
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 18 October 2021
Sec. Cardiac Electrophysiology
Volume 12 - 2021 | https://doi.org/10.3389/fphys.2021.606931
This article is part of the Research TopicPerturbations in Metabolic Cues: Implications for Adverse Cardiac Function Leading to Sudden Cardiac DeathView all 12 articles
 Hiroyuki Yamakawa1,2
Hiroyuki Yamakawa1,2 Tomoko S. Kato3Jaeduk Yoshimura Noh4
Tomoko S. Kato3Jaeduk Yoshimura Noh4 Shinsuke Yuasa1Akio Kawamura3
Shinsuke Yuasa1Akio Kawamura3 Keiichi Fukuda1
Keiichi Fukuda1 Yoshiyasu Aizawa3*
Yoshiyasu Aizawa3*Thyroid hormones (THs) are synthesized in the thyroid gland, and they circulate in the blood to regulate cells, tissues, and organs in the body. In particular, they exert several effects on the cardiovascular system. It is well known that THs raise the heart rate and cardiac contractility, improve the systolic and diastolic function of the heart, and decrease systemic vascular resistance. In the past 30 years, some researchers have studied the molecular pathways that mediate the role of TH in the cardiovascular system, to better understand its mechanisms of action. Two types of mechanisms, which are genomic and non-genomic pathways, underlie the effects of THs on cardiomyocytes. In this review, we summarize the current knowledge of the action of THs in the cardiac function, the clinical manifestation and parameters of their hemodynamics, and treatment principles for patients with hyperthyroid- or hypothyroid-associated heart disease. We also describe the cardiovascular drugs that induce thyroid dysfunction and explain the mechanism underlying the thyroid toxicity of amiodarone, which is considered the most effective antiarrhythmic agent. Finally, we discuss the recent reports on the involvement of thyroid hormones in the regulation of myocardial regeneration and metabolism in the adult heart.
The thyroid gland secretes two thyroid hormones (THs), 3,5,3′-triiodothyronine (T3) and 3,5,3′,5′−tetraiodothyronine (T4 also known as thyroxine). Moreover, THs are synthesized using iodine, influence metabolism, and biosynthesize proteins in the body. These THs are regulated by thyroid stimulating hormone (TSH), which is secreted by the anterior pituitary gland. In turn, TSH is regulated by the hypothalamus via thyrotropin-releasing hormone (TRH). Thyroid hormones exhibit a variety of effects on the heart and peripheral vascular system It is well known that they raise the heart rate and cardiac contractility, improve the systolic and diastolic function of the heart, and decrease the systemic vascular resistance (SVR) in resting condition (Klein and Ojamaa, 2001).
Thyroid dysfunction, which causes hyperthyroidism and hypothyroidism, is associated with increased cardiovascular risk factors (Klein and Ojamaa, 2001; Rodondi et al., 2010; Collet et al., 2012). For example, hyperthyroidism increases the risk of atrial fibrillation (AF), cardiovascular disease (CVD), and heart failure (HF) (Biondi, 2012). Conversely, hypothyroidism is associated with hypertension and dyslipidemia and also causes CVD (Rodondi et al., 2010; Pearce, 2012). In particular, the intracellular effects of THs in cardiomyocytes occur via two types of mechanisms, genomic and non-genomic, with the genomic pathway predominating (Khan et al., 2020). The details of the mechanisms are described in Section 3.
There has been a long controversy regarding whether increased cardiovascular risk is related to thyroid dysfunction (Langén et al., 2018) and whether there is an association between thyroid disorder and the risk of sudden cardiac death (SCD) (Chaker et al., 2016). Sudden cardiac death is an unexpected death or arrest from a cardiovascular cause that occurs outside a hospital or in the emergency room (Lopshire and Zipes, 2006). The major cause of SCDs is lethal ventricular arrhythmias in patients with underlying coronary heart disease (Weisfeldt et al., 2011; Hayashi et al., 2015). Moreover, SCD also develops within 1 h. Over 50% of SCD cases are due to coronary hearts diseases (Hayashi et al., 2015), and these account for almost 20% of the total mortality.
Several groups have reported a relationship between thyroid function and SCD. Charker et al. concluded that elevated free T4 levels might increase the risk of SCD in patients with euthyroid thyroid disease, studied in a prospective population-based cohort (Chaker et al., 2016). Mitchell et al. investigated whether patients with HF with a reduced ejection fraction (HFrEF) and a thyroid functional disorder had an increased risk of SCD (Mitchell et al., 2013). They concluded that a thyroid dysfunction in patients with symptomatic HF and an ejection fraction ≤ 35% had a strong positive correlation with risk of death. Similar results were obtained after adjusting for known mortality predictors [Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT)] (Mitchell et al., 2013). Langén et al. reported that thyroid dysfunction could be related to an increased overall mortality and risk of SCD and that large-scale randomized control trials are essential to decide whether to treat patients with mild thyroid insufficiency (Langén et al., 2018).
In this review, we summarize the effects of TH on the heart (Klein and Ojamaa, 2001) and the clinical symptoms of thyroid dysfunction from the viewpoint of the cardiology (Klein and Danzi, 2007). In addition, we discuss the changes in and the mechanisms of TH metabolism that have an influence on arrhythmias and congestive HF (Danzi and Klein, 2014). Further, we specify the cardiovascular drugs that induce thyroid dysfunction and explain the mechanism underlying the thyroid toxicity of amiodarone, which is considered the most effective antiarrhythmic agent.
In the thyroid gland, two main iodinated hormones, T3 (triiodothyronine) and T4, (tetraiodothyronine; also known as thyroxine) are secreted. By binding to thyroid hormone receptors (TRs), T3 and T4 exert biological activity in responsive tissues. T3 is regarded as a biologically active hormone (Jabbar et al., 2017), and T4 has a few documented non-genomic effects but is largely regarded as a prohormone. Most T4 is deiodinated to T3 in the liver, kidneys, and skeletal muscle (Klein and Danzi, 2007). T3 is carried through blood circulation to each target tissue and organ such as the heart and peripheral blood vessels. Then, these tissues and organs are regulated by serum levels of T3 solely or preponderantly (Danzi and Klein, 2014).
Symptoms of hyperthyroidism include cardiac and hemodynamic symptoms, such as palpitations, widened pulse pressure, dyspnea on exertion, tachycardia, exercise intolerance, and AF (Table 1) (Dahl et al., 2008). Cardiac contractility, as well as resting heart rate, is increased by THs. Cardiac output can increase by 50–300% under hyperthyroidism compared to that in normal conditions. This enhancement of cardiac output is based on synergistic effects of a raised heart rate, increased cardiac contractility, and dilation of peripheral blood vessels (Biondi et al., 2002).
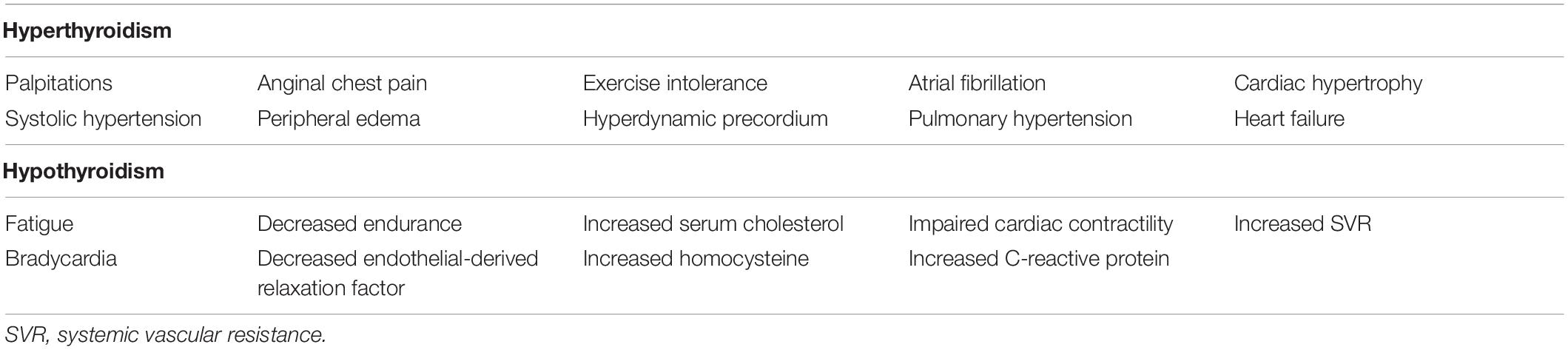
Table 1. Cardiovascular clinical manifestations and laboratory findings associated with hyperthyroidism and hypothyroidism (Danzi and Klein, 2014).
The cardiovascular symptoms of hypothyroidism are not obvious, in contrast to the distinct clinical manifestations of hyperthyroidism. Multiple characteristic phenotypes of hypothyroidism have been described, including bradycardia, diastolic hypertension, narrow pulse pressure, fatigue, myalgia, elevated cholesterol, and a feeling of puffiness (Table 1), but the serum TSH level is an accurate diagnostic indicator of hypothyroidism (Dahl et al., 2008). The cardiovascular effects of hypothyroidism significantly differ from those of hyperthyroidism; for example, cardiac output can be reduced by 30 to 50%, compared to that in a normal state (Danzi and Klein, 2004). However, it is significant that the treatment of hypothyroidism can normalize cardiovascular hemodynamics with a slight change in the resting heart rate (Crowley et al., 1977).
The effects of THs at the cardiac intracellular level are divided into genomic and non-genomic pathways (Khan et al., 2020). In the genomic pathway, THs regulate the expression of target genes by binding to nuclear receptors in cardiomyocytes. In contrast, the non-genomic pathway includes effects on ion channels of the cardiomyocytes and effects of THs on the peripheral circulation, which regulate hemodynamics and the cardiac ejection fraction (Klein and Ojamaa, 2001; Cooper and Biondi, 2012).
Thyroid hormones (THs) regulate the expression of genes coding for cardiac proteins. T3 binds to TRs in the cardiomyocyte nucleus, and this regulates transcription by binding to thyroid hormone response elements (TREs) in regulatory regions of target genes (Kahaly and Dillmann, 2005). Thyroid hormone responses (TRs) are members of the superfamily of steroid hormone receptors. An important feature of their activity is that they bind to TREs with or without ligand, which is distinct from other steroid hormone receptors. TRs bind to TREs as retinoid X receptors (RXRa, RXRb, or RXRg) (Lazar and Chin, 1990; Giammanco et al., 2020).
Table 2 shows the regulation by TH of genes coding for cardiac proteins (Klein and Ojamaa, 2001). One effect of TH in cardiomyocytes is to control cardiac contractility and ejection fraction. THs upregulate the expression of genes encoding sodium/potassium-transporting ATPases (Na+/K+ ATPase), α-myosin heavy chain (myosin heavy chain 6; encoded by MYH6), and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2; encoded by ATP2A2) and downregulate the transcription of β−myosin heavy chain (myosin heavy chain 7; encoded by MYH7) and phospholamban (PLN; encoded by PLN) (He et al., 1997; Kaasik et al., 1997; Holt et al., 1999).
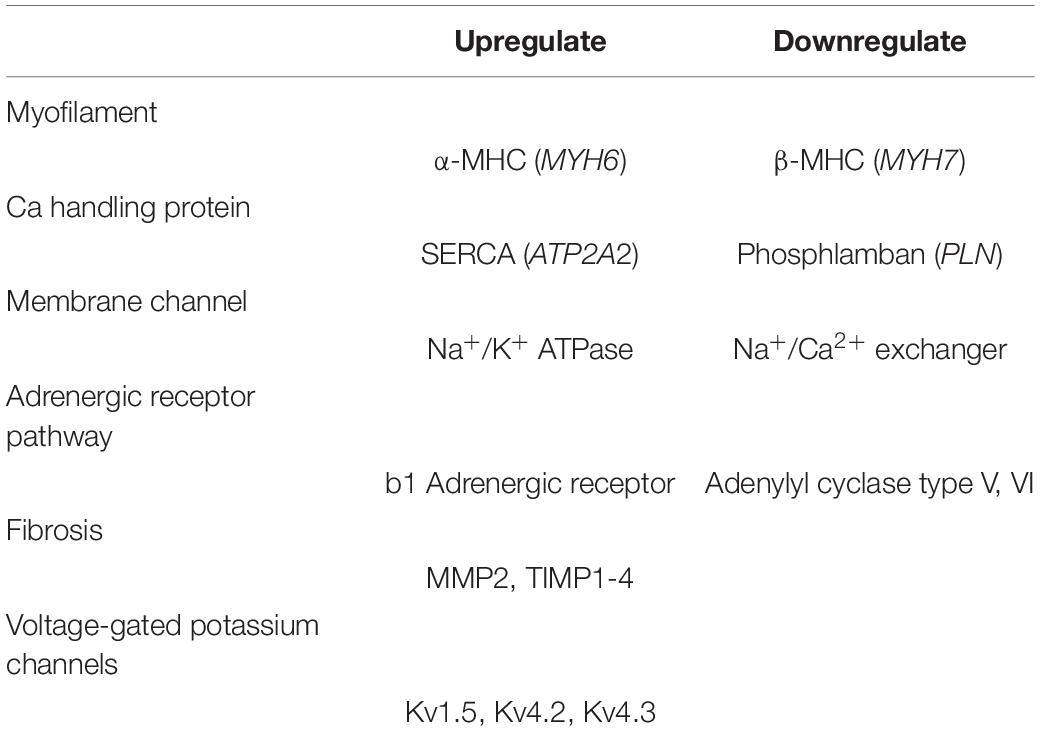
Table 2. Regulation of the genes coding for cardiac proteins by THs [modified from reference (Klein and Ojamaa, 2001)].
α-myosin and β-myosin heavy chains are major components of the cardiomyocyte contractile structures called sarcomeres (Nadal-Ginard and Mahdavi, 1989). The upregulation of SERCA2 and the downregulation of PLN increase calcium concentrations in cardiomyocytes and enhance systolic contraction. Thyroid hormones can increase the protein expression of SERCA2 and decrease the protein expression of PLN in the sarcoplasmic reticulum, leading to improved ventricular relaxation (Kiss et al., 1994; Kranias and Hajjar, 2012). Thyroid hormones (especially T3) also have a direct inotropic effect on cardiomyocytes by upregulating the expression of the β1-adrenergic receptors (Hoit et al., 1997). On the other hand, T3 has been reported to suppress the expression of adenylate cyclase (Klein and Danzi, 2007, 2016). In another study, the authors hypothesized that T3 primarily regulates genes that control cardiac pacemaker cells, exerting a positive chronotropic effect in these cells (Klein and Danzi, 2016).
Chen et al. reported that treatment with a T4 antagonist decreases the collagen fibers in the left ventricular non-infarcted area, with increasing expression of matrix metalloproteinase-2 (MMP2) and tissue inhibitor of matrix metalloproteinases 1 to 4 (MMP1 to 4). Paradoxically, atrial fibrosis is also decreased by T4 (Chen et al., 2013; Zhang et al., 2014), and stimulation by various THs increased cardiac angiogenesis as well as cardiomyocyte growth (von Hafe et al., 2019). In addition, THs regulate several plasma membrane and ion transporters, for example Na+/K+-ATPase, Na+/Ca2+ exchanger (NCX) and voltage-gated potassium channels, including Kv1.5, Kv4.2, and Kv4.3, at both the transcriptional and post-transcriptional levels. Consequently, THs regulate cardiomyocyte excitation-contraction coupling (EC coupling) responses (Gick et al., 1990; Ojamaa et al., 1999).
Thyroid hormones (THs) have two major non-genomic effects, on cardiomyocytes of several membrane ion channels, such as Na+, K+, and Ca2+ channels (Klein and Ojamaa, 2001; Giammanco et al., 2020). In neonatal rat cardiomyocytes, they regulate phosphatidylinositol 3-kinase (PI3K) or serine/threonine-protein kinase (AKT) signaling pathways (Kuzman et al., 2005). Thyroid hormone prevents cell death by AKT pathway in the vascular smooth muscle (Ojamaa et al., 1996).
Other than these pathways, PI3K/AKT-induced physiological hypertrophy is regulated by insulin-like growth factor-1 (IGF-1) (Fujio et al., 2000), p85α (Kenessey and Ojamaa, 2006), angiotensin-1 receptor (Diniz et al., 2009), ubiquitin proteasomes (Rajagopalan et al., 2013), epidermal growth factor receptor (Rajagopalan et al., 2008), and extracellular signal-regulated kinases (Pantos et al., 2007). THs also influence cardiac mitochondrial function (Marín-García, 2010) and regulate impaired myocardial bioenergetic status and function (Madathil et al., 2015).
In addition, plasma membrane-bound sites include integrin αvβ3, a member of a family of proteins that mediate bidirectional interactions between cells and the extracellular matrix (ECM) and regulate tissue organization and cell migration processes. The integrin αvβ3, which is a member of a family of proteins that interactions between cell and the ECM and regulate migration processes, dependent pathway is primary T4-sensitive and effective some intracellular signals, such as protein kinase B (PKB/AKT) and mitogen-activated protein kinases (MAPKs) that phosphorylate intracellular proteins. In addition, some of them can regulate the nucleus and transcription (Cayrol et al., 2019; Davis et al., 2019; Hercbergs, 2019).
The mitochondrial isoforms of other hormone receptors, including mtRXR (mitochondrial RXR), have also been identified (Casas et al., 2003). In addition of the localization of TR (thyroid receptor) in mitochondria there may be TH-mediated pathway between the nuclear and mitochondrial genome (Wirth and Meyer, 2017).
Hyperthyroidism is commonly affected by stimulation of the TSH receptors by autoantibodies [Graves’ disease (GD)] or as a result of the autonomous production of THs by thyroid nodules (Cooper and Biondi, 2012). In general, the prevalence of hyperthyroidism is approximately 0.5% (Cooper and Biondi, 2012). It predominantly affects women aged 30–50 and is caused by GD in 70% of cases. Graves’ disease with TSH receptor antibodies is characterized by a diffuse goiter, exophthalmos, and pretibial myxedema. Aside from GD, 20% of patients with hyperthyroidism show autonomous production of THs by a nodular goiter (Toft and Boon, 2000).
The cardiac effects in thyroid hormones upregulate resting heart rate, blood volume, and myocardial contractility compared to normal. However, as shown in Table 1, exacerbation of hyperthyroidism can also be detrimental to cardiac function (von Hafe et al., 2019). In patients with hyperthyroidism, exercise intolerance is caused by impaired ability to further increase the heart rate and cardiac contraction and to lower the SVR (Forfar et al., 1982). In a consecutive case series study of 24 patients, Duyff et al. reported that 16 patients showed objective signs or symptoms of neuromuscular dysfunction (Duyff et al., 2000). Cardiac output is markedly elevated in patients with hyperthyroidism. The hyperthyroidism has been implicated in a 16% increase in the risk of major cardiovascular events, as well as involvement in an increase in cardiovascular death (Selmer et al., 2014).
Cardiac arrhythmias or electrocardiogram (ECG) abnormalities, which include sinus tachycardia, AF, and shortened PR and QT intervals, are sometimes observed in patients with hyperthyroidism (Kahaly and Dillmann, 2005). Although it is rare, atrio-ventricular blockage might be observed in patients with GD (Mohr-Kahaly et al., 1996). In almost all patients with hyperthyroidism, the most common rhythm disturbance is sinus tachycardia (Nordyke et al., 1988; Biondi et al., 2000).
Atrial fibrillation (AF) is recognized as the most common supraventricular arrhythmia in patients with thyrotoxicosis (Nakazawa et al., 2000). In patients with hyperthyroidism, the prevalence of AF ranges between 2 and 20%, and their risk of AF is approximately six-fold higher than that of healthy people (Klein and Danzi, 2007). The primary consideration for the management of AF is to control heart rate. β-blockers are one of the widely used drugs in the treatment of AF in cases of hyperthyroidism (Klein and Danzi, 2007). These drugs can bring down the ventricular rate and stabilize the rapid symptoms, but they have little effect on converting AF to sinus rhythm or on hyperthyroidism. Therefore, treatment of hyperthyroidism is optimal for long-term AF management. This normally employs radioiodine treatment or antithyroid drugs (ATDs), which can restore sinus rhythms within a few months in the majority of hyperthyroidism patients (Nakazawa et al., 2000). One prospective study in middle-aged and elderly people in Rotterdam (Rotterdam Study) reported that the risk of AF, sudden cardiac death, and decreased life expectancy is associated with elevated free T4 levels, even if thyroid function is within normal limits (Bano et al., 2017; Razvi et al., 2018).
While the major arrythmias in patients with hyperthyroidism are atrial, ventricular arrhythmias are rare and occur about as often as in healthy people (Osman et al., 2002). Ventricular tachycardia (VT) is one of the major causes of death in patients with CVD. A cardiac electrical storm (ES) is defined as electrical instability involving hemodynamic disturbances with significant VT, occurring in at least three or more episodes within 24 h, and requiring direct current cardioversion (Dorian and Cass, 1997). This is likely in patients with hyperthyroidism, in whom VT is typically severe (Colzani et al., 2001; Jao et al., 2004). Ventricular arrhythmia is seen in thyrotoxicosis patients undergoing antithyroid therapy, though only rarely (von Olshausen et al., 1989; Osman et al., 2002). Ventricular tachycardia usually occurs in association with underlying structural heart diseases or HF from various etiologies (Polikar et al., 1993; Marrakchi et al., 2015).
Hypothyroidism and hyperthyroidism can both lead to HF (Schmidt-Ott and Ascheim, 2006), and the prognosis for patients with hyperthyroidism even at a mild level is poor, as these patients suffer from arrythmia, as well as HF. When these patients do not receive treatment, hyperthyroidism might lead to HF because of arrhythmias, cardiac hypertrophy, and increased blood volumes (Biondi, 2012). Furthermore, in a case-based study, patients with hyperthyroidism who did not get proper medical treatment had a higher mortality risk with CVD (Franklyn et al., 2005; Vale et al., 2019). Patients with severe hyperthyroidism can suffer from “high-output HF”. Precisely, high-output HF is defined as congestive HF with increasing cardiac output (DeGroot and Leonard, 1970); resulting “tachycardia-induced cardiomyopathy” depends on the duration of high-output HF with hyperthyroidism (Cruz et al., 1990).
In young patients with hyperthyroidism, this thyrotoxicosis is not associated with underlying heart disease, and therefore, the heart is not damaged. However, symptoms of HF occur in cases of enlarged cardiac output and low SVR, and enlarged blood flow volumes caused by chronic stimulation of the renin angiotensin aldosterone system can be present. The symptoms of patients with high-output HF are breathlessness at rest, fatigability, and the accumulation of fluid with peripheral edema, pleural effusion, pulmonary hypertension, and hepatic congestion (Biondi, 2012).
Siu et al. have estimated that the risk of low-output HF is 6–15% in patients with hyperthyroidism (Siu et al., 2007). Elderly patients with hyperthyroidism might suffer from HF with a reduced ejection fraction. These low-output HF patients have low cardiac output, increasing the SVR, a reduction in ventricular contractility, and impaired left ventricular filling, without increased blood volume. The risk of HF with reduced ejection fraction is increased in patients with hyperthyroidism suffering from cardiac disorders such as ischemic heart disease, hypertensive heart, valvular disease and/or AF (Biondi, 2012). Clinically apparent hyperthyroidism with a hyperdynamic state increases the risk of AF (Cooper and Biondi, 2012). The synergistic effects of reducing SVR, increasing contractility, and increasing the heart rate augments cardiac output (Cooper and Biondi, 2012).
It has been reported that cardiovascular diseases related to thyroid function can be further improved by treating the thyroid gland (Barreto-Chaves et al., 2020). Muthukumar et al. reported that patients with cardiovascular dysfunction related to hyperthyroidism could improve their cardiac function by bringing the thyroid to normal levels with thyroid medication (Muthukumar et al., 2016). And they reported that these aforementioned patients could also have their cardiac function completely restored after total thyroidectomy (Muthukumar et al., 2016). In fact, Saad et al. have reported that treatment of the thyroid gland significantly prevented cardiac dysfunction in a mouse model of T4-induced cardiac dysfunction (Saad et al., 2017). Furthermore, reversibility of heart disease was observed after 2 weeks of T4 treatment, including cardiac hypertrophy (Saad et al., 2017).
In SardiNIA study, Delitala et al. demonstrated that serum FT4 levels are associated with carotid-femoral artery PWV (pulse wave velocity), and high levels of free T4 was is one aggravating factor of aortic stiffness. And they considered that T4 may contribute to the atherosclerosis and the aging process in the vascular system (Delitala et al., 2015; Vale et al., 2019).
Importantly, death from heart failure is the major cause of cardiovascular death in both hyperthyroidism and subclinical hyperthyroidism (Selmer et al., 2014).
Subclinical hyperthyroidism is defined as a condition in which serum TSH levels are below the lower limit of normal, but the width of serum T3 and T4 concentrations is within the normal range (Surks et al., 2004; Biondi and Cooper, 2008; Bahn et al., 2011). Subclinical hyperthyroidism has two major causes. The first encompasses exogenous factors such as an excessive dosage of thyroid hormone replacement drugs, high-dose glucocorticoids, and others. The second is an endogenous factor, namely underlying thyroid disease that causes the overactivity of THs. A considerable proportion (15–20%) of patients who take levothyroxine have a low TSH serum level (Canaris et al., 2000; Vadiveloo et al., 2011; Taylor et al., 2014). In the USA, the prevalence of endogenous subclinical hyperthyroidism varies and depends on age, sex, and iodine intake. Cappola et al. reported that the prevalence of subclinical hyperthyroidism in iodine-sufficient cases is almost 2% (Cappola et al., 2006).
Several observational clinical studies have reported a relationship between subclinical hyperthyroidism and incident CVD (Parle et al., 2001; Iervasi et al., 2007), AF (Cappola et al., 2006; Collet et al., 2012), HF (Rodondi et al., 2008; Biondi et al., 2015), and cardiovascular mortality (Collet et al., 2012). It is not clear whether subclinical hyperthyroidism is associated with high-risk cardiovascular morbidity and mortality. However, the European Thyroid Association guidelines recommend that older patients with hyperthyroidism should have a serum TSH < 0.1 mU/L (Biondi et al., 2015).
In this section, we review the medical history of GD, which is a major cause of hyperthyroidism. Graves’ disease is treated by ATDs, which decrease TH synthesis, by radioactive iodine (RAI) therapy, or total thyroidectomy (Smith and Hegedüs, 2016; Kahaly et al., 2018). Antithyroid drugs is the major treatment in Europe, United States, and Asia (Brito et al., 2016). The main ATDs are thionamides, for example propylthiouracil (PTU), carbimazole (CBZ), and methimazole (MMI). Propylthiouracil controls the conversion from T4 to T3 by inhibiting enzymatic activity in the peripheral organs such as the liver or kidney. As a result, PTU further reduces the density of blood T3. Carbimazole acts by obstructing hormone generation in the thyroid gland, and more significantly, by affecting the hyperstimulation of the thyroid gland at a level prior to biosynthesis. Carbimazole must be decarboxylated to produce MMI in the liver. Methimazole, as well as PTU, is absorbed immediately and accumulates at a high density in the thyroid gland, restraining the synthesis process of TH. All thionamides inhibit the coupling of iodothyronines and reduce the biosynthesis of THs (Cappola and Ladenson, 2003). As a result, these drugs inhibit the production of TH by iodide peroxidase. Specifically, iodide peroxidase oxidizes an iodide ion to iodine and iodinates a tyrosine residue of the thyroglobulin, and this process is indispensable in the production of T4. ATD has been recommended as a first choice drug for the treatment of GD, particularly for short-period GD treatment prior to thyroidectomy or RAI therapy (Bartalena, 2013; Smith and Hegedüs, 2016). Higher doses of PTU inhibit the deiodination of T4 to T3 (Cooper, 2005) and have severe side effects, such as severe hepatic disorder and anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis. However, the half-life of PTU (75 min–150 min) is much shorter than that of MMI (6 h) (Kahaly et al., 2018). Hyperthyroidism is linked to risk of enlargement in CVD and mortality in the first year following radioiodine treatment, but early diagnosis and proper treatment of hyperthyroidism along with cardiac treatment might reduce mortality (Toft and Boon, 2000).
In general, hypothyroidism is diagnosed when serum TSH levels are high (usually > 10 mU/L) and serum-free T4 levels are low (< 9–10 pmol/L). Clinically apparent hypothyroidism can be seen in 0.2–2.0% of non-pregnant adults (Canaris et al., 2000). Symptomatic thyroid dysfunction is found in 1–2% of the population and is more frequent in women. Except for previous radioiodine treatment or thyroidectomy for GD, the most common cause is autoimmunity of the thyroid gland or Hashimoto’s thyroiditis (Hashimoto’s disease). Hashimoto’s thyroiditis can often be accompanied by hard goiter, in which the thyroid gland becomes atrophied causing progressive fibrosis during the course of the disease, and resulting in diminished function of the thyroid gland. Distinct from hyperthyroidism, there is a link between low serum concentrations of HTs (T3 and T4) and a reduction in cardiac output, heart rate, stroke volume, and myocardial contractility (Toft and Boon, 2000).
One of the most important cardiac dysfunctions, especially among patients with hypothyroidism, is diastolic dysfunction. This diastolic dysfunction is observed not only at rest, but also during exercise. This exacerbates the symptoms and worsens the prognosis of potential heart failure patients in hypothyroid patients (Selmer et al., 2014; von Hafe et al., 2019). It is well known that hypothyroidism is associated with chronic heart failure. However, in rare cases, hypothyroidism may also be associated with pericardial effusion and cardiac tamponade (Grais, 2010; Patil et al., 2011).
In addition to the abovementioned clinical symptoms, remarkable changes in modifiable atherosclerotic risk factors are also observed in clinically apparent hypothyroidism, including hypercholesterolemia, diastolic hypertension, carotid intima-media thickening, and reduced production of endothelial-derived relaxation factor (nitric oxide) (Cappola and Ladenson, 2003). All of these clinical manifestations are improved by TH replacement therapy (Cappola and Ladenson, 2003).
It is generally accepted that typical hypothyroid ECG changes include bradycardia, long-PQ segment, low voltage of the QRS complex, and flattening or T-wave inversion. However, it is important, though not well known, that hypothyroidism induces atrioventricular blockage and acquired long QT syndrome (Marrakchi et al., 2015). In the case of supraventricular arrhythmias, hypothyroidism or subclinical hypothyroidism has a certain clinical impact. For example, patients with thyroid dysfunction have a higher probability of having AF than healthy people (Sawin, 1995; Baumgartner et al., 2017). Klemperer et al. reported that perioperative T3 treatment reduces the incidence or necessity of postoperative AF in patients with normal thyroid function during cardiopulmonary bypass surgery. However, experiments on the mechanism underlying this discovery have not yet been reproduced (Klemperer et al., 1996). Kim et al. confirmed that hypothyroidism has no relationship with 10-year risk of incident AF from the famous cardiovascular cohort of the Framingham Heart Study (Kim et al., 2014).
Regarding ventricular arrhythmias, some groups have reported that patients with hypothyroidism might experience life-threatening arrhythmia, for example a torsade de pointes type ventricular tachycardia (TdP type ventricular tachycardia) and VT due to prolonged QT syndrome (Chojnowski et al., 2007). That study also reported that hypothyroidism decreases the expression of protein T3 in cardiomyocytes and that it can cause reduced cardiac contractility and heart rate, as well as delayed conduction of electrical stimulation in the heart. This might be the reason for bradycardia and elongation of the QT interval and consequent fatal arrhythmia, such as TdP-type ventricular tachycardia. In this case, the causes of long QT syndrome and shock include the decreased T3 expression and disorders of the electrolyte balance, for example hypokalemia and hypocalcemia (Chojnowski et al., 2007). For patients with hypothyroidism, it is necessary to monitor the effectiveness of amiodarone for the prevention of ventricular arrhythmic recurrence. It has been reported that lidocaine (class IB antiarrhythmic drug) or bretylium tosylate (class III antiarrhythmic drug) might be useful to prevent these paroxysmal ventricular tachycardias and endocavitary electrode stimulation, in place of amiodarone (Chess-Williams and Coker, 1989).
It is thought that TH deficiency raises the risk of developing and exacerbates HF (Rodondi et al., 2008). Basic experiments have reported that hypothyroidism suppresses myosin heavy chain 6 protein expression and enhances myosin heavy chain 7 protein expression and that hypothyroidism induces cardiac atrophy as a result. In addition, hypothyroidism is associated with the increased dilation of ventricular chambers and reduced myocardial perfusion (Liu et al., 2008; Biondi, 2012). Congestive HF and myxedema have been recognized in patients with hypothyroidism (Schwimmer et al., 1947), and their HF and myxedema symptoms improved with treatment for hypothyroidism. More recently, it was reported that patients with hypothyroidism were among patients with reversible dilated cardiomyopathy (Khochtali et al., 2011). Recent clinical studies have reported that patients with cardiovascular disease who have reduced T3 levels have a higher risk of death from heart failure (Wang et al., 2017; Neves et al., 2019). Rezvi et al. reviewed thyroid hormone supplementation in HF (Neves et al., 2020). Studies in patients without HF as well as HFrEF suggest an effect of thyroid hormone supplementation in improving diastolic function (Pingitore et al., 2008). In the HFrEF animal model, it has been suggested that thyroid hormone supplementation in HF improves cardiac function (Vale et al., 2019).
It is interesting that the metabolism and serum levels of THs are changed by conditions of HF, myocardial infarction, and cardiac surgery. In these situations, the conversion of T4 to T3 decreases. Diseases with normal serum levels of TSH and no symptoms suggestive of hypothyroidism despite a decrease in blood thyroid hormone are called euthyroid sick syndrome or non-thyroidal illness. Among them, those with only low T3 levels are called low T3 syndrome (LT3S) (Nagayo, 2018).
Amin et al. reported that the Patients with heart failure with HFrEF (left ventricular ejection fraction < 40%) and with LT3S take the oral T3 supplements (Liothyronine) for 1.5 months, and that the patients can be found to improve cardiac function considering the blood laboratory data and echocardiographic data (Liothyronine group N = 25, Placebo grouper N = 25) (Amin et al., 2015). Further, LT3S impairs cardiac dysfunction and as a result induces heart disease. Patients with advanced heart disease and LT3S have increased mortality (Pingitore et al., 2005; Gerdes and Iervasi, 2010; Mourouzis et al., 2011).
Pingitore et al. conducted a clinical study to determine whether T3 administration improves cardiac function in patients with low T3 syndrome (LT3S) who have suffered an acute myocardial infarction (AMI) (LT3S/AMI) (Pingitore et al., 2019). Pingitore et al. concluded that the patients with LTS3S/AMI had improved cardiac functions, which were assessed using cardiac MRI to evaluate various parameters (for example infarct sizes, and cardiac function), after 6 months of treatment with liothyronine (T3) therapy [The THIRST Study (Thyroid Hormone Replacement Therapy in ST elevation myocardial infarction); Phase II study] (T3 supplement group N = 19, Placebo grouper N = 18) (Pingitore et al., 2019; Lisco et al., 2020). Some researchers reported that the changes in gene expression associated with cardiac dysfunction are similar to those induced in hypothyroidism, suggesting that TH dysfunction might be one aggravating factor for HF (Kinugawa et al., 2001; Biondi, 2012).
THs are involved in lipid metabolism (Cappola and Ladenson, 2003). Hyperthyroidism is not an exacerbating factor for the lipid profile. For several years, hypothyroidism was thought to be linked to hyperlipidemia. In fact, many patients with clinical hypothyroidism show obvious clinical symptoms (Klein and Danzi, 2007; Jabbar et al., 2017).
There are two aspects of the association between hypothyroidism and coronary disease. First, hypothyroidism has an influence on hypertension and hypercholesterolemia in these patients. In particular, this hypertension and hypercholesterolemia due to hypothyroidism accelerate atherosclerosis. Second, it is thought that hypothyroidism reduces cardiac oxygen requirements and decreases their effective use and that this process induces CVD (Kahaly and Dillmann, 2005). It is notable that low serum T4 levels are associated with increased low-density lipoprotein-cholesterol and that hypothyroidism has also been implicated in hyper-triglyceridemia and low free fatty acid levels.
Thyroid hormone replacement is desirable in patients with hypothyroidism, even in patients with myocardial infarction. In fact, this is because thyroid hormone is said to be an important factor in regulating the structure and function of the left ventricle in the late post-myocardial infarction period (Jankauskienė et al., 2016; von Hafe et al., 2019). In molecular biology, low T3 levels can induce oxidative stress and apoptosis, which may exacerbate ventricular dysfunction (Jankauskienė et al., 2016).
Zhang et al. reported that in a rat model of cardiac reperfusion injury, the addition of T3 enhances the gene expression of transcription factor Hypoxia-Inducible Factor alpha (HIF-1α). It thereby regulates mitochondrial opening and protects cardiomyocytes (Zhang et al., 2018).
Subclinical hypothyroidism is defined as being associated with a serum TSH level above the normal range, but with normal TH levels. Subclinical hypothyroidism is classified into two types according to the level of TSH. Mild subclinical hypothyroidism is diagnosed by a mildly high TSH level (between 4.0–4.5 and 10.0 mU/L). Severe subclinical hypothyroidism is diagnosed by a TSH level > 10.0 mU/L. However, the upper limit of normal TSH has not been clearly defined. For the diagnosis of subclinical hypothyroidism, it is necessary to consider both clinical symptoms and the level of TSH (Hamilton et al., 2008; Razvi et al., 2018). In general, the prevalence of subclinical hypothyroidism is 4–20% in adults (Biondi and Cooper, 2008; Abreu et al., 2017). There are many causes for this wide range of reported prevalence. Specifically, differences in age, sex, race, body mass index, dietary iodine intake, and serum TSH measurements at different diagnostic institutions might contribute to this. The prevalence of a high serum level of TSH is higher in Caucasians than in African-American people (Hollowell et al., 2002). It is also thought that at least 10% of older women (aged 60 and older) are diagnosed with subclinical hypothyroidism (Parle et al., 1991). This wide prevalence suggests that patients with more cardiovascular risk factors than expected are present. The cardiac function of patients with subclinical hypothyroidism shows abnormalities including extended isovolumic relaxation time and impaired ventricular filling (Monzani et al., 2001). To date, several studies of systolic dysfunction in patients with subclinical hypothyroidism have been conducted. Ripoli et al. reported that there is an association between subclinical hypothyroidism and systolic dysfunction and that thyroxine replacement therapy improves cardiac contractility in patients with hypothyroidism (Ripoli et al., 2005). In patients with subclinical hypothyroidism during exercise, both diastolic and systolic function are impaired and as a result, exercise tolerance is reduced in these patients (Brenta et al., 2003).
It is not currently known whether there is an association between the severity of subclinical hypothyroidism and increased risk of CVD. According to meta-analyses of observational studies, patients aged < 65 years (Razvi et al., 2008) with severe hypothyroidism, defined as a TSH level > 10 mU/L, do have a high risk of CVD (Rodondi et al., 2010). According to the guidelines of the European Thyroid Association, treatment for hypothyroidism is recommended for patients with severe hypothyroid disease (serum THS > 10 mU/L), symptoms of hypothyroidism, an age < 70 years, and elevated risk of CVD (Pearce et al., 2013).
One favored therapy involves taking levothyroxine (L-thyroxine) with solid preparation on an empty stomach. Patients who not only have clinical manifestations of hypothyroidism, but also a diagnosis based on biochemical examinations, are recommended treatment. Levothyroxine is marketed by several pharmaceutical companies, but switching to generic levothyroxines is not recommended for patients who are stable with one regimen (Jonklaas et al., 2014; Chaker et al., 2017). In patients with clinically manifesting hypothyroidism, 1.5–1.8 mg/kg/day of levothyroxine is desirable as a daily optimal dose (Pearce et al., 2013; Jonklaas et al., 2014). Generally, in patients with CVD, the initial dose is 12.5–25.0 mg/day. Based on symptoms and the serum TSH level, it is desirable to gradually increase the dose afterward (Jonklaas et al., 2014).
Thyroid hormone replacement therapy improves diastolic function in patients with hypothyroidism and also in patients with heart failure with preserved ejection fraction (HFpEF). Thyroid hormone is said to improve cardiac diastolic function, both pathophysiologically and through gene expression. However, further preclinical and clinical studies are needed to clarify the role of thyroid hormones in the treatment of HFpEF (Neves et al., 2020). In patients with HFpEF, it is advisable to use beta-blockers in combination with thyroid hormones when they are used. The risk of cardiovascular events due to sympathetic hyperactivity in thyroid hormones can be inhibited by the combined use of beta-blockers. As a result, the combination of the two drugs can improve cardiac function while foreshadowing arrhythmias, cardiac hypertrophy, and cardiac dysfunction (Ortiz et al., 2019).
For some CVD patients with hypothyroidism, cardiac function is improved by hypothyroidism treatment. Furthermore, treatment can improve prognostic factors of thyroid function and the cardiovascular system (Crowley et al., 1977). However, it has not been clarified whether patients treated for hypothyroidism have exacerbated heart disease when the treatment is discontinued (Klein and Danzi, 2007). The clinical implications of low T3 levels in patients with normal TSH levels have also not yet been determined. Measurement of T3 does not correlate with therapeutic efficacy (Abdalla and Bianco, 2014).
The interrelationship between thyroid hormones and the heart is a very important focus area. Thus far, we have described how changes in THs cause cardiovascular dysfunction. In this section, we summarize the ways in which cardiac medications routinely used by cardiologists affect thyroid hormones.
As mentioned above, among drugs used commonly by many cardiologists, there are some that alter TH levels in patients with normal thyroid function. Several of these can alter the levels of THs in euthyroid patients. Fadel et al. summarized the effects of cardiovascular drugs on thyroid function [Table 3, (Fadel et al., 2000)]. When cardiovascular drugs are used, TH levels must be checked every 3–4 months. The most widely used drug that affects thyroid function is amiodarone. Approximately 50% of patients taking amiodarone for a long period have elevated serum T4 levels. However, the serum levels of T3 and TSH are within a normal range (Harjai and Licata, 1997). One of the main effects of amiodarone on thyroid function is the inhibition of the conversion of T4 to T3 by amiodarone and its major active metabolite, desethylamiodarone (Burch, 2019). Ruzieh et al. reported that the likelihood of experiencing an amiodarone-related adverse event was greater than with placebo. (relative risk about 4.44 versus placebo) (Ruzieh et al., 2019). In the following sections (6.2 to 6.4), we summarize the effect of amiodarone on thyroid function in detail.
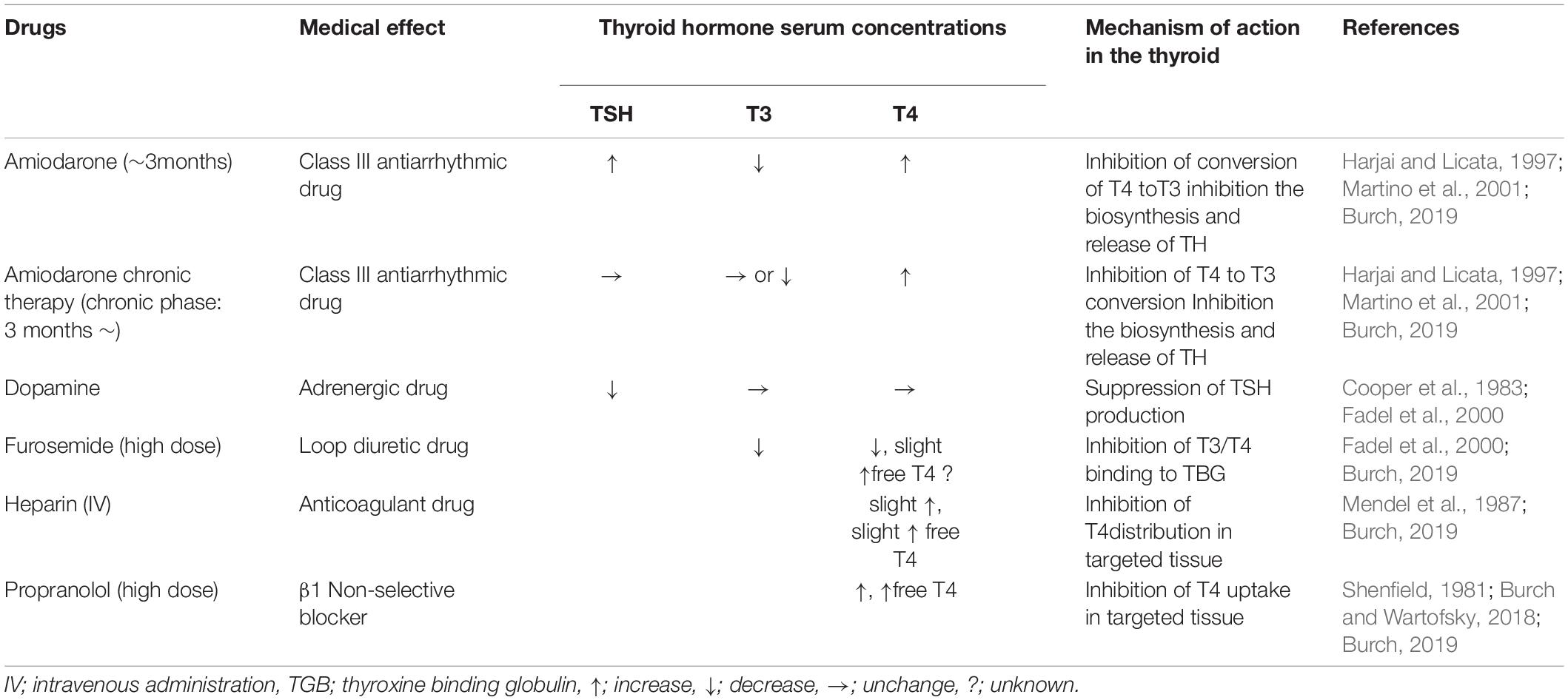
Table 3. Effect of cardiovascular drugs on thyroid hormone function [modified from reference (Fadel et al., 2000; Burch, 2019)].
This section summarizes the relation between some drugs in cardiology and thyroid function.
Dopamine suppressed TSH secretion in about 50% of humans. Administration of the dopamine receptor antagonists metoclopramide or domperidone increases TSH in primary hypothyroidism. Normal level of THs suppresses TSH elevation, but hypothyroidism does not respond to this suppression, resulting in an elevation response (Cooper et al., 1983). High dose fulosemide is said to attenuate the effects of thyroid hormones by binding to TGB (thyroxine binding globulin), which is one of transporter portein of THs (Fadel et al., 2000; Burch, 2019). However, there are few documents that explain the above in pharmacokinetics, and it is necessary to examine it in detail in the future. Heparin increases T3 and T4 from binding proteins indirectly by increasing free fatty acids (Mendel et al., 1987; Burch, 2019). Propranolol in high volume inhibits the conversion of T4 to T3. As a result, it increases T4 and free T4 (Burch and Wartofsky, 2018; Burch, 2019). In hyperthyroidism, the effect of propranolol is enhanced (Shenfield, 1981), and vice versa in hypothyroidism (Burch, 2019).
Amiodarone is regarded as the most effective antiarrhythmic drug but has various toxicities, and cardiologists need to exercise caution (Trohman et al., 2019). The combination of amiodarone and a beta-blocker is the preferred treatment for ES (Nademanee et al., 2000). According to the 2017 AHA/ACC/HRS Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death, amiodarone is recommended as a Class IIb indication for the acute treatment of hemodynamically stable VT (Al-Khatib et al., 2018). Amiodarone is effective at suppressing AF, as well as ventricular arrhythmia. In particular, it is thought to be the most effective drug for maintaining sinus rhythms against paroxysmal and persistent AF for a long duration. It is also recognized as the first-choice agent for patients with AF due to HF or symptomatic AF (Singh, 2008; Al-Khatib et al., 2018).
The numerous side effects of amiodarone might involve the skin, eyes, brain, lungs, liver, and peripheral nervous system (Trohman et al., 2019). The major side effect is the production of corneal microdeposits (> 90% of treated patients). These are very similar in appearance to vortex keratopathy, which is associated with Fabry disease (D’Amico et al., 1981; Wasielica-Poslednik et al., 2011).
It is important for clinical physicians, as well as cardiologists, to keep in mind that the use of amiodarone affects thyroid function. That is, some patients using amiodarone suffer from hypothyroidism (5% to 10% of treated patients) and some suffer from hyperthyroidism (0.9–10% of treated patients) (Harjai and Licata, 1997; Trohman et al., 2019). The iodine richness of amiodarone is the recognized cause of its thyrotoxic side effects.
Changes in serum TSH, T4, and T3 levels are seen in patients receiving amiodarone treatment. The pattern of TH levels differs between the acute phase (∼ 3 months) and the chronic phase (after 3 months) after the start of amiodarone use. In both the acute and chronic phases, the serum T4 level rises and the serum T3 level decreases, though the serum reverse T3 (rT3) level rises. Interestingly, the serum TSH levels are elevated in the acute phase, but return to a normal range during the chronic phase (Martino et al., 2001). There are two reasons why serum TH levels change during these two phases, specifically (1) the unique effects of amiodarone and (2) the effects of its constituent iodine (Basaria and Cooper, 2005).
Amiodarone can cause hyperthyroidism (amiodarone-induced thyrotoxicosis: AIT), which is subdivided into two main forms. Type I AIT is usually a predisposing factor for goiter abnormalities (for example asymptomatic GD) and results from the over-synthesis and over-release of THs. The self-regulatory mechanism of THs regulates iodine metabolism in the thyroid gland according to iodine content (Harjai and Licata, 1997). Typical cases of type I AIT occur in patients with a background of non-toxic goiter or asymptomatic GD with an overdose of iodine (Trohman et al., 2019). The prevalence of type I AIT is higher in areas with low iodine intake. Treatment for AIT type I is similar to that for normal hyperthyroidism (Hamilton et al., 2020).
Type II AIT is a thyrotoxicosis cause by destructive thyroiditis and is characterized by acute or subacute destruction of the thyroid gland and massive leakage of THs in patients with no underlying thyroid disease while taking amiodarone (Tsang and Houlden, 2009). The prevalence of type II AIT is higher in areas with high iodine intake. Mild thyroiditis of type II AIT (with mild elevation of free T4 and free T3) is likely to improve spontaneously with follow-up alone. In severe type II AIT, the use of glucocorticoid is beneficial even under amiodarone administration (Daniels, 2001; Hamilton et al., 2020).
Sometimes we find the patients who have a combination of the two types of AIT. In such cases, it is advisable to treat them in both types of AIT. Specifically, type 1 should be treated with antithyroid therapy and type 2 with steroids (Osuna et al., 2017; Omidi et al., 2020).
Amiodarone can also cause hypothyroidism, which inhibits the biosynthesis and release of THs. This is called amiodarone-induced hypothyroidism. This is because large amounts of iodine are released during amiodarone metabolism (Harjai and Licata, 1997). Intrathyroid iodine organization resumes with the normal synthesis of T4 and T3 (Martino et al., 1984). The development of hypothyroidism with amiodarone has been associated with previous Hashimoto’s thyroiditis. However, the use of amiodarone is not contraindicated even if serum TSH levels are elevated before or during treatment, because thyroid dysfunction can be easily treated with T4 (Toft and Boon, 2000). After discontinuing amiodarone treatment, thyroid function might recover, but persistent reduced function has been observed. In particular, many patients for whom thyroid function does not improve even after the discontinuation of oral amiodarone administration are considered to have underlying autoimmune thyroid (Martino et al., 2001).
In this manuscript, we discussed how TH plays an important role in cardiovascular disease at the molecular level via genomic and non-genomic pathways. Serious cardiac complications such as arrythmia, congestive HF, and angina pectoris might arise in patients with hyperthyroidism or hypothyroidism, and their treatment requires control of the underlying TH levels. Judging from previous clinical observational studies and small-scale intervention studies, which have evaluated the association between THs and risk factors for CVD, it can be concluded that among patients who should be treated for thyroid function, those with severe CVD should be given priority thyroid treatment (Biondi and Cooper, 2008). Abnormal levels of THs might also serve as a prognostic marker (Jabbar et al., 2017).
Since the discovery of iPS cells by Takahashi and Yamanaka (2006), the study of myocardial regeneration has attracted the attention of scientists worldwide (Takahashi and Yamanaka, 2006). Several hormones have been suggested to be involved in myocardial regeneration. One of these is TH, which was recently reported to affect both the metabolic profile and the mitotic cycle in cardiomyocytes (Nakada et al., 2017; Hirose et al., 2019; Tan et al., 2019; Bogush et al., 2020).
In mouse cardiomyocytes, prolonged inhibition of TH with propylthiouracil prolongs the proliferative period of cardiomyocytes during cardiac development (Hirose et al., 2019). Transgenic mice with cardiac-specific suppression of TRα exhibited a higher number of cardiomyocytes and higher expression of cell cycle markers compared to normal mice during cardiac development. In addition, adult TRα transgenic mice demonstrated a 10-fold increase in the number of proliferating diploid cardiomyocytes, as well as improved recovery of cardiac function after ischemia-reperfusion injury (Hirose et al., 2019).
Two recent reports have shown that exogenous administration of thyroid hormone (T3) increases the number of cardiomyocytes in neonatal mouse hearts (Tan et al., 2019; Bogush et al., 2020). Tan et al. determined that T3 activates mitochondria-generated H2O2 (mH2O2), which in turn activates c-Jun N-terminal kinase-2α2 (JNK2α2) via peroxiredoxin-1 (Tan et al., 2019) to promote cardiomyocyte proliferation. Exogenous T3 stimulates proliferative ERK1/2 signaling and affects cardiomyocytes in the cardiac apex, but is suppressed in P8 by the expression of DUSP5, a nuclear phosphho-ERK1/2-specific dual-specificity phosphatase (Bogush et al., 2020).
It is important to maintain optimal levels of THs in the cardiac tissue, as well as suitable serum TH levels, to stabilize homeostasis. Further, basic studies (such as molecular biology and omics-based analysis), and clinical studies (such as cohort studies and randomized clinical trials) of THs will help us understand their effects on CVD. Moreover, we can elucidate the mechanisms of the demonstrated effects via induced arrhythmia or myocyte remodeling and dysfunction.
HY and YA: conceptualization and funding acquisition. HY: writing – original draft preparation. TK and YA: writing – review and editing. HY, SY, and KF: supervision. All authors contributed to the article and approved the submitted version.
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI) [Grant Nos. 17K09524 (YA) and 18K08047 (HY)], the Fukuda Foundation for Medical Technology, the Daiwa Securities Health Foundation, and the Miyata Cardiac Research Promotion Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to John Martin for editing this manuscript.
AF, atrial fibrillation; AIT, amiodarone-induced thyrotoxicosis; AKT, serine/threonine-protein kinase; AMI, acute myocardial infarction; ANCA, anti-neutrophil cytoplasmic antibodies; ATD, antithyroid drugs; CBZ, carbimazole; CVD, cardiovascular disease; DUSP5, specific dual-specificity phosphatase 5; EC coupling, excitation-contraction coupling; ECG, electrocardiogram; ECM, extracellular matrix; ERK, extracellular signal-regulated kinase; ES, electrical storm; GD, Graves’ disease; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; HIF-1α, Hypoxia-Inducible Factor alpha; IGF-1, insulin-like growth factor-1; IV, intravenous administration; JNK2α2, c-JunN-terminal kinase-2 α 2; LT3S, low T3 syndrome; MAPK, mitogen-activated protein kinase; MMI, methimazole; MMP, matrix metalloproteinase; mH2O2, mitochondria-generated H2O2; mtRXR, mitochondriala RXR; MYH, myosin heavy chain; NCX, Na + /Ca2 + exchanger; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PLN, phospholamban; PTU, propylthiouracil; PWV, pluse wave velocity; RAI, radioactive iodine; RXR, retinoid X receptor; SCD, sudden cardiac death; SERCA2, sarcoplasmic/endoplasmic reticulum calcium ATPase 2; SVR, systemic vascular resistance; SVR, synergistic effects of reducing; TdP, torsade de pointes; TGB, thyroxine binding globulin; TH, thyroid hormone; TR, thyroid hormone receptor; TRE, thyroid hormone response element; TRH, thyrotropin-releasing hormone; VT, Ventricular tachycardia.
Abdalla, S. M., and Bianco, A. C. (2014). Defending plasma T3 is a biological priority. Clin. Endocrinol. 81, 633–641. doi: 10.1111/cen.12538
Abreu, I. M., Lau, E., de Sousa Pinto, B., and Carvalho, D. (2017). Subclinical hypothyroidism: to treat or not to treat, that is the question! A systematic review with meta-analysis on lipid profile. Endocr. Connect. 6, 188–199. doi: 10.1530/ec-17-0028
Al-Khatib, S. M., Stevenson, W. G., Ackerman, M. J., Bryant, W. J., Callans, D. J., Curtis, A. B., et al. (2018). 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Executive summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm 15, e190–e252. doi: 10.1016/j.hrthm.2017.10.035
Amin, A., Chitsazan, M., Taghavi, S., and Ardeshiri, M. (2015). Effects of triiodothyronine replacement therapy in patients with chronic stable heart failure and low-triiodothyronine syndrome: a randomized, double-blind, placebo-controlled study. ESC Heart Fail 2, 5–11. doi: 10.1002/ehf2.12025
Bahn, R. S., Burch, H. B., Cooper, D. S., Garber, J. R., Greenlee, M. C., Klein, I., et al. (2011). Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Endocr. Pract. 17, 456–520. doi: 10.4158/ep.17.3.456
Bano, A., Dhana, K., Chaker, L., Kavousi, M., Ikram, M. A., Mattace-Raso, F. U. S., et al. (2017). Association of Thyroid Function With Life Expectancy With and Without Cardiovascular Disease: The Rotterdam Study. JAMA Intern. Med. 177, 1650–1657. doi: 10.1001/jamainternmed.2017.4836
Barreto-Chaves, M. L., Senger, N., Fevereiro, M., Parletta, A. C., and Takano, A. (2020). Impact of hyperthyroidism on cardiac hypertrophy. Endocr. Connect 9, R59–R69. doi: 10.1530/ec-19-0543
Bartalena, L. (2013). Diagnosis and management of Graves disease: a global overview. Nat. Rev. Endocrinol. 9, 724–734. doi: 10.1038/nrendo.2013.193
Basaria, S., and Cooper, D. S. (2005). Amiodarone and the thyroid. Am. J. Med. 118, 706–714. doi: 10.1016/j.amjmed.2004.11.028
Baumgartner, C., da Costa, B. R., Collet, T. H., Feller, M., Floriani, C., Bauer, D. C., et al. (2017). Thyroid Function Within the Normal Range, Subclinical Hypothyroidism, and the Risk of Atrial Fibrillation. Circulation 136, 2100–2116. doi: 10.1161/circulationaha.117.028753
Biondi, B. (2012). Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur. J. Endocrinol. 167, 609–618. doi: 10.1530/eje-12-0627
Biondi, B., and Cooper, D. S. (2008). The clinical significance of subclinical thyroid dysfunction. Endocr. Rev. 29, 76–131. doi: 10.1210/er.2006-0043
Biondi, B., Bartalena, L., Cooper, D. S., Hegedüs, L., Laurberg, P., and Kahaly, G. J. (2015). The 2015 European Thyroid Association Guidelines on Diagnosis and Treatment of Endogenous Subclinical Hyperthyroidism. Eur. Thyroid J. 4, 149–163. doi: 10.1159/000438750
Biondi, B., Palmieri, E. A., Fazio, S., Cosco, C., Nocera, M., Saccà, L., et al. (2000). Endogenous subclinical hyperthyroidism affects quality of life and cardiac morphology and function in young and middle-aged patients. J. Clin. Endocrinol. Metab. 85, 4701–4705. doi: 10.1210/jcem.85.12.7085
Biondi, B., Palmieri, E. A., Lombardi, G., and Fazio, S. (2002). Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J. Clin. Endocrinol. Metab. 87, 968–974. doi: 10.1210/jcem.87.3.8302
Bogush, N., Tan, L., Naib, H., Faizullabhoy, E., Calvert, J. W., Iismaa, S. E., et al. (2020). DUSP5 expression in left ventricular cardiomyocytes of young hearts regulates thyroid hormone (T3)-induced proliferative ERK1/2 signaling. Sci. Rep. 10:21918. doi: 10.1038/s41598-020-78825-x
Brenta, G., Mutti, L. A., Schnitman, M., Fretes, O., Perrone, A., and Matute, M. L. (2003). Assessment of left ventricular diastolic function by radionuclide ventriculography at rest and exercise in subclinical hypothyroidism, and its response to L-thyroxine therapy. Am. J. Cardiol. 91, 1327–1330. doi: 10.1016/s0002-9149(03)00322-9
Brito, J. P., Schilz, S., Singh Ospina, N., Rodriguez-Gutierrez, R., Maraka, S., Sangaralingham, L. R., et al. (2016). Antithyroid Drugs-The Most Common Treatment for Graves’ Disease in the United States: A Nationwide Population-Based Study. Thyroid 26, 1144–1145. doi: 10.1089/thy.2016.0222
Burch, H. B. (2019). Drug Effects on the Thyroid. N. Engl. J. Med. 381, 749–761. doi: 10.1056/NEJMra1901214
Burch, H. B., and Wartofsky, L. (2018). Life-Threatening Thyrotoxicosis: Thyroid Storm. Endocr. Metabol. Med. Emergenc. Clinic. Guide 22, 262–283.
Canaris, G. J., Manowitz, N. R., Mayor, G., and Ridgway, E. C. (2000). The Colorado thyroid disease prevalence study. Arch. Intern. Med. 160, 526–534. doi: 10.1001/archinte.160.4.526
Cappola, A. R., and Ladenson, P. W. (2003). Hypothyroidism and atherosclerosis. J. Clin. Endocrinol. Metab. 88, 2438–2444. doi: 10.1210/jc.2003-030398
Cappola, A. R., Fried, L. P., Arnold, A. M., Danese, M. D., Kuller, L. H., Burke, G. L., et al. (2006). Thyroid status, cardiovascular risk, and mortality in older adults. JAMA 295, 1033–1041. doi: 10.1001/jama.295.9.1033
Casas, F., Daury, L., Grandemange, S., Busson, M., Seyer, P., Hatier, R., et al. (2003). Endocrine regulation of mitochondrial activity: involvement of truncated RXRalpha and c-Erb Aalpha1 proteins. FASEB J. 17, 426–436. doi: 10.1096/fj.02-0732com
Cayrol, F., Sterle, H. A., Díaz Flaqué, M. C., Barreiro Arcos, M. L., and Cremaschi, G. A. (2019). Non-genomic Actions of Thyroid Hormones Regulate the Growth and Angiogenesis of T Cell Lymphomas. Front. Endocrinol. 10:63. doi: 10.3389/fendo.2019.00063
Chaker, L., Bianco, A. C., Jonklaas, J., and Peeters, R. P. (2017). Hypothyroidism. Lancet 390, 1550–1562. doi: 10.1016/s0140-6736(17)30703-1
Chaker, L., van den Berg, M. E., Niemeijer, M. N., Franco, O. H., Dehghan, A., Hofman, A., et al. (2016). Thyroid Function and Sudden Cardiac Death: A Prospective Population-Based Cohort Study. Circulation 134, 713–722. doi: 10.1161/circulationaha.115.020789
Chen, Y. F., Weltman, N. Y., Li, X., Youmans, S., Krause, D., and Gerdes, A. M. (2013). Improvement of left ventricular remodeling after myocardial infarction with eight weeks L-thyroxine treatment in rats. J. Transl. Med. 11:40. doi: 10.1186/1479-5876-11-40
Chess-Williams, R., and Coker, S. J. (1989). Ventricular fibrillation is reduced in hypothyroid rats with enhanced myocardial alpha-adrenoceptor responsiveness. Br. J. Pharmacol. 98, 95–100. doi: 10.1111/j.1476-5381.1989.tb16867.x
Chojnowski, K., Bielec, A., Czarkowski, M., Dmowska-Chalaba, J., Kochanowski, J., and Wasowska, A. (2007). Repeated ventricular. Cardiol. J. 14, 198–201.
Collet, T. H., Gussekloo, J., Bauer, D. C., den Elzen, W. P., Cappola, A. R., Balmer, P., et al. (2012). Subclinical hyperthyroidism and the risk of coronary heart disease and mortality. Arch. Intern. Med. 172, 799–809. doi: 10.1001/archinternmed.2012.402
Colzani, R. M., Emdin, M., Conforti, F., Passino, C., Scarlattini, M., and Iervasi, G. (2001). Hyperthyroidism is associated with lengthening of ventricular repolarization. Clin. Endocrinol. 55, 27–32. doi: 10.1046/j.1365-2265.2001.01295.x
Cooper, D. S., and Biondi, B. (2012). Subclinical thyroid disease. Lancet 379, 1142–1154. doi: 10.1016/s0140-6736(11)60276-6
Cooper, D. S., Klibanski, A., and Ridgway, E. C. (1983). Dopaminergic modulation of TSH and its subunits: in vivo and in vitro studies. Clin. Endocrinol. 18, 265–275. doi: 10.1111/j.1365-2265.1983.tb03211.x
Crowley, W. F. Jr., Ridgway, E. C., Bough, E. W., Francis, G. S., Daniels, G. H., Kourides, I. A., et al. (1977). Noninvasive evaluation of cardiac function in hypothyroidism. Response to gradual thyroxine replacement. N. Engl. J. Med. 296, 1–6. doi: 10.1056/nejm197701062960101
Cruz, F. E., Cheriex, E. C., Smeets, J. L., Atié, J., Peres, A. K., Penn, O. C., et al. (1990). Reversibility of tachycardia-induced cardiomyopathy after cure of incessant supraventricular tachycardia. J. Am. Coll. Cardiol. 16, 739–744. doi: 10.1016/0735-1097(90)90368-y
Dahl, P., Danzi, S., and Klein, I. (2008). Thyrotoxic cardiac disease. Curr. Heart Fail Rep. 5, 170–176. doi: 10.1007/s11897-008-0026-9
D’Amico, D. J., Kenyon, K. R., and Ruskin, J. N. (1981). Amiodarone keratopathy: drug-induced lipid storage disease. Arch. Ophthalmol. 99, 257–261. doi: 10.1001/archopht.1981.03930010259007
Daniels, G. H. (2001). Amiodarone-induced thyrotoxicosis. J. Clin. Endocrinol. Metab. 86, 3–8. doi: 10.1210/jcem.86.1.7119
Danzi, S., and Klein, I. (2004). Thyroid hormone and the cardiovascular system. Minerva Endocrinol. 29, 139–150.
Danzi, S., and Klein, I. (2014). Thyroid disease and the cardiovascular system. Endocrinol. Metab. Clin. North Am. 43, 517–528. doi: 10.1016/j.ecl.2014.02.005
Davis, P. J., Ashur-Fabian, O., Incerpi, S., and Mousa, S. A. (2019). Editorial: Non Genomic Actions of Thyroid Hormones in Cancer. Front. Endocrinol. 10:847. doi: 10.3389/fendo.2019.00847
DeGroot, W. J., and Leonard, J. J. (1970). Hyperthyroidism as a high cardiac output state. Am. Heart J. 79, 265–275. doi: 10.1016/0002-8703(70)90318-2
Delitala, A. P., Orrù, M., Filigheddu, F., Pilia, M. G., Delitala, G., Ganau, A., et al. (2015). Serum free thyroxine levels are positively associated with arterial stiffness in the SardiNIA study. Clin. Endocrinol. 82, 592–597. doi: 10.1111/cen.12532
Diniz, G. P., Carneiro-Ramos, M. S., and Barreto-Chaves, M. L. (2009). Angiotensin type 1 receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3beta/mTOR signaling pathway. Basic Res. Cardiol. 104, 653–667. doi: 10.1007/s00395-009-0043-1
Dorian, P., and Cass, D. (1997). An overview of the management of electrical storm. Can. J. Cardiol. 13(Suppl. A), 13a–17a.
Duyff, R. F., Van den Bosch, J., Laman, D. M., van Loon, B. J., and Linssen, W. H. (2000). Neuromuscular findings in thyroid dysfunction: a prospective clinical and electrodiagnostic study. J. Neurol. Neurosurg. Psychiatry 68, 750–755. doi: 10.1136/jnnp.68.6.750
Fadel, B. M., Ellahham, S., Ringel, M. D., Lindsay, J. Jr., Wartofsky, L., and Burman, K. D. (2000). Hyperthyroid heart disease. Clin. Cardiol. 23, 402–408. doi: 10.1002/clc.4960230605
Forfar, J. C., Muir, A. L., Sawers, S. A., and Toft, A. D. (1982). Abnormal left ventricular function in hyperthyroidism: evidence for a possible reversible cardiomyopathy. N. Engl. J. Med. 307, 1165–1170. doi: 10.1056/nejm198211043071901
Franklyn, J. A., Sheppard, M. C., and Maisonneuve, P. (2005). Thyroid function and mortality in patients treated for hyperthyroidism. JAMA 294, 71–80. doi: 10.1001/jama.294.1.71
Fujio, Y., Nguyen, T., Wencker, D., Kitsis, R. N., and Walsh, K. (2000). Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 101, 660–667. doi: 10.1161/01.cir.101.6.660
Gerdes, A. M., and Iervasi, G. (2010). Thyroid replacement therapy and heart failure. Circulation 122, 385–393. doi: 10.1161/circulationaha.109.917922
Giammanco, M., Di Liegro, C. M., Schiera, G., and Di Liegro, I. (2020). Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. Int. J. Mol. Sci. 21:ijms21114140. doi: 10.3390/ijms21114140
Gick, G. G., Melikian, J., and Ismail-Beigi, F. (1990). Thyroidal enhancement of rat myocardial Na,K-ATPase: preferential expression of alpha 2 activity and mRNA abundance. J. Membr. Biol. 115, 273–282. doi: 10.1007/bf01868642
Grais, I. M. (2010). Bedside skills: a 50-year personal retrospective. Tex. Heart Inst. J. 37, 629–632.
Hamilton, D. Sr., Nandkeolyar, S., Lan, H., Desai, P., Evans, J., Hauschild, C., et al. (2020). Amiodarone: A Comprehensive Guide for Clinicians. Am. J. Cardiovasc. Drugs 20, 549–558. doi: 10.1007/s40256-020-00401-5
Hamilton, T. E., Davis, S., Onstad, L., and Kopecky, K. J. (2008). Thyrotropin levels in a population with no clinical, autoantibody, or ultrasonographic evidence of thyroid disease: implications for the diagnosis of subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 93, 1224–1230. doi: 10.1210/jc.2006-2300
Harjai, K. J., and Licata, A. A. (1997). Effects of amiodarone on thyroid function. Ann. Intern. Med. 126, 63–73. doi: 10.7326/0003-4819-126-1-199701010-00009
Hayashi, M., Shimizu, W., and Albert, C. M. (2015). The spectrum of epidemiology underlying sudden cardiac death. Circ. Res. 116, 1887–1906. doi: 10.1161/circresaha.116.304521
He, H., Giordano, F. J., Hilal-Dandan, R., Choi, D. J., Rockman, H. A., McDonough, P. M., et al. (1997). Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J. Clin. Invest. 100, 380–389. doi: 10.1172/jci119544
Hercbergs, A. (2019). Clinical Implications and Impact of Discovery of the Thyroid Hormone Receptor on Integrin αvβ3-A Review. Front. Endocrinol. 10:565. doi: 10.3389/fendo.2019.00565
Hirose, K., Payumo, A. Y., Cutie, S., Hoang, A., Zhang, H., Guyot, R., et al. (2019). Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 364, 184–188. doi: 10.1126/science.aar2038
Hoit, B. D., Khoury, S. F., Shao, Y., Gabel, M., Liggett, S. B., and Walsh, R. A. (1997). Effects of thyroid hormone on cardiac beta-adrenergic responsiveness in conscious baboons. Circulation 96, 592–598. doi: 10.1161/01.cir.96.2.592
Hollowell, J. G., Staehling, N. W., Flanders, W. D., Hannon, W. H., Gunter, E. W., Spencer, C. A., et al. (2002). Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J. Clin. Endocrinol. Metab. 87, 489–499. doi: 10.1210/jcem.87.2.8182
Holt, E., Sjaastad, I., Lunde, P. K., Christensen, G., and Sejersted, O. M. (1999). Thyroid hormone control of contraction and the Ca(2+)-ATPase/phospholamban complex in adult rat ventricular myocytes. J. Mol. Cell Cardiol. 31, 645–656. doi: 10.1006/jmcc.1998.0900
Iervasi, G., Molinaro, S., Landi, P., Taddei, M. C., Galli, E., Mariani, F., et al. (2007). Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch. Intern. Med. 167, 1526–1532. doi: 10.1001/archinte.167.14.1526
Jabbar, A., Pingitore, A., Pearce, S. H., Zaman, A., Iervasi, G., and Razvi, S. (2017). Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 14, 39–55. doi: 10.1038/nrcardio.2016.174
Jankauskienė, E., Orda, P., Barauskienė, G., Mickuvienė, N., Brožaitienė, J., Vaškelytė, J. J., et al. (2016). Relationship between left ventricular mechanics and low free triiodothyronine levels after myocardial infarction: a prospective study. Intern. Emerg. Med. 11, 391–398. doi: 10.1007/s11739-015-1370-x
Jao, Y. T., Chen, Y., Lee, W. H., and Tai, F. T. (2004). Thyroid storm and ventricular tachycardia. South Med. J. 97, 604–607. doi: 10.1097/00007611-200406000-00020
Jonklaas, J., Bianco, A. C., Bauer, A. J., Burman, K. D., Cappola, A. R., Celi, F. S., et al. (2014). Guidelines for the treatment of hypothyroidism: prepared by the american thyroid association task force on thyroid hormone replacement. Thyroid 24, 1670–1751. doi: 10.1089/thy.2014.0028
Kaasik, A., Paju, K., Vetter, R., and Seppet, E. K. (1997). Thyroid hormones increase the contractility but suppress the effects of beta-adrenergic agonist by decreasing phospholamban expression in rat atria. Cardiovasc. Res. 35, 106–112. doi: 10.1016/s0008-6363(97)00069-2
Kahaly, G. J., and Dillmann, W. H. (2005). Thyroid hormone action in the heart. Endocr. Rev. 26, 704–728. doi: 10.1210/er.2003-0033
Kahaly, G. J., Bartalena, L., Hegedüs, L., Leenhardt, L., Poppe, K., and Pearce, S. H. (2018). 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. Eur. Thyroid J. 7, 167–186. doi: 10.1159/000490384
Kenessey, A., and Ojamaa, K. (2006). Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J. Biol. Chem. 281, 20666–20672. doi: 10.1074/jbc.M512671200
Khan, R., Sikanderkhel, S., Gui, J., Adeniyi, A. R., O’Dell, K., Erickson, M., et al. (2020). Thyroid and Cardiovascular Disease: A Focused Review on the Impact of Hyperthyroidism in Heart Failure. Cardiol. Res. 11, 68–75. doi: 10.14740/cr1034
Khochtali, I., Hamza, N., Harzallah, O., Hamdi, S., Saad, J., Golli, M., et al. (2011). Reversible dilated cardiomyopathy caused by hypothyroidism. Int. Arch. Med. 4:20. doi: 10.1186/1755-7682-4-20
Kim, E. J., Lyass, A., Wang, N., Massaro, J. M., Fox, C. S., Benjamin, E. J., et al. (2014). Relation of hypothyroidism and incident atrial fibrillation (from the Framingham Heart Study). Am. Heart J. 167, 123–126. doi: 10.1016/j.ahj.2013.10.012
Kinugawa, K., Minobe, W. A., Wood, W. M., Ridgway, E. C., Baxter, J. D., Ribeiro, R. C., et al. (2001). Signaling pathways responsible for fetal gene induction in the failing human heart: evidence for altered thyroid hormone receptor gene expression. Circulation 103, 1089–1094. doi: 10.1161/01.cir.103.8.1089
Kiss, E., Jakab, G., Kranias, E. G., and Edes, I. (1994). Thyroid hormone-induced alterations in phospholamban protein expression. Regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. Circ. Res. 75, 245–251. doi: 10.1161/01.res.75.2.245
Klein, I., and Danzi, S. (2007). Thyroid disease and the heart. Circulation 116, 1725–1735. doi: 10.1161/circulationaha.106.678326
Klein, I., and Danzi, S. (2016). Thyroid Disease and the Heart. Curr. Probl. Cardiol. 41, 65–92. doi: 10.1016/j.cpcardiol.2015.04.002
Klein, I., and Ojamaa, K. (2001). Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 344, 501–509. doi: 10.1056/nejm200102153440707
Klemperer, J. D., Klein, I. L., Ojamaa, K., Helm, R. E., Gomez, M., Isom, O. W., et al. (1996). Triiodothyronine therapy lowers the incidence of atrial fibrillation after cardiac operations. Ann. Thorac. Surg. 61, 1323–1327. doi: 10.1016/0003-4975(96)00102-6
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/circresaha.111.259754
Kuzman, J. A., Gerdes, A. M., Kobayashi, S., and Liang, Q. (2005). Thyroid hormone activates Akt and prevents serum starvation-induced cell death in neonatal rat cardiomyocytes. J. Mol. Cell Cardiol. 39, 841–844. doi: 10.1016/j.yjmcc.2005.07.019
Langén, V. L., Niiranen, T. J., Puukka, P., Lehtonen, A. O., Hernesniemi, J. A., Sundvall, J., et al. (2018). Thyroid-stimulating hormone and risk of sudden cardiac death, total mortality and cardiovascular morbidity. Clin. Endocrinol. 88, 105–113. doi: 10.1111/cen.13472
Lazar, M. A., and Chin, W. W. (1990). Nuclear thyroid hormone receptors. J. Clin. Invest. 86, 1777–1782. doi: 10.1172/jci114906
Lisco, G., De Tullio, A., Iacoviello, M., and Triggiani, V. (2020). Congestive Heart Failure and Thyroid Dysfunction: The Role of the Low T3 Syndrome and Therapeutic Aspects. Endocr. Metab. Immune Disord. Drug Targets 20, 646–653. doi: 10.2174/1871530319666191119112950
Liu, Y., Redetzke, R. A., Said, S., Pottala, J. V., de Escobar, G. M., and Gerdes, A. M. (2008). Serum thyroid hormone levels may not accurately reflect thyroid tissue levels and cardiac function in mild hypothyroidism. Am. J. Physiol. Heart Circ. Physiol. 294, H2137–H2143. doi: 10.1152/ajpheart.01379.2007
Lopshire, J. C., and Zipes, D. P. (2006). Sudden cardiac death: better understanding of risks, mechanisms, and treatment. Circulation 114, 1134–1136. doi: 10.1161/circulationaha.106.647933
Madathil, A., Hollingsworth, K. G., Blamire, A. M., Razvi, S., Newton, J. L., Taylor, R., et al. (2015). Levothyroxine improves abnormal cardiac bioenergetics in subclinical hypothyroidism: a cardiac magnetic resonance spectroscopic study. J. Clin. Endocrinol. Metab. 100, E607–E610. doi: 10.1210/jc.2014-2942
Marín-García, J. (2010). Thyroid hormone and myocardial mitochondrial biogenesis. Vascul. Pharmacol. 52, 120–130. doi: 10.1016/j.vph.2009.10.008
Marrakchi, S., Kanoun, F., Idriss, S., Kammoun, I., and Kachboura, S. (2015). Arrhythmia and thyroid dysfunction. Herz 40(Suppl. 2), 101–109. doi: 10.1007/s00059-014-4123-0
Martino, E., Bartalena, L., Bogazzi, F., and Braverman, L. E. (2001). The effects of amiodarone on the thyroid. Endocr. Rev. 22, 240–254. doi: 10.1210/edrv.22.2.0427
Martino, E., Safran, M., Aghini-Lombardi, F., Rajatanavin, R., Lenziardi, M., Fay, M., et al. (1984). Environmental iodine intake and thyroid dysfunction during chronic amiodarone therapy. Ann. Intern. Med. 101, 28–34. doi: 10.7326/0003-4819-101-1-28
Mendel, C. M., Frost, P. H., Kunitake, S. T., and Cavalieri, R. R. (1987). Mechanism of the heparin-induced increase in the concentration of free thyroxine in plasma. J. Clin. Endocrinol. Metab. 65, 1259–1264. doi: 10.1210/jcem-65-6-1259
Mitchell, J. E., Hellkamp, A. S., Mark, D. B., Anderson, J., Johnson, G. W., Poole, J. E., et al. (2013). Thyroid function in heart failure and impact on mortality. JACC Heart Fail 1, 48–55. doi: 10.1016/j.jchf.2012.10.004
Mohr-Kahaly, S., Kahaly, G., and Meyer, J. (1996). [Cardiovascular effects of thyroid hormones]. Z. Kardiol. 85(Suppl. 6), 219–231.
Monzani, F., Di Bello, V., Caraccio, N., Bertini, A., Giorgi, D., Giusti, C., et al. (2001). Effect of levothyroxine on cardiac function and structure in subclinical hypothyroidism: a double blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 86, 1110–1115. doi: 10.1210/jcem.86.3.7291
Mourouzis, I., Forini, F., Pantos, C., and Iervasi, G. (2011). Thyroid hormone and cardiac disease: from basic concepts to clinical application. J. Thyroid Res. 2011:958626. doi: 10.4061/2011/958626
Muthukumar, S., Sadacharan, D., Ravikumar, K., Mohanapriya, G., Hussain, Z., and Suresh, R. V. (2016). A prospective study on cardiovascular dysfunction in patients with hyperthyroidism and its reversal after surgical cure. World J. Surg. 40, 622–628.
Nadal-Ginard, B., and Mahdavi, V. (1989). Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J. Clin. Invest. 84, 1693–1700. doi: 10.1172/jci114351
Nademanee, K., Taylor, R., Bailey, W. E., Rieders, D. E., and Kosar, E. M. (2000). Treating electrical storm : sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation 102, 742–747. doi: 10.1161/01.cir.102.7.742
Nagayo, T. (2018). Low T3 syndrome, Low T4 syndrome (euthyroido sick syndrome). Nihon Rinsho Jap. J. Clin. Med. 2018(Suppl. 1), 504–507.
Nakada, Y., Canseco, D. C., Thet, S., Abdisalaam, S., Asaithamby, A., Santos, C. X., et al. (2017). Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227. doi: 10.1038/nature20173
Nakazawa, H., Lythall, D. A., Noh, J., Ishikawa, N., Sugino, K., Ito, K., et al. (2000). Is there a place for the late cardioversion of atrial fibrillation? A long-term follow-up study of patients with post-thyrotoxic atrial fibrillation. Eur. Heart J. 21, 327–333. doi: 10.1053/euhj.1999.1956
Neves, J. S., Leitão, L., Baeta Baptista, R., Bigotte Vieira, M., Magriço, R., Viegas Dias, C., et al. (2019). Lower free triiodothyronine levels within the reference range are associated with higher cardiovascular mortality: An analysis of the NHANES. Int. J. Cardiol. 285, 115–120.
Neves, J. S., Vale, C., von Hafe, M., Borges-Canha, M., Leite, A. R., Almeida-Coelho, J., et al. (2020). Thyroid hormones and modulation of diastolic function: a promising target for heart failure with preserved ejection fraction. Ther. Adv. Endocrinol. Metab. 11:2042018820958331. doi: 10.1177/2042018820958331
Nordyke, R. A., Gilbert, F. I. Jr., and Harada, A. S. (1988). Graves’ disease. Influence of age on clinical findings. Arch. Intern. Med. 148, 626–631. doi: 10.1001/archinte.148.3.626
Ojamaa, K., Klemperer, J. D., and Klein, I. (1996). Acute effects of thyroid hormone on vascular smooth muscle. Thyroid 6, 505–512. doi: 10.1089/thy.1996.6.505
Ojamaa, K., Sabet, A., Kenessey, A., Shenoy, R., and Klein, I. (1999). Regulation of rat cardiac Kv1.5 gene expression by thyroid hormone is rapid and chamber specific. Endocrinology 140, 3170–3176. doi: 10.1210/endo.140.7.6776
Omidi, N., Khorgami, M., Tajrishi, F. Z., Seyedhoseinpour, A., and Pasbakhsh, P. (2020). The Role of Thyroid Diseases and their Medications in Cardiovascular Disorders: A Review of the Literature. Curr. Cardiol. Rev. 16, 103–116. doi: 10.2174/1573403x15666191008111238
Ortiz, V. D., Türck, P., Teixeira, R., Lima-Seolin, B. G., Lacerda, D., Fraga, S. F., et al. (2019). Carvedilol and thyroid hormones co-administration mitigates oxidative stress and improves cardiac function after acute myocardial infarction. Eur. J. Pharmacol. 854, 159–166.
Osman, F., Gammage, M. D., Sheppard, M. C., and Franklyn, J. A. (2002). Clinical review 142: cardiac dysrhythmias and thyroid dysfunction: the hidden menace? J. Clin. Endocrinol. Metab. 87, 963–967. doi: 10.1210/jcem.87.3.8217
Osuna, P. M., Udovcic, M., and Sharma, M. D. (2017). Hyperthyroidism and the Heart. Methodist Debakey Cardiovasc. J. 13, 60–63. doi: 10.14797/mdcj-13-2-60
Pantos, C., Mourouzis, I., Markakis, K., Dimopoulos, A., Xinaris, C., Kokkinos, A. D., et al. (2007). Thyroid hormone attenuates cardiac remodeling and improves hemodynamics early after acute myocardial infarction in rats. Eur. J. Cardiothorac. Surg. 32, 333–339. doi: 10.1016/j.ejcts.2007.05.004
Parle, J. V., Franklyn, J. A., Cross, K. W., Jones, S. C., and Sheppard, M. C. (1991). Prevalence and follow-up of abnormal thyrotrophin (TSH) concentrations in the elderly in the United Kingdom. Clin. Endocrinol. 34, 77–83. doi: 10.1111/j.1365-2265.1991.tb01739.x
Parle, J. V., Maisonneuve, P., Sheppard, M. C., Boyle, P., and Franklyn, J. A. (2001). Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet 358, 861–865. doi: 10.1016/s0140-6736(01)06067-6
Patil, V. C., Patil, H. V., Agrawal, V., and Patil, S. (2011). Cardiac tamponade in a patient with primary hypothyroidism. Ind. J. Endocr. Metab. 15, S144–S146. doi: 10.4103/2230-8210.83358
Pearce, E. N. (2012). Update in lipid alterations in subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 97, 326–333. doi: 10.1210/jc.2011-2532
Pearce, S. H., Brabant, G., Duntas, L. H., Monzani, F., Peeters, R. P., Razvi, S., et al. (2013). 2013 ETA Guideline: Management of Subclinical Hypothyroidism. Eur. Thyroid J. 2, 215–228. doi: 10.1159/000356507
Pingitore, A., Galli, E., Barison, A., Iervasi, A., Scarlattini, M., Nucci, D., et al. (2008). Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 93, 1351–1358. doi: 10.1210/jc.2007-2210
Pingitore, A., Landi, P., Taddei, M. C., Ripoli, A., L’Abbate, A., and Iervasi, G. (2005). Triiodothyronine levels for risk stratification of patients with chronic heart failure. Am. J. Med. 118, 132–136. doi: 10.1016/j.amjmed.2004.07.052
Pingitore, A., Mastorci, F., Piaggi, P., Aquaro, G. D., Molinaro, S., Ravani, M., et al. (2019). Usefulness of Triiodothyronine Replacement Therapy in Patients With ST Elevation Myocardial Infarction and Borderline/Reduced Triiodothyronine Levels (from the THIRST Study). Am. J. Cardiol. 123, 905–912. doi: 10.1016/j.amjcard.2018.12.020
Polikar, R., Burger, A. G., Scherrer, U., and Nicod, P. (1993). The thyroid and the heart. Circulation 87, 1435–1441. doi: 10.1161/01.cir.87.5.1435
Rajagopalan, V., Zhao, M., Reddy, S., Fajardo, G., Wang, X., Dewey, S., et al. (2013). Altered ubiquitin-proteasome signaling in right ventricular hypertrophy and failure. Am. J. Physiol. Heart Circ. Physiol. 305, H551–H562. doi: 10.1152/ajpheart.00771.2012
Rajagopalan, V., Zucker, I. H., Jones, J. A., Carlson, M., and Ma, Y. J. (2008). Cardiac ErbB-1/ErbB-2 mutant expression in young adult mice leads to cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol. 295, H543–H554. doi: 10.1152/ajpheart.91436.2007
Razvi, S., Jabbar, A., Pingitore, A., Danzi, S., Biondi, B., Klein, I., et al. (2018). Thyroid Hormones and Cardiovascular Function and Diseases. J. Am. Coll. Cardiol. 71, 1781–1796. doi: 10.1016/j.jacc.2018.02.045
Razvi, S., Shakoor, A., Vanderpump, M., Weaver, J. U., and Pearce, S. H. (2008). The influence of age on the relationship between subclinical hypothyroidism and ischemic heart disease: a metaanalysis. J. Clin. Endocrinol. Metab. 93, 2998–3007. doi: 10.1210/jc.2008-0167
Ripoli, A., Pingitore, A., Favilli, B., Bottoni, A., Turchi, S., Osman, N. F., et al. (2005). Does subclinical hypothyroidism affect cardiac pump performance? Evidence from a magnetic resonance imaging study. J. Am. Coll. Cardiol. 45, 439–445. doi: 10.1016/j.jacc.2004.10.044
Rodondi, N., Bauer, D. C., Cappola, A. R., Cornuz, J., Robbins, J., Fried, L. P., et al. (2008). Subclinical thyroid dysfunction, cardiac function, and the risk of heart failure. The Cardiovascular Health study. J. Am. Coll. Cardiol. 52, 1152–1159. doi: 10.1016/j.jacc.2008.07.009
Rodondi, N., den Elzen, W. P., Bauer, D. C., Cappola, A. R., Razvi, S., Walsh, J. P., et al. (2010). Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA 304, 1365–1374. doi: 10.1001/jama.2010.1361
Ruzieh, M., Moroi, M. K., Aboujamous, N. M., Ghahramani, M., Naccarelli, G. V., Mandrola, J., et al. (2019). Meta-Analysis Comparing the Relative Risk of Adverse Events for Amiodarone Versus Placebo. Am. J. Cardiol. 124, 1889–1893. doi: 10.1016/j.amjcard.2019.09.008
Saad, N. S., Repas, S. J., Floyd, K., Janssen, P. M. L., and Elnakish, M. T. (2017). Recovery following Thyroxine Treatment Withdrawal, but Not Propylthiouracil, Averts In Vivo and Ex Vivo Thyroxine-Provoked Cardiac Complications in Adult FVB/N Mice. BioMed Res. Int. 2017:6071031. doi: 10.1155/2017/6071031
Schmidt-Ott, U. M., and Ascheim, D. D. (2006). Thyroid hormone and heart failure. Curr. Heart Fail Rep. 3, 114–119. doi: 10.1007/s11897-006-0010-1
Schwimmer, D., Vogel, M., and Mc, G. T. (1947). Clinical manifestations in forty cases of myxedema. J. Clin. Endocrinol. Metab. 7:468.
Selmer, C., Olesen, J. B., Hansen, M. L., Kappelgaard, L. M. V., Madsen, J. C., Hansen, P. R., et al. (2014). Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: a large population study. J. Clin. Endocr. Metabol. 99, 2372–2382. doi: 10.1210/jc.2013-4184
Shenfield, G. M. (1981). Influence of Thyroid Dysfunction on Drug Pharmacokinetics. Clin. Pharmacokinet. 6, 275–297. doi: 10.2165/00003088-198106040-00003
Singh, B. N. (2008). Amiodarone as paradigm for developing new drugs for atrial fibrillation. J. Cardiovasc. Pharmacol. 52, 300–305. doi: 10.1097/FJC.0b013e31818914b6
Siu, C. W., Yeung, C. Y., Lau, C. P., Kung, A. W., and Tse, H. F. (2007). Incidence, clinical characteristics and outcome of congestive heart failure as the initial presentation in patients with primary hyperthyroidism. Heart 93, 483–487. doi: 10.1136/hrt.2006.100628
Smith, T. J., and Hegedüs, L. (2016). Graves’ Disease. N. Engl. J. Med. 375, 1552–1565. doi: 10.1056/NEJMra1510030
Surks, M. I., Ortiz, E., Daniels, G. H., Sawin, C. T., Col, N. F., Cobin, R. H., et al. (2004). Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. JAMA 291, 228–238. doi: 10.1001/jama.291.2.228
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tan, L., Bogush, N., Naib, H., Perry, J., Calvert, J. W., Martin, D. I. K., et al. (2019). Redox activation of JNK2α2 mediates thyroid hormone-stimulated proliferation of neonatal murine cardiomyocytes. Sci. Rep. 9:17731. doi: 10.1038/s41598-019-53705-1
Taylor, P. N., Iqbal, A., Minassian, C., Sayers, A., Draman, M. S., Greenwood, R., et al. (2014). Falling threshold for treatment of borderline elevated thyrotropin levels-balancing benefits and risks: evidence from a large community-based study. JAMA Intern. Med. 174, 32–39. doi: 10.1001/jamainternmed.2013.11312
Toft, A. D., and Boon, N. A. (2000). Thyroid disease and the heart. Heart 84, 455–460. doi: 10.1136/heart.84.4.455
Trohman, R. G., Sharma, P. S., McAninch, E. A., and Bianco, A. C. (2019). Amiodarone and thyroid physiology, pathophysiology, diagnosis and management. Trends Cardiovasc. Med. 29, 285–295. doi: 10.1016/j.tcm.2018.09.005
Tsang, W., and Houlden, R. L. (2009). Amiodarone-induced thyrotoxicosis: a review. Can. J. Cardiol. 25, 421–424. doi: 10.1016/s0828-282x(09)70512-4
Vadiveloo, T., Donnan, P. T., Cochrane, L., and Leese, G. P. (2011). The Thyroid Epidemiology, Audit, and Research Study (TEARS): morbidity in patients with endogenous subclinical hyperthyroidism. J. Clin. Endocrinol. Metab. 96, 1344–1351. doi: 10.1210/jc.2010-2693
Vale, C., Neves, J. S., von Hafe, M., Borges-Canha, M., and Leite-Moreira, A. (2019). The Role of Thyroid Hormones in Heart Failure. Cardiovasc. Drugs Ther. 33, 179–188. doi: 10.1007/s10557-019-06870-4
von Hafe, M., Neves, J. S., Vale, C., Borges-Canha, M., and Leite-Moreira, A. (2019). The impact of thyroid hormone dysfunction on ischemic heart disease. Endocr. Connect. 8, R76–R90. doi: 10.1530/ec-19-0096
von Olshausen, K., Bischoff, S., Kahaly, G., Mohr-Kahaly, S., Erbel, R., Beyer, J., et al. (1989). Cardiac arrhythmias and heart rate in hyperthyroidism. Am. J. Cardiol. 63, 930–933. doi: 10.1016/0002-9149(89)90142-2
Wang, B., Liu, S., Li, L., Yao, Q., Song, R., Shao, X., et al. (2017). Non-thyroidal illness syndrome in patients with cardiovascular diseases: A systematic review and meta-analysis. Int. J. Cardiol. 226, 1–10.
Wasielica-Poslednik, J., Pfeiffer, N., Reinke, J., and Pitz, S. (2011). Confocal laser-scanning microscopy allows differentiation between Fabry disease and amiodarone-induced keratopathy. Graefes Arch. Clin. Exp. Ophthalmol. 249, 1689–1696. doi: 10.1007/s00417-011-1726-5
Weisfeldt, M. L., Everson-Stewart, S., Sitlani, C., Rea, T., Aufderheide, T. P., Atkins, D. L., et al. (2011). Ventricular tachyarrhythmias after cardiac arrest in public versus at home. N. Engl. J. Med. 364, 313–321. doi: 10.1056/NEJMoa1010663
Wirth, E. K., and Meyer, F. (2017). Neuronal effects of thyroid hormone metabolites. Mol. Cell Endocrinol. 458, 136–142. doi: 10.1016/j.mce.2017.01.007
Zhang, K., Tang, Y. D., Zhang, Y., Ojamaa, K., Li, Y., Saini, A. S., et al. (2018). Comparison of Therapeutic Triiodothyronine Versus Metoprolol in the Treatment of Myocardial Infarction in Rats. Thyroid 28, 799–810. doi: 10.1089/thy.2017.0544
Zhang, Y., Dedkov, E. I., Lee, B. III, Li, Y., Pun, K., and Gerdes, A. M. (2014). Thyroid hormone replacement therapy attenuates atrial remodeling and reduces atrial fibrillation inducibility in a rat myocardial infarction-heart failure model. J. Card. Fail 20, 1012–1019. doi: 10.1016/j.cardfail.2014.10.003
Keywords: thyroid hormone, hyperthyroidism, hypothyroidism, cardiovascular disease, genomic pathways, non-genomic pathways, amiodarone, cardiac regeneration
Citation: Yamakawa H, Kato TS, Noh JY, Yuasa S, Kawamura A, Fukuda K and Aizawa Y (2021) Thyroid Hormone Plays an Important Role in Cardiac Function: From Bench to Bedside. Front. Physiol. 12:606931. doi: 10.3389/fphys.2021.606931
Received: 16 September 2020; Accepted: 28 September 2021;
Published: 18 October 2021.
Edited by:
Brian P. Delisle, University of Kentucky, United StatesReviewed by:
Félix Vargas, University of Granada, SpainCopyright © 2021 Yamakawa, Kato, Noh, Yuasa, Kawamura, Fukuda and Aizawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshiyasu Aizawa, eW9zaGl5YXN1NjEyQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.