 Laia Gifre-Renom
Laia Gifre-Renom Elizabeth A. V. Jones
Elizabeth A. V. Jones
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 25 February 2021
Sec. Vascular Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.639645
This article is part of the Research Topic Mechanisms of Vessel Development: From a Primitive Draft to a Mature Vasculature View all 14 articles
From developmental stages until adulthood, the circulatory system remodels in response to changes in blood flow in order to maintain vascular homeostasis. Remodeling processes can be driven by de novo formation of vessels or angiogenesis, and by the restructuration of already existing vessels, such as vessel enlargement and regression. Notably, vessel enlargement can occur as fast as in few hours in response to changes in flow and pressure. The high plasticity and responsiveness of blood vessels rely on endothelial cells. Changes within the bloodstream, such as increasing shear stress in a narrowing vessel or lowering blood flow in redundant vessels, are sensed by endothelial cells and activate downstream signaling cascades, promoting behavioral changes in the involved cells. This way, endothelial cells can reorganize themselves to restore normal circulation levels within the vessel. However, the dysregulation of such processes can entail severe pathological circumstances with disturbances affecting diverse organs, such as human hereditary telangiectasias. There are different pathways through which endothelial cells react to promote vessel enlargement and mechanisms may differ depending on whether remodeling occurs in the adult or in developmental models. Understanding the molecular mechanisms involved in the fast-adapting processes governing vessel enlargement can open the door to a new set of therapeutical approaches to be applied in occlusive vascular diseases. Therefore, we have outlined here the latest advances in the study of vessel enlargement in physiology and pathology, with a special insight in the pathways involved in its regulation.
During development, vascular beds often form as honeycombed shaped capillary plexus that then become perfused and remodel to form a hierarchical vessel structure. One of the most visible changes that occur in this process is the enlargement of both arteries and veins. Vessel enlargement can occur remarkably quickly. In the embryo, this occurs within a single day after the onset of blood flow. As such, vessel enlargement has therapeutic potential in occlusive vascular diseases that slower processes, such as angiogenesis, lack. However, so far, most research on therapeutic strategies has been focused on angiogenesis and significantly less advancement has been made to exploit vessel enlargement as a potential therapy in ischemic diseases. In strokes and heart attacks, for example, local collateral vessels immediately dilate to restore blood flow, but angiogenesis is a latter process in the body’s attempt to restore proper perfusion (Schaper and Ito, 1996). Vessel can dilate on the short term, but can also undergo outward remodeling such that vessel diameter changes are permanent (Silver and Vita, 2006). As such, it is primarily vessel enlargement that prevents excessive cell death.
Though occlusive vascular diseases are important pathologies where inducing vessel enlargement could provide therapeutic benefits, when vessels enlarge ectopically, there can be devastating negative consequences. Arterial-venous malformations (AVMs) are a form of anomalous vessel enlargement where enlarged shunts bypass the capillary bed and directly connect arteries and veins. Because the venous network is then exposed to arterial blood pressure, AVMs are prone to hemorrhage, which can be fatal depending on the organ where they occur. For instance, cerebral AVMs account for 50% of childhood strokes (Meyer-Heim and Boltshauser, 2003). Though AVMs are the most serious example of pathological vessel enlargement, these are not the only example of such a process. Varicose veins also represent a form of pathological vessel enlargement (Jacobs et al., 2017) that can cause itching and discomfort, and are one of the most common reported medical conditions in Western countries (Beebe-Dimmer et al., 2005).
Despite the importance of vessel enlargement, we are just beginning to understand how diameter changes occur. It was initially assumed that vessels enlarged by proliferation and while this may be true in some vascular beds, proliferation is a slow process and therefore could not restore blood flow on the timescales needed after stroke or heart attack. Vessel dilation, followed by outward remodeling can, at least partially, mitigate the slow process of cell proliferation. More recently, however, migration and capillary fusion have been proposed as methods by which a vascular network can increase the diameter of vessels. It is important to note, however, that it is unlikely that any of these processes happen in isolation. As such, vascular networks likely use a combination of means to increase vessel diameters. We therefore review here the processes and pathways by which vessels enlarge, and highlight differences and similarities between vascular development, vasculopathy and enlargement in adult vascular beds.
Four mechanisms of vessel enlargement have been identified, though the prevalence or relative importance of each of these mechanisms is still not clear. First, endothelial cell migration can lead to the accumulation of cells in one region, leading to regional enlargement of a vessel. Second, smaller vessels can fuse with each other, thereby rapidly increasing the diameter. Vessels can also increase in diameter because of either local proliferation of endothelial cells (third mechanism), or by local hypertrophy of endothelial cells (fourth). Vessel enlargement occurring in developmental vascular networks refers to the enlargement of capillaries to form arterioles/venules. In the adult, vessel enlargement has mostly only been studied with respect to enlargement of larger vessels (arterioles and venules) and of collateral vessels. Differences in the type of vessel which is enlarging can also account for differences in mechanism highlighted below.
Migration is currently the most well accepted mechanism for vessel enlargement in developmental models. In this mechanism, vessel enlargement occurs by a reshuffling of existing vessels guided by changes in shear stress. Shear stress is the force per unit area exerted by flow, expressed either in N/m2 (i.e., Pascals) or in dyne/cm2. Shear stress can be thought of as friction against the endothelium. It relates both to the speed of the blood flow and vessel geometry. In the migration paradigm of vessel enlargement, endothelial cells migrate against flow when shear stress levels are decreased compared to physiological normal levels (which are 15 dyn/cm2 in humans or 30 dyn/cm2 in mouse) and stop migrating in the presence of physiological levels (Franco et al., 2015; Tabibian et al., 2020). Therefore, vessels with the lowest flow rates regress increasing the pool of available endothelial cells. Furthermore, because endothelial cells stop migrating in high shear stress vessels, they accumulate in regions of high shear stress (Figure 1). This idea was first proposed approximately 20 years ago (Hughes and Chang-Ling, 2000) but gained significantly more interest as imaging technology improved, allowing individual endothelial cell tracking (Udan et al., 2013; Franco et al., 2015).
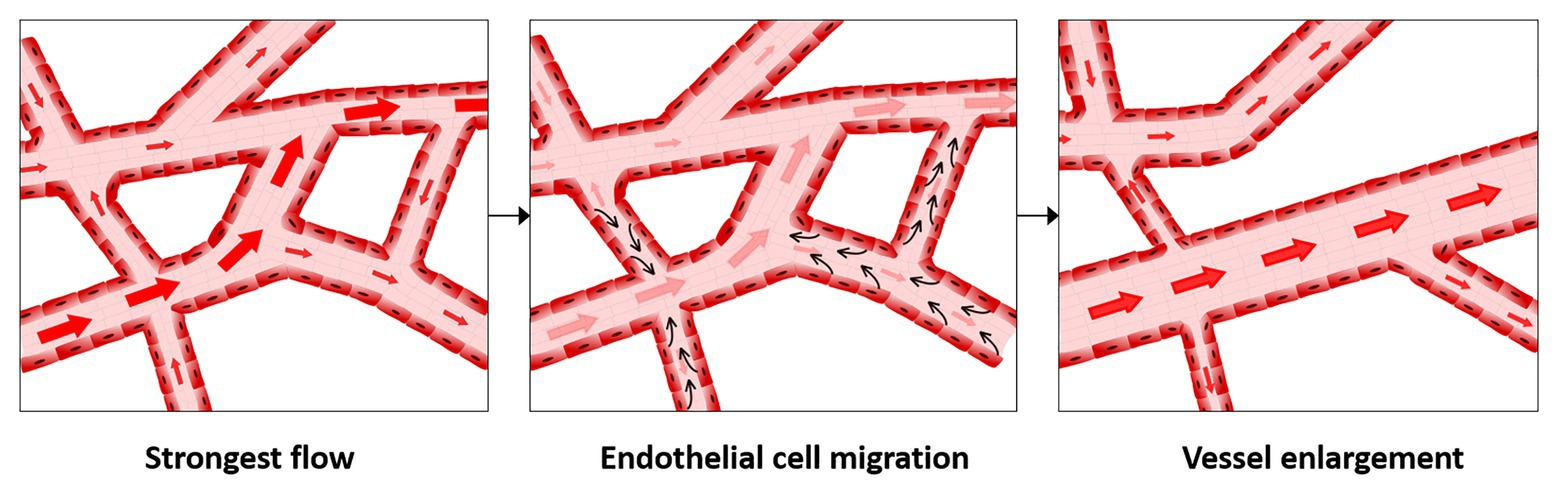
Figure 1. Migration-induced vessel enlargement. Under low shear stress, endothelial cells migrate against flow and stop or reduce migration when exposed to shear stresses at or above their shear stress physiological levels. In this way, endothelial cells accumulate in vessels under higher shear stress, leading to vessel enlargement.
In support of this mechanism, multiple groups have shown that there is very little proliferation or apoptosis during vessel remodeling and vessel enlargement, either in the retina, in zebrafish embryo or in mouse embryo (Hughes and Chang-Ling, 2000; Udan et al., 2013; Franco et al., 2015). Myosin light chain 2a (MLC2a)−/− embryos, which have severely reduced flow and do not undergo vessel enlargement, have the same levels of endothelial cell proliferation and apoptosis as control wild-type embryos (Udan et al., 2013). Furthermore, time-lapse microscopy of developing embryos has shown that there is a stunning amount of endothelial cell migration occurring during vascular remodeling (Sato et al., 2010; Cui et al., 2013). Using a quail embryo that has Yellow Fluorescent Protein (YFP)-labeled endothelial cells, Cui et al. (2013) imaged vitelline artery formation (see especially movie S4). Endothelial cells can be observed migrating against flow, towards the embryo proper, as a mass collective movement. Similarly, using time-lapse microscopy of whole mouse embryos between embryonic day (E) E8.5 and E9.5, Udan et al. (2013) showed that endothelial cells leave low flow capillaries towards enlarging vessels, whereas in the MLC2a−/− mutant embryos, endothelial cells lose their ability to undergo directional migration.
Our group has recently used the migration-induced mechanism of vessel enlargement to develop computational models of remodeling and vessel enlargement (Tabibian et al., 2020). In vitro, we found that there is indeed a bell-shaped pattern of migration with respect to shear stress. Endothelial cells do not migrate in static or at extremely low shear stress levels. Endothelial cells exposed to shear stress levels between 0.5 and 5 dyn/cm2 show the highest level of migration, with a peak at 1 dyn/cm2. Higher shear stress levels return migration rates to static levels (Tabibian et al., 2020). This information was then used to build the computational model where shear-stress levels defined the speed of migration, but the direction of migration was influenced both by the direction of the flow and a requirement for collective cell movement. This alone gave modest predictive ability, which was improved by the addition of growth of avascular regions and, more surprisingly, by the addition of endothelial cell elongation in the direction of flow. Although the role of cell elongation in remodeling is currently unexplored, studies on mice where endothelial cells cannot elongate did not report vessel enlargement defects (Baeyens et al., 2014; Corti et al., 2019). Those results, however, also found that shear stress magnitude is sensed apart from shear stress direction (Baeyens et al., 2014), consistent with our computational results.
In order to migrate against flow, endothelial cells are first polarized against the direction of flow (Franco et al., 2015). Labeling the Golgi and nucleus allows endothelial cell polarization to be visualized. By comparing polarization to predicted flow patterns, endothelial cells were shown to be polarized against flow in the developing retina (Franco et al., 2015). A key player in the maintenance of the endothelial polarization, first identified in the retina, is the primary cilium of endothelial cells (Vion et al., 2018). Primary cilia are present on endothelial cells when exposed to low shear stress, whereas higher levels of shear stress often cause disassembly of the cilia (Iomini et al., 2004; Egorova et al., 2011; Ten Dijke et al., 2012). By a genetic deletion of the essential cilia component intraflagellar transport protein 88 (IFT88), Vion et al. (2018) described a random and premature regression of blood vessels related to an increase in migration and a reduced polarization of the endothelial cells.
The Bone Morphogenetic Protein/Activin receptor-like kinase 1/Endoglin (BMP/ALK1/ENG) pathway is also required for polarized migration in response to flow (Figure 2). Mutations in the ENG and ACVRL1 genes (encoding for ENG and ALK1) cause Hereditary Hemorrhagic Telangiectasia (HHT), which is a genetic form of AVM development (McAllister et al., 1994; Johnson et al., 1996). Loss of directional migration has been reported for ENG (Jin et al., 2017) and ALK1 (Rochon et al., 2016) in mouse ENG-knockouts and in a zebrafish AVM model lacking ALK1, respectively. Recently, our group has identified that, under shear stress, SMAD1/5/9 (transcription factors in this pathway) control the expression of the gene (GJA4) for the gap junction protein Cx37 (Connexin37; Peacock et al., 2020). We also found that Cx37 has a critical role in the maintenance of the directionality of endothelial cell migration under flow. Interestingly, mechanotransduction by the primary cilium also functions through the ALK1 pathway, with the primary cilium increasing BMP9 responsiveness of endothelial cells under low shear stress, thereby decreasing their migration speed. This may prevent premature vascular regression under low shear stress processes, such as during the initial remodeling of the retinal vascular network (Vion et al., 2018).
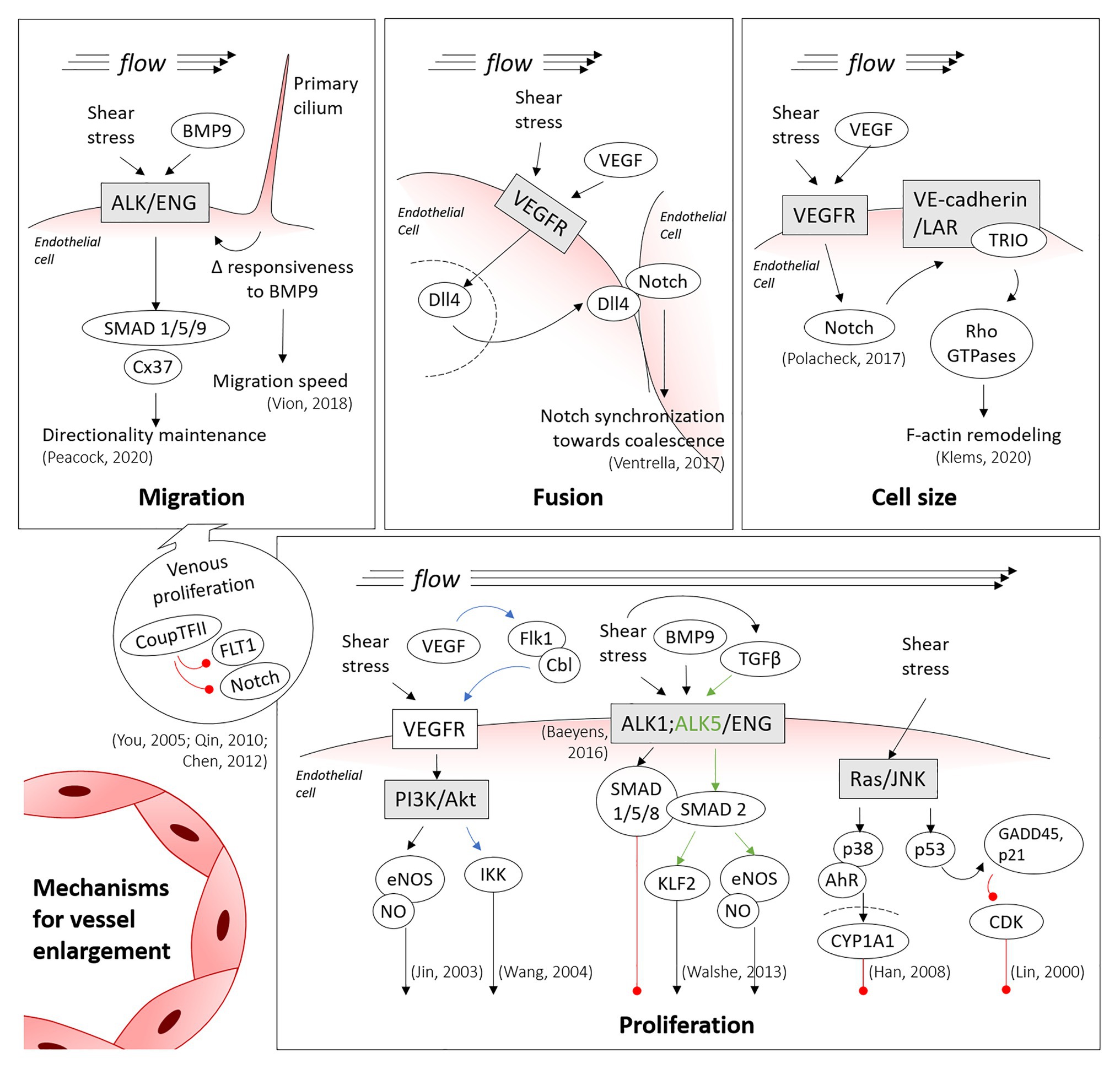
Figure 2. Main mechanisms involved in vessel enlargement. Summary of the shear-inducible mechanotransduction pathways involved in migration, fusion, hypertrophy and proliferation of endothelial cells, the processes directing vessel enlargement. Blue and green arrows are colored to clarify the trigger of specific downstream pathways, being shear stress involved in both cases. Red lines denote inhibition (thus, cell cycle arrest, in the proliferation panel). Discontinued lines denote the nuclei membranes.
Though regressing cells are a source of endothelial cells for enlarging vessels, the venous vascular bed also contributes cells. Notably, proliferation is higher in venous endothelial cells than in arterial endothelial cells (Red-Horse et al., 2010; Ehling et al., 2013) and, therefore, venous cells provide a source of cells for the migration model of vessel enlargement (Figure 2). Coup-TFII, which is one of the most important venous transcription factors, can repress the expression of Fms Related Receptor Tyrosine Kinase 1 (FLT1; also known as VEGFR1, Vascular Endothelial Growth Factor Receptor 1) and Notch, thereby promoting endothelial cell proliferation (You et al., 2005; Qin et al., 2010; Chen et al., 2012). In the mouse embryonic heart, for instance, coronary arteries have been shown to form from venous endothelial cells (Su et al., 2018). In the mouse retina, labeling tip cells permitted to observe the integration of the labeled cells in growing arteries, but rarely into the venous vascular bed (Xu et al., 2014; Pitulescu et al., 2017). This has led to the proposal that endothelial cells proliferate in veins, migrate from low shear stress veins through the capillary bed and then ultimately stop migrating in the arterial vascular bed because of high shear stress levels. They therefore accumulate in this region, inducing vessel enlargement (Red-Horse and Siekmann, 2019).
Vessel fusion was first described over three decades ago but has received less attention than other mechanisms of vessel enlargement. Drake and Little (1995) were first to describe this process. By time-lapsing dorsal aorta development in avian embryos, they showed that a capillary bed initially formed along the length of the embryo proper and that with the onset of blood flow, these capillaries merge together forming larger and larger vessels (Figure 3A; Rupp et al., 2004). These observations were further confirmed with the development of transgenic quail embryos that allowed clear visualizations of the forming dorsal aorta [see movies S3 and S6, (Sato et al., 2010)]. Moreover, both the vitelline artery and vitelline vein were also reported to form by fusion of smaller capillaries in these transgenic quails (Sato et al., 2010).
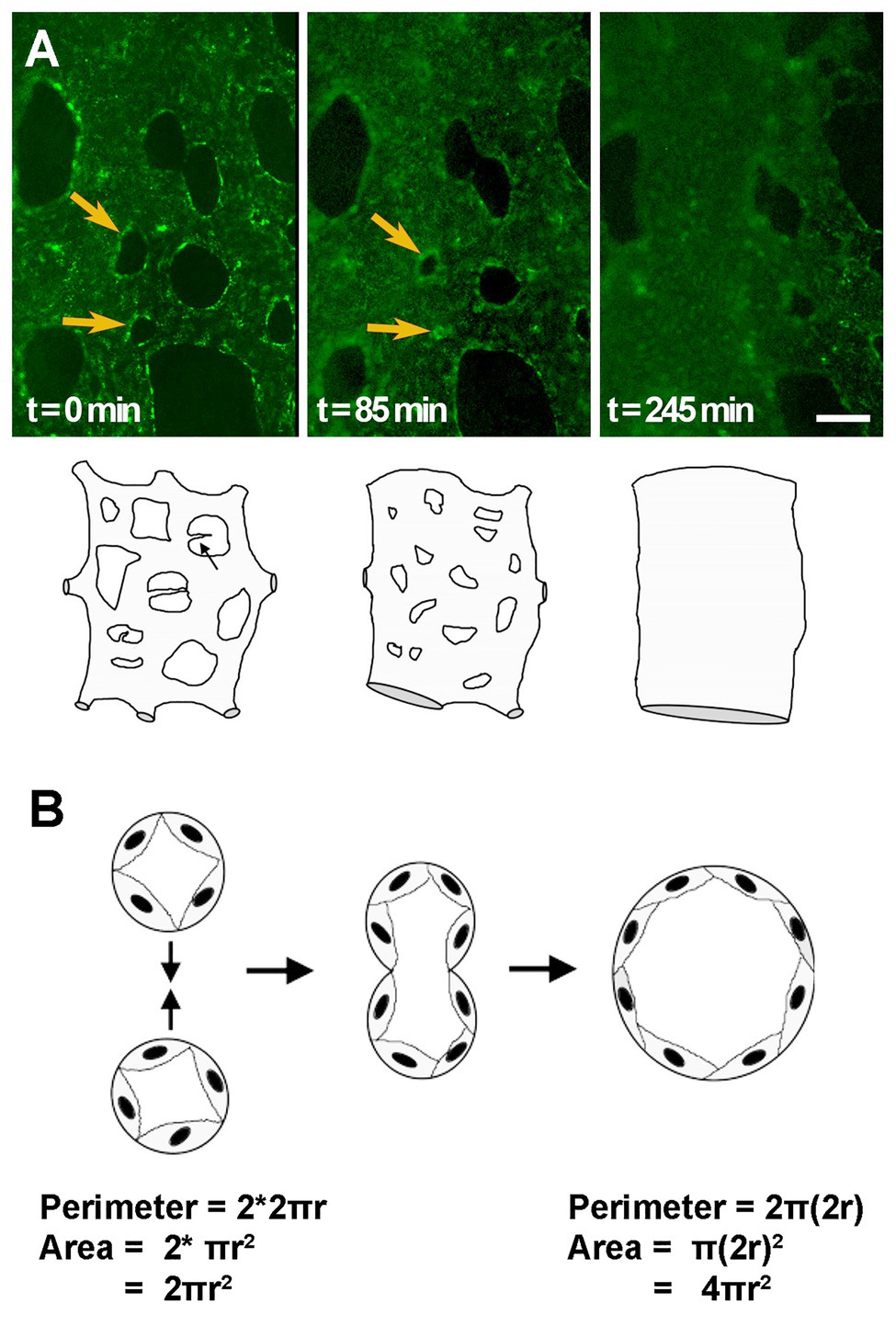
Figure 3. Vessel enlargement by fusion. (A) Capillary plexus vessels merge to form a larger vessel, as shown by time-lapse microscopy of AcLDL labeled vitelline vessels in quail embryos. Yellow arrows indicate avascular regions that become smaller and smaller until two adjacent vessels fuse. Cartoon exemplifies changes that can be observed by time-lapse microscopy. (B) Schematic of the fusion process in transverse view. The same number of cells, or perimeter length, leads to a doubling in the area for flow after fusion. Cartoons adapted from (Drake and Little, 1999).
Though the initial reports on fusion mainly focused on the dorsal aorta in avian embryos, this process was later shown to also occur in other models and other vascular beds. In mouse, time-lapse microscopy showed that fusion is the main mechanism by which yolk sac vessel enlarge, leading to periodic jumps in vessel diameter rather than smooth linear increases in vessel diameter (Udan et al., 2013). Here, fusion processes could be identified in the formation of both the vitelline artery and the vitelline vein in mouse. Quantification of sprouting/regression/fusion/splitting events during remodeling of the yolk sac vasculature shows that fusion is more common than vessel regression in the embryo, but slightly less common than angiogenesis (Chouinard-Pelletier et al., 2013). The overall number of fusion events is on the same order of magnitude as sprouting and regression events. The coalesce of a capillary bed along the embryonic midline has never been reported for the mouse dorsal aorta formation. However, the Semaphorin 3E (SEMA3E)−/− embryo, involved in repulsive endothelial cell guidance, shows a transient phenotype whereby a plexus is present that eventually coalesces to paired dorsal aortae. This suggests that vessel coalescence may be retarded in these mutants and, therefore, observable (Meadows et al., 2013). Vessel enlargement in other organs has also been reported to occur by fusion, or coalescence, of smaller vessels, such as the central pancreatic duct, which develops from smaller capillary vessels joining and fusing into one large diameter vessel (Azizoglu et al., 2016).
Fusion is a much faster process to enlarge a vessel than either migration or proliferation. Within hours, two small vessels can merge leading to a doubling in the radius of the vessel, but a 4-fold increase in the cross-sectional area for flow (Figure 3B). This may explain why reports of fusion have been limited to embryonic vascular beds, where remodeling must occur much more rapidly than in more mature systems. It is also possible, thus, that difficulties in detecting fusion prevent its identification in other vascular beds. Morphologically, as vessels fuse, the avascular region between the two vessels becomes smaller and smaller, eventually creating “pillars” of avascular tissue that are identical to the pillars present in intussusceptive angiogenesis. As such, static images cannot differentiate fusion from intussusceptive angiogenesis. Indeed, fusion may only have been identified in embryonic vasculatures because these are the vascular beds where time-lapse microscopy at the resolution of capillaries is possible.
Just as for migration, flow is essential for fusion to occur. In the aforementioned SEMA3E−/− mutants, the timing of resolution of the unfused phenotype correlates to the onset of blood flow (Meadows et al., 2013). In normal vascular development, fusion occurs in regions with the highest flow, such as the region where the vitelline artery and vein are forming (Sato et al., 2010; Chouinard-Pelletier et al., 2013; Udan et al., 2013). Unexpectedly, if flow patterns are altered to reduce shear stress, an increased number of fusion events is observed (Chouinard-Pelletier et al., 2013). If shear stress levels are increased instead, the opposite occurs, and less fusion is present. Though these results may appear paradoxical, the increased flow in the region of the forming vitelline artery and vein do not necessarily mean that increased shear stress drives fusion. Shear stress relates not only to the flow velocity but also to geometry of the vessels. If an avascular region between two fusing vessels acts as an obstruction in the middle of a fast-flowing stream, then, as the velocity of that stream increases, shear stress will be increased on the upstream side of the avascular region but decreased on the downstream side (Figure 4). As such, gradients of shear stress may drive fusion events rather than just increases in the total amount or velocity of flow.
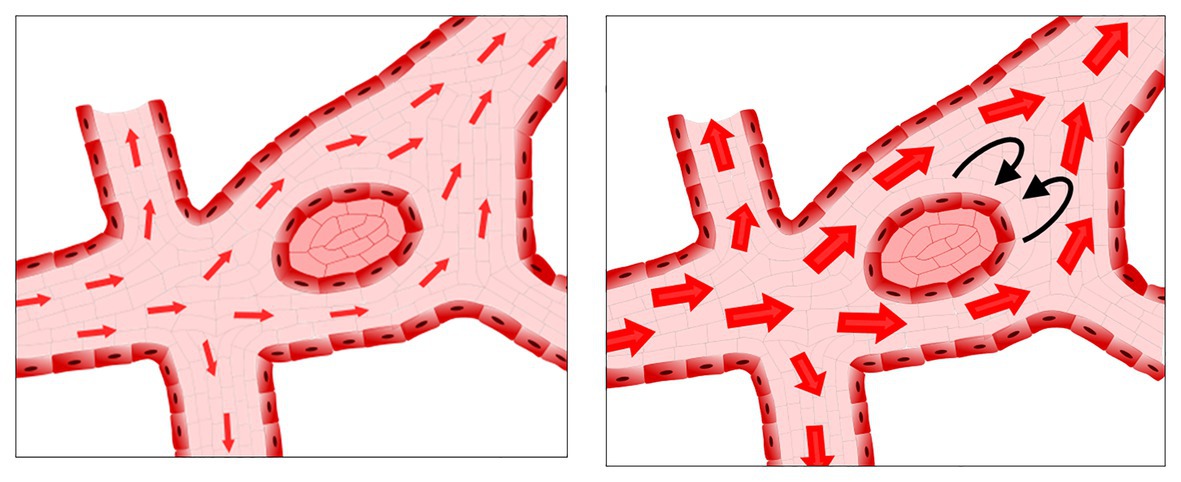
Figure 4. Possible low shear stress regions in the presence of increased flow rates. As flow increases, avascular regions can act as obstacles to the flow, resulting in regions of low shear stress and/or recirculation downstream from the avascular region.
Given the difficulties in identifying fusion events, very little is known about the mechanism of fusion. There are various phenotypes, however, that present with a hyperfused vascular plexus. Inhibition of Notch in the embryonic yolk sac results in increased number of fusion events (Chouinard-Pelletier et al., 2013; Caolo et al., 2018). The increased amount of fusion observed during Notch inhibition can be rescued by increasing shear stress levels (Caolo et al., 2018). In the chick embryo yolk sac, exogenous vascular endothelial growth factor (VEGF) induced increased vascular fusion (Drake and Little, 1995). Sema3E signaling induces the expression of sFLT1 (Zygmunt et al., 2011) and, as such, the SEMA3E−/− should have reduced VEGF signaling. Though the interplay of VEGF and Notch signaling is well studied for sprouting angiogenesis (Eichmann and Simons, 2012), why they would induce fusion in other situations is a mystery. The key may lie in the role of VEGF in synchronizing Notch signaling (Figure 2). In somitogenesis, Notch is involved in synchronizing cells such that all cells cycle together and express the same genes together (Jiang et al., 2000). In endothelial cells, levels of Notch targeting genes also oscillate, but in an unsynchronized manner. Under higher levels of VEGF, however, Notch-induced gene expression synchronizes endothelial cells, favoring vessel enlargement rather than extension (Ubezio et al., 2016). The act of favoring “self” (staying together) rather than “other” (extending a sprout) has previously been proposed for the role of Notch target genes in boundary formation (Ventrella et al., 2017). In this model, Notch synchronization would create a situation where vessel coalesce with each other (i.e., fuse), as they prefer adhering to one another rather than remaining separate.
Though no significant proliferation is observed during vessel enlargement in developmental models, whether the retina or the embryo, this is not the case for vessel enlargement in adult vascular beds. Chronic changes in flow in the adult lead to expansion of collateral blood vessels that restore normal blood flow levels. This process was termed arteriogenesis (Scholz et al., 2001). After partial coronary ligation model, endothelial cell proliferation is observed in enlarging vessels, peaking 3 weeks after implantation of the constrictor (Schaper et al., 1971). In hind limb ischemia models, significant endothelial cell and smooth muscle cell proliferation is observed in the enlarging vessels within 2–3 days of ligation (Scholz et al., 2000). Vessel enlargement by proliferation occurs over a period of days and weeks, not hours. Though endothelial cells are clearly proliferating within the growing arterioles (Scholz et al., 2000), this does not exclude a role for migration in collateral enlargement.
Vascular endothelial growth factor is most well known as a mitogen for endothelial cells proliferation. VEGF acts by binding to VEGF receptors, and this phosphorylates protein kinases that activate downstream Phosphoinositide 3-kinase (PI3K) and Mitogen-activated protein kinase (MAPK) pathways promoting proliferation (Figure 2). VEGF expression is triggered by hypoxia. The proliferation process involved in arteriole enlargement, however, does not respond to hypoxia (Deindl et al., 2001). In fact, although experimentally inhibited VEGF in femoral artery ligation (an occlusion model) results in inhibited arteriogenesis (Jacobi et al., 2004; Lloyd et al., 2005; Toyota et al., 2005), VEGF is not expressed in the tissue near the growing collaterals (Lee et al., 2004). Thus, although endothelial cells clearly proliferate within the growing arterioles (Scholz et al., 2000), the role of VEGF is unlikely to be needed for this local proliferation. Instead, VEGF may be needed to induce proliferation distal from the site of arteriogenesis, though it is also possible that VEGF could be responsible for the release of blood-marrow derived cells.
In the case of flow-induced remodeling, VEGF itself is also not a necessary ligand to activate PI3K/MAPK signaling pathways. Laminar flow induces a transphosphorylation of VEGF receptor 2 (VEGFR2) that activates the downstream PI3K pathway in a ligand-independent manner (Jin et al., 2003). This leads to the phosphorylation of Akt that has pleiotropic effects on proliferation. Akt phosphorylation by flow is highest at physiological shear stress levels between 10 and 20 dyn/cm2 (Dimmeler et al., 1998; Li et al., 2009), however, these levels of shear stress are known to induce quiescence and not proliferation in endothelial cells. VEGFR2 and Akt phosphorylation are, however, short-lived events that occur within the first 1–2 h of a change in shear stress (Shay-Salit et al., 2002; Guo et al., 2007). Oscillatory flow, which in contrast to unidirectional flow does induce increased proliferation, leads to prolonged VEGFR2 and Akt phosphorylation (Guo et al., 2007). Furthermore, unidirectional flow also activates other factors such as AMP-activated protein kinase (AMPK), which counteract the pro-proliferative signals (Guo et al., 2007).
Shear stress can also modulate endothelial cell proliferation through other signaling pathways. The phosphorylation of endothelial Nitric Oxide Synthase (eNOS) and its increased activity induced by shear stress was one of the first studied effects of mechanotransduction (Buga et al., 1991; Kuchan and Frangos, 1994). Nitric oxide (NO) not only induces vasodilation but also induces proliferation. In vasodilation, NO is produced by endothelial cells, diffuses to smooth muscle cells where it activates soluble guanylyl cyclase by binding to the heme group (Zhao et al., 1999). Estimates vary concerning the concentration at which this occurs, however, most publications have placed this between 5 and 100 nM (Chen et al., 2008). As a pro-proliferative compound, NO acts by controlling protein activation by reacting with cysteine residues to induce S-nitrosylation. NO leads to S-nitrosylation of MAPK phosphatase 7 (MKP7), rendering it inactive which then prevents the inactivation of c-Jun N-terminal Kinase 3 (JNK3; Pi et al., 2009). The concentrations at which NO induce proliferation are extremely low, in the pico to nanomolar ranges that occur due to release of NO by macrophages and endothelial cells (Ridnour et al., 2005; Thomas et al., 2008; Figure 2). At concentrations in the micromolar range, NO inhibits proliferation and induces cell cycle arrest in several cell types (Gooch et al., 1997; Heller et al., 1999).
Though the ALK1/ENG complex affects endothelial cell migration, this signaling pathway also has a key role in regulating the proliferation of endothelial cells (Goumans et al., 2002; David et al., 2007). The ALK1/ENG complex is, thus, a key component through which shear stress can block endothelial cells from entering the cell cycle (Baeyens et al., 2016; Figure 2). By recognizing both BMP9 and flow, the ALK1/ENG complex allows the modulation of vascular morphogenesis in response to flow (Baeyens et al., 2016). Mutations in SMAD4 and in the Growth differentiation factor 2 (GDF2) genes (encoding for SMAD4 and BMP9) had been also reported later on to cause variants of HHT. Both the endothelial-specific ablation of SMAD4 (a transcription factor in the ALK1/ENG pathway) and of ENG show increased proliferation of endothelial cells within the developing shunt (Ola et al., 2018; Tual-Chalot et al., 2020). The later, however, has been described to involve the VEGF signaling pathway (Tual-Chalot et al., 2020).
Transforming Growth Factor-β (TGF-β) is involved in the maintenance of the endothelium in a nonactivated state (Walshe et al., 2009) and protecting it from aberrant permeability and perfusion, and from apoptosis (Walshe et al., 2011). Interestingly, experiments on HUVECs demonstrated that shear stress activates TGF-β, leading to downstream activation of Krüppel-Like Factor 2 (KLF2) and eNOS in an ALK5 dependent manner (Walshe et al., 2013; Figure 2). Moreover, TGF-β malfunction through the SMAD signaling pathway has been linked to diverse cerebrovascular diseases related to aberrant endothelial cell proliferation (including HHT), as reviewed by Zhang and Yang (2020).
Flow can also modulate the endothelial cell cycle through other pathways. For example, in bovine aortic endothelial cells, 24 h of laminar shear stress (3–12 dyn/cm2) activated the phosphorylation of p53 protein through the JNK pathway. The increased levels of p53 in turn activated Growth Arrest and DNA Damage 45 (GADD45) and p21 proteins, inhibiting the Cyclin-Dependent Kinase (CDK) and, thus, arresting endothelial cell proliferation (Lin et al., 2000; Figure 2). Transcription factor Aryl hydrocarbon receptor (AhR) is also sensitive to shear stress. In this case, laminar shear stress between 6 and 15 dyn/cm2 induced –likely also through the JNK/p38 pathway– the expression and the translocation of AhR into the nucleus. In the nucleus, AhR promotes an increase in Cytochrome P450 Family 1 Subfamily A Member 1 (CYP1A1) expression and the subsequent shear stress-induced arrest of the cell cycle (Han et al., 2008; Figure 2).
Endothelial cells have an amazing ability to change their cell size. Vessel enlargement by hypertrophy results in an increase in the size of individual endothelial cells and can extremely rapidly increase vessel diameter. Endothelial cell density in vessels is very high, meaning that there is a large potential for growth purely by altering their size.
In normal embryonic development, no differences in cell density are observed along vessel growth based on somite stage. Nonetheless, between large and small vessels a 30% reduction in endothelial cell density was reported (Udan et al., 2013). The difference, however, was not large enough to explain the difference in vessel diameter, suggesting that size of endothelial cells might be involved. Similarly, in the retina there is no overall change in endothelial cells density as large vessels form, but in this case, the endothelial cell density was not compared between large and small vessels (Franco et al., 2015). In transgenic zebrafish embryos with constitutive or inducible expression of Placental Growth Factor (PLGF) under control of a somite muscle-specific promoter (PLGFmusc), cell size was found to contribute to vessel enlargement but, importantly, alone could not account for the diameter increase (Klems et al., 2020). As such, in normal vascular development as well, endothelial cell hypertrophy appears to contribute to vessel enlargement but never acts alone.
Though endothelial cell size increases appear to play a lesser role in developmental vessel enlargement, this process could still contribute to pathological vessel enlargement. In an AVM model of constitutive expression of active Notch4, pathological vessel enlargement occurs not due to an increase in endothelial cell density nor proliferation but, instead, due to an increased size of individual endothelial cells (Murphy et al., 2014). In zebrafish embryos, TRIO (Trio Rho Guanine Nucleotide Exchange Factor) activation, which in turn activates Ras homologous (Rho) GTPases, also leads to increased cell size causing an enlargement in arterial caliber (Klems et al., 2020). Interestingly, these results may be linked because shear-induced Notch activation has been shown to regulate and activate the assembly of a VE-Cadherin/LAR (leukocyte antigen-related protein tyrosine phosphatase)/TRIO complex (Polacheck et al., 2017; Figure 2). Other AVM models, such as the endothelial cell specific knockout of ENG (Choi et al., 2014), also present with an increase in endothelial cell size, both in zebrafish and mouse embryos (Sugden et al., 2017). Overall, however, the contribution of cell size changes to vessel growth is rarely assessed.
The process of vessel enlargement is referred to as arteriogenesis when it occurs in the mature arterial vascular network, but veins can also increase in diameter. The process of increasing venous diameter is so understudied that it lacks a name, though it is occasionally referred to as venogenesis.
In vascular development, fusion has been reported as the predominant mechanism of venous vessel enlargement in both mouse and chicken embryos (le Noble et al., 2004; Udan et al., 2013). In the chick embryo, the vitelline vein arises from a region that is genetically arterial before vessel enlargement begins (le Noble et al., 2004). In the developing retina, endothelial specific ablation of Cell Division Control protein 42 (CDC42 homolog) impairs migration, and enlarged veins and capillaries form without arterial enlargement (Lavina et al., 2018). This increased diameter was attributed to the presence of an increase in the number of endothelial cells per vessel length, without an increase in venous proliferation.
Understanding the process of vein enlargement requires mutants that specifically show changes in the diameter of veins. As such, somatic mutations that lead to venous malformations can be especially informative. Venous malformations are enlarged veins that present with few mural cells. The most well studied somatic mutations in venous malformation are the ones related to the Tyrosine-protein kinase (Tie2) receptor (Limaye et al., 2009; Soblet et al., 2013). Constitutive ablation of TIE2 leads to embryonic lethality at E10.5 (Sato et al., 1995). If TIE2 is ablated at later stages, however, arteries continue to form but veins do not (Chu et al., 2016). This is associated with a loss of venous markers [EphB4 (Ephrin type-B receptor 4), APJ (apelin receptor)] without any increase in arterial markers, indicating that this may be a defect in venous specification rather than vessel enlargement. Constitutively active forms of Tie2 that replicate somatic TIE2 mutations found in venous malformations, cause increased migration but loss of proper polarization in endothelial cells in vitro (Cai et al., 2019). However, it is not clear whether loss of proper cell identity or improper migration lead to vessel enlargement in venous malformations.
Though venous malformations arise from veins, some argue that all AVMs originate from venous endothelial cells. In biopsies from patients with telangiectasias, the enlargement of post-capillary venules precedes AVM formation (Braverman et al., 1990). In mouse models, exogenous expression of activated Notch4 induces AVM formation (Murphy et al., 2014). However, when this expression is limited to arteries, no AVMs form (Murphy et al., 2014). Deletion of the ENG only in venous and capillary endothelial cells results in the same rate of AVM formation as for deletion in all endothelial cells (Singh et al., 2020). Conversely, in retinas of an endothelial-specific ENG knockout model, imaging of developing AVMs showed that these initiated in arterioles and grew toward the venous vasculature (Jin et al., 2017). Although this would suggest an arterialization of the capillaries, in mouse models of AVM formation, the AVMs themselves express venous markers and downregulate arterial marker (Ola et al., 2018).
Another venous malformation that leads to increased venogenesis is varicose vein development. Though most research on vessel enlargement focuses on the role of shear stress as the stimuli for enlargement, varicose vein development occurs due to defective valves leading to an increase in hydrodynamic pressure (Welkie et al., 1992; Nicolaides et al., 1993). It should be noted, however, that the increased pressure in veins also leads to altered shear stress. On a cellular level, varicose vein development involves activation of endothelial cells leading to immune cell recruitment, increased vessel leakiness and loss of smooth muscle cells (Segiet et al., 2015). Varicose veins have upregulated Notch pathway genes like Delta Like Canonical Notch Ligand 4 (DLL4), Hairy/enhancer-of-split related with YRPW motif protein 2 (HEY2), and EPHB2 (Surendran et al., 2016). Smooth muscle cells become enlarged and surrounded by an increased amount of extracellular matrix, suggesting that they de-differentiate into a synthetic phenotype (Wali and Eid, 2001). Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs) play an important role in the development of varicose veins. Increases in TIMP-1 levels and in the TIMP/MMP-2 ratio lead to an increase in extracellular matrix deposition and a decrease in degradation processes (Badier-Commander et al., 2000). The mechanism of enlargement for varicose vein is, therefore, much more akin to collateral vessel enlargement than to vessel enlargement in developmental models.
Vessel enlargement occurs during embryonic development but continues to occur in the adult vascular networks. Any time the vasculature is exposed to a chronic change in flow, the vasculature adapts through either enlargement or inward remodeling to accommodate the altered flow. The most common experimental model for vessel enlargement in the adult is the remodeling of collateral vessels after occlusion, whether by partial occlusion of a coronary artery or by ligation of the femoral or middle cerebral artery. Other models, such as arteriovenous fistulas, have also been used to study the mechanisms of vessel enlargement. Vessel enlargement post-natally is inherently different than in developmental models, such as the retina, since post-natal enlargement requires the degradation of an existing basement membrane as well as the detachment and proliferation of mural cells. Furthermore, the remodeling of adult vessels, such as the collaterals, is initiated by endothelial cell activation, leukocyte invasion, and proliferation of vascular cells (Ma and Bai, 2020). This leads to the question, what parallels exist between vessel enlargement in developmental models and in adult models? And though there may be more than one way in which a vessel enlarges, it is likely that there will be common components from which we can gain significant insight.
In femoral artery ligation, arteriogenesis occurs far away from where ischemia is occurring (Resnick et al., 2003; Pipp et al., 2004). As such, just as with developmental angiogenesis, it is a process driven by shear stress. Just after coronary occlusion, the endothelial cells in the collateral vessel lose alignment with flow and take on a bulged appearance (Cai and Schaper, 2008). These cells increase DNA synthesis (Schaper et al., 1971; Pasyk et al., 1982) and proliferation, as indicated by Bromodeoxyuridine (BrdU) incorporation or Ki67 staining (Arras et al., 1998b; Wolf et al., 1998). Interestingly, these are all behaviors associated with low or recirculating shear stress patterns and not with increased shear stress levels. Adult endothelial cells, however, are adapted to the flow to which they are exposed and become activated in response to altered flow (Ward et al., 2020). Furthermore, though physiological shear stress reduces activation and proliferation, when shear levels are extremely elevated (above 30 dyn/cm2 in humans), the response is outward remodeling (Dolan et al., 2013). Thus, difference between developmental and adult remodeling may arise from either one of the following facts. On the one hand, developmental vasculature is naïve and therefore, it is not adapting to its “expected” flow; on the other hand, the stimulus for remodeling is a physiological level of shear stress (i.e., 15 dyn/cm2) for vessel enlargement during development, but an acute non-physiological level of shear stress (above 30 dyn/cm2) in models of vessel enlargement in the adult.
The activated endothelium produces NO that is essential for collateral growth. Both eNOS and iNOS (respectively, endothelial and inducible NOS) are upregulated in remodeling collateral vessel (Cai et al., 2004; Yang et al., 2015). When NO production is inhibited with L-NAME [N(G)-nitro L-arginine methyl ester], there is an almost complete inhibition of collateral enlargement (Eitenmuller et al., 2006; Park et al., 2010). It is not clear, however, whether this is due to a true inhibition of growth or related to increased vasoconstriction (Cai and Schaper, 2008). Endothelial NOS itself is involved in maintaining collateral vessel under physiological conditions. Mice that lack eNOS are born with a normal number of collateral vessels in the brain, but the number of these vessels decreases over the first 6 months of life as compared to age-matched controls (Dai and Faber, 2010). The primary role of NO in vessel enlargement is in the recruitment of immune cells (Park et al., 2010). Delivery of NO donors induces VE-Cadherin disassembly that is necessary for immune cell recruitment. Conversely, NO inhibitor L-NAME prevents increased vessel permeability and immune cell recruitment after vessel occlusion (Yang et al., 2015). Though NO is critical in vessel enlargement in post-natal stages, it has not been extensively investigated during development. The triple knockout of NOS enzymes is born at normal mendelian frequency with no reported vascular defects at birth (Morishita et al., 2005). Conversely, however, culture of E8.5 mouse embryos with the NO inhibitor L-NMMA [N(G)-monomethyl L-arginine] prevents the formation of large vessels in the yolk sac vasculature (Nath et al., 2004). NO influences endothelial cell proliferation (Morbidelli et al., 1996) and migration (Noiri et al., 1998). As such, NO could have a role in several of the mechanisms by which vessel enlarge.
Another difference between adult and developmental vessel enlargement is the involvement of immune cells. Immune cells, especially monocytes and macrophages, are essential for adult collateral growth and vessel enlargement in general (Arras et al., 1998a; Voskuil et al., 2003). Inhibiting monocyte recruitment in the adult impairs arteriogenesis during collateral remodeling (Heil et al., 2002, 2004). Recruited monocytes produce Tumor necrosis factor-α (TNF-α) and VEGF, which induces endothelial and smooth muscle cell mitoses (Schaper and Ito, 1996).
Though essential when vessel enlarge post-natally, functional immune cells may not be as present in vessel enlargement occurring just after the onset of flow in the embryo. The first immune cells form at E8.5 in the form of erythromyeloid progenitor and primitive macrophages (Gomez Perdiguero et al., 2015), and no functional role for these progenitors has been established until much later in development. At Hamburger Hamilton stage 18 in avian embryos (equivalent to E12.5–13.5 in mouse), circulating phagocytic cells are recruited to sites of vascular remodeling (Al-Roubaie et al., 2012). In the zebrafish embryo, depletion of myeloid cells using a pu.1 morpholino inhibits collateral growth in the gridlock mutant embryo (Gray et al., 2007). However, both these reports were for embryos at stages much older than the ones that gave the results showing vessel enlargement by migration and/or fusion (Chouinard-Pelletier et al., 2013). The retinal vasculature does form post-natally, when resident myeloid cells are present in the retina (Haupt et al., 2019). Ablation of macrophages using chlodronate liposomes results in a dramatic loss of vascular density, which makes it difficult to assess whether vessel enlargement itself is affected (Checchin et al., 2006).
One of the roles of immune cells in arteriogenesis is to degrade the basement membrane. During arteriogenesis, the elastic lamina is broken down by MMPs to give the vessels room to expand (Haas et al., 2007; Dodd et al., 2011). Inflammatory cells are also an important source of MMPs, as well as of other proteases. Macrophages secrete cytokines that induce MMP expression by endothelial cells (Galis et al., 1994a). Vessel enlargement in response to arteriovenous fistula induces a more than 1700-fold increase in MMP-2, along with a 12–60-fold increase in MMP-9, Membrane-type 1 MMP (MT1-MMP), and TIMP-2 (Sho et al., 2002). These increases correlate to the timing of elastic lamina degradation (Sho et al., 2002). Notably, MMPs are produced as inactive zymogens and, in addition, TIMPs can inhibit their activity. Therefore, increased expression of MMPs does not necessarily indicate increased activity. Indeed, many MMPs are constitutively expressed by endothelial cells and smooth muscle cells (Hanemaaijer et al., 1993; Galis et al., 1994a) but show no enzymatic activity until activated by disease (Galis et al., 1994b, 1995). Though essential in adult remodeling, no single mutant of MMPs has shown defects during embryogenesis. The double mutant of MMP2 and MT1-MMP does die perinatally, with a defect in the formation of vessels with wider diameters (Oh et al., 2004). This is, however, a very late stage of vascular development, after initial vascular remodeling has occurred.
One of the effects of immune cell recruitment and matrix degradation is an increase in permeability. Middle cerebral artery occlusion leads to an increase in permeability in the blood-brain-barrier to large molecules such as fibrinogen, Immunoglobulin G (IgG) or nanoparticles within 2–4 h of the occlusion (Okada et al., 1994; Fischer et al., 2002). Degradation is necessary for this increase in permeability, as inhibiting MMPs with BB-1101 prevents permeability increases immediately after middle cerebral artery occlusion (Rosenberg et al., 1998).
The presence of this extensive basement membrane is one of the main reasons that post-natal vessel enlargement is unlike to occur by vascular fusion. The presence of extensive matrix and mural cells from the arterioles onward would be a physical barrier for fusion. Hence, vascular fusion could only occur on the capillary level allowing arterioles to grow, which would then have to further increase in diameter by combination with another mechanism.
Vessel enlargement plays a critical role both during development as well as in the adult vasculature, with a high capacity for adapting to flow changes. Through an extremely fast responsiveness and interconnected processes such as fusion, endothelial cell migration and proliferation, vessels reshape in order to accommodate changes in flow rates such as to restore physiological levels. Because these processes are so much present along all the vasculature lifetime, dysregulations may entail critical pathologies. However, more and more pathways and molecular interconnections are being uncovered, shedding light to a better understanding and control over these pathologies.
LG-R and EAVJ contributed to writing, editing, and making figures. All authors contributed to the article and approved the submitted version.
This work was supported by the Fonds Wetenschappelijk Onderzoek (G091018N and G0B5920N) and by internal funding from the KU Leuven (IDN/19/031 and C14/19/095). This project has also received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No 848109.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Al-Roubaie, S., Hughes, J. H., Filla, M. B., Lansford, R., Lehoux, S., and Jones, E. A. (2012). Time-lapse microscopy of macrophages during embryonic vascular development. Dev. Dyn. 241, 1423–1431. doi: 10.1002/dvdy.23835
Arras, M., Ito, W. D., Scholz, D., Winkler, B., Schaper, J., and Schaper, W. (1998a). Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Invest. 101, 40–50. doi: 10.1172/JCI119877
Arras, M., Strasser, R., Mohri, M., Doll, R., Eckert, P., Schaper, W., et al. (1998b). Tumor necrosis factor-alpha is expressed by monocytes/macrophages following cardiac microembolization and is antagonized by cyclosporine. Basic Res. Cardiol. 93, 97–107. doi: 10.1007/s003950050069
Azizoglu, D. B., Chong, D. C., Villasenor, A., Magenheim, J., Barry, D. M., Lee, S., et al. (2016). Vascular development in the vertebrate pancreas. Dev. Biol. 420, 67–78. doi: 10.1016/j.ydbio.2016.10.009
Badier-Commander, C., Verbeuren, T., Lebard, C., Michel, J. B., and Jacob, M. P. (2000). Increased TIMP/MMP ratio in varicose veins: a possible explanation for extracellular matrix accumulation. J. Pathol. 192, 105–112. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH670>3.0.CO;2-1
Baeyens, N., Larrivee, B., Ola, R., Hayward-Piatkowskyi, B., Dubrac, A., Huang, B., et al. (2016). Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 214, 807–816. doi: 10.1083/jcb.201603106
Baeyens, N., Mulligan-Kehoe, M. J., Corti, F., Simon, D. D., Ross, T. D., Rhodes, J. M., et al. (2014). Syndecan 4 is required for endothelial alignment in flow and atheroprotective signaling. Proc. Natl. Acad. Sci. U. S. A. 111, 17308–17313. doi: 10.1073/pnas.1413725111
Beebe-Dimmer, J. L., Pfeifer, J. R., Engle, J. S., and Schottenfeld, D. (2005). The epidemiology of chronic venous insufficiency and varicose veins. Ann. Epidemiol. 15, 175–184. doi: 10.1016/j.annepidem.2004.05.015
Braverman, I. M., Keh, A., and Jacobson, B. S. (1990). Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J. Invest. Dermatol. 95, 422–427. doi: 10.1111/1523-1747.ep12555569
Buga, G. M., Gold, M. E., Fukuto, J. M., and Ignarro, L. J. (1991). Shear stress-induced release of nitric oxide from endothelial cells grown on beads. Hypertension 17, 187–193. doi: 10.1161/01.hyp.17.2.187
Cai, W. J., Kocsis, E., Luo, X., Schaper, W., and Schaper, J. (2004). Expression of endothelial nitric oxide synthase in the vascular wall during arteriogenesis. Mol. Cell. Biochem. 264, 193–200. doi: 10.1023/b:mcbi.0000044388.27953.a0
Cai, W., and Schaper, W. (2008). Mechanisms of arteriogenesis. Acta Biochim. Biophys. Sin. 40, 681–692. doi: 10.1093/abbs/40.8.681
Cai, Y., Schrenk, S., Goines, J., Davis, G. E., and Boscolo, E. (2019). Constitutive active mutant TIE2 induces enlarged vascular lumen formation with loss of Apico-basal polarity and Pericyte recruitment. Sci. Rep. 9:12352. doi: 10.1038/s41598-019-48854-2
Caolo, V., Peacock, H. M., Kasaai, B., Swennen, G., Gordon, E., Claesson-Welsh, L., et al. (2018). Shear stress and VE-cadherin. Arterioscler. Thromb. Vasc. Biol. 38, 2174–2183. doi: 10.1161/ATVBAHA.118.310823
Checchin, D., Sennlaub, F., Levavasseur, E., Leduc, M., and Chemtob, S. (2006). Potential role of microglia in retinal blood vessel formation. Invest. Ophthalmol. Vis. Sci. 47, 3595–3602. doi: 10.1167/iovs.05-1522
Chen, K., Pittman, R. N., and Popel, A. S. (2008). Nitric oxide in the vasculature: where does it come from and where does it go? A quantitative perspective. Antioxid. Redox Signal. 10, 1185–1198. doi: 10.1089/ars.2007.1959
Chen, X., Qin, J., Cheng, C. M., Tsai, M. J., and Tsai, S. Y. (2012). COUP-TFII is a major regulator of cell cycle and notch signaling pathways. Mol. Endocrinol. 26, 1268–1277. doi: 10.1210/me.2011-1305
Choi, E. J., Chen, W., Jun, K., Arthur, H. M., Young, W. L., and Su, H. (2014). Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One 9:e88511. doi: 10.1371/journal.pone.0088511
Chouinard-Pelletier, G., Jahnsen, E. D., and Jones, E. A. (2013). Increased shear stress inhibits angiogenesis in veins and not arteries during vascular development. Angiogenesis 16, 71–83. doi: 10.1007/s10456-012-9300-2
Chu, M., Li, T., Shen, B., Cao, X., Zhong, H., Zhang, L., et al. (2016). Angiopoietin receptor Tie2 is required for vein specification and maintenance via regulating COUP-TFII. elife 5:e21032. doi: 10.7554/eLife.21032
Corti, F., Wang, Y., Rhodes, J. M., Atri, D., Archer-Hartmann, S., Zhang, J., et al. (2019). N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat. Commun. 10:1562. doi: 10.1038/s41467-019-09605-z
Cui, C., Filla, M. B., Jones, E. A., Lansford, R., Cheuvront, T., Al-Roubaie, S., et al. (2013). Embryogenesis of the first circulating endothelial cells. PLoS One 8:e60841. doi: 10.1371/journal.pone.0060841
Dai, X., and Faber, J. E. (2010). Endothelial nitric oxide synthase deficiency causes collateral vessel rarefaction and impairs activation of a cell cycle gene network during arteriogenesis. Circ. Res. 106, 1870–1881. doi: 10.1161/CIRCRESAHA.109.212746
David, L., Mallet, C., Mazerbourg, S., Feige, J. J., and Bailly, S. (2007). Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953–1961. doi: 10.1182/blood-2006-07-034124
Deindl, E., Buschmann, I., Hoefer, I. E., Podzuweit, T., Boengler, K., Vogel, S., et al. (2001). Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 89, 779–786. doi: 10.1161/hh2101.098613
Dimmeler, S., Assmus, B., Hermann, C., Haendeler, J., and Zeiher, A. M. (1998). Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ. Res. 83, 334–341.
Dodd, T., Jadhav, R., Wiggins, L., Stewart, J., Smith, E., Russell, J. C., et al. (2011). MMPs 2 and 9 are essential for coronary collateral growth and are prominently regulated by p38 MAPK. J. Mol. Cell. Cardiol. 51, 1015–1025. doi: 10.1016/j.yjmcc.2011.08.012
Dolan, J. M., Kolega, J., and Meng, H. (2013). High wall shear stress and spatial gradients in vascular pathology: a review. Ann. Biomed. Eng. 41, 1411–1427. doi: 10.1007/s10439-012-0695-0
Drake, C. J., and Little, C. D. (1995). Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc. Natl. Acad. Sci. U. S. A. 92, 7657–7661. doi: 10.1073/pnas.92.17.7657
Drake, C. J., and Little, C. D. (1999). VEGF and vascular fusion: implications for normal and pathological vessels. J. Histochem. Cytochem. 47, 1351–1356.
Egorova, A. D., Khedoe, P. P., Goumans, M. J., Yoder, B. K., Nauli, S. M., Ten Dijke, P., et al. (2011). Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res. 108, 1093–1101. doi: 10.1161/CIRCRESAHA.110.231860
Ehling, M., Adams, S., Benedito, R., and Adams, R. H. (2013). Notch controls retinal blood vessel maturation and quiescence. Development 140, 3051–3061. doi: 10.1242/dev.093351
Eichmann, A., and Simons, M. (2012). VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 24, 188–193. doi: 10.1016/j.ceb.2012.02.002
Eitenmuller, I., Volger, O., Kluge, A., Troidl, K., Barancik, M., Cai, W. J., et al. (2006). The range of adaptation by collateral vessels after femoral artery occlusion. Circ. Res. 99, 656–662. doi: 10.1161/01.RES.0000242560.77512.dd
Fischer, S., Wobben, M., Marti, H. H., Renz, D., and Schaper, W. (2002). Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc. Res. 63, 70–80. doi: 10.1006/mvre.2001.2367
Franco, C. A., Jones, M. L., Bernabeu, M. O., Geudens, I., Mathivet, T., Rosa, A., et al. (2015). Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 13:e1002125. doi: 10.1371/journal.pbio.1002125
Galis, Z. S., Muszynski, M., Sukhova, G. K., Simon-Morrissey, E., Unemori, E. N., Lark, M. W., et al. (1994a). Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ. Res. 75, 181–189.
Galis, Z. S., Sukhova, G. K., Lark, M. W., and Libby, P. (1994b). Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest. 94, 2493–2503.
Galis, Z. S., Sukhova, G. K., and Libby, P. (1995). Microscopic localization of active proteases by in situ zymography: detection of matrix metalloproteinase activity in vascular tissue. FASEB J. 9, 974–980.
Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L., et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. doi: 10.1038/nature13989
Gooch, K. J., Dangler, C. A., and Frangos, J. A. (1997). Exogenous, basal, and flow-induced nitric oxide production and endothelial cell proliferation. J. Cell. Physiol. 171, 252–258.
Goumans, M. J., Valdimarsdottir, G., Itoh, S., Rosendahl, A., Sideras, P., and Ten Dijke, P. (2002). Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21, 1743–1753. doi: 10.1093/emboj/21.7.1743
Gray, C., Packham, I. M., Wurmser, F., Eastley, N. C., Hellewell, P. G., Ingham, P. W., et al. (2007). Ischemia is not required for arteriogenesis in zebrafish embryos. Arterioscler. Thromb. Vasc. Biol. 27, 2135–2141. doi: 10.1161/ATVBAHA.107.143990
Guo, D., Chien, S., and Shyy, J. Y. (2007). Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ. Res. 100, 564–571. doi: 10.1161/01.RES.0000259561.23876.c5
Haas, T. L., Doyle, J. L., Distasi, M. R., Norton, L. E., Sheridan, K. M., and Unthank, J. L. (2007). Involvement of MMPs in the outward remodeling of collateral mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 293, H2429–H2437. doi: 10.1152/ajpheart.00100.2007
Han, Z., Miwa, Y., Obikane, H., Mitsumata, M., Takahashi-Yanaga, F., Morimoto, S., et al. (2008). Aryl hydrocarbon receptor mediates laminar fluid shear stress-induced CYP1A1 activation and cell cycle arrest in vascular endothelial cells. Cardiovasc. Res. 77, 809–818. doi: 10.1093/cvr/cvm095
Hanemaaijer, R., Koolwijk, P., Le Clercq, L., de Vree, W. J., and van Hinsbergh, V. W. (1993). Regulation of matrix metalloproteinase expression in human vein and microvascular endothelial cells. Effects of tumour necrosis factor alpha, interleukin 1 and phorbol ester. Biochem. J. 296, 803–809. doi: 10.1042/bj2960803
Haupt, F., Krishnasamy, K., Napp, L. C., Augustynik, M., Limbourg, A., Gamrekelashvili, J., et al. (2019). Retinal myeloid cells regulate tip cell selection and vascular branching morphogenesis via notch ligand Delta-like 1. Sci. Rep. 9:9798. doi: 10.1038/s41598-019-46308-3
Heil, M., Ziegelhoeffer, T., Pipp, F., Kostin, S., Martin, S., Clauss, M., et al. (2002). Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Heart Circ. Physiol. 283, H2411–H2419. doi: 10.1152/ajpheart.01098.2001
Heil, M., Ziegelhoeffer, T., Wagner, S., Fernandez, B., Helisch, A., Martin, S., et al. (2004). Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ. Res. 94, 671–677. doi: 10.1161/01.RES.0000122041.73808.B5
Heller, R., Polack, T., Grabner, R., and Till, U. (1999). Nitric oxide inhibits proliferation of human endothelial cells via a mechanism independent of cGMP. Atherosclerosis 144, 49–57.
Hughes, S., and Chang-Ling, T. (2000). Roles of endothelial cell migration and apoptosis in vascular remodeling during development of the central nervous system. Microcirculation 7, 317–333. doi: 10.1080/mic.7.5.317.333
Iomini, C., Tejada, K., Mo, W., Vaananen, H., and Piperno, G. (2004). Primary cilia of human endothelial cells disassemble under laminar shear stress. J. Cell Biol. 164, 811–817. doi: 10.1083/jcb.200312133
Jacobi, J., Tam, B. Y., Wu, G., Hoffman, J., Cooke, J. P., and Kuo, C. J. (2004). Adenoviral gene transfer with soluble vascular endothelial growth factor receptors impairs angiogenesis and perfusion in a murine model of hindlimb ischemia. Circulation 110, 2424–2429. doi: 10.1161/01.CIR.0000145142.85645.EA
Jacobs, B. N., Andraska, E. A., Obi, A. T., and Wakefield, T. W. (2017). Pathophysiology of varicose veins. J. Vasc. Surg. Venous Lymphat. Disord. 5, 460–467. doi: 10.1016/j.jvsv.2016.12.014
Jiang, Y. J., Aerne, B. L., Smithers, L., Haddon, C., Ish-Horowicz, D., and Lewis, J. (2000). Notch signalling and the synchronization of the somite segmentation clock. Nature 408, 475–479. doi: 10.1038/35044091
Jin, Y., Muhl, L., Burmakin, M., Wang, Y., Duchez, A. C., Betsholtz, C., et al. (2017). Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 19, 639–652. doi: 10.1038/ncb3534
Jin, Z. G., Ueba, H., Tanimoto, T., Lungu, A. O., Frame, M. D., and Berk, B. C. (2003). Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ. Res. 93, 354–363. doi: 10.1161/01.RES.0000089257.94002.96
Johnson, D. W., Berg, J. N., Baldwin, M. A., Gallione, C. J., Marondel, I., Yoon, S. J., et al. (1996). Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13, 189–195.
Klems, A., van Rijssel, J., Ramms, A. S., Wild, R., Hammer, J., Merkel, M., et al. (2020). The GEF Trio controls endothelial cell size and arterial remodeling downstream of Vegf signaling in both zebrafish and cell models. Nat. Commun. 11:5319. doi: 10.1038/s41467-020-19008-0
Kuchan, M. J., and Frangos, J. A. (1994). Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am. J. Phys. 266, C628–C636.
Lavina, B., Castro, M., Niaudet, C., Cruys, B., Alvarez-Aznar, A., Carmeliet, P., et al. (2018). Defective endothelial cell migration in the absence of Cdc42 leads to capillary-venous malformations. Development 145:dev161182. doi: 10.1242/dev.161182
Le Noble, F., Moyon, D., Pardanaud, L., Yuan, L., Djonov, V., Matthijsen, R., et al. (2004). Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 131, 361–375. doi: 10.1242/dev.00929
Lee, C. W., Stabile, E., Kinnaird, T., Shou, M., Devaney, J. M., Epstein, S. E., et al. (2004). Temporal patterns of gene expression after acute hindlimb ischemia in mice: insights into the genomic program for collateral vessel development. J. Am. Coll. Cardiol. 43, 474–482. doi: 10.1016/j.jacc.2003.09.033
Li, M., Stenmark, K. R., Shandas, R., and Tan, W. (2009). Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J. Vasc. Res. 46, 561–571. doi: 10.1159/000226224
Limaye, N., Wouters, V., Uebelhoer, M., Tuominen, M., Wirkkala, R., Mulliken, J. B., et al. (2009). Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat. Genet. 41, 118–124. doi: 10.1038/ng.272
Lin, K., Hsu, P. P., Chen, B. P., Yuan, S., Usami, S., Shyy, J. Y., et al. (2000). Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. U. S. A. 97, 9385–9389. doi: 10.1073/pnas.170282597
Lloyd, P. G., Prior, B. M., Li, H., Yang, H. T., and Terjung, R. L. (2005). VEGF receptor antagonism blocks arteriogenesis, but only partially inhibits angiogenesis, in skeletal muscle of exercise-trained rats. Am. J. Physiol. Heart Circ. Physiol. 288, H759–H768. doi: 10.1152/ajpheart.00786.2004
Ma, T., and Bai, Y. P. (2020). The hydromechanics in arteriogenesis. Aging Med. 3, 169–177. doi: 10.1002/agm2.12101
McAllister, K. A., Grogg, K. M., Johnson, D. W., Gallione, C. J., Baldwin, M. A., Jackson, C. E., et al. (1994). Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 8, 345–351.
Meadows, S. M., Ratliff, L. A., Singh, M. K., Epstein, J. A., and Cleaver, O. (2013). Resolution of defective dorsal aortae patterning in Sema3E-deficient mice occurs via angiogenic remodeling. Dev. Dyn. 242, 580–590. doi: 10.1002/dvdy.23949
Meyer-Heim, A. D., and Boltshauser, E. (2003). Spontaneous intracranial haemorrhage in children: aetiology, presentation and outcome. Brain Dev. 25, 416–421. doi: 10.1016/s0387-7604(03)00029-9
Morbidelli, L., Chang, C. H., Douglas, J. G., Granger, H. J., Ledda, F., and Ziche, M. (1996). Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am. J. Phys. 270, H411–H415.
Morishita, T., Tsutsui, M., Shimokawa, H., Sabanai, K., Tasaki, H., Suda, O., et al. (2005). Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc. Natl. Acad. Sci. U. S. A. 102, 10616–10621. doi: 10.1073/pnas.0502236102
Murphy, P. A., Kim, T. N., Huang, L., Nielsen, C. M., Lawton, M. T., Adams, R. H., et al. (2014). Constitutively active Notch4 receptor elicits brain arteriovenous malformations through enlargement of capillary-like vessels. Proc. Natl. Acad. Sci. U. S. A. 111, 18007–18012. doi: 10.1073/pnas.1415316111
Nath, A. K., Enciso, J., Kuniyasu, M., Hao, X. Y., Madri, J. A., and Pinter, E. (2004). Nitric oxide modulates murine yolk sac vasculogenesis and rescues glucose induced vasculopathy. Development 131, 2485–2496. doi: 10.1242/dev.01131
Nicolaides, A. N., Hussein, M. K., Szendro, G., Christopoulos, D., Vasdekis, S., and Clarke, H. (1993). The relation of venous ulceration with ambulatory venous pressure measurements. J. Vasc. Surg. 17, 414–419.
Noiri, E., Lee, E., Testa, J., Quigley, J., Colflesh, D., Keese, C. R., et al. (1998). Podokinesis in endothelial cell migration: role of nitric oxide. Am. J. Phys. 274, C236–C244.
Oh, J., Takahashi, R., Adachi, E., Kondo, S., Kuratomi, S., Noma, A., et al. (2004). Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene 23, 5041–5048. doi: 10.1038/sj.onc.1207688
Okada, Y., Copeland, B. R., Fitridge, R., and Koziol, J. A., and Del Zoppo, G. J. (1994). Fibrin contributes to microvascular obstructions and parenchymal changes during early focal cerebral ischemia and reperfusion. Stroke 25, 1847–1853.
Ola, R., Kunzel, S. H., Zhang, F., Genet, G., Chakraborty, R., Pibouin-Fragner, L., et al. (2018). SMAD4 prevents flow induced arteriovenous malformations by inhibiting casein kinase 2. Circulation 138, 2379–2394. doi: 10.1161/CIRCULATIONAHA.118.033842
Park, B., Hoffman, A., Yang, Y., Yan, J., Tie, G., Bagshahi, H., et al. (2010). Endothelial nitric oxide synthase affects both early and late collateral arterial adaptation and blood flow recovery after induction of hind limb ischemia in mice. J. Vasc. Surg. 51, 165–173. doi: 10.1016/j.jvs.2009.08.045
Pasyk, S., Schaper, W., Schaper, J., Pasyk, K., Miskiewicz, G., and Steinseifer, B. (1982). DNA synthesis in coronary collaterals after coronary artery occlusion in conscious dog. Am. J. Phys. 242, H1031–H1037.
Peacock, H. M., Tabibian, A., Criem, N., Caolo, V., Hamard, L., Deryckere, A., et al. (2020). Impaired SMAD1/5 Mechanotransduction and Cx37 (Connexin37) expression enable pathological vessel enlargement and shunting. Arterioscler. Thromb. Vasc. Biol. 40, e87–e104. doi: 10.1161/ATVBAHA.119.313122
Pi, X., Wu, Y., Ferguson, J. E. 3rd, Portbury, A. L., and Patterson, C. (2009). SDF-1alpha stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc. Natl. Acad. Sci. U. S. A. 106, 5675–5680. doi: 10.1073/pnas.0809568106
Pipp, F., Boehm, S., Cai, W. J., Adili, F., Ziegler, B., Karanovic, G., et al. (2004). Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 24, 1664–1668. doi: 10.1161/01.ATV.0000138028.14390.e4
Pitulescu, M. E., Schmidt, I., Giaimo, B. D., Antoine, T., Berkenfeld, F., Ferrante, F., et al. (2017). Dll4 and notch signalling couples sprouting angiogenesis and artery formation. Nat. Cell Biol. 19, 915–927. doi: 10.1038/ncb3555
Polacheck, W. J., Kutys, M. L., Yang, J., Eyckmans, J., Wu, Y., Vasavada, H., et al. (2017). A non-canonical notch complex regulates adherens junctions and vascular barrier function. Nature 552, 258–262. doi: 10.1038/nature24998
Qin, J., Chen, X., Yu-Lee, L. Y., Tsai, M. J., and Tsai, S. Y. (2010). Nuclear receptor COUP-TFII controls pancreatic islet tumor angiogenesis by regulating vascular endothelial growth factor/vascular endothelial growth factor receptor-2 signaling. Cancer Res. 70, 8812–8821. doi: 10.1158/0008-5472.CAN-10-0551
Red-Horse, K., and Siekmann, A. F. (2019). Veins and arteries build hierarchical branching patterns differently: bottom-up versus top-down. BioEssays 41:e1800198. doi: 10.1002/bies.201800198
Red-Horse, K., Ueno, H., Weissman, I. L., and Krasnow, M. A. (2010). Coronary arteries form by developmental reprogramming of venous cells. Nature 464, 549–553. doi: 10.1038/nature08873
Resnick, N., Einav, S., Chen-Konak, L., Zilberman, M., Yahav, H., and Shay-Salit, A. (2003). Hemodynamic forces as a stimulus for arteriogenesis. Endothelium 10, 197–206. doi: 10.1080/10623320390246289
Ridnour, L. A., Isenberg, J. S., Espey, M. G., Thomas, D. D., Roberts, D. D., and Wink, D. A. (2005). Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. U. S. A. 102, 13147–13152. doi: 10.1073/pnas.0502979102
Rochon, E. R., Menon, P. G., and Roman, B. L. (2016). Alk1 controls arterial endothelial cell migration in lumenized vessels. Development 143, 2593–2602. doi: 10.1242/dev.135392
Rosenberg, G. A., Estrada, E. Y., and Dencoff, J. E. (1998). Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke 29, 2189–2195.
Rupp, P. A., Czirok, A., and Little, C. D. (2004). alphavbeta3 integrin-dependent endothelial cell dynamics in vivo. Development 131, 2887–2897. doi: 10.1242/dev.01160
Sato, Y., Poynter, G., Huss, D., Filla, M. B., Czirok, A., Rongish, B. J., et al. (2010). Dynamic analysis of vascular morphogenesis using transgenic quail embryos. PLoS One 5:e12674. doi: 10.1371/journal.pone.0012674
Sato, T. N., Tozawa, Y., Deutsch, U., Wolburg-Buchholz, K., Fujiwara, Y., Gendron-Maguire, M., et al. (1995). Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 376, 70–74.
Schaper, W., De Brabander, M., and Lewi, P. (1971). DNA synthesis and mitoses in coronary collateral vessels of the dog. Circ. Res. 28, 671–679.
Schaper, W., and Ito, W. D. (1996). Molecular mechanisms of coronary collateral vessel growth. Circ. Res. 79, 911–919.
Scholz, D., Cai, W. J., and Schaper, W. (2001). Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis 4, 247–257. doi: 10.1023/a:1016094004084
Scholz, D., Ito, W., Fleming, I., Deindl, E., Sauer, A., Wiesnet, M., et al. (2000). Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. 436, 257–270. doi: 10.1007/s004280050039
Segiet, O. A., Brzozowa-Zasada, M., Piecuch, A., Dudek, D., Reichman-Warmusz, E., and Wojnicz, R. (2015). Biomolecular mechanisms in varicose veins development. Ann. Vasc. Surg. 29, 377–384. doi: 10.1016/j.avsg.2014.10.009
Shay-Salit, A., Shushy, M., Wolfovitz, E., Yahav, H., Breviario, F., Dejana, E., et al. (2002). VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 99, 9462–9467. doi: 10.1073/pnas.142224299
Sho, E., Sho, M., Singh, T. M., Nanjo, H., Komatsu, M., Xu, C., et al. (2002). Arterial enlargement in response to high flow requires early expression of matrix metalloproteinases to degrade extracellular matrix. Exp. Mol. Pathol. 73, 142–153. doi: 10.1006/exmp.2002.2457
Silver, A. E., and Vita, J. A. (2006). Shear-stress-mediated arterial remodeling in atherosclerosis: too much of a good thing? Circulation 113, 2787–2789. doi: 10.1161/CIRCULATIONAHA.106.634378
Singh, E., Redgrave, R. E., Phillips, H. M., and Arthur, H. M. (2020). Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis 23, 559–566. doi: 10.1007/s10456-020-09731-z
Soblet, J., Limaye, N., Uebelhoer, M., Boon, L. M., and Vikkula, M. (2013). Variable somatic TIE2 mutations in half of sporadic venous malformations. Mol. Syndromol. 4, 179–183. doi: 10.1159/000348327
Su, T., Stanley, G., Sinha, R., D’amato, G., Das, S., Rhee, S., et al. (2018). Single-cell analysis of early progenitor cells that build coronary arteries. Nature 559, 356–362. doi: 10.1038/s41586-018-0288-7
Sugden, W. W., Meissner, R., Aegerter-Wilmsen, T., Tsaryk, R., Leonard, E. V., Bussmann, J., et al. (2017). Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat. Cell Biol. 19, 653–665. doi: 10.1038/ncb3528
Surendran, S., Ramegowda, K. S., Suresh, A., Binil Raj, S. S., Lakkappa, R. K., Kamalapurkar, G., et al. (2016). Arterialization and anomalous vein wall remodeling in varicose veins is associated with upregulated FoxC2-Dll4 pathway. Lab. Investig. 96, 399–408. doi: 10.1038/labinvest.2015.167
Tabibian, A., Ghaffari, S., Vargas, D. A., Van Oosterwyck, H., and Jones, E. A. V. (2020). Simulating flow induced migration in vascular remodelling. PLoS Comput. Biol. 16:e1007874. doi: 10.1371/journal.pcbi.1007874
Ten Dijke, P., Egorova, A. D., Goumans, M. J., Poelmann, R. E., and Hierck, B. P. (2012). TGF-beta signaling in endothelial-to-mesenchymal transition: the role of shear stress and primary cilia. Sci. Signal. 5:pt2. doi: 10.1126/scisignal.2002722
Thomas, D. D., Ridnour, L. A., Isenberg, J. S., Flores-Santana, W., Switzer, C. H., Donzelli, S., et al. (2008). The chemical biology of nitric oxide: implications in cellular signaling. Free Radic. Biol. Med. 45, 18–31. doi: 10.1016/j.freeradbiomed.2008.03.020
Toyota, E., Warltier, D. C., Brock, T., Ritman, E., Kolz, C., O’malley, P., et al. (2005). Vascular endothelial growth factor is required for coronary collateral growth in the rat. Circulation 112, 2108–2113. doi: 10.1161/CIRCULATIONAHA.104.526954
Tual-Chalot, S., Garcia-Collado, M., Redgrave, R. E., Singh, E., Davison, B., Park, C., et al. (2020). Loss of endothelial endoglin promotes high-output heart failure through peripheral arteriovenous shunting driven by VEGF signaling. Circ. Res. 126, 243–257. doi: 10.1161/CIRCRESAHA.119.315974
Ubezio, B., Blanco, R. A., Geudens, I., Stanchi, F., Mathivet, T., Jones, M. L., et al. (2016). Synchronization of endothelial Dll4-notch dynamics switch blood vessels from branching to expansion. elife 5:e12167. doi: 10.7554/eLife.12167
Udan, R. S., Vadakkan, T. J., and Dickinson, M. E. (2013). Dynamic responses of endothelial cells to changes in blood flow during vascular remodeling of the mouse yolk sac. Development 140, 4041–4050. doi: 10.1242/dev.096255
Ventrella, R., Kaplan, N., and Getsios, S. (2017). Asymmetry at cell-cell interfaces direct cell sorting, boundary formation, and tissue morphogenesis. Exp. Cell Res. 358, 58–64. doi: 10.1016/j.yexcr.2017.03.024
Vion, A. C., Alt, S., Klaus-Bergmann, A., Szymborska, A., Zheng, T., Perovic, T., et al. (2018). Primary cilia sensitize endothelial cells to BMP and prevent excessive vascular regression. J. Cell Biol. 217, 1651–1665. doi: 10.1083/jcb.201706151
Voskuil, M., Van Royen, N., Hoefer, I. E., Seidler, R., Guth, B. D., Bode, C., et al. (2003). Modulation of collateral artery growth in a porcine hindlimb ligation model using MCP-1. Am. J. Physiol. Heart Circ. Physiol. 284, H1422–H1428. doi: 10.1152/ajpheart.00506.2002
Wali, M. A., and Eid, R. A. (2001). Smooth muscle changes in varicose veins: an ultrastructural study. J. Smooth Muscle Res. 37, 123–135. doi: 10.1540/jsmr.37.123
Walshe, T. E., Dela Paz, N. G., and D’amore, P. A. (2013). The role of shear-induced transforming growth factor-beta signaling in the endothelium. Arterioscler. Thromb. Vasc. Biol. 33, 2608–2617. doi: 10.1161/ATVBAHA.113.302161
Walshe, T. E., Dole, V. S., Maharaj, A. S., Patten, I. S., Wagner, D. D., and D’amore, P. A. (2009). Inhibition of VEGF or TGF-{beta} signaling activates endothelium and increases leukocyte rolling. Arterioscler. Thromb. Vasc. Biol. 29, 1185–1192. doi: 10.1161/ATVBAHA.109.186742
Walshe, T. E., Leach, L. L., and D’amore, P. A. (2011). TGF-beta signaling is required for maintenance of retinal ganglion cell differentiation and survival. Neuroscience 189, 123–131. doi: 10.1016/j.neuroscience.2011.05.020
Ward, A. O., Angelini, G. D., Caputo, M., Evans, P. C., Johnson, J. L., Suleiman, M. S., et al. (2020). NF-kappaB inhibition prevents acute shear stress-induced inflammation in the saphenous vein graft endothelium. Sci. Rep. 10:15133. doi: 10.1038/s41598-020-71781-6
Welkie, J. F., Comerota, A. J., Kerr, R. P., Katz, M. L., Jayheimer, E. C., and Brigham, R. A. (1992). The hemodynamics of venous ulceration. Ann. Vasc. Surg. 6, 1–4.
Wolf, C., Cai, W. J., Vosschulte, R., Koltai, S., Mousavipour, D., Scholz, D., et al. (1998). Vascular remodeling and altered protein expression during growth of coronary collateral arteries. J. Mol. Cell. Cardiol. 30, 2291–2305.
Xu, C., Hasan, S. S., Schmidt, I., Rocha, S. F., Pitulescu, M. E., Bussmann, J., et al. (2014). Arteries are formed by vein-derived endothelial tip cells. Nat. Commun. 5:5758. doi: 10.1038/ncomms6758
Yang, B., Cai, B., Deng, P., Wu, X., Guan, Y., Zhang, B., et al. (2015). Nitric oxide increases arterial endotheial permeability through mediating VE-cadherin expression during arteriogenesis. PLoS One 10:e0127931. doi: 10.1371/journal.pone.0127931
You, L. R., Lin, F. J., Lee, C. T., Demayo, F. J., Tsai, M. J., and Tsai, S. Y. (2005). Suppression of notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435, 98–104. doi: 10.1038/nature03511
Zhang, Y., and Yang, X. (2020). The roles of TGF-beta signaling in cerebrovascular diseases. Front. Cell Dev. Biol. 8:567682. doi: 10.3389/fcell.2020.567682
Zhao, Y., Brandish, P. E., Ballou, D. P., and Marletta, M. A. (1999). A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc. Natl. Acad. Sci. U. S. A. 96, 14753–14758.
Keywords: venogenesis, migration, vascular fusion, mechanotransduction, collateral growth, arteriogenesis, vessel enlargement, arterial venous malformation
Citation: Gifre-Renom L and Jones EAV (2021) Vessel Enlargement in Development and Pathophysiology. Front. Physiol. 12:639645. doi: 10.3389/fphys.2021.639645
Edited by:
Anna Rita Cantelmo, Université Lille Nord de France, FranceReviewed by:
Anthony Wayne Orr, Louisiana State University Health Shreveport, United StatesCopyright © 2021 Gifre-Renom and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth A. V. Jones, bGl6LmpvbmVzQGt1bGV1dmVuLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.