (2016). Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 96, 253–305. doi: 10.1152/physrev.00007.2015
Altamirano, F., Valladares, D., Henriquez-Olguin, C., Casas, M., Lopez, J. R., Allen, P. D., et al. (2013). Nifedipine treatment reduces resting calcium concentration, oxidative and apoptotic gene expression, and improves muscle function in dystrophic mdx mice. PLoS ONE 8:e81222. doi: 10.1371/journal.pone.0081222
Anderson, D. M., Anderson, K. M., Chang, C. L., Makarewich, C. A., Nelson, B. R., McAnally, J. R., et al. (2015). A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160, 595–606. doi: 10.1016/j.cell.2015.01.009
Andrews, N. W., Almeida, P. E., and Corrotte, M. (2014). Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 24, 734–742. doi: 10.1016/j.tcb.2014.07.008
Arvanitis, D. A., Vafiadaki, E., Sanoudou, D., and Kranias, E. G. (2011). Histidine-rich calcium binding protein: the new regulator of sarcoplasmic reticulum calcium cycling. J. Mol. Cell. Cardiol. 50, 43–49. doi: 10.1016/j.yjmcc.2010.08.021
Avila, G., de la Rosa, J. A., Monsalvo-Villegas, A., and Montiel-Jaen, M. G. (2019). Ca(2+) channels mediate bidirectional signaling between sarcolemma and sarcoplasmic reticulum in muscle cells. Cells 9:55. doi: 10.3390/cells9010055
Avila-Medina, J., Mayoral-Gonzalez, I., Dominguez-Rodriguez, A., Gallardo-Castillo, I., Ribas, J., Ordonez, A., et al. (2018). The complex role of store operated calcium entry pathways and related proteins in the function of cardiac, skeletal and vascular smooth muscle cells. Front. Physiol. 9:257. doi: 10.3389/fphys.2018.00257
Babu, G. J., Bhupathy, P., Carnes, C. A., Billman, G. E., and Periasamy, M. (2007). Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J. Mol. Cell. Cardiol. 43, 215–222. doi: 10.1016/j.yjmcc.2007.05.009
Barthelemy, F., Wang, R. T., Hsu, C., Douine, E. D., Marcantonio, E. E., Nelson, S. F., et al. (2019). Targeting RyR activity boosts antisense exon 44 and 45 skipping in human DMD skeletal or cardiac muscle culture models. Mol. Ther. Nucleic Acids 18, 580–589. doi: 10.1016/j.omtn.2019.09.020
Bartoli, M., and Richard, I. (2005). Calpains in muscle wasting. Int. J. Biochem. Cell Biol. 37, 2115–2133. doi: 10.1016/j.biocel.2004.12.012
Beard, N. A., Laver, D. R., and Dulhunty, A. F. (2004). Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 85, 33–69. doi: 10.1016/j.pbiomolbio.2003.07.001
Bella, P., Farini, A., Banfi, S., Parolini, D., Tonna, N., Meregalli, M., et al. (2020). Blockade of IGF2R improves muscle regeneration and ameliorates Duchenne muscular dystrophy. EMBO Mol. Med. 12:e11019. doi: 10.15252/emmm.201911019
Bellinger, A. M., Reiken, S., Carlson, C., Mongillo, M., Liu, X., Rothman, L., et al. (2009). Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330. doi: 10.1038/nm.1916
Bhattacharya, S. K., Palmieri, G. M., Bertorini, T. E., and Nutting, D. F. (1982). The effects of diltiazem in dystrophic hamsters. Muscle Nerve 5, 73–78. doi: 10.1002/mus.880050114
Bhupathy, P., Babu, G. J., and Periasamy, M. (2007). Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J. Mol. Cell. Cardiol. 42, 903–911. doi: 10.1016/j.yjmcc.2007.03.738
Boittin, F. X., Shapovalov, G., Hirn, C., and Ruegg, U. T. (2010). Phospholipase A2-derived lysophosphatidylcholine triggers Ca2+ entry in dystrophic skeletal muscle fibers. Biochem. Biophys. Res. Commun. 391, 401–406. doi: 10.1016/j.bbrc.2009.11.070
Briston, T., Selwood, D. L., Szabadkai, G., and Duchen, M. R. (2019). Mitochondrial permeability transition: a molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 40, 50–70. doi: 10.1016/j.tips.2018.11.004
Burdi, R., Didonna, M. P., Pignol, B., Nico, B., Mangieri, D., Rolland, J. F., et al. (2006). First evaluation of the potential effectiveness in muscular dystrophy of a novel chimeric compound, BN 82270, acting as calpain-inhibitor and anti-oxidant. Neuromuscul. Disord. 16, 237–248. doi: 10.1016/j.nmd.2006.01.013
Burr, A. R., Millay, D. P., Goonasekera, S. A., Park, K. H., Sargent, M. A., Collins, J., et al. (2014). Na+ dysregulation coupled with Ca2+ entry through NCX1 promotes muscular dystrophy in mice. Mol. Cell. Biol. 34, 1991–2002. doi: 10.1128/MCB.00339-14
Burr, A. R., and Molkentin, J. D. (2015). Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 22, 1402–1412. doi: 10.1038/cdd.2015.65
Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L., et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93. doi: 10.1016/S1474-4422(09)70271-6
Calderon, J. C., Bolanos, P., and Caputo, C. (2014). The excitation-contraction coupling mechanism in skeletal muscle. Biophys. Rev. 6, 133–160. doi: 10.1007/s12551-013-0135-x
Capogrosso, R. F., Mantuano, P., Uaesoontrachoon, K., Cozzoli, A., Giustino, A., Dow, T., et al. (2018). Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: proof-of-concept study and independent validation of efficacy. FASEB J. 32, 1025–1043. doi: 10.1096/fj.201700182RRR
Capote, J., DiFranco, M., and Vergara, J. L. (2010). Excitation-contraction coupling alterations in mdx and utrophin/dystrophin double knockout mice: a comparative study. Am. J. Physiol. Cell Physiol. 298, C1077–C1086. doi: 10.1152/ajpcell.00428.2009
Cea, L. A., Puebla, C., Cisterna, B. A., Escamilla, R., Vargas, A. A., Frank, M., et al. (2016). Fast skeletal myofibers of mdx mouse, model of Duchenne muscular dystrophy, express connexin hemichannels that lead to apoptosis. Cell. Mol. Life Sci. 73, 2583–2599. doi: 10.1007/s00018-016-2132-2
Cea, L. A., Riquelme, M. A., Cisterna, B. A., Puebla, C., Vega, J. L., Rovegno, M., et al. (2012). Connexin- and pannexin-based channels in normal skeletal muscles and their possible role in muscle atrophy. J. Membr. Biol. 245, 423–436. doi: 10.1007/s00232-012-9485-8
Chakkalakal, J. V., Thompson, J., Parks, R. J., and Jasmin, B. J. (2005). Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB J. 19, 880–891. doi: 10.1096/fj.04-1956rev
Chan, S., and Head, S. I. (2011). The role of branched fibres in the pathogenesis of Duchenne muscular dystrophy. Exp. Physiol. 96, 564–571. doi: 10.1113/expphysiol.2010.056713
Chen, Y., Csordas, G., Jowdy, C., Schneider, T. G., Csordas, N., Wang, W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 111, 863–875. doi: 10.1161/CIRCRESAHA.112.266585
Cheng, X., Zhang, X., Gao, Q., Ali Samie, M., Azar, M., Tsang, W. L., et al. (2014). The intracellular Ca(2)(+) channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 20, 1187–1192. doi: 10.1038/nm.3611
Childers, M. K., Bogan, J. R., Bogan, D. J., Greiner, H., Holder, M., Grange, R. W., et al. (2011). Chronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of duchenne muscular dystrophy. Front. Pharmacol. 2:89. doi: 10.3389/fphar.2011.00089
Church, J. E., Trieu, J., Sheorey, R., Chee, A. Y., Naim, T., Baum, D. M., et al. (2014). Functional beta-adrenoceptors are important for early muscle regeneration in mice through effects on myoblast proliferation and differentiation. PLoS ONE 9:e101379. doi: 10.1371/journal.pone.0101379
Collet, C., Allard, B., Tourneur, Y., and Jacquemond, V. (1999). Intracellular calcium signals measured with indo-1 in isolated skeletal muscle fibres from control and mdx mice. J. Physiol. 520 (Pt 2), 417–429.
Connolly, A. M., Florence, J. M., Cradock, M. M., Malkus, E. C., Schierbecker, J. R., Siener, C. A., et al. (2013). Motor and cognitive assessment of infants and young boys with Duchenne Muscular Dystrophy: results from the Muscular Dystrophy Association DMD Clinical Research Network. Neuromuscul. Disord. 23, 529–539. doi: 10.1016/j.nmd.2013.04.005
Conti, A., Gorza, L., and Sorrentino, V. (1996). Differential distribution of ryanodine receptor type 3 (RyR3) gene product in mammalian skeletal muscles. Biochem. J. 316 (Pt 1), 19–23. doi: 10.1042/bj3160019
Cooper, S. T., and Head, S. I. (2015). Membrane injury and repair in the muscular dystrophies. Neuroscientist 21, 653–668. doi: 10.1177/1073858414558336
Csordas, G., Weaver, D., and Hajnoczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 28, 523–540. doi: 10.1016/j.tcb.2018.02.009
Culligan, K., Banville, N., Dowling, P., and Ohlendieck, K. (2002). Drastic reduction of calsequestrin-like proteins and impaired calcium binding in dystrophic mdx muscle. J. Appl. Physiol. 92, 435–445. doi: 10.1152/japplphysiol.00903.2001
Cully, T. R., and Rodney, G. G. (2020). Nox4 - RyR1 - Nox2: regulators of micro-domain signaling in skeletal muscle. Redox Biol. 36:101557. doi: 10.1016/j.redox.2020.101557
Danialou, G., Comtois, A. S., Dudley, R., Karpati, G., Vincent, G., Des Rosiers, C., et al. (2001). Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 15, 1655–1657. doi: 10.1096/fj.01-0030fje
Dayton, W. R., Reville, W. J., Goll, D. E., and Stromer, M. H. (1976). A Ca2+-activated protease possibly involved in myofibrillar protein turnover. Partial characterization of the purified enzyme. Biochemistry 15, 2159–2167.
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
Derler, I., Jardin, I., and Romanin, C. (2016). Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 310, C643–C662. doi: 10.1152/ajpcell.00007.2016
Deval, E., Levitsky, D. O., Marchand, E., Cantereau, A., Raymond, G., and Cognard, C. (2002). Na(+)/Ca(2+) exchange in human myotubes: intracellular calcium rises in response to external sodium depletion are enhanced in DMD. Neuromuscul. Disord. 12, 665–673. doi: 10.1016/s0960-8966(02)00022-6
DiFranco, M., Woods, C. E., Capote, J., and Vergara, J. L. (2008). Dystrophic skeletal muscle fibers display alterations at the level of calcium microdomains. Proc. Natl. Acad. Sci. U.S.A. 105, 14698–14703. doi: 10.1073/pnas.0802217105
Divet, A., and Huchet-Cadiou, C. (2002). Sarcoplasmic reticulum function in slow- and fast-twitch skeletal muscles from mdx mice. Pflugers Arch. 444, 634–643. doi: 10.1007/s00424-002-0854-5
Dowling, P., Lohan, J., and Ohlendieck, K. (2003). Comparative analysis of Dp427-deficient mdx tissues shows that the milder dystrophic phenotype of extraocular and toe muscle fibres is associated with a persistent expression of beta-dystroglycan. Eur. J. Cell Biol. 82, 222–230. doi: 10.1078/0171-9335-00315
Duan, D. (2015). Duchenne muscular dystrophy gene therapy in the canine model. Hum. Gene Ther. Clin. Dev. 26, 57–69. doi: 10.1089/humc.2015.006
Duan, D., Goemans, N., Takeda, S. I., Mercuri, E., and Aartsma-Rus, A. (2021). Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 7:13. doi: 10.1038/s41572-021-00248-3
Edwards, J. N., Friedrich, O., Cully, T. R., von Wegner, F., Murphy, R. M., and Launikonis, B. S. (2010). Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am. J. Physiol. Cell Physiol. 299, C42–C50. doi: 10.1152/ajpcell.00524.2009
Eisner, D. A., Caldwell, J. L., Kistamas, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. doi: 10.1161/CIRCRESAHA.117.310230
Endo, M. (2009). Calcium-induced calcium release in skeletal muscle. Physiol. Rev. 89, 1153–1176. doi: 10.1152/physrev.00040.2008
Ervasti, J. M., and Campbell, K. P. (1993). A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809–823.
Farini, A., Sitzia, C., Cassinelli, L., Colleoni, F., Parolini, D., Giovanella, U., et al. (2016). Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling mediates delayed myogenesis in Duchenne muscular dystrophy fetal muscle. Development 143, 658–669. doi: 10.1242/dev.126193
Fauconnier, J., Thireau, J., Reiken, S., Cassan, C., Richard, S., Matecki, S., et al. (2010). Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 107, 1559–1564. doi: 10.1073/pnas.0908540107
Feno, S., Butera, G., Vecellio Reane, D., Rizzuto, R., and Raffaello, A. (2019). Crosstalk between calcium and ROS in pathophysiological conditions. Oxid. Med. Cell. Longev. 2019:9324018. doi: 10.1155/2019/9324018
Ferretti, R., Marques, M. J., Pertille, A., and Santo Neto, H. (2009). Sarcoplasmic-endoplasmic-reticulum Ca2+-ATPase and calsequestrin are overexpressed in spared intrinsic laryngeal muscles of dystrophin-deficient mdx mice. Muscle Nerve 39, 609–615. doi: 10.1002/mus.21154
Filippin, L., Magalhaes, P. J., Di Benedetto, G., Colella, M., and Pozzan, T. (2003). Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 278, 39224–39234. doi: 10.1074/jbc.M302301200
Finkel, T., Menazza, S., Holmstrom, K. M., Parks, R. J., Liu, J., Sun, J., et al. (2015). The ins and outs of mitochondrial calcium. Circ. Res. 116, 1810–1819. doi: 10.1161/CIRCRESAHA.116.305484
Finsterer, J. (2006). Cardiopulmonary support in duchenne muscular dystrophy. Lung 184, 205–215. doi: 10.1007/s00408-005-2584-x
Flis, V. V., and Daum, G. (2013). Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 5:a013235. doi: 10.1101/cshperspect.a013235
Franzini-Armstrong, C. (2018). The relationship between form and function throughout the history of excitation-contraction coupling. J. Gen. Physiol. 150, 189–210. doi: 10.1085/jgp.201711889
Friedrich, O., Both, M., Gillis, J. M., Chamberlain, J. S., and Fink, R. H. (2004). Mini-dystrophin restores L-type calcium currents in skeletal muscle of transgenic mdx mice. J. Physiol. 555(Pt 1), 251–265. doi: 10.1113/jphysiol.2003.054213
Friedrich, O., von Wegner, F., Chamberlain, J. S., Fink, R. H., and Rohrbach, P. (2008). L-type Ca2+ channel function is linked to dystrophin expression in mammalian muscle. PLoS ONE 3:e1762. doi: 10.1371/journal.pone.0001762
Gailly, P. (2002). New aspects of calcium signaling in skeletal muscle cells: implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta 1600, 38–44. doi: 10.1016/s1570-9639(02)00442-9
Gao, Q. Q., and McNally, E. M. (2015). The dystrophin complex: structure, function, and implications for therapy. Compr. Physiol. 5, 1223–1239. doi: 10.1002/cphy.c140048
Garcia-Perez, C., Hajnoczky, G., and Csordas, G. (2008). Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J. Biol. Chem. 283, 32771–32780. doi: 10.1074/jbc.M803385200
Garcia-Perez, C., Schneider, T. G., Hajnoczky, G., and Csordas, G. (2011). Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am. J. Physiol. Heart Circ. Physiol. 301, H1907–H1915. doi: 10.1152/ajpheart.00397.2011
Gehlert, S., Bloch, W., and Suhr, F. (2015). Ca2+-dependent regulations and signaling in skeletal muscle: from electro-mechanical coupling to adaptation. Int. J. Mol. Sci. 16, 1066–1095. doi: 10.3390/ijms16011066
Gehrig, S. M., van der Poel, C., Sayer, T. A., Schertzer, J. D., Henstridge, D. C., Church, J. E., et al. (2012). Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 484, 394–398. doi: 10.1038/nature10980
Gervasio, O. L., Whitehead, N. P., Yeung, E. W., Phillips, W. D., and Allen, D. G. (2008). TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J. Cell Sci. 121(Pt 13), 2246–2255. doi: 10.1242/jcs.032003
Gillis, J. M. (1996). Membrane abnormalities and Ca homeostasis in muscles of the mdx mouse, an animal model of the Duchenne muscular dystrophy: a review. Acta Physiol. Scand. 156, 397–406. doi: 10.1046/j.1365-201X.1996.201000.x
Gillis, J. M. (1999). Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J. Muscle Res. Cell Motil. 20, 605–625. doi: 10.1023/a:1005545325254
Gonzalez, D. R., Treuer, A. V., Lamirault, G., Mayo, V., Cao, Y., Dulce, R. A., et al. (2014). NADPH oxidase-2 inhibition restores contractility and intracellular calcium handling and reduces arrhythmogenicity in dystrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 307, H710–H721. doi: 10.1152/ajpheart.00890.2013
Gonzalez, J. P., Ramachandran, J., Himelman, E., Badr, M. A., Kang, C., Nouet, J., et al. (2018). Normalization of connexin 43 protein levels prevents cellular and functional signs of dystrophic cardiomyopathy in mice. Neuromuscul. Disord. 28, 361–372. doi: 10.1016/j.nmd.2018.01.012
Goonasekera, S. A., Lam, C. K., Millay, D. P., Sargent, M. A., Hajjar, R. J., Kranias, E. G., et al. (2011). Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044–1052. doi: 10.1172/JCI43844
Gorecki, D. C. (2019). P2X7 purinoceptor as a therapeutic target in muscular dystrophies. Curr. Opin. Pharmacol. 47, 40–45. doi: 10.1016/j.coph.2019.02.003
Gorlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Grounds, M. D., Radley, H. G., Lynch, G. S., Nagaraju, K., and De Luca, A. (2008). Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 31, 1–19. doi: 10.1016/j.nbd.2008.03.008
Head, S. I. (2010). Branched fibres in old dystrophic mdx muscle are associated with mechanical weakening of the sarcolemma, abnormal Ca2+ transients and a breakdown of Ca2+ homeostasis during fatigue. Exp. Physiol. 95, 641–656. doi: 10.1113/expphysiol.2009.052019
Himelman, E., Lillo, M. A., Nouet, J., Gonzalez, J. P., Zhao, Q., Xie, L. H., et al. (2020). Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J. Clin. Invest. 130, 1713–1727. doi: 10.1172/JCI128190
Hirn, C., Shapovalov, G., Petermann, O., Roulet, E., and Ruegg, U. T. (2008). Nav1.4 deregulation in dystrophic skeletal muscle leads to Na+ overload and enhanced cell death. J. Gen. Physiol. 132, 199–208. doi: 10.1085/jgp.200810024
Hopf, F. W., Turner, P. R., Denetclaw, W. F. Jr., Reddy, P., and Steinhardt, R. A. (1996). A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am. J. Physiol. 271(4 Pt 1), C1325–C1339. doi: 10.1152/ajpcell.1996.271.4.C1325
Horakova, L., Strosova, M. K., Spickett, C. M., and Blaskovic, D. (2013). Impairment of calcium ATPases by high glucose and potential pharmacological protection. Free Radic. Res. 47(Suppl. 1), 81–92. doi: 10.3109/10715762.2013.807923
Houang, E. M., Haman, K. J., Filareto, A., Perlingeiro, R. C., Bates, F. S., Lowe, D. A., et al. (2015). Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther. Methods Clin. Dev. 2:15042. doi: 10.1038/mtm.2015.42
Hoxha, M. (2019). Duchenne muscular dystrophy: focus on arachidonic acid metabolites. Biomed. Pharmacother. 110, 796–802. doi: 10.1016/j.biopha.2018.12.034
Hussain, T., Mangath, H., Sundaram, C., and Anandaraj, M. P. J. S. (2000). Expression of the gene for large subunit of m-calpain is elevated in skeletal muscle from Duchenne muscular dystrophy patients. J. Genet. 79, 77–80. doi: 10.1007/Bf02728949
Ismail, H. M., Scapozza, L., Ruegg, U. T., and Dorchies, O. M. (2014). Diapocynin, a dimer of the NADPH oxidase inhibitor apocynin, reduces ROS production… and prevents force loss in eccentrically contracting dystrophic muscle. PLoS ONE 9:e110708. doi: 10.1371/journal.pone.0110708
Iwata, Y., Katanosaka, Y., Arai, Y., Komamura, K., Miyatake, K., and Shigekawa, M. (2003). A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J. Cell Biol. 161, 957–967. doi: 10.1083/jcb.200301101
Iwata, Y., Katanosaka, Y., Arai, Y., Shigekawa, M., and Wakabayashi, S. (2009). Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum. Mol. Genet. 18, 824–834. doi: 10.1093/hmg/ddn408
Iwata, Y., Katanosaka, Y., Hisamitsu, T., and Wakabayashi, S. (2007). Enhanced Na+/H+ exchange activity contributes to the pathogenesis of muscular dystrophy via involvement of P2 receptors. Am. J. Pathol. 171, 1576–1587. doi: 10.2353/ajpath.2007.070452
Jiao, Q., Takeshima, H., Ishikawa, Y., and Minamisawa, S. (2012). Sarcalumenin plays a critical role in age-related cardiac dysfunction due to decreases in SERCA2a expression and activity. Cell Calcium 51, 31–39. doi: 10.1016/j.ceca.2011.10.003
Jorgensen, L. H., Blain, A., Greally, E., Laval, S. H., Blamire, A. M., Davison, B. J., et al. (2011). Long-term blocking of calcium channels in mdx mice results in differential effects on heart and skeletal muscle. Am. J. Pathol. 178, 273–283. doi: 10.1016/j.ajpath.2010.11.027
Jung, C., Martins, A. S., Niggli, E., and Shirokova, N. (2008). Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 77, 766–773. doi: 10.1093/cvr/cvm089
Kang, C., Badr, M. A., Kyrychenko, V., Eskelinen, E. L., and Shirokova, N. (2018). Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc. Res. 114, 90–102. doi: 10.1093/cvr/cvx201
Kho, C., Lee, A., Jeong, D., Oh, J. G., Gorski, P. A., Fish, K., et al. (2015). Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 6:7229. doi: 10.1038/ncomms8229
Kim, J. H., Kwak, H. B., Thompson, L. V., and Lawler, J. M. (2013). Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 34, 1–13. doi: 10.1007/s10974-012-9330-9
Kirschner, J., Schessl, J., Schara, U., Reitter, B., Stettner, G. M., Hobbiebrunken, E., et al. (2010). Treatment of Duchenne muscular dystrophy with ciclosporin A: a randomised, double-blind, placebo-controlled multicentre trial. Lancet Neurol. 9, 1053–1059. doi: 10.1016/S1474-4422(10)70196-4
Koenig, M., Monaco, A. P., and Kunkel, L. M. (1988). The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53, 219–228.
Koenig, X., Rubi, L., Obermair, G. J., Cervenka, R., Dang, X. B., Lukacs, P., et al. (2014). Enhanced currents through L-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am. J. Physiol. Heart Circ. Physiol. 306, H564–H573. doi: 10.1152/ajpheart.00441.2013
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/CIRCRESAHA.111.259754
Kuo, I. Y., and Ehrlich, B. E. (2015). Signaling in muscle contraction. Cold Spring Harb. Perspect. Biol. 7:a006023. doi: 10.1101/cshperspect.a006023
Kuznetsov, A. V., Winkler, K., Wiedemann, F. R., von Bossanyi, P., Dietzmann, K., and Kunz, W. S. (1998). Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 183, 87–96. doi: 10.1023/a:1006868130002
Kwon, H. W., Kwon, B. S., Kim, G. B., Chae, J. H., Park, J. D., Bae, E. J., et al. (2012). The effect of enalapril and carvedilol on left ventricular dysfunction in middle childhood and adolescent patients with muscular dystrophy. Korean Circ. J. 42, 184–191. doi: 10.4070/kcj.2012.42.3.184
Kwong, J. Q., and Molkentin, J. D. (2015). Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 21, 206–214. doi: 10.1016/j.cmet.2014.12.001
Kyrychenko, S., Kyrychenko, V., Badr, M. A., Ikeda, Y., Sadoshima, J., and Shirokova, N. (2015a). Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 108, 324–334. doi: 10.1093/cvr/cvv238
Kyrychenko, V., Polakova, E., Janicek, R., and Shirokova, N. (2015b). Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 58, 186–195. doi: 10.1016/j.ceca.2015.04.006
Lamb, G. D. (2000). Excitation-contraction coupling in skeletal muscle: comparisons with cardiac muscle. Clin. Exp. Pharmacol. Physiol. 27, 216–224. doi: 10.1046/j.1440-1681.2000.03224.x
Lancel, S., Qin, F., Lennon, S. L., Zhang, J., Tong, X., Mazzini, M. J., et al. (2010). Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ. Res. 107, 228–232. doi: 10.1161/CIRCRESAHA.110.217570
Lanner, J. T., Georgiou, D. K., Joshi, A. D., and Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2:a003996. doi: 10.1101/cshperspect.a003996
Law, M. L., Prins, K. W., Olander, M. E., and Metzger, J. M. (2018). Exacerbation of dystrophic cardiomyopathy by phospholamban deficiency mediated chronically increased cardiac Ca(2+) cycling in vivo. Am. J. Physiol. Heart Circ. Physiol. 315, H1544–H1552. doi: 10.1152/ajpheart.00341.2018
Lee, E. H. (2010). Ca2+ channels and skeletal muscle diseases. Prog. Biophys. Mol. Biol. 103, 35–43. doi: 10.1016/j.pbiomolbio.2010.05.003
Lindahl, M., Backman, E., Henriksson, K. G., Gorospe, J. R., and Hoffman, E. P. (1995). Phospholipase A2 activity in dystrophinopathies. Neuromuscul. Disord. 5, 193–199. doi: 10.1016/0960-8966(94)00045-b
Loehr, J. A., Stinnett, G. R., Hernandez-Rivera, M., Roten, W. T., Wilson, L. J., Pautler, R. G., et al. (2016). Eliminating Nox2 reactive oxygen species production protects dystrophic skeletal muscle from pathological calcium influx assessed in vivo by manganese-enhanced magnetic resonance imaging. J. Physiol. 594, 6395–6405. doi: 10.1113/JP272907
Lohan, J., and Ohlendieck, K. (2004). Drastic reduction in the luminal Ca2+ -binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim. Biophys. Acta 1689, 252–258. doi: 10.1016/j.bbadis.2004.04.002
Lopez, J. R., Uryash, A., Faury, G., Esteve, E., and Adams, J. A. (2020). Contribution of TRPC channels to intracellular Ca(2 +) dyshomeostasis in smooth muscle from mdx mice. Front. Physiol. 11:126. doi: 10.3389/fphys.2020.00126
Lyon, A. R., Babalis, D., Morley-Smith, A. C., Hedger, M., Suarez Barrientos, A., Foldes, G., et al. (2020). Investigation of the safety and feasibility of AAV1/SERCA2a gene transfer in patients with chronic heart failure supported with a left ventricular assist device - the SERCA-LVAD TRIAL. Gene Ther. 27, 579–590. doi: 10.1038/s41434-020-0171-7
MacLennan, D. H., and Kranias, E. G. (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577. doi: 10.1038/nrm1151
MacLennan, P. A., McArdle, A., and Edwards, R. H. (1991). Effects of calcium on protein turnover of incubated muscles from mdx mice. Am. J. Physiol. 260(4 Pt 1), E594–E598. doi: 10.1152/ajpendo.1991.260.4.E594
Makarewich, C. A., Bezprozvannaya, S., Gibson, A. M., Bassel-Duby, R., and Olson, E. N. (2020). Gene therapy with the DWORF micropeptide attenuates cardiomyopathy in mice. Circ. Res. 127, 1340–1342. doi: 10.1161/CIRCRESAHA.120.317156
Makarewich, C. A., Munir, A. Z., Schiattarella, G. G., Bezprozvannaya, S., Raguimova, O. N., Cho, E. E., et al. (2018). The DWORF micropeptide enhances contractility and prevents heart failure in a mouse model of dilated cardiomyopathy. Elife 7:e38319. doi: 10.7554/eLife.38319
Mareedu, S., Dwivedi, S., Niranjan, N., and Babu, G. J. (2019). “Sarcolipin knockdown therapy for duchenne muscular dystrophy,” in Muscle Gene Therapy, eds D. Duan and J. R. Mendell (Cham: Springer International Publishing), 405–415.
Mareedu, S., Pachon, R. E., Jayapalraj, T., Fefelova, N., Balakrishnan, R., Niranjan, N., et al. (2021). Sarcolipin haploinsufficiency prevents dystrophic cardiomyopathy in mdx mice. Am. J. Physiol. Heart Circ. Physiol. 320, H200–H210. doi: 10.1152/ajpheart.00601.2020
Markham, B. E., Kernodle, S., Nemzek, J., Wilkinson, J. E., and Sigler, R. (2015). Chronic dosing with membrane sealant poloxamer 188 NF improves respiratory dysfunction in dystrophic Mdx and Mdx/Utrophin-/- mice. PLoS ONE 10:e0134832. doi: 10.1371/journal.pone.0134832
Matsumura, C. Y., Pertille, A., Albuquerque, T. C., Santo Neto, H., and Marques, M. J. (2009). Diltiazem and verapamil protect dystrophin-deficient muscle fibers of MDX mice from degeneration: a potential role in calcium buffering and sarcolemmal stability. Muscle Nerve 39, 167–176. doi: 10.1002/mus.21188
Matsumura, C. Y., Taniguti, A. P., Pertille, A., Santo Neto, H., and Marques, M. J. (2011). Stretch-activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am. J. Physiol. Cell Physiol. 301, C1344–C1350. doi: 10.1152/ajpcell.00056.2011
Mazala, D. A., Pratt, S. J. P., Chen, D., Molkentin, J. D., Lovering, R. M., and Chin, E. R. (2015). SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol 308, C699–C709. doi: 10.1152/ajpcell.00341.2014
Mendell, J. R., and Lloyd-Puryear, M. (2013). Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 48, 21–26. doi: 10.1002/mus.23810
Millay, D. P., Sargent, M. A., Osinska, H., Baines, C. P., Barton, E. R., Vuagniaux, G., et al. (2008). Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 14, 442–447. doi: 10.1038/nm1736
Miyatake, S., Shimizu-Motohashi, Y., Takeda, S., and Aoki, Y. (2016). Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 10, 2745–2758. doi: 10.2147/DDDT.S110163
Moens, P., Baatsen, P. H., and Marechal, G. (1993). Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 14, 446–451.
Monaco, A. P., Neve, R. L., Colletti-Feener, C., Bertelson, C. J., Kurnit, D. M., and Kunkel, L. M. (1986). Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 323, 646–650. doi: 10.1038/323646a0
Mondin, L., Balghi, H., Constantin, B., Cognard, C., and Sebille, S. (2009). Negative modulation of inositol 1,4,5-trisphosphate type 1 receptor expression prevents dystrophin-deficient muscle cells death. Am. J. Physiol. Cell Physiol. 297, C1133–C1145. doi: 10.1152/ajpcell.00048.2009
Moore, T. M., Lin, A. J., Strumwasser, A. R., Cory, K., Whitney, K., Ho, T., et al. (2020). Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 11:690. doi: 10.3389/fphys.2020.00690
Morgan, J. E., Prola, A., Mariot, V., Pini, V., Meng, J., Hourde, C., et al. (2018). Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 9:3655. doi: 10.1038/s41467-018-06057-9
Morine, K. J., Sleeper, M. M., Barton, E. R., and Sweeney, H. L. (2010). Overexpression of SERCA1a in the mdx diaphragm reduces susceptibility to contraction-induced damage. Hum. Gene Ther. 21, 1735–1739. doi: 10.1089/hum.2010.077
Mule, F., D'Angelo, S., Tabacchi, G., Amato, A., and Serio, R. (1999). Mechanical activity of small and large intestine in normal and mdx mice: a comparative analysis. Neurogastroenterol. Motil. 11, 133–139. doi: 10.1046/j.1365-2982.1999.00142.x
Mule, F., and Serio, R. (2001). Increased calcium influx is responsible for the sustained mechanical tone in colon from dystrophic (mdx) mice. Gastroenterology 120, 1430–1437. doi: 10.1053/gast.2001.24054
Murphy, R. M., Larkins, N. T., Mollica, J. P., Beard, N. A., and Lamb, G. D. (2009). Calsequestrin content and SERCA determine normal and maximal Ca2+ storage levels in sarcoplasmic reticulum of fast- and slow-twitch fibres of rat. J. Physiol. 587, 443–460. doi: 10.1113/jphysiol.2008.163162
Nakada, T., Kashihara, T., Komatsu, M., Kojima, K., Takeshita, T., and Yamada, M. (2018). Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 115, 4507–4512. doi: 10.1073/pnas.1716649115
Nelson, B. R., Makarewich, C. A., Anderson, D. M., Winders, B. R., Troupes, C. D., Wu, F., et al. (2016). A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351, 271–275. doi: 10.1126/science.aad4076
Niranjan, N., Mareedu, S., Tian, Y., Kodippili, K., Fefelova, N., Voit, A., et al. (2019). Sarcolipin overexpression impairs myogenic differentiation in Duchenne muscular dystrophy. Am. J. Physiol. Cell Physiol. 317, C813–C824. doi: 10.1152/ajpcell.00146.2019
Olthoff, J. T., Lindsay, A., Abo-Zahrah, R., Baltgalvis, K. A., Patrinostro, X., Belanto, J. J., et al. (2018). Loss of peroxiredoxin-2 exacerbates eccentric contraction-induced force loss in dystrophin-deficient muscle. Nat. Commun. 9:5104. doi: 10.1038/s41467-018-07639-3
Pal, R., Palmieri, M., Loehr, J. A., Li, S., Abo-Zahrah, R., Monroe, T. O., et al. (2014). Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 5:4425. doi: 10.1038/ncomms5425
Pan, Z., Brotto, M., and Ma, J. (2014). Store-operated Ca2+ entry in muscle physiology and diseases. BMB Rep. 47, 69–79. doi: 10.5483/bmbrep.2014.47.2.015
Pant, M., Sopariwala, D. H., Bal, N. C., Lowe, J., Delfin, D. A., Rafael-Fortney, J., et al. (2015). Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of duchenne muscular dystrophy. PLoS ONE 10:e0123875. doi: 10.1371/journal.pone.0123875
Patergnani, S., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., et al. (2011). Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 9:19. doi: 10.1186/1478-811X-9-19
Pathak, T., and Trebak, M. (2018). Mitochondrial Ca(2+) signaling. Pharmacol. Ther. 192, 112–123. doi: 10.1016/j.pharmthera.2018.07.001
Periasamy, M., and Kalyanasundaram, A. (2007). SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve 35, 430–442. doi: 10.1002/mus.20745
Pertille, A., de Carvalho, C. L., Matsumura, C. Y., Neto, H. S., and Marques, M. J. (2010). Calcium-binding proteins in skeletal muscles of the mdx mice: potential role in the pathogenesis of Duchenne muscular dystrophy. Int. J. Exp. Pathol. 91, 63–71. doi: 10.1111/j.1365-2613.2009.00688.x
Petrof, B. J., Shrager, J. B., Stedman, H. H., Kelly, A. M., and Sweeney, H. L. (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 90, 3710–3714.
Plant, D. R., and Lynch, G. S. (2003). Depolarization-induced contraction and SR function in mechanically skinned muscle fibers from dystrophic mdx mice. Am. J. Physiol. Cell Physiol. 285, C522–C528. doi: 10.1152/ajpcell.00369.2002
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440–1445. doi: 10.1126/science.1202768
Protasi, F. (2002). Structural interaction between RYRs and DHPRs in calcium release units of cardiac and skeletal muscle cells. Front. Biosci. 7, d650–d658. doi: 10.2741/A801
Quinlan, J. G., Wong, B. L., Niemeier, R. T., McCullough, A. S., Levin, L., and Emanuele, M. (2006). Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul. Disord. 16, 855–864. doi: 10.1016/j.nmd.2006.09.016
Raturi, A., and Simmen, T. (2013). Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 1833, 213–224. doi: 10.1016/j.bbamcr.2012.04.013
Reeve, J. L., McArdle, A., and Jackson, M. J. (1997). Age-related changes in muscle calcium content in dystrophin-deficient mdx mice. Muscle Nerve 20, 357–360. doi: 10.1002/(SICI)1097-4598(199703)20:3<357::AID-MUS14>3.0.CO;2-Y
Reutenauer, J., Dorchies, O. M., Patthey-Vuadens, O., Vuagniaux, G., and Ruegg, U. T. (2008). Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 155, 574–584. doi: 10.1038/bjp.2008.285
Rios, E. (2018). Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J. Gen. Physiol. 150, 521–537. doi: 10.1085/jgp.201711959
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi: 10.1126/science.280.5370.1763
Robert, V., Massimino, M. L., Tosello, V., Marsault, R., Cantini, M., Sorrentino, V., et al. (2001). Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 276, 4647–4651. doi: 10.1074/jbc.M006337200
Robin, G., Berthier, C., and Allard, B. (2012). Sarcoplasmic reticulum Ca2+ permeation explored from the lumen side in mdx muscle fibers under voltage control. J. Gen. Physiol. 139, 209–218. doi: 10.1085/jgp.201110738
Rybalka, E., Timpani, C. A., Cooke, M. B., Williams, A. D., and Hayes, A. (2014). Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 9:e115763. doi: 10.1371/journal.pone.0115763
Sabourin, J., Lamiche, C., Vandebrouck, A., Magaud, C., Rivet, J., Cognard, C., et al. (2009). Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J. Biol. Chem. 284, 36248–36261. doi: 10.1074/jbc.M109.012872
Santulli, G., Nakashima, R., Yuan, Q., and Marks, A. R. (2017). Intracellular calcium release channels: an update. J. Physiol. 595, 3041–3051. doi: 10.1113/JP272781
Schiavone, M., Zulian, A., Menazza, S., Petronilli, V., Argenton, F., Merlini, L., et al. (2017). Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol Res. 125(Pt B), 122–131. doi: 10.1016/j.phrs.2017.09.001
Schneider, J. S., Shanmugam, M., Gonzalez, J. P., Lopez, H., Gordan, R., Fraidenraich, D., et al. (2013). Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 34, 349–356. doi: 10.1007/s10974-013-9350-0
Selsby, J., Pendrak, K., Zadel, M., Tian, Z., Pham, J., Carver, T., et al. (2010). Leupeptin-based inhibitors do not improve the mdx phenotype. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1192–R1201. doi: 10.1152/ajpregu.00586.2009
Shaikh, S. A., Sahoo, S. K., and Periasamy, M. (2016). Phospholamban and sarcolipin: are they functionally redundant or distinct regulators of the Sarco(Endo)Plasmic Reticulum Calcium ATPase? J. Mol. Cell. Cardiol. 91, 81–91. doi: 10.1016/j.yjmcc.2015.12.030
Shanmuga Sundaram, J., Mohana Rao, V., Meena, A. K., and Anandaraj, M. P. (2006). Altered expression, intracellular distribution and activity of lymphocyte calpain II in Duchenne muscular dystrophy. Clin. Chim. Acta 373, 82–87. doi: 10.1016/j.cca.2006.05.004
Shannon, T. R. (2009). Ryanodine receptor Ca2+ sensitivity and excitation-contraction coupling in muscular dystrophy and heart failure: similar and yet different. Am. J. Physiol. Heart Circ. Physiol 297, H1965–H1966. doi: 10.1152/ajpheart.00945.2009
Shin, J. H., Bostick, B., Yue, Y., Hajjar, R., and Duan, D. (2011). SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J. Transl. Med. 9:132. doi: 10.1186/1479-5876-9-132
Shirokova, N., and Niggli, E. (2013). Cardiac phenotype of Duchenne Muscular Dystrophy: insights from cellular studies. J. Mol. Cell. Cardiol. 58, 217–224. doi: 10.1016/j.yjmcc.2012.12.009
Silva, M. T., Wensing, L. A., Brum, P. C., Camara, N. O., and Miyabara, E. H. (2014). Impaired structural and functional regeneration of skeletal muscles from beta2-adrenoceptor knockout mice. Acta Physiol. 211, 617–633. doi: 10.1111/apha.12329
Simpson, P. B., Mehotra, S., Lange, G. D., and Russell, J. T. (1997). High density distribution of endoplasmic reticulum proteins and mitochondria at specialized Ca2+ release sites in oligodendrocyte processes. J. Biol. Chem. 272, 22654–22661. doi: 10.1074/jbc.272.36.22654
Simpson, P. B., and Russell, J. T. (1997). Role of sarcoplasmic/endoplasmic-reticulum Ca2+-ATPases in mediating Ca2+ waves and local Ca2+-release microdomains in cultured glia. Biochem. J. 325 (Pt 1), 239–247. doi: 10.1042/bj3250239
Skura, C. L., Fowler, E. G., Wetzel, G. T., Graves, M., and Spencer, M. J. (2008). Albuterol increases lean body mass in ambulatory boys with Duchenne or Becker muscular dystrophy. Neurology 70, 137–143. doi: 10.1212/01.WNL.0000287070.00149.a9
Smani, T., Zakharov, S. I., Csutora, P., Leno, E., Trepakova, E. S., and Bolotina, V. M. (2004). A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 6, 113–120. doi: 10.1038/ncb1089
Smani, T., Zakharov, S. I., Leno, E., Csutora, P., Trepakova, E. S., and Bolotina, V. M. (2003). Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J. Biol. Chem. 278, 11909–11915. doi: 10.1074/jbc.M210878200
Smith, W. N., Dirks, A., Sugiura, T., Muller, S., Scarpace, P., and Powers, S. K. (2002). Alteration of contractile force and mass in the senescent diaphragm with beta(2)-agonist treatment. J. Appl. Physiol. 92, 941–948. doi: 10.1152/japplphysiol.00576.2001
Spencer, M. J., Croall, D. E., and Tidball, J. G. (1995). Calpains are activated in necrotic fibers from mdx dystrophic mice. J. Biol. Chem. 270, 10909–10914. doi: 10.1074/jbc.270.18.10909
Spencer, M. J., and Mellgren, R. L. (2002). Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum. Mol. Genet. 11, 2645–2655. doi: 10.1093/hmg/11.21.2645
Sperl, W., Skladal, D., Gnaiger, E., Wyss, M., Mayr, U., Hager, J., et al. (1997). High resolution respirometry of permeabilized skeletal muscle fibers in the diagnosis of neuromuscular disorders. Mol. Cell. Biochem. 174, 71–78.
Spinazzola, J. M., and Kunkel, L. M. (2016). Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 4, 1179–1194. doi: 10.1080/21678707.2016.1240613
Spurney, C. F., Knoblach, S., Pistilli, E. E., Nagaraju, K., Martin, G. R., and Hoffman, E. P. (2008). Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul. Disord. 18, 371–381. doi: 10.1016/j.nmd.2008.03.008
Sudevan, S., Takiura, M., Kubota, Y., Higashitani, N., Cooke, M., Ellwood, R. A., et al. (2019). Mitochondrial dysfunction causes Ca(2+) overload and ECM degradation-mediated muscle damage in C. elegans. FASEB J. 33, 9540–9550. doi: 10.1096/fj.201802298R
Szabadkai, G., Bianchi, K., Varnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Tanihata, J., Nagata, T., Ito, N., Saito, T., Nakamura, A., Minamisawa, S., et al. (2018). Truncated dystrophin ameliorates the dystrophic phenotype of mdx mice by reducing sarcolipin-mediated SERCA inhibition. Biochem. Biophys. Res. Commun. 505, 51–59. doi: 10.1016/j.bbrc.2018.09.039
Terry, R. L., Kaneb, H. M., and Wells, D. J. (2014). Poloxamer [corrected] 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS ONE 9:e91221. doi: 10.1371/journal.pone.0091221
Tidball, J. G., Albrecht, D. E., Lokensgard, B. E., and Spencer, M. J. (1995). Apoptosis precedes necrosis of dystrophin-deficient muscle. J. Cell Sci. 108 (Pt 6), 2197–2204.
Touyz, R. M., Anagnostopoulou, A., Rios, F., Montezano, A. C., and Camargo, L. L. (2019). NOX5: molecular biology and pathophysiology. Exp. Physiol. 104, 605–616. doi: 10.1113/EP086204
Townsend, D., Turner, I., Yasuda, S., Martindale, J., Davis, J., Shillingford, M., et al. (2010). Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Invest. 120, 1140–1150. doi: 10.1172/JCI41329
Turner, P. R., Fong, P. Y., Denetclaw, W. F., and Steinhardt, R. A. (1991). Increased calcium influx in dystrophic muscle. J. Cell Biol. 115, 1701–1712. doi: 10.1083/jcb.115.6.1701
Turner, P. R., Westwood, T., Regen, C. M., and Steinhardt, R. A. (1988). Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 335, 735–738. doi: 10.1038/335735a0
Valladares, D., Utreras-Mendoza, Y., Campos, C., Morales, C., Diaz-Vegas, A., Contreras-Ferrat, A., et al. (2018). IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta 1864, 3685–3695. doi: 10.1016/j.bbadis.2018.08.042
Vance, J. E. (2015). Phospholipid synthesis and transport in mammalian cells. Traffic 16, 1–18. doi: 10.1111/tra.12230
Vandebrouck, A., Sabourin, J., Rivet, J., Balghi, H., Sebille, S., Kitzis, A., et al. (2007). Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 21, 608–617. doi: 10.1096/fj.06-6683com
Vandebrouck, C., Martin, D., Colson-Van Schoor, M., Debaix, H., and Gailly, P. (2002). Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 158, 1089–1096. doi: 10.1083/jcb.200203091
Viola, H. M., Davies, S. M., Filipovska, A., and Hool, L. C. (2013). L-type Ca(2+) channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Heart Circ. Physiol. 304, H767–H775. doi: 10.1152/ajpheart.00700.2012
Voit, A., Patel, V., Pachon, R., Shah, V., Bakhutma, M., Kohlbrenner, E., et al. (2017). Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 8:1068. doi: 10.1038/s41467-017-01146-7
Wang, Q., Wang, W., Wang, G., Rodney, G. G., and Wehrens, X. H. (2015). Crosstalk between RyR2 oxidation and phosphorylation contributes to cardiac dysfunction in mice with Duchenne muscular dystrophy. J. Mol. Cell Cardiol. 89(Pt B), 177–184. doi: 10.1016/j.yjmcc.2015.11.009
Wasala, N. B., Yue, Y., Lostal, W., Wasala, L. P., Niranjan, N., Hajjar, R. J., et al. (2020). Single SERCA2a therapy ameliorated dilated cardiomyopathy for 18 months in a mouse model of duchenne muscular dystrophy. Mol. Ther. 28, 845–854. doi: 10.1016/j.ymthe.2019.12.011
Whitehead, N. P., Yeung, E. W., and Allen, D. G. (2006). Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 33, 657–662. doi: 10.1111/j.1440-1681.2006.04394.x
Williams, G. S., Boyman, L., Chikando, A. C., Khairallah, R. J., and Lederer, W. J. (2013). Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U.S.A. 110, 10479–10486. doi: 10.1073/pnas.1300410110
Williams, I. A., and Allen, D. G. (2007a). Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Heart Circ. Physiol. 292, H846–H855. doi: 10.1152/ajpheart.00688.2006
Williams, I. A., and Allen, D. G. (2007b). The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 293, H1969–H1977. doi: 10.1152/ajpheart.00489.2007
Woods, C. E., Novo, D., DiFranco, M., and Vergara, J. L. (2004). The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J. Physiol. 557(Pt 1), 59–75. doi: 10.1113/jphysiol.2004.061291
Yasuda, S., Townsend, D., Michele, D. E., Favre, E. G., Day, S. M., and Metzger, J. M. (2005). Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 436, 1025–1029. doi: 10.1038/nature03844
Yeung, E. W., Head, S. I., and Allen, D. G. (2003). Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J. Physiol. 552(Pt 2), 449–458. doi: 10.1113/jphysiol.2003.047373
Yeung, E. W., Whitehead, N. P., Suchyna, T. M., Gottlieb, P. A., Sachs, F., and Allen, D. G. (2005). Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 562(Pt 2), 367–380. doi: 10.1113/jphysiol.2004.075275
Young, C. N., Brutkowski, W., Lien, C. F., Arkle, S., Lochmuller, H., Zablocki, K., et al. (2012). P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: relationship to pathology and potential target for treatment. J. Cell. Mol. Med. 16, 1026–1037. doi: 10.1111/j.1582-4934.2011.01397.x
Young, C. N. J., Chira, N., Rog, J., Al-Khalidi, R., Benard, M., Galas, L., et al. (2018). Sustained activation of P2X7 induces MMP-2-evoked cleavage and functional purinoceptor inhibition. J. Mol. Cell Biol. 10, 229–242. doi: 10.1093/jmcb/mjx030
Yu, L., Zhang, X., Yang, Y., Li, D., Tang, K., Zhao, Z., et al. (2020). Small-molecule activation of lysosomal TRP channels ameliorates Duchenne muscular dystrophy in mouse models. Sci. Adv. 6:eaaz2736. doi: 10.1126/sciadv.aaz2736
Zanou, N., Iwata, Y., Schakman, O., Lebacq, J., Wakabayashi, S., and Gailly, P. (2009). Essential role of TRPV2 ion channel in the sensitivity of dystrophic muscle to eccentric contractions. FEBS Lett. 583, 3600–3604. doi: 10.1016/j.febslet.2009.10.033
Zsebo, K., Yaroshinsky, A., Rudy, J. J., Wagner, K., Greenberg, B., Jessup, M., et al. (2014). Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ. Res. 114, 101–108. doi: 10.1161/CIRCRESAHA.113.302421
Keywords: Duchenne muscular dystrophy, calcium, sarco(endo)plasmic reticulum calcium ATPase, sarcolipin, ryanodine receptor, sarcolemma, dystrophin, mitochondria
Citation: Mareedu S, Million ED, Duan D and Babu GJ (2021) Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 12:647010. doi: 10.3389/fphys.2021.647010
Received: 28 December 2020; Accepted: 02 March 2021;
Published: 09 April 2021.
Edited by:
Helen Cristina Miranda, Case Western Reserve University, United StatesReviewed by:
Stewart Ian Head, Western Sydney University, AustraliaCopyright © 2021 Mareedu, Million, Duan and Babu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gopal J. Babu, YmFidWdvQG5qbXMucnV0Z2Vycy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.