
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 09 March 2021
Sec. Vascular Physiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.632260
This article is part of the Research TopicDiabetes, Hypertension and Cardiovascular DiseasesView all 14 articles
 Sara Vázquez-Sánchez1
Sara Vázquez-Sánchez1 Jonay Poveda1†
Jonay Poveda1† José Alberto Navarro-García1†
José Alberto Navarro-García1† Laura González-Lafuente1
Laura González-Lafuente1 Elena Rodríguez-Sánchez1
Elena Rodríguez-Sánchez1 Luis M. Ruilope1,2,3
Luis M. Ruilope1,2,3 Gema Ruiz-Hurtado1,2*
Gema Ruiz-Hurtado1,2*Fibroblast growth factor-23 (FGF)-23 is a phosphaturic hormone involved in mineral bone metabolism that helps control phosphate homeostasis and reduces 1,25-dihydroxyvitamin D synthesis. Recent data have highlighted the relevant direct FGF-23 effects on the myocardium, and high plasma levels of FGF-23 have been associated with adverse cardiovascular outcomes in humans, such as heart failure and arrhythmias. Therefore, FGF-23 has emerged as a novel biomarker of cardiovascular risk in the last decade. Indeed, experimental data suggest FGF-23 as a direct mediator of cardiac hypertrophy development, cardiac fibrosis and cardiac dysfunction via specific myocardial FGF receptor (FGFR) activation. Therefore, the FGF-23/FGFR pathway might be a suitable therapeutic target for reducing the deleterious effects of FGF-23 on the cardiovascular system. More research is needed to fully understand the intracellular FGF-23-dependent mechanisms, clarify the downstream pathways and identify which could be the most appropriate targets for better therapeutic intervention. This review updates the current knowledge on both clinical and experimental studies and highlights the evidence linking FGF-23 to cardiovascular events. The aim of this review is to establish the specific role of FGF-23 in the heart, its detrimental effects on cardiac tissue and the possible new therapeutic opportunities to block these effects.
Cardiovascular disease remains the leading cause of morbidity and mortality worldwide, and heart failure (HF) has become a major public health problem in the industrialised world (Members et al., 2012). The prevalence of coronary artery disease in the general population is 7.3% (Enbergs et al., 2000), while it is around 15–20% for left ventricular hypertrophy (LVH) (Weber, 1991) and 3.9% for HF (Mosterd et al., 1999). Recent research provides evidence that a high plasma level of fibroblast growth factor-23 (FGF)-23 is a hallmark of cardiac damage resulting in deleterious remodelling and the induction of cardiac hypertrophy. In the last few years, FGF-23 has been identified as a new trigger of cardiac dysfunction (Seiler et al., 2011; Leifheit-Nestler et al., 2018b; Navarro-García et al., 2019). Therefore, it is essential to not only understand the mechanisms whereby FGF-23 signals are transduced but also the variables associated with the increase in FGF-23 so new therapeutic strategies can be developed and applied in the near future (Chonchol et al., 2016; Rodelo-Haad et al., 2018).
FGF-23 is a hormone mainly synthesised by osteocytes and osteoblasts in long bones, although it can also be produced and secreted by different tissues, such as liver or even heart tissue under stress conditions (Hu et al., 2013). FGF-23 is involved in phosphate homeostasis and promotes renal phosphate excretion (Wolf, 2012; Mace et al., 2015), downregulation of 1,25-dihydroxyvitamin D (vitamin D) synthesis (Krajisnik et al., 2007; Mace et al., 2015; Navarro-García et al., 2018) and a decrease in parathyroid hormone (PTH) secretion (Navarro-García et al., 2018; von Jeinsen et al., 2019). FGF-23 is a 32 kDa protein encoded by the gene fgf23 located in chromosome 12 and belongs to the FGF subfamily FGF19, which corresponds to the endocrine FGFs (Hu et al., 2013). It appears in the circulating blood in two forms: intact full-length proteins and C-terminal proteins resulting from the rupture of the entire molecule by furin-like enzymes (Tagliabracci et al., 2014). FGF receptors (FGFRs) mediate the effects of FGF-23 and have tyrosine-kinase activity (Farrell and Breeze, 2018). There are four isoforms (FGFR1–4), but FGF-23 has a low affinity for all FGFRs and requires a cofactor to promote its union (Kurosu and Kuro-o, 2009). FGFRs are widely expressed in various tissues, including bone, kidney and heart tissues. In the kidneys, FGF-23 binds to FGFR1 using protein Klotho as a cofactor (Kurosu and Kuro-o, 2009); however, in the heart, FGF-23 binds to FGFR4 independently of Klotho (Grabner et al., 2015; Faul, 2017). Therefore, whether FGF-23 exerts its effect on the heart through an unknown cofactor or it does not need the cofactor link remains unclear (Urakawa et al., 2006).
FGF-23 was originally identified through a gene mutation in patients with autosomal dominant hypophosphatemic rickets or X-linked hypophosphatemic rickets that results in elevated serum levels of FGF-23 (White et al., 2000). The discovery of FGF-23 has revolutionised our understanding of mineral metabolism. Multilevel feedback and the kinetics of hormone actions within the bone–kidney–parathyroid loops system allow the controlled expression or action of the mineral–bone components, such as phosphates, PTH and vitamin D (Blau and Collins, 2015). Changes in any of these components lead to a misbalance in the feedback loops, altering the circulating levels of all these components and affecting several organs, including off-target organs such as the heart (Navarro-García et al., 2018). For instance, in chronic kidney disease (CKD), as kidney excretion capacity decreases, serum phosphate levels increase and stimulate FGF-23 synthesis, causing a rise of PTH and a deficiency of vitamin D, which can lead to adverse cardiovascular events like LVH and HF (Faul et al., 2011; Mathew et al., 2014; Navarro-García et al., 2018).
Recent evidence from human and experimental studies points to a key role of FGF-23 in the development and progression of cardiac disease. Significant changes in FGF-23 secretion and its action on the heart may have broad health implications that need to be elucidated. This review summarises our current understanding of FGF-23 and focuses on emerging areas of FGF-23 in human cardiac events and its intracellular pathways in cardiomyocytes and fibroblasts.
Systemic levels of FGF-23 are commonly analysed as an important biomarker for the diagnosis and prognosis of mineral bone disorders (MBD), and its measurement is recommended for patients with CKD. Currently, the extra-renal effects of this protein are becoming even more relevant, finding other targets such as the parathyroid gland and the cardiovascular and immune systems (Haffner and Leifheit-Nestler, 2017). The role of FGF-23 in cardiac disturbances is not clear or defined, especially the effects related not only to structural changes in the myocardium but also those related to cardiac dysfunction. High circulating levels of FGF-23 have been associated with cardiovascular pathophysiologic events, such as LVH, endothelial dysfunction and atherosclerosis (Lutsey et al., 2014). In addition, it remains unknown whether FGF-23 plays a detrimental role in cardiovascular events or if high plasma levels of FGF-23 are a simple consequence of cardiac disturbances. In this review, we identify the agreements and contradictions between clinical studies and put forward a scenario in which the specific role of FGF-23 in cardiovascular diseases can also be significantly harmful. Moreover, another unknown aspect that has arisen is the variability in FGF-23 levels according to different demographic variables, such as age, gender, ethnicity and clinical background. This issue is described in the following section.
High plasma levels of FGF-23 are associated with older age groups (>60 years) (Ix et al., 2012; Kestenbaum et al., 2014; Lutsey et al., 2014; Wright et al., 2014; Masson et al., 2015; Panwar et al., 2015; Speer et al., 2015; Souma et al., 2016; Ter Maaten et al., 2018; Huo et al., 2019), although some studies in younger populations have not found a significant correlation with age (Parker et al., 2010; Agarwal et al., 2014; di Giuseppe et al., 2014; Poelzl et al., 2014; Di Giuseppe et al., 2015; Chonchol et al., 2016; Takahashi et al., 2018). As age increases, metabolic disturbances are more prevalent; therefore, the lack of correlation with age may be due to the lack of metabolic changes. Another reason could be the low Klotho levels observed in older populations. Klotho is an anti-ageing factor, mainly expressed in the kidneys, which functions as a coreceptor of FGF-23 to increase the renal excretion of phosphates (Bian et al., 2014). As Klotho expression declines, serum phosphates increase and stimulate bone FGF-23 production, e.g., in CKD patients (Drew et al., 2017).
Interestingly, although the cardiovascular risk is higher in men, elevated FGF-23 levels are more commonly correlated with women (Parker et al., 2010; Seiler et al., 2011; Ix et al., 2012; Agarwal et al., 2014; di Giuseppe et al., 2014; Wright et al., 2014; Di Giuseppe et al., 2015; Masson et al., 2015; Panwar et al., 2015; Speer et al., 2015; Souma et al., 2016; Reindl et al., 2017; Ter Maaten et al., 2018; Cheng et al., 2020) than men (Kestenbaum et al., 2014; Chonchol et al., 2016; Huo et al., 2019). Nevertheless, some authors suggest that levels of FGF-23 could be independent of sex (Shibata et al., 2013; Lutsey et al., 2014; Poelzl et al., 2014; Koller et al., 2015). This discordance could be explained by the fact that most studies were carried out with older patients, including post-menopausal women. It is known that oestrogens diminish plasma FGF-23 levels, which might explain the increased FGF-23 levels in post-menopausal women compared to men (Ix et al., 2011). The physiological reason for the oestrogen effect on FGF-23 levels is that the oestrogen receptor regulates urinary phosphorus excretion at the proximal tubule, so in post-menopausal women, serum phosphorus levels are increased, thereby increasing FGF-23 secretion (Ix et al., 2011).
In contrast, the relationship between ethnicity and high plasma levels of FGF-23 is not yet clear. Some studies have associated greater levels of FGF-23 with Caucasian ethnicity (Ix et al., 2012; Chonchol et al., 2016; Patel et al., 2020), others with African American or Hispanic ethnicities (Lutsey et al., 2014; Wright et al., 2014), and others with none of them (Parker et al., 2010; Kestenbaum et al., 2014; Panwar et al., 2015; Robinson-Cohen et al., 2020). For instance, studies carried out in very diverse populations did not find any correlation with ethnicity variables (Kestenbaum et al., 2014). In this sense, Gutiérrez et al. (2010) demonstrated that a high level of serum phosphates, which leads to increased FGF-23 levels, is associated with low socioeconomic status independently of ethnicity. Some studies have even pointed to a relationship between FGF-23 levels and industrialisation, possibly due to different phosphate dietary intake through the addition of phosphates as food preservatives (Yuen et al., 2016). Therefore, future research and clinical studies should incorporate all these variables to achieve an intersectional perspective of the levels of FGF-23 in the general population (Table 1).

Table 1. Demographic and clinical variables correlated with high plasma levels of FGF-23.
FGF-23 is an early and complementary predictor of adverse cardiac events and could be suitable for improving risk assessment in vulnerable patients with HF or reduced ejection fraction (Koller et al., 2015). FGF-23 is positively correlated with but does not directly depend on the classical biomarkers of cardiac damage, such as N-terminal-pro-B-type natriuretic peptide (NT-proBNP) (Seiler et al., 2011; Shibata et al., 2013; Kestenbaum et al., 2014; Poelzl et al., 2014; Koller et al., 2015; Masson et al., 2015; Panwar et al., 2015; Speer et al., 2015; Wohlfahrt et al., 2015; Ter Maaten et al., 2018; von Jeinsen et al., 2019; Song et al., 2021), high-sensitive cardiac troponin T (hs-cTnT) (Kestenbaum et al., 2014; Masson et al., 2015; Song et al., 2021) and C-reactive protein (CRP) (Parker et al., 2010; Seiler et al., 2011; Ix et al., 2012; Lutsey et al., 2014; Panwar et al., 2015; Speer et al., 2015; Reindl et al., 2017; Song et al., 2021). Furthermore, it has already been demonstrated that the predictive value of the combination of these biomarkers on cardiovascular risk assessment is significantly greater than any of them alone (Wohlfahrt et al., 2015). In specific clinical circumstances, however, several authors have noted the relevance of FGF-23 as an independent biomarker. In this sense, Speer et al. (2015) demonstrated that the predictive value of FGF-23 for mortality and complications after cardiac surgery is comparable and even greater than the European System for Cardiac Operative Risk Evaluation (EuroSCORE II) and as good as that of NT-proBNP.
Elevated systemic FGF-23 levels have been strongly associated with an increased risk of mortality, including cardiovascular mortality (Parker et al., 2010; Ix et al., 2012; Ärnlöv et al., 2013b; Poelzl et al., 2014; Masson et al., 2015; Speer et al., 2015; Chonchol et al., 2016; Ter Maaten et al., 2018; von Jeinsen et al., 2019). Indeed, several studies have shown that patients with high plasma levels of FGF-23 have a higher incidence of cardiovascular events and less survival (Parker et al., 2010; Masson et al., 2015; Speer et al., 2015). This fact could explain the increased mortality risk in these patients. In this sense, several authors have associated high plasma levels of FGF-23 with a greater risk of cardiovascular death than other causes of death (Ärnlöv et al., 2013a; Lutsey et al., 2014; Souma et al., 2016; Silva et al., 2019). Marthi et al. (2018) carried out a meta-analysis that looked at thirty-four studies: 17 with the general population, nine with CKD non-dialysis patients and eight with the dialysis population. Overall, when comparing participants classified by FGF-23 quartiles, in the top versus bottom third of baseline FGF-23 concentration there was a 70% increased risk of all-cause mortality and 42% increased risk of cardiovascular mortality (Marthi et al., 2018).
In general terms, the increased mortality risk associated with FGF-23 may be a consequence of the augmented incidence of cardiovascular events in patients with higher serum FGF-23 levels. LVH, HF, arrhythmias, myocardial infarction (MI) and vascular alterations such as stroke and vascular calcification are the most common adverse and fatal cardiac events studied in relation to FGF-23 (Table 2). It is important to highlight, however, that higher levels of FGF-23 have been associated not only with cardiovascular mortality but also with a significant increase in the risk of non-cardiovascular causes of death. Therefore, FGF-23 should be considered a relevant and specific predictor of mortality, and the relevance of FGF-23 as a predictor of mortality will most likely increase in the coming years.

Table 2. Risk of cardiac events and high plasma levels of FGF-23.
FGF-23 was associated with the development of LVH in CKD patients for the first time in 2009 by Gutiérrez et al. (2009), and this was corroborated by Faul et al. (2011). Currently, several studies have associated high plasma levels of FGF-23 with increased left ventricular mass (LVM), and consequently, with the degree of cardiac hypertrophy developed in the general population (Gutiérrez et al., 2008; Mirza et al., 2009; Seiler et al., 2011; Shibata et al., 2013; Agarwal et al., 2014; Kestenbaum et al., 2014; Masson et al., 2015; Panwar et al., 2015; Reindl et al., 2017; Silva et al., 2019; Cheng et al., 2020; Patel et al., 2020). In particular, FGF-23 has been linked to a greater risk of concentric hypertrophy, which is apparently a compensated cardiac hypertrophy that results in increased wall thickness without ventricular dilation (Mirza et al., 2009; Silva et al., 2019). In addition, it has been demonstrated that serum FGF-23 levels are associated with LVM and LVH in the older population (Mirza et al., 2009) and with less left ventricular ejection fraction and LVM in cardiology inpatients (Shibata et al., 2013). Therefore, the relationship between FGF-23 and cardiac hypertrophy, as well as the former’s direct effect on cardiac remodelling through molecular pathways, has been well established and is discussed in the experimental section (section 3.2).
It is well established that LVH causes pathologic changes in heart structure and function, increasing the risk of adverse cardiovascular events such as arrhythmias, coronary disease and HF. Indeed, elevations in circulating FGF-23 levels have been shown to have a strong relationship with HF (Parker et al., 2010; Kestenbaum et al., 2014; Lutsey et al., 2014; Ferreira et al., 2019, 2020; Cheng et al., 2020; Robinson-Cohen et al., 2020). HF is a clinical syndrome caused by a structural and/or functional cardiac abnormality and is characterised by the inability of the heart to supply the peripheral tissue with enough blood and oxygen (Atherton et al., 2016). It is one of the most common causes of morbidity and mortality (Ziaeian and Fonarow, 2016). Andersen et al. (2016) analysed serum FGF-23 in patients with HF versus healthy subjects and showed that FGF-23 levels were significantly higher in HF patients (1526 ± 1601 versus 55 ± 20 RU/mL, p = 0.007). Focusing on myocardial tissue, these authors also compared intracardiac FGF-23 expression levels between HF patients and healthy subjects but no differences were found (Andersen et al., 2016). These results suggest that increased plasma, but not cardiac FGF-23 levels might play an important role in the development of HF. However, as FGF-23 can be synthesised at the intracardiac level under pathological or stress circumstances such as MI, the contribution of cardiac FGF-23 should be further studied (Andrukhova et al., 2015).
In addition, Di Giuseppe et al. (2015) demonstrated that the relationship between FGF-23 levels and the development of HF was independent of other established risk factor markers, such as NT-proBNP. Ix et al. (2012) also showed that this relationship was stronger when kidney function was impaired, possibly due to the extremely high serum FGF-23 levels found in patients with kidney dysfunction (Wolf, 2010, 2012). Nevertheless, the meta-analysis by Marthi et al. (2018) did not find a trend across different levels of kidney function in the association between high FGF-23 and HF. From a cardiac functional point of view, many studies have correlated high plasma levels of FGF-23 with a reduced left ventricular ejected fraction (LVEF; <40%), which indicates that high FGF-23 levels are directly linked to systolic dysfunction (Seiler et al., 2011; Shibata et al., 2013; Agarwal et al., 2014; Poelzl et al., 2014; von Jeinsen et al., 2019; Song et al., 2021). Furthermore, high plasma levels of FGF-23 are also associated with albuminuria in CKD which is strongly associated with Heart Failure with reduced Ejection Fraction (HFrEF), but not with Heart Failure with preserved Ejection Fraction (HFpEF) (Nayor et al., 2017). However, several articles have shown that increased FGF-23 levels are associated with both types of HF, HFpEF (Almahmoud et al., 2018; van de Wouw et al., 2019; Kanagala et al., 2020; Roy et al., 2020) and HFrEF (Koller et al., 2015; Gruson et al., 2017). Thus, further studies are required to clarify the role of FGF-23 in each HF subtype. HF symptoms are graduated in the New York Heart Association (NYHA) functional classification based on the limitations to the physical activity of the patients caused by cardiac symptoms (Atherton et al., 2016). Importantly, elevations in FGF-23 levels have been associated with the most severe classes on the NYHA scale (Poelzl et al., 2014; Koller et al., 2015; Wohlfahrt et al., 2015; Ter Maaten et al., 2018; von Jeinsen et al., 2019) and, therefore, with more severe cardiac functional impairment and more serious stages of HF.
FGF-23 has been related to heart rate disturbances causing arrhythmias, which can be classified as extra beats, supraventricular tachycardia including atrial fibrillation (AF), ventricular arrhythmias and bradyarrhythmias. The vast majority of the clinical studies have correlated high plasma levels of FGF-23 with AF (Seiler et al., 2011; Alonso et al., 2014; Mathew et al., 2014; Masson et al., 2015; Panwar et al., 2015; Ter Maaten et al., 2018; Chua et al., 2019; Dong et al., 2019). According to Mathew et al. (2014), each two-fold increase in circulating FGF-23 increased the risk of AF by 41% in their multi-ethnic study of atherosclerosis and by 30% in their cardiovascular health study. There are several mechanisms by which high plasma levels of FGF-23 could cause AF. FGF-23 induces changes in the cardiac structure such as LVH and is also linked to vascular calcification (Mathew et al., 2014). Both factors modify filling pressures, thus increasing atrial size, the main risk factor for AF (Mathew et al., 2014). Evidence of other types of rhythm alterations, such as those related to ventricular arrhythmia, is yet to be provided in a clinical setting, however.
Myocardial infarction is associated with severe complications, such as HF, pericarditis, arrhythmias, ventricular aneurysm and septal defects (Fox et al., 1979). The relationship between high plasma levels of FGF-23 and MI is controversial and requires further research to conclude whether high plasma levels of FGF-23 increase MI risk or not. Some authors have found a significant increase in the levels of FGF-23 after an MI (Ärnlöv et al., 2013b; Lutsey et al., 2014; Di Giuseppe et al., 2015; Takahashi et al., 2018; Ferreira et al., 2019); however, other authors have described a weak increase (Marthi et al., 2018) and some did not find any variation in FGF-23 at all (Parker et al., 2010; Taylor et al., 2011; Ix et al., 2012). At present, circulating FGF-23 levels in patients who have suffered an MI are being studied in three clinical trials that will shed light on this issue and lead to important future conclusions (reported at www.clinicaltrials.gov: NCT01971619, NCT02548364, and NCT03405207).
FGF-23 not only has a harmful effect on the heart but also on the vasculature, as shown by its relation to strokes (Parker et al., 2010; Ärnlöv et al., 2013b; Wright et al., 2014; Di Giuseppe et al., 2015; Marthi et al., 2018). Although Kestenbaum et al. (2014) found a non-significant relationship between elevated FGF-23 and all-cause stroke, Wright et al. (2014) found that elevated FGF-23 was a risk factor independent of CKD for overall stroke in a racially and ethnically diverse urban community. It is important to note that there are different types of strokes depending on the triggering cause: ischemic strokes, which include thrombotic strokes and cardioembolic strokes; haemorrhagic strokes; transient ischemic attacks; and cryptogenic strokes. A relationship has only been demonstrated between FGF-23 and haemorrhagic stroke (Di Giuseppe et al., 2015) and FGF-23 and cardioembolic stroke (Panwar et al., 2015), however. Therefore, although stroke is beyond the scope of this review, the harmful effects of FGF-23 on the vasculature deserve to be mentioned.
Vascular calcification is a major risk factor for cardiovascular disease which contributes to the high incidence of cardiovascular mortality (Pescatore et al., 2019). FGF-23, as a mineral metabolism factor, plays a direct role in calcification of tissues by modulating disturbances in calcium and phosphate balance (Donate-Correa et al., 2019). The relationship between increased systemic FGF-23 levels and vascular calcification has been shown experimentally in in vitro approaches (Jimbo et al., 2014), in patients with established cardiovascular disease (Donate-Correa et al., 2019), in pre-dialysis diabetic (Silva et al., 2015) and in CKD patients (Dzgoeva et al., 2016). Hence, FGF-23 outstands as a promising prognostic biomarker and therapeutic target in vascular calcification.
Consequently, FGF-23 may be considered as a biomarker and predictor of several important cardiac adverse events, but the mechanisms by which FGF-23 could have a harmful effect directly or indirectly on the heart remain misunderstood.
Finally, several clinical studies have linked the baseline characteristics of individuals with high plasma levels of FGF-23, discovering a direct correlation with bad health habits, MBD and CKD and with comorbidities such as diabetes and hypertension (Table 3).

Table 3. Other disorders and comorbidities correlated with high plasma levels of FGF-23.
Many authors have found a relationship between high plasma levels of FGF-23 and smoking (Parker et al., 2010; Seiler et al., 2011; Ix et al., 2012; Agarwal et al., 2014; di Giuseppe et al., 2014; Di Giuseppe et al., 2015; Masson et al., 2015; Panwar et al., 2015; Souma et al., 2016; Patel et al., 2020), and only a few authors have failed to find this relationship (Ärnlöv et al., 2013a; Kestenbaum et al., 2014). Furthermore, patients with low levels of physical activity seem to have greater levels of FGF-23 (Ix et al., 2012; Kestenbaum et al., 2014; Masson et al., 2015). Gardinier et al. (2016) also demonstrated a significant increase in FGF-23 levels during exercise, mainly due to the response in osteocytes to the increased expression of PTH. Other studies have not found any changes in FGF-23 levels during exercise, however, even after demonstrating significantly increased Klotho levels and decreased PTH levels (Fakhrpour et al., 2020). A recent study comparing master athletes with age-matched controls showed a significantly better redox balance in athletes, with significantly higher Klotho levels possibly explaining the lower FGF-23 levels in people with higher physical activity (Rosa et al., 2020). It would be interesting to separate physical activity from inadequate food intake, however, as this could also influence FGF-23 levels. In this sense, food preserved with phosphate additives is associated with higher FGF-23 levels (Yuen et al., 2016; Pool et al., 2020). Thus, future studies should further explore the relationship between chronic dietary phosphate intake, FGF-23 levels and their specific association with cardiovascular risk. As smoking, low levels of physical activity and inadequate food intake are cardiovascular risk factors, these factors would enhance the role of FGF-23 in cardiovascular outcomes.
Mineral bone disorders are usually the consequence of electrolyte imbalances, i.e., hyperphosphatemia, hyperkalaemia, or hypercalcemia (Navarro-García et al., 2018), which have been associated with high plasma levels of FGF-23. Indeed, elevated serum levels of FGF-23 are related to increased levels of phosphate (Parker et al., 2010; Seiler et al., 2011; Taylor et al., 2011; Shibata et al., 2013; Agarwal et al., 2014; Brandenburg et al., 2014; Kestenbaum et al., 2014; Lutsey et al., 2014; Poelzl et al., 2014; Wright et al., 2014; Panwar et al., 2015; Souma et al., 2016; Reindl et al., 2017), calcium (Ca2+) (Parker et al., 2010; Wright et al., 2014; Di Giuseppe et al., 2015; Panwar et al., 2015; Reindl et al., 2017; Patel et al., 2020) and PTH (Taylor et al., 2011; Brandenburg et al., 2014; di Giuseppe et al., 2014; Lutsey et al., 2014; Poelzl et al., 2014; Wright et al., 2014; Di Giuseppe et al., 2015; Souma et al., 2016; Sharma et al., 2020) and low levels of vitamin D (Shibata et al., 2013; Poelzl et al., 2014; Chonchol et al., 2016; Souma et al., 2016). Although it should also be noted that some authors did not find a relationship with Ca2+ (Agarwal et al., 2014), PTH (Shibata et al., 2013) or vitamin D (Taylor et al., 2011; Kestenbaum et al., 2014; Masson et al., 2015). It is well known that there is a complex network between FGF-23, PTH, vitamin D, calcium, phosphates and Klotho levels. This physiological network consists of a feedback mechanism through which disturbances in the level of one of these substances leads to changes in the levels of all the others (Navarro-García et al., 2018). Therefore, increased levels of PTH or vitamin D stimulate the synthesis of FGF-23 in the long bones and reduced renal Klotho expression increases serum phosphate load, which also induces FGF-23 synthesis. These mineral disturbances are classic complications in CKD, but in the last few decades, it has been shown that they also have a huge impact on cardiac tissue (Figure 1) because they have direct and indirect cardiotoxic effects, such as the development of LVH, HF or arrhythmias like AF (Navarro-García et al., 2018).
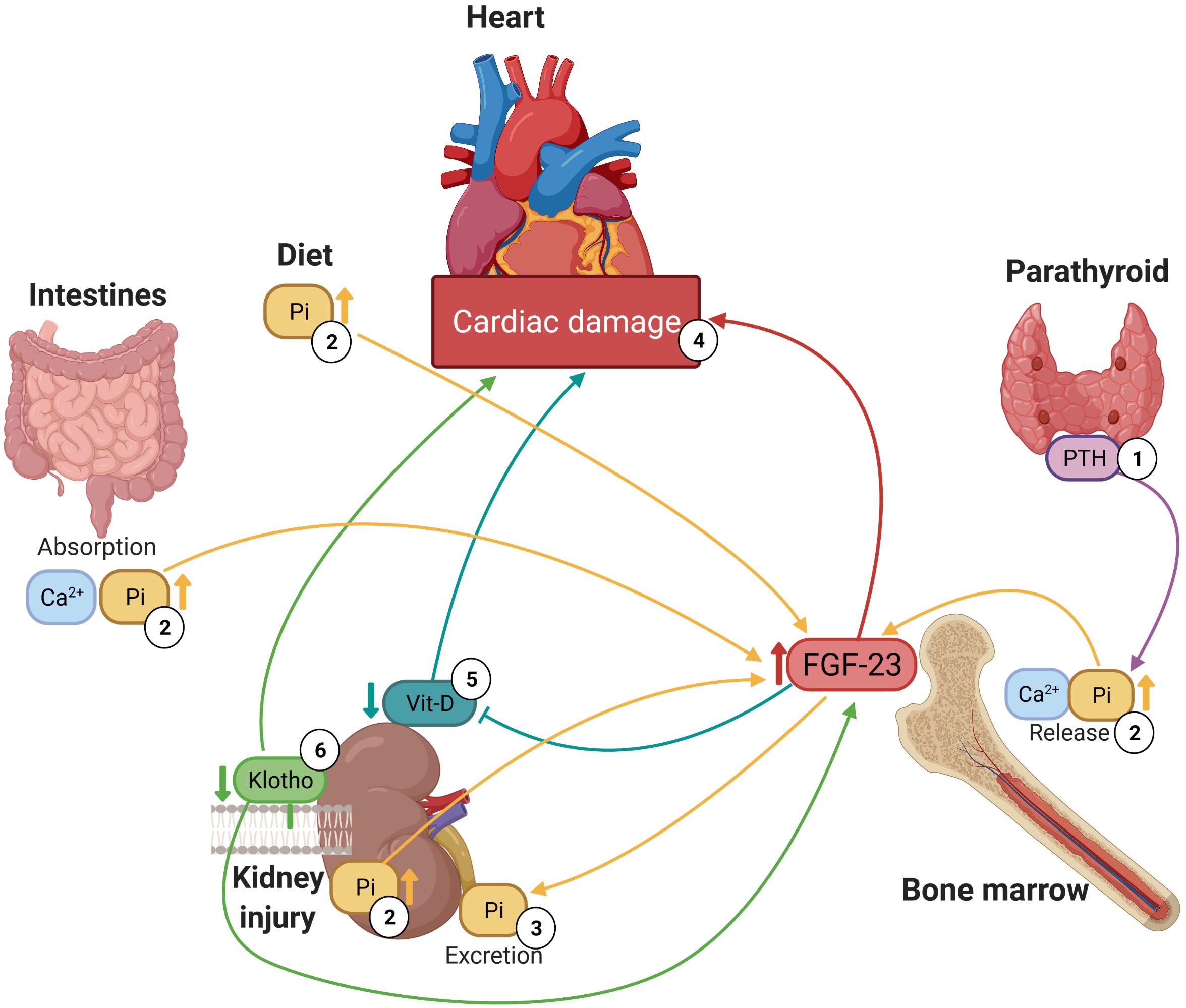
Figure 1. Relationship between high plasma levels of FGF-23, mineral bone metabolism components and cardiac damage. (1) Parathyroid stimulates the release of phosphate and calcium from large calcium stores in the bones into the bloodstream. (2) High plasma levels of phosphate promote FGF-23 synthesis in bone marrow. (3) FGF-23 enhances the excretion of phosphate in kidneys. (4) High circulating FGF-23 levels are associated with several forms as cardiac damage including hypertrophy and HF. (5) FGF-23 reduces circulating 1,25(OH)2D, active form of vitamin D, and deficiency of vitamin D is associated with cardiac damage. (6) Low levels of Klotho are linked to both high plasma levels of FGF-23 and cardiac damage.
In relation to kidney disease, high plasma levels of FGF-23 have also been associated with impaired kidney function as represented by an estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 (Parker et al., 2010; Seiler et al., 2011; Ix et al., 2012; Shibata et al., 2013; Ärnlöv et al., 2013a, b; Agarwal et al., 2014; Brandenburg et al., 2014; Kestenbaum et al., 2014; Lutsey et al., 2014; Poelzl et al., 2014; Wright et al., 2014; Koller et al., 2015; Panwar et al., 2015; Speer et al., 2015; Wohlfahrt et al., 2015; Souma et al., 2016; Ter Maaten et al., 2018; von Jeinsen et al., 2019; Patel et al., 2020; Sharma et al., 2020) and high levels of creatinine (Taylor et al., 2011; Ix et al., 2012; Shibata et al., 2013; Ärnlöv et al., 2013a; Agarwal et al., 2014; Kestenbaum et al., 2014; Wright et al., 2014; Koller et al., 2015; Masson et al., 2015; Panwar et al., 2015; Speer et al., 2015; Souma et al., 2016; Sharma et al., 2020; Song et al., 2021). di Giuseppe et al. (2014); Andersen et al. (2016), and Reindl et al. (2017) did not find evidence of this association; however, the number of subjects they studied was limited. CKD is characterised by excessive plasma levels of phosphate; therefore, increased synthesis of FGF-23 is a compensatory mechanism that preserves phosphate homeostasis by increasing urinary excretion. FGF-23 decreases the activity of the sodium-phosphate cotransporter in the renal proximal tubule and reduces dietary absorption by down-regulating the expression of the enzyme responsible for synthesising calcitriol, 1α-hydroxylase (Gutiérrez et al., 2008; Wolf, 2012). Therefore, high plasma levels of FGF-23 are a good biomarker for impaired renal function.
Two of the major comorbidities that contribute to the development of cardiovascular disease are diabetes and hypertension, both of which now have a high prevalence (Ruilope, 2012; Strain and Paldanius, 2018). Recent studies have associated elevated plasma levels of FGF-23 with a higher prevalence of diabetes mellitus (Parker et al., 2010; Ix et al., 2012; Agarwal et al., 2014; Kestenbaum et al., 2014; Lutsey et al., 2014; Wright et al., 2014; Panwar et al., 2015; Speer et al., 2015; Wohlfahrt et al., 2015; Sharma et al., 2020; Song et al., 2021) and hypertension (Ix et al., 2012; Agarwal et al., 2014; Kestenbaum et al., 2014; Lutsey et al., 2014; Wright et al., 2014; Masson et al., 2015; Patel et al., 2020; Sharma et al., 2020). Some studies have demonstrated a reduction in soluble Klotho levels in patients with impaired fasting glucose (Zhang and Liu, 2018), together with increased levels of FGF-23 (Zaheer et al., 2017) in diabetic patients compared to healthy volunteers, even with preserved kidney function. In the field of hypertension, Liu et al. and other studies have found an association between FGF-23 and arterial stiffness after renal transplantation when renal function is restored (Feng et al., 2020; Liu et al., 2020). These comorbidities contribute to an increased risk of mortality and adverse outcomes and worsen the quality of life of patients and might be explained by the cardiovascular effects of FGF-23. Some studies did not support this association (Parker et al., 2010; Ärnlöv et al., 2013a; Masson et al., 2015), however. Further studies are needed to clarify the independence of hypertension and diabetes in elevating systemic levels of FGF-23.
In conclusion, high plasma levels of FGF-23 are associated with certain demographics, such as age and sex; with cardiovascular diseases like LVH, HF and AF; with clinical disturbances such as low eGFR, high phosphate and NT-pro-BNP; and with comorbidities like diabetes and hypertension. All these studies point to FGF-23 as an important and independent biomarker involved in several pathologic processes, including cardiovascular disease. This idea must be interpreted with caution, however, as contradictions remain in the literature and a clear causal relationship is yet to be established, at least in a clinical setting. Hence, there is a need to experimentally address the relationship between high plasma levels of FGF-23 and cardiac disturbances to shed light on the origin of FGF-23 as a cause or consequence of cardiac damage and identify the specific FGF-23 downstream molecular pathways involved.
As already mentioned, over the last decade, many studies have shown a significant and independent relationship between high plasma levels of FGF-23 and detrimental cardiac outcomes in a clinical setting (Table 2). In this cardiovascular setting, an urgent roadmap for further research has emerged, and experiments aimed at unravelling the role of FGF-23 in cardiac pathology are needed. To date, research studies have demonstrated that FGF-23 might induce cardiac dysfunction, hypertrophy and fibrosis in the heart.
Correct heart function depends directly on the functional cells of the heart, that is, on the precise and coordinated contraction and relaxation cycle of cardiomyocytes (Fozzard, 1991). During contraction, intracellular Ca2+ levels of cardiomyocytes increase due to changes in the cardiomyocyte membrane action potential (AP) (Bers, 2002). AP is the reversible change in membrane potential as a consequence of the opening of different ion channels and leads to cardiomyocyte contraction in a process called excitation–contraction (EC) coupling, which represents the translation of electrical stimulation into mechanical work (Bers, 2002). Ca2+ is a key player in cardiac EC coupling (Fabiato and Fabiato, 1975) as a rise in free cytosolic Ca2+ is essential for the contraction of the cardiomyocytes (Bers, 2002). L-type calcium channels at the sarcolemma open due to the depolarisation of the AP, allowing the first Ca2+ influx into the cytoplasm (Fabiato and Fabiato, 1975). This Ca2+ influx activates ryanodine receptors (RyRs), triggering a greater amount of Ca2+ release from the sarcoplasmic reticulum (SR) to the cytoplasm (Bers, 2006). The intracellular Ca2+ concentration increases and Ca2+ binds to troponin C at the myofilaments, starting the cardiomyocyte contraction (Bers, 2006).
To allow physiological relaxation, the Ca2+ levels should decrease in the same amount as the previous increase (Eisner et al., 2017). This reduction of cytosolic Ca2+ mostly takes place via two different processes: 92% of cytosolic Ca2+ is pumped back to the SR by the action of the SR–Ca2+–adenosine triphosphatase 2a (SERCA) and, to a lesser extent (7%), Ca2+ is extruded from the cytoplasm to the extracellular medium by the Na+–Ca2+ exchanger (Bers, 2006). This Ca2+ handling in the cardiomyocytes is a complex physiological process perfectly regulated by different proteins, and it is well known that calmodulin kinase II (CaMKII) and cAMP-dependent protein kinase (PKA) regulate the open probability of RyRs through several phosphorylations at different sites, increasing their activity (Bovo et al., 2017). In addition, CaMKII and PKA can also phosphorylate phospholamban (PLB), a SR membrane protein that regulates SERCA activity. PLB binds to SERCA and inhibits its activity in situations of dephosphorylation (Kranias and Hajjar, 2012).
In this context, it has been shown that FGF-23 is involved in this Ca2+ handling by increasing phosphorylation of regulator proteins such as CaMKII (Kao et al., 2014; Navarro-García et al., 2019), which could drive cardiomyocytes to develop a cellular phenotype related to contractile dysfunction and predisposition to arrhythmias (Curran et al., 2010) (Figure 2). Recent works have demonstrated that FGF-23 might induce pro-arrhythmogenic Ca2+ events by itself in isolated adult rat ventricular cardiomyocytes due to alterations in Ca2+ handling (Navarro-García et al., 2019). Direct acute perfusion of 100 ng/mL FGF-23 reduced systolic Ca2+ release and increased the spontaneous diastolic Ca2+ leak, increasing the incidence of arrhythmogenic Ca2+ events (Navarro-García et al., 2019). This phenotype is frequently observed in failing cardiomyocytes (Ruiz-Hurtado et al., 2015; Alvarado et al., 2017; Chen et al., 2020), which might explain the relationship between FGF-23 levels and HF incidence described previously in this review. The events observed in cells are strongly influenced by Ca2+ mishandling and could be induced by RyRs hyperactivity and a reduction in SERCA activity, both of which are responsible for reduced SR Ca2+ load (Navarro-García et al., 2019). These alterations would lead to impaired cardiomyocyte contraction, represented by less cell shortening (Navarro-García et al., 2019).
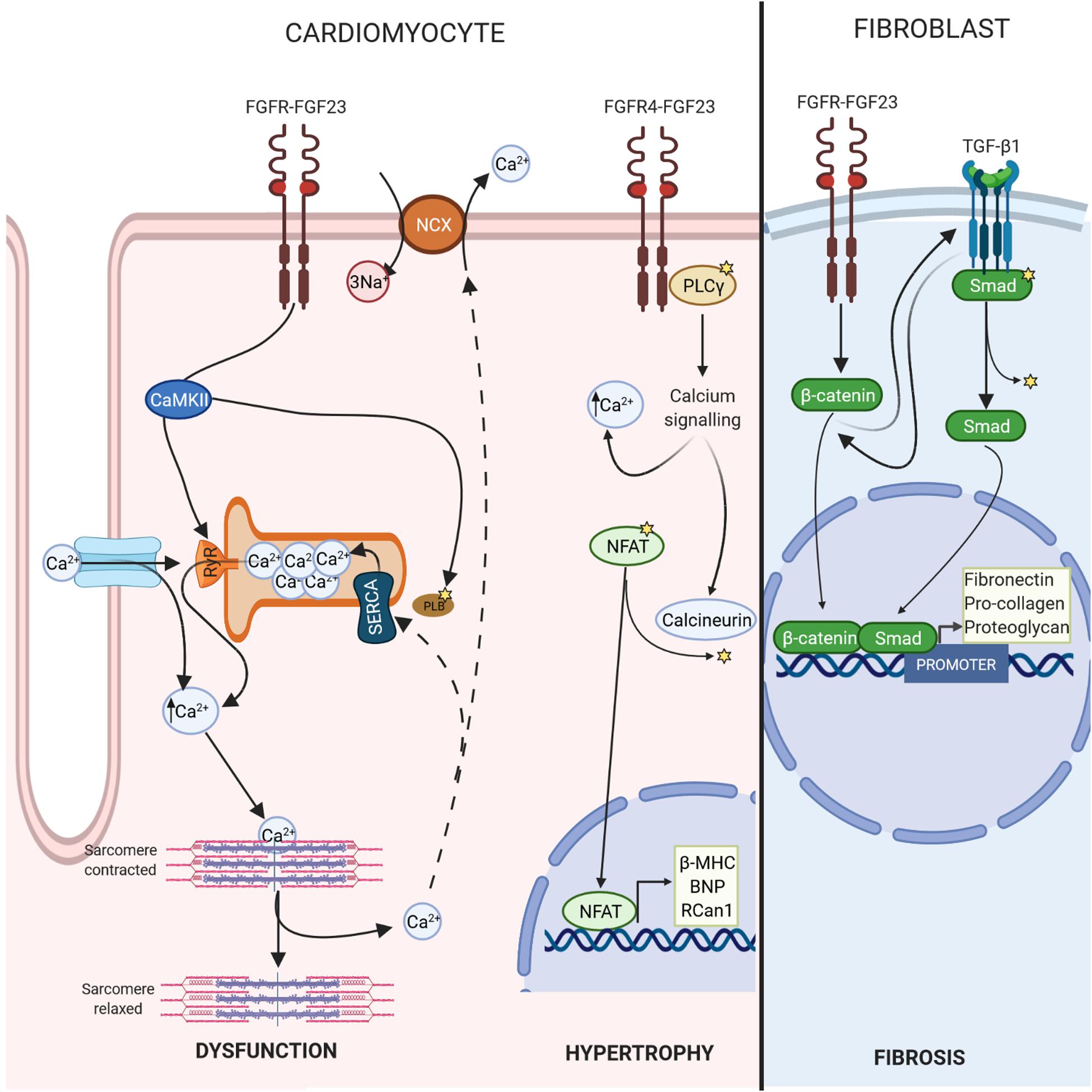
Figure 2. FGF-23-FGFR signalling. FGF-23 binds to the extracellular domain of its receptor, causing autophosphorylation. In cardiomyocytes, this change leads to phosphorylate calcium regulatory proteins, such as CAMKII, which promotes Ca2+ mishandling and cardiac dysfunction. In addition, FGF-23/FGFR4 triggers calcium cascade and the activation of calcineurin, which dephosphorylate NFAT inducing transcription of pro-hypertrophic genes such as β-MHC, BNP, or Rcan1. In fibroblasts, FGF-23 promotes fibrosis through activation of β-catenin.
Similar results related to cardiac dysfunction and predisposition to ventricular arrhythmia were found in nephrectomised mice and homozygous Klotho-hypomorphic (kl/kl) mice, both of which are experimental animal models that have middle (∼300 pg/mL) or very high (∼400.000 pg/mL) circulating FGF-23 levels (Navarro-García et al., 2020). Interestingly, when these mice are treated with recombinant Klotho, cardiac dysfunction and pro-arrhythmogenic Ca2+ release events are prevented (Navarro-García et al., 2020). This highlights the possible protection of Klotho against the deleterious effects of FGF-23. Moreover, other studies reported that the cellular HL-1 line of atrial murine cardiomyocytes had a greater L-type Ca2+ current and greater Ca2+ transients and SR Ca2+ content after maintained FGF-23 (25 ng/mL) exposure during 24 h of treatment compared to control cells (Kao et al., 2014). This higher SR Ca2+ content may be a consequence of increased SERCA activity due to phosphorylation of PLB at the CaMKII-specific site (Thr17) and not due to an intrinsic increase in the protein expression of SERCA or PLB (Kao et al., 2014). FGF-23 also dysregulated Ca2+ homeostasis, triggering delayed after depolarizations that have been related to the initiation of ventricular arrhythmias (Kao et al., 2014).
It is important to note that although both the cellular studies described above showed apparently different effects of FGF-23, the first showed the acute effect of FGF-23 on ventricular cardiomyocytes from a rat, while the second was carried out on an atrial cell line exposed to FGF-23 for longer periods. In both cases, the conclusion was that FGF-23 led to a greater predisposition to arrhythmias. Another effect of FGF-23 was described using mice ventricular muscle strips and showed that FGF-23 (9 ng/mL) was able to induce a significant increase in the peak isometric tension, slope and area of contractile waveforms (Touchberry et al., 2013). Therefore, in addition to the effect of FGF-23 on predisposition to cellular arrhythmia, it also could promote important alterations in cardiac function that regulate the contraction and relaxation process. It is well known that the main action of FGF-23, its phosphaturic action in the kidney, takes place via FGFR-binding; however, the FGFR isoform responsible for this effect of FGF-23 on cardiac function is yet to be identified, although some research has pointed to FGFR1 (Touchberry et al., 2013; Schumacher et al., 2019) or FGFR4 (Faul et al., 2011; Faul, 2017; Grabner et al., 2017).
Electrocardiogram (ECG) and echocardiography are two non-invasive methods used to estimate in vivo cardiac function. Ca2+ mishandling causes important changes in EC coupling, and this could also lead to pro-arrhythmogenic activity of cardiomyocytes, which is associated with abnormal heart rhythms. Indeed, a single-dose FGF-23 injection (40 μg/kg) in healthy mice was able to induce a significant increase (60%) in the occurrence of premature ventricular contractions recorded by ECG (Navarro-García et al., 2019). In addition, echocardiography allows the LV dimensions during systole and diastole, fractional shortening and ejection fraction to be measured. In general, mice with high circulating levels of FGF-23 had worse cardiac function than control mice, showing a lower percentage of fractional shortening, larger LV internal diameter in diastole, less cardiac output or higher LV diastolic pressure (Faul et al., 2011; Touchberry et al., 2013; Andrukhova et al., 2015; Hao et al., 2016; Han et al., 2020). Hence, FGF-23 could be an important factor causing changes in cardiac function and heart rhythm that can lead to the onset and worsening of cardiac disease.
Ca2+ abnormalities, usually those related to Ca2+ overload, precede cardiac dysfunction, while LVH develops later (Li et al., 2019). LVH, which is present in 15–20% of the general population, is one of the main risk factors for the development of a variety of adverse cardiac events and is related to pathological cardiac remodelling (Weber, 1991). It has already been demonstrated that FGF-23 promotes cardiac hypertrophy directly by acting on FGFR4, which subsequently triggers the phospholipase (PLC)γ/calcineurin/nuclear factor of activated T-cells (NFAT) signalling pathway (Faul et al., 2011; Grabner et al., 2015; Leifheit-Nestler et al., 2018b; Han et al., 2020). FGF-23 binds to FGFR4, causing its auto-phosphorylation, which then induces PLCγ activation through the binding of PLCγ to a specific phosphorylated tyrosine residue within the FGFR4 cytoplasmic sequence (Vainikka et al., 1994). Activated PLCγ increases cytoplasmic Ca2+ levels and activates calcineurin, the main regulator of intracellular Ca2+ (Eswarakumar et al., 2005), with the subsequent dephosphorylation of NFAT (Molkentin, 2004). Dephosphorylated NFAT translocates to the nucleus, where it promotes the transcription of pro-hypertrophic genes, including regulator of calcineurin 1 (Rcan1), brain natriuretic peptide (BNP), atrial natriuretic peptide (ANP) and β-myosin heavy chain (β-MHC) (Han et al., 2020) (Figure 2).
The hypertrophic effect of FGF-23 has been proven in in vitro and in vivo experiments. HL-1 atrial cells incubated with recombinant FGF-23 (rFGF-23) exhibited a higher cross-sectional surface area and an increase in pro-hypertrophic genes (Touchberry et al., 2013). Similarly, murine models with high levels of circulating FGF-23 showed greater mRNA levels of pro-hypertrophic genes, greater heart weight/body weight or heart weight/tibial length ratios and greater LV thickness (Faul et al., 2011; Andrukhova et al., 2015; Hao et al., 2016; Slavic et al., 2017; Leifheit-Nestler et al., 2018b; Han et al., 2020). Faul et al. (2011) published a large study where they demonstrated the hypertrophic effect of FGF-23. Cell surface area and expression of pro-hypertrophic genes in isolated neonatal rat ventricular myocytes (NRVM) increased after 48 h of treatment with FGF-23 (10, 25, and 100 ng/mL), while direct administration of rFGF-23 into the LV myocardium in mice caused cardiac hypertrophy after 14 days of treatment, even without an increase in circulating FGF-23 levels or other changes in mineral metabolism (Faul et al., 2011). Mice injected with FGF-23 showed greater heart weight/tibial length ratios and LV wall thickness in contrast to vehicle mice (Faul et al., 2011). This cardiac remodelling was also observed in mice after rFGF-23 (40 μg/kg) intravenous injections twice daily for 5 days, after which the immunohistochemistry and RT-PCR of pro-hypertrophic genes in the heart showed the presence of LVH development (Faul et al., 2011). Han et al. (2020) reported similar results in mice after intraperitoneal administration of rFGF-23 at 75 ng/g twice daily for 5 days. Leifheit-Nestler et al. (2018b) studied two different mouse strains: kl/kl mice and an experimental model for human X-linked hypophosphatemia with a mutation that inactivates the endopeptidases of the X chromosome (Hyp) mice. Both models showed high endogenous FGF-23 synthesis and elevated circulating FGF-23 levels compared to wild-type (WT) controls. kl/kl mice exhibited adverse cardiac remodelling, their heart weight/body weight ratios and cross-sectional areas of myocytes were higher than those of WT mice and the cardiac mRNA of their pro-hypertrophic genes was also increased. In the same way, experimental models of cardiac damage, such as post MI, ischemia reperfusion (I/R) and transversal aortic constriction, have been used to demonstrate that LVH is related to a profound increase in circulating levels of FGF-23, higher cardiac mRNA and protein expression of FGF-23 (Andrukhova et al., 2015; Hao et al., 2016; Slavic et al., 2017). All these data point to FGF-23 as a direct cause of LVH.
Furthermore, several studies have provided evidence that FGF-23 activates the PLCγ/calcineurin/NFAT signalling pathway. In vivo, healthy mice treated with rFGF-23 showed upregulated PLCγ protein levels compared to vehicle-treated mice (Han et al., 2020). In addition, in vitro treatment of rFGF-23 in NRVM activates PLCγ, and incubation with the PLCγ inhibitor U73122 blocks the hypertrophic effect of FGF-23 (Faul et al., 2011). Other evidence has been reported by Faul et al. (2011) and Grabner et al. (2015), who analysed NFAT activity after incubation with rFGF-23 in C2C12 myoblast (an immortalised mouse myoblast cell line) and in NRVM, respectively, and found that cells treated with FGF-23 showed a rise in NFAT activity compared to vehicle-treated cells. Also, Grabner et al. (2015) showed that this effect of FGF-23 was blocked by the calcineurin inhibitor cyclosporine A. In a study by Leifheit-Nestler et al. (2018b), qRT-PCR analysis revealed that cardiac FGFR4 mRNA expression was upregulated in both kl/kl and Hyp mice compared to their respective WT controls, but especially in kl/kl mice. In contrast, calcineurin protein levels were increased and NFAT phosphorylation was reduced only in kl/kl mice, therefore, the pro-hypertrophic genes were upregulated in kl/kl mice but not Hyp mice. There are two likely reasons for the differences observed between mouse strains. First, the circulating levels of FGF-23 are much higher in kl/kl mice. Second, the differences in Ca2+ and phosphate levels: kl/kl mice present with hyperphosphatemia and hypercalcemia, while Hyp mice have hypophosphatemia and normal levels of Ca2+. Interestingly, this hypertrophic effect is also influenced by the absence of Klotho, as kl/kl mice, which is the experimental model with the highest systemic FGF-23 levels, show a greater degree of hypertrophy in a Klotho-deficient context. It is also important to mention that soluble Klotho could act as a circulating lure for FGF-23, impeding its binding to cardiac FGFRs; act as a coreceptor in cardiomyocytes, causing a switch in or even the blockade of the FGF-23 signalling pathway; or interact with an unknown receptor activating the cardiac protective signalling pathway. Thus, Klotho could block or reduce the hypertrophic effect of FGF-23 on the heart, as it does with the deleterious effect of FGF-23 on cardiac function (Navarro-García et al., 2019, 2020). Indeed, several authors have shown the cardioprotective action of recombinant supplementation with Klotho in several experimental models of direct or indirect cardiac damage (Xie et al., 2012; Hu et al., 2017; Liu et al., 2019; Navarro-García et al., 2019, 2020; Wright et al., 2019; Han et al., 2020).
FGFR4 expression is upregulated when FGF-23 levels are elevated, therefore, FGF-23 is considered to be an inducer of cardiac hypertrophy through FGFR4 (Grabner et al., 2015). Immunoprecipitated PLCγ-bound FGFR4 was elevated in isolated NRVM treated for 30 min with FGF-23 at 25 ng/mL compared to control cells and cells co-treated with FGF-23 plus anti-FGFR4 antibodies (Grabner et al., 2015). Although a high phosphate (2%) diet is known to induce LVH and FGF-23 synthesis (Hu et al., 2015), FGFR4 knockout (KO) mice did not show LVH after 12 weeks of a high phosphate diet. In this way, blocking FGFR4 could be potentially useful for avoiding deleterious FGF-23 cardiac actions in a clinical setting (Grabner et al., 2015). Grabner et al. (2017) attributed a substantial role in LVH pathogenesis to FGFR4 and claimed that FGF-23/FGFR4-mediated LVH could be reversible, at least in rodents. To validate this hypothesis, they carried out three types of experiments. The first found that the hypertrophic effect was reversible after incubation of NRVM with FGF-23 for 24 h and then with anti-FGFR4 antibodies. In addition, pre-treatment with anti-FGFR4 antibodies prevented FGF-23-induced hypertrophy. In the second experiment, LVH induced by 5/6 nephrectomy in rats, a model that presents high FGF-23 levels, did not progress in animals treated with anti-FGFR4 antibodies for 4 weeks after surgery compared with untreated nephrectomised rats. In the third experiment, 18-month-old FGFR4 KO mice were protected from LVH (despite having high plasma levels of FGF-23) compared with WT mice, which, in normal conditions, at this age develop LVH (Grabner et al., 2017). Finally, a recent study using inducible FGFR4 KO mice in which FGFR4 was depleted only in the heart by tamoxifen treatment, found that they did not develop LVH after intraperitoneal injections of rFGF-23 at 75 ng/g twice daily for 5 days, unlike WT mice given the same treatment (Han et al., 2020). All these experimental studies show that FGF-23 activates cardiac PLCγ/calcineurin/NFAT signalling and development of LVH through FGFR4 (Figure 2).
Fibrosis is a response aimed at protecting an organ from an injurious event; however, it leads to massive deposition of the extracellular matrix (ECM), disrupting the normal tissue architecture and inducing destruction of the parenchyma (Franklin, 1997). Tissue fibrosis is responsible for increased organ size; therefore, an increase in the heart’s mass can be due to cellular hypertrophy or cardiac fibrosis or both. The cellular hypertrophic effect of FGF-23 has already been established (see section 3.2); however, the role of circulating and/or cardiac FGF-23 in the progression of cardiac fibrosis is not yet clear. Hao et al. (2016) suggested that FGF-23 promotes myocardial fibrosis through the activation of β-catenin, which is a profibrotic factor that cross-talks with transforming growth factor-β1 (TGF-β1) signalling, also an important fibrogenic agent. TGF-β1 receptors/Smad complexes stimulate chemotaxis in fibroblasts and increase the expression of collagen, fibronectin, and proteoglycans, the main components of the ECM (Leifheit-Nestler and Haffner, 2018). TGF-β1 signalling can induce the expression of β-catenin pathway members and vice versa (Guo et al., 2012). When β-catenin translocases into the nucleus, Smads and β-catenin form a complex at the promoter to coregulate specific gene expression and stimulate the transcription of fibrosis-related genes (Guo et al., 2012) (Figure 2). In adult myocardial fibroblasts, rFGF-23 induces proliferation in a dose-dependent manner (15, 25, and 50 ng/mL) through overexpression of profibrotic genes, such as β-catenin and procollagen I and II (Hao et al., 2016). Furthermore, FGF-23 increases the proliferation of neonatal rat cardiofibroblasts and expression of TGF-β1, TGF-β1 receptor/Smad complexes, connective tissue growth factor and collagen I (Leifheit-Nestler et al., 2018a; Böckmann et al., 2019). It has been shown that FGF-23 also intensifies the effect of TGF-B1 via FGFR1 promoting fibrosis, whereas cotreatment with an inhibitor of FGFR1 avoids this fibrosis (Kuga et al., 2020).
Böckmann et al. (2019) also hypothesised that FGF-23 might activate the local renin–angiotensin–aldosterone system, promoting fibrosis through increased cardiac mRNA expression levels of angiotensinogen, angiotensin-converting enzyme and angiotensin II receptor type 1. This hypothesis has already been demonstrated via the cotreatment of neonatal rat cardiofibroblasts with antihypertensive drugs like spironolactone, which prevented fibrotic development (Böckmann et al., 2019). Therefore, further studies are needed to unravel the different pathways through which FGF-23 induces cardiac fibrosis.
FGF-23-induced cardiac fibrosis has also been observed using in vivo experimental approaches. Leifheit-Nestler et al. (2018b) showed that kl/kl mice developed myocardial fibrosis due to a significant rise in the expression of collagen I and TGF-β1. Furthermore, Hu et al. (2015) showed that TGF-β1 is responsible for activating the extracellular signal-regulated kinase 1/2 pathway in kl/kl mice, which leads to cardiac hypertrophy and fibrosis. It has been demonstrated that direct damage to the myocardium raises cardiac levels of FGF-23 (Schumacher et al., 2019). In an MI mice model, FGF-23 and FGFR1 increased early in the myocardium (Schumacher et al., 2019). This cardiac FGF-23 could result from an increased uptake from the circulation or may be synthesised intrinsically in the heart (Schumacher et al., 2019). The main sources of FGF-23 in the heart are local fibroblasts, which increase fibroblast migration and the expression of fibronectin and collagen I (Schumacher et al., 2019). Hao et al. (2016) used adeno-associated virus-carrying FGF-23 injections in mice followed by MI or I/R. The treated mice showed significantly increased cardiac fibrosis compared to negative controls, and this effect was prevented by the inhibition of β-catenin (Hao et al., 2016). FGF-23 might have a positive role in healing after an MI by stimulating the fibroblasts to fill the empty area and produce ECM components (Schumacher et al., 2019); however, it induces pathologic fibrosis in the heart after long exposures and can also influence cardiac hypertrophy development.
Various therapeutic approaches are currently being considered to avoid the harmful effects of FGF-23. The key aspects are listed here as follows: FGF-23 antibodies, blockers of FGFRs and phosphate binders. In addition, Klotho as a potential therapy has been included based on the relevant cardio-protection showed by the exogenous recombinant Klotho supplementation examined in several experimental studies.
FGF-23 antibodies could be considered as the first therapeutic option; however, it is important to point out that FGF-23 is a pleiotropic factor that regulates many physiological processes, such as phosphate excretion. These processes would also be blocked with the use of these antibodies. For example, chronic treatment with FGF-23 antibodies for 6 weeks in an experimental CKD rat model improved secondary hyperparathyroidism by decreasing PTH and increasing vitamin D, but phosphate levels, aortic calcification and even mortality were significantly increased (Shalhoub et al., 2012). Therefore, this therapeutic option can be dismissed as the negative impact of the side effects might be greater than the clinical benefits as in situations of renal dysfunction. Currently, these antibodies have only been validated for the treatment of X-linked hypophosphatemic rickets caused by high plasma levels of FGF-23 (Carpenter et al., 2014). There have been several studies in humans (Carpenter et al., 2014; Imel et al., 2015) and Hyp mice (Yamazaki et al., 2008; Aono et al., 2009, 2011) that support the efficacy of FGF-23 antibodies in ameliorating biochemical, morphological, histological and clinical abnormalities associated with X-linked hypophosphatemia, but the safety of this treatment is still under investigation (Fukumoto, 2016) (reported at www.clinicaltrials.gov: NCT02163577 and NCT02312687).
FGF-23 exerts its action by binding to FGFRs 1–4, and several studies have proposed FGFR4 as the main FGFR isoform that mediates the effect of FGF-23 on the heart (Grabner et al., 2015, 2017; Leifheit-Nestler et al., 2016). Therefore, a useful therapeutic option currently being studied to impede FGF-23 effects on the heart consists of the specific blockage of FGFR4 (Grabner et al., 2017). This receptor has been related to the deleterious cardiovascular effect mediated by FGF-23, such as LVH inducing the overexpression of cardiac remodelling genes, including BNP and β-MHC (Leifheit-Nestler et al., 2018b). Moreover, the phosphaturic action of FGF-23 is mediated by FGFR1 in the kidneys. Therefore, blocking FGFR4 could prevent the adverse effects of FGF-23 on the heart without interfering in the other physiological functions of FGF-23, such as increased phosphate excretion in situations where the renal function has declined (Taylor et al., 2019). In support of this, Grabner et al. (2017) showed in vitro in NRVM that the hypertrophic effect of FGF-23 can be prevented by pre-treating these cells with anti-FGFR4. Furthermore, in the same study, LVH did not progress in 5/6 nephrectomised rats treated with anti-FGFR4 antibodies for 4 weeks after surgery compared with the control group (in which a further 28% increase in LVM was observed) (Grabner et al., 2017). These experimental results make FGFR4 an interesting therapeutic target, the safety of which has already been proven in clinical trials in cardiovascular diseases (reported at www.clinicaltrials.gov: NCT02476019). In addition, FGFR4 blockade is currently used in oncology trials as neoadjuvant chemotherapy to improve patients’ response, since it has a beneficial anti-angiogenic effect (Xin et al., 2018; Gluz et al., 2020).
High plasma levels of FGF-23 are correlated with high levels of phosphate, which are also considered to be a cause of cardiovascular adverse events (Zoccali et al., 2013). In this way, decreasing phosphate levels could improve the prognosis of patients with cardiovascular disease who have high plasma levels of FGF-23. There are three common types of phosphorus binders: Ca2+-containing binders, aluminium-containing binders and non-Ca2+ or Ca2+-free phosphate binders (Chan et al., 2017). These drugs are commonly used in the CKD population in combination with a dietary phosphate restriction (Ketteler and Biggar, 2013). Sharon et al. showed that patients with CKD and hyperparathyroidism treated with cinacalcet for 20 weeks had a 30% decrease in their levels of FGF-23 compared with the placebo group, and this was associated with a lower risk of cardiovascular events and mortality (Moe et al., 2015). In another study, Gonzalez-Parra et al. (2011) showed that treatment with the phosphate binder lanthanum carbonate in stage 3 CKD patients was associated with a decrease in serum FGF-23 levels and a decrease in the risk of adverse cardiovascular events. Nonetheless, the effect of these treatments is modest, and the levels of FGF-23 remain above the normal range despite the reported decreases (Zoccali et al., 2013).
Recent experimental research has pointed to Klotho as a potentially useful treatment for reducing or avoiding the harmful effects of FGF-23 on the heart. Indeed, Han et al. (2020) showed that the FGF-23/FGFR4 signalling is attenuated by intraperitoneal administration of soluble Klotho in mice, preventing FGF-23-induced LVH. Moreover, Hu et al. (2015) and Navarro-García et al. (2020) used a kl/kl mice to show that Klotho deficiency is a novel intermediate mediator of pathologic cardiac remodelling and dysfunction induced by high plasma levels of FGF-23. There are three hypothetical actions by which Klotho could prevent the effects of FGF-23 on the heart: by being a soluble lure for FGF-23 and impeding its binding to cardiac FGFR4; by acting as a coreceptor in cardiomyocytes, causing a switch in or even inhibition of the FGF-23 signalling pathway; or by interacting with an unknown receptor and activating the cardiac protective signalling pathway. To date, Klotho therapy has been studied only at the experimental level. Further research is needed to determine the ability of Klotho therapy to prevent the detrimental effects of FGF-23 in patients while maintaining the physiological phosphaturic FGF-23 action.
In summary, more experimental research and clinical data are required to identify which is the best option for controlling the deleterious effects of FGF-23, especially on the heart, while maintaining its physiological phosphaturic action. This option could involve blocking the FGF-23/FGFR4 axis or it could involve the promising therapies that use recombinant Klotho.
In conclusion, FGF-23 has a direct association with the development and progression of several events related to high cardiovascular risk, such as LVH, HF and arrhythmias, and its value as a cardiovascular biomarker in humans has been clinically demonstrated. Furthermore, a variety of in vitro and in vivo experimental approaches have indicated that FGF-23/FGFR axis signalling, especially that of the FGF-23/FGFR4 axis, provides an important base for the development of cardiac dysfunction through Ca2+ mishandling and cardiac hypertrophy and cardiac fibrosis through increased expression of target genes and proteins. Therefore, this axis represents an excellent therapeutic target for interrupting the harmful cardiac effects of FGF-23. Indeed, diverse neutralising anti-FGF-23 or anti-FGFR blocking antibodies are being tested in human clinical trials to assess their utility and safety. Whether FGF-23/FGFR signalling per se can initiate changes in cardiac function and structure or whether it only modulates cardiac remodelling in concert with other cardio-toxic factors remains unclear, however. Further research is needed to define whether FGF-23 effects are only deleterious or are dose- and/or time-dependent profitable effects, whether cardiomyocytes may themselves produce FGF-23, and the role of FGF-23 in myocardial damage. Therefore, it is essential to identify all the FGF-23-dependent pathways and their mediators that may have potential diagnostic, prognostic and therapeutic significance to cardiovascular diseases.
SV-S, JP, JAN-G, and GR-H wrote the manuscript with the contribution of LG-L, ER-S, and LR. All authors have read and agreed to the published version of the manuscript.
This work was supported by the Fundación Renal Íñigo Álvarez de Toledo (FRIAT) and by several projects at the Instituto de Salud Carlos III (PI17/01093, PI17/01193, and PI20/00763), and was co-funded by the European Regional Development Fund (Fondos FEDER).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
SV-S is an assistant researcher from the Education and Research Council of Madrid (PEJ-2019-AI/SAL-13521), ER-S is a PFIS Ph.D. student (FI18/00261), JP is a Sara Borrell researcher from ISCIII (CD19/00029) and GR-H is a Miguel Servet researcher from ISCIII (CP15/00129 and CPII20/00022).
Agarwal, I., Ide, N., Ix, J. H., Kestenbaum, B., Lanske, B., Schiller, N. B., et al. (2014). Fibroblast growth factor–23 and cardiac structure and function. J. Am. Heart Assoc. 3:e000584.
Almahmoud, M. F., Soliman, E. Z., Bertoni, A. G., Kestenbaum, B., Katz, R., Lima, J. A. C., et al. (2018). Fibroblast growth factor-23 and heart failure with reduced versus preserved ejection fraction: MESA. J. Am. Heart Assoc. 7:e008334.
Alonso, A., Misialek, J. R., Eckfeldt, J. H., Selvin, E., Coresh, J., Chen, L. Y., et al. (2014). Circulating fibroblast growth factor-23 and the incidence of atrial fibrillation: the atherosclerosis risk in Communities study. J. Am. Heart Assoc. 3:e001082.
Alvarado, F. J., Chen, X., and Valdivia, H. H. (2017). Ablation of the cardiac ryanodine receptor phospho-site Ser2808 does not alter the adrenergic response or the progression to heart failure in mice. Elimination of the genetic background as critical variable. J. Mol. Cell Cardiol. 103, 40–47. doi: 10.1016/j.yjmcc.2017.01.001
Andersen, I. A., Huntley, B. K., Sandberg, S. S., Heublein, D. M., and Burnett, J. C. Jr. (2016). Elevation of circulating but not myocardial FGF23 in human acute decompensated heart failure. Nephrol. Dial. Transplant. 31, 767–772. doi: 10.1093/ndt/gfv398
Andrukhova, O., Slavic, S., Odörfer, K. I., and Erben, R. G. (2015). Experimental myocardial infarction upregulates circulating fibroblast growth factor-23. J. Bone Mineral Res. 30, 1831–1839. doi: 10.1002/jbmr.2527
Aono, Y., Hasegawa, H., Yamazaki, Y., Shimada, T., Fujita, T., Yamashita, T., et al. (2011). Anti-FGF-23 neutralizing antibodies ameliorate muscle weakness and decreased spontaneous movement of Hyp mice. J. Bone Mineral Res. 26, 803–810. doi: 10.1002/jbmr.275
Aono, Y., Yamazaki, Y., Yasutake, J., Kawata, T., Hasegawa, H., Urakawa, I., et al. (2009). Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J. Bone Mineral Res. 24, 1879–1888. doi: 10.1359/jbmr.090509
Ärnlöv, J., Carlsson, A. C., Sundström, J., Ingelsson, E., Larsson, A., Lind, L., et al. (2013a). Higher fibroblast growth factor-23 increases the risk of all-cause and cardiovascular mortality in the community. Kidney Int. 83, 160–166. doi: 10.1038/ki.2012.327
Ärnlöv, J., Carlsson, A. C., Sundström, J., Ingelsson, E., Larsson, A., Lind, L., et al. (2013b). Serum FGF23 and risk of cardiovascular events in relation to mineral metabolism and cardiovascular pathology. Clin. J. Am. Soc. Nephrol. 8, 781–786. doi: 10.2215/cjn.09570912
Atherton, J. J., Bauersachs, J., Carerj, S., Ceconi, C., and Coca, A. (2016). Erol Ç, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. J. Heart Fail. 18, 891–975.
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology 21, 380–387. doi: 10.1152/physiol.00019.2006
Bian, A., Xing, C., and Hu, M. C. (2014). Alpha Klotho and phosphate homeostasis. J. Endocrinol. Invest. 37, 1121–1126. doi: 10.1007/s40618-014-0158-6
Blau, J. E., and Collins, M. T. (2015). The PTH-Vitamin D-FGF23 axis. Rev. Endocr. Metab. Disord. 16, 165–174.
Böckmann, I., Lischka, J., Richter, B., Deppe, J., Rahn, A., Fischer, D.-C., et al. (2019). FGF23-mediated activation of local RAAS promotes cardiac hypertrophy and fibrosis. Int. J.Mol. Sci. 20:4634. doi: 10.3390/ijms20184634
Bortnick, A. E., Xu, S., Kim, R. S., Kestenbaum, B., Ix, J. H., Jenny, N. S., et al. (2019). Biomarkers of mineral metabolism and progression of aortic valve and mitral annular calcification: the multi-ethnic study of atherosclerosis. Atherosclerosis 285, 79–86. doi: 10.1016/j.atherosclerosis.2019.04.215
Bovo, E., Huke, S., Blatter, L. A., and Zima, A. V. (2017). The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J. Mol. Cell. Cardiol. 104, 9–16. doi: 10.1016/j.yjmcc.2017.01.015
Brandenburg, V. M., Kleber, M. E., Vervloet, M. G., Tomaschitz, A., Pilz, S., Stojakovic, T., et al. (2014). Fibroblast growth factor 23 (FGF23) and mortality: the ludwigshafen risk and cardiovascular health study. Atherosclerosis 237, 53–59. doi: 10.1016/j.atherosclerosis.2014.08.037
Carpenter, T. O., Imel, E. A., Ruppe, M. D., Weber, T. J., Klausner, M. A., Wooddell, M. M., et al. (2014). Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J. Clin. Invest. 124, 1587–1597. doi: 10.1172/jci72829
Chan, S., Au, K., Francis, R. S., Mudge, D. W., Johnson, D. W., and Pillans, P. I. (2017). Phosphate binders in patients with chronic kidney disease. Austr. Prescriber. 40, 10–14.
Chen, J., Xu, S., Zhou, W., Wu, L., Wang, L., and Li, W. (2020). Exendin-4 reduces ventricular arrhythmia activity and calcium sparks-mediated sarcoplasmic reticulum Ca Leak in Rats with Heart failure. Int. Heart J. 61, 145–152. doi: 10.1536/ihj.19-327
Cheng, N., He, Y., Dang, A., Lv, N., Wang, X., and Li, H. (2020). Association between plasma fibroblast growth factor 23 and left ventricular mass index in patients with Takayasu arteritis. Clin. Rheumatol. 39, 1591–1599. doi: 10.1007/s10067-019-04895-6
Chonchol, M., Greene, T., Zhang, Y., Hoofnagle, A. N., and Cheung, A. K. (2016). Low vitamin D and high fibroblast growth factor 23 serum levels associate with infectious and cardiac deaths in the HEMO study. J. Am. Soc.Nephrol. 27, 227–237. doi: 10.1681/asn.2014101009
Chua, W., Purmah, Y., Cardoso, V. R., Gkoutos, G. V., Tull, S. P., Neculau, G., et al. (2019). Data-driven discovery and validation of circulating blood-based biomarkers associated with prevalent atrial fibrillation. Eur. Heart J. 40, 1268–1276. doi: 10.1093/eurheartj/ehy815
Curran, J., Brown, K. H., Santiago, D. J., Pogwizd, S., Bers, D. M., and Shannon, T. R. (2010). Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J. Mol. Cell. Cardiol. 49, 25–32. doi: 10.1016/j.yjmcc.2010.03.013
di Giuseppe, R., Buijsse, B., Hirche, F., Wirth, J., Arregui, M., Westphal, S., et al. (2014). Plasma fibroblast growth factor 23, parathyroid hormone, 25-hydroxyvitamin D3, and risk of heart failure: a prospective, case-cohort study. J. Clin. Endocrinol. Metab. 99, 947–955. doi: 10.1210/jc.2013-2963
Di Giuseppe, R., Kühn, T., Hirche, F., Buijsse, B., Dierkes, J., Fritsche, A., et al. (2015). Plasma fibroblast growth factor 23 and risk of cardiovascular disease: results from the EPIC-Germany case-cohort study. Eur. J. Epidemiol. 30, 131–141. doi: 10.1007/s10654-014-9982-4
Donate-Correa, J., Martín-Núñez, E., Hernández-Carballo, C., Ferri, C., Tagua, V. G., Delgado-Molinos, A., et al. (2019). Fibroblast growth factor 23 expression in human calcified vascular tissues. Aging 11, 7899–7913. doi: 10.18632/aging.102297
Dong, Q., Li, S., Wang, W., Han, L., Xia, Z., Wu, Y., et al. (2019). FGF23 regulates atrial fibrosis in atrial fibrillation by mediating the STAT3 and SMAD3 pathways. J. Cell Physiol. 234, 19502–19510. doi: 10.1002/jcp.28548
Drew, D. A., Katz, R., Kritchevsky, S., Ix, J., Shlipak, M., Gutiérrez, O. M., et al. (2017). Association between soluble klotho and change in kidney function: the health aging and body composition study. J. Am. Soc. Nephrol. 28, 1859–1866. doi: 10.1681/asn.2016080828
Dzgoeva, F. U., Sopoev, M. Y., Bestaeva, T. L., Khamitsaeva, O. V., Ktsoeva, F. A., and Sageeva, R. O. (2016). [Role of fibroblast growth factor 23 in the development of cardiovascular diseases in patients with end-stage renal failure on programmed hemodialysis]. Ter Arkh. 88, 51–56. doi: 10.17116/terarkh2016881251-56
Eisner, D. A., Caldwell, J. L., Kistamás, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. doi: 10.1016/b978-012436570-4/50007-8
Enbergs, A., Bürger, R., Reinecke, H., Borggrefe, M., Breithardt, G., and Kerber, S. (2000). Prevalence of coronary artery disease in a general population without suspicion of coronary artery disease: angiographic analysis of subjects aged 40 to 70 years referred for catheter ablation therapy. Eur. Heart J. 21, 45–52. doi: 10.1053/euhj.1999.1763
Eswarakumar, V., Lax, I., and Schlessinger, J. (2005). Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 16, 139–149.
Fabiato, A., and Fabiato, F. (1975). Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol. 249, 469–495. doi: 10.1113/jphysiol.1975.sp011026
Fakhrpour, R., Hamid Tayebi Khosroshahi, H., Ebrahim, K., Ahmadizad, S., Abbasnejad, M., Mesgari Abbasi, M., et al. (2020). Effect of sixteen weeks combined training on FGF-23, klotho, and fetuin-a levels in patients on maintenance hemodialysis. Iran J. Kidney Dis. 14, 212–218.
Farrell, B., and Breeze, A. L. (2018). Structure, activation and dysregulation of fibroblast growth factor receptor kinases: perspectives for clinical targeting. Biochem. Soc. Trans. 46, 1753–1770. doi: 10.1042/bst20180004
Faul, C. (2017). Cardiac actions of fibroblast growth factor 23. Bone 100, 69–79. doi: 10.1016/j.bone.2016.10.001
Faul, C., Amaral, A. P., Oskouei, B., Hu, M.-C., Sloan, A., Isakova, T., et al. (2011). FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 121, 4393–4408.
Feng, S., Wang, H., Yang, J., Hu, X., Wang, W., Liu, H., et al. (2020). Kidney transplantation improves arterial stiffness in patients with end-stage renal disease. Int. Urol. Nephrol. 52, 877–884. doi: 10.1007/s11255-020-02376-3
Ferreira, J. P., Sharma, A., Mehta, C., Bakris, G., Rossignol, P., White, W. B., et al. (2020). Multi-proteomic approach to predict specific cardiovascular events in patients with diabetes and myocardial infarction: findings from the EXAMINE trial. Clin. Res. Cardiol. [Epub ahead of print].
Ferreira, J. P., Verdonschot, J., Collier, T., Wang, P., Pizard, A., Bär, C., et al. (2019). Proteomic Bioprofiles and mechanistic pathways of progression to heart failure. Circ. Heart Fail. 12:e005897.
Fox, A. C., Glassman, E., and Isom, O. W. (1979). Surgically remediable complications of myocardial infarction. Prog. Cardiovasc. Dis. 21, 461–484. doi: 10.1016/0033-0620(79)90026-4
Fozzard, H. A. (1991). Excitation-Contraction Coupling in the Heart. Cellular and Molecular Mechanisms in Hypertension. Berlin: Springer, 135–142.
Franklin, T. (1997). Therapeutic approaches to organ fibrosis. Int. J. Biochem. Cell Biol. 29, 79–89. doi: 10.1016/s1357-2725(96)00121-5
Fukumoto, S. (2016). FGF23-FGF receptor/Klotho pathway as a new drug target for disorders of bone and mineral metabolism. Calcified Tissue Int. 98, 334–340. doi: 10.1007/s00223-015-0029-y
Gardinier, J. D., Al-Omaishi, S., Morris, M. D., and Kohn, D. H. (2016). PTH signaling mediates perilacunar remodeling during exercise. Matrix Biol. 52-54, 162–175. doi: 10.1016/j.matbio.2016.02.010
Gluz, O., Kolberg-Liedtke, C., Prat, A., Christgen, M., Gebauer, D., Kates, R., et al. (2020). Efficacy of deescalated chemotherapy according to PAM50 subtypes, immune and proliferation genes in triple-negative early breast cancer: primary translational analysis of the WSG-ADAPT-TN trial. Int. J. Cancer 146, 262–271. doi: 10.1002/ijc.32488
Gonzalez-Parra, E., Gonzalez-Casaus, M. L., Galán, A., Martinez-Calero, A., Navas, V., Rodriguez, M., et al. (2011). Lanthanum carbonate reduces FGF23 in chronic kidney disease Stage 3 patients. Nephrol. Dial. Transplant. 26, 2567–2571. doi: 10.1093/ndt/gfr144
Grabner, A., Amaral, A. P., Schramm, K., Singh, S., Sloan, A., Yanucil, C., et al. (2015). Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 22, 1020–1032.
Grabner, A., Schramm, K., Silswal, N., Hendrix, M., Yanucil, C., Czaya, B., et al. (2017). FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci. Rep. 7, 1–12.
Gruson, D., Ferracin, B., Ahn, S. A., and Rousseau, M. F. (2017). Head to head comparison of intact and C-terminal fibroblast growth factor 23 in heart failure patients with reduced ejection fraction. Int. J, Cardiol. 248, 270–273. doi: 10.1016/j.ijcard.2017.06.129
Guo, Y., Xiao, L., Sun, L., and Liu, F. (2012). Wnt/β-catenin signaling: a promising new target for fibrosis diseases. Physiol. Res. 61, 337–346. doi: 10.33549/physiolres.932289
Gutiérrez, O. M., Anderson, C., Isakova, T., Scialla, J., Negrea, L., Anderson, A. H., et al. (2010). Low socioeconomic status associates with higher serum phosphate irrespective of race. J. Am. Soc. Nephrol. 21, 1953–1960. doi: 10.1681/asn.2010020221
Gutiérrez, O. M., Januzzi, J. L., Isakova, T., Laliberte, K., Smith, K., Collerone, G., et al. (2009). Fibroblast growth factor-23 and left ventricular hypertrophy in chronic kidney disease. Circulation 119, 2545–2552. doi: 10.1161/circulationaha.108.844506
Gutiérrez, O. M., Mannstadt, M., Isakova, T., Rauh-Hain, J. A., Tamez, H., Shah, A., et al. (2008). Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 359, 584–592.
Haffner, D., and Leifheit-Nestler, M. (2017). Extrarenal effects of FGF23. Pediatr. Nephrol. 32, 753–765. doi: 10.1007/s00467-016-3505-3
Han, X., Cai, C., Xiao, Z., and Quarles, L. D. (2020). FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice. J. Mol. Cell. Cardiol. 138, 66–74. doi: 10.1016/j.yjmcc.2019.11.149
Hao, H., Li, X., Li, Q., Lin, H., Chen, Z., Xie, J., et al. (2016). FGF23 promotes myocardial fibrosis in mice through activation of β-catenin. Oncotarget 7, 64649–64664. doi: 10.18632/oncotarget.11623
Hu, M. C., Shi, M., Cho, H. J., Adams-Huet, B., Paek, J., Hill, K., et al. (2015). Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J. Am. Soc. Nephrol. 26, 1290–1302. doi: 10.1681/asn.2014050465
Hu, M. C., Shi, M., Gillings, N., Flores, B., Takahashi, M., Kuro-o, M., et al. (2017). Recombinant α-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 91, 1104–1114. doi: 10.1016/j.kint.2016.10.034
Hu, M. C., Shiizaki, K., Kuro-o, M., and Moe, O. W. (2013). Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Revi. Physiol. 75, 503–533. doi: 10.1146/annurev-physiol-030212-183727
Huo, Y. Y., Bai, X. J., Han, L. L., Wang, N., Han, W., and Sun, X. F. (2019). [Association of fibroblast growth factor 23 with age-related cardiac diastolic function subclinical state in a healthy Chinese population]. Zhonghua Yi Xue Za Zhi. 99, 1390–1396.
Imel, E. A., Zhang, X., Ruppe, M. D., Weber, T. J., Klausner, M. A., Ito, T., et al. (2015). Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J. Clin. Endocrinol. Metab. 100, 2565–2573. doi: 10.1210/jc.2015-1551
Ix, J. H., Chonchol, M., Laughlin, G. A., Shlipak, M. G., and Whooley, M. A. (2011). Relation of sex and estrogen therapy to serum fibroblast growth factor 23, serum phosphorus, and urine phosphorus: the Heart and Soul Study. Am. J. Kidney Dis. 58, 737–745. doi: 10.1053/j.ajkd.2011.06.011
Ix, J. H., Katz, R., Kestenbaum, B. R., de Boer, I. H., Chonchol, M., Mukamal, K. J., et al. (2012). Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J. Am. Coll. Cardiol. 60, 200–207. doi: 10.1016/j.jacc.2012.03.040
Jimbo, R., Kawakami-Mori, F., Mu, S., Hirohama, D., Majtan, B., Shimizu, Y., et al. (2014). Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 85, 1103–1111. doi: 10.1038/ki.2013.332
Kanagala, P., Arnold, J. R., Khan, J. N., Singh, A., Gulsin, G. S., Eltayeb, M., et al. (2020). Fibroblast-growth-factor-23 in heart failure with preserved ejection fraction: relation to exercise capacity and outcomes. ESC Heart Fail. 7, 4089–4099. doi: 10.1002/ehf2.13020
Kao, Y. H., Chen, Y. C., Lin, Y. K., Shiu, R. J., Chao, T. F., Chen, S. A., et al. (2014). FGF-23 dysregulates calcium homeostasis and electrophysiological properties in HL-1 atrial cells. Eur. J. Clin. Inves. 44, 795–801. doi: 10.1111/eci.12296
Kestenbaum, B., Sachs, M. C., Hoofnagle, A. N., Siscovick, D. S., Ix, J. H., Robinson-Cohen, C., et al. (2014). Fibroblast growth factor-23 and cardiovascular disease in the general population: the multi-ethnic study of atherosclerosis. Circ. Heart Fail. 7, 409–417. doi: 10.1161/circheartfailure.113.000952
Ketteler, M., and Biggar, P. H. (2013). Use of phosphate binders in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 22, 413–420. doi: 10.1097/mnh.0b013e32836214d4
Koller, L., Kleber, M. E., Brandenburg, V. M., Goliasch, G., Richter, B., Sulzgruber, P., et al. (2015). Fibroblast growth factor 23 is an independent and specific predictor of mortality in patients with heart failure and reduced ejection fraction. Circ. Heart Fail. 8, 1059–1067. doi: 10.1161/circheartfailure.115.002341
Krajisnik, T., Björklund, P., Marsell, R., Ljunggren, O., Akerström, G., Jonsson, K. B., et al. (2007). Fibroblast growth factor-23 regulates parathyroid hormone and 1-hydroxylase expression in cultured bovine parathyroid cells. J. Endocrinol. 195, 125–131. doi: 10.1677/joe-07-0267
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/circresaha.111.259754
Kuga, K., Kusakari, Y., Uesugi, K., Semba, K., Urashima, T., Akaike, T., et al. (2020). Fibrosis growth factor 23 is a promoting factor for cardiac fibrosis in the presence of transforming growth factor-β1. PLoS One 15:e0231905. doi: 10.1371/journal.pone.0231905
Kurosu, H., and Kuro-o, M. (2009). The Klotho gene family as a regulator of endocrine fibroblast growth factors. Mol. Cell. Endocrinol. 299, 72–78. doi: 10.1016/j.mce.2008.10.052
Leifheit-Nestler, M., Große Siemer, R., Flasbart, K., Richter, B., Kirchhoff, F., Ziegler, W. H., et al. (2016). Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol. Dial Transplant. 31, 1088–1099. doi: 10.1093/ndt/gfv421
Leifheit-Nestler, M., and Haffner, D. (2018). Paracrine effects of FGF23 on the heart. Front. Endocrinol. 9:278. doi: 10.3389/fendo.2018.00278
Leifheit-Nestler, M., Kirchhoff, F., Nespor, J., Richter, B., Soetje, B., Klintschar, M., et al. (2018a). Fibroblast growth factor 23 is induced by an activated renin–angiotensin–aldosterone system in cardiac myocytes and promotes the pro-fibrotic crosstalk between cardiac myocytes and fibroblasts. Nephrol. Dial. Transplant. 33, 1722–1734. doi: 10.1093/ndt/gfy006
Leifheit-Nestler, M., Richter, B., Basaran, M., Nespor, J., Vogt, I., Alesutan, I., et al. (2018b). Impact of altered mineral metabolism on pathological cardiac remodeling in elevated fibroblast growth factor 23. Front. Endocrinol. 9:333. doi: 10.3389/fendo.2018.00333
Li, J., Kemp, B. A., Howell, N. L., Massey, J., Miñczuk, K., Huang, Q., et al. (2019). Metabolic changes in spontaneously hypertensive rat hearts precede cardiac dysfunction and left ventricular hypertrophy. J. Am. Heart Assoc. 8:e010926.
Liu, X., Niu, Y., Zhang, X., Zhang, Y., Yu, Y., Huang, J., et al. (2019). Recombinant α-klotho protein alleviated acute cardiorenal injury in a mouse model of lipopolysaccharide-induced septic cardiorenal syndrome type 5. Anal. Cell. Pathol. 2019:5853426.
Liu, Y. C., Tsai, J. P., Wang, L. H., Lee, M. C., and Hsu, B. G. (2020). Positive correlation of serum fibroblast growth factor 23 with peripheral arterial stiffness in kidney transplantation patients. Clin. Chim. Acta 505, 9–14. doi: 10.1016/j.cca.2020.02.014
Lutsey, P. L., Alonso, A., Selvin, E., Pankow, J. S., Michos, E. D., Agarwal, S. K., et al. (2014). Fibroblast growth factor-23 and incident coronary heart disease, heart failure, and cardiovascular mortality: the atherosclerosis risk in communities study. J. Am. Heart Assoc. 3:e000936.
Mace, M. L., Gravesen, E., Hofman-Bang, J., Olgaard, K., and Lewin, E. (2015). Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. 88, 1304–1313. doi: 10.1038/ki.2015.231
Marthi, A., Donovan, K., Haynes, R., Wheeler, D. C., Baigent, C., Rooney, C. M., et al. (2018). Fibroblast growth factor-23 and risks of cardiovascular and noncardiovascular diseases: a meta-analysis. J. Am. Soc. Nephrol. 29, 2015–2027. doi: 10.1681/asn.2017121334
Masson, S., Agabiti, N., Vago, T., Miceli, M., Mayer, F., Letizia, T., et al. (2015). The fibroblast growth factor-23 and Vitamin D emerge as nontraditional risk factors and may affect cardiovascular risk. J. Int. Med. 277, 318–330. doi: 10.1111/joim.12232
Mathew, J. S., Sachs, M. C., Katz, R., Patton, K. K., Heckbert, S. R., Hoofnagle, A. N., et al. (2014). Fibroblast growth factor-23 and incident atrial fibrillation: the Multi-Ethnic Study of Atherosclerosis (MESA) and the Cardiovascular Health Study (CHS). Circulation 130, 298–307. doi: 10.1161/circulationaha.113.005499
Members, W. G., Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Benjamin, E. J., Berry, J. D., et al. (2012). Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125, e2–e220.
Mirza, M. A., Larsson, A., Melhus, H., Lind, L., and Larsson, T. E. (2009). Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 207, 546–551. doi: 10.1016/j.atherosclerosis.2009.05.013
Moe, S. M., Chertow, G. M., Parfrey, P. S., Kubo, Y., Block, G. A., Correa-Rotter, R., et al. (2015). Cinacalcet, fibroblast growth factor-23, and cardiovascular disease in hemodialysis: the evaluation of cinacalcet HCl therapy to lower cardiovascular events (EVOLVE) trial. Circulation 132, 27–39. doi: 10.1161/circulationaha.114.013876
Molkentin, J. D. (2004). Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 63, 467–475. doi: 10.1016/j.cardiores.2004.01.021
Mosterd, A., Hoes, A., De Bruyne, M., Deckers, J., Linker, D., Hofman, A., et al. (1999). Prevalence of heart failure and left ventricular dysfunction in the general population. The Rotterdam Study. Eur. Heart J. 20, 447–455. doi: 10.1053/euhj.1998.1239
Navarro-García, J. A., Delgado, C., Fernández-Velasco, M., Val-Blasco, A., Rodríguez-Sánchez, E., Aceves-Ripoll, J., et al. (2019). Fibroblast growth factor-23 promotes rhythm alterations and contractile dysfunction in adult ventricular cardiomyocytes. Nephrol. Dial. Transplant. 34, 1864–1875. doi: 10.1093/ndt/gfy392
Navarro-García, J. A., Rueda, A., Romero-García, T., Aceves-Ripoll, J., Rodríguez-Sánchez, E., González-Lafuente, L., et al. (2020). Enhanced Klotho availability protects against cardiac dysfunction induced by uraemic cardiomyopathy by regulating Ca. Br. J. Pharmacol. 177, 4701–4719. doi: 10.1111/bph.15235
Navarro-García, J. A., Fernández-Velasco, M., Delgado, C., Delgado, J. F., Kuro-O, M., Ruilope, L. M., et al. (2018). PTH, vitamin D, and the FGF-23–klotho axis and heart: Going beyond the confines of nephrology. Eur. J. Clin. Investigat. 48:e12902. doi: 10.1111/eci.12902
Nayor, M., Larson, M. G., Wang, N., Santhanakrishnan, R., Lee, D. S., Tsao, C. W., et al. (2017). The association of chronic kidney disease and microalbuminuria with heart failure with preserved vs. reduced ejection fraction. Eur. J. Heart Fail. 19, 615–623. doi: 10.1002/ejhf.778
Panwar, B., Jenny, N. S., Howard, V. J., Wadley, V. G., Muntner, P., Kissela, B. M., et al. (2015). Fibroblast growth factor 23 and risk of incident stroke in community-living adults. Stroke 46, 322–328. doi: 10.1161/strokeaha.114.007489
Parker, B. D., Schurgers, L. J., Brandenburg, V. M., Christenson, R. H., Vermeer, C., Ketteler, M., et al. (2010). The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Anna. Int. Med. 152, 640–648. doi: 10.7326/0003-4819-152-10-201005180-00004
Patel, R. B., Ning, H., de Boer, I. H., Kestenbaum, B., Lima, J. A. C., Mehta, R., et al. (2020). Fibroblast growth factor 23 and long-term cardiac function: the multi-ethnic study of atherosclerosis. Circ. Cardiovasc. Imaging 13:e011925.
Pescatore, L. A., Gamarra, L. F., and Liberman, M. (2019). Multifaceted mechanisms of vascular calcification in aging. Arterioscler. Thromb. Vasc. Biol. 39, 1307–1316. doi: 10.1161/atvbaha.118.311576
Poelzl, G., Trenkler, C., Kliebhan, J., Wuertinger, P., Seger, C., Kaser, S., et al. (2014). FGF 23 is associated with disease severity and prognosis in chronic heart failure. Eur. J. Clin. Invest. 44, 1150–1158.
Pool, L. R., Kershaw, K. N., Gordon-Larsen, P., Gutiérrez, O. M., Reis, J. P., Isakova, T., et al. (2020). Racial differences in the associations between food insecurity and fibroblast growth factor 23 in the coronary artery risk development in young adults study. J. Ren. Nutr. 30, 509–517. doi: 10.1053/j.jrn.2020.01.020
Reindl, M., Reinstadler, S. J., Feistritzer, H.-J., Mueller, L., Koch, C., Mayr, A., et al. (2017). Fibroblast growth factor 23 as novel biomarker for early risk stratification after ST-elevation myocardial infarction. Heart 103, 856–862. doi: 10.1136/heartjnl-2016-310520
Robinson-Cohen, C., Shlipak, M., Sarnak, M., Katz, R., Peralta, C., Young, B., et al. (2020). Impact of race on the association of mineral metabolism with heart failure: the Multi-Ethnic Study of Atherosclerosis. J. Clin. Endocrinol. Metab. 105, e1144–e1151.
Rodelo-Haad, C., Rodríguez-Ortiz, M. E., Martin-Malo, A., Pendon-Ruiz de Mier, M. V., Agüera, M. L., Muñoz-Castañeda, J. R., et al. (2018). Phosphate control in reducing FGF23 levels in hemodialysis patients. PLoS One 13:e0201537. doi: 10.1371/journal.pone.0201537
Rosa, T. S., Neves, R. V. P., Deus, L. A., Sousa, C. V., da Silva Aguiar, S., de Souza, M. K., et al. (2020). Sprint and endurance training in relation to redox balance, inflammatory status and biomarkers of aging in master athletes. Nitric Oxide 102, 42–51. doi: 10.1016/j.niox.2020.05.004
Roy, C., Lejeune, S., Slimani, A., de Meester, C., Ahn As, S. A., Rousseau, M. F., et al. (2020). Fibroblast growth factor 23: a biomarker of fibrosis and prognosis in heart failure with preserved ejection fraction. ESC Heart Fail. 7, 2494–2507. doi: 10.1002/ehf2.12816
Ruilope, L. M. (2012). Current challenges in the clinical management of hypertension. Nat. Rev. Cardiol. 9, 267–275. doi: 10.1038/nrcardio.2011.157
Ruiz-Hurtado, G., Li, L., Fernández-Velasco, M., Rueda, A., Lefebvre, F., Wang, Y., et al. (2015). Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. J. Gen. Physiol. 146, 295–306. doi: 10.1085/jgp.201511366
Schumacher, D., Alampour-Rajabi, S., Ponomariov, V., Curaj, A., Wu, Z., Staudt, M., et al. (2019). Cardiac FGF23: New insights into the role and function of FGF23 after acute myocardial infarction. Cardiovasc. Pathol. 40, 47–54. doi: 10.1016/j.carpath.2019.02.001
Seiler, S., Cremers, B., Rebling, N. M., Hornof, F., Jeken, J., Kersting, S., et al. (2011). The phosphatonin fibroblast growth factor 23 links calcium–phosphate metabolism with left-ventricular dysfunction and atrial fibrillation. Eur. Heart J. 32, 2688–2696. doi: 10.1093/eurheartj/ehr215
Shalhoub, V., Shatzen, E. M., Ward, S. C., Davis, J., Stevens, J., Bi, V., et al. (2012). FGF23 neutralization improves chronic kidney disease–associated hyperparathyroidism yet increases mortality. J. Clin. Invest. 122, 2543–2553. doi: 10.1172/jci61405
Sharma, S., Katz, R., Bullen, A. L., Chaves, P. H. M., de Leeuw, P. W., Kroon, A. A., et al. (2020). Intact and C-Terminal FGF23 assays-do kidney function, inflammation, and low iron influence relationships with outcomes? J. Clin. Endocrinol. Metab. 105, e4875–e4885.
Shibata, K., Fujita, S.-I., Morita, H., Okamoto, Y., Sohmiya, K., Hoshiga, M., et al. (2013). Association between circulating fibroblast growth factor 23, α-Klotho, and the left ventricular ejection fraction and left ventricular mass in cardiology inpatients. PLoS One 8:e73184. doi: 10.1371/journal.pone.0073184
Silva, A. P., Gundlach, K., Büchel, J., Jerónimo, T., Fragoso, A., Silva, C., et al. (2015). Low magnesium levels and FGF-23 dysregulation predict mitral valve calcification as well as intima media thickness in predialysis diabetic patients. Int. J. Endocrinol. 2015:308190.
Silva, A. P., Mendes, F., Carias, E., Gonçalves, R. B., Fragoso, A., Dias, C., et al. (2019). Plasmatic Klotho and FGF23 Levels as Biomarkers of CKD-Associated Cardiac Disease in Type 2 Diabetic Patients. Int. J. Mol. Sci. 20:1536. doi: 10.3390/ijms20071536
Slavic, S., Ford, K., Modert, M., Becirovic, A., Handschuh, S., Baierl, A., et al. (2017). Genetic ablation of Fgf23 or klotho does not modulate experimental heart hypertrophy induced by pressure overload. Sci. Rep. 7, 1–12.
Song, T., Fu, Y., Wang, Y., Li, W., Zhao, J., Wang, X., et al. (2021). FGF-23 correlates with endocrine and metabolism dysregulation, worse cardiac and renal function, inflammation level, stenosis degree, and independently predicts in-stent restenosis risk in coronary heart disease patients underwent drug-eluting-stent PCI. BMC Cardiovasc. Disord. 21:24. doi: 10.1186/s12872-020-01839-w
Souma, N., Isakova, T., Lipiszko, D., Sacco, R. L., Elkind, M. S., DeRosa, J. T., et al. (2016). Fibroblast growth factor 23 and cause-specific mortality in the general population: the Northern Manhattan Study. J. Clin. Endocrinol. Metab. 101, 3779–3786. doi: 10.1210/jc.2016-2215
Speer, T., Groesdonk, H. V., Zapf, B., Buescher, V., Beyse, M., Duerr, L., et al. (2015). A single preoperative FGF23 measurement is a strong predictor of outcome in patients undergoing elective cardiac surgery: a prospective observational study. Crit. Care 19:190.
Strain, W. D., and Paldanius, P. (2018). Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 17:57.
Tagliabracci, V. S., Engel, J. L., Wiley, S. E., Xiao, J., Gonzalez, D. J., Appaiah, H. N., et al. (2014). Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. U.S.A. 111, 5520–5525. doi: 10.1073/pnas.1402218111
Takahashi, H., Ozeki, M., Fujisaka, T., Morita, H., Fujita, S. I., Takeda, Y., et al. (2018). Changes in serum fibroblast growth factor 23 in patients with acute myocardial infarction. Circ. J. 82, 767–774. doi: 10.1253/circj.cj-17-0826
Taylor, A., Yanucil, C., Musgrove, J., Shi, M., Ide, S., Souma, T., et al. (2019). FGFR4 does not contribute to progression of chronic kidney disease. Sci. Rep. 9, 1–6.
Taylor, E. N., Rimm, E. B., Stampfer, M. J., and Curhan, G. C. (2011). Plasma fibroblast growth factor 23, parathyroid hormone, phosphorus, and risk of coronary heart disease. Am. Heart J. 161, 956–962. doi: 10.1016/j.ahj.2011.02.012
Ter Maaten, J. M., Voors, A. A., Damman, K., van der Meer, P., Anker, S. D., Cleland, J. G., et al. (2018). Fibroblast growth factor 23 is related to profiles indicating volume overload, poor therapy optimization and prognosis in patients with new-onset and worsening heart failure. Int. J. Cardiol. 253, 84–90. doi: 10.1016/j.ijcard.2017.10.010
Touchberry, C. D., Green, T. M., Tchikrizov, V., Mannix, J. E., Mao, T. F., Carney, B. W., et al. (2013). FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 304, E863–E873.
Urakawa, I., Yamazaki, Y., Shimada, T., Iijima, K., Hasegawa, H., Okawa, K., et al. (2006). Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774. doi: 10.1038/nature05315
Vainikka, S., Joukov, V., Wennström, S., Bergman, M., Pelicci, P. G., and Alitalo, K. (1994). Signal transduction by fibroblast growth factor receptor-4 (FGFR-4). Comparison with FGFR-1. J. Biol. Chem. 269, 18320–18326. doi: 10.1016/s0021-9258(17)32309-8
van de Wouw, J., Broekhuizen, M., Sorop, O., Joles, J. A., Verhaar, M. C., Duncker, D. J., et al. (2019). Chronic kidney disease as a risk factor for heart failure with preserved ejection fraction: a focus on microcirculatory factors and therapeutic targets. Front. Physiol. 10:1108. doi: 10.3389/fphys.2019.01108
von Jeinsen, B., Sopova, K., Palapies, L., Leistner, D. M., Fichtlscherer, S., Seeger, F. H., et al. (2019). Bone marrow and plasma FGF-23 in heart failure patients: novel insights into the heart–bone axis. ESC Heart Fail. 6, 536–544. doi: 10.1002/ehf2.12416
Weber, J. R. (1991). Left ventricular hypertrophy: its prevalence, etiology, and significance. Clin. Cardiol. 14(Suppl. 3), 13–17. doi: 10.1002/clc.4960140704
White, K. E., Evans, W. E., O’Riordan, J. L., Speer, M. C., Econs, M. J., Lorenz-Depiereux, B., et al. (2000). Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 26, 345–348. doi: 10.1038/81664
Wohlfahrt, P., Melenovsky, V., Kotrc, M., Benes, J., Jabor, A., Franekova, J., et al. (2015). Association of fibroblast growth factor-23 levels and angiotensin-converting enzyme inhibition in chronic systolic heart failure. JACC: Heart Fail. 3, 829–839. doi: 10.1016/j.jchf.2015.05.012
Wolf, M. (2010). Forging forward with 10 burning questions on FGF23 in kidney disease. J. Am. Soc. Nephrol. 21, 1427–1435. doi: 10.1681/asn.2009121293
Wolf, M. (2012). Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 82, 737–747. doi: 10.1038/ki.2012.176
Wright, C. B., Dong, C., Stark, M., Silverberg, S., Rundek, T., Elkind, M. S., et al. (2014). Plasma FGF23 and the risk of stroke: the Northern Manhattan Study (NOMAS). Neurology 82, 1700–1706. doi: 10.1212/wnl.0000000000000410
Wright, J. D., An, S. W., Xie, J., Lim, C., and Huang, C. L. (2019). Soluble klotho regulates TRPC6 calcium signaling via lipid rafts, independent of the FGFR-FGF23 pathway. FASEB J. 33, 9182–9193. doi: 10.1096/fj.201900321r
Xie, J., Cha, S.-K., An, S.-W., Kuro-o, M., Birnbaumer, L., and Huang, C.-L. (2012). Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 3, 1–11.
Xin, Z., Song, X., Jiang, B., Gongsun, X., Song, L., Qin, Q., et al. (2018). Blocking FGFR4 exerts distinct anti-tumorigenic effects in esophageal squamous cell carcinoma. Thorac Cancer 9, 1687–1698. doi: 10.1111/1759-7714.12883
Yamazaki, Y., Tamada, T., Kasai, N., Urakawa, I., Aono, Y., Hasegawa, H., et al. (2008). Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J. Bone Mineral Res. 23, 1509–1518. doi: 10.1359/jbmr.080417
Yuen, S. N., Kramer, H., Luke, A., Bovet, P., Plange-Rhule, J., Forrester, T., et al. (2016). Fibroblast growth factor-23 (FGF-23) levels differ across populations by degree of industrialization. J. Clin. Endocrinol. Metab. 101, 2246–2253. doi: 10.1210/jc.2015-3558
Zaheer, S., de Boer, I. H., Allison, M., Brown, J. M., Psaty, B. M., Robinson-Cohen, C., et al. (2017). Fibroblast growth factor 23, mineral metabolism, and adiposity in normal kidney function. J. Clin. Endocrinol. Metab. 102, 1387–1395. doi: 10.1210/jc.2016-3563
Zhang, L., and Liu, T. (2018). Clinical implication of alterations in serum Klotho levels in patients with type 2 diabetes mellitus and its associated complications. J. Diabetes Complications 32, 922–930. doi: 10.1016/j.jdiacomp.2018.06.002
Ziaeian, B., and Fonarow, G. C. (2016). Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 13, 368–378.
Keywords: FGF-23, FGFR, heart, LVH, heart failure – pharmacological treatment – systolic dysfunction
Citation: Vázquez-Sánchez S, Poveda J, Navarro-García JA, González-Lafuente L, Rodríguez-Sánchez E, Ruilope LM and Ruiz-Hurtado G (2021) An Overview of FGF-23 as a Novel Candidate Biomarker of Cardiovascular Risk. Front. Physiol. 12:632260. doi: 10.3389/fphys.2021.632260
Received: 22 November 2020; Accepted: 15 February 2021;
Published: 09 March 2021.
Edited by:
Modar Kassan, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Ebba Brakenhielm, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2021 Vázquez-Sánchez, Poveda, Navarro-García, González-Lafuente, Rodríguez-Sánchez, Ruilope and Ruiz-Hurtado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gema Ruiz-Hurtado, Z2VtYXJ1aXpAaDEyby5lcw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.