 Xinmin Yang1†Linbo Yao1†
Xinmin Yang1†Linbo Yao1† Xianghui Fu2Rajarshi Mukherjee3Qing Xia1
Xianghui Fu2Rajarshi Mukherjee3Qing Xia1 Monika A. Jakubowska4
Monika A. Jakubowska4 Pawel E. Ferdek5
Pawel E. Ferdek5 Wei Huang1*
Wei Huang1*- 1Department of Integrated Traditional Chinese Medicine and Western Medicine, Sichuan Provincial Pancreatitis Center and West China-Liverpool Biomedical Research Center, West China Hospital, Sichuan University, Chengdu, China
- 2Division of Endocrinology and Metabolism, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center of Biotherapy, Chengdu, China
- 3Liverpool Pancreatitis Research Group, Liverpool University Hospitals National Health Service Foundation Trust and Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, United Kingdom
- 4Malopolska Center of Biotechnology, Jagiellonian University, Krakow, Poland
- 5Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Krakow, Poland
Acute pancreatitis is a potentially severe inflammatory disease that may be associated with a substantial morbidity and mortality. Currently there is no specific treatment for the disease, which indicates an ongoing demand for research into its pathogenesis and development of new therapeutic strategies. Due to the unpredictable course of acute pancreatitis and relatively concealed anatomical site in the retro-peritoneum, research on the human pancreas remains challenging. As a result, for over the last 100 years studies on the pathogenesis of this disease have heavily relied on animal models. This review aims to summarize different animal models of acute pancreatitis from the past to present and discuss their main characteristics and applications. It identifies key studies that have enhanced our current understanding of the pathogenesis of acute pancreatitis and highlights the instrumental role of animal models in translational research for developing novel therapies.
Introduction
Acute pancreatitis (AP) is an inflammatory disorder of the pancreas, which ranges from mild, self-limiting disease to a severe form that is associated with multiple organ dysfunction syndrome (MODS), high morbidity, and mortality (Hines and Pandol, 2019). Although in the last two decades advances have been made in the supportive management of AP, specific, and effective drug treatment is still lacking due to the poorly understood pathobiology of the disease (Moggia et al., 2017). Ideally, studies on the etiology, pathogenesis, and treatment of AP should be carried out on the human pancreas. However, the unpredictable nature of the disease, heterogeneity of disease presentations, and limited access to human samples, make research on human tissues impractical and often very difficult. For these reasons, experimental models have been widely used to study AP for more than a century (Gorelick and Lerch, 2017). In recent years, the most commonly used AP models are carried out on rodents (rats and mice), which are relatively inexpensive to maintain, easy to handle, accessible, and allow induction of moderate to severe pancreatic injury. These experimental models not only provide an opportunity for mechanistic studies but also enable development of therapeutic strategies.
What makes a perfect animal model? Ideally it should reflect the etiology, pathobiology, histopathology, the clinical course, and outcome of the disease in humans. However, these aims are impossible to reproduce all in one model. Our knowledge of mechanisms relevant to the human condition, the multitude of genetic and environmental factors that are likely to influence the risk of disease development and the natural history of the disease remains lacking (Gorelick and Lerch, 2017). Hence, the crucial consideration for researchers selecting a model for research studies is “What is the specific research question being asked by the study?.” Experimental AP models can be divided into in vivo and in vitro models. Further in vivo experimental AP models can be generally sub-divided into non-invasive and invasive models. Although these categories describe the logistic differences in inducing each respective model, certain models may have greater utility over others dependent on which mechanisms they focus on, which animal species they are induced in, and which disease outcomes are reproduced. Here we comprehensively review the history, development, and current use of important experimental AP models as well as explore their mechanisms, advantages, limitations, clinical relevance, and the scope for future work.
Caerulein/Cholecystokinin
Early in 1895, Mouret (1895) found that excessive cholinergic stimulation causes vacuolization and necrosis of the pancreas, which are typical features of AP. Later in 1929, Villaret et al. (1929) reported the first secretagogue hyperstimulation-induced AP model by injection of acetylcholine, a cholinergic agonist, into the canine pancreas and this was later reproduced in a rat model (Leblond and Sergeyeva, 1944). Subsequently, cholecystokinin octapeptide (CCK-8) and its analog caerulein (Lampel and Kern, 1977), as well as carbamylcholine (Adler et al., 1983), anticholinesterase (Dressel et al., 1982), and scorpion toxin (Gallagher et al., 1981; Pantoja et al., 1983; Novaes et al., 1989) have been shown to induce AP.
CCK was named after its main function related to promoting contraction of gallbladder smooth muscle and bile discharge (Ivy, 1929). Later, it was found that it can act on the pancreas to stimulate the secretion of pancreatic digestive enzymes (Harper and Raper, 1943) and insulin (Kuntz et al., 2004). The fundamental mechanism of pancreatic pathology induced by CCK and its analogs is based on the action of these chemicals on CCK receptors, which in turn leads to activation of second messenger pathways related to secretion of pancreatic enzymes (e.g., amylase) in pancreatic acinar cells (PACs) like phospholipase C-inositol trisphosphate-calcium (Ca2+). There are also protein-protein interaction pathways that mainly regulate non-secretory processes, including biosynthesis and growth, such as three major mitogen-activated protein kinase pathways (ERK, JNK, and p38 MAPK) and several other pathways that are still unknown. While CCK-8 is most well-studied, CCK-58 is the main circulating form in humans and dogs, and the only endocrine form of CCK-58 in rats (Reeve et al., 2004). CCK-8 and CCK-58 have the same effect on Ca2+ signaling, zymogen activation, and cell death in PACs at high and low agonist concentrations in vitro (Criddle et al., 2009). A recent review has summarized the regulation of the CCK pathway in PACs in detail (Williams, 2019).
Caerulein, a CCK analog, was first isolated from skin extracts of the Australian green tree frog (Litoria caerulea) and was immediately acknowledged for its physiological activity mimicking natural hormones (Anastasi et al., 1968). CCK and caerulein have a very similar amino acid sequence, but compared to the CCK, caerulein is a decapeptide that has methionine substituted to threonine and two additional N-terminal residues (Figure 1A). Both peptides show almost the same potency in vitro, but caerulein is more potent to induce AP in vivo (Figure 1B). The increased biological activity is related to the additional N-terminal residues, and a result of the substitution of methionine for threonine (Shorrock et al., 1991). To date, caerulein remains the most widely used compound to induce AP (CER-AP) in rodents.
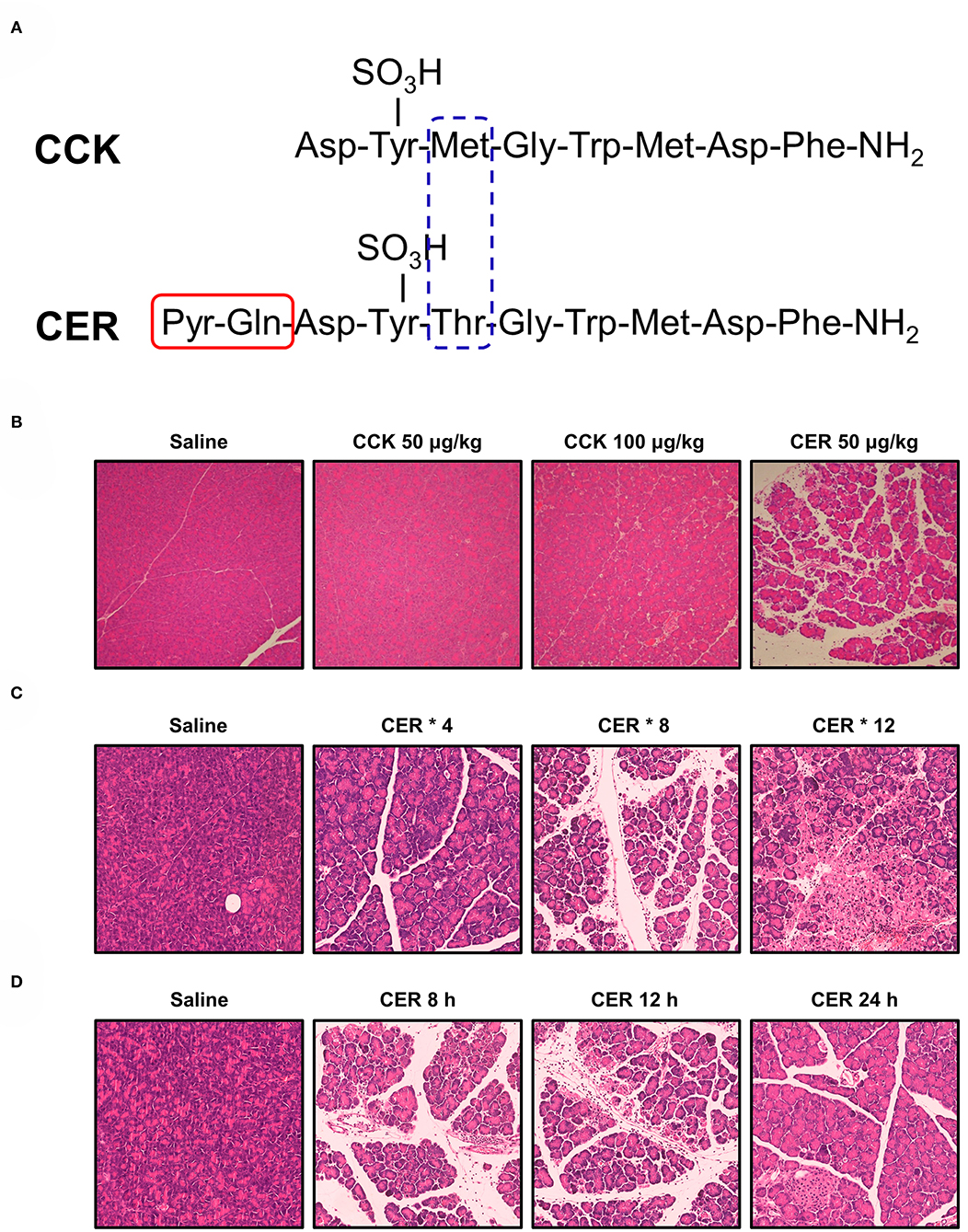
Figure 1. Structure of cholecystokinin and caerulein and their effects on pancreas. (A) Structure of cholecystokinin (CCK) and caerulein (CER). Pyr, pyroglutamic acid. (B) Comparison of the potency of CCK and CER in inducing acute pancreatitis (AP). Mice received seven intraperitoneal (i.p.) injections of CCK (50 or 100 μg/kg; MW: 1142.20), CER (50 μg/kg; MW: 1,352.40), or normal saline at hourly intervals. Mice were sacrificed 12 h after the first injection. While CER induced typical features of AP (edema, vacuolization, inflammatory cell infiltration, and necrosis), CCK at both doses did not cause disenable pancreas histopathological changes. (C) Comparison of the efficacy of different injection regimens of CER in inducing AP. Mice received i.p. injection of CER (50 μg/kg) or normal saline at 1 h apart. The mice were killed 12 h after the first injection. After 4 injections of CER, there were focally increased edema between lobules, periductal neutrophil infiltration, and minimal acinar cell necrosis; after 8 injections of CER, there were diffusely increased edema, parenchyma neutrophil infiltration, and periductal and focal acinar cell necrosis; after 12 injections of CER, there were disrupted and separated acini structure, marked parenchyma neutrophil infiltration, and focal and diffuse parenchymal necrosis. (D) Comparison pancreatic changes at different time after AP induced by 8 injections of CER. Mice received 8 times i.p. injection of CER (50 μg/kg) or normal saline at 1 h apart. At 8 h, there were marked pancreas histopathological changes, which peaked at 12 h and started to recover at 24 h. All experiments used C57BL/6J mice and the images were at magnification of × 200.
There is a clear dose-response relationship between the structural and biochemical changes of the pancreas in response to caerulein administration (Bieger et al., 1976b). Continuous infusion of maximal physiological doses of caerulein (0.25 μg/kg/h) causes rapid degranulation of the exocrine pancreas in rats (Bieger et al., 1976a). Administration with a supramaximal dose leads to vacuolization within the acinar cells, followed by regeneration of the pancreas (Tardini et al., 1971). At an even higher dose, caerulein causes pancreatic interstitial edema and inflammatory cell infiltration together with a significant increase of the pancreatic enzyme levels in the blood (Willemer et al., 1992). Based on the above findings, in 1977 Lampel and Kern (1977) described a non-lethal CER-AP model in rats, after which CER-AP was successfully reproduced in mice (Niederau et al., 1985).
CCK and its analogs were shown to induce pancreatic injury in a time-dependent manner in addition to a dose-dependent manner (Watanabe et al., 1984). One of the early consequences of hyperstimulation with caerulein is the formation of pancreatic oedema. This may be due to increased vascular permeability and hydrostatic pressure (Lerch et al., 1995b; Weidenbach et al., 1995). However, the exact mechanism leading to the formation of extensive oedema is still not fully understood as it does not reflect the extent of damage to PACs. This model is now widely used for the analysis of early intracellular events in AP. Although the pancreatic injury can be controlled by appropriate dosage and frequency of injections (Figure 1C) and the pancreas begins to recover after reaching its peak over time (Figure 1D), this model is generally self-limiting without MODS and lethality, which may be its biggest limitation.
To address this limitation, caerulein is often combined with other compounds to achieve increased severity of CER-AP. For example, lipopolysaccharide (LPS) in CER-AP (Sugita et al., 1997; Yamaguchi et al., 1999; Chao et al., 2006) exaggerates the inflammatory response and MODS, mimicking AP-associated sepsis. Infusion of rats with enterokinase (EK) after caerulein administration causes pancreatic necrosis, hemorrhage, and high mortality rates (Hartwig et al., 2007). Similar effects can also be replicated in mice (Hartwig et al., 2008), making it possible to undertake transgenic studies. Therefore, the “two-hit” model induced by caerulein with LPS or EK, is a useful tool to study inflammatory changes, sepsis, and bacterial translocation in AP (Xue et al., 2009). Whereas, one must remember that although the additional administration of LPS or EK results in a greater immune response in this setting, AP, at least on initiation, is primarily a sterile inflammation and this aspect of the disease is quite difficult to model.
The caerulein/CCK model exhibits the closest parallel with clinical AP induced by scorpion venom or organophosphate insecticides. It is non-invasive, easy to conduct, highly reproducible, reflects a vast number of in vitro studies, making it a favorable model for AP. It is also compatible with other models, sharing histopathological changes consistent with early phases of human AP (Rifai et al., 2008). All these factors explain why CER-AP is so widely accepted and commonly used by pancreatic investigators (Saluja et al., 2007; Lerch and Gorelick, 2013).
Basic Amino Acids
Intraperitoneal (i.p.) administration of excessive doses of certain amino acids, such as L-arginine (Toma et al., 2000), L-ornithine (Rakonczay et al., 2008), L-lysine (Biczo et al., 2011b), and L-histidine (Zhang et al., 2018), causes necrotising AP in rats and/or mice. Why these particular basic amino acids induce AP has not yet been fully clarified, but is likely related to shared metabolic pathways in vivo determined by the structural similarities.
L-Arginine
L-arginine-induced AP (ARG-AP) is currently the most commonly used amino acid-induced AP model in rats and mice. The effect of L-arginine on the pancreas has been extensively investigated since 1984, when single i.p. injection of L-arginine at 5 g/kg led to long lasting PAC and adipose tissue necrosis in the rat pancreas, without affecting islets and other organs (Mizunuma et al., 1984). These findings were further confirmed in the same year (Kishino and Kawamura, 1984). Higher doses (7.5 g/kg) of L-arginine can be lethal for experimental animal, whereas a dose of 2.5 g/kg only caused mild pancreatic injury. In addition, severe AP can be induced in mice by i.p. injection of L-arginine in two doses of 4 g/kg each, at 1 h apart (Dawra et al., 2007). Subsequently, in various studies either single or double injections of L-arginine were applied at different doses to induce necrotising AP in rats or mice. It is worth noting that when using the AP model induced by L-arginine, the dose, concentration, pH of L-arginine, and the strain of mice must be taken into account (Kui et al., 2015) and that D-arginine has no effect on the pancreas (Dawra et al., 2007). Severe acute inflammation of the mouse pancreas induced by L-arginine is classically followed by lung injury. There is a certain failure rate and mortality rate in this model (Hegyi et al., 2004). ARG-AP model is rarely fatal in rats, but it has a mortality rate of 5–7% in mice (Dawra and Saluja, 2012). In our laboratory, we induced AP in C57BL/6J mice (~25 g) with 4 g/kg × 2 of L-arginine, and the mortality was 1~3%. Moreover, the mortality usually occurs within a few hours after the second injection, and before AP occurs, it may be caused by metabolic disorder caused by excessive amino acids.
The mechanisms of AP induced by L-arginine remain unclear. Amino acid imbalance (Mizunuma et al., 1984), reactive oxygen species (Czako et al., 1998; Rakonczay et al., 2003), inflammatory mediators (Czako et al., 2000; Takacs et al., 2002b; Rakonczay et al., 2003), nitric oxide (Takacs et al., 2002a), cytoskeletal changes (Tashiro et al., 2001), intracellular Ca2+ signaling (Zhang et al., 2018), mitochondrial dysfunction (Biczo et al., 2018; Zhang et al., 2018) and endoplasmic reticulum stress (Kubisch et al., 2006) have all been postulated to be involved in this process.
L-Ornithine
Rakonczay et al. reported a simple, non-invasive model of necrotising AP induced by i.p. injection of 3 g/kg L-ornithine, which is even more effective than L-arginine in rats (Rakonczay et al., 2008). Since L-ornithine is a product of L-arginine metabolism in vivo (in the urea cycle), it is inferred that large doses of L-arginine cause pancreatic injury at least partially through L-ornithine (Figure 2A). Biczo et al. found that pancreatic polyamine catabolism was activated in L-ornithine-induced AP, and tried to ameliorate it with metabolically stable polyamine analogs, which turned out ineffective (Biczo et al., 2010). We also tried to induce AP with L-ornithine (2 × 4 g/kg) in mice, but found the model to be excessively severe and mice were dead within few hours (Zhang et al., 2018).
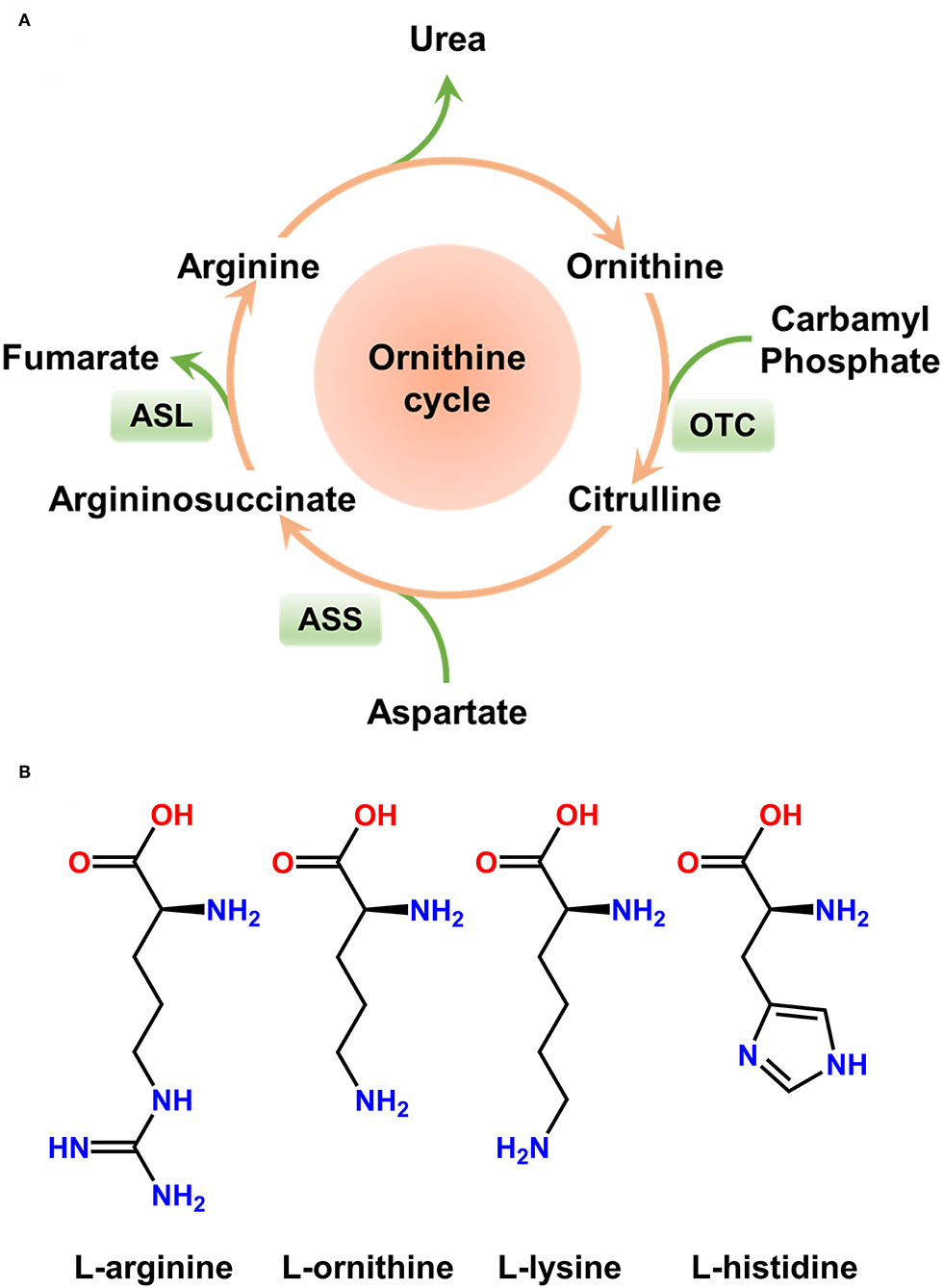
Figure 2. Ornithine cycle and molecular structure of basic amino acids. (A) Ornithine cycle. Arginine is partially metabolized into ornithine in vivo. (B) Molecular structure of basic amino acids inducing acute pancreatitis in rodents. Large doses of L-arginine, L-ornithine, L-lysine, and L-histidine have been proven to induce severe acute pancreatitis.
L-Lysine
In vivo administration of L-lysine causes early mitochondrial damage as evidenced by degenerated mitochondria shown by electron microscopy and impaired ATP synthase activity in mitochondria of isolated PACs (Biczo et al., 2011b). The mitochondrial injury appears prior to the activation of trypsinogen and nuclear factor kappa-B (NF-κB) (Biczo et al., 2011b) which suggests that the aliphatic amino acid might be more likely to cause AP due to its positive charge and L-lysine induces mitochondria injury.
L-Histidine
Although previous studies showed that i.p. injection of 3 g/kg L-histidine had no effect on the rat pancreas (Biczo et al., 2011a), our group reported for the first time that i.p. administration of 2 × 4 g/kg L-histidine (1 h apart) to mice can induce AP (Zhang et al., 2018). L-histidine at a dose of >3 g/kg could also likely trigger AP in rats, but this has not yet been tested. One of the potential limitations is the relatively low solubility of L-histidine in physiological saline, which requires larger amounts of liquid for injections of higher concentrations of the amino acid.
The effects of different amino acids on the pancreas are summarized in Table 1. These AP models have a well-defined, gradually progressive pancreatic necrosis, and associated lung injury. Therefore, they are suitable for addressing the molecular mechanisms and regenerative processes in necrotising AP. We used L-arginine, L-ornithine, and L-histidine and found significant differences in the mechanism of pancreatic injury induced by different basic amino acids (Zhang et al., 2018). Our data indicate that the metabolism of L-arginine to L-ornithine is involved in the pathogenesis of AP induced by L-arginine (Zhang et al., 2018). It is noteworthy that basic amino acids with aliphatic side chains (Figure 2B) appear to be more effective inducers of AP. It is well-known that hereditary diseases of branched chain amino acid metabolism will greatly increase the risk of AP in human beings (Lerch et al., 2006). Therefore, the model of AP induced by amino acids may also have links to human disease. However, it must be remembered that in clinical settings human AP caused by overdose of amino acids is rare (Saka et al., 2004).
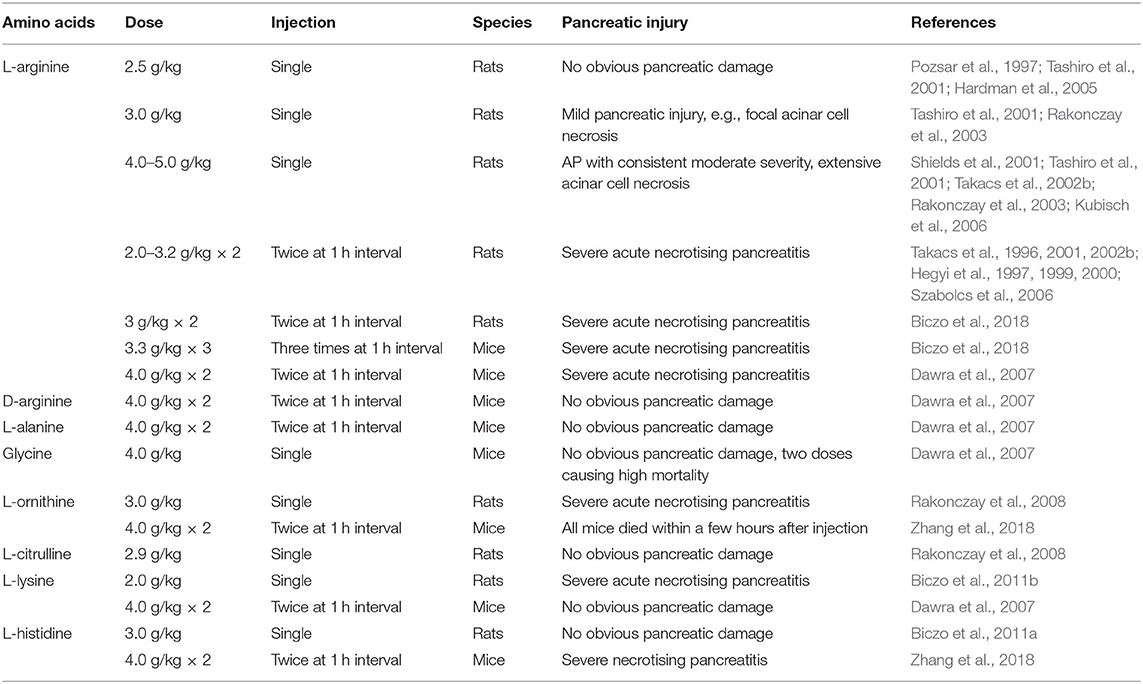
Table 1. Effect of different amino acids on the pancreas in vivo.
Ethanol and Free Fatty Acids
Alcohol is the second leading cause of AP worldwide (Petrov and Yadav, 2019). However, the mechanisms whereby ethanol exerts its deleterious effects are still relatively poorly understood (Clemens et al., 2016). Early animal experimental findings from acute ethanol administration via various routes have shown that alcohol increases pancreatic duct permeability, reduces pancreatic blood flow and microcirculation (Friedman et al., 1983; Foitzik et al., 1998), decreases pancreatic oxygen consumption (Foitzik et al., 1995), and induces oxidative stress (Weber et al., 1995). Whereas, ethanol alone fails to cause AP, which is in line with the human disease: since <5% alcoholics will develop clinical AP (Forsmark et al., 2016). Instead, it appears that the pancreas is sensitized to injury by alcohol consumption, and an additional factor trigger initiation of the alcohol-associated pancreatic injury. There is a dose-dependent sensitization of ethanol to CCK (or caerulein)- or bile acid-mediated PAC injury in vitro (Katz et al., 1996; Lu et al., 2002) and pancreatic damage in vivo (Foitzik et al., 1994; Andrzejewska et al., 1998; Pandol et al., 1999). A recent report showed that although alcohol feeding does not cause experimental AP, endoplasmic reticulum stress, and cell death of PACs can be triggered upon simultaneous exposure to ethanol and cigarette smoke (Lugea et al., 2017a). As a consequence, ethanol has been considered a key aggravating factor in the development of AP.
On the other hand, ethanol metabolites by both oxidative and non-oxidative pathways (Figure 3A) have been shown to cause a number of changes that can predispose the pancreas to AP (Laposata and Lange, 1986). The oxidative metabolism of ethanol is catalyzed by alcohol dehydrogenase and cytochrome P450 2E1, resulting in the production of reactive oxygen species and acetaldehyde (Wilson and Apte, 2003; Cederbaum, 2012). Oxidative metabolism of ethanol has also been shown to sensitize pancreatic mitochondria to activate mitochondrial permeability transition pore, leading to mitochondrial failure (Shalbueva et al., 2013).
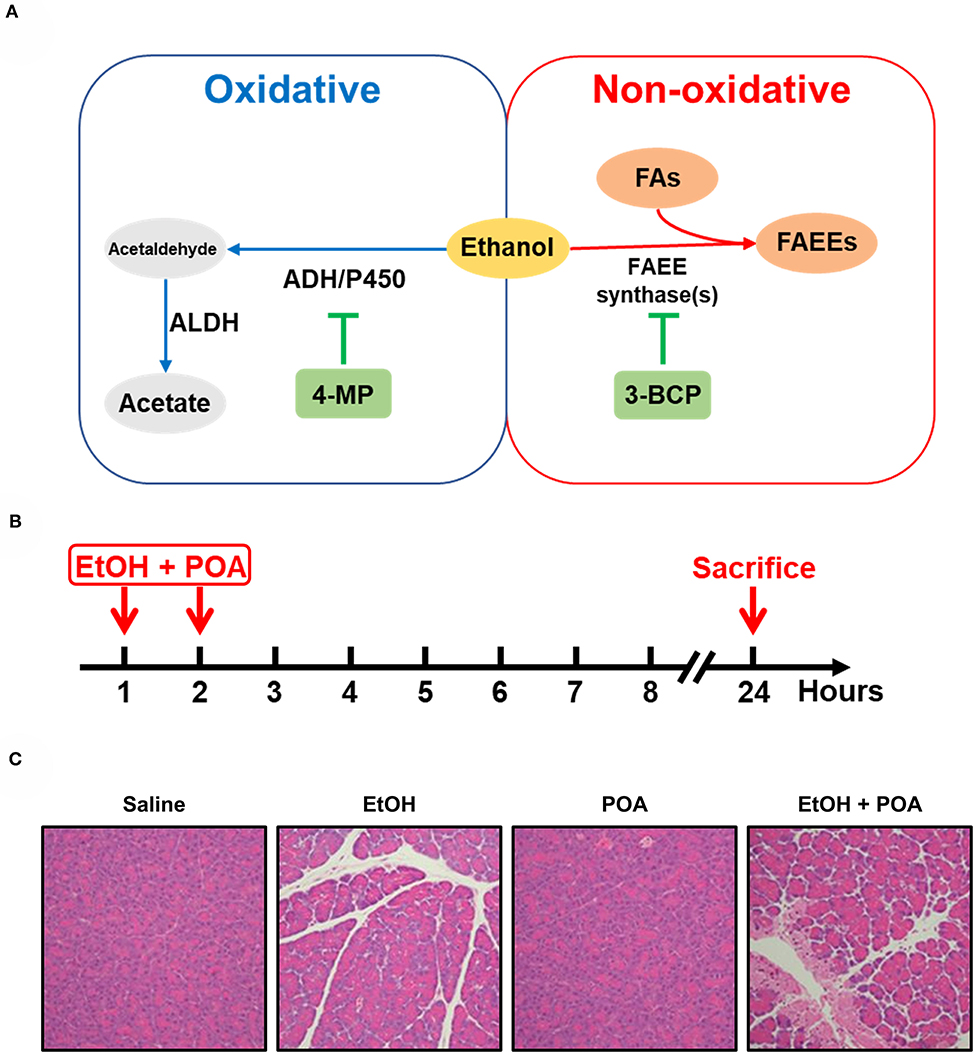
Figure 3. Ethanol metabolism and fatty acid ethyl ester-induced acute pancreatitis. (A) Non-oxidative and oxidative ethanol metabolism pathways. AHD, alcohol dehydrogenase; P450, cytochrome P450; ALDH, aldehyde dehydrogenase; 4-MP, 4-methylpyrazole; FAs, fatty acids; FAEEs, fatty acid ethyl esters; 3-BCP, 3-benzyl-6-chloro-2-pyrone. (B) A schematic of fatty acid ethyl ester-induced acute pancreatitis (FAEE-AP) in mice. The model was induced by concomitant administration of ethanol (1.35 g/kg) and palmitoleic acid (POA, 150 mg/kg). (C) Representative H&E images of pancreas histopathological slides of FAEE-AP (Huang et al., 2014).
Non-oxidative ethanol metabolism is accomplished by a diverse group of enzymes known as fatty acid ethyl ester (FAEE) synthases, which combines free fatty acids (FFAs) with ethanol generating FAEEs (Wilson and Apte, 2003; Zelner et al., 2013). Alcohol consumption is associated with elevated plasma triglycerides, impaired lipolysis, and increased FFAs fluxes from adipose tissue to the liver (Klop et al., 2013). And high concentrations of FFAs are noted in the serum and pancreatic necrosis debridement fluid of patients with AP (Sztefko and Panek, 2001; Navina et al., 2011; Patel et al., 2015; Noel et al., 2016). The pancreas is particularly rich in FAEE synthases, hence the relatively high accumulation of FAEEs in this organ (Laposata and Lange, 1986; Gukovskaya et al., 2002). Direct evidence that FAEEs cause some features of AP comes from Werner et al. (2002). They found that when an ethanol infusion was combined with inhibitors of the oxidative pathway of alcohol metabolism, the injury to the pancreas was exacerbated compared to ethanol alone, and the extent of pancreatic injury was dependent on the formation of FAEEs (Werner et al., 2002). This observation fits in with the previous post-mortem findings demonstrating the presence of high concentrations of FAEEs in the pancreas of patients with acute alcohol intoxication at the time of death (Ishii et al., 1986). Direct intravenous infusion of FAEEs also induces pancreatic oedema, pancreatic trypsinogen activation, and vacuolisation in the pancreas (Werner et al., 1997). Subsequently, in vitro data from our group have shown that FAEEs at relatively low concentrations (10–100 μM) cause prominent cytosolic Ca2+ rises, leading to the impairment of the mitochondrial functions and subsequent necrosis of PACs (Criddle et al., 2006).
Based on the available literature from in vitro and in vivo studies, we postulated that a more etiologically relevant model could be developed by concomitant administration of fatty acids and non-toxic concentrations of ethanol (Huang et al., 2014). To study the effect of FAEEs on AP initiation, we i.p. injected ethanol (1.35 g/kg) and palmitoleic acid (150 mg/kg), a monounsaturated fatty acid, for two times (Figure 3B), and successfully induced pancreatic injury in mice, including marked pancreatic oedema, neutrophil infiltration, and local necrosis (Figure 3C). This model also causes thickening of alveolar membrane and infiltration of inflammatory cells in the lung, but there is no or very little effect on the liver, kidney or heart (Huang et al., 2014). Other groups reproduced this model with different FFAs, such as ethanol (1.32 g/kg) and palmitoleic acid (2 mg/kg) in mice (Vigna et al., 2014), ethanol (1.75 g/kg), and palmitic acid (750 mg/kg) in mice (Maleth et al., 2015), ethanol (1.35 g/kg), and palmitoleic acid (2 or 150 mg/kg) in hamsters (Wang et al., 2016).
The lack of a widely accepted alcohol-induced AP model in pancreatology remains a drawback in the study of alcoholic AP. The existing animal models are to date the best research tools, amongst which the Lieber-DeCarli method (Bertola et al., 2013) is the most widely used experimental model to study alcoholic diseases in rodents. The fact that AP does not occur in all patients with alcohol abuse in a clinical environment strongly suggests other factors must play a role in determining individual susceptibility. Future models based on transgenic mice harboring genetic predisposing variants combined with the administration of alcohol or its metabolites may prove to be more useful.
Bile Acids
Although a variety of experimental AP models have been described, the clinical relevance of these models might be questioned due to the fact that they do not depend on the replication of events that are considered clinically triggering AP. One of such events is biliary reflux into the pancreas through the pancreatic duct, which is believed to be the most common cause of AP in humans (Lerch and Aghdassi, 2010).
After the pancreatic duct infusion-induced AP (PDI-AP) report by Bernard (1856), bile salts such as sodium chenodeoxycholate, sodium taurocholate (NaTC), sodium glycodeoxycholic acid, taurodeoxycholate, and taurolithocholic acid 3-sulfate (TLCS) have been used to induce PDI-AP in different species. In these models, pancreatic injury develops rapidly and is limited to the pancreatic head and body, while the pancreatic tail is much less affected. Since this model requires retrograde pancreatic duct injection, the degree of pancreatic damage is closely related to the pressure in the pancreatic duct, the type of inducer and the injection speed. Manual injection may not be steady enough, and micro-dose liquid medicine injection pump should be used instead. The frequently used PDI-AP animal models induced by bile acids in rodents are summarized in Table 2 and the recent advances have been reviewed elsewhere (Wan et al., 2012).
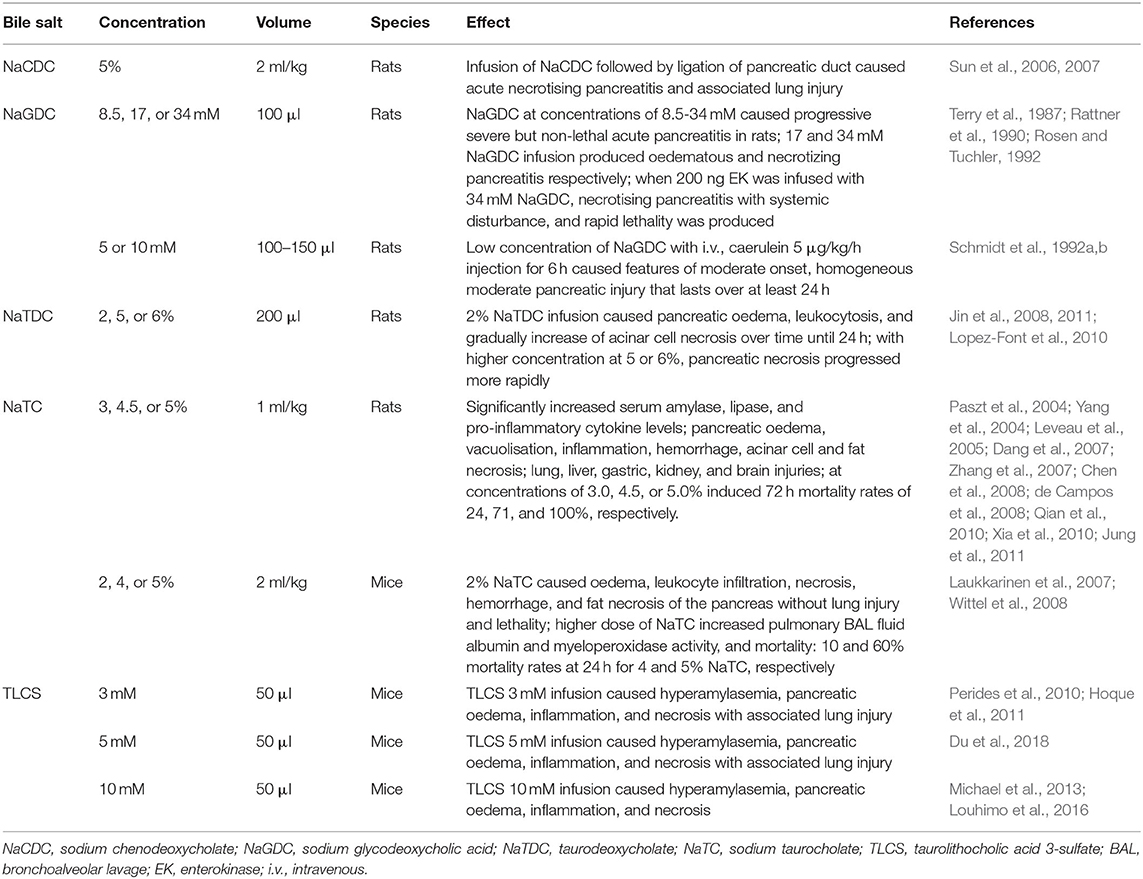
Table 2. Bile salt-induced experimental acute pancreatitis models and frequently used protocols in rodents.
Among these bile salts, the taurine-conjugated bile salt NaTC is the most widely used and best characterized thus far for inducing PDI-AP. The rat model of NaTC infusion provides a well-defined tool to research MODS in severe AP, mirroring the human condition. This model has high mortality rates of 24–100% (Silva-Vaz et al., 2019) and the mortality increases with the amount of NaTC injected. Similarly to the caerulein/LPS model, this model is suitable for studying bacterial translocation when combined with LPS (Yamanel et al., 2005). When the NaTC/LPS model is supplemented with trypsin, even more severe MODS is produced (Yamano et al., 1998).
A non-lethal PDI-AP model (Laukkarinen et al., 2007) produced necrotising AP in the head of the pancreas without associated lung injury. This validated mouse model (Wittel et al., 2008; Ziegler et al., 2011) allows for standardizing pancreas histopathological stimuli on many levels from the isolated mitochondria (Odinokova et al., 2009) to whole animal (Mareninova et al., 2009), especially in genetically engineered mice (Mukherjee et al., 2016). Importantly, TLCS which has been extensively characterized in vitro, is generally preferred over NaTC for inducing PDI-AP, especially after the identification of the G protein-coupled bile acid receptor1 (Gpbar1) present on the apical pole of PACs (Perides et al., 2010). Mice lacking this receptor (Gpbar1−/−) were completely protected against AP induced by TLCS in vivo as well as treatment with 500 μM TLCS of PACs isolated from these mice did not result in pathophysiological Ca2+ responses, intrapancreatic trypsinogen activation, and cell death that are normally seen in wild type PACs (Perides et al., 2010). Subsequently, a variety of AP signaling pathways, including intracellular Ca2+ overload, have been verified on this model. The severity of this model is similar to that of human diseases. Paradoxically, while this is an excellent biliary AP model, it is not an ideal AP-MODS model because it requires surgical operation.
Pancreatic Duct Ligation
The pancreatic duct ligation AP (PDL-AP) model is an attempt to experimentally recreate the “common channel,” or the common biliopancreatic duct obstruction, postulated by Opie (1901) as the mechanistic explanation for biliary AP following a gallstone lodging at the Ampulla of Vater, causing bile reflux along the pancreatic duct. Churg and Richter (1971) first linked this model to changes in the pancreatic exocrine function. Meyerholz and Samuel (2007) demonstrated that early changes in the duct and PACs can be observed at 1–5 h after ductal ligation, and 24 and 48 h were the best time points to capture interstitial oedema and inflammatory changes of the pancreas. Therefore, several researchers use this model in rats to investigate the early stages of the disease pathogenesis (Lerch et al., 1992). Studies have shown that apoptosis is the main mechanism of cell death in the rat PDL-AP model (Walker et al., 1992).
Recent advances (Samuel et al., 2010; Yuan et al., 2011; Le et al., 2015) allow the application of PDL techniques in mice (Meyerholz et al., 2008). PDL in mice causes AP with systemic inflammation and MODS, whereas biliary duct ligation and sham surgery does not (Samuel et al., 2010). The 4-day mortality of mice in the PDL group was shown to be 100%, whereas no mortality occurred in the sham operation and biliary duct ligation groups at 15 days (Samuel et al., 2010). This model appears to mimic gallstone obstruction-induced AP with a high mortality, thus it could potentially be used for investigation of the pathogenesis of severe AP and testing new therapies. The PDL-AP model may also be suitable for studying bacterial disorders, because biliary obstruction is associated with intestinal bacterial overgrowth and translocation (Nieuwenhuijs et al., 2000). The PDL-AP model has the advantage of avoiding of the systemic application of chemical inducers and thus undesirable side effects, but it requires surgery, which can be challenging particularly in small animals.
Endoscopy
Since its first description in the late 1960s as a diagnostic technique (McCune et al., 1968), endoscopic retrograde cholangiopancreatography (ERCP) has evolved over the years to a predominantly therapeutic tool (Cotton, 2012). Compared with other endoscopic examinations, ERCP carries a higher potential for complications (Andriulli et al., 2007). Post-ERCP pancreatitis (PEP) is one of the most frequent complications of ERCP with an incidence of 1.5–15% (Parekh et al., 2017). However, the exact etiology as to why AP develops in some patients is unknown, some risk factors are gender, age, physician experience, and previous history of PEP (Radadiya et al., 2019). There are several potential underlying mechanisms of pancreatic injury during ERCP, including mechanical, thermal, chemical, hydrostatic, enzymatic, and microbiologic insults (Parekh et al., 2017).
Early in 1979, Kivisaari et al. imitated the process of ERCP by retrograde infusion of meglumine in various concentrations for 30 s at a pressure of 20 to 50 mmHg, which proved sufficient to produce evidence of both gross and microscopic AP in rats after 4 days such as oedema, leukocytosis, necrosis, and hemorrhage, atrophy, and early fibrosis (Kivisaari, 1979). Since then, different drugs, including interleukin-10, gabexate mesylate, heparin or somatostatin, nitrate derivates or diclofenac have been tested to reduce the incidence, and severity of PEP (He et al., 2003; Hackert et al., 2004; Folch-Puy et al., 2006; Xiong et al., 2007; Haciahmetoglu et al., 2008; Jin et al., 2015). He et al. described a model of PEP based on the hypothesis that a constant, relatively high pressure of an intraductal injection should cause AP through disruption of the ductal integrity (He et al., 2003). Contributing factors to the injury may include chemical toxicity from the contrast agent, disruption of pancreatic ducts, and even a rupture of acinar lobules as a result of forceful injection of contrast material. Increased pressure within the pancreatic duct has been indirectly implicated as a cause of PEP as multiple studies have shown that placement of a pancreatic stent following ERCP in high-risk patients reduced the incidence of PEP (Freeman and Guda, 2004). The model was carried out in rats by retrograde infusion of meglumine into the common biliopancreatic duct at a high pressure (50 mmHg) thus inducing typical histopathological changes of AP with significant increases of serum amylase and pancreatic myeloperoxidase activity at 24 h (He et al., 2003; Xiong et al., 2007). The model mimicked the procedure of ERCP with meglumine manifesting as edema, inflammation, necrosis, and hemorrhage. This was consistent with clinical PEP, which usually is relatively mild. However, the role of applied pressure alone has been debated after Hackert et al. (2004) and Folch-Puy et al. (2006) suggested that the contrast itself may be playing a role in the development of PEP. Hackert's work showed the inflammatory reaction develops in the pancreatic tissue when duct ligation is combined with intraductal infusion of the contrast medium (Merriam et al., 1996; Hackert et al., 2004).
In addition, other studies show that intraductal regulation of pH (Noble et al., 2008) and the mechanical damage caused by direct papillary trauma (Bozkurt et al., 2013) were found to affect the onset of AP, and might be an important factor in PEP. What is more, some studies show that no correlation was detected with increasing pressure or with the use of contrast agent (Haciahmetoglu et al., 2008). Recently, Jin et al. (2015) and Wen et al. (2018) have developed a novel model of PEP in mice. Their major benefit is the ability to complement pharmacological inhibition of calcineurin along with the use of calcineurin knockouts (Wen et al., 2018, 2020). Besides, pancreatic injury after retrograde injection of contrast material has also been described in larger animals such as dogs. The frequently used PEP animal models induced by different pressure and contrast in rodents are summarized in Table 3. These models ultimately represent a combination of pancreatic ductal pressure and exposure to contrast agents, both potentiating outcomes (Figure 4).
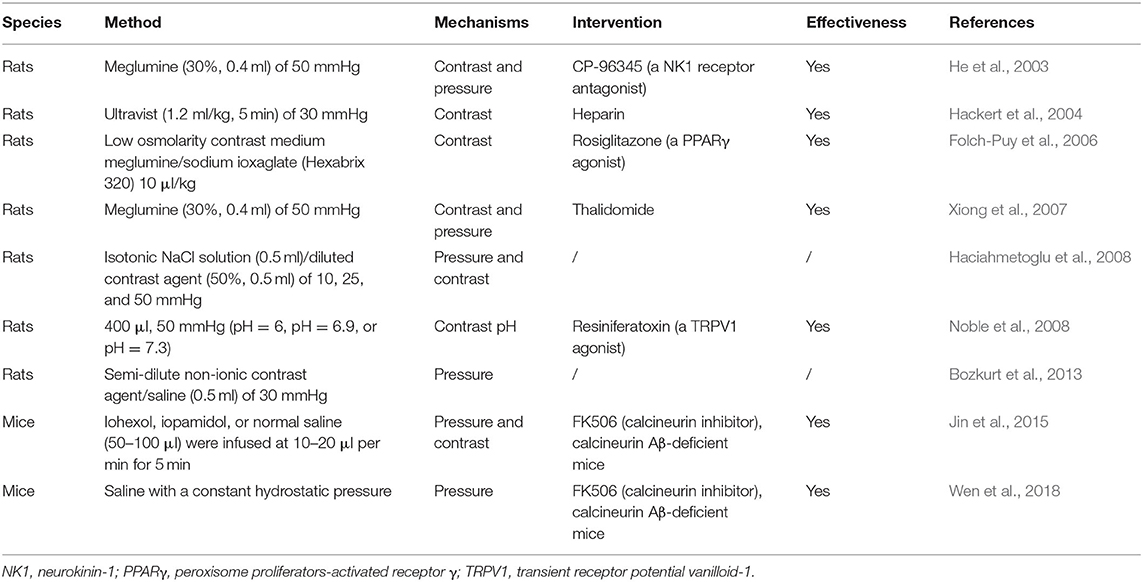
Table 3. Post-ERCP pancreatitis models.
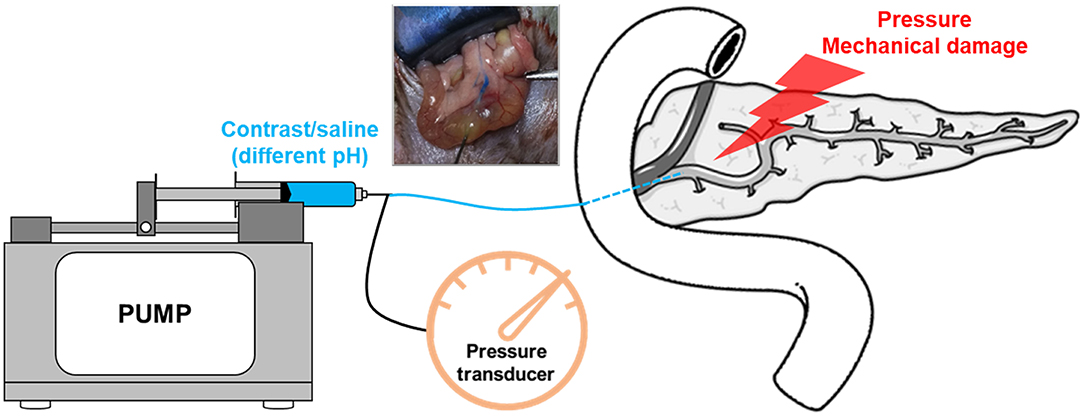
Figure 4. A schematic of experimental post-ERCP pancreatitis model. The model is induced by performing retrograde biliopancreatic ductal infusion, and the pump is used to control rate or pressure. Intraductal pressures can be monitored using a pressure transducer.
Other Experimental AP Models
In recent years, pancreatic genetics has made great progress, and the identification of mutations in genes involved in the trypsin-dependent pathway is a significant milestone in understanding pancreatitis onset (Mayerle et al., 2019). Genetic experimental animal models, based on genetic techniques (transgenic, knock out, knock in, or knock down), have provided in vivo compelling evidence of concept of premature intrapancreatic activation of digestive proteases (Geisz and Sahin-Toth, 2018; Jancso et al., 2018; Sendler et al., 2018; Geisz et al., 2019; Gui et al., 2020; Jancso and Sahin-Toth, 2020). For example, an innovative new model has recently been developed based on the T7D23A knock-in mouse, which carries a heterozygous p.D23A mutation in mouse cationic trypsinogen (isoform T7) (Geisz and Sahin-Toth, 2018). T7D23A mice develop spontaneous AP with pancreatic edema, inflammation, and necrosis with serum amylase elevation at an early age progressing to features of chronic pancreatitis, with clinical relevance to hereditary pancreatitis (Geisz and Sahin-Toth, 2018). Using genetically manipulated mice, the mis-folding–dependent, ductal, NF-κB, and cytokine signaling pathways have also been extensively studied (Bi and Ji, 2016; Mayerle et al., 2019).
Other AP models that have been reported in the literature generally have not gained prominence due to their respective limitations. Choline-deficient, 0.5% DL-ethionine supplemented diet-induced AP was commonly used before, but it is gradually eliminated because of some limited conditions (sex, age, and non-specific pancreatic toxicity) and clinical relevance (Lombardi and Rao, 1975). The immune-induced model may represent immune factor-induced AP in humans, however, the difficulty to set it up, its limited reproducibility and other limitations have thus far hampered its use (Janigan et al., 1975; Seelig and Seelig, 1975; Nevalainen et al., 1977; Nevalainen, 1978; Jancar et al., 1988; Letko et al., 1990; Kanno et al., 1992; Axelsson et al., 2008). A haemolysis-induced model has been used to mimic AP patients under haemolysis (Saruc et al., 2003). Dibutyltin dichloride- (Merkord and Hennighausen, 1989) and coxsackievirus- (De Palma et al., 2008, 2009) induced models are also not very widely used due to the lack of major clinical relevance. As for drug-induced AP (Forsmark et al., 2016; Meczker et al., 2020), different responses to drugs between animals and humans make this particularly difficult to model.
In vitro and Ex vivo Models
In vitro and Ex vivo
Besides animal models, in vitro experimental AP models primary PACs, acinar carcinoma cell lines AR42J (rat) (Lugea et al., 2017a) and 266-6 (mouse) (Siveke et al., 2008), and isolated pancreatic lobules (Won et al., 2011) are often used. In 1978, Williams et al. (1978) first developed a technique to isolate PACs and tested the effects of supramaximal doses of CCK and caerulein. Mouse PACs express CCK receptors, of both high and low affinity, which can be activated by low and high concentrations of caerulein/CCK further activating intracellular signaling pathways (summarized in Supplementary Table 1) (Williams, 2001, 2002). Low physiological doses of CCK (or caerulein) bind to high-affinity CCK receptors and mediate pancreatic secretion and growth, whereas high or supra-physiological doses (a concentration exceeding the dose at which maximal amylase secretion is observed) bind to low affinity receptors and inhibit pancreatic secretion, resulting in zymogen activation and PAC injury (Williams, 2001, 2002; Saluja et al., 2007). Grady et al. (1998) and Saluja et al. (1999) reported that no enzyme activation occurred at CCK concentrations below 10−10 M, whereas supramaximal stimulation by CCK (concentration >10−10 M) results in zymogen activation and PACs injury. Although CCK receptors are abundant in mouse PACs, it is controversial whether CCK receptors exist in human PACs. In 2001, Ji et al. reported that CCK does not cause any changes in human PAC function as was assessed by Ca2+ release, amylase secretion, and ERK phosphorylation (Ji et al., 2001). Subsequently, some researchers have reported that CCK can cause Ca2+ oscillation and stimulate enzyme secretion in human PACs (Murphy et al., 2008; Wen et al., 2015; Mukherjee et al., 2016; Lugea et al., 2017a,b; Waldron et al., 2019). We have summarized some key information about these studies in Supplementary Table 2.
Subsequent studies carried out in animal models that simulate the human disease suggested that the PACs were the initial site of morphological damage (Lerch et al., 1992). The latest reviews summarizes the effects of CCK on PACs (Williams, 2019) and early acinar events in the pathogenesis of AP (Saluja et al., 2019). The most notable limitation of primary PACs is their in vitro viability is relatively short and thus they cannot be used for long-term experiments or subculturing, an advantage that acinar carcinoma cell lines can offer. However, one must be note that while acinar carcinoma cell lines retain some phenotypes of primary PACs (i.e., containing a multitude of digestive enzyme mRNAs and responding to CCK), they may have changed enzyme activities and receptors or start to express other specific receptors.
In contrast to the methods that yield single PACs, isolation of the pancreatic lobules (Won et al., 2011; Gryshchenko et al., 2016) offers a more physiological ex vivo model to study signaling events in the exocrine pancreatic tissue. Since the lobules preserve most of the spatial characteristics of the intact acini and contain non-PACs such as such as pancreatic stellate cells (PSCs), they can be used to study the role of interactions between different cell types in the development AP. Lobules of size up to 1 mm3 may be manually cut from a saline-injected mouse pancreas (Won et al., 2011), whereas much smaller lobules can be isolated from the organ using a modified protocol of collagenase digestion (Ferdek et al., 2016; Gryshchenko et al., 2016; Jakubowska et al., 2016). Loading the lobules with fluorescent Ca2+ indicators has made it possible to investigate how different populations of pancreatic cells orchestrate physiological and pathological Ca2+ signals (Won et al., 2011; Ferdek et al., 2016; Gryshchenko et al., 2016; Jakubowska et al., 2016). This model was successfully used to show that Ca2+ signal evoked in cells of one phenotype is not transmitted to different cells (Won et al., 2011). Using the multiphoton imaging and Fluo-4 AM Ca2+ indicator, Won et al. (2011) confirmed that, even in a close vicinity, different cell populations may form entirely separated signaling units. Gryshchenko et al. (2016) later confirmed that blockade of the bradykinin B2 receptor with antagonist WIN64338 reduces PAC necrosis in the lobules, elicited by common toxins of the pancreas: ethanol, FAEE, or bile acids. Further analyses of the lobular signaling pointed toward more toxic effects of certain bile acids in PSCs than in PACs (Ferdek et al., 2016): this study showed that sodium bile salts, cholate, and taurocholate, elicit noxious Ca2+ signals in PSCs, and induce considerable levels of PSC necrosis in the lobules, whereas TLCS mainly induces pathological Ca2+ signals and necrotic cell death in PACs. Importantly, development of the real-time method to simultaneously record Ca2+ signals (Fura-2 indicator) and nitric oxide signals (DAF-2 fluorescent dye) in the lobules, confirmed the interplay between these signaling pathways solely in PSCs, and their absence in PACs, in response to stimulation of the lobules with bradykinin (Jakubowska et al., 2016). PSCs generated Ca2+ responses in form of Ca2+ oscillations or transients, accompanied by the intracellular increase in nitric oxide, in response to bradykinin, cholate, and taurocholate, but not TLCS. In a mouse model of alcohol-induced AP, lobular PSCs were desensitized to the signaling mediated by bradykinin B2 receptor, but sensitized to the signaling mediated by B1 receptor (Gryshchenko et al., 2018). This sensitivity switch is a general feature of inflammatory diseases (Petho and Reeh, 2012). The aforementioned studies demonstrate that pancreatic lobules are a valuable alternative to single cell type in vitro models, particularly in the early stages of investigation. This model could potentially be developed further to investigate signaling events in pancreatic lobules of human origin.
Normal exocrine functions of the pancreas center around pancreatic juice secretion (Hegyi et al., 2011; Hegyi and Petersen, 2013). In order to form this enzyme-rich alkaline fluid, cells of the acinar compartment cooperate with the epithelia of pancreatic ducts. PACs secrete enzymes capable of hydrolysis of the proteins, polysaccharides, lipids, and nucleic acids, whereas pancreatic ductal cells (PDCs) release isotonic solution rich in bicarbonate that aids the transport of the enzymes into the duodenum and neutralizes the gastric acid (Lee et al., 2012). Not only PACs but also PDCs play their roles in the development of AP. For example, obstruction of the pancreatic duct can influence the trafficking of PAC membrane (Lerch et al., 1995a); and inducers of AP, such as bile acids, reduce the intracellular pH of PDCs (Venglovecz et al., 2008). The technique of PAC isolation via enzymatic digestion of the pancreas has been widely applied in the past few decades to study the physiology and pathophysiology of these cells (Thorn et al., 1993). Similarly, the protocol of pancreatic duct isolation was initially developed in the 80's and it is also based on enzymatic digestion of the tissue (Argent et al., 1986). However, this enzymatic processing could affect the physiology of ductal epithelia and thus may require additional steps facilitating ductal regeneration, which extends and complicates the entire procedure (Gal et al., 2019). In order to address this limitation, Gal et al. recently proposed a novel in situ approach that aims to preserve the physiological environment and the ductal structure of the mouse pancreatic tissue (Gal et al., 2019). By injecting (post-mortem) low melting point agarose into the pancreatic duct and a subsequent cooling of the whole organ, agarose settles in the pancreatic ductal system and helps to maintain its three dimensional architecture. Importantly, by using a vibratome, slices of the agarose-reinforced pancreatic tissue can be cut and used for the subsequent analyses, such as measurements of Ca2+ signals in real-time (Gal et al., 2019). The authors showed that treatment of PDCs with 1 mM chenodeoxycholic acid evokes robust transient increases in intracellular Ca2+ and triggers cell movement that precedes the development of Ca2+ responses in these cells (Gal et al., 2019). This new in situ model is likely to be a very useful tool for testing the effects of the common inducers of AP on PDCs and may add to our knowledge about the roles these cells play in pancreatic pathologies (Supplementary Table 3).
While models of the isolated mouse pancreatic ducts offer means for studying the functional characteristics of the epithelial surfaces, they are not designed to investigate ducts as structures that form fluid-filled cavities. This potential limitation can be addressed by employing pancreatic ductal organoid cultures (Boj et al., 2015) for analyzing ion secretion (Molnar et al., 2020). Ductal epithelia can be grown as organoids with a hollow center (the lumen), which preserves the planar cell polarity and the physiological pattern of the membrane transporters. In order to measure real time changes in the intraluminal pH, concentration of chloride anions or the processes of bicarbonate secretion, the lumen can be injected with an appropriate fluorescent indicator (Molnar et al., 2020). In the near future, organoid cultures might aid the preclinical studies on the disrupted physiological processes that drive the development of pancreatic disorders.
Comparison of Commonly Used Models in Rodents
Not all animal models of AP developed in the past are equally popular among researchers today. It is very difficult to compare models of AP due to the differences between animals and strains used, the microbiome and the environmental cues, or reagents/experimental conditions. There is a growing trend toward the use of PDI-AP and PDL-AP models particularly in genetic studies. L-arginine and other amino acids-induced animal models in rodents are now undergoing thorough investigation. Current animal models of AP are classified for severity on the basis of an inducer/etiology that causes pancreatic necrosis (Lerch and Gorelick, 2013). In contrast, the presence of necrosis in the human disease does not automatically translate into poorer outcomes. This is a significant limitation since severity in human AP is unrelated to pancreatic necrosis or etiology, with the exception of hypertriglyceridemia-associated AP. The comparison of currently used models is summarized in Table 4.
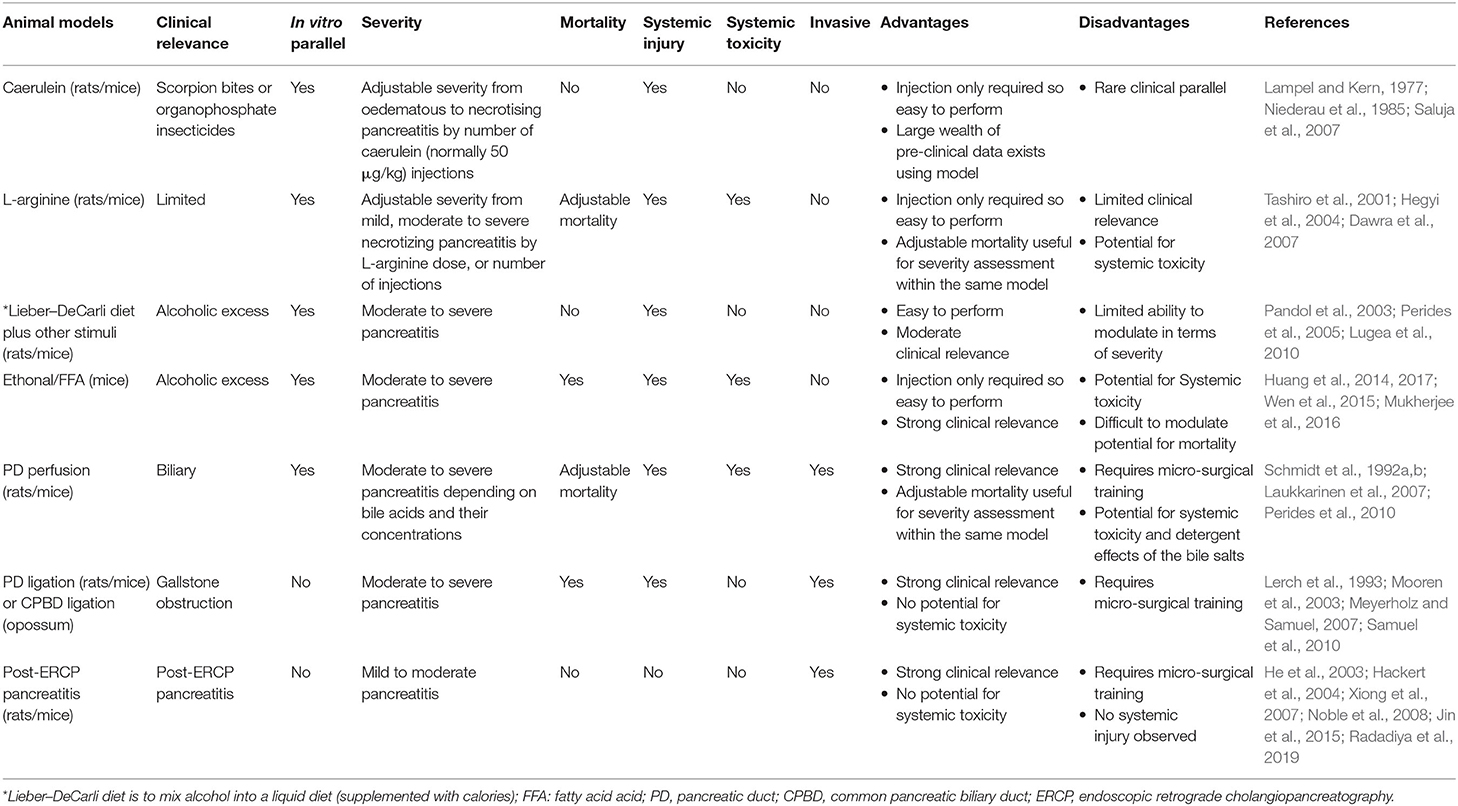
Table 4. Comparison of commonly used acute pancreatitis animal models in rodents.
Experimental AP Models in Translational Research
In recent years, there has been an increasing number of medical studies in the pancreatic field (Mukherjee et al., 2019). Animal models play here an indispensable role by bridging basic science with translational research. Animal models allow detailed investigation of the crucial events in the pathophysiology of the disease and are thus important in establishing causality. The names, targets, models used and whether they are effective for system damage caused by AP are summarized in Table 5. However, it pays to remember that even very promising findings made on animal models, particularly those regarding novel therapeutic agents, may not always show efficacy in clinical trials (Moggia et al., 2017). The latter caveat may be improved by understanding critical thresholds for cellular events (Barreto et al., 2020) and assessing potential pharmacological therapeutics in different models including in vivo and in vitro with multiple biochemical, immunological and histopathological indices, before designing appropriate clinical trials. Some drugs with good curative effect on animal models that successfully transformed into clinical trials are shown in Table 6. It is also crucial to understand that in animal studies the therapeutic agent is most often administered prophylactically i.e., before or simultaneously with the administration of AP. However, in majority of clinical trials treatment can only be instituted after the onset of symptoms. Hence the main focus of animal studies has been toward understanding the mechanistic pathways involved in the pathogenesis of disease. To date, there is no perfect model that shares all typical characteristics of human AP and the failure of anti-inflammatory (Kingsnorth et al., 1995; Kingsnorth, 1996; Johnson et al., 2001), antioxidants (Siriwardena et al., 2007) and antibiotics (Dellinger et al., 2007; Bai et al., 2008; Garcia-Barrasa et al., 2009) to ameliorate AP in human clinical trials, despite their success in animal models, demonstrates well the difficulties in translating the results from animal studies to the clinic. Therefore, there is a long way to go from animal experiment to clinical transformation. Considering these factors it is vital that looking to the future we embrace advances in cellular technologies and in particular human organoids that may provide improved and more representative models (Kim et al., 2020).
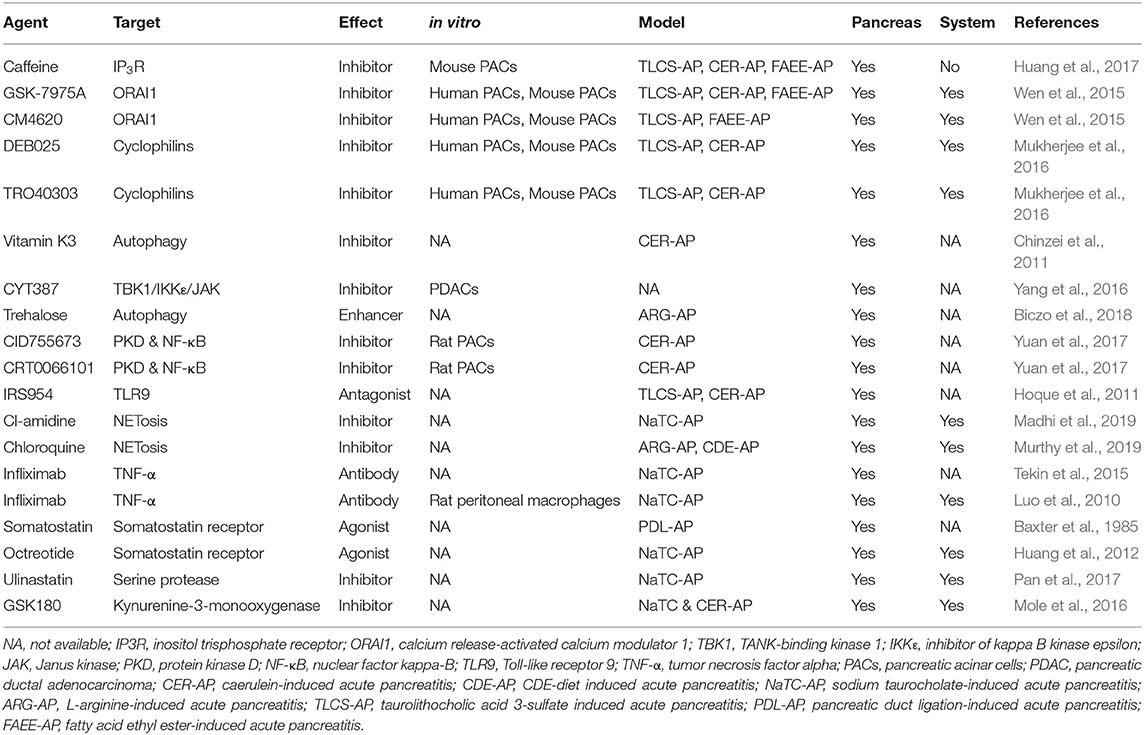
Table 5. Pharmacological therapy tested in pre-clinical AP models.
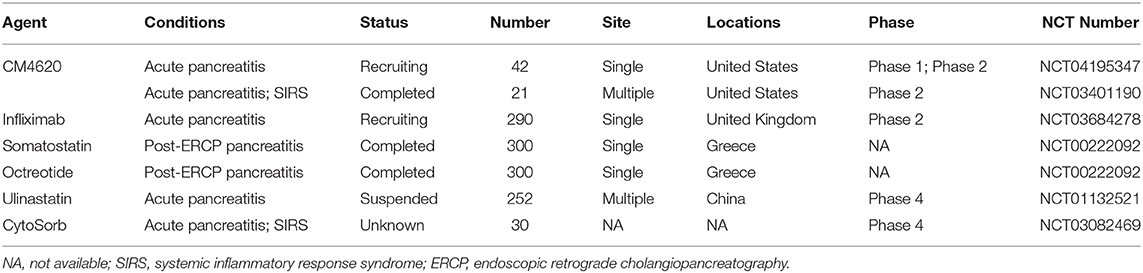
Table 6. Pharmacological therapy for AP in currently clinical trials.
Summary
Animal models of AP, whether they are invasive or non-invasive, carried out on large or small animals, wild type or transgenic animals, have provided and continue to provide key insights into the etiology and pathogenesis of AP as well as aid the identification of new therapeutic targets or biomarkers useful for the treatment of the disease. They remain an indispensable tool for the study of AP. As our understanding of the disease continues to improve, it is likely that new and more relevant models will be developed in the near future.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
XY and LY performed literature search and drafted the manuscript. PF, MJ, and RM reviewed and critically revised the manuscript. XF and QX obtained funding and had important intelligent input. WH obtained funding, conceptualized the study, supervised students, drafted, and critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Nature Science Foundation of China (Grant No. 81973632, WH; Grant No. 81774120, QX; Grant No. 81970561, XF) and the Ministry of Science and Technology of China (Grant No. 2018ZX09201018-005, XF); the National Science Center of Poland (Narodowe Centrum Nauki, NCN, No. 2019/33/B/NZ3/02578, PF), HOMING Programme of the Foundation for Polish Science (Fundacja na rzecz Nauki Polskiej, FNP, No. HOMING/2017-4/31, PF; No, HOMING/2017-3/23, MJ), co-financed by the European Union under the European Regional Development Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.614591/full#supplementary-material
References
Adler, G., Gerhards, G., Schick, J., Rohr, G., and Kern, H. F. (1983). Effects of in vivo cholinergic stimulation of rat exocrine pancreas. Am. J. Physiol. 244, G623–629. doi: 10.1152/ajpgi.1983.244.6.G623
Anastasi, A., Erspamer, V., and Endean, R. (1968). Isolation and amino acid sequence of caerulein, the active decapeptide of the skin of hyla caerulea. Arch. Biochem. Biophys. 125, 57–68. doi: 10.1016/0003-9861(68)90638-3
Andriulli, A., Loperfido, S., Napolitano, G., Niro, G., Valvano, M. R., Spirito, F., et al. (2007). Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am. J. Gastroenterol. 102, 1781–1788. doi: 10.1111/j.1572-0241.2007.01279.x
Andrzejewska, A., Dlugosz, J. W., and Jurkowska, G. (1998). The effect of antecedent acute ethanol ingestion on the pancreas ultrastructure in taurocholate pancreatitis in rats. Exp. Mol. Pathol. 65, 64–77. doi: 10.1006/exmp.1998.2226
Argent, B. E., Arkle, S., Cullen, M. J., and Green, R. (1986). Morphological, biochemical and secretory studies on rat pancreatic ducts maintained in tissue culture. Q. J. Exp. Physiol. 71, 633–648. doi: 10.1113/expphysiol.1986.sp003023
Axelsson, J., Norrman, G., Malmstrom, A., Westrom, B., and Andersson, R. (2008). Initiation of acute pancreatitis by heparan sulphate in the rat. Scand. J. Gastroenterol. 43, 480–489. doi: 10.1080/00365520701733814
Bai, Y., Gao, J., Zou, D. W., and Li, Z. S. (2008). Prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in acute necrotizing pancreatitis: evidence from a meta-analysis of randomized controlled trials. Am. J. Gastroenterol. 103, 104–110. doi: 10.1111/j.1572-0241.2007.01575.x
Barreto, S. G., Habtezion, A., Gukovskaya, A., Lugea, A., Jeon, C., Yadav, D., et al. (2020). Critical thresholds: key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut 1–10. doi: 10.1136/gutjnl-2020-322163
Baxter, J. N., Jenkins, S. A., Day, D. W., Roberts, N. B., Cowell, D. C., Mackie, C. R., et al. (1985). Effects of somatostatin and a long-acting somatostatin analogue on the prevention and treatment of experimentally induced acute pancreatitis in the rat. Br. J. Surg. 72, 382–385. doi: 10.1002/bjs.1800720516
Bertola, A., Mathews, S., Ki, S. H., Wang, H., and Gao, B. (2013). Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637. doi: 10.1038/nprot.2013.032
Bi, Y., and Ji, B. (2016). Spontaneous Pancreatitis in Genetically Modified Animal Strains. Pancreapedia: Exocrine Pancreas Knowledge Base. doi: 10.3998/panc.2016.8
Biczo, G., Hegyi, P., Dosa, S., Balla, Z., Venglovecz, V., Ivanyi, B., et al. (2011a). Aliphatic, but not imidazole, basic amino acids cause severe acute necrotizing pancreatitis in rats. Pancreas 40, 486–487. doi: 10.1097/MPA.0b013e31820a598a
Biczo, G., Hegyi, P., Dosa, S., Shalbuyeva, N., Berczi, S., Sinervirta, R., et al. (2011b). The crucial role of early mitochondrial injury in L-lysine-induced acute pancreatitis. Antioxid. Redox Signal. 15, 2669–2681. doi: 10.1089/ars.2011.4065
Biczo, G., Hegyi, P., Sinervirta, R., Berczi, S., Dosa, S., Siska, A., et al. (2010). Characterization of polyamine homeostasis in l-ornithine-induced acute pancreatitis in rats. Pancreas 39, 1047–1056. doi: 10.1097/MPA.0b013e3181d3cdf0
Biczo, G., Vegh, E. T., Shalbueva, N., Mareninova, O. A., Elperin, J., Lotshaw, E., et al. (2018). Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 154, 689–703. doi: 10.1053/j.gastro.2017.10.012
Bieger, W., Martin-Achard, A., Bassler, M., and Kern, H. F. (1976a). Studies on intracellular transport of secretory proteins in the rat exocrine pancreas. IV. Stimulation by in vivo infusion of caerulein. Cell Tissue Res. 165, 435–453. doi: 10.1007/BF00224474
Bieger, W., Seybold, J., and Kern, H. F. (1976b). Studies on intracellular transport of secretory proteins in the rat exocrine pancreas. V. Kinetic studies on accelerated transport following caerulein infusion in vivo. Cell Tissue Res. 170, 203–219. doi: 10.1007/BF00224299
Boj, S. F., Hwang, C. I., Baker, L. A., Chio, I. I., Engle, D. D., Corbo, V., et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. doi: 10.1016/j.cell.2014.12.021
Bozkurt, S., Guner, A., Kadioglu, H., Kece, C., Reis, E., and Coskun, H. (2013). The effects of different mechanisms on the development of post-ERCP pancreatitis in an ERCP model in rats. Turk. J. Gastroenterol. 24, 469–475. doi: 10.4318/tjg.2013.0654
Cederbaum, A. I. (2012). Alcohol metabolism. Clin. Liver Dis. 16, 667–685. doi: 10.1016/j.cld.2012.08.002
Chao, K. C., Chao, K. F., Chuang, C. C., and Liu, S. H. (2006). Blockade of interleukin 6 accelerates acinar cell apoptosis and attenuates experimental acute pancreatitis in vivo. Br. J. Surg. 93, 332–338. doi: 10.1002/bjs.5251
Chen, X., Li, S. L., Wu, T., and Liu, J. D. (2008). Proteasome inhibitor ameliorates severe acute pancreatitis and associated lung injury of rats. World J. Gastroenterol. 14, 3249–3253. doi: 10.3748/wjg.14.3249
Chinzei, R., Masuda, A., Nishiumi, S., Nishida, M., Onoyama, M., Sanuki, T., et al. (2011). Vitamin K3 attenuates cerulein-induced acute pancreatitis through inhibition of the autophagic pathway. Pancreas 40, 84–94. doi: 10.1097/MPA.0b013e3181f69fc9
Churg, A., and Richter, W. R. (1971). Early changes in the exocrine pancreas of the dog and rat after ligation of the pancreatic duct. A light and electron microscopic study. Am. J. Pathol. 63, 521–546.
Clemens, D. L., Schneider, K. J., Arkfeld, C. K., Grode, J. R., Wells, M. A., and Singh, S. (2016). Alcoholic pancreatitis: new insights into the pathogenesis and treatment. World J. Gastrointest. Pathophysiol. 7, 48–58. doi: 10.4291/wjgp.v7.i1.48
Cotton, P. B. (2012). Endoscopic retrograde cholangiopancreatography: maximizing benefits and minimizing risks. Gastrointest. Endosc. Clin. N. Am. 22, 587–599. doi: 10.1016/j.giec.2012.05.002
Criddle, D. N., Booth, D. M., Mukherjee, R., McLaughlin, E., Green, G. M., Sutton, R., et al. (2009). Cholecystokinin-58 and cholecystokinin-8 exhibit similar actions on calcium signaling, zymogen secretion, and cell fate in murine pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G1085–1092. doi: 10.1152/ajpgi.00119.2009
Criddle, D. N., Murphy, J., Fistetto, G., Barrow, S., Tepikin, A. V., Neoptolemos, J. P., et al. (2006). Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130, 781–793. doi: 10.1053/j.gastro.2005.12.031
Czako, L., Takacs, T., Varga, I. S., Hai, D. Q., Tiszlavicz, L., Hegyi, P., et al. (2000). The pathogenesis of L-arginine-induced acute necrotizing pancreatitis: inflammatory mediators and endogenous cholecystokinin. J. Physiol. Paris 94, 43–50. doi: 10.1016/S0928-4257(99)00104-7
Czako, L., Takacs, T., Varga, I. S., Tiszlavicz, L., Hai, D. Q., Hegyi, P., et al. (1998). Involvement of oxygen-derived free radicals in L-arginine-induced acute pancreatitis. Dig. Dis. Sci. 43, 1770–1777. doi: 10.1023/A:1018839821176
Dang, S. C., Zhang, J. X., Qu, J. G., Wang, X. Q., and Fan, X. (2007). Ligustrazine alleviates gastric mucosal injury in a rat model of acute necrotizing pancreatitis. Hepatobiliary Pancreat Dis. Int. 6, 213–218. doi: 10.1111/j.1523-5378.2007.00489.x
Dawra, R., and Saluja, A. K. (2012). L-arginine-Induced Experimental Acute Pancreatitis. Pancreapedia: Exocrine Pancreas Knowledge Base. doi: 10.3998/panc.2012.6
Dawra, R., Sharif, R., Phillips, P., Dudeja, V., Dhaulakhandi, D., and Saluja, A. K. (2007). Development of a new mouse model of acute pancreatitis induced by administration of L-arginine. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1009–1018. doi: 10.1152/ajpgi.00167.2006
de Campos, T., Deree, J., Martins, J. O., Loomis, W. H., Shenvi, E., Putnam, J. G., et al. (2008). Pentoxifylline attenuates pulmonary inflammation and neutrophil activation in experimental acute pancreatitis. Pancreas 37, 42–49. doi: 10.1097/MPA.0b013e3181612d19
De Palma, A. M., Thibaut, H. J., Li, S., Van Aelst, I., Dillen, C., Swinnen, M., et al. (2009). Inflammatory rather than infectious insults play a role in exocrine tissue damage in a mouse model for coxsackievirus B4-induced pancreatitis. J. Pathol. 217, 633–641. doi: 10.1002/path.2501
De Palma, A. M., Verbeken, E., Van Aelst, I., Van den Steen, P. E., Opdenakker, G., and Neyts, J. (2008). Increased gelatinase B/matrix metalloproteinase 9 (MMP-9) activity in a murine model of acute coxsackievirus B4-induced pancreatitis. Virology 382, 20–27. doi: 10.1016/j.virol.2008.08.046
Dellinger, E. P., Tellado, J. M., Soto, N. E., Ashley, S. W., Barie, P. S., Dugernier, T., et al. (2007). Early antibiotic treatment for severe acute necrotizing pancreatitis: a randomized, double-blind, placebo-controlled study. Ann. Surg. 245, 674–683. doi: 10.1097/01.sla.0000250414.09255.84
Dressel, T. D., Goodale, R. L, Jr., Zweber, B., and Borner, J. W. (1982). The effect of atropine and duct decompression on the evolution of diazinon-induced acute canine pancreatitis. Ann. Surg. 195, 424–434. doi: 10.1097/00000658-198204000-00008
Du, D., Yao, L., Zhang, R., Shi, N., Shen, Y., Yang, X., et al. (2018). Protective effects of flavonoids from Coreopsis tinctoria Nutt. on experimental acute pancreatitis via Nrf-2/ARE-mediated antioxidant pathways. J. Ethnopharmacol. 224, 261–272. doi: 10.1016/j.jep.2018.06.003
Ferdek, P. E., Jakubowska, M. A., Gerasimenko, J. V., Gerasimenko, O. V., and Petersen, O. H. (2016). Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium-driven bile uptake. J. Physiol. 594, 6147–6164. doi: 10.1113/JP272774
Foitzik, T., Fernandez-del Castillo, C., Rattner, D. W., Klar, E., and Warshaw, A. L. (1995). Alcohol selectively impairs oxygenation of the pancreas. Arch. Surg. 130, 357–360. doi: 10.1001/archsurg.1995.01430040019001
Foitzik, T., Hotz, H. G., Hot, B., Kirchengast, M., and Buhr, H. J. (1998). Endothelin-1 mediates the alcohol-induced reduction of pancreatic capillary blood flow. J. Gastrointest. Surg. 2, 379–384. doi: 10.1016/S1091-255X(98)80078-4
Foitzik, T., Lewandrowski, K. B., Fernandez-del Castillo, C., Rattner, D. W., Klar, E., and Warshaw, A. L. (1994). Exocrine hyperstimulation but not pancreatic duct obstruction increases the susceptibility to alcohol-related pancreatic injury. Arch. Surg. 129, 1081–1085. doi: 10.1001/archsurg.1994.01420340095018
Folch-Puy, E., Granell, S., Iovanna, J. L., Barthet, M., and Closa, D. (2006). Peroxisome proliferator-activated receptor gamma agonist reduces the severity of post-ERCP pancreatitis in rats. World J. Gastroenterol. 12, 6458–6463. doi: 10.3748/wjg.v12.i40.6458
Forsmark, C. E., Vege, S. S., and Wilcox, C. M. (2016). Acute pancreatitis. N. Engl. J. Med. 375, 1972–1981. doi: 10.1056/NEJMra1505202
Freeman, M. L., and Guda, N. M. (2004). Prevention of post-ERCP pancreatitis: a comprehensive review. Gastrointest. Endosc. 59, 845–864. doi: 10.1016/S0016-5107(04)00353-0
Friedman, H. S., Lowery, R., Shaughnessy, E., and Scorza, J. (1983). The effects of ethanol on pancreatic blood flow in awake and anesthetized dogs. Proc. Soc. Exp. Biol. Med. 174, 377–382. doi: 10.3181/00379727-174-41751
Gal, E., Dolensek, J., Stozer, A., Pohorec, V., Ebert, A., and Venglovecz, V. (2019). A novel in situ approach to studying pancreatic ducts in mice. Front. Physiol. 10:938. doi: 10.3389/fphys.2019.00938
Gallagher, S., Sankaran, H., and Williams, J. A. (1981). Mechanism of scorpion toxin-induced enzyme secretion in rat pancreas. Gastroenterology 80, 970–973. doi: 10.1016/0016-5085(81)90067-6
Garcia-Barrasa, A., Borobia, F. G., Pallares, R., Jorba, R., Poves, I., Busquets, J., et al. (2009). A double-blind, placebo-controlled trial of ciprofloxacin prophylaxis in patients with acute necrotizing pancreatitis. J. Gastrointest. Surg. 13, 768–774. doi: 10.1007/s11605-008-0773-7
Geisz, A., Jancso, Z., Nemeth, B. C., Hegyi, E., and Sahin-Toth, M. (2019). Natural single-nucleotide deletion in chymotrypsinogen C gene increases severity of secretagogue-induced pancreatitis in C57BL/6 mice. JCI Insight 4:e129717. doi: 10.1172/jci.insight.129717
Geisz, A., and Sahin-Toth, M. (2018). A preclinical model of chronic pancreatitis driven by trypsinogen autoactivation. Nat. Commun. 9:5033. doi: 10.1038/s41467-018-07347-y
Gorelick, F. S., and Lerch, M. M. (2017). Do animal models of acute pancreatitis reproduce human disease? Cell Mol. Gastroenterol. Hepatol. 4, 251–262. doi: 10.1016/j.jcmgh.2017.05.007
Grady, T., Mah'Moud, M., Otani, T., Rhee, S., Lerch, M. M., and Gorelick, F. S. (1998). Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am. J. Physiol. 275, G1010–1017. doi: 10.1152/ajpgi.1998.275.5.G1010
Gryshchenko, O., Gerasimenko, J. V., Gerasimenko, O. V., and Petersen, O. H. (2016). Ca2+ signals mediated by bradykinin type 2 receptors in normal pancreatic stellate cells can be inhibited by specific Ca2+ channel blockade. J. Physiol. 594, 281–293. doi: 10.1113/JP271468
Gryshchenko, O., Gerasimenko, J. V., Peng, S., Gerasimenko, O. V., and Petersen, O. H. (2018). Calcium signalling in the acinar environment of the exocrine pancreas: physiology and pathophysiology. J. Physiol. 596, 2663–2678. doi: 10.1113/JP275395
Gui, F., Zhang, Y., Wan, J., Zhan, X., Yao, Y., Li, Y., et al. (2020). Trypsin activity governs increased susceptibility to pancreatitis in mice expressing human PRSS1R122H. J. Clin. Invest. 130, 189–202. doi: 10.1172/JCI130172
Gukovskaya, A. S., Mouria, M., Gukovsky, I., Reyes, C. N., Kasho, V. N., Faller, L. D., et al. (2002). Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology 122, 106–118. doi: 10.1053/gast.2002.30302
Haciahmetoglu, T., Ertekin, C., Dolay, K., Yanar, F., Yanar, H., and Kapran, Y. (2008). The effects of contrast agent and intraductal pressure changes on the development of pancreatitis in an ERCP model in rats. Langenbecks. Arch. Surg. 393, 367–372. doi: 10.1007/s00423-007-0214-1
Hackert, T., Werner, J., Gebhard, M. M., and Klar, E. (2004). Effects of heparin in experimental models of acute pancreatitis and post-ERCP pancreatitis. Surgery 135, 131–138. doi: 10.1016/j.surg.2003.08.001
Hardman, J., Shields, C., Schofield, D., McMahon, R., Redmond, H. P., and Siriwardena, A. K. (2005). Intravenous antioxidant modulation of end-organ damage in L-arginine-induced experimental acute pancreatitis. Pancreatology 5, 380–386. doi: 10.1159/000086538
Harper, A. A., and Raper, H. S. (1943). Pancreozymin, a stimulant of the secretion of pancreatic enzymes in extracts of the small intestine. J. Physiol. 102, 115–125. doi: 10.1113/jphysiol.1943.sp004021
Hartwig, W., Kolvenbach, M., Hackert, T., Fortunato, F., Schneider, L., Buchler, M. W., et al. (2007). Enterokinase induces severe necrosis and rapid mortality in cerulein pancreatitis: characterization of a novel noninvasive rat model of necro-hemorrhagic pancreatitis. Surgery 142, 327–336. doi: 10.1016/j.surg.2007.04.023
Hartwig, W., Schimmel, E., Hackert, T., Fortunato, F., Bergmann, F., Baczako, A., et al. (2008). A novel animal model of severe pancreatitis in mice and its differences to the rat. Surgery 144, 394–403. doi: 10.1016/j.surg.2008.04.006
He, Z. J., Winston, J. H., Yusuf, T. E., Micci, M. A., Elfert, A., Xiao, S. Y., et al. (2003). Intraductal administration of an NK1 receptor antagonist attenuates the inflammatory response to retrograde infusion of radiological contrast in rats: implications for the pathogenesis and prevention of ERCP-induced pancreatitis. Pancreas 27, e13–17. doi: 10.1097/00006676-200307000-00018
Hegyi, P., Czako, L., Takacs, T., Szilvassy, Z., and Lonovics, J. (1999). Pancreatic secretory responses in L-arginine-induced pancreatitis: comparison of diabetic and nondiabetic rats. Pancreas 19, 167–174. doi: 10.1097/00006676-199908000-00010
Hegyi, P., Pandol, S., Venglovecz, V., and Rakonczay, Z, Jr. (2011). The acinar-ductal tango in the pathogenesis of acute pancreatitis. Gut 60, 544–552. doi: 10.1136/gut.2010.218461
Hegyi, P., and Petersen, O. H. (2013). The exocrine pancreas: the acinar-ductal tango in physiology and pathophysiology. Rev. Physiol. Biochem. Pharmacol. 165, 1–30. doi: 10.1007/112_2013_14
Hegyi, P., Rakonczay, Z, Jr., Sari, R., Gog, C., Lonovics, J., Takacs, T., et al. (2004). L-arginine-induced experimental pancreatitis. World J. Gastroenterol. 10, 2003–2009. doi: 10.3748/wjg.v10.i14.2003
Hegyi, P., Takacs, T., Jarmay, K., Nagy, I., Czako, L., and Lonovics, J. (1997). Spontaneous and cholecystokinin-octapeptide-promoted regeneration of the pancreas following L-arginine-induced pancreatitis in rat. Int. J. Pancreatol. 22, 193–200.
Hegyi, P., Takacs, T., Tiszlavicz, L., Czako, L., and Lonovics, J. (2000). Recovery of exocrine pancreas six months following pancreatitis induction with L-arginine in streptozotocin-diabetic rats. J. Physiol. Paris 94, 51–55. doi: 10.1016/S0928-4257(99)00103-5
Hines, O. J., and Pandol, S. J. (2019). Management of severe acute pancreatitis. BMJ 367:l6227. doi: 10.1136/bmj.l6227
Hoque, R., Sohail, M., Malik, A., Sarwar, S., Luo, Y., Shah, A., et al. (2011). TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 141, 358–369. doi: 10.1053/j.gastro.2011.03.041
Huang, W., Booth, D. M., Cane, M. C., Chvanov, M., Javed, M. A., Elliott, V. L., et al. (2014). Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63, 1313–1324. doi: 10.1136/gutjnl-2012-304058
Huang, W., Cane, M. C., Mukherjee, R., Szatmary, P., Zhang, X., Elliott, V., et al. (2017). Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut 66, 301–313. doi: 10.1136/gutjnl-2015-309363
Huang, Y. X., Li, W. D., Jia, L., Qiu, J. H., Jiang, S. M., Ou, Y., et al. (2012). Infliximab enhances the therapeutic effectiveness of octreotide on acute necrotizing pancreatitis in rat model. Pancreas 41, 849–854. doi: 10.1097/MPA.0b013e31823fbdc3
Ishii, H., Salem, H. H., Bell, C. E., Laposata, E. A., and Majerus, P. W. (1986). Thrombomodulin, an endothelial anticoagulant protein, is absent from the human brain. Blood 67, 362–365. doi: 10.1182/blood.V67.2.362.362
Ivy, A. C. (1929). A hormone mechanism for gall-bladder contraction & evacuation. Am. J. Surg. 7, 455–459. doi: 10.1016/S0002-9610(29)90551-1
Jakubowska, M. A., Ferdek, P. E., Gerasimenko, O. V., Gerasimenko, J. V., and Petersen, O. H. (2016). Nitric oxide signals are interlinked with calcium signals in normal pancreatic stellate cells upon oxidative stress and inflammation. Open Biol. 6:160149. doi: 10.1098/rsob.160149
Jancar, S., De Giaccobi, G., Mariano, M., Mencia-Huerta, J. M., Sirois, P., and Braquet, P. (1988). Immune complex induced pancreatitis: effect of BN 52021, a selective antagonist of platelet-activating factor. Prostaglandins 35, 757–770. doi: 10.1016/0090-6980(88)90148-7
Jancso, Z., Hegyi, E., and Sahin-Toth, M. (2018). Chymotrypsin reduces the severity of secretagogue-induced pancreatitis in mice. Gastroenterology 155, 1017–1021. doi: 10.1053/j.gastro.2018.06.041
Jancso, Z., and Sahin-Toth, M. (2020). Mutation that promotes activation of trypsinogen increases severity of secretagogue-induced pancreatitis in mice. Gastroenterology 158, 1083–1094. doi: 10.1053/j.gastro.2019.11.020
Janigan, D. T., Nevalainen, T. J., MacAulay, M. A., and Vethamany, V. G. (1975). Foreign serum-induced pancreatitis in mice. I. A new model of acute pancreatitis. Lab. Invest. 33, 591–607.
Ji, B., Bi, Y., Simeone, D., Mortensen, R. M., and Logsdon, C. D. (2001). Human pancreatic acinar cells lack functional responses to cholecystokinin and gastrin. Gastroenterology 121, 1380–1390. doi: 10.1053/gast.2001.29557
Jin, H. T., Lamsa, T., Hyvonen, M. T., Sand, J., Raty, S., Grigorenko, N., et al. (2008). A polyamine analog bismethylspermine ameliorates severe pancreatitis induced by intraductal infusion of taurodeoxycholate. Surgery 144, 49–56. doi: 10.1016/j.surg.2008.03.029
Jin, H. T., Lamsa, T., Nordback, P. H., Hyvonen, M. T., Raty, S., Nordback, I., et al. (2011). Polyamine catabolism in relation to trypsin activation and apoptosis in experimental acute pancreatitis. Pancreatology 11, 83–91. doi: 10.1159/000327260
Jin, S., Orabi, A. I., Le, T., Javed, T. A., Sah, S., Eisses, J. F., et al. (2015). Exposure to radiocontrast agents induces pancreatic inflammation by activation of nuclear factor-kappaB, calcium signaling, and calcineurin. Gastroenterology 149, 753–764.e711. doi: 10.1053/j.gastro.2015.05.004
Johnson, C. D., Kingsnorth, A. N., Imrie, C. W., McMahon, M. J., Neoptolemos, J. P., McKay, C., et al. (2001). Double blind, randomised, placebo controlled study of a platelet activating factor antagonist, lexipafant, in the treatment and prevention of organ failure in predicted severe acute pancreatitis. Gut 48, 62–69. doi: 10.1136/gut.48.1.62
Jung, K. H., Song, S. U., Yi, T., Jeon, M. S., Hong, S. W., Zheng, H. M., et al. (2011). Human bone marrow-derived clonal mesenchymal stem cells inhibit inflammation and reduce acute pancreatitis in rats. Gastroenterology 140, 998–1008. doi: 10.1053/j.gastro.2010.11.047
Kanno, H., Nose, M., Itoh, J., Taniguchi, Y., and Kyogoku, M. (1992). Spontaneous development of pancreatitis in the MRL/Mp strain of mice in autoimmune mechanism. Clin. Exp. Immunol. 89, 68–73. doi: 10.1111/j.1365-2249.1992.tb06879.x
Katz, M., Carangelo, R., Miller, L. J., and Gorelick, F. (1996). Effect of ethanol on cholecystokinin-stimulated zymogen conversion in pancreatic acinar cells. Am. J. Physiol. 270, G171–175. doi: 10.1152/ajpgi.1996.270.1.G171
Kim, J., Koo, B. K., and Knoblich, J. A. (2020). Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584. doi: 10.1038/s41580-020-0259-3
Kingsnorth, A. N. (1996). Platelet-activating factor. Scand. J. Gastroenterol. Suppl. 219, 28–31. doi: 10.3109/00365529609104996
Kingsnorth, A. N., Galloway, S. W., and Formela, L. J. (1995). Randomized, double-blind phase II trial of lexipafant, a platelet-activating factor antagonist, in human acute pancreatitis. Br. J. Surg. 82, 1414–1420. doi: 10.1002/bjs.1800821039
Kishino, Y., and Kawamura, S. (1984). Pancreatic damage induced by injecting a large dose of arginine. Virchows Arch. B. Cell Pathol. 47, 147–155. doi: 10.1007/BF02890197
Kivisaari, L. (1979). Contrast absorption and pancreatic inflammation following experimental ERCP. Invest. Radiol. 14, 493–497. doi: 10.1097/00004424-197911000-00008
Klop, B., do Rego, A. T., and Cabezas, M. C. (2013). Alcohol and plasma triglycerides. Curr. Opin. Lipidol. 24, 321–326. doi: 10.1097/MOL.0b013e3283606845
Kubisch, C. H., Sans, M. D., Arumugam, T., Ernst, S. A., Williams, J. A., and Logsdon, C. D. (2006). Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G238–245. doi: 10.1152/ajpgi.00471.2005
Kui, B., Balla, Z., Vasas, B., Vegh, E. T., Pallagi, P., Kormanyos, E. S., et al. (2015). New insights into the methodology of L-arginine-induced acute pancreatitis. PLoS ONE 10:e0117588. doi: 10.1371/journal.pone.0117588
Kuntz, E., Pinget, M., and Damge, P. (2004). Cholecystokinin octapeptide: a potential growth factor for pancreatic beta cells in diabetic rats. JOP 5, 464–475.
Lampel, M., and Kern, H. F. (1977). Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch. A Pathol. Anat. Histol. 373, 97–117. doi: 10.1007/BF00432156
Laposata, E. A., and Lange, L. G. (1986). Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 231, 497–499. doi: 10.1126/science.3941913
Laukkarinen, J. M., Van Acker, G. J., Weiss, E. R., Steer, M. L., and Perides, G. (2007). A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut 56, 1590–1598. doi: 10.1136/gut.2007.124230
Le, T., Eisses, J. F., Lemon, K. L., Ozolek, J. A., Pociask, D. A., Orabi, A. I., et al. (2015). Intraductal infusion of taurocholate followed by distal common bile duct ligation leads to a severe necrotic model of pancreatitis in mice. Pancreas 44, 493–499. doi: 10.1097/MPA.0000000000000285
Leblond, C. P., and Sergeyeva, M. A. (1944). Vacuolation of the acinar cells in the pancreas of the rat after treatment with thyroxine or acetylcholine. Anat. Rec. 90, 235–242. doi: 10.1002/ar.1090900308
Lee, M. G., Ohana, E., Park, H. W., Yang, D., and Muallem, S. (2012). Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 92, 39–74. doi: 10.1152/physrev.00011.2011
Lerch, M. M., and Aghdassi, A. A. (2010). The role of bile acids in gallstone-induced pancreatitis. Gastroenterology 138, 429–433. doi: 10.1053/j.gastro.2009.12.012
Lerch, M. M., and Gorelick, F. S. (2013). Models of acute and chronic pancreatitis. Gastroenterology 144, 1180–1193. doi: 10.1053/j.gastro.2012.12.043
Lerch, M. M., Saluja, A. K., Dawra, R., Ramarao, P., Saluja, M., and Steer, M. L. (1992). Acute necrotizing pancreatitis in the opossum: earliest morphological changes involve acinar cells. Gastroenterology 103, 205–213. doi: 10.1016/0016-5085(92)91114-J
Lerch, M. M., Saluja, A. K., Runzi, M., Dawra, R., Saluja, M., and Steer, M. L. (1993). Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology 104, 853–861. doi: 10.1016/0016-5085(93)91022-A
Lerch, M. M., Saluja, A. K., Runzi, M., Dawra, R., and Steer, M. L. (1995a). Luminal endocytosis and intracellular targeting by acinar cells during early biliary pancreatitis in the opossum. J. Clin. Invest. 95, 2222–2231. doi: 10.1172/JCI117912
Lerch, M. M., Weidenbach, H., Gress, T. M., and Adler, G. (1995b). Effect of kinin inhibition in experimental acute pancreatitis. Am. J. Physiol. 269, G490–499. doi: 10.1152/ajpgi.1995.269.4.G490
Lerch, M. M., Zenker, M., Turi, S., and Mayerle, J. (2006). Developmental and metabolic disorders of the pancreas. Endocrinol. Metab. Clin. North Am. 35, 219–241. doi: 10.1016/j.ecl.2006.02.004
Letko, G., Mantke, R., Sokolowski, A., and Spormann, H. (1990). Enzymatic and histologic investigations into the course of pancreatic alterations induced by anti-acinar-cell-antiserum. Int. Surg. 75, 254–258.
Leveau, P., Wang, X., Sun, Z., Borjesson, A., Andersson, E., and Andersson, R. (2005). Severity of pancreatitis-associated gut barrier dysfunction is reduced following treatment with the PAF inhibitor lexipafant. Biochem. Pharmacol. 69, 1325–1331. doi: 10.1016/j.bcp.2005.01.023
Lombardi, B., and Rao, N. K. (1975). Acute hemorrhagic pancreatic necrosis in mice. Influence of the age and sex of the animals and of dietary ethionine, choline, methionine, and adenine sulfate. Am. J. Pathol. 81, 87–100.
Lopez-Font, I., Gea-Sorli, S., de-Madaria, E., Gutierrez, L. M., Perez-Mateo, M., and Closa, D. (2010). Pancreatic and pulmonary mast cells activation during experimental acute pancreatitis. World J. Gastroenterol. 16, 3411–3417. doi: 10.3748/wjg.v16.i27.3411
Louhimo, J., Steer, M. L., and Perides, G. (2016). Necroptosis is an important severity determinant and potential therapeutic target in experimental severe pancreatitis. Cell Mol. Gastroenterol. Hepatol. 2, 519–535. doi: 10.1016/j.jcmgh.2016.04.002
Lu, Z., Karne, S., Kolodecik, T., and Gorelick, F. S. (2002). Alcohols enhance caerulein-induced zymogen activation in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G501–507. doi: 10.1152/ajpgi.00388.2001
Lugea, A., Gerloff, A., Su, H. Y., Xu, Z., Go, A., Hu, C., et al. (2017a). The combination of alcohol and cigarette smoke induces endoplasmic reticulum stress and cell death in pancreatic acinar cells. Gastroenterology 153, 1674–1686. doi: 10.1053/j.gastro.2017.08.036
Lugea, A., Gong, J., Nguyen, J., Nieto, J., French, S. W., and Pandol, S. J. (2010). Cholinergic mediation of alcohol-induced experimental pancreatitis. Alcohol. Clin. Exp. Res. 34, 1768–1781. doi: 10.1111/j.1530-0277.2010.01264.x
Lugea, A., Waldron, R. T., Mareninova, O. A., Shalbueva, N., Deng, N., Su, H. Y., et al. (2017b). Human pancreatic acinar cells: proteomic characterization, physiologic responses, and organellar disorders in ex vivo pancreatitis. Am. J. Pathol. 187, 2726–2743. doi: 10.1016/j.ajpath.2017.08.017
Luo, S., Wang, R., Jiang, W., Lin, X., Qiu, P., and Yan, G. (2010). A novel recombinant snake venom metalloproteinase from Agkistrodon acutus protects against taurocholate-induced severe acute pancreatitis in rats. Biochimie 92, 1354–1361. doi: 10.1016/j.biochi.2010.06.018
Madhi, R., Rahman, M., Taha, D., Morgelin, M., and Thorlacius, H. (2019). Targeting peptidylarginine deiminase reduces neutrophil extracellular trap formation and tissue injury in severe acute pancreatitis. J. Cell. Physiol. 234, 11850–11860. doi: 10.1002/jcp.27874
Maleth, J., Balazs, A., Pallagi, P., Balla, Z., Kui, B., Katona, M., et al. (2015). Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology 148, 427–439.e416. doi: 10.1053/j.gastro.2014.11.002
Mareninova, O. A., Hermann, K., French, S. W., O'Konski, M. S., Pandol, S. J., Webster, P., et al. (2009). Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Invest. 119, 3340–3355. doi: 10.1172/JCI38674
Mayerle, J., Sendler, M., Hegyi, E., Beyer, G., Lerch, M. M., and Sahin-Toth, M. (2019). Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology 156, 1951–1968.e1951. doi: 10.1053/j.gastro.2018.11.081
McCune, W. S., Shorb, P. E., and Moscovitz, H. (1968). Endoscopic cannulation of the ampulla of vater: a preliminary report. Ann. Surg. 167, 752–756. doi: 10.1097/00000658-196805000-00013
Meczker, A., Hanak, L., Parniczky, A., Szentesi, A., Eross, B., Hegyi, P., et al. (2020). Analysis of 1060 cases of drug-induced acute pancreatitis. Gastroenterology 159, 1958–1961.e8. doi: 10.1053/j.gastro.2020.07.016
Merkord, J., and Hennighausen, G. (1989). Acute pancreatitis and bile duct lesions in rat induced by dibutyltin dichloride. Exp. Pathol. 36, 59–62. doi: 10.1016/S0232-1513(89)80113-6
Merriam, L. T., Wilcockson, D., Samuel, I., and Joehl, R. J. (1996). Ligation-induced acute pancreatitis increases pancreatic circulating trypsinogen activation peptides. J. Surg. Res. 60, 417–421. doi: 10.1006/jsre.1996.0068
Meyerholz, D. K., and Samuel, I. (2007). Morphologic characterization of early ligation-induced acute pancreatitis in rats. Am. J. Surg. 194, 652–658. doi: 10.1016/j.amjsurg.2007.07.014
Meyerholz, D. K., Williard, D. E., Grittmann, A. M., and Samuel, I. (2008). Murine pancreatic duct ligation induces stress kinase activation, acute pancreatitis, and acute lung injury. Am. J. Surg. 196, 675–682. doi: 10.1016/j.amjsurg.2008.07.009
Michael, E. S., Kuliopulos, A., Covic, L., Steer, M. L., and Perides, G. (2013). Pharmacological inhibition of PAR2 with the pepducin P2pal-18S protects mice against acute experimental biliary pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G516–526. doi: 10.1152/ajpgi.00296.2012
Mizunuma, T., Kawamura, S., and Kishino, Y. (1984). Effects of injecting excess arginine on rat pancreas. J. Nutr. 114, 467–471. doi: 10.1093/jn/114.3.467
Moggia, E., Koti, R., Belgaumkar, A. P., Fazio, F., Pereira, S. P., Davidson, B. R., et al. (2017). Pharmacological interventions for acute pancreatitis. Cochrane Database Syst. Rev. 4:CD011384. doi: 10.1002/14651858.CD011384.pub2
Mole, D. J., Webster, S. P., Uings, I., Zheng, X., Binnie, M., Wilson, K., et al. (2016). Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 22, 202–209. doi: 10.1038/nm.4020
Molnar, R., Madacsy, T., Varga, A., Nemeth, M., Katona, X., Gorog, M., et al. (2020). Mouse pancreatic ductal organoid culture as a relevant model to study exocrine pancreatic ion secretion. Lab. Invest. 100, 84–97. doi: 10.1038/s41374-019-0300-3
Mooren, F., Hlouschek, V., Finkes, T., Turi, S., Weber, I. A., Singh, J., et al. (2003). Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J. Biol. Chem. 278, 9361–9369. doi: 10.1074/jbc.M207454200
Mouret, J. (1895). Contribution à l'étude des cellules glandulaires (pancreas). J. Anat. Physiol. 31, 221–236.
Mukherjee, R., Mareninova, O. A., Odinokova, I. V., Huang, W., Murphy, J., Chvanov, M., et al. (2016). Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut 65, 1333–1346. doi: 10.1136/gutjnl-2014-308553
Mukherjee, R., Nunes, Q., Huang, W., and Sutton, R. (2019). Precision medicine for acute pancreatitis: current status and future opportunities. Precis. Clin. Med. 2, 81–86. doi: 10.1093/pcmedi/pbz010
Murphy, J. A., Criddle, D. N., Sherwood, M., Chvanov, M., Mukherjee, R., McLaughlin, E., et al. (2008). Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135, 632–641. doi: 10.1053/j.gastro.2008.05.026
Murthy, P., Singhi, A. D., Ross, M. A., Loughran, P., Paragomi, P., Papachristou, G. I., et al. (2019). Enhanced neutrophil extracellular trap formation in acute pancreatitis contributes to disease severity and is reduced by chloroquine. Front. Immunol. 10:28. doi: 10.3389/fimmu.2019.00028
Navina, S., Acharya, C., DeLany, J. P., Orlichenko, L. S., Baty, C. J., Shiva, S. S., et al. (2011). Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci. Transl. Med. 3:107ra110. doi: 10.1126/scitranslmed.3002573
Nevalainen, T. J. (1978). Pancreatic injury caused by intraductal injection of foreign serum in rat. Virchows Arch. B Cell Pathol. 27, 89–98.
Nevalainen, T. J., Fowlie, F. E., and Janigan, D. T. (1977). Foreign serum-induced pancreatitis in mice. II. Secretory disturbances of acinar cells. Lab. Invest. 36, 469–473.
Niederau, C., Ferrell, L. D., and Grendell, J. H. (1985). Caerulein-induced acute necrotizing pancreatitis in mice: protective effects of proglumide, benzotript, and secretin. Gastroenterology 88, 1192–1204. doi: 10.1016/S0016-5085(85)80079-2
Nieuwenhuijs, V. B., van Dijk, J. E., Gooszen, H. G., and Akkermans, L. M. (2000). Obstructive jaundice, bacterial translocation and interdigestive small-bowel motility in rats. Digestion 62, 255–261. doi: 10.1159/000007824
Noble, M. D., Romac, J., Vigna, S. R., and Liddle, R. A. (2008). A pH-sensitive, neurogenic pathway mediates disease severity in a model of post-ERCP pancreatitis. Gut 57, 1566–1571. doi: 10.1136/gut.2008.148551
Noel, P., Patel, K., Durgampudi, C., Trivedi, R. N., de Oliveira, C., Crowell, M. D., et al. (2016). Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut 65, 100–111. doi: 10.1136/gutjnl-2014-308043
Novaes, G., Cabral, A. P., de Falco, C. N., and de Queiroz, A. C. (1989). Acute pancreatitis induced by scorpion toxin, tityustoxin. Histopathological study in rats. Arq. Gastroenterol. 26, 9–12.
Odinokova, I. V., Sung, K. F., Mareninova, O. A., Hermann, K., Evtodienko, Y., Andreyev, A., et al. (2009). Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut 58, 431–442. doi: 10.1136/gut.2007.147207
Opie, E. L. (1901). The etiology of acute hemorrhagic pancreatitis. Bull. Johns Hopkins Hosp. 12, 182–190.
Pan, Y., Fang, H. Z., Lu, F. C., Pan, M. G., Chen, F., Xiong, P., et al. (2017). Ulinastatin ameliorates tissue damage of severe acute pancreatitis through modulating regulatory T cells. J. Inflamm. Lond. 14:7. doi: 10.1186/s12950-017-0154-7
Pandol, S. J., Gukovsky, I., Satoh, A., Lugea, A., and Gukovskaya, A. S. (2003). Animal and in vitro models of alcoholic pancreatitis: role of cholecystokinin. Pancreas 27, 297–300. doi: 10.1097/00006676-200311000-00004
Pandol, S. J., Periskic, S., Gukovsky, I., Zaninovic, V., Jung, Y., Zong, Y., et al. (1999). Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 117, 706–716. doi: 10.1016/S0016-5085(99)70465-8
Pantoja, J. L., Renner, I. G., Abramson, S. B., and Edmondson, H. A. (1983). Production of acute hemorrhagic-pancreatitis in the dog using venom of the scorpion, buthus-quinquestriatus. Dig. Dis. Sci. 28, 429–439. doi: 10.1007/BF02430532
Parekh, P. J., Majithia, R., Sikka, S. K., and Baron, T. H. (2017). The “scope” of post-ERCP pancreatitis. Mayo Clin. Proc. 92, 434–448. doi: 10.1016/j.mayocp.2016.10.028
Paszt, A., Takacs, T., Rakonczay, Z., Kaszaki, J., Wolfard, A., Tiszlavicz, L., et al. (2004). The role of the glucocorticoid-dependent mechanism in the progression of sodium taurocholate-induced acute pancreatitis in the rat. Pancreas 29, 75–82. doi: 10.1097/00006676-200407000-00059
Patel, K., Trivedi, R. N., Durgampudi, C., Noel, P., Cline, R. A., DeLany, J. P., et al. (2015). Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am. J. Pathol. 185, 808–819. doi: 10.1016/j.ajpath.2014.11.019
Perides, G., Laukkarinen, J. M., Vassileva, G., and Steer, M. L. (2010). Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138, 715–725. doi: 10.1053/j.gastro.2009.10.052
Perides, G., Tao, X., West, N., Sharma, A., and Steer, M. L. (2005). A mouse model of ethanol dependent pancreatic fibrosis. Gut 54, 1461–1467. doi: 10.1136/gut.2004.062919
Petho, G., and Reeh, P. W. (2012). Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol. Rev. 92, 1699–1775. doi: 10.1152/physrev.00048.2010
Petrov, M. S., and Yadav, D. (2019). Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 16, 175–184. doi: 10.1038/s41575-018-0087-5
Pozsar, J., Schwab, R., Simon, K., Fekete, L., Orgovan, G., and Pap, A. (1997). Effect of endotoxin administration on the severity of acute pancreatitis in two experimental models. Int. J. Pancreatol. 22, 31–37. doi: 10.1007/BF02803902
Qian, M., Fang, L., and Cui, Y. (2010). Expression of NOD2 in a rat model of acute pancreatitis. Pancreas 39, 1034–1040. doi: 10.1097/MPA.0b013e3181da0f1d
Radadiya, D., Devani, K., Arora, S., Charilaou, P., Brahmbhatt, B., Young, M., et al. (2019). Peri-procedural aggressive hydration for post endoscopic retrograde cholangiopancreatography (ERCP) pancreatitis prophylaxsis: meta-analysis of randomized controlled trials. Pancreatology 19, 819–827. doi: 10.1016/j.pan.2019.07.046
Rakonczay, Z, Jr., Hegyi, P., Dosa, S., Ivanyi, B., Jarmay, K., Biczo, G., et al. (2008). A new severe acute necrotizing pancreatitis model induced by L-ornithine in rats. Crit. Care Med. 36, 2117–2127. doi: 10.1097/CCM.0b013e31817d7f5c
Rakonczay, Z, Jr., Jarmay, K., Kaszaki, J., Mandi, Y., Duda, E., Hegyi, P., et al. (2003). NF-kappaB activation is detrimental in arginine-induced acute pancreatitis. Free Radic. Biol. Med. 34, 696–709. doi: 10.1016/S0891-5849(02)01373-4
Rattner, D. W., Napolitano, L. M., Corsetti, J., Compton, C., Stanford, G. G., Warshaw, A. L., et al. (1990). Hypocalcemia in experimental pancreatitis occurs independently of changes in serum nonesterified fatty acid levels. Int. J. Pancreatol. 6, 249–262.
Reeve, J. R, Jr., Wu, S. V., Keire, D. A., Faull, K., Chew, P., Solomon, T. E., et al. (2004). Differential bile-pancreatic secretory effects of CCK-58 and CCK-8. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G395–402. doi: 10.1152/ajpgi.00020.2003
Rifai, Y., Elder, A. S., Carati, C. J., Hussey, D. J., Li, X., Woods, C. M., et al. (2008). The tripeptide analog feG ameliorates severity of acute pancreatitis in a caerulein mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1094–1099. doi: 10.1152/ajpgi.00534.2007
Rosen, H. R., and Tuchler, H. (1992). Pulmonary injury in acute experimental pancreatitis correlates with elevated levels of free fatty acids in rats. HPB Surg. 6, 79–90. doi: 10.1155/1992/92916
Saka, M., Tuzun, A., Ates, Y., Bagci, S., Karaeren, N., and Dagalp, K. (2004). Acute pancreatitis possibly due to arginine use: a case report. Turk. J. Gastroenterol. 15, 56–58.
Saluja, A., Dudeja, V., Dawra, R., and Sah, R. P. (2019). Early Intra-acinar events in pathogenesis of pancreatitis. Gastroenterology 156, 1979–1993. doi: 10.1053/j.gastro.2019.01.268
Saluja, A. K., Bhagat, L., Lee, H. S., Bhatia, M., Frossard, J. L., and Steer, M. L. (1999). Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am. J. Physiol. 276, G835–842. doi: 10.1152/ajpgi.1999.276.4.G835
Saluja, A. K., Lerch, M. M., Phillips, P. A., and Dudeja, V. (2007). Why does pancreatic overstimulation cause pancreatitis? Annu. Rev. Physiol. 69, 249–269. doi: 10.1146/annurev.physiol.69.031905.161253
Samuel, I., Yuan, Z., Meyerholz, D. K., Twait, E., Williard, D. E., and Kempuraj, D. (2010). A novel model of severe gallstone pancreatitis: murine pancreatic duct ligation results in systemic inflammation and substantial mortality. Pancreatology 10, 536–544. doi: 10.1159/000320776
Saruc, M., Yuceyar, H., Turkel, N., Ozutemiz, O., Tuzcuoglu, I., Yuce, G., et al. (2003). An experimental model of hemolysis-induced acute pancreatitis. Braz. J. Med. Biol. Res. 36, 879–886. doi: 10.1590/S0100-879X2003000700008
Schmidt, J., Lewandrowsi, K., Warshaw, A. L., Compton, C. C., and Rattner, D. W. (1992a). Morphometric characteristics and homogeneity of a new model of acute pancreatitis in the rat. Int. J. Pancreatol. 12, 41–51.
Schmidt, J., Rattner, D. W., Lewandrowski, K., Compton, C. C., Mandavilli, U., Knoefel, W. T., et al. (1992b). A better model of acute pancreatitis for evaluating therapy. Ann. Surg. 215, 44–56. doi: 10.1097/00000658-199201000-00007
Seelig, R., and Seelig, H. P. (1975). The possible role of serum complement system in the formal pathogenesis of acute pancreatitis II. Cobra venom factor pancreatitis–sodiumtaurocholate and deoxycholate pancreatitis. Acta Hepatogastroenterol. 22, 335–346.
Sendler, M., Weiss, F. U., Golchert, J., Homuth, G., van den Brandt, C., Mahajan, U. M., et al. (2018). Cathepsin B-mediated activation of trypsinogen in endocytosing macrophages increases severity of pancreatitis in mice. Gastroenterology 154, 704–718.e710. doi: 10.1053/j.gastro.2017.10.018
Shalbueva, N., Mareninova, O. A., Gerloff, A., Yuan, J., Waldron, R. T., Pandol, S. J., et al. (2013). Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 144, 437–446.e436. doi: 10.1053/j.gastro.2012.10.037
Shields, C. J., Sookhai, S., Winter, D. C., Dowdall, J. F., Kingston, G., Parfrey, N., et al. (2001). Attenuation of pancreatitis-induced pulmonary injury by aerosolized hypertonic saline. Surg. Infect. 2, 215–223. doi: 10.1089/109629601317202696
Shorrock, K., Austen, B. M., and Hermon-Taylor, J. (1991). Hyperstimulation pancreatitis in mice induced by cholecystokinin octapeptide, caerulein, and novel analogues: effect of molecular structure on potency. Pancreas 6, 404–406. doi: 10.1097/00006676-199107000-00005
Silva-Vaz, P., Abrantes, A. M., Castelo-Branco, M., Gouveia, A., Botelho, M. F., and Tralhao, J. G. (2019). Murine models of acute pancreatitis: a critical appraisal of clinical relevance. Int. J. Mol. Sci. 20:2794. doi: 10.3390/ijms20112794
Siriwardena, A. K., Mason, J. M., Balachandra, S., Bagul, A., Galloway, S., Formela, L., et al. (2007). Randomised, double blind, placebo controlled trial of intravenous antioxidant (n-acetylcysteine, selenium, vitamin C) therapy in severe acute pancreatitis. Gut 56, 1439–1444. doi: 10.1136/gut.2006.115873
Siveke, J. T., Lubeseder-Martellato, C., Lee, M., Mazur, P. K., Nakhai, H., Radtke, F., et al. (2008). Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology 134, 544–555. doi: 10.1053/j.gastro.2007.11.003
Sugita, H., Yamaguchi, Y., Ikei, S., Yamada, S., and Ogawa, M. (1997). Enhanced expression of cytokine-induced neutrophil chemoattractant (CINC) by bronchoalveolar macrophages in cerulein-induced pancreatitis rats. Dig. Dis. Sci. 42, 154–160. doi: 10.1023/A:1018809810561
Sun, W., Watanabe, Y., Toki, A., and Wang, Z. Q. (2007). Beneficial effects of hydrocortisone in induced acute pancreatitis of rats. Chin. Med. J. 120, 1757–1761. doi: 10.1097/00029330-200710020-00005
Sun, W., Watanabe, Y., and Wang, Z. Q. (2006). Expression and significance of ICAM-1 and its counter receptors LFA-1 and Mac-1 in experimental acute pancreatitis of rats. World J. Gastroenterol. 12, 5005–5009. doi: 10.3748/wjg.v12.i31.5005
Szabolcs, A., Reiter, R. J., Letoha, T., Hegyi, P., Papai, G., Varga, I., et al. (2006). Effect of melatonin on the severity of L-arginine-induced experimental acute pancreatitis in rats. World J. Gastroenterol. 12, 251–258. doi: 10.3748/wjg.v12.i2.251
Sztefko, K., and Panek, J. (2001). Serum free fatty acid concentration in patients with acute pancreatitis. Pancreatology 1, 230–236. doi: 10.1159/000055816
Takacs, T., Czako, L., Jarmay, K., Farkas, G, Jr., Mandi, Y., and Lonovics, J. (1996). Cytokine level changes in L-arginine-induced acute pancreatitis in rat. Acta Physiol. Hung. 84, 147–156.
Takacs, T., Czako, L., Morschl, E., Laszlo, F., Tiszlavicz, L., Rakonczay, Z, Jr., et al. (2002a). The role of nitric oxide in edema formation in L-arginine-induced acute pancreatitis. Pancreas 25, 277–282. doi: 10.1097/00006676-200210000-00010
Takacs, T., Hegyi, P., Jarmay, K., Czako, L., Gog, C., Rakonczay, Z, Jr., et al. (2001). Cholecystokinin fails to promote pancreatic regeneration in diabetic rats following the induction of experimental pancreatitis. Pharmacol. Res. 44, 363–372. doi: 10.1006/phrs.2001.0843
Takacs, T., Rakonczay, Z, Jr., Varga, I. S., Ivanyi, B., Mandi, Y., Boros, I., et al. (2002b). Comparative effects of water immersion pretreatment on three different acute pancreatitis models in rats. Biochem. Cell Biol. 80, 241–251. doi: 10.1139/o02-006
Tardini, A., Anversa, P., Bordi, C., Bertaccini, G., and Impicciatore, M. (1971). Ultrastructural and biochemical changes after marked caerulein stimulation of the exocrine pancreas in the dog. Am. J. Pathol. 62, 35–56.
Tashiro, M., Schafer, C., Yao, H., Ernst, S. A., and Williams, J. A. (2001). Arginine induced acute pancreatitis alters the actin cytoskeleton and increases heat shock protein expression in rat pancreatic acinar cells. Gut 49, 241–250. doi: 10.1136/gut.49.2.241
Tekin, S. O., Teksoz, S., Terzioglu, D., Arikan, A. E., Ozcevik, H., and Uslu, E. (2015). Use of infliximab in treatment of acute pancreatitis. Bratisl. Lek. Listy. 116, 167–172. doi: 10.4149/BLL_2015_034
Terry, T. R., Grant, D. A., and Hermon-Taylor, J. (1987). Intraduct enterokinase is lethal in rats with experimental bile-salt pancreatitis. Br. J. Surg. 74, 40–43. doi: 10.1002/bjs.1800740113
Thorn, P., Lawrie, A. M., Smith, P. M., Gallacher, D. V., and Petersen, O. H. (1993). Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell 74, 661–668. doi: 10.1016/0092-8674(93)90513-P
Toma, H., Winston, J., Micci, M. A., Shenoy, M., and Pasricha, P. J. (2000). Nerve growth factor expression is up-regulated in the rat model of L-arginine-induced acute pancreatitis. Gastroenterology 119, 1373–1381. doi: 10.1053/gast.2000.19264
Venglovecz, V., Rakonczay, Z, Jr., Ozsvari, B., Takacs, T., Lonovics, J., Varro, A., et al. (2008). Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 57, 1102–1112. doi: 10.1136/gut.2007.134361
Vigna, S. R., Shahid, R. A., and Liddle, R. A. (2014). Ethanol contributes to neurogenic pancreatitis by activation of TRPV1. FASEB J. 28, 891–896. doi: 10.1096/fj.13-236208
Villaret, M., Justin-Besancon, L., and Even, R. (1929). Effects de l'acetylcholine sur la secretion pancreatique. C R Soc Biol. 101:70.
Waldron, R. T., Chen, Y., Pham, H., Go, A., Su, H. Y., Hu, C., et al. (2019). The Orai Ca2+ channel inhibitor CM4620 targets both parenchymal and immune cells to reduce inflammation in experimental acute pancreatitis. J. Physiol. 597, 3085–3105. doi: 10.1113/JP277856
Walker, N. I., Winterford, C. M., and Kerr, J. F. (1992). Ultrastructure of the rat pancreas after experimental duct ligation. II. Duct and stromal cell proliferation, differentiation, and deletion. Pancreas 7, 420–434. doi: 10.1097/00006676-199207000-00002
Wan, M. H., Huang, W., Latawiec, D., Jiang, K., Booth, D. M., Elliott, V., et al. (2012). Review of experimental animal models of biliary acute pancreatitis and recent advances in basic research. HPB 14, 73–81. doi: 10.1111/j.1477-2574.2011.00408.x
Wang, Y., Kayoumu, A., Lu, G., Xu, P., Qiu, X., Chen, L., et al. (2016). Experimental models in syrian golden hamster replicate human acute pancreatitis. Sci. Rep. 6:28014. doi: 10.1038/srep28014
Watanabe, O., Baccino, F. M., Steer, M. L., and Meldolesi, J. (1984). Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am. J. Physiol. 246, G457–467. doi: 10.1152/ajpgi.1984.246.4.G457
Weber, H., Merkord, J., Jonas, L., Wagner, A., Schroder, H., Kading, U., et al. (1995). Oxygen radical generation and acute pancreatitis: effects of dibutyltin dichloride/ethanol and ethanol on rat pancreas. Pancreas 11, 382–388. doi: 10.1097/00006676-199511000-00010
Weidenbach, H., Lerch, M. M., Gress, T. M., Pfaff, D., Turi, S., and Adler, G. (1995). Vasoactive mediators and the progression from oedematous to necrotising experimental acute pancreatitis. Gut 37, 434–440. doi: 10.1136/gut.37.3.434
Wen, L., Javed, T. A., Dobbs, A. K., Brown, R., Niu, M., Li, L., et al. (2020). The protective effects of calcineurin on pancreatitis in mice depend on the cellular source. Gastroenterology 159, 1036–1050.e8. doi: 10.1053/j.gastro.2020.05.051
Wen, L., Javed, T. A., Yimlamai, D., Mukherjee, A., Xiao, X., and Husain, S. Z. (2018). Transient high pressure in pancreatic ducts promotes inflammation and alters tight junctions via calcineurin signaling in mice. Gastroenterology 155, 1250–1263.e1255. doi: 10.1053/j.gastro.2018.06.036
Wen, L., Voronina, S., Javed, M. A., Awais, M., Szatmary, P., Latawiec, D., et al. (2015). Inhibitors of ORAI1 prevent cytosolic calcium-associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology 149, 481–492.e487. doi: 10.1053/j.gastro.2015.04.015
Werner, J., Laposata, M., Fernandez-del Castillo, C., Saghir, M., Iozzo, R. V., Lewandrowski, K. B., et al. (1997). Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology 113, 286–294. doi: 10.1016/S0016-5085(97)70106-9
Werner, J., Saghir, M., Warshaw, A. L., Lewandrowski, K. B., Laposata, M., Iozzo, R. V., et al. (2002). Alcoholic pancreatitis in rats: injury from nonoxidative metabolites of ethanol. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G65–73. doi: 10.1152/ajpgi.00419.2001
Willemer, S., Elsasser, H. P., and Adler, G. (1992). Hormone-induced pancreatitis. Eur. Surg. Res. 24 (Suppl. 1), 29–39. doi: 10.1159/000129237
Williams, J. A. (2001). Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu. Rev. Physiol. 63, 77–97. doi: 10.1146/annurev.physiol.63.1.77
Williams, J. A. (2002). Receptor biology and intracellular regulatory mechanisms in pancreatic acinar cells. Curr. Opin. Gastroenterol. 18, 529–535. doi: 10.1097/00001574-200209000-00002
Williams, J. A. (2019). Cholecystokinin (CCK) regulation of pancreatic acinar cells: physiological actions and signal transduction mechanisms. Compr. Physiol. 9, 535–564. doi: 10.1002/cphy.c180014
Williams, J. A., Korc, M., and Dormer, R. L. (1978). Action of secretagogues on a new preparation of functionally intact, isolated pancreatic acini. Am. J. Physiol. 235, 517–524. doi: 10.1152/ajpendo.1978.235.5.E517
Wilson, J. S., and Apte, M. V. (2003). Role of alcohol metabolism in alcoholic pancreatitis. Pancreas 27, 311–315. doi: 10.1097/00006676-200311000-00007
Wittel, U. A., Wiech, T., Chakraborty, S., Boss, B., Lauch, R., Batra, S. K., et al. (2008). Taurocholate-induced pancreatitis: a model of severe necrotizing pancreatitis in mice. Pancreas 36, e9–21. doi: 10.1097/MPA.0b013e3181575103
Won, J. H., Zhang, Y., Ji, B., Logsdon, C. D., and Yule, D. I. (2011). Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol. Biol. Cell 22, 421–436. doi: 10.1091/mbc.e10-10-0807
Xia, X. M., Wang, F. Y., Wang, Z. K., Wan, H. J., Xu, W. A., and Lu, H. (2010). Emodin enhances alveolar epithelial barrier function in rats with experimental acute pancreatitis. World J. Gastroenterol. 16, 2994–3001. doi: 10.3748/wjg.v16.i24.2994
Xiong, G. S., Wu, S. M., Wang, Z. H., Mo, J. Z., and Xiao, S. D. (2007). Effects of thalidomide in experimental models of post-endoscopic retrograde cholangiopancreatography pancreatitis. J. Gastroenterol. Hepatol. 22, 371–376. doi: 10.1111/j.1440-1746.2006.04552.x
Xue, P., Deng, L. H., Zhang, Z. D., Yang, X. N., Wan, M. H., Song, B., et al. (2009). Infectious complications in patients with severe acute pancreatitis. Dig. Dis. Sci. 54, 2748–2753. doi: 10.1007/s10620-008-0668-1
Yamaguchi, Y., Okabe, K., Liang, J., Matsumura, F., Akizuki, E., Matsuda, T., et al. (1999). The novel carboxamide derivative IS-741 reduces neutrophil chemoattractant production by bronchoalveolar macrophages in rats with cerulein-induced pancreatitis complicated by sepsis. Digestion 60 (Suppl. 1), 52–56. doi: 10.1159/000051454
Yamanel, L., Mas, M. R., Comert, B., Isik, A. T., Aydin, S., Mas, N., et al. (2005). The effect of activated protein C on experimental acute necrotizing pancreatitis. Crit. Care 9, R184–190. doi: 10.1186/cc3485
Yamano, M., Umeda, M., Miyata, K., and Yamada, T. (1998). Protective effect of the combined treatment of pancreatic and neutrophil elastase inhibitors on acute pancreatitis elicited by lipopolysaccharide in rats given intraductal injection of taurocholate plus trypsin. Naunyn schmiedebergs. Arch. Pharmacol. 357, 558–564. doi: 10.1007/PL00005208
Yang, S., Imamura, Y., Jenkins, R. W., Canadas, I., Kitajima, S., Aref, A., et al. (2016). Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol. Res. 4, 520–530. doi: 10.1158/2326-6066.CIR-15-0235
Yang, Y. L., Li, J. P., Li, K. Z., and Dou, K. F. (2004). Tumor necrosis factor alpha antibody prevents brain damage of rats with acute necrotizing pancreatitis. World J. Gastroenterol. 10, 2898–2900. doi: 10.3748/wjg.v10.i19.2898
Yuan, J., Tan, T., Geng, M., Tan, G., Chheda, C., and Pandol, S. J. (2017). Novel small molecule inhibitors of protein kinase D suppress NF-kappaB activation and attenuate the severity of rat cerulein pancreatitis. Front. Physiol. 8:1014. doi: 10.3389/fphys.2017.01014
Yuan, Z., Meyerholz, D. K., Twait, E. C., Kempuraj, D., Williard, D. E., and Samuel, I. (2011). Systemic inflammation with multiorgan dysfunction is the cause of death in murine ligation-induced acute pancreatitis. J. Gastrointest. Surg. 15, 1670–1678. doi: 10.1007/s11605-011-1643-2
Zelner, I., Matlow, J. N., Natekar, A., and Koren, G. (2013). Synthesis of fatty acid ethyl esters in mammalian tissues after ethanol exposure: a systematic review of the literature. Drug Metab. Rev. 45, 277–299. doi: 10.3109/03602532.2013.795584
Zhang, X., Jin, T., Shi, N., Yao, L., Yang, X., Han, C., et al. (2018). Mechanisms of pancreatic injury induced by basic amino acids differ between L-arginine, L-ornithine, and L-histidine. Front. Physiol. 9:1922. doi: 10.3389/fphys.2018.01922
Zhang, X. P., Ye, Q., Jiang, X. G., Ma, M. L., Zhu, F. B., Zhang, R. P., et al. (2007). Preparation method of an ideal model of multiple organ injury of rat with severe acute pancreatitis. World J. Gastroenterol. 13, 4566–4573. doi: 10.3748/wjg.v13.i34.4566
Keywords: acute pancreatitis, animal models, pancreatic acinar cells, pancreatic stellate cells, pancreatic ductal cells, translational research
Citation: Yang X, Yao L, Fu X, Mukherjee R, Xia Q, Jakubowska MA, Ferdek PE and Huang W (2020) Experimental Acute Pancreatitis Models: History, Current Status, and Role in Translational Research. Front. Physiol. 11:614591. doi: 10.3389/fphys.2020.614591
Received: 06 October 2020; Accepted: 30 November 2020;
Published: 23 December 2020.
Edited by:
Richard T. Waldron, Cedars Sinai Medical Center, United StatesReviewed by:
József Maléth, University of Szeged, HungaryMatthias Sendler, Universitätsmedizin Greifswald, Germany
Copyright © 2020 Yang, Yao, Fu, Mukherjee, Xia, Jakubowska, Ferdek and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Huang, ZHJfd2VpX2h1YW5nQHNjdS5lZHUuY24=
†These authors have contributed equally to this work