 Simone Regina Potje
Simone Regina Potje Tiago Dal-Cin Paula
Tiago Dal-Cin Paula Lusiane Maria Bendhack
Lusiane Maria Bendhack- Faculty of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, Ribeirão Preto, Brazil
This review highlights recent findings about the role that endothelial glycocalyx and caveolae play in vascular homeostasis. We describe the structure, synthesis, and function of glycocalyx and caveolae in vascular cells under physiological and pathophysiological conditions. Special focus will be given in glycocalyx and caveolae that are associated with impaired production of nitric oxide (NO) and generation of reactive oxygen species (ROS). Such alterations could contribute to the development of cardiovascular diseases, such as atherosclerosis, and hypertension.
Introduction
The role that endothelium plays in modulating the vascular tone includes the synthesis and release of several vasoactive substances, especially the vasodilator nitric oxide (NO) (Cahill and Redmond, 2016). Endothelial NO synthase (eNOS) is responsible for the synthesis of most of the NO that is produced in endothelial cells (ECs) (Zhao et al., 2015). eNOS is localized on domains named caveolae, which are spread over the entire ECs surface (Shaul, 2003). The glycocalyx is a polysaccharide-rich layer, which underlies mechano-transduction and mediates the physiological activation of NO synthesis by shear stress (Pahakis et al., 2007). More specifically, the glycocalyx components transform mechanical signals into biochemical signals, to activate eNOS (Florian et al., 2003; Pahakis et al., 2007), thereby contributing to vascular homeostasis (Alphonsus and Rodseth, 2014).
Shedding of glycocalyx and changes in the structure of caveolae decreases eNOS activity, which reduces NO bioavailability and generates reactive oxygen species (ROS) (Kumagai et al., 2009; Potje et al., 2019). Both consequences are associated with cardiovascular diseases such as atherosclerosis and hypertension. Therefore, the organization and function of glycocalyx and caveolae might be altered in atherosclerosis and hypertension, which results in release of deleterious ROS that contribute to these pathological conditions.
This review aims to highlight recent findings about the activation of glycocalyx and caveolar enzymes that participate in the synthesis and release of NO and ROS and alterations that could impair the proper function of glycocalyx and caveolae in pathological conditions like atherosclerosis and hypertension.
Structure and Synthesis of Endothelial Glycocalyx
As reviewed by different groups, the endothelial glycocalyx is mainly composed of glycosaminoglycans, proteoglycans, and glycoproteins. Heparan sulfate, chondroitin sulfate, and hyaluronic acid chains constitute glycosaminoglycans. Proteoglycans include core protein families such as perlecan, syndecans-1, -2, -3, -4, and glypican-1. Lastly, glycoproteins consist of sialic acid oligosaccharides (Uchimido et al., 2019; Möckl, 2020).
Heparan sulfate is the predominant constituent (from 50 to 90%) of glycocalyx (Reitsma et al., 2007). The syndecan family can contain three to eight potential heparan sulfate or chondroitin sulfate attachment sites depending on the specific syndecan member. These sites are located close to the syndecan NH2-terminal ectodomain or adjacent to the transmembrane domain near the syndecan COOH terminal. Glypican-1 is the only proteoglycan that is expressed exclusively in ECs. It binds specifically to the heparan sulfate chain and is localized in lipid rafts (caveolae) through a C-terminal glycosylphosphatidylinositol (GPI) anchor (Uchimido et al., 2019; Möckl, 2020).
Biosynthesis of glycosaminoglycans is a complex process that is initiated by chain polymerization and which depends on various stepwise reactions like sulfation and epimerization. This process happens in many cellular components including endoplasmic reticulum and Golgi apparatus, which are responsible for the secretory pathway (Uchimido et al., 2019; Möckl, 2020). On the other hand, hyaluronic acid is directly assembled in the membrane by hyaluronan synthases, and it is secreted into the extracellular space (Agarwal et al., 2019).
Vascular Protective Effects of Glycocalyx
Glycocalyx functions as a vascular protector because it participates in angiogenesis, exerts an anticoagulant effect, prevents leukocyte adhesion, acts as a selective permeable barrier and filter, operates as a mechano-transducer of shear stress, and contributes to maintaining the vascular tone.
Glycocalyx, specifically heparan sulfate, regulates angiogenesis by playing a proangiogenic role (Fuster and Wang, 2010). 6-O-sulfation of heparan sulfate is an essential regulator of vascular morphogenesis in zebrafish (Chen et al., 2005). In addition, decreased heparan sulfate N-sulfation impairs recruitment of pericytes and development of vasculature in N-deacetylase/N-sulfotransferase (Ndst)-1 knockout mice (Abramsson et al., 2007). Moreover, complete loss of heparan sulfate chains in mural cells causes embryonic death in the late stages of vascular morphogenesis and stability (Stenzel et al., 2009). In this way, glycocalyx contributes to angiogenesis process.
Antithrombin III is the main anticoagulant molecule that can bind to specific sites of heparan sulfate; it also inhibits coagulant factors and inactivates factors IX and X (Shimada et al., 1991; Quinsey et al., 2004). Likewise, tissue factor pathway inhibitor (TFPI) can also bind to heparan sulfates and block the initial steps of blood coagulation by inhibiting factors VIIa and Xa (Kato, 2002). Additionally, dermathan sulfate in glycocalyx can activate heparin cofactor II, which inhibits thrombin (Tovar et al., 2005). Furthermore, degradation of endothelial glycocalyx induced by hyperglycemia activates coagulation in healthy subjects (Nieuwdorp et al., 2006b). Therefore, glycocalyx has anticoagulant and antithrombotic effects.
The glycocalyx layer has consistency and anti-adhesive character, promoting resistance to penetration of circulating leukocytes and preventing leukocyte-endothelial adhesion in vascular smooth muscle cells (VSMCs). Besides, degradation of the glycocalyx layer provoked by heparitinase in mouse cremaster venules increases leukocyte adhesion in a dose-dependent manner (Constantinescu et al., 2003). In addition, enzymatic degradation of glycocalyx promoted increased of ICAM-1 expression, which was associated to a de-regulation in NF-κB activity in response to flow and leukocyte adhesion (McDonald et al., 2016). Moreover, endotoxemia stimulated in mice by tumor necrosis factor-α (TNF-α) rapidly degrades pulmonary microvascular glycocalyx, which contributes to neutrophil adhesion (Schmidt et al., 2012). Consequently, the damage of glycocalyx favors the adherence of leukocyte on the ECs.
Tumor necrosis factor-α treatment increases porosity and permeation due to glycocalyx shedding with enhanced intraluminal volume (Henry and Duling, 2000). Patients with type 1 diabetes show glycocalyx damage in sublingual capillaries, which is associated with microalbuminuria (Nieuwdorp et al., 2006a). Furthermore, degradation of heparan sulfate by heparanase promotes injury in porcine aortic ECs, which was associated to apoptosis and cell death (Han et al., 2005). Thus, glycocalyx works as a barrier and filter, besides protecting vascular cells.
Shear stress on ECs is a frictional force (mechanical signals) per unit area created by laminar blood flow (Pohl et al., 1986). Heparan sulfate is important to detect the direction of shear stress because degradation of this substance prevents shear stress-induced directional migration of ECs and inhibits recruitment of phosphorylated focal adhesion kinase in the flow direction (Moon et al., 2005). Nevertheless, remodeling of glycocalyx in response to short and long periods of shear stress has been reported (Liu et al., 2016). Moreover, reorganization of actin cytoskeleton and focal adhesions in response to fluid shear stress has been shown in rat fat-pad ECs in various flow media (Thi et al., 2004). Similarly, changes in the actin cytoskeleton and caveolae have been demonstrated after long-term shear stress (24 h), which also redistributes and restores heparan sulfate, syndecan-1, and glypican-1 on the apical surface of ECs (Zeng and Tarbell, 2014). In this way, the actin cytoskeleton contributes to the structural stability of glycocalyx under shear stress (Li and Wang, 2018).
Mechano-transduction is the conversion of mechanical signals induced by shear stress into biochemical signals inside ECs (Dabagh et al., 2017). Endothelial glycocalyx has been described as the primary sensor activating the mechano-transduction process, creating an immediate response to shear stress stimulus and producing NO. Removing heparan sulfate and other glycocalyx components through a selective enzyme that degrades endothelial glycocalyx constituents blocks shear-induced NO production in ECs (Florian et al., 2003; Pahakis et al., 2007; Yen et al., 2015; Dragovich et al., 2016). Furthermore, NO production mediated by glycocalyx is associated with calcium influx mediated by endothelial transient receptor potential (TRP) channels. Under stimulation, the proteoglycans promote tension in the lipid bilayer, which spreads through ECs due to its interaction with cytoskeleton, and then both proteoglycans and cytoskeleton may activate a diversity of mechanically sensitive ion channels, such as TRP channels (Dragovich et al., 2016). In addition, reduced NO production induced by flow has been reported in isolated canine femoral arteries treated with hyaluronidase, which degrades the hyaluronic acid GAG (Mochizuki et al., 2003). These results show that intact glycocalyx, mainly heparan sulfate chains, are needed to activate eNOS and thus produce NO.
Glypican-1 seems to be the main heparan sulfate proteoglycan that is associated with NO production, along with eNOS, both resides in caveolae. First, glypican-1 knockdown blocks eNOS activation under shear stress stimulus (Ebong et al., 2014). Additionally, glypican-1 removal significantly suppresses eNOS activation mediated by several steady shear stress magnitudes (Zeng and Liu, 2016). Besides that, atomic force microscopy (AFM) selectively applied on glypican-1 for a limited time significantly increases NO production, whereas pulling on syndecan-1, CD44, and hyaluronic acid does not change NO concentration (Bartosch et al., 2017). Furthermore, disturbed flow (DF) reduces caveolin-1 (Cav-1) expression and impairs its co-localization with eNOS, consequently reducing eNOS phosphorylation at Serine1177 (Harding et al., 2018). Taken together, these results indicate that glypican-1 is a primary mechano-sensor for shear stress-induced NO production, and that the glypican-1-caveolae-eNOS-NO pathway is essential for vascular tone maintenance.
Formation of Caveolae
Lipid rafts (also known as lipid microdomains) and caveolae are domains of the plasma membrane that share the same composition, such as cholesterol, sphingolipids, and glycosyl-phosphatidylinositol GPI-anchored proteins. However, the caveolae structure is an invagination at the membrane. On the other hand, lipids rafts are flat areas of the membrane (Bieberich, 2018). Caveolae were first described in the 1950s by using an electron microscope. Due to lack of experimental approaches and technologies, the caveolar functions remained mostly unclear until the 1990s (Anderson, 1998). Now, caveolae are defined as 60–80-nm-wide pits in the plasma membrane that contain oligomeric caveolin (Parton and Simons, 2007). Caveolae are predominantly expressed in vascular ECs, but they are also present in VSMCs (Gratton et al., 2004). Molecular understanding of caveolar formation is advancing rapidly, and we now know that sculpting the membrane to generate the characteristic bulb-shaped caveolar pit involves coordinated action of integral membrane proteins and peripheral membrane coat proteins in a process that depends on their multiple interactions with membrane lipids (Parton et al., 2018).
Three mammalian caveolins exist: Cav-1, Cav-2, and Cav-3. Cav-1 and Cav-2 are generally expressed together in different types of cells other than muscle cells, whereas Cav-3 is predominantly expressed in muscle cells (Razani and Lisanti, 2001). Some cells, including smooth muscle cells and cardiomyocytes, can express Cav-1, Cav-2, and Cav-3, (Head et al., 2006; Robenek et al., 2008). Each caveola has estimated 140–150 Cav-1 molecules (Pelkmans and Zerial, 2005). Cav-1 loss results in complete absence of caveolae (Drab et al., 2001). Moreover, Cav-1 expression in cells without caveolae causes caveolae to form (Vogel et al., 2019). Therefore, Cav-1 is crucial for caveolar formation.
Identifying the family of cytoplasmic proteins that cooperatively work with caveolins for caveolar formation and function has expanded our understanding of caveolar biology. Liu et al. (2008) described that Cavins are cytoplasmic proteins with amino-terminal coiled-coil domains that play a role as protein component of caveolae, where they form large heteromeric complexes that are recruited by caveolins in cells expressing caveolae (Bastiani et al., 2009). The Cavin family includes Cavin-1 (also known as polymerase I and transcript release factor, PTRF), Cavin-2 (SDPR, serum deprivation response protein), Cavin-3 (also known as related gene product that binds to c-kinase - SRBC), and Cavin-4 (also known as muscle-restricted coiled-coil protein, MURC). Cavin knockout mice are viable, but they present a lipodystrophic phenotype with high triglyceride levels, glucose intolerance, and hyperinsulinemia (Liu et al., 2008). In addition, caveolae are completely absent in Cavin knockout mice in specific tissues like lung epithelium, intestinal smooth muscle, skeletal muscle, and ECs. In this way, formation of caveolae requires Cavin-1 (Liu et al., 2008). Cavin-2 and Cavin-3 have been identified as protein kinase C (PKC) substrates and have been suggested to target PKC for caveolae. Cavin-2 has been associated with caveolar membrane curvature, and Cavin-3 affects formation of caveolar endocytic carriers (Hill et al., 2008). Cavin-4, which is predominantly expressed in cardiac and skeletal muscles, has been related to myogenesis and muscle hypertrophy via RHOA–RHO-associated kinase (ROCK), ERK1, and ERK2, as well as to regulation of atrial natriuretic peptide transcription in cardiac muscle (Ogata et al., 2008; Bastiani et al., 2009).
After trafficking to the plasma membrane, caveolin oligomers are stabilized by the complex of Cavins (Hayer et al., 2010). Lipids and/or membrane lipid order may also be important for this interaction. The four members of the Cavin family bind to phosphatidylserine, which is abundant on the cytoplasmic face of the plasma membrane, particularly in areas that are rich in caveolae (Fairn et al., 2011). Cav-1 peptides can generate phosphatidylserine domains in liposomes, so membrane lipid reorganization by caveolins might also contribute to a stable interaction in the plasma membrane (Wanaski et al., 2003). In this way, Cavins and caveolins preserve the stable coat around the bulb of caveolae (Hill et al., 2008). Additionally, a protein called Eps15 homology domain protein 2 (EHD2) is involved in mediating caveolar stabilization in the plasma membrane (Stoeber et al., 2012). Moreover, the protein pacsin2 participates in membrane bending to form caveolae and to release NO from eNOS-expressing cells (Hansen et al., 2011), thus it reduces vascular tone ex vivo and lowers blood pressure in mice (Bernatchez et al., 2011).
The way through which caveolae suffer endocytosis has been a subject of controversy for many years. However, a consensus has emerged that dynamin drives caveolar budding from the surface. In ECs, caveolae have been proposed to bud in from the luminal surface and to fuse with the abluminal surface, to mediate efficient trans-endothelial transport from the blood stream to the underlying tissues. In other cell types, caveolae fuse with early endosomes (Rippe et al., 2002; Oh et al., 2007).
Caveolae and Signal Transduction Molecules
Recently, the structures of caveolae and caveolin proteins have been discovered to play an important role in cellular physiological functions, particularly functions related to cholesterol transport, endocytosis, tumor suppression, and cell signal transduction (Lian et al., 2019).
Signal transduction is promoted by neurotransmitters, circulating hormones, and growth factors that are critical for the regulation of vasculature. Many such regulators act by interacting with plasma membrane receptors and subsequently perturbation pathways that modulate metabolic activity, growth, death, and differentiated functions of the target cells (Insel and Patel, 2009).
Membrane rafts and caveolae concentrate a subset of membrane constituents, including proteins and other components involved in transport and signal transduction (Allen et al., 2007; Patel et al., 2008). Lipid rafts have non-homogeneously organized signaling, which facilitates temporally and spatially efficient cellular regulation by extracellular hormones and growth factors. The interior of cells has gradients of second messengers and effectors (cyclic AMP, Ca2þ, protein kinases/phosphatases, and phosphodiesterases) that participate in vascular signaling (Bauman et al., 2006; Echarri et al., 2007). Membrane rafts that lack caveolins also concentrate signaling molecules, implying that other factors (e.g., binding to lipids) contribute to the interaction of signaling entities with rafts and caveolins (Patel et al., 2008).
In several types of cells, including ECs and VSMCs, mediators of Ca2+ signaling such as Ca2+-ATPase, inositol 1,4,5-trisphosphate receptors, Ca2+ pumps, L-type Ca2+ channels, large-conductance Ca2+-activated K+ channels, calmodulin, and TRP channels are localized in cholesterol-rich membrane domains (Wang et al., 2005). Moreover, in VSMCs, caveolae are closely associated with peripheral sarcoplasmic reticulum, a major site for Ca2+ release that has been postulated to be the preferred site of Ca2+ entry in response to Ca2+ depletion (Shaw et al., 2006). These observations suggest that membrane rafts and caveolae have a role in Ca2+ signaling.
According to Durr et al. (2004), various proteins like G-protein-coupled receptor (GPCR) and downstream signaling enzymes such as eNOS are specifically enriched in caveolae in ECs. Additionally, caveolae contribute to GPCR desensitization and internalization (Chini and Parenti, 2004). For example, the stimulation with angiotensin II (Ang II) promotes rapid translocation of AT1 receptor (AT1R) to caveolae, then AT1R bind to Cav-1, which delays AT1R reactivation after prolonged stimulus with Ang II (Ishizaka et al., 1998; Czikora et al., 2015).
Contribution of eNOS and Cav-1 to NO Generation
Controlling eNOS activation falls under a complex regulatory mechanism that includes tonic inhibitory interaction with Cav-1 (Ju et al., 1997) and post-translational modifications like myristoylation, palmitoylation, phosphorylation, and stimulatory responses, to raise intracellular Ca2+ concentrations (Sessa, 2004).
Endothelial NO synthase remains associated with Cav-1, which is the major component of caveolae. eNOS requires palmitoylation and myristoylation to be targeted to the caveolar microdomains. The interactions between Cav-1 and eNOS have been shown to regulate NO release negatively (Grayson et al., 2012). In this way, Cav-1 over-expression decreases basal NO production in a “control” cellular state. Moreover, under agonist activation, eNOS translocates away from caveolae, thereby removing tonic Cav-1 inhibition (Frank et al., 2003). Feron et al. (1998) identified that, after agonist-dependent eNOS activation, removal of tonic inhibition between eNOS and Cav-1 coincides with de-palmitoylation concomitant with eNOS translocation to the non-caveolar fraction, which indicates increased NO biosynthesis. Conversely, when eNOS returns to the membrane/caveolae, it is re-palmitoylated, and its inhibitory interaction with Cav-1/eNOS is reasserted.
A model for activation of eNOS bound to Cav-1 considers that, under stimulation with Ca2+-mobilizing agonists, the inhibitory scaffold of Cav-1 is relieved via calcium-regulated binding of calmodulin and Hsp90 to displace eNOS from Cav-1, thus allowing efficient NO production (Sessa, 2004). Evidence supporting the inhibition model includes enhanced NO-dependent vascular function in blood vessels from Cav-1 knockout mice and increased NO production in ECs isolated from Cav-1 knockout mice, an effect that is rescued by Cav-1 reintroduction (Drab et al., 2001; Razani et al., 2001; Murata et al., 2007). Besides, transduction of cells or blood vessels with Cavtratin, a synthetic cell permeable Cav-1 CSD peptide, reduces NO release and inflammation in vivo (Bucci et al., 2006). Alanine scanning of this scaffolding region demonstrated that the threonine residues 90 and 91 (T90, T91) and phenylalanine 92 (F92) underlie eNOS inhibition. This is supported by lack of eNOS inhibition by the F92A–Cav-1 mutant in reconstituted cells and a Cavtratin-derived peptide with the T90/91 and F92 substitutions (a peptide called Cavnoxin) as revealed by studies in vitro and in vivo (Bernatchez et al., 2005).
Sowa (2012) showed that Cav-1 in caveolae but not in lipid rafts can inhibit eNOS under basal conditions. Although Cav-1 in caveolae keeps eNOS inactive, the specific localization of Cav-1 in this cell organelle is necessary for its activation (Chen et al., 2012). In addition, Cav-1/eNOS interaction is necessary to prevent inadequate NO production under basal conditions and to facilitate integration of extracellular stimuli with intracellular NO signals (Rath et al., 2009).
Oxidative Stress in Cardiovascular Diseases
Reactive oxygen species are a group of heterogeneous molecules that are characterized by highly reactive oxygen atoms, short half-life, and strong capacity to engage in oxidation reactions (Vara and Pula, 2014). They are essential for homeostasis of the cardiovascular system and play a role in signaling pathways in different cells. An imbalance in antioxidant and oxidant systems promotes ROS overproduction, which culminates in oxidative stress, a well-known and important hallmark of cardiovascular diseases (Panth et al., 2016). When ROS levels overtake the cellular defenses, protein, lipids, and DNA can undergo oxidation, which can lead to cellular damage, tissue injury, and inflammation (Sena et al., 2018). ROS are produced by distinct enzymatic sources like xantine oxidase, NADPH-oxidase (NOX), cyclooxygenase (COX), lipoxygenase (LOX), monomeric eNOS (uncoupled eNOS), myeloperoxidase, and also by the respiratory chain in mitochondria (Vara and Pula, 2014; Sena et al., 2018). The chemical species anion superoxide (O2–), peroxynitrite (ONOO–), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) underlie deleterious effects of oxidative stress. However, ROS present not only deleterious, but also physiological effects on vascular tone in VSMCs and on ECs motility, proliferation, and permeability (Vara and Pula, 2014). For example, O2– induces protein kinase-dependent contraction in VSMCs under high pressure (Ungvari et al., 2003), whereas H2O2 upregulates vascular endothelial growth factor receptor 2 (González-Pacheco et al., 2006).
Each oxidant chemical species can be removed from the cellular environment by different enzymes that make up the antioxidant system, including superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase, thioredoxin, peroxiredoxin, and glutathione transferase. SOD dismutates O2– in H2O2, which is broken down into O2 and H2O by CAT and glutathione peroxidase (Birben et al., 2012). The radical species can also activate the nuclear factor erythroid 2-like 2 (Nrf-2), a transcription factor that is involved in the dynamic regulation of the antioxidant system, thereby activating the expression of promoters containing the antioxidant response element (Satta et al., 2017).
In the vascular system, both ECs and VSMCs can be either producers or targets of ROS. H2O2 is produced by NOX4 in ECs (Burtenshaw et al., 2019) and it can elicit different responses in VSMCs depending on its concentration (Gil-Longo and González-Vázquez, 2005). Importantly, H2O2 seems to be increased in aortas from hypertensive rats under stimulus (Silva et al., 2013). O2– can be produced by NOX isoforms expressed in the membrane and in the intracellular compartment of ECs (Li and Shah, 2002) and by mitochondria, which are considered the major source of O2– in ECs (Li et al., 2016). In cardiovascular diseases, O2– significantly contributes to endothelial dysfunction because it rapidly reacts with NO, to produce the highly oxidizing ONOO– (Radi, 2018), thus decreasing NO bioavailability. In addition, ROS can induce conversion of ECs to myofibroblasts, losing its endothelial properties (Montorfano et al., 2014).
Endothelial dysfunction is characterized by reduced endothelial response to different stimulus that release NO and other chemical mediators related to vasodilation or higher levels of endothelial chemical mediators associated with vasoconstriction (Vanhoutte et al., 2017). Therefore, exacerbated oxidative stress in ECs modifies the response of endothelial NO, and ROS produced by ECs can induce response in VSMCs.
In VSMCs, ROS production is mediated especially by NOX1 and NOX4 and produces O2– and H2O2, respectively (Burtenshaw et al., 2019). Increased ROS in VSMCs is a common feature of different models of hypertension such as AngII-infused model (Rajagopalan et al., 1996;Zhou et al., 2020), spontaneously hypertensive rats (SHR) (Graton et al., 2019), renovascular hypertension (2K-1C) (Castro et al., 2012;Oliveira-Paula et al., 2016), and Doca-Salt rats (Amaral et al., 2015). Physiologically, contraction mediated by activation of α-1 adrenoceptors can partially depend on O2– (Tsai and Jiang, 2010). The hypercontractile profile of VSMCs of hypertensive rats seems to depend on ROS production (Camargo et al., 2018). Additionally, ROS produced by VSMCs can reduce NO bioavailability.
Oxidative Stress in Caveolae and Caveolin
Cav-1 seems to be involved in the process involving ROS as target or controlling ROS production. Oxidative stress mediated by H2O2 degrades Cav-1 in skeletal muscle cells (Mougeolle et al., 2015). H2O2 is increased in aorta of renal hypertensive rats (Silva et al., 2013), and the total number of caveolae is reduced in aorta of hypertensive rats (Rodrigues et al., 2010; Potje et al., 2019). Thus, H2O2 overproduction can be important to reduce Cav-1 levels and to disrupt the function of caveolae in hypertension.
Cav-1 is related to the AngII (AT1) receptor because the AT1-R⋅caveolin complex requires an intact caveolin scaffolding domain, but not co-localization in the caveolae (Wyse et al., 2003). Exposure to the agonist Ang II changes the Cav-1 levels in VSMCs (Ishizaka et al., 1998). Interestingly, Cav-1 loss increases NOX activity and ROS production in VSMCs (Zuo et al., 2005; Chen et al., 2014). In contrast, Cav-1 deletion can prevent remodeling induced by Ang II (Forrester et al., 2017). As stated before, Nrf-2 is an important element that controls the levels of antioxidant enzymes. ROS can activate Nrf-2 migration to the nucleus, thereby raising the expression of antioxidant enzymes and leading to detoxification of the cells (Satta et al., 2017). Curiously, Cav-1 seems to repress this migration given that Cav-1 knockout mice constantly present high Nrf-2 levels in nucleus (Volonte et al., 2013). Changes in Nrf-2 levels can alter the physiology of normal cells due to upstream and downstream of molecules that are associated with a defective Nrf2 signaling system (Satta et al., 2017). Thus, most of the common features of hypertensive vessels, e.g., endothelial dysfunction and hypercontractile VSMCs, can result from ROS actions, Cav-1 levels, and caveolar functions.
Effect of ROS on Endothelial Glycocalyx Degradation
Specific enzymes, named sheddases, as well as metalloproteinases (MMPs), heparanase, and hyaluronidase degrade glycocalyx. Sheddases are activated by pro-inflammatory cytokines such as TNFα (Ramnath et al., 2014), interleukin-1beta (Haywood-Watson et al., 2011), interleukin-6, and interleukin-8 and also by shear stress, hypoxia, and ROS (Lipowsky and Lescanic, 2013). In this review, we focus only on how ROS affect endothelial glycocalyx given that ROS cleave and destabilize the glycocalyx structure (Sieve et al., 2018).
MMPs modify the constituents of glycocalyx, thus disrupting glycocalyx in pathological conditions (Lipowsky, 2011). MMPs can cleave the protein core of syndecan, promoting shedding of the syndecan family and consequent thrombosis, destabilization of vascular walls, endothelial dysfunction, and inflammation (Fitzgerald et al., 2000; Chen et al., 2017). In, addition, MMP-2 was associated to direct chondroitin sulfate cleavage (Hsu et al., 2006), while MMP-7 was responsible for cleavage of perlecan and heparan sulfate proteoglycans (Grindel et al., 2014) and MMP-9 mediated disruption of syndecan-4 (Ramnath et al., 2014). ROS also decrease the levels of the tissue inhibitors of metalloproteinases (TIMPs), thereby increasing the activity of MMPs (Siwik and Colucci, 2004). The inhibition of MMPs was able to restore the shedding of syndecan-4 in early diabetic disease (Ramnath et al., 2020).
Heparanase is an endoglycosidase that cleaves the side chains of heparan sulfate present in the syndecan and glypican families through hydrolysis, to disrupt glycocalyx (Garsen et al., 2014). Degradation of heparan sulfate reduces extracellular SOD (ecSOD), which remains attached to the heparan sulfate portion. ecSOD protects vascular cells from oxidative stress and its overexpression can attenuate heparanase expression, which suggests a prophylactic effect to prevent glycocalyx degradation (Kumagai et al., 2009).
Hyaluronidase degrades hyaluronic acid into fragments via hydrolysis of the disaccharides at hexosaminidic β (1–4) linkages (Wang et al., 2020). In addition, hyaluronic acid degradation products can produce ROS, which triggers several vascular disease processes (Soltés et al., 2006). Moreover, other ROS derived from O2– and nitrogen monoxide (⋅NO), including H2O2, ONOO–, and hypochlorous acid, depolymerize hyaluronic acid (Uchiyama et al., 1990).
Alterations in Caveolar Function and Glycocalyx in Atherosclerosis and Hypertension
Atherosclerosis and hypertension are multifactorial diseases. Atherosclerosis and its consequences represent the major cause of cardiovascular mortality. This disease is characterized by endothelial dysfunction, increased platelet adhesion, leukocyte recruitment, and accumulation of lipoproteins that evade phagocytosis (Eckardt et al., 2019). Endothelial dysfunction in the areas where atherosclerosis develops occurs through entry of lipoproteins, which is followed by lesions, leading to production of proinflammatory cytokines, migration of monocytes, and accumulation of macrophages (Gimbrone and Garcia-Cardeña, 2016). In turn, hypertension is a multifactorial disease that is associated with endothelial dysfunction, exacerbated oxidative stress, and inflammation in blood vessels (Dharmashankar and Widlansky, 2010). ECs play a pivotal role in vessel balance and pathophysiological conditions because they are exposed to inflammatory mediators that can impair or even destroy the endothelial layer and its components.
Caveolae and Cav-1 seem to play a part in atherosclerosis development. Numerous atherogenic proteins colocalize with caveolae in ECs, and caveolae are involved with transcytosis of low-density lipoprotein (LDL) particles (Frank et al., 2004; Sowa, 2012). Additionally, Cav-1 protein seems to have an atherosclerotic role. First, Cav-1 overexpression in ECs can increase atherosclerosis progression in apolipoprotein E-deficient mice (Fernández-Hernando et al., 2010). Moreover, the absence of Cav-1 promotes atheroprotection in vessels of Cav-1 knockout mice (Zhang et al., 2020). As proposed by Zhang et al. (2020), activation of endothelial autophagy by Cav-1 deficiency protects against atherosclerosis progression. In brief, autophagy is described as an evolutionarily conserved subcellular process that mediates degradation of proteins and damaged organelles via lysosomes (Mizushima and Komatsu, 2011).
As reported by Milovanova et al. (2008), apart from modulating NO production by eNOS, Cav-1 can modulate ROS production by NOX. In pulmonary hypertension, Cav-1 is a negative regulator of ROS derived from NO since lack of Cav-1 expression in pulmonary hypertension increases NOX activity and enhances ROS production (Chen et al., 2014). Furthermore, Cav-1 deletion prevents transactivation of hypertensive vascular remodeling and contributes to increased mitochondrial ROS levels in a model of AngII-induced hypertension (Forrester et al., 2017). On the other hand, lipid rafts and caveolae structural disruption with cholesterol disassembly drugs, increased ROS production in a different way than NOX. Recently, we have shown that caveolar structural disruption with methyl-β-cyclodextrin uncouples eNOS and raises ROS levels in Wistar normotensive rats and SHR aortas and mesenteric arteries (Potje et al., 2019). Besides that, the number of caveolae is reduced in renal hypertensive (2K-1C) and SHR rats as compared to normotensive rats, which impairs acetylcholine-induced endothelium-dependent relaxation and NO production (Rodrigues et al., 2010; Potje et al., 2019). The smaller number of caveolae could account for impaired NO donor-induced relaxation in 2K-1C rat aortas as compared to normotensive rat aortas (Rodrigues et al., 2007).
The literature contains controversial data about the role that Cav-1 has in the determination of arterial pressure. Whereas several authors do not report increased arterial pressure in Cav-1 knockout mice as compared to control wild-type (WT) mice, other authors describe lower arterial pressure in Cav-1 knockout mice than in WT mice (for a review, see Rahman and Sward, 2009). Cav-1 knockout mice present increased circulating NO levels and vasodilation, but the arterial pressure values measured by telemetry in awake mice and WT mice are similar (Desjardins et al., 2008). As proposed by Insel and Patel (2007), chronic Cav-1 deficiency could be compensated by other vascular mechanisms, to maintain the arterial pressure. Also, eNOS could be uncoupled in hypertensive vessels.
In physiological conditions, arterial ECs submitted to uniform flow (UF) release NO constantly (Noris et al., 1995). As described by Eckardt et al. (2019), the pathogenesis of atherosclerosis is associated with alterations in vascular glycocalyx. Glycocalyx degradation stimulates lipid flux, increasing lipid deposition in the arterial walls. This is associated with reduced eNOS expression, which decreases NO production and impairs vasodilation (Mitra et al., 2017). In addition, in most cases of atherosclerosis, plaques appear in the carotid bifurcation and aortic arch, which are regions with DF (Gimbrone and Garcia-Cardeña, 2013), thereby suggesting a relationship between hemodynamics and atherosclerosis progression. Cav-1 expression is reduced in DF as compared to UF, which indicates that Cav-1 regulation depends on the flow (Harding et al., 2018). Furthermore, as described by Harding et al. (2018), expression of active eNOS phosphorylated at Ser1177 is 50% lower in DF aortic arch than in UF abdominal aorta.
In this way, caveolae, Cav-1, and glycocalyx play an important role in vascular homeostasis, contributing to adequate NO production. However, atherosclerosis and hypertension impair NO bioavailability due to lower eNOS expression, eNOS inactivation, changes in Cav-1 expression or NOX4 activity, and eNOS uncoupling, leading to deleterious ROS overproduction.
Discussion
In this review, we discuss recent findings about the physiological role of glycocalyx and caveolae, to maintenance of vascular tone, as well as alterations in these structures that are associated with the development of atherosclerosis and hypertension.
Even the glycocalyx has been reported as the primary sensor to mechano-transduction, a study demonstrated that caveolae show a unique molecular topography (Schnitzer et al., 1995) and may act as either mechano-sensors or transducers (Uittenbogaard et al., 2000; Gratton et al., 2004). Therefore, it could exist a relationship between glycocalyx and caveolae that is sensitive to feel mechanical forces and start the mechano-transduction process, and to promote an effective control of vascular tone.
During the first 30 min of exposition to shear stress, aortic and vein ECs (BAEC, bovine aortic EC; HUVEC, human umbilical vein EC) presented an accumulation of heparan sulfate and glypican-1 in the cell junctions. In contrast, there were no movement from chondroitin sulfate, syndecan-1, and Cav-1, indicating that these components and particularly the caveolae structure are anchored sufficiently to resist against initial exposure to shear stress (Zeng et al., 2013). On the other hand, the chronic shear stress (6 h) stimulated by changes in flow intensity in perfused lung microvessels was able to increase fivefold Cav-1 expression and sixfold caveolae density at the luminal surface compared with no-flow control, which contributed to enhanced mechano-sensivity in cultured ECs (Rizzo et al., 2003). Moreover, the glypican-1 inhibition, but not syndecan-1, blocked eNOS activation induced by shear stress in mammalian epithelial cells (Ebong et al., 2014). These studies clarify the activation of glypican-1-caveolae-eNOS-NO pathway under mechanical stimulus. In this way, there is a relationship between glycocalyx and caveolae, where they are exchanging information all the time, and both are susceptible to reorganization underlie different stimulus to regulate vascular tone and promote vascular homeostasis.
Furthermore, the relationship between glycocalyx and caveolae is not only observed during shear stress and mechano-transduction. Catestatin is a peptide derived from glycoprotein chromogranin A, which is expressed in neuroendocrine and cardiac cells. Catestatin acts in several organs/systems, including the cardiovascular system. The catestatin was applied to BAECs, where it co-localizes with heparan sulfate proteoglycans, promoting endocytosis of caveolae and inducing Cav-1 internalization, followed by eNOS phosphorylation at Serine1179 (Fornero et al., 2014). Therefore, the glycocalyx and caveolae collaborate with each other during the catestatin-dependent eNOS-activation.
Glycocalyx contributes to maintaining vascular homeostasis, and it protects the EC surface. Thus, its disruption and shedding contribute to the development of cardiovascular diseases. Therefore, preventing its degradation is important. In a review, Becker et al. (2010) brought together the pharmacological options to avoid glycocalyx shedding and perturbation, which included hydrocortisone application, use of antithrombin III, and infusion of human plasma albumin, which seems to be the effective treatment. Apart from that, rat fat pad ECs supplemented with heparan sulfate or sphingosine 1-phosphate regenerate glycocalyx (Mensah et al., 2017). Another agent, sulodexide, which is a mixture of heparan sulfate and dermatan sulfate (Coccheri and Mannello, 2013), also reconstitutes glycocalyx in patients (Broekhuizen et al., 2010). Nuclear magnetic resonance analysis demonstrated that Krüppel-like Factor 2 (KLF2) inhibits endothelial glycolysis and contributes to hexosamine and glucuronic acid biosynthesis (Wang et al., 2020). In addition, inflammatory cytokines like TNF-α, interleukin-1β, interleukin-6, and interleukin-8, as well as ROS can activate heparanase, MMPs, and hyaluronidase, which are enzymes that cleave chains of glycocalyx constituents (Uchimido et al., 2019). Hence, antioxidant drugs and direct inhibition of cytokines may be another option to prevent glycocalyx degradation.
In the last 20 years, many studies have evidenced the relevance of caveolins by using Cav-1 knockout mice with cardiovascular abnormalities (Li et al., 2005; Lian et al., 2019). As suggested by Forrester et al. (2017), Cav-1 may be the therapeutic target to treat hypertension and atherosclerosis. However, the role of Cav-1 is controversial in the literature. It has dual action: Cav-1 impairs vascular functions in specific cases and at the same time, it seems to be essential to maintain vascular homeostasis. Hypertension induced by AngII in Cav-1 knockout mice does not develop vascular remodeling, which means that Cav-1 deletion attenuates vascular hypertrophy and perivascular fibrosis (Forrester et al., 2017). On the other hand, on the basis of the mouse hypoxia model, reduced Cav-1 expression increases ROS production, and macrophages isolated from Cav-1 knockout mice and Cav-1 knockdown siRNA in human lung fibroblasts enhances ROS production. The absence of Cav-1 negatively regulates NOX-mediated ROS production (Chen et al., 2014). Additionally, Cav1-deficient mice exhibit pulmonary hypertension, impairment of left ventricular diastolic function, increased pulmonary vascular remodeling, and right ventricle hypertrophy and decreased contractility (Zhao et al., 2002). Furthermore, the lack of Cav-1 improves NO-dependent vascular function and produces higher levels of NO (Drab et al., 2001; Razani et al., 2001; Murata et al., 2007), which suggests that Cav-1 impairs vascular function and contributes to the development of cardiovascular diseases. Notwithstanding, various studies have shown that the presence of Cav-1 is mandatory for eNOS activation (Chen et al., 2012).
Navarro et al. (2014) suggested gene or cell therapy as antisense and siRNA approaches to target Cav-1 directly or to modulate caveolar and lipid levels as an alternative intervention either to increase or to decrease Cav-1 expression. Moreover, activation of some GPCRs would allow to control or to re-program Cav-1 expression levels to explore therapeutic outcomes in cardiovascular diseases. Besides, the pathway glypican-1/caveolin-1/eNOS/NO should be better explored for better understanding of this path and possible therapeutic treatments.
Conclusion
The structure and function of both glycocalyx and caveolae are essential for maintenance of vascular homeostasis. Under pathological conditions, that are associated with ROS synthesis and release, the glycocalyx and caveolae structure and function could change, leading to impairment of their physiological function, which are the hallmark of cardiovascular diseases (see Figure 1).
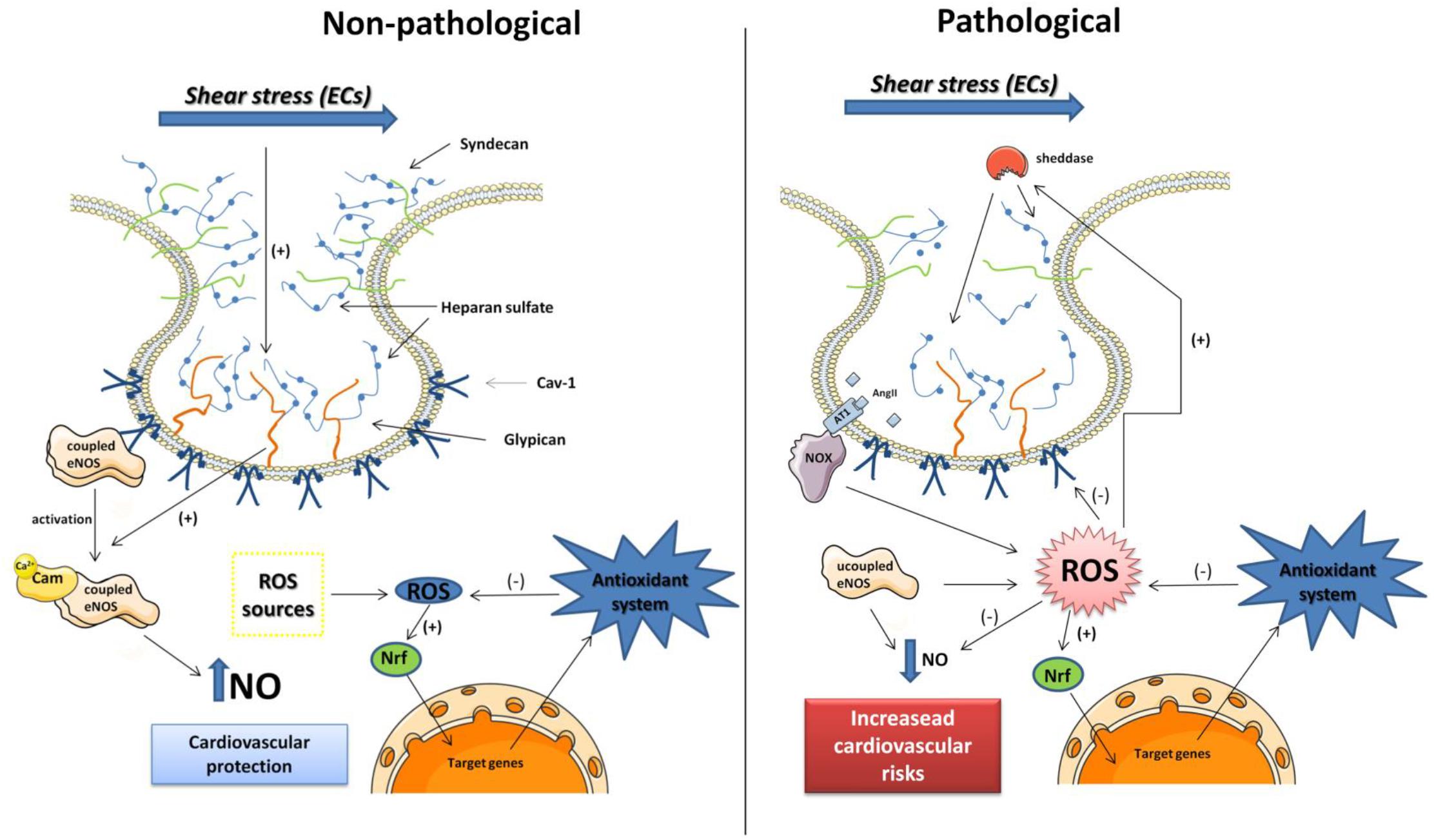
Figure 1. Schematic representation of caveola and glycocalyx proteins in endothelial cells (ECs) under non-pathological and pathological conditions. In non-pathological conditions, the glycocalyx proteins are intact and the coupled eNOS (dimeric form) is inactivated linked to Cav-1 protein. The eNOS is activated by shear stress mediated by Glypican-1 mechano-transduction as well as increase in calcium levels. The NO bioavailability can protect the cardiovascular system, and ROS produced by different sources stimulates the antioxidant system that also protects the cells of oxidative damage. In pathological conditions, the rise in ROS production mediated by pro-oxidant molecules as Ang II overtakes the antioxidant defenses. The ROS can degrade Cav-1, the eNOS is uncoupled (monomeric) that can be source of ROS, and NO bioavailability is reduced. ROS also stimulates the sheddases activity clivating the heparan sulfate from glypican and syndecan losing its ability to induce eNOS activation mediated by shear stress. Those frames with low NO levels can potentiate the cardiovascular risks.
Author Contributions
SRP and LMB conceived the original scope of this manuscript. SRP, TP, MP, and LMB wrote specific sections. TP made the schematic representation of Figure 1. SRP and LMB critically reviewed and revised the final manuscript. All the authors have equally made a substantial intellectual contribution to this work in organizing and writing the manuscript, and all of them have approved it for submission.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) 305601/2018-0.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramsson, A., Kurup, S., Busse, M., Yamada, S., Lindblom, P., Schallmeiner, E., et al. (2007). Defective N-sulfation of heparan sulfate proteoglycans limits PDGF-BB binding and pericyte recruitment in vascular development. Genes Dev. 21, 316–331. doi: 10.1101/gad.398207
Agarwal, G., Krishnan, V. K., Prasad, S. B., Bhaduri, A., and Jayaraman, G. (2019). Biosynthesis of hyaluronic acid polymer: dissecting the role of sub structural elements of hyaluronan synthase. Sci. Rep. 9:12510. doi: 10.1038/s41598-019-48878-8
Allen, J. A., Halverson-Tamboli, R. A., and Rasenick, M. M. (2007). Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140. doi: 10.1038/nrn2059
Alphonsus, C. S., and Rodseth, R. N. (2014). The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia 69, 777–784. doi: 10.1111/anae.12661
Amaral, J. H., Ferreira, G. C., Pinheiro, L. C., Montenegro, M. F., and Tanus-Santos, J. E. (2015). Consistent antioxidant and antihypertensive effects of oral sodium nitrite in DOCA-salt hypertension. Redox Biol. 5, 340–346. doi: 10.1016/j.redox.2015.06.009
Anderson, R. G. (1998). The caveolae membrane system. Annu. Rev. Biochem. 67, 199–225. doi: 10.1146/annurev.biochem.67.1.199
Bartosch, A. M. W., Mathews, R., and Tarbell, J. M. (2017). Endothelial glycocalyx-mediated nitric oxide production in response to selective AFM pulling. Biophys. J. 113, 101–108. doi: 10.1016/j.bpj.2017.05.033
Bastiani, M., Liu, L., Hill, M. M., Jedrychowski, M. P., Nixon, S. J., Lo, H. P., et al. (2009). MURC/cavin-4 and cavin family members form tissue-specific caveolar complexes. J. Cell Biol. 185, 1259–1273. doi: 10.1083/jcb.200903053
Bauman, A. L., Soughayer, J., Nguyen, B. T., Willoughby, D., Carnegie, G. K., Wong, W., et al. (2006). Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol. Cell. 23, 925–931. doi: 10.1016/j.molcel.2006.07.025
Becker, B. F., Chappell, D., Bruegger, D., Annecke, T., and Jacob, M. (2010). Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc. Res. 87, 300–310. doi: 10.1093/cvr/cvq137
Bernatchez, P., Sharma, A., Bauer, P. M., Marin, E., and Sessa, W. C. (2011). A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J. Clin. Invest. 121, 3747–3755. doi: 10.1172/JCI44778
Bernatchez, P. N., Bauer, P. M., Yu, J., Prendergast, J. S., He, P., and Sessa, W. C. (2005). Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell permeable peptides. Proc. Natl. Acad. Sci. U.S.A. 102, 761–766. doi: 10.1073/pnas.0407224102
Bieberich, E. (2018). Sphingolipids and lipid rafts: novel concepts and methods of analysis. Chem. Phys. Lipids 216, 114–131. doi: 10.1016/j.chemphyslip.2018.08.003
Birben, E., Sahiner, U. M., Sackesen, C., Erzurum, S., and Kalayci, O. (2012). Oxidative stress and antioxidant defense. World Allergy Organ. J. 5, 9–19. doi: 10.1097/WOX.0b013e3182439613
Broekhuizen, L. N., Lemkes, B. A., Mooij, H. L., Meuwese, M. C., Verberne, H., Holleman, F., et al. (2010). Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 53, 2646–2655. doi: 10.1007/s00125-010-1910-x
Bucci, M., Gratton, J. P., Rudic, R. D., Acevedo, L., Roviezzo, F., Cirino, G., et al. (2006). In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 6, 1362–1367. doi: 10.1038/82176
Burtenshaw, D., Kitching, M., Redmond, E. M., Megson, I. L., and Cahill, P. A. (2019). Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front. Cardiovasc. Med. 6:89. doi: 10.3389/fcvm.2019.00089
Cahill, P. A., and Redmond, E. M. (2016). Vascular endothelium – Gatekeeper of vessel health. Atherosclerosis 248, 97–109. doi: 10.1016/j.atherosclerosis.2016.03.007
Camargo, L. L., Harvey, A. P., Rios, F. J., Tsiropoulou, S., Da Silva, R. N. O., Cao, Z., et al. (2018). Vascular Nox (NADPH Oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension 72, 235–246. doi: 10.1161/HYPERTENSIONAHA.118.10824
Castro, M. M., Rizzi, E., Ceron, C. S., Guimaraes, D. A., Rodrigues, G. J., Bendhack, L. M., et al. (2012). Doxycycline ameliorates 2K-1C hypertension-induced vascular dysfunction in rats by attenuating oxidative stress and improving nitric oxide bioavailability. Nitric Oxide 26, 162–168. doi: 10.1016/j.niox.2012.01.009
Chen, E., Stringer, S. E., Rusch, M. A., Selleck, S. B., and Ekker, S. C. (2005). A unique role for 6-O sulfation modification in zebrafish vascular development. Dev. Biol. 284, 364–376. doi: 10.1016/j.ydbio.2005.05.032
Chen, F., Barman, S., Yu, Y., Haigh, S., Wang, Y., Black, S. M., et al. (2014). Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Rad. Biol. Med. 73, 201–213. doi: 10.1016/j.freeradbiomed.2014.04.029
Chen, Y., Peng, W., Raffetto, J. D., and Khalil, R. A. (2017). Matrix metalloproteinases in remodeling of lower extremity veins and chronic venous disease. Prog. Mol. Biol. Transl. Sci. 147, 267–299. doi: 10.1016/bs.pmbts.2017.02.003
Chen, Z., Bakhshi, F. R., Shajahan, A. N., Sharma, T., Mao, M., Trane, A., et al. (2012). Nitric oxide-dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol. Biol. Cell 23, 1388–1398. doi: 10.1091/mbc.E11-09-0811
Chini, B., and Parenti, M. (2004). G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J. Mol. Endocrinol. 32, 325–338. doi: 10.1677/jme.0.0320325
Coccheri, S., and Mannello, F. (2013). Development and use of sulodexide in vascular diseases: implications for treatment. Drug Des. Devel. Ther. 8, 49–65. doi: 10.2147/DDDT.S6762
Constantinescu, A. A., Vink, H., and Spaan, J. A. (2003). Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 23, 1541–1547. doi: 10.1161/01.ATV.0000085630.24353.3D
Czikora, I., Feher, A., Lucas, R., Fulton, D. J., and Bagi, Z. (2015). Caveolin-1 prevents sustained angiotensin II-induced resistance artery constriction and obesity-induced high blood pressure. Am. J. Physiol Heart Circ. Physiol. 308, H376–H385. doi: 10.1152/ajpheart.00649.2014
Dabagh, M., Jalali, P., Butler, P. J., Randles, A., and Tarbell, J. M. (2017). Mechanotransmission in endothelial cells subjected to oscillatory and multi-directional shear flow. J. R. Soc. Interface 14:20170185. doi: 10.1098/rsif.2017.0185
Desjardins, F., Lobysheva, I., Pelat, M., Gallez, B., Feron, O., Dessy, C., et al. (2008). Control of blood pressure variability in caveolin-1-deficient mice: role of nitric oxide identified in vivo through spectral analysis. Cardiovasc. Res. 79, 527–536. doi: 10.1093/cvr/cvn080
Dharmashankar, K., and Widlansky, M. E. (2010). Vascular endothelial function and hypertension: insights and directions. Curr. Hypertens. Rep. 12, 448–455. doi: 10.1007/s11906-010-0150-2
Drab, M., Verkade, P., Elger, M., Kasper, M., Lohn, M., Lauterbach, B., et al. (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449–2452. doi: 10.1126/science.1062688
Dragovich, M. A., Chester, D., Fu, B. M., Wu, C., Xu, Y., Goligorsky, M. S., et al. (2016). Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am. J. Physiol. Cell Physiol. 311, C846–C853. doi: 10.1152/ajpcell.00288.2015
Durr, E., Yu, J., Krasinska, K. M., Carver, L. A., Yates, J. R., Testa, J. E., et al. (2004). Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat. Biotechnol. 22, 985–992. doi: 10.1038/nbt993
Ebong, E. E., Lopez-Quintero, S. V., Rizzo, V., Spray, D. C., and Tarbell, J. M. (2014). Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr. Biol. 6, 338–347. doi: 10.1039/c3ib40199e
Echarri, A., Muriel, O., and Del Pozo, M. A. (2007). Intracellular trafficking of raft/caveolae domains: insights from integrin signaling. Semin. Cell Dev. Biol. 18, 627–637. doi: 10.1016/j.semcdb.2007.08.004
Eckardt, V., Weber, C., and Von Hundelshausen, P. (2019). Glycans and glycan-binding proteins in atherosclerosis. Thromb. Haemost. 119, 1265–1273. doi: 10.1055/s-0039-1692720
Fairn, G. D., Schieber, N. L., Ariotti, N., Murphy, S., Kuerschner, L., Webb, R. I., et al. (2011). High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell. Biol. 194, 257–275. doi: 10.1083/jcb.201012028
Fernández-Hernando, C., Yu, J., Dávalos, A., Prendergast, J., and Sessa, W. C. (2010). Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 177, 998–1003. doi: 10.2353/ajpath.2010.091287
Feron, O., Saldana, F., Michel, J. B., and Michel, T. (1998). The endothelial nitric-oxide synthase-caveolin regulatory cycle. J. Biol. Chem. 273, 3125–3128. doi: 10.1074/jbc.273.6.3125
Fitzgerald, M. L., Wang, Z., Park, P. W., Murphy, G., and Bernfield, M. (2000). Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell. Biol. 148, 811–824. doi: 10.1083/jcb.148.4.811
Florian, J. A., Kosky, J. R., Ainslie, K., Pang, Z., Dull, R. O., and Tarbell, J. M. (2003). Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 93, e136–e142. doi: 10.1161/01.RES.0000101744.47866.D5
Fornero, S., Bassino, E., Ramella, R., Gallina, C., Mahata, S. K., Tota, B., et al. (2014). Obligatory role for endothelial heparan sulphate proteoglycans and caveolae internalization in catestatin-dependent eNOS activation. Biomed. Res. Int. 2014:783623. doi: 10.1155/2014/783623
Forrester, S. J., Elliott, K. J., Kawai, T., Obama, T., Boyer, M. J., Preston, K. J., et al. (2017). Caveolin-1 deletion prevents hypertensive vascular remodeling induced by angiotensin II. Hypertension 69, 79–86. doi: 10.1161/HYPERTENSIONAHA.116.08278
Frank, P. G., Lee, H., Park, D. S., Tandon, N. N., Scherer, P. E., and Lisanti, M. P. (2004). Genetic ablation of caveolin-1 confers protection against atherosclerosis. Atheroscler. Thromb. Vasc. Biol. 24, 98–105. doi: 10.1161/01.ATV.0000101182.89118.E5
Frank, P. G., Woodman, S. E., Park, D. S., and Lisanti, M. P. (2003). Caveolin, caveolae, and endothelial cell function. Arterioscler. Thromb. Vasc. Biol. 23, 1161–1168. doi: 10.1161/01.ATV.0000070546.16946.3A
Fuster, M. M., and Wang, L. (2010). Endothelial heparan sulfate in angiogenesis. Prog Mol. Biol. Transl. Sci. 93, 179–212. doi: 10.1016/S1877-1173(10)93009-3
Garsen, M., Rops, A. L., Rabelink, T. J., Berden, J. H., and van der Vlag, J. (2014). The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrol. Dial. Transplant. 29, 49–55. doi: 10.1093/ndt/gft410
Gil-Longo, J., and González-Vázquez, C. (2005). Characterization of four different effects elicited by H2O2 in rat aorta. Vasc. Pharmacol. 43, 128–138. doi: 10.1016/j.vph.2005.06.001
Gimbrone, M. A. Jr., and Garcia-Cardeña, G. (2013). Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 22, 9–15. doi: 10.1016/j.carpath.2012.06.006
Gimbrone, M. A. Jr., and Garcia-Cardeña, G. (2016). Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118, 620–636. doi: 10.1161/CIRCRESAHA.115.306301
González-Pacheco, F. R., Deudero, J. J., Castellanos, M. C., Castilla, M. A., Alvarez-Arroyo, M. V., Yagüe, S., et al. (2006). Mechanisms of endothelial response to oxidative aggression: protective role of autologous VEGF and induction of VEGFR2 by H2O2. Am. J. Physiol. Heart Circ. Physiol. 291, H1395–H1401. doi: 10.1152/ajpheart.01277.2005
Graton, M. E., Potje, S. R., Troiano, J. A., Vale, G. T., Perassa, L. A., Nakamune, A. C. M. S., et al. (2019). Apocynin alters redox signaling in conductance and resistance vessels of spontaneously hypertensive rats. Free Rad. Biol. Med. 134, 53–63. doi: 10.1016/j.freeradbiomed.2018.12.026
Gratton, J. P., Bernatchez, P., and Sessa, W. C. (2004). Caveolae and caveolins in the cardiovascular system. Circ. Res. 94, 1408–1417. doi: 10.1161/01.RES.0000129178.56294.17
Grayson, T. H., Chadha, P. S., Bertrand, P. P., Chen, H., Morris, M. J., Senadheera, S., et al. (2012). Increased caveolae density and caveolin-1 expression accompany impaired NO-mediated vasorelaxation in diet-induced obesity. Histochem. Cell Biol. 139, 309–321. doi: 10.1007/s00418-012-1032-2
Grindel, B. J., Martinez, J. R., Pennington, C. L., Muldoon, M., Stave, J., Chung, L. W., et al. (2014). Matrilysin/matrix metalloproteinase-7 (MMP7) cleavage of perlecan/HSPG2 creates a molecular switch to alter prostate cancer cell behavior. Matrix Biol. 36, 64–76. doi: 10.1016/j.matbio.2014.04.005
Han, J., Mandal, A. K., and Hiebert, L. M. (2005). Endothelial cell injury by high glucose and heparanase is prevented by insulin, heparin and basic fibroblast growth factor. Cardiovasc. Diabetol. 4:12. doi: 10.1186/1475-2840-4-12
Hansen, C. G., Howard, G., and Nichols, B. J. (2011). Pacsin 2 is recruited to caveolae and functions in caveolar biogenesis. J. Cell Sci. 124(Pt 16), 2777–2785. doi: 10.1242/jcs.084319
Harding, I. C., Mitra, R., Mensah, S. A., Herman, I. M., and Ebong, E. E. (2018). Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J. Transl. Med. 16:364. doi: 10.1186/s12967-018-1721-2
Hayer, A., Stoeber, M., Bissig, C., and Helenius, A. (2010). Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic 11, 361–382. doi: 10.1111/j.1600-0854.2009.01023.x
Haywood-Watson, R. J., Holcomb, J. B., Gonzalez, E. A., Peng, Z., Pati, S., Park, P. W., et al. (2011). Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS One 6:e23530. doi: 10.1371/journal.pone.0023530
Head, B. P., Patel, H. H., Roth, D. M., Murray, F., Swaney, J. S., Niesman, I. R., et al. (2006). Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 281, 26391–26399. doi: 10.1074/jbc.M602577200
Henry, C. B., and Duling, B. R. (2000). TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 279, H2815–H2823. doi: 10.1152/ajpheart.2000.279.6.H2815
Hill, M. M., Bastiani, M., Luetterforst, R., Kirkham, M., Kirkham, A., Nixon, S. J., et al. (2008). PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124. doi: 10.1016/j.cell.2007.11.042
Hsu, J. Y., McKeon, R., Goussev, S., Werb, Z., Lee, J. U., Trivedi, A., et al. (2006). Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J. Neurosci. 26, 9841–9850. doi: 10.1523/JNEUROSCI.1993-06.2006
Insel, P. A., and Patel, H. H. (2007). Do studies in caveolin- knockouts teach us about physiology and pharmacology or instead, the ways mice compensate the “lost proteins”? Br. J. Pharmacol. 150, 251–254. doi: 10.1038/sj.bjp.0706981
Insel, P. A., and Patel, H. H. (2009). Membrane rafts and caveolae in cardiovascular signaling. Curr. Opin. Nephrol. Hypertens 18, 50–56. doi: 10.1097/MNH.0b013e3283186f82
Ishizaka, N., Griendling, K. K., Lassègue, B., and Alexander, R. W. (1998). Angiotensin II type 1 receptor: relationship with caveolae and caveolin after initial agonist stimulation. Hypertension 32, 459–466. doi: 10.1161/01.hyp.32.3.459
Ju, H., Zou, R., Venema, V. J., and Venema, R. C. (1997). Direct interaction of endothelial nitric oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272, 18522–18525. doi: 10.1074/jbc.272.30.18522
Kato, H. (2002). Regulation of functions of vascular wall cells by tissue factor pathway inhibitor: basic and clinical aspects. Arterioscler. Thromb. Vasc. Biol. 22, 539–548. doi: 10.1161/01.atv.0000013904.40673.cc
Kumagai, R., Lu, X., and Kassab, G. S. (2009). Role of glycocalyx in flow-induced production of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 47, 600–607. doi: 10.1016/j.freeradbiomed.2009.05.034
Li, J. M., and Shah, A. M. (2002). Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J. Biol. Chem. 277, 19952–19960. doi: 10.1074/jbc.M110073200
Li, W., and Wang, W. (2018). Structural alteration of the endothelial glycocalyx: contribution of the actin cytoskeleton. Biomech. Model. Mechanobiol. 17, 147–158. doi: 10.1007/s10237-017-0950-2
Li, X., Fang, P., Li, Y., Kuo, Y. M., Andrews, A. J., Nanayakkara, G., et al. (2016). Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler. Thromb. Vasc. Biol. 36, 1090–1100. doi: 10.1161/ATVBAHA.115.306964
Li, X. A., Everson, W. V., and Smart, E. J. (2005). Caveolae, lipid rafts, and vascular disease. Trends Cardiovasc. Med. 15, 92–96. doi: 10.1016/j.tcm.2005.04.001
Lian, X., Matthaeus, C., Kaβmann, M., Daumke, O., and Gollasch, M. (2019). Pathophysiological Role of caveolae in hypertension. Front. Med. 6:153. doi: 10.3389/fmed.2019.00153
Lipowsky, H. H. (2011). Protease activity and the role of the endothelial glycocalyx in inflammation. Drug Discov. Today Dis. Models 8, 57–62. doi: 10.1016/j.ddmod.2011.05.004
Lipowsky, H. H., and Lescanic, A. (2013). The effect of doxycycline on shedding of the glycocalyx due to reactive oxygen species. Microvasc. Res. 90, 80–85. doi: 10.1016/j.mvr.2013.07.004
Liu, J. X., Yan, Z. P., Zhang, Y. Y., Wu, J., Liu, X. H., and Zeng, Y. (2016). Hemodynamic shear stress regulates the transcriptional expression of heparan sulfate proteoglycans in human umbilical vein endothelial cell. Cell. Mol. Biol. 62, 28–34. doi: 10.14715/cmb/2016.62.8.5
Liu, L., Brown, D., McKee, M., LeBrasseur, N. K., Yang, D., Albrecht, K. H., et al. (2008). Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia and glucose intolerance. Cell Metab. 8, 310–317. doi: 10.1016/j.cmet.2008.07.008
McDonald, K. K., Cooper, S., Danielzak, L., and Leask, R. L. (2016). Glycocalyx degradation induces a proinflammatory phenotype and increased leukocyte adhesion in cultured endothelial cells under Flow. PLoS One 11:e0167576. doi: 10.1371/journal.pone.0167576
Mensah, S. A., Cheng, M. J., Homayoni, H., Plouffe, B. D., Coury, A. J., and Ebong, E. E. (2017). Regeneration of glycocalyx by heparan sulfate and sphingosine 1-phosphate restores inter-endothelial communication. PLoS One 12:e0186116. doi: 10.1371/journal.pone.0186116
Milovanova, T., Chatterjee, S., Hawkinsy, B. J., Hong, N., Sorokina, E. M., Debolt, K., et al. (2008). Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim. Biophys. Acta 1783, 1866–1875. doi: 10.1016/j.bbamcr.2008.05.010
Mitra, R., O’Neil, G. L., Harding, I. C., Cheng, M. J., Mensah, S. A., and Ebong, E. E. (2017). Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr. Atheroscler. Rep. 19, 63–75. doi: 10.1007/s11883-017-0691-9
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mochizuki, S., Vink, H., Hiramatsu, O., Kajita, T., Shigeto, F., Spaan, J. A. E., et al. (2003). Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am. J. Physiol. Heart Circ. Physiol. 285, H722–H726. doi: 10.1152/ajpheart.00691
Möckl, L. (2020). The emerging role of the mammalian glycocalyx in functional membrane organization and immune system regulation. Front. Cell. Dev. Biol. 8:253. doi: 10.3389/fcell.2020.00253
Montorfano, I., Becerra, A., Cerro, R., Echeverría, C., Sáez, E., Morales, M. G., et al. (2014). Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β1 and TGF-β2-dependent pathway. Lab. Invest. 94, 1068–1082. doi: 10.1038/labinvest.2014.100
Moon, J. J., Matsumoto, M., Patel, S., Lee, L., Guan, J. L., and Li, S. (2005). Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J. Cell Physiol. 203, 166–176. doi: 10.1002/jcp.20220
Mougeolle, A., Poussard, S., Decossas, M., Lamaze, C., Lambert, O., and Dargelos, E. (2015). Oxidative stress induces caveolin 1 degradation and impairs caveolae functions in skeletal muscle cells. PLoS One 10:e0122654. doi: 10.1371/journal.pone.0122654
Murata, T., Lin, M. I., Huang, Y., Yu, J., Bauer, P. M., Giordano, F. J., et al. (2007). Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J. Exp. Med. 204, 2373–2382. doi: 10.1084/jem.20062340
Navarro, G., Borroto-Escuela, D. O., Fuxe, K., and Franco, R. (2014). Potential of caveolae in the therapy of cardiovascular and neurological diseases. Front. Physiol. 5:370. doi: 10.3389/fphys.2014.00370
Nieuwdorp, M., Mooij, H. L., Kroon, J., Atasever, B., Spaan, J. A., Ince, C., et al. (2006a). Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes 55, 1127–1132. doi: 10.2337/diabetes.55.04.06.db05-1619
Nieuwdorp, M., van Haeften, T. W., Gouverneur, M. C., Mooij, H. L., van Lieshout, M. H., Levi, M., et al. (2006b). Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes 5, 480–486. doi: 10.2337/diabetes.55.02.06.db05-1103
Noris, M., Morigi, M., Donadelli, R., Ariello, S., Foppolo, M., Todeschini, M., et al. (1995). Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ. Res. 76, 536–543. doi: 10.1161/01.res.76.4.536
Ogata, T., Ueyama, T., Isodono, K., Tagawa, M., Takehara, N., Kawashima, T., et al. (2008). MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol. Cell. Biol. 28, 3424–3436. doi: 10.1128/MCB.02186-07
Oh, P., Borgström, P., Witkiewicz, H., Li, Y., Borgström, B. J., Chrastina, A., et al. (2007). Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotech. 25, 327–337. doi: 10.1038/nbt1292
Oliveira-Paula, G. H., Pinheiro, L. C., Guimaraes, D. A., Tella, S. O., Blanco, A. L., Angelis, C. D., et al. (2016). Tempol improves xanthine oxidoreductase-mediated vascular responses to nitrite in experimental renovascular hypertension. Redox Biol. 8, 398–406. doi: 10.1016/j.redox.2016.04.001
Pahakis, M. Y., Kosky, J. R., Dull, R. O., and Tarbell, J. M. (2007). The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 355, 228–233. doi: 10.1016/j.bbrc.2007.01.137
Panth, N., Paudel, K. R., and Parajuli, K. (2016). Reactive oxygen species: a key hallmark of cardiovascular disease. Adv. Med. 2016:9152732. doi: 10.1155/2016/9152732
Parton, R. G., and Simons, K. (2007). The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194. doi: 10.1038/nrm2122
Parton, R. G., Tillu, V. A., and Collins, B. M. (2018). Caveolae. Curr. Biol. 28, R402–R405. doi: 10.1016/j.cub.2017.11.075
Patel, H. H., Murray, F., and Insel, P. A. (2008). Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol. Toxicol. 48, 359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841
Pelkmans, L., and Zerial, M. (2005). Kinase-regulated quantal assemblies and kiss-and-run recycling of caveolae. Nature 436, 128–133. doi: 10.1038/nature03866
Pohl, U., Holtz, J., Busse, R., and Bassenge, E. (1986). Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension 8, 37–44. doi: 10.1161/01.hyp.8.1.37
Potje, S. R., Grando, M. D., Chignalia, A. Z., Antoniali, C., and Bendhack, L. M. (2019). Reduced caveolae density in arteries of SHR contributes to endothelial dysfunction and ROS production. Sci. Rep. 9:6696. doi: 10.1038/s41598-019-43193-8
Quinsey, N. S., Greedy, A. L., Bottomley, S. P., Whisstock, J. C., and Pike, R. N. (2004). Antithrombin: in control of coagulation. Int. J. Biochem. Cell Biol. 36, 386–389. doi: 10.1016/s1357-2725(03)00244-9
Radi, R. (2018). Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. U.S.A. 115, 5839–5848. doi: 10.1073/pnas.1804932115
Rahman, A., and Sward, K. (2009). The role of caveolin-1 in cardiovascular regulation. Acta Physiol. 195, 231–245. doi: 10.1111/j.1748-1716.2008.01907.x
Rajagopalan, S., Kurz, S., Münzel, T., Tarpey, M., Freeman, B. A., Griendling, K. K., et al. (1996). Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 97, 1916–1923. doi: 10.1172/JCI118623
Ramnath, R., Foster, R. R., Qiu, Y., Cope, G., Butler, M. J., Salmon, A. H., et al. (2014). Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor α: a contributor to endothelial cell glycocalyx dysfunction. FASEB J. 28, 4686–4699. doi: 10.1096/fj.14-252221
Ramnath, R. D., Butler, M. J., Newman, G., Desideri, S., Russell, A., Lay, A. C., et al. (2020). Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. 97, 951–965. doi: 10.1016/j.kint.2019.09.035
Rath, G., Dessy, C., and Feron, O. (2009). Caveolae, caveolin and control of vascular tone: nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J. Physiol. Pharmacol. 60(Suppl. 4), 105–109.
Razani, B., Engelman, J. A., Wang, X. B., Schubert, W., Zhang, X. L., Marks, C. B., et al. (2001). Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276, 38121–38138. doi: 10.1074/jbc.M105408200
Razani, B., and Lisanti, M. P. (2001). Caveolin-deficient mice: insights into caveolar function human disease. J. Clin. Invest. 108, 1553–1561. doi: 10.1172/JCI14611
Reitsma, S., Slaaf, D. W., Vink, H., van Zandvoort, M. A., and de Egbrink, M. G. (2007). The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 454, 345–359. doi: 10.1007/s00424-007-0212-8
Rippe, B., Rosengren, B. I., Carlsson, O., and Venturoli, D. (2002). Transendothelial transport: the vesicle controversy. J. Vasc. Res. 39, 375–390. doi: 10.1159/000064521
Rizzo, V., Morton, C., DePaola, N., Schnitzer, J. E., and Davies, P. F. (2003). Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am. J. Physiol. Heart Circ. Physiol. 285, H1720–H1729. doi: 10.1152/ajpheart.00344.2002
Robenek, H., Weissen-Plenz, G., and Severs, N. J. (2008). Freeze-fracture replica immunolabelling reveals caveolin-1 in the human cardiomyocyte plasma membrane. J. Cell. Mol. Med. 12, 2519–2521. doi: 10.1111/j.1582-4934.2008.00498.x
Rodrigues, G. J., Restini, C. B., Lunardi, C. N., Moreira, J. E., Lima, R. G., da Silva, R. S., et al. (2007). Caveolae dysfunction contributes to impaired relaxation induced by nitric oxide donor in aorta from renal hypertensive rats. J. Pharmacol. Exp. Ther. 323, 831–837. doi: 10.1124/jpet.107.127241
Rodrigues, G. J., Restini, C. B., Lunardi, C. N., Neto, M. A., Moreira, J. E., and Bendhack, L. M. (2010). Decreased number of caveolae in endothelial cells impairs the relaxation induced by acetylcholine in hypertensive rat aortas. Eur. J. Pharmacol. 627, 251–257. doi: 10.1016/j.ejphar.2009.11.010
Satta, S., Mahmoud, A. M., Wilkinson, F. L., Yvonne-Alexander, M., and White, S. J. (2017). The role of Nrf2 in cardiovascular function and disease. Oxid. Med. Cell. Longev. 2017:9237263. doi: 10.1155/2017/9237263
Schmidt, E. P., Yang, Y., Janssen, W. J., Gandjeva, A., Perez, M. J., Barthel, L., et al. (2012). The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 18, 1217–1223. doi: 10.1038/nm.2843
Schnitzer, J. E., Oh, P., Jacobson, B. S., and Dvorak, A. M. (1995). Caveolae from luminal plasmalemma of rat lung endothelium: microdomains enriched in caveolin, Ca (2+)-ATPase, and inositol trisphosphate receptor. Proc. Natl. Acad. Sci. U.S.A. 92, 1759–1763. doi: 10.1073/pnas.92.5.1759
Sena, C. M., Leandro, A., Azul, L., Seiça, R., and Perry, G. (2018). Vascular oxidative stress: impact and therapeutic approaches. Front. Physiol. 9:1668. doi: 10.3389/fphys.2018.01668
Shaul, P. W. (2003). Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J. Physiol. 547(Pt 1), 21–33. doi: 10.1113/jphysiol.2002.031534
Shaw, L., Sweeney, M. A., O’Neill, S. C., Jones, C. J. P., Austin, C., and Taggart, M. J. (2006). Caveolae and sarcoplasmic reticular coupling in smooth muscle cells of pressurized arteries: the relevance for Ca2+ oscillations and tone. Cardiovasc. Res. 69, 825–835. doi: 10.1016/j.cardiores.2005.12.016
Shimada, K., Kobayashi, M., Kimura, S., Nishinaga, M., Takeuchi, K., and Ozawa, T. (1991). Anticoagulant heparin-like glycosaminoglycans on endothelial cell surface. Jpn. Circ. J. 55, 1016–1021. doi: 10.1253/jcj.55.1016
Sieve, I., Münster-Kühnel, A. K., and Hilfiker-Kleiner, D. (2018). Regulation and function of endothelial glycocalyx layer in vascular diseases. Vasc. Pharmacol. 100, 26–33. doi: 10.1016/j.vph.2017.09.002
Silva, B. R., Pernomian, L., Grando, M. D., Amaral, J. H., Tanus-Santos, J. E., and Bendhack, L. M. (2013). Hydrogen peroxide modulates phenylephrine-induced contractile response in renal hypertensive rat aorta. Eur. J. Pharmacol. 721, 193–200. doi: 10.1016/j.ejphar.2013.09.036
Siwik, D. A., and Colucci, W. S. (2004). Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail. Rev. 9, 43–51. doi: 10.1023/B:HREV.0000011393.40674.13
Soltés, L., Mendichi, R., Kogan, G., Schiller, J., Stankovska, M., and Arnhold, J. (2006). Degradative action of reactive oxygen species on hyaluronan. Biomacromolecules 7, 659–668. doi: 10.1021/bm050867v
Sowa, G. (2012). Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front. Physiol. 2:120. doi: 10.3389/fphys.2011.00120
Stenzel, D., Nye, E., Nisancioglu, M., Adams, R. H., Yamaguchi, Y., and Gerhardt, H. (2009). Peripheral mural cell recruitment requires cell-autonomous heparan sulfate. Blood 114, 915–924. doi: 10.1182/blood-2008-10-186239
Stoeber, M., Stoeck, I. K., Hanni, C., Bleck, C. K. E., Balistreri, G., and Helenius, A. (2012). Oligomers of the ATPase EHD2 confine caveolae to the plasma membrane through association with actin. EMBO J. 31, 2350–2364. doi: 10.1038/emboj.2012.98
Thi, M. M., Tarbel, l J M, Weinbaum, S., and Spray, D. C. (2004). The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc. Natl. Acad. Sci. U.S.A. 101, 16483–16488. doi: 10.1073/pnas.0407474101
Tovar, A. M., de Mattos, D. A., Stelling, M. P., Sarcinelli-Luz, B. S., Nazareth, R. A., and Mourão, P. A. (2005). Dermatan sulfate is the predominant antithrombotic glycosaminoglycan in vessel walls: implications for a possible physiological function of heparin cofactor II. Biochim. Biophys. Acta. 1740, 45–53. doi: 10.1016/j.bbadis.2005.02.008
Tsai, M. H., and Jiang, M. J. (2010). Reactive oxygen species are involved in regulating alpha1-adrenoceptor-activated vascular smooth muscle contraction. J. Biomed. Sci. 17:67. doi: 10.1186/1423-0127-17-67
Uchimido, R., Schmidt, E. P., and Shapiro, N. I. (2019). The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit. Care 23:16. doi: 10.1186/s13054-018-2292-6
Uchiyama, H., Dobashi, Y., Ohkouchi, K., and Nagasawa, K. (1990). Chemical change involved in the oxidative reductive depolymerization of hyaluronic acid. J. Biol. Chem. 265, 7753–7759.
Uittenbogaard, A., Shaul, P. W., Yuhanna, I. S., Blair, A., and Smart, E. J. (2000). High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J. Biol. Chem. 275, 11278–11283. doi: 10.1074/jbc.275.15.11278
Ungvari, Z., Csiszar, A., Huang, A., Kaminski, P. M., Wolin, M. S., and Koller, A. (2003). High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation 108, 1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D
Vanhoutte, P. M., Shimokawa, H., Félétou, M., and Tang, E. H. C. (2017). Endothelial dysfunction and vascular disease – a 30th anniversary update. Acta Physiol. 219, 22–96. doi: 10.1111/apha.12646
Vara, D., and Pula, G. (2014). Reactive oxygen species: physiological roles in the regulation of vascular cells. Curr. Mol. Med. 14, 1103–1125. doi: 10.2174/1566524014666140603114010
Vogel, E. R., Manlove, L. J., Kuipers, I., Thompson, M. A., Fang, Y. H., Freeman, M. R., et al. (2019). Caveolin-1 scaffolding domain peptide prevents hyperoxia-induced airway remodeling in a neonatal mouse model. Am. J. Physiol. Lung Cell. Mol. Physiol. 317, L99–L108. doi: 10.1152/ajplung.00111.2018
Volonte, D., Liu, Z., Musille, P. M., Stoppani, E., Wakabayashi, N., Di, Y. P., et al. (2013). Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Mol. Biol. Cell. 24, 1852–1862. doi: 10.1091/mbc.E12-09-0666
Wanaski, S. P., Ng, B. K., and Glaser, M. (2003). Caveolin scaffolding region and the membrane binding region of SRC form lateral membrane domains. Biochemistry 42, 42–56. doi: 10.1021/bi012097n
Wang, G., Kostidis, S., Tiemeier, G. L., Sol, W. M. P. J., de Vries, M. R., Giera, M., et al. (2020). Shear stress regulation of endothelial glycocalyx structure is determined by glucobiosynthesis. Arterioscler. Thromb. Vasc. Biol. 40, 350–364. doi: 10.1161/ATVBAHA.119.313399
Wang, X. L., Ye, D., Peterson, T. E., Cao, S., Shah, V. H., Katusic, Z. S., et al. (2005). Caveolae targeting and regulation of large conductance Ca (2+)-activated K+ channels in vascular endothelial cells. J. Biol. Chem. 280, 11656–11664. doi: 10.1074/jbc.M410987200
Wyse, B. D., Prior, I. A., Qian, H., Morrow, I. C., Nixon, S., Muncke, C., et al. (2003). Caveolin interacts with the angiotensin II type 1 receptor during exocytic transport but not at the plasma membrane. J. Biol. Chem. 278, 23738–23746. doi: 10.1074/jbc.M212892200
Yen, W., Cai, B., Yang, J., Zhang, L., Zeng, M., Tarbell, J. M., et al. (2015). Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS One 10:e0117133. doi: 10.1371/journal.pone.0117133
Zeng, Y., and Liu, J. (2016). Role of glypican-1 in endothelial NOS activation under various steady shear stress magnitudes. Exp. Cell Res. 348, 184–189. doi: 10.1016/j.yexcr.2016.09.017
Zeng, Y., and Tarbell, J. M. (2014). The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS One 9:e86249. doi: 10.1371/journal.pone.0086249
Zeng, Y., Waters, M., Andrews, A., Honarmandi, P., Ebong, E. E., Rizzo, V., et al. (2013). Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am. J. Physiol. Heart Circ. Physiol. 305, H811–H820. doi: 10.1152/ajpheart.00764.2012
Zhang, X., Ramírez, C. M., Aryal, B., Madrigal-Matute, J., Liu, X., Diaz, A., et al. (2020). Cav-1 (Caveolin-1) deficiency increases autophagy in the endothelium and attenuates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 40, 1510–1522. doi: 10.1161/ATVBAHA.120.314291
Zhao, Y., Vanhoutte, P. M., and Leung, S. W. (2015). Vascular nitric oxide: beyond eNOS. J. Pharmacol. Sci. 129, 83–94. doi: 10.1016/j.jphs.2015.09.002
Zhao, Y. Y., Liu, Y., Stan, R. V., Fan, L., Gu, Y., Dalton, N., et al. (2002). Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. U.S.A. 99, 11375–11380. doi: 10.1073/pnas.172360799
Zhou, B., Qiu, Y., Wu, N., Chen, A. D., Zhou, H., Chen, Q., et al. (2020). FNDC5 attenuates oxidative stress and NLRP3 inflammasome activation in vascular smooth muscle cells via activating the AMPK-SIRT1 signal pathway. Oxid. Med. Cell. Longev. 2020:6384803. doi: 10.1155/2020/6384803
Zuo, L., Ushio-Fukai, M., Ikeda, S., Hilenski, L., Patrushev, N., and Alexander, R. W. (2005). Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler. Thromb. Vasc. Biol. 25, 1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18
Keywords: glycocalyx, caveolae, vascular tone, ROS, hypertension, atherosclerosis, endothelial cells, eNOS
Citation: Potje SR, Paula TD, Paulo M and Bendhack LM (2021) The Role of Glycocalyx and Caveolae in Vascular Homeostasis and Diseases. Front. Physiol. 11:620840. doi: 10.3389/fphys.2020.620840
Received: 23 October 2020; Accepted: 15 December 2020;
Published: 13 January 2021.
Edited by:
Ana Paula Davel, Campinas State University, BrazilReviewed by:
Virginia Soares Lemos, Federal University of Minas Gerais, BrazilKristina Kusche-Vihrog, University of Lübeck, Germany
Copyright © 2021 Potje, Paula, Paulo and Bendhack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lusiane Maria Bendhack, YmVuZGhhY2tAdXNwLmJy