 Caijuan Dong
Caijuan Dong Ya Wang1
Ya Wang1 Tingzhong Wang
Tingzhong Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 17 December 2020
Sec. Membrane Physiology and Membrane Biophysics
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.609733
This article is part of the Research TopicInherited Arrhythmias of the Cardiac Sodium Channel Nav1.5View all 8 articles
Cardiac voltage-gated sodium channel NaV1.5, encoded by SCN5A, is crucial for the upstroke of action potential and excitation of cardiomyocytes. NaV1.5 undergoes complex processes before it reaches the target membrane microdomains and performs normal functions. A variety of protein partners are needed to achieve the balance between SCN5A transcription and mRNA decay, endoplasmic reticulum retention and export, Golgi apparatus retention and export, selective anchoring and degradation, activation, and inactivation of sodium currents. Subtle alterations can impair NaV1.5 in terms of expression or function, eventually leading to NaV1.5-associated diseases such as lethal arrhythmias and cardiomyopathy.
The cardiac voltage-gated sodium channel NaV1.5 is made up of a 220-kDa pore-forming α-subunit and 30-kDa auxiliary β-subunits. For the convenience of reading, NaV1.5 in the following parts refers to the α-subunit of NaV1.5. The SCN5A gene, found on chromosome 3p21, encodes the NaV1.5 channel, which includes four homologous domains (DI-DIV), a C-terminus, and an N-terminus (Figure 1). Each domain of the NaV1.5 channel is composed of six membrane-spanning segments (S1-S6). Specifically, the S4 transmembrane segment acts as a voltage sensor. The S5 and S6 transmembrane segments form a pore with the intermembrane P-loop, which determines ion selectivity and influx (Jiang et al., 2020). The NaV1.5 channel mediates the rapid influx of sodium ions (Na+), which triggers cardiac action potential (AP), induces rapid depolarization, and initiates the excitation-contraction coupling cascades in the cardiomyocytes (Han et al., 2018). The sodium current (INa) generated by NaV1.5 is determined by the channel density on the plasma membrane and channel gating. NaV1.5 is traditionally regarded as a monomer. However, it is well accepted that NaV1.5 is assembled as a dimer and exhibits coupled gating properties (Clatot et al., 2017). β1-β4 subunits, encoded by the SCN1b-4b genes, consist of an extracellular N-terminus, a transmembrane α-helix, and an intracellular C terminus (Salvage et al., 2020). The β-subunits act as gating modulators of NaV1.5 and facilitate NaV1.5 residence at the intercalated disk.
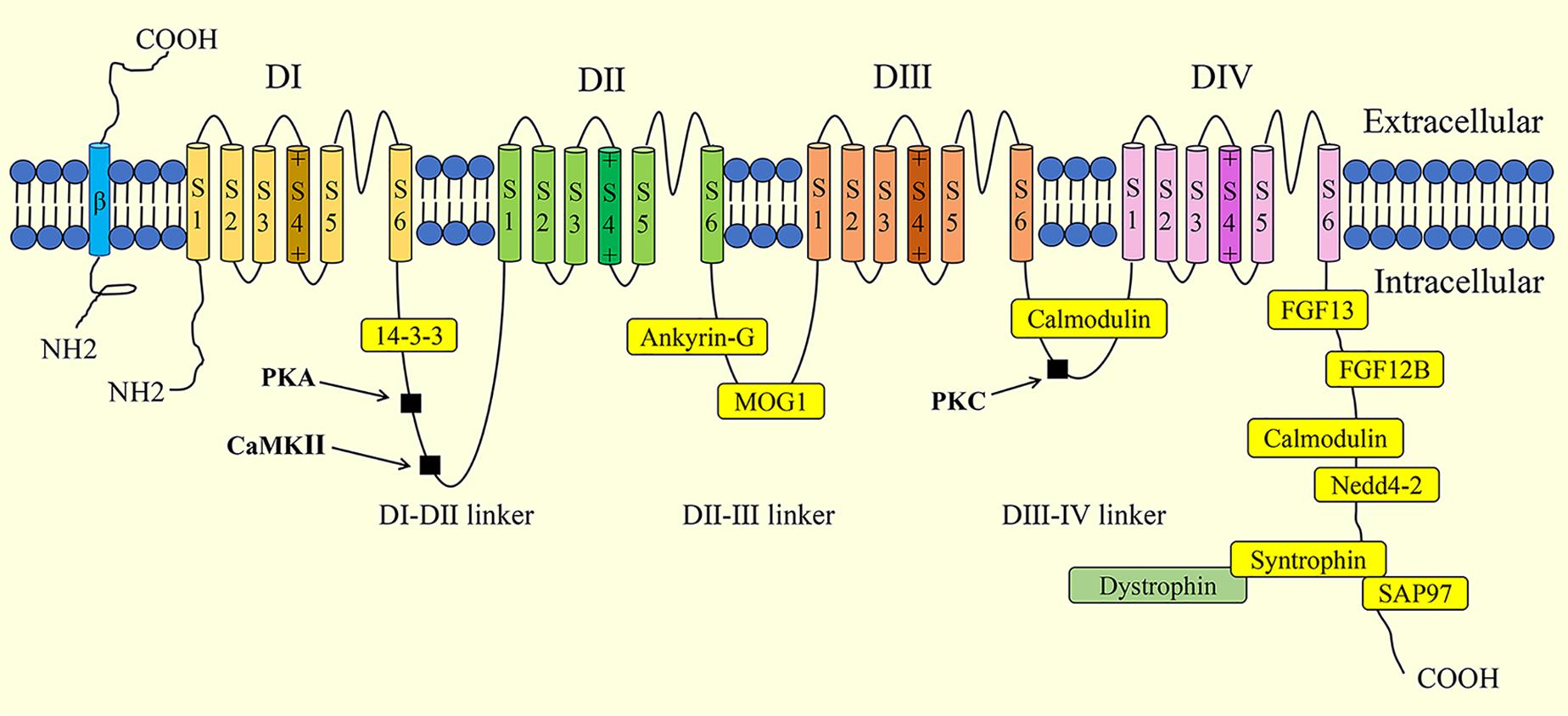
Figure 1. Schematic illustration of the structure of cardiac sodium channel (NaV,1.5) and its interaction proteins. Phosphorylation sites are indicated in black squares. PKA, protein kinase A; CaMKII, Ca2+/Calmodulin-dependent kinase II; PKC, protein kinase C; FGF, fibroblast growth factor; SAP97, synapse-associated protein 97.
The life cycle of NaV1.5 begins with the transcription of the SCN5A gene and RNA processing. Then, NaV1.5 proteins are synthesized at the rough endoplasmic reticulum (ER) and follow the secretory pathway to reach the plasma membrane where the proteins exert their respective function. Various proteins are reported to be involved in the biosynthesis, trafficking, anchoring, post-transcriptional modification, gating regulation, and degradation of NaV1.5 by directly or indirectly interacting with the different motifs and domains of NaV1.5.
The aim of this review is to provide an updated account of the life cycle of NaV1.5, while summarizing SCN5A-related diseases and novel therapeutic strategies targeting NaV1.5.
Gene expression in eukaryotes starts with transcription, the transition of a DNA sequence into an RNA transcript (known as precursor mRNA) assisted with RNA polymerase and transcription factors. The precursor mRNA is the copying of the coding and noncoding genes which then need to undergo splicing and post-transcriptional modifications to produce mature mRNA and ultimately translate into polypeptides (Figure 2). The human SCN5A gene spans approximately 8,000 bp and consists of 28 exons, which can be spliced together to generate the full-length NaV1.5 protein (about 2016 amino acids). Exons 2–28 encode the protein-coding sequence. Exon 1 and part of exon 2 encode the mRNA 5′-untranslated region (5′-UTR). Exon 28 encodes the mRNA 3′-UTR (Wang et al., 1996). The transcription of the SCN5A gene is regulated by various transcription factors. Positive transcription factors such as TBX5 and NF-κB increase SCN5A mRNA abundance and further increase the expression and current of NaV1.5, while negative transcription factors such as FOXO1 and Snail show the opposite effects (Table 1). MicroRNAs (miRNAs) regulate gene expression through translational repression or mRNA degradation. miR-192-5p is reported to negatively regulate the expression of NaV1.5 and reduce INa density by interacting with the 3′-UTR of human SCN5A mRNA (Zhao et al., 2015).
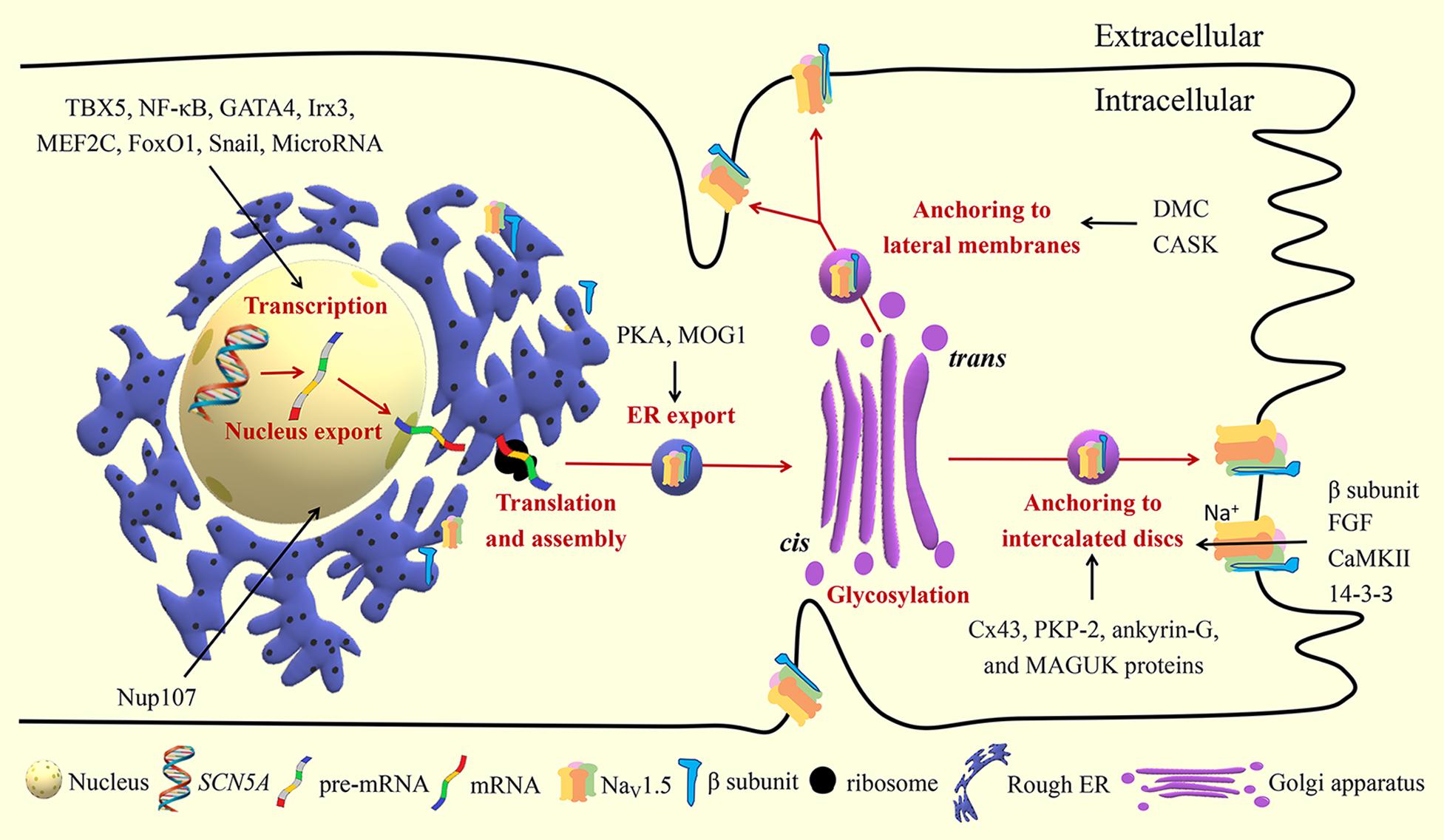
Figure 2. Biosynthesis and trafficking of NaV1.5. TBX5, T-box transcription factor 5; NF-κB, nuclear factor kappa-B; Irx3, Iroquois homeobox 3; MEF2C, myocyte enhancer factor-2C; Forkhead box O1; Nup107, nucleoporin 107; PKA, protein kinase A; Cx43, connexin 43; PKP-2, plakophilin 2; MAGUK, membrane-associated guanylate kinase; DMC, dystrophin-syntrophin multiprotein complex; CASK, calcium/calmodulin-dependent serine protein kinase; FGF, fibroblast growth factor; CaMKII, Ca2 +/ calmodulin-dependent kinase II.
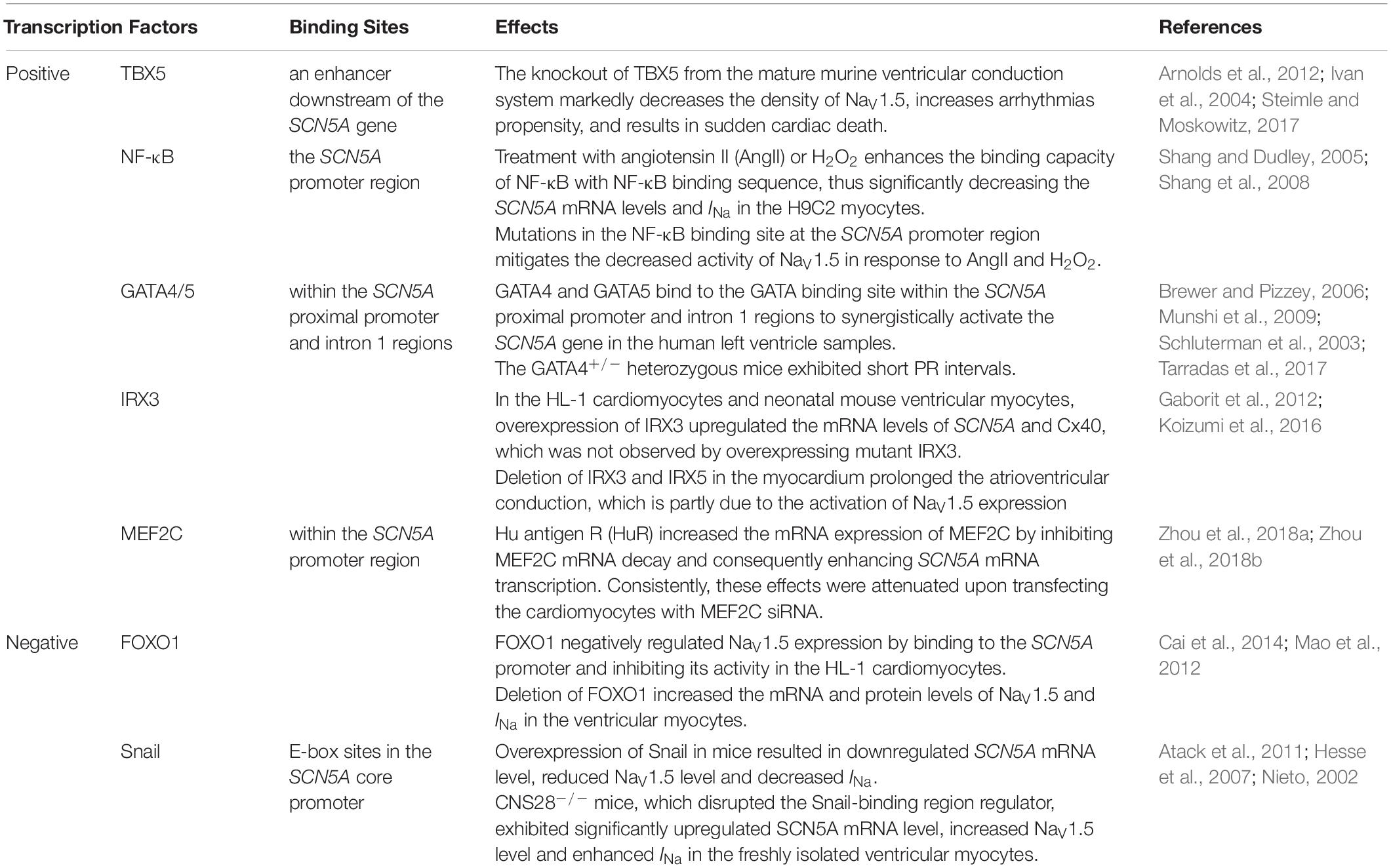
Table 1. Transcription factors of NaV1.5 and their binding sites.
Alternative splicing creates multiple variants of NaV1.5 including functional and nonfunctional types (Schroeter et al., 2010). Compared to full-length NaV1.5, spliced NaV1.5 variants like NaV1.5a, NaV1.5d, and NaV1.5e present altered electrophysiological kinetic properties; NaV1.5c show unchanged electrophysiological properties; NaV1.5b, NaV1.5f, and truncated variants E28B-E28D do not generate currents (Schroeter et al., 2010). The splicing machinery might explain the inconsistent genotype-phenotype outcomes. For instance, the LQT3-associated mutation SCN5A-T1620K showed electrophysiological properties similar to those of hNaV1.5, hNaV1.5a, and hNaV1.5c, but different effects in the hNaV1.5d background (Gaetano et al., 2011). Nevertheless, the exact mechanisms of NaV1.5 splicing regulation are still unclear but involve species-dependent, tissue-specific, and developmental factors.
NaV1.5 splice variants are reported to be involved in the pathophysiology of heart failure (HF), SCN5A channelopathies, and myotonic dystrophy. With angiotensin II (AngII) or hypoxia stimuli (signals common to HF), activated splice factors RBM25 and hLuc7A increase NaV1.5 splice variants E28C and E28D. Increased E28C and E28D variants cause a reading frame shift and encode truncated Na+ channels before DIV of the channel due to premature stop codons (Gao et al., 2011). The nonfunctional channels are trapped in the endoplasmic reticulum (ER) and trigger the unfolded protein response (UPR). UPR activation further leads to destabilization of the remaining full-length SCN5A mRNA, exacerbating the reduction of INa (Gao et al., 2013; Noyes et al., 2017). Studies also found that SCN5A splicing in cardiomyocytes and white blood cell (WBC) changes in a correlated manner during HF. If altered WBCs SCN5A variants were proven to increase the risk of sudden death or other HF-associated arrhythmias, a blood examination to predict HF-associated arrhythmic risk would be possible (Gao and Dudley, 2013).
SCN5A mRNA exported from the nucleus is translated to protein and assembled in the rough ER. Next, the protein is translocated to the Golgi apparatus for further modification. After exiting the Golgi apparatus, NaV1.5 settles in the plasma membrane to exert its functions.
Nuclear pore complexes, also known as nucleoporins, mediate the exchange of molecules between the nucleoplasm and cytoplasm. Nucleoporin 107 (Nup107) selectively facilitates the export of SCN5A mRNA from the nucleus to cytoplasm without affecting the SCN5A mRNA level (Guan et al., 2019). Additionally, increased Nup107 and NaV1.5 were observed in the cardiomyocytes and heart tissues under hypoxic and oxidative stress conditions. This suggests that Nup107 is a protein that reacts rapidly to the insults and can be developed as a novel molecular therapeutic target for myocardial ischemic damage.
As of writing, it is not clear whether there are other nucleoporins that take part in the nucleus-cytoplasmic trafficking of SCN5A mRNA. MOG1 is found to interact with Ran GTP, a small protein that mediates the nucleus import and export of proteins and RNA (Steggerda and Paschal, 2001). MOG1 is shown to promote intracellular trafficking of NaV1.5 from the ER (Yu et al., 2018). Thus, MOG1 may also play a regulatory role in the nucleus-cytoplasmic trafficking of SCN5A mRNA.
ER is the first station of the secretory pathway where the processes of protein folding, quality control, and protein complex assembly take place. Several ER retention and export motifs have been identified in ion channels (Mikosch and Homann, 2009; Benyair et al., 2011). ER export motifs are crucial for efficient ER export of mature and properly folded proteins through coat protein II (COPII) vesicles. COPII-coated vesicles in association with Sec23/24, Sec13/31, Sar1, and Sec12 mediate anterograde protein trafficking between the ER and Golgi (Kurokawa and Nakano, 2019). MOG1 facilitates the export of NaV1.5 from the ER and improves NaV1.5 expression on the cell surface, probably by interacting with Sar1A and Sar1B (Chakrabarti et al., 2013; Wang et al., 2018; Yu et al., 2018; Kurokawa and Nakano, 2019). It is now recognized that ER retention signals can also play an important role in ER export of many plasma membrane proteins. After masking the ER retention signals, the proteins seem to receive an “exit card” and are released from the ER. The DI-II linker region of NaV1.5 contains three putative RXR-type ER retention motifs (479RKR481, 533RRR535, and 659RQR661). Protein kinase A (PKA)-mediated phosphorylation of NaV1.5 masks the retention signals in the DI-II linker region of NaV1.5, and promotes the export of NaV1.5 from the ER to the Golgi (Zhou et al., 2002; Rook et al., 2012).
Nevertheless, the ER export mechanism of NaV1.5 is still poorly understood. Rab GTPases, the largest members of the Ras superfamily, play an important role in protein trafficking between intracellular compartments in eukaryotes (Li and Marlin, 2018). Rab1 and Rab2 are reported to regulate protein transport between the ER and Golgi, but their role in NaV1.5 trafficking is uncertain and needs to be tested in further studies.
Selective Golgi export of proteins from the Golgi to the plasma membrane is another key step in protein trafficking, which has been studied in inward-rectifying potassium channels (Li et al., 2016). However, little is known about the regulation of NaV1.5 transport from the Golgi. It is speculated that Golgi retention and export motifs possibly modulate NaV1.5 exit from the Golgi (Gao et al., 2014).
Cardiac NaV1.5 channels are arranged in clusters, also called pools, in the following membrane microdomains of cardiomyocytes: intercalated discs (ID), T-tubules, costameres, and caveolae. ID mainly consist of three intercellular adhesion structures namely, gap junctions, adherens junctions, and desmosomes (Clark et al., 2002). ID is responsible for electromechanical coupling. The upstroke of AP caused by Na+ influx through NaV1.5 channels can spread quickly through IDs to ensure the coordinated excitation and contraction among cardiomyocytes. T-tubules are tubular invaginations of the lateral sarcolemma that link the sarcoplasmic reticulum and initiate excitation-contraction coupling. AP mediated by NaV1.5 is transmitted along the T-tubules, induces calcium release, and further triggers the contraction of cardiomyocytes. Costameres are sarcolemmal transverse rib-like structures that adhere adjacent cardiomyocytes to the extracellular matrix and maintain the three-dimensional structure of the myocardium (Balse and Eichel, 2018). Caveolae are 50–100 nm cave-like invaginations of the plasma membrane that are made up of essential scaffolding proteins called caveolins. Besides sodium channels (NaV1.5), L-type calcium channels, potassium channels (KV1.5), sodium-calcium exchanger, and Na+/K+ ATPase have been subcellularly localized to caveolae and may allow these channels to be integrated into macromolecular complexes to exert their functions and to be regulated in a unified manner (Balijepalli and Kamp, 2008). T-tubules, costameres, and caveolae are all located in the lateral membranes of cardiomyocytes. NaV1.5 channels are located in different membrane microdomains of cardiomyocytes, which are determined by their specific partners, such as cytoskeleton-binding proteins, gap junctional proteins, and desmosomal proteins (Table 2; Balse and Eichel, 2018).
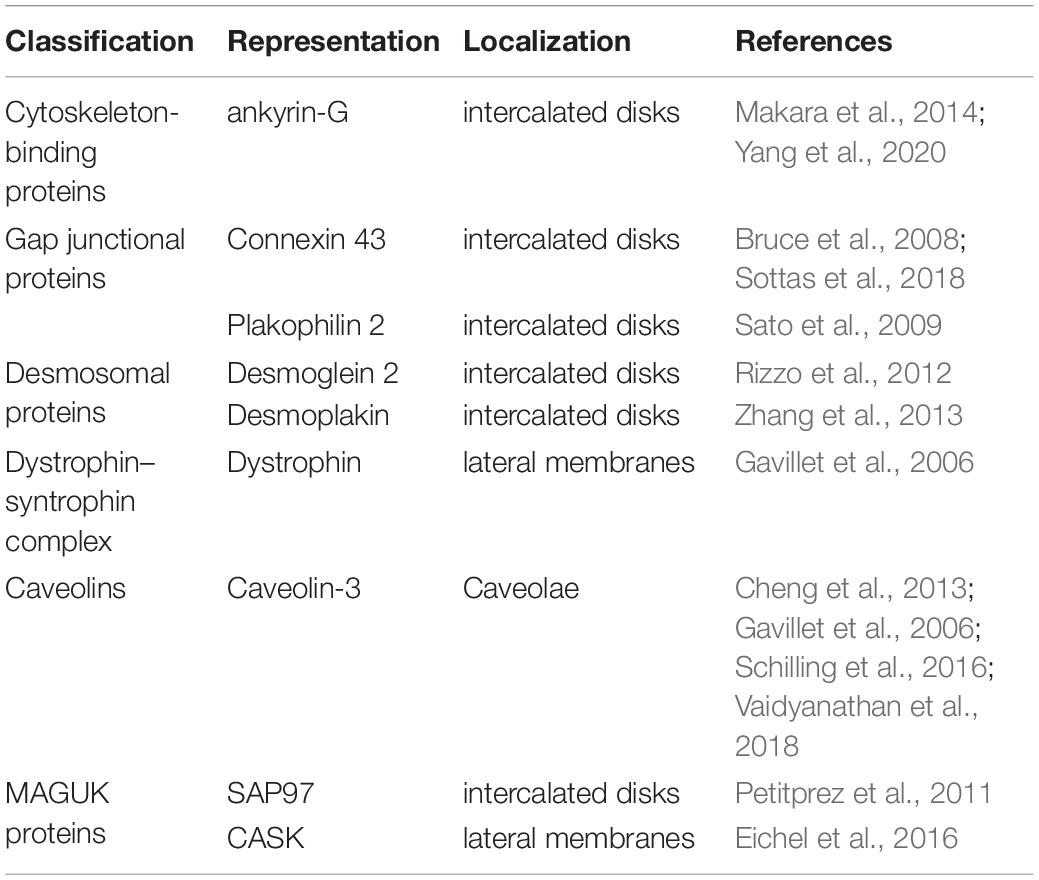
Table 2. Protein partners associated with NaV1.5 localization in the cardiomyocytes.
To our knowledge, a sorting process at the early stages possibly exists, which affects the final anchoring of membrane proteins toward specific microdomains. To better understand the mechanism of NaV1.5 anchoring, it is of great value to investigate the complex processes of sorting, identification by protein chaperones, and organization of microdomains, both in physiological and pathological conditions.
The last step of the NaV1.5 channel life cycle is degradation, which can be considered as a form of retrograde trafficking (Figure 3). There are two major pathways for the degradation of NaV1.5: the proteasome and autophagic degradation pathways.
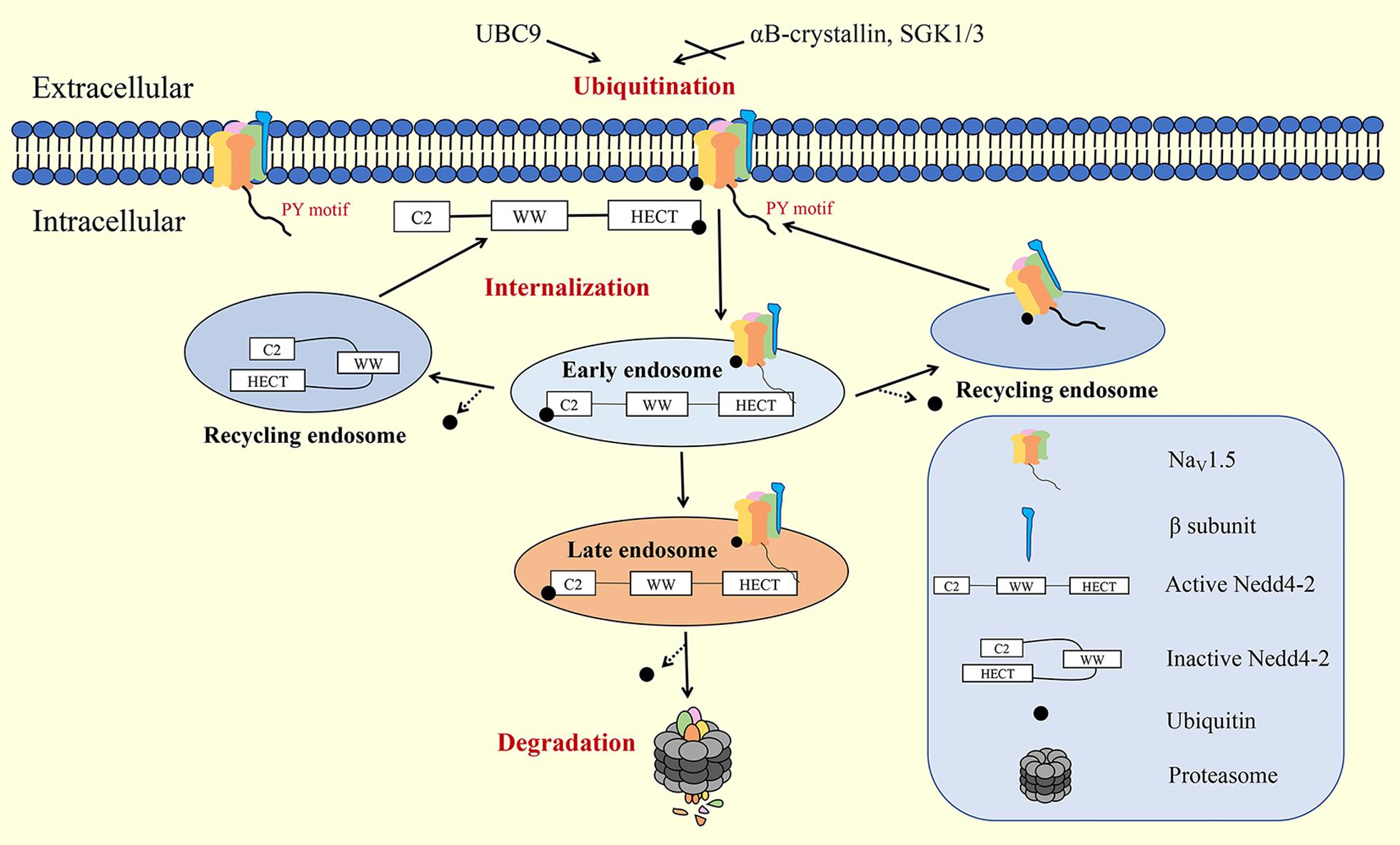
Figure 3. Internalization and degradation of NaV1.5 on the plasma membrane. Nedd4-2, neural precursor cell-expressed developmentally downregulated 4 type 2; SGK, serum- and glucocorticoid-inducible kinase.
Ubiquitination is a common signal for protein internalization and degradation. E3 ubiquitin ligase Nedd4-2 contains WW domains, an N-terminal calcium/protein binding C2 domain, and a C-terminal HECT domain. When the C2 domain binds to the HECT domain, Nedd4-2 presents an inactive state. When Ca2+ binds to the C2 domain, the HECT domain is exposed and Nedd4-2 is activated. Activated Nedd4-2 binds to the PY motifs of target proteins through its WW domains for ubiquitination (Lamothe and Zhang, 2016). Ion channels tagged with ubiquitin are usually compartmentalized into vesicles destined for degradation (by the lysosomal or proteasomal system) or recycled back to the membrane via small GTPase Rabs. Nedd4-2 binds to the PY motif of NaV1.5 through WW domains for ubiquitination, which leads to the rapid internalization and degradation of NaV1.5 (van Bemmelen et al., 2004). Calcium-dependent Nedd4-2 upregulation accelerates the degradation of NaV1.5 and decreases the INac density on the plasma membrane (Luo et al., 2017). αB-crystallin (Huang et al., 2016), serum- and glucocorticoid-inducible kinase (SGK) (Boehmer et al., 2003), and UBC9 (Tang et al., 2019) are reported to interact with Nedd4-2 to regulate the degradation of NaV1.5.
Adenosine 5′-monophosphate-activated protein kinase (AMPK) plays a major role in mediating the autophagic degradation of NaV1.5 during cardiac ischemia and reperfusion (I/R) injury. I/R stimulation was reported to downregulate NaV1.5 abundance in rat hearts. An in vitro study showed that under conditions of I/R stress, NaV1.5 was degraded through the AMPK-mediated autophagic pathway rather than through the proteasome pathway (Tang et al., 2019). AMPK assists NaV1.5 to bind to the autophagic adapter protein and microtubule-associated protein 1 light chain 3 (LC3) by phosphorylating NaV1.5 at threonine (T) 101, and enhances the subsequent degradation through the autophagic pathway (Liu et al., 2019).
NaV1.5 activation depends on the S4 segment, which contains multiple positively charged amino acids that move across the membrane in response to changes in membrane potential. The voltage-sensing domain (VSD), formed by S1-S4, rotates when membrane depolarization moves the S4 segment within DI–DIII outward relative to other channel segments. This allows Na+ to flow inwardly through the pores composed of the S5 and S6 segments and the P loop. Early INa induces rapid AP upstroke, leading to further channel activation. NaV1.5 inactivation involves two distinct processes: fast and slow inactivation. Fast inactivation is coupled to the activation process and occurs within milliseconds. This process may involve an isoleucine-phenylalanine methionine (IFM) motif, which is a highly conserved amino acid triplet in the III–IV linker (Chadda et al., 2017). Unlike fast inactivation, slow inactivation is not coupled to the activation process and can last for as long as a few seconds. Conformational modifications in the following three domains drive the slow inactivation process: the trans-membrane segment S4/D4, the loop between S5-S6/DII (P-loop/DII), and segment S6/DII (Detta et al., 2015). When NaV1.5 channels do not completely inactivate, a lasting current less than 0.5% of the peak current can be detected in the cardiomyocytes, which is known as late INa (INa–L). Because of the short duration of the AP plateau, INa–L can result in situations where the Na+ “load” becomes twice as much as peak INa–P over time, and this plays an important role in angina, arrhythmia, and HF (Makielski, 2016). Many proteins have been identified to modulate the gating properties of NaV1.5, such as β-subunits, FGF, CaMKII, and 14-3-3. Their interacting domains and modulation effects are summarized in Table 3.
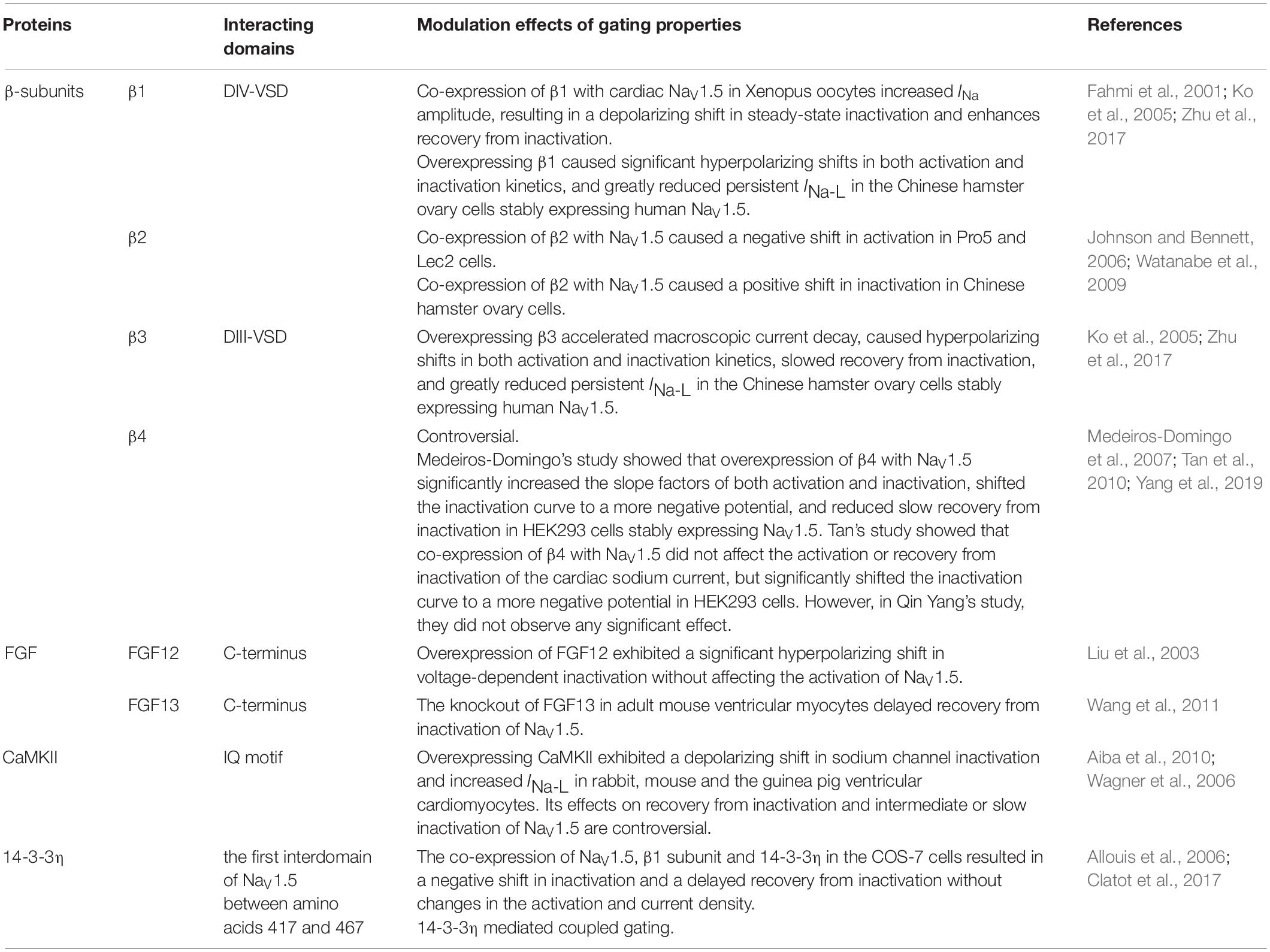
Table 3. Proteins and their modulation effects on the gating properties of NaV1.5 channels.
Interacting proteins modify the structure and properties of NaV1.5 through post-transcriptional modifications (PTMs), such as glycosylation, phosphorylation, methylation, acetylation, and ubiquitylation. PTMs, such as PKA- and CaMKII-mediated phosphorylation and Nedd4-2-mediated ubiquitylation have been discussed in the former sections. We will put more emphasis on the glycosylation, SUMOylation, methylation, and acetylation of NaV1.5 in the following sections.
Many ion channels are known to contain glycan moieties oriented toward the outside of the membrane, which are anchored to residues, such as serine, threonine (O-linked glycosylation), or asparagine (N-linked glycosylation) (Marionneau and Abriel, 2015). Sialic acids, which are negatively charged terminal residues, are subsequently linked with N-glycans or O-glycans through trans-Golgi sialyltransferase activity and contribute to the extracellular surface potential. The NaV is a heavily N-glycosylated protein with carbohydrates that make up 20 to 30% of total mass. However, NaV1.5 is an exception, with approximately 5% by weight of carbohydrates (Mercier et al., 2015). N-glycosylation, which is initiated in the ER and terminated in the Golgi apparatus, is reported to be essential for membrane localization and gating properties. The D25N mutation of the β1 subunit resulted in decreased targeting of NaV1.5 to the plasma membrane due to a maturation (glycosylation) defect (Baroni et al., 2018). Ventricular NaV from ST3Gal4 (a glycogene responsible for glycoprotein sialylation) deficient mice is reported to exhibit gating at higher depolarized potentials, slower inactivation, and faster recovery than in control mice (Ednie et al., 2013).
Arginine methylation within the interdomain linker between DI and DII of NaV1.5 is catalyzed by protein arginine methyltransferases (PRMT), which transfer methyl groups from the cofactor S-adenosyl-L-methionine (SAM) to the protein. Arginine methylation is an important PTM in voltage-gated ion channels. PRMT-3 and -5 methylate NaV1.5 and increase NaV1.5 current density by increasing the cell surface expression in HEK293 cells (Beltran-Alvarez et al., 2013). Arginine methylation of NaV1.5 is also detected in the human heart tissue of patients with end-stage HF. Additionally, R526 methylation is identified as the major NaV1.5 methylation site in patients with end-stage HF (Beltran-Alvarez et al., 2014). Moreover, the methylation-phosphorylation crosstalk of NaV1.5 has been previously reported. NaV1.5 methylation at R513 significantly decreased S516 phosphorylation, which consequently hindered R513 methylation (Beltran-Alvarez et al., 2015).
N-terminal acetylation, which is irreversibly catalyzed by ribosome-associated N-terminal acetyltransferases (NATs), is a major PTM of eukaryotic proteins that regulates protein–protein interactions and membrane targeting. Recently, N-terminal acetylation has been reported to be a degradation signal that inhibits protein targeting to the secretory pathway (Arnesen, 2011). N-terminal acetylation of NaV1.5 at the initial 30 alanine residues was discovered in the hearts of patients with end-stage HF (Beltran-Alvarez et al., 2014). In contrast, N-lysine acetylation, which is catalyzed by histone acetyltransferases (HATs), can be reversed by histone deacetylases and sirtuins. Sirtuin1 deacetylase-mediated acetylation at lysine 1479 of NaV1.5 results in enhanced trafficking to the surface of the plasma membrane and increased INa (Yoon et al., 2017).
SUMOylation, a PTM, is analogous to ubiquitylation involving an enzymatic cascade. In contrast to ubiquitylation, SUMOylation involves conjugating the proteins to small ubiquitin-like modifiers (SUMOs). SUMOylation can affect protein structure and subcellular localization. Recently, NaV1.5 was reported to be SUMOylated only at lysine 442. The mutation of lysine 442 or application of a deSUMOylating enzyme inhibits hypoxic reopening in response to hypoxia. SUMOylation of NaV1.5 is associated with an increase in INa–L (Plant et al., 2020).
Recently, novel post-transcriptional modifications have been reported. Zhang et al. found that an endogenous “lactate clock” turns on gene expression to promote homeostasis in bacterially challenged M1 macrophages (Zhang et al., 2019). The presence of histone lactylation represents a new avenue for understanding the pathophysiological mechanism of many diseases, including ischemic cardiovascular disease. It has been established that NaV1.5 channels are significantly downregulated under conditions of ischemia and hypoxia, which is accompanied by the accumulation of lactic acid. Whether lactylation is involved in the regulation of NaV1.5 under pathological conditions has not yet been revealed.
NaV1.5 is associated with the pathogenesis of various cardiovascular diseases, such as arrhythmia, ischemic cardiomyopathy, heart failure, and sudden cardiac death. Most cases of cardiac arrhythmia and sudden cardiac death are caused by SCN5A-related mutations, which change NaV1.5 expression and function. The gain-of-function mutations in SCN5A, which lead to increased INa–L, can lead to long QT syndrome 3 (LQT3). Loss-of-function mutations in SCN5A, which lead to decreased INa, can cause Brugada syndrome (BrS), sick sinus node syndrome, and atrial fibrillation (Li et al., 2018). To date, the reported BrS-associated SCN5A mutations and LQT3-associated mutations are over 300 and over 80, respectively. Previous studies have demonstrated that NaV1.5 expression is downregulated in HF (Luo et al., 2017). The upregulated expression of Nedd4-2, co-localization of Nedd4-2 with NaV1.5, and increased diastolic calcium concentrations were observed in the volume-overloaded HF rat model, suggesting that Nedd4-2-mediated ubiquitination plays an important role in the downregulation of NaV1.5 in HF (Luo et al., 2017). SCN5A variants are now recognized to be involved in the pathogenesis of HF. A1180V, an SCN5A variant, decreased INa and moderately increased INa–L. A1180V carriers without cardiac dysfunction initially exhibit deteriorated cardiac functions and progress to HF or atrioventricular block during follow-up (Ge et al., 2008). Although some studies have elucidated the correlation between NaV1.5 and HF, the specific mechanisms are not yet known.
Considering the important role of NaV1.5 in these diseases, novel therapeutic strategies targeting the biosynthesis, trafficking, and sodium current of NaV1.5 have been developed.
Several studies have proposed that promoting the process of translational readthrough can enable ribosomes to neglect the stop codon to produce full-length proteins and reduce the harmful results caused by nonsense mutations to a certain degree. Pharmacological strategies to enhance translational readthrough include aminoglycosides, small-interfering RNA (siRNA) that target eukaryotic release factors (eRF), and suppressor tRNAs that recognize stop codons (Carnes et al., 2003). This idea was tested in the nonsense mutation SCN5A-W822X by using aminoglycosides and siRNA against eRF3a and proved effective, which might be a novel treatment strategy for nonsense SCN5A mutation carriers (Teng et al., 2009). However, its application in clinical practice is largely limited due to safety concerns, such as introducing sequence changes to restored full-length proteins.
H558R, a common SCN5A polymorphism, restores the defective trafficking of BrS-associated SCN5A mutations R282H, S216L, K317N, and D1690N by promoting proper protein folding (Marangoni et al., 2011; Shinlapawittayatorn et al., 2011; Nunez et al., 2013). The mechanisms by which H558R rescues SCN5A mutations are controversial. Decreased SCN5A promoter methylation by H588R is one of the mechanisms involved (Matsumura et al., 2017). In an in vitro study, small peptides that span the H558R polymorphism, have been shown to be sufficient in restoring the trafficking defect of BrS-associated SCN5A mutations-R282H, indicating that it might be possible to use R558-containing peptides as a novel strategy tailored to specific BrS mutants (Shinlapawittayatorn et al., 2011). However, the rescue effects in vivo still need to be investigated.
Ranolazine, eleclazine, amiodarone, mexiletine, flecainide, and quinidine targeting INa–L have been widely studied for the treatment of LQT3 patients (Makielski, 2016). For example, we take mexiletine and ranolazine. Ranolazine, which is approved for the treatment of angina pectoris, preferentially reduces INa–L. To date, Ranolazine has been investigated with respect to SCN5A mutations such as ΔKPQ, Y1767C, R1623Q, and D1790G (Moss et al., 2008; Rajamani et al., 2009; Huang et al., 2011; Chorin et al., 2016). Mexiletine, an oral class Ib anti-arrhythmic agent, targets cardiac NaV1.5, and preferentially inhibits INa–L. It has been reported that mexiletine can effectively shorten the QT interval in a subset of patients with LQT3, while other patients are insensitive to the drug (Mazzanti et al., 2016). The conformation of the less activated DIII voltage-sensing domain (DIII-VSD) of NaV1.5 is hypothesized to be the cause for mexiletine insensitivity in these patients (Zhu et al., 2019). A combination of mexiletine with a drug that can promote DIII-VSD activation can be a novel therapeutic strategy for these mexiletine-insensitive patients.
The life cycle of NaV1.5 begins with the transcription and translation of the SCN5A gene. NaV1.5 is then trafficked from the ER to the Golgi apparatus and subsequently to the sarcolemma and cytoskeleton. Finally, NaV1.5 is internalized and degraded. In the past decades, great achievements have been made in the regulation of NaV1.5 from biosynthesis and trafficking to degradation. The knowledge we have generated helps in the understanding of the pathogenesis of SCN5A-related diseases and management of these patients. However, the complete life cycle of NaV1.5 has not been fully mapped. In the end of each section, we have pointed out the corresponding unsolved problems. All these problems can be summarized in two sections: regulation of NaV1.5 under physiological conditions and in pathological conditions. We believe that with joint efforts, we will achieve a deeper understanding of the life cycle of NaV1.5 and explore more effective treatment strategies to reduce mortality and improve the quality of life of patients suffering from SCN5A-related diseases.
CD and YW drafted the manuscript. AM and TW conceived and supervised the review. All authors contributed to the article and approved the submitted version.
This project was supported by the National Natural Science Foundation of China (81870256 to TW and 81600272 to AM) and the Key Program of International Cooperation and Exchanges of Shaanxi, China (2018KW-061 to TW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the highly qualified native English-speaking editors at Wiley Editing Services for editing the English.
Aiba, T., Hesketh, G. G., Liu, T., Carlisle, R., Villa-Abrille, M. C., O’Rourke, B., et al. (2010). Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 85, 454–463. doi: 10.1093/cvr/cvp324
Allouis, M., Le Bouffant, F., Wilders, R., Peroz, D., Schott, J. J., Noireaud, J., et al. (2006). 14-3-3 Is a regulator of the cardiac voltage-gated sodium channel NaV1.5. Circ. Res. 98, 1538–1546. doi: 10.1161/01.RES.0000229244.97497.2c
Arnesen, T. (2011). Towards a functional understanding of protein N-terminal acetylation. PLoS Biol. 9:e1001074. doi: 10.1371/journal.pbio.1001074
Arnolds, D. E., Liu, F., Fahrenbach, J. P., Kim, G. H., Schillinger, K. J., Smemo, S., et al. (2012). TBX5 drives SCN5A expression to regulate cardiac conduction system function. J. Clin. Invest. 122, 2509–2518. doi: 10.1172/JCI62617
Atack, T. C., Stroud, D. M., Watanabe, H., Yang, T., Hall, L., Hipkens, S. B., et al. (2011). Informatic and functional approaches to identifying a regulatory region for the cardiac sodium channel. Circ. Res. 109, 38–46. doi: 10.1161/CIRCRESAHA.110.235630
Balijepalli, R. C., and Kamp, T. J. (2008). Caveolae, ion channels and cardiac arrhythmias. Prog. Biophys. Mol. Biol. 98, 149–160. doi: 10.1016/j.pbiomolbio.2009.01.012
Balse, E., and Eichel, C. (2018). The cardiac sodium channel and its protein partners. Handb. Exp. Pharmacol. 246, 73–99. doi: 10.1007/164_2017_45
Baroni, D., Picco, C., and Moran, O. (2018). A mutation ofSCN1B associated with GEFS+ causes functional and maturation defects of the voltage-dependent sodium channel. Hum. Mutat. 39, 1402–1415. doi: 10.1002/humu.23589
Beltran-Alvarez, P., Espejo, A., Schmauder, R., Beltran, C., Mrowka, R., Linke, T., et al. (2013). Protein arginine methyl transferases-3 and -5 increase cell surface expression of cardiac sodium channel. FEBS Lett. 587, 3159–3165. doi: 10.1016/j.febslet.2013.07.043
Beltran-Alvarez, P., Feixas, F., Osuna, S., Díaz-Hernández, R., Brugada, R., and Pagans, S. (2015). Interplay between R513 methylation and S516 phosphorylation of the cardiac voltage-gated sodium channel. Amino Acids 472, 429–434. doi: 10.1007/s00726-014-1890-0
Beltran-Alvarez, P., Tarradas, A., Chiva, C., Perez-Serra, A., Batlle, M., Perez-Villa, F., et al. (2014). Identification of N-terminal protein acetylation and arginine methylation of the voltage-gated sodium channel in end-stage heart failure human heart. J. Mol. Cell Cardiol. 76, 126–129. doi: 10.1016/j.yjmcc.2014.08.014
Benyair, R., Ron, E., and Lederkremer, G. Z. (2011). Protein quality control, retention, and degradation at the endoplasmic reticulum. Int. Rev. Cell Mol. Biol. 292, 197–280. doi: 10.1016/B978-0-12-386033-0.00005-0
Boehmer, C., Wilhelm, V., Palmada, M., Wallisch, S., Henke, G., Brinkmeier, H., et al. (2003). Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc. Res. 57, 1079–1084. doi: 10.1016/s0008-6363(02)00837-4
Brewer, A., and Pizzey, J. (2006). GATA factors in vertebrate heart development and disease. Exp. Rev. Mol. Med. 8, 1–20. doi: 10.1017/S1462399406000093
Bruce, A. F., Rothery, S., Dupont, E., and Severs, N. J. (2008). Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc. Res. 77, 757–765. doi: 10.1093/cvr/cvm083
Cai, B., Wang, N., Mao, W., You, T., Lu, Y., Li, X., et al. (2014). Deletion of FoxO1 leads to shortening of QRS by increasing Na+ channel activity through enhanced expression of both cardiac NaV1.5 and β3 subunit. J. Mol. Cell Cardiol. 74, 297–306. doi: 10.1016/j.yjmcc.2014.06.006
Carnes, J., Jacobson, M., Leinwand, L., and Yarus, M. (2003). Stop codon suppression via inhibition of eRF1 expression. RNA 9, 648–653. doi: 10.1261/rna.5280103
Chadda, K. R., Jeevaratnam, K., Lei, M., and Huang, C. L. (2017). Sodium channel biophysics, late sodium current and genetic arrhythmic syndromes. Pflug. Arch. 469, 629–641. doi: 10.1007/s00424-017-1959-1
Chakrabarti, S., Wu, X., Yang, Z., Wu, L., Yong, S. L., Zhang, C., et al. (2013). MOG1 rescues defective trafficking of NaV1.5 mutations in Brugada syndrome and sick sinus syndrome. Circ. Arrhythm Electrophysiol. 6, 392–401. doi: 10.1161/CIRCEP.111.000206
Cheng, J., Valdivia, C. R., Vaidyanathan, R., Balijepalli, R. C., Ackerman, M. J., and Makielski, J. C. (2013). Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J. Mol. Cell Cardiol. 61, 102–110. doi: 10.1016/j.yjmcc.2013.03.013
Chorin, E., Hu, D., Antzelevitch, C., Hochstadt, A., Belardinelli, L., Zeltser, D., et al. (2016). Ranolazine for congenital Long-QT Syndrome Type III: experimental and long-term clinical data. Circ. Arrhythm Electrophysiol. 9:e004370. doi: 10.1161/CIRCEP.116.004370
Clark, K. A., McElhinny, A. S., Beckerle, M. C., and Gregorio, C. C. (2002). Striated muscle cytoarchitecture: an intricate web of form and function. Annu. Rev. Cell Dev. Biol. 18, 637–706. doi: 10.1146/annurev.cellbio.18.012502.105840
Clatot, J., Hoshi, M., Wan, X. P., Liu, H. Y., Jain, A., Shinlapawittayatorn, K., et al. (2017). Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 8:2077. doi: 10.1038/s41467-017-02262-0
Detta, N., Frisso, G., and Salvatore, F. (2015). The multi-faceted aspects of the complex cardiac NaV1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta 1854, 1502–1509. doi: 10.1016/j.bbapap.2015.07.009
Ednie, A. R., Horton, K. K., Wu, J., and Bennett, E. S. (2013). Expression of the sialyltransferase, ST3Gal4, impacts cardiac voltage-gated sodium channel activity, refractory period and ventricular conduction. J. Mol. Cell Cardiol. 59, 117–127. doi: 10.1016/j.yjmcc.2013.02.013
Eichel, C. A., Beuriot, A., Chevalier, M. Y. E., Rougier, J.-S., Louault, F., Dilanian, G., et al. (2016). Lateral membrane-specific MAGUK CASK down-regulates NaV1.5 channel in cardiac myocytes. Circ. Res. 119, 544–556. doi: 10.1161/CIRCRESAHA.116.309254
Fahmi, A. I., Patel, M., Stevens, E. B., Fowden, A. L., John, I. I. I. J. E., Lee, K., et al. (2001). The sodium channel b-subunit SCN3b modulates the kinetics of SCN5A and is expressed heterogeneously in sheep heart. J. Physiol. 537, 693–700. doi: 10.1111/j.1469-7793.2001.00693.x
Gaborit, N., Sakuma, R., Wylie, J. N., Kim, K. H., Zhang, S. S., Hui, C. C., et al. (2012). Cooperative and antagonistic roles for Irx3 and Irx5 in cardiac morphogenesis and postnatal physiology. Development 139, 4007–4019. doi: 10.1242/dev.081703
Gaetano, C., Walzik, S., Schroeter, A., Benndorf, K., and Zimmer, T. (2011). Alternative splicing of the cardiac sodium channel creates multiple variants of mutant T1620K channels. PLoS One 6:e0019188. doi: 10.1371/journal.pone.0019188
Gao, C., Cai, Y., Wang, Y., Kang, B. H., Aniento, F., Robinson, D. G., et al. (2014). Retention mechanisms for ER and Golgi membrane proteins. Trends Plant Sci. 19, 508–515. doi: 10.1016/j.tplants.2014.04.004
Gao, G., and Dudley, S. C. Jr. (2013). SCN5A splicing variants and the possibility of predicting heart failure-associated arrhythmia. Exp. Rev. Cardiovasc. Ther. 11, 117–119. doi: 10.1586/erc.12.180
Gao, G., Xie, A., Huang, S. C., Zhou, A., Zhang, J., Herman, A. M., et al. (2011). Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation 124, 1124–1131. doi: 10.1161/CIRCULATIONAHA.111.044495
Gao, G., Xie, A., Zhang, J., Herman, A. M., Jeong, E. M., Gu, L., et al. (2013). Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ. Arrhythm Electrophysiol. 6, 1018–1024. doi: 10.1161/CIRCEP.113.000274
Gavillet, B., Rougier, J. S., Domenighetti, A. A., Behar, R., Boixel, C., Ruchat, P., et al. (2006). Cardiac sodium channel NaV1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 99, 407–414. doi: 10.1161/01.RES.0000237466.13252.5e
Ge, J., Sun, A., Paajanen, V., Wang, S., Su, C., Yang, Z., et al. (2008). Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ. Arrhythm. Electrophysiol. 1, 83–92. doi: 10.1161/CIRCEP.107.750752
Guan, Y., Gao, X., Tang, Q., Huang, L., Gao, S., Yu, S., et al. (2019). Nucleoporin 107 facilitates the nuclear export ofSCN5A mRNA to regulate cardiac bioelectricity. J. Cell Mol. Med. 23, 1448–1457. doi: 10.1111/jcmm.14051
Han, D., Tan, H., Sun, C., and Li, G. (2018). Dysfunctional NaV1.5 channels due to SCN5A mutations. Exp. Biol. Med. 243, 852–863. doi: 10.1177/1535370218777972
Hesse, M., Kondo, C. S., Clark, R. B., Su, L., Allen, F. L., Geary-Joo, C. T. M., et al. (2007). Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel SCN5A. Cardiovasc. Res. 75, 498–509. doi: 10.1016/j.cardiores.2007.04.009
Huang, H., Priori, S. G., Napolitano, C., O’Leary, M. E., and Chahine, M. (2011). Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of NaV1.5 channels. Am. J. Physiol. Heart Circ. Physiol. 300, H288–H299. doi: 10.1152/ajpheart.00539.2010
Huang, Y., Wang, Z., Liu, Y., Xiong, H., Zhao, Y., Wu, L., et al. (2016). αB-Crystallin interacts with NaV1.5 and regulates ubiquitination and internalization of cell surface Nav1.5. J. Biol. Chem. 291, 11030–11041. doi: 10.1074/jbc.M115.695080
Ivan, P. G. M., Pizard, A., Patel, V. V., Bruneau, B. G., Kim, J. B., Kupershmidt, S., et al. (2004). The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 131, 4107–4116. doi: 10.1242/dev.01265
Jiang, D., Shi, H., Tonggu, L., Gamal El-Din, T. M., Lenaeus, M. J., Zhao, Y., et al. (2020). Structure of the cardiac sodium channel. Cell 180, 122–134. doi: 10.1016/j.cell.2019.11.041
Johnson, D., and Bennett, E. S. (2006). Isoform-specific effects of the beta2 subunit on voltage-gated sodium channel gating. J. Biol. Chem. 281, 25875–25881. doi: 10.1074/jbc.M605060200
Ko, S. H., Lenkowski, P. W., Lee, H. C., Mounsey, J. P., and Patel, M. K. (2005). Modulation of Na(v)1.5 by beta1– and beta3-subunit co-expression in mammalian cells. Pflug. Arch. 449, 403–412. doi: 10.1007/s00424-004-1348-4
Koizumi, A., Sasano, T., Kimura, W., Miyamoto, Y., Aiba, T., Ishikawa, T., et al. (2016). Genetic defects in a His-Purkinje system transcription factor. IRX3, cause lethal cardiac arrhythmias. Eur. Heart J. 37, 1469–1475. doi: 10.1093/eurheartj/ehv449
Kurokawa, K., and Nakano, A. (2019). The ER exit sites are specialized ER zones for the transport of cargo proteins from the ER to the Golgi apparatus. J. Biochem. 165, 109–114. doi: 10.1093/jb/mvy080
Lamothe, S. M., and Zhang, S. (2016). Chapter Five - Ubiquitination of ion channels and transporters. Prog. Mol. Biol. Transl. Sci. 141, 161–223. doi: 10.1016/bs.pmbts.2016.02.005
Li, G., and Marlin, M. C. (2018). Rab Family of GTPases. Methods Mol. Biol. 1298, 1–15. doi: 10.1007/978-1-4939-2569-8_1
Li, W., Yin, L., Shen, C., Hu, K., Ge, J., and Sun, A. (2018). SCN5A variants: association with cardiac disorders. Front. Physiol. 9:1372. doi: 10.3389/fphys.2018.01372
Li, X., Ortega, B., Kim, B., and Welling, P. A. (2016). A common signal patch drives AP-1 Protein-dependent Golgi export of inwardly rectifying potassium channels. J. Biol. Chem. 291, 14963–14972. doi: 10.1074/jbc.M116.729822
Liu, C.-J., Dib-Hajj, S. D., Renganathan, M., Cummins, T. R., and Waxman, S. G. (2003). Modulation of the cardiac sodium channel NaV1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 278, 1029–1036. doi: 10.1074/jbc.M207074200
Liu, X. H., Chen, Z., Han, Z. L., Liu, Y., Wu, X., Peng, Y. Z., et al. (2019). AMPK-mediated degradation of NaV1.5 through autophagy. FASEB J. 33, 5366–5376. doi: 10.1096/fj.201801583RR
Luo, L., Ning, F., Du, Y., Song, B., Yang, D., Salvage, S. C., et al. (2017). Calcium-dependent Nedd4-2 upregulation mediates degradation of the cardiac sodium channel NaV1.5: implications for heart failure. Acta Physiol. 221, 44–58. doi: 10.1111/apha.12872
Makara, M. A., Curran, J., Little, S. C., Musa, H., Polina, I., Smith, S. A., et al. (2014). Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 115, 929–938. doi: 10.1161/CIRCRESAHA.115.305154
Makielski, J. C. (2016). Late sodium current: a mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc. Med. 26, 115–122. doi: 10.1016/j.tcm.2015.05.006
Mao, W., You, T., Ye, B., Li, X., Dong, H. H., Hill, J. A., et al. (2012). Reactive oxygen species suppress cardiac NaV1.5 expression through Foxo1. PLoS One 7:e32738. doi: 10.1371/journal.pone.0032738
Marangoni, S., Di Resta, C., Rocchetti, M., Barile, L., Rizzetto, R., Summa, A., et al. (2011). A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: standard and dynamic characterization. Cardiovasc. Res. 91, 606–616. doi: 10.1093/cvr/cvr142
Marionneau, C., and Abriel, H. (2015). Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell Cardiol. 82, 36–47. doi: 10.1016/j.yjmcc.2015.02.013
Matsumura, H., Nakano, Y., Ochi, H., Onohara, Y., Sairaku, A., Tokuyama, T., et al. (2017). H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. J Biomed. Sci. 24:91. doi: 10.1186/s12929-017-0397-x
Mazzanti, A., Maragna, R., Faragli, A., Monteforte, N., Bloise, R., Memmi, M., et al. (2016). Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J. Am. Coll. Cardiol. 67, 1053–1058. doi: 10.1016/j.jacc.2015.12.033106
Medeiros-Domingo, A., Kaku, T., Tester, D. J., Iturralde-Torres, P., Itty, A., Ye, B., et al. (2007). SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134–142. doi: 10.1161/CIRCULATIONAHA.106.659086
Mercier, A. L., Clément, R., Harnois, T., Bourmeyster, N., Bois, P., and Chatelier, A. (2015). NaV1.5 channels can reach the plasma membrane through distinct N-glycosylation states. BBA Gen Subj. 1850, 1215–1223.
Mikosch, M., and Homann, U. (2009). How do ER export motifs work on ion channel trafficking? Curr. Opin. Plant Biol. 12, 685–689. doi: 10.1016/j.pbi.2009.09.020
Moss, A. J., Zareba, W., Schwarz, K. Q., Rosero, S., McNitt, S., and Robinson, J. L. (2008). Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J. Cardiovasc. Electrophysiol. 19, 1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x
Munshi, N. V., McAnally, J., Bezprozvannaya, S., Berry, J. M., Richardson, J. A., Hill, J. A., et al. (2009). Cx30.2 enhancer analysis identifies Gata4 as a novel regulator of atrioventricular delay. Development 136, 2665–2674. doi: 10.1242/dev.038562
Nieto, M. A. (2002). The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 3, 155–166. doi: 10.1038/nrm757
Noyes, A. M., Zhou, A., Gao, G., Gu, L., Day, S., Andrew Wasserstrom, J., et al. (2017). Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy. Int. J. Cardiol. 249, 282–286. doi: 10.1016/j.ijcard.2017.08.071
Nunez, L., Barana, A., Amoros, I., de la Fuente, M. G., Dolz-Gaiton, P., Gomez, R., et al. (2013). p.D1690N NaV1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm 10, 264–272. doi: 10.1016/j.hrthm.2012.10.025
Petitprez, S., Zmoos, A. F., Ogrodnik, J., Balse, E., Raad, N., El-Haou, S., et al. (2011). SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels NaV1.5 in cardiomyocytes. Circ. Res. 108, 294–304. doi: 10.1161/CIRCRESAHA.110.228312
Plant, L. D., Xiong, D., Romero, J., Dai, H., and Goldstein, S. A. N. (2020). Hypoxia produces pro-arrhythmic late sodium current in cardiac myocytes by SUMOylation of NaV1.5 Channels. Cell Rep. 30, 2225–2236. doi: 10.1016/j.celrep.2020.01.025
Rajamani, S., El-Bizri, N., Shryock, J. C., Makielski, J. C., and Belardinelli, L. (2009). Use-dependent block of cardiac late Na(+) current by ranolazine. Heart Rhythm 6, 1625–1631. doi: 10.1016/j.hrthm.2009.07.042
Rizzo, S., Lodder, E. M., Verkerk, A. O., Wolswinkel, R., Beekman, L., Pilichou, K., et al. (2012). Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 95, 409–418. doi: 10.1093/cvr/cvs219
Rook, M. B., Evers, M. M., Vos, M. A., and Bierhuizen, M. F. (2012). Biology of cardiac sodium channel NaV1.5 expression. Cardiovasc. Res. 93, 12–23. doi: 10.1093/cvr/cvr252
Salvage, S. C., Huang, C. L., and Jackson, A. P. (2020). Cell-adhesion properties of beta-subunits in the regulation of cardiomyocyte sodium channels. Biomolecules 10:989. doi: 10.3390/biom10070989
Sato, P. Y., Musa, H., Coombs, W., Guerrero-Serna, G., Patino, G. A., Taffet, S. M., et al. (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 105, 523–526. doi: 10.1161/CIRCRESAHA.109.201418
Schilling, J. M., Horikawa, Y. T., Zemljic-Harpf, A. E., Vincent, K. P., Tyan, L., Yu, J. K., et al. (2016). Electrophysiology and metabolism of caveolin-3-overexpressing mice. Basic Res. Cardiol. 111:28. doi: 10.1007/s00395-016-0542-9
Schluterman, M. K., Butler, C. A., Cohen, J. C., King, I. N., Srivastava, D., Eapen, R. S., et al. (2003). GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424, 443–447. doi: 10.1038/nature01827
Schroeter, A., Walzik, S., Blechschmidt, S., Haufe, V., Benndorf, K., and Zimmer, T. (2010). Structure and function of splice variants of the cardiac voltage-gated sodium channel Na(v)1.5. J. Mol. Cell Cardiol. 49, 16–24. doi: 10.1016/j.yjmcc.2010.04.004
Shang, L. L., and Dudley, S. C. (2005). Tandem promoters and developmentally regulated 5’- and 3’-mRNA untranslated regions of the mouse SCN5A cardiac sodium channel. J. Biol. Chem. 280, 933–940. doi: 10.1074/jbc.M409977200
Shang, L. L., Sanyal, S., Pfahnl, A. E., Jiao, Z., Allen, J., Liu, H., et al. (2008). NF-κB-dependent transcriptional regulation of the cardiac SCN5A sodium channel by angiotensin II. Am. J. Physiol. Cell Ph 294, 372–379. doi: 10.1152/ajpcell.00186.2007
Shinlapawittayatorn, K., Dudash, L. A., Du, X. X., Heller, L., Poelzing, S., Ficker, E., et al. (2011). A novel strategy using cardiac sodium channel polymorphic fragments to rescue trafficking-deficient SCN5A mutations. Circ. Cardiovasc. Genet. 4, 500–509. doi: 10.1161/CIRCGENETICS.111.960633
Sottas, V., Wahl, C. M., Trache, M. C., Bartolf-Kopp, M., Cambridge, S., Hecker, M., et al. (2018). Improving electrical properties of iPSC-cardiomyocytes by enhancing Cx43 expression. J. Mol. Cell Cardiol. 120, 31–41. doi: 10.1016/j.yjmcc.2018.05.010
Steggerda, S. M., and Paschal, B. M. (2001). Identification of a conserved loop in mog1 that releases GTP from Ran. Traffic 211, 804–811. doi: 10.1034/j.1600-0854.2001.21109.x
Steimle, J. D., and Moskowitz, I. P. (2017). TBX5: a key regulator of heart development. Curr. Top. Dev. Biol. 122, 195–221. doi: 10.1016/bs.ctdb.2016.08.008
Tan, B. H., Pundi, K. N., Van Norstrand, D. W., Valdivia, C. R., Tester, D. J., Medeiros-Domingo, A., et al. (2010). Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm 7, 771–778. doi: 10.1016/j.hrthm.2010.01.032
Tang, B., Hu, Y., Wang, Z., Cheng, C., Wang, P., Liang, L., et al. (2019). UBC9 regulates cardiac sodium channel NaV1.5 ubiquitination, degradation and sodium current density. J. Mol. Cell Cardiol. 129, 79–91. doi: 10.1016/j.yjmcc.2019.02.007
Tarradas, A., Pinsach-Abuin, M. L., Mackintosh, C., Llora-Batlle, O., Perez-Serra, A., Batlle, M., et al. (2017). Transcriptional regulation of the sodium channel gene (SCN5A) by GATA4 in human heart. J. Mol. Cell Cardiol. 102, 74–82. doi: 10.1016/j.yjmcc.2016.10.013
Teng, S., Gao, L., Paajanen, V., Pu, J., and Fan, Z. (2009). Readthrough of nonsense mutation W822X in the SCN5A gene can effectively restore expression of cardiac Na+ channels. Cardiovasc. Res. 83, 473–480. doi: 10.1093/cvr/cvp116
Vaidyanathan, R., Reilly, L., and Eckhardt, L. L. (2018). Caveolin-3 microdomain: arrhythmia implications for potassium inward rectifier and cardiac sodium channel. Front. Physiol. 9:1548. doi: 10.3389/fphys.2018.01548
van Bemmelen, M. X., Rougier, J.-S., Gavillet, B., Apothéloz, F., Daidié, D., Tateyama, M., et al. (2004). Cardiac voltage-gated sodium channel NaV1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ. Res. 95, 284–291. doi: 10.1161/01.RES.0000136816.05109.89
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/Jci26620
Wang, C., Hennessey, J. A., Kirkton, R. D., Wang, C., Graham, V., Puranam, R. S., et al. (2011). Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 109, 775–782. doi: 10.1161/CIRCRESAHA.111.247957
Wang, Q., Li, Z., Shen, J., and Keating, M. T. (1996). Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics 34, 9–16. doi: 10.1006/geno.1996.0236
Wang, Z., Yu, G., Liu, Y., Liu, S., Aridor, M., Huang, Y., et al. (2018). Small GTPases SAR1A and SAR1B regulate the trafficking of the cardiac sodium channel NaV1.5. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3672–3684. doi: 10.1016/j.bbadis.2018.09.003
Watanabe, H., Darbar, D., Kaiser, D. W., Jiramongkolchai, K., Chopra, S., Donahue, B. S., et al. (2009). Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275. doi: 10.1161/CIRCEP.108.779181
Yang, H.-Q., Pérez-Hernández, M., Sanchez-Alonso, J., Shevchuk, A., Gorelik, J., Rothenberg, E., et al. (2020). Ankyrin-G mediates targeting of both Na and K channels to the rat cardiac intercalated disc. eLife 9:e52373. doi: 10.7554/eLife.52373
Yang, Q., Xiong, H., Xu, C., Huang, Y., Tu, X., Wu, G., et al. (2019). Identification of rare variants in cardiac sodium channel beta4-subunit gene SCN4B associated with ventricular tachycardia. Mol. Genet. Genom. 294, 1059–1071. doi: 10.1007/s00438-019-01567-7
Yoon, J. Y., Vikram, A., London, B., and Irani, K. (2017). Reversible lysine acetylation: another layer of post-translational regulation of the cardiac sodium channel. Channels 11, 360–361. doi: 10.1080/19336950.2017.1340015
Yu, G., Liu, Y., Qin, J., Wang, Z., Hu, Y., Wang, F., et al. (2018). Mechanistic insights into the interaction of the MOG1 protein with the cardiac sodium channel Nav 1.5 clarify the molecular basis of Brugada syndrome. J. Biol. Chem. 293, 18207–18217. doi: 10.1074/jbc.RA118.003997
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., Weng, Y., et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. doi: 10.1038/s41586-019-1678-1
Zhang, Q., Deng, C., Rao, F., Modi, R. M., Zhu, J., Liu, X., et al. (2013). Silencing of desmoplakin decreases connexin43/NaV1.5 expression and sodium current in HL1 cardiomyocytes. Mol. Med. Rep. 8, 780–786. doi: 10.3892/mmr.2013.1594
Zhao, Y., Huang, Y., Li, W., Wang, Z., Zhan, S., Zhou, M., et al. (2015). Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim. Biophys. Acta 1852, 2024–2034. doi: 10.1016/j.bbadis.2015.07.016
Zhou, A., Shi, G., Kang, G. J., Xie, A., Liu, H., Jiang, N., et al. (2018a). RNA binding protein, HuR, regulates SCN5A expression through stabilizing MEF2C transcription factor mRNA. J. Am. Heart Assoc. 7:e007802. doi: 10.1161/JAHA.117.007802
Zhou, A., Xie, A., Kim, T. Y., Liu, H., Shi, G., Kang, G.-J., et al. (2018b). HuR-mediated SCN5A mRNA stability reduces arrhythmic risk in heart failure. Heart Rhythm 15, 1072–1080. doi: 10.1016/j.hrthm.2018.02.018
Zhou, J., Shin, H. G., Yi, J., Shen, W., Williams, C. P., and Murray, K. T. (2002). Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ. Res. 91, 540–546. doi: 10.1161/01.res.0000033598.00903.27
Zhu, W., Mazzanti, A., Voelker, T. L., Hou, P., Moreno, J. D., Angsutararux, P., et al. (2019). Predicting patient response to the antiarrhythmic mexiletine based on genetic variation. Circ. Res. 124, 539–552. doi: 10.1161/CIRCRESAHA.118.314050
Keywords: NaV1.5, SCN5A, biosynthesis, trafficking, anchoring, degradation, gating modulation, post-transcriptional modification
Citation: Dong C, Wang Y, Ma A and Wang T (2020) Life Cycle of the Cardiac Voltage-Gated Sodium Channel NaV1.5. Front. Physiol. 11:609733. doi: 10.3389/fphys.2020.609733
Received: 25 September 2020; Accepted: 12 November 2020;
Published: 17 December 2020.
Edited by:
Elena V. Zaklyazminskaya, Russian National Research Center of Surgery named after B.V. Petrovsky, RussiaReviewed by:
Viacheslav Nikolaev, University of Göttingen, GermanyCopyright © 2020 Dong, Wang, Ma and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiqun Ma, bWFhaXF1bjExMDhAMTI2LmNvbQ==; Tingzhong Wang, dGluZ3pob25nLndhbmdAeGp0dS5lZHUuY24=
†ORCID: Tingzhong Wang, orcid.org/0000-0002-6390-7967
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.