
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 08 December 2020
Sec. Craniofacial Biology and Dental Research
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.606889
This article is part of the Research TopicCranial Placodes and Neural Crest Interactions in Craniofacial DevelopmentView all 15 articles
 Elizabeth N. Schock1
Elizabeth N. Schock1 Carole LaBonne1,2*
Carole LaBonne1,2*Sox transcription factors play many diverse roles during development, including regulating stem cell states, directing differentiation, and influencing the local chromatin landscape. Of the twenty vertebrate Sox factors, several play critical roles in the development the neural crest, a key vertebrate innovation, and the subsequent formation of neural crest-derived structures, including the craniofacial complex. Herein, we review the specific roles for individual Sox factors during neural crest cell formation and discuss how some factors may have been essential for the evolution of the neural crest. Additionally, we describe how Sox factors direct neural crest cell differentiation into diverse lineages such as melanocytes, glia, and cartilage and detail their involvement in the development of specific craniofacial structures. Finally, we highlight several SOXopathies associated with craniofacial phenotypes.
Since the hallmark discovery of the SRY gene, the master regulator of male sex determination (Gubbay et al., 1990; Lovell-Badge, 2010), twenty mammalian SRY-related HMG box containing (SOX) transcription factors have been identified. A significant number of important developmental functions have been described for Sox transcription factors. These range from maintaining stem cell states to promoting differentiation (reviewed in Kamachi and Kondoh, 2013). The growing number of developmental disorders associated with mutations in SOX genes underscores the importance of these factors during development (Angelozzi and Lefebvre, 2019). In this review we focus on the Sox factors that have roles in the formation of the neural crest as well as those important for the development of cell types within and components of the craniofacial complex. Finally, we highlight the “SOXopathies” that are associated with a variety of craniofacial phenotypes.
The twenty mammalian Sox factors are divided into nine subfamilies (A, B1, B2-H) based mainly upon the homology of a 79 amino acid DNA binding region termed the High Mobility Group (HMG) domain (Figure 1A; Bowles et al., 2000). Duplication events, slow divergence, and co-option of functional elements are hypothesized to have driven Sox family evolution (Bowles et al., 2000). Consistent with this, members of the same subfamily often having overlapping expression patterns and various degrees of functional redundancy (Figure 1B; Heenan et al., 2016). SOX factors bind DNA at C[A/T]TTG[T/A][T/A] sequences or similar motifs. Notably, the HMG domain binds DNA at the minor groove causing the DNA to bend. This facilitates local chromatin modifications and can increase DNA accessibility for partner factor binding (Hou et al., 2017). Furthermore, some SOX factors (SOX2 and SOX9) have been shown to engage at regions of condensed chromatin and are considered pioneer factors (Adam et al., 2015; Soufi et al., 2015; Julian et al., 2017). SOX2, through the HMG domain, recognizes a degenerate Sox motif on nucleosomal DNA (Soufi et al., 2015). At these degenerate sites less DNA bending occurs which facilitates SOX2 binding on the minor groove of nucleosomal DNA (Soufi et al., 2015). At some of these sites, binding may, in part, be facilitated or stabilized by chromatin-associated proteins such as poly(ADP-ribose) polymerase-1 (PARP-1) (Liu and Kraus, 2017). The extent to which SOX9’s pioneer activity mechanistically mimics that of SOX2 is not known and is an area ripe for future investigation. In order to activate gene expression, SOX factors generally require cooperation with a partner factor (reviewed in Kondoh and Kamachi, 2010). Examples of this include SOX10-MITF pairing during melanocyte specification, SOX2-OCT3/4 pairing in embryonic stem cells, and SOX2-BRN2 pairing in neural progenitors (Ambrosetti et al., 1997; Jiao et al., 2004; Tanaka et al., 2004; Takahashi and Yamanaka, 2006). Sox factors, other than the SoxB2 subfamily, were initially characterized as transcriptional activators, however, it has since been shown that many Sox factors can function as either activators or repressors in a context-dependent manner (reviewed in Chew and Gallo, 2009).

Figure 1. Sox transcription factor subfamiles and their functional domains. (A) Diagram of human SOX proteins grouped by subfamily. Major protein functional domains are depicted as colored boxes: High-mobility group (HMG) domain (light blue), transactivation domain (TAD; green), transrepression domain (TRD; orange), SoxB homology domain (dark blue), coiled-coil (CC) domain (yellow), and dimerization (DIM) domain (purple). (B) Phylogenetic tree depicting Sox subfamily members across key organisms. The emergence of the neural crest (green) in vertebrates coincided with duplication events among various Sox subfamilies, most notably SoxC and SoxE.
Within a Sox subfamily, the structural domains of the proteins outside of the HMG domain are similar, but not identical. These domains include coiled-coiled, dimerization, and transactivation/transrepression domains (Figure 1A). SoxD factors harbor a coiled-coil domain that mediates homo- or hetero-dimerization with other SoxD factors, stabilizing binding to adjacent HMG sites on DNA (Lefebvre et al., 1998). SoxE factors possess a 40 amino acid dimerization (DIM) domain upstream of the HMG domain (Peirano and Wegner, 2000). Their dimerization (homo- or hetero-) is DNA dependent and reliant upon the presence of a palindromic DNA binding sequence (Peirano and Wegner, 2000; Peirano et al., 2000; Schlierf et al., 2002; Huang et al., 2015). While SOXE factors can form heterodimers, they do not appear to dimerize with non-SoxE proteins (Huang et al., 2015; Hou et al., 2017). SoxE factors additionally possess both a C-terminal transactivation domain (TAD) and a second transactivation domain in the middle of the protein (TAM, or K2 domain) (Schreiner et al., 2007). Recent work suggests that these two domains synergize, resulting in a SOXE factor bipartite transactivation mechanism (Haseeb and Lefebvre, 2019). The members of other Sox subfamilies possess a single transactivation/repression domain. Interestingly, Sox transcriptional activity can depend upon whether the transcriptional partner is an activator or repressor (Kamachi and Kondoh, 2013). For example, SOX2-OCT3/4 synergistically activate Fgfr4 expression and the SOX9-SOX5/6 complex activates Col2a1 during chondrogenesis (Ambrosetti et al., 1997; Liu and Lefebvre, 2015). In contrast, SOX9-GLI2/3 represses Col10a1 in non-hypertrophic chondrocytes (Leung et al., 2011). SOX proteins also associate with non-DNA binding cofactors, such as Groucho co-repressors. SOX2-GRG5 represses neural differentiation markers and SOX9-GRG4 represses Dct expression during melanocyte development (Lee et al., 2012; Liu et al., 2014). These findings highlight the importance of cellular context and partner protein/cofactor availability for Sox function.
Sox factors have diverse roles during development and typically members of the same subfamily have similar or redundant functions. The contributions of SoxB1 factors to maintaining pluripotency have been intensely studied (reviewed in Abdelalim et al., 2014), and SOX2 is one of the four Yamanaka factors able to reprogram somatic cells to a pluripotent state (Takahashi and Yamanaka, 2006). Interestingly, other subfamilies of Sox factors are capable of replacing SoxB1 function either during the reprogramming process or in stem cells. SOX15 and SOX18 can substitute for SOX2 during the reprogramming process, but are less efficient (Nakagawa et al., 2008). While SOX17 is not an effective substitute for SOX2, a reengineered SOX17 (SOX17 E122K) reprograms cells with high efficiency (Jauch et al., 2011). Likewise, the reprogramming efficiency of SOX18 increases when it is reengineered to have an analogous point mutation within the HMG domain and the C-terminal of the protein is swapped for the SOX17 C-terminal (Aksoy et al., 2013). The point mutation within the SOX18 HMG domain alone was not sufficient for reprogramming nor was swapping the C-terminal for that of SOX2 (Aksoy et al., 2013). With respect to regulating a stem cell state in embryos, morpholino mediated knockdown of Sox2 and Sox3 in Xenopus leads to a loss of pluripotency and this can be partially rescued by expression of SoxE factors (Buitrago-Delgado et al., 2018). Together these data suggest that while specific subfamilies of Sox factors may be optimized for particular developmental roles, other Sox subfamilies may be able to serve as a substitute, albeit less efficiently. This paradigm is particularly interesting in the context of the neural crest and the retention of embryonic potential in those cells (discussed below; Buitrago-Delgado et al., 2015).
The neural crest is a vertebrate specific population of cells that contribute significantly to the vertebrate body plan, including much of the craniofacial complex. In addition to giving rise to the bone and cartilage of the face, the neural crest also give rise to melanocytes, the majority of the peripheral nervous system, and contribute directly to facial structures such as the tongue, teeth, and palate (Chai et al., 2000; Haldin and LaBonne, 2010; Bronner and LeDouarin, 2012; Prasad et al., 2012). Embryonically, neural crest cells (and cranial placodes) arise at the neural plate border which lies between the neural plate and non-neural ectoderm. Gradients of BMP, FGF, and WNT signaling have all been implicated in the establishment of these three regions. The initial formation of the neural plate border occurs in cells with intermediate levels of BMP signaling and high WNT signaling. A unique signature of transcription factors (pax3/7, zic1/2, msx1, myc) define the neural plate border region and these factors subsequently activate the neural crest gene regulatory network (GRN), which includes several Sox factors (Simoes-Costa and Bronner, 2015). Sox2, sox3, sox8, sox9, sox11, and sox15 are expressed within the neural plate border (Figure 2; Spokony et al., 2002; Wakamatsu et al., 2004; O’Donnell et al., 2006; Roellig et al., 2017). SOX2 expressing cells within the neural plate border can contribute to both the neural crest and the non-neural ectoderm, and modulating levels of SOX2 can impact the balance between neural and neural crest domains (Roellig et al., 2017). The observed expression of sox8 and sox9 correlates with the formation of definitive neural crest cells within the neural plate border.
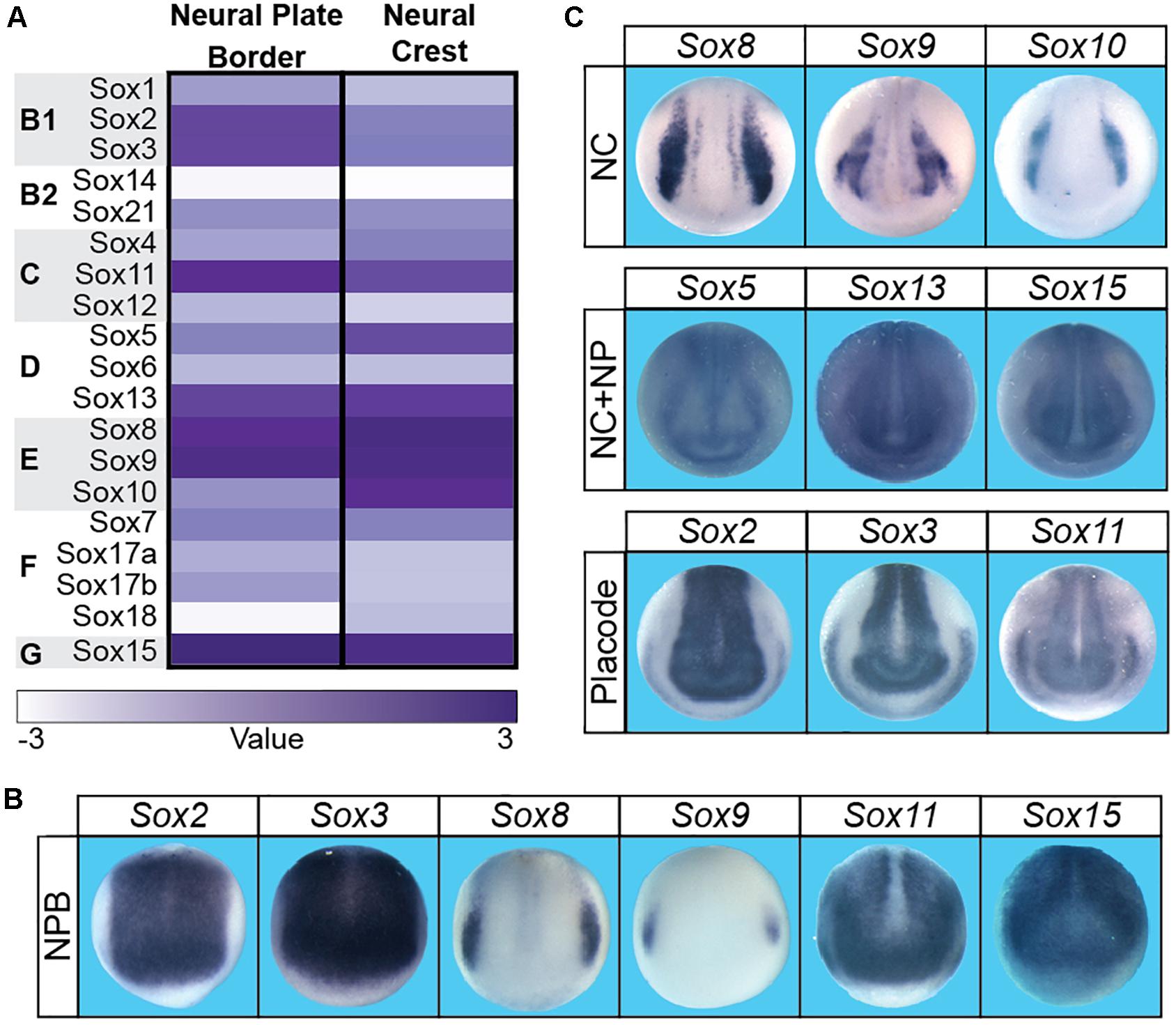
Figure 2. There were boxes subdividing the three sections (A–C) in the submitted figure. These boxes are not present in this proof. Sox factor expression during neural plate border and neural crest formation in Xenopus laevis. (A) Heatmap of Sox gene expression in neural plate border and neural crest cells in Xenopus laevis. High levels of expression are associated with a dark purple while low levels are depicted as white. (B) In situ hybridizations in early neurula Xenopus laevis embryos for sox2, sox3, sox8, sox9, sox11, and sox15. Each of these factors has neural plate border expression (domain most clearly defined by sox8 expression). (C) In situ hybridizations in late neurula Xenopus laevis embryos for sox factors expressed in the neural crest (sox8, sox9, sox10), neural crest + neural plate (sox5, sox13, sox15), and neural plate + placodes (sox2, sox3, sox11).
Sox transcription factors play important roles in controlling the developmental potential of the neural crest progenitor population as well as in their subsequent lineage decisions. Members of the SoxC, SoxG, SoxD, and SoxE families are expressed in the pre-migratory neural crest and functional roles for most of these factors have been reported (Figure 2). By contrast, SoxB1 factors are expressed in the neural plate and pre-placodal ectoderm and can inhibit neural crest formation (Wakamatsu et al., 2004; Roellig et al., 2017; Buitrago-Delgado et al., 2018). SoxC factors (sox4/11/12) are expressed in the neural crest and the neural plate of Xenopus and lamprey (Petromyzon marinus) embryos and seem to have evolutionarily conserved functions. Loss of function experiments for SoxC factors resulted in failed neural crest formation in both species (Uy et al., 2015). SoxC factors are also expressed in migrating neural crest, but a requirement for these factors during migration or subsequent lineage diversification has yet to be described (Cizelsky et al., 2013; Uy et al., 2015). Similarly, the SoxG family member, Sox15, is expressed in the pre-migratory neural crest, but a role for Sox15 in neural crest development has yet to be reported. Interestingly, Sox15 is expressed in mouse embryonic stem cells and, like SOXB1 proteins, can associate with OCT3/4 (Maruyama et al., 2005). While Sox15-null mice are viable, it is possible that SOX15 function is redundant to that of SOXB1 factors in ES cells. As neural crest cells retain stem-cell like potential, it would be interesting to investigate if SOX15 plays a role in controlling the developmental potential of these cells (Buitrago-Delgado et al., 2015).
Of the three SoxD family members, Sox5 and Sox13 are expressed in the forming neural crest; however, only a role for SOX5 has been reported in these cells. SOX5 is expressed in the pre-migratory neural crest in both chick and Xenopus embryos. Loss of function experiments demonstrated that Sox5 is necessary for neural crest, placode, and neural plate border formation. Interestingly, increasing Sox5 levels phenocopies these effects (Nordin and LaBonne, 2014), suggesting that maintaining the correct level of sox5 expression is key to proper neural crest formation. Sox5 can serve as an effector of BMP signaling in the ectoderm (and in other biological contexts). Through its central coiled-coil domain, Sox5 physically interacts with BMP R-Smad complexes and promotes activation of BMP target genes (Nordin and LaBonne, 2014). Since BMP signaling is essential for neural plate border/neural crest formation, Sox5 likely aids in activation of BMP targets in neural plate border/neural crest cells. Expression of sox5 persists as neural crest cells migrate, and overexpression of sox5 results in both a delay in migration and an increase in the total number of neural crest cells (Perez-Alcala et al., 2004; Nordin and LaBonne, 2014). Whether this increase is at the expense of other cell types in the ectoderm remains to be determined.
The evolutionary emergence of the neural crest correlated with the duplication, diversification, and neofunctionalization of a single ancestral SoxE gene (Tai et al., 2016). Where it has been examined, vertebrates possess two/three SoxE paralogs (Tai et al., 2016). SoxE factors have been shown to be required for neural crest formation across multiple species (Spokony et al., 2002; Honore et al., 2003; Hong and Saint-Jeannet, 2005; Taylor and LaBonne, 2005; O’Donnell et al., 2006; Buitrago-Delgado et al., 2018). Interestingly, the temporal order of SoxE gene expression within the neural crest differs across species. In Xenopus, sox8 is expressed first followed by sox9 then sox10 (O’Donnell et al., 2006; Buitrago-Delgado et al., 2018). In chick and mouse, Sox9 is the first SoxE factor to be expressed within the neural crest followed by Sox10 then Sox8 (Cheung and Briscoe, 2003). In zebrafish, sox9a and sox9b expression precedes that of sox10 in the neural crest while sox8 is not expressed in these cells (Dutton et al., 2001; Li et al., 2002; Yan et al., 2005). The varied timing of Sox expression is consistent with a high degree of functional redundancy among these factors. In the agnathan, Petromyzon marinus, SoxE1 and SoxE2 are expressed in the neural folds and migrating neural crest while SoxE3, the ortholog to Sox9 in gnathostomes, lacks early embryonic expression (McCauley and Bronner-Fraser, 2006). The differences in order of expression and function between agnathan and gnathostome SoxE factors may suggest independent divergence from the ancestral SoxE (Lakiza et al., 2011).
Evidence across multiple organisms suggests that while SoxE factors are required for neural crest formation, the individual factors may be functionally redundant. Loss of function studies in Xenopus demonstrated that sox8, sox9, and sox10 are necessary for neural crest formation and proper migration (Spokony et al., 2002; Honore et al., 2003; Taylor and LaBonne, 2005; O’Donnell et al., 2006). The neural crest can be rescued in Sox8 morphants by any of the SoxE factors suggesting there is functional redundancy between the SoxE factors (Taylor and LaBonne, 2005; O’Donnell et al., 2006). By contrast, murine SoxE mutants do not have obvious early neural crest defects. Sox8 null mice are viable with no obvious morphological defects suggesting that during mammalian development the other SoxE factors are able to compensate for loss of Sox8 (Sock et al., 2001). Additionally, the neural crest in Sox9fl/fl;Wnt1Cre embryos migrates normally to the craniofacial complex; this suggests that neural crest formation is not altered in these conditional mutants; however, increased apoptosis has been observed in the trunk neural crest of Sox9–/– mice (Cheung et al., 2005). The Sox9fl/fl;Wnt1Cre mice do, however, have major craniofacial defects, as discussed below (Mori-Akiyama et al., 2003). In Sox10lacZ/Sox10lacZ mice and Sox10 hypomorphs, neural crest formation and migration is not altered (Britsch et al., 2001; Schreiner et al., 2007). Again, this suggests that the other SoxE factors can compensate for the loss of a single SoxE factor during neural crest formation. In contrast, during neural crest cell differentiation, each SoxE factor may have unique functions. This is evidenced by Sox10Sox8ki/Sox8ki embryos, where Sox8 has been inserted into the Sox10 locus. These mice still have severe enteric nervous system defects and lack melanocytes which phenotypically parallel a loss of Sox10 (Kellerer et al., 2006).
SOX proteins undergo a number of post-translational modifications, including: acetylation, methylation, phosphorylation, SUMOylation, and ubiquitination (Williams et al., 2019). Several of these modifications have been shown to impact neural crest development (Huang et al., 2000; Taylor and LaBonne, 2005, 2007; Sakai et al., 2006; Lee et al., 2012; Liu et al., 2013). Phosphorylation of SOX9 at serine 64 and serine 211, conserved residues in amniotes, by PKA increased SOX9 DNA-binding affinity at a Col2a1 enhancer element and promoted reporter activation (Huang et al., 2000). Furthermore, phosphorylation of the analogous serine residues in chick is required for delamination of trunk neural crest cells (Liu et al., 2013). Delamination is mediated through SOX9-SNAI2 interaction, and phosphorylation of SOX9 is required for this pairing. Additionally, SOX9 serine 64 and serine 181 phosphorylation (in chick) promotes SOX9 SUMOylation (Liu et al., 2013). SOX9 SUMOylation is not required for trunk delamination, but SUMOylation state impacts neural crest cell formation (Taylor and LaBonne, 2005; Liu et al., 2013). Blocking Sox9 SUMOylation promotes neural crest formation whereas constitutively SUMOylated Sox9 represses neural crest formation (Taylor and LaBonne, 2005). Sox9 inhibition of the neural crest state is mediated through SUMO-dependent recruitment of the Groucho family protein Grg4, which is a transcriptional co-repressor (Lee et al., 2012). The SUMOylation state of Sox9 also influences inner ear development. A constitutively SUMOylated form of Sox9 promotes otic vesicle formation whereas a form of Sox9 that cannot be SUMOylated inhibits ear formation, but promotes ectopic melanocytes (Taylor and LaBonne, 2005).
Likewise, SOX10 is SUMOylated at three conserved lysine residues (Taylor and LaBonne, 2005; Girard and Goossens, 2006). SUMOylation state of SOX10 does not impact nuclear localization or the ability of SOX10 to bind DNA; however, it inhibits the transcriptional activation of target genes such as MITF (Girard and Goossens, 2006). Furthermore, it was demonstrated that SUMOylation interferes with the ability of SOX10 to synergize with PAX3 to activate MITF gene expression. This was also true for SOX10 and ERG2 and their target GJB1 in Schwann cells (Girard and Goossens, 2006). Whether this relationship between SOX10 SUMOylation state and transcriptional activation is true for all target genes is yet to be determined. It is likely, however, that this relationship is more complex and context dependent as observed with other transcription factors (Long et al., 2004; Taylor and LaBonne, 2005; Rosonina et al., 2017). Finally, SoxE factors also have putative acetylation and methylation sites (Williams et al., 2019); however, functional roles for these modifications during neural crest development remains largely unknown. One study demonstrated that Sox9 is acetylated by Tip60; however, the acetylation state did not impact the ability of Sox9 to activate Col2a1 expression (Hattori et al., 2008). Whether Sox9 acetylation state affects transcription of other target genes or if other Sox9 post-translational modifications regulate Col2a1 expression are questions that remain to be answered.
The functional relationship between SoxE factors and other transcription factors essential for neural crest formation has been examined in several systems. Studies in Xenopus have shown that Snail1 promotes sox10 expression and knockdown of sox9 leads to reduced twist, snail1 and pax3 expression in the neural crest, whereas sox9 or sox10 gain-of-function expands the domains of foxD3, snail2, and sox10 expression at the neural plate border (Spokony et al., 2002; Honore et al., 2003; Buitrago-Delgado et al., 2018). In chick, overexpression of SOX9 is not sufficient to induce PAX3/7, but can induce SNAI2, FOXD3, and SOX10 (Cheung and Briscoe, 2003). Additionally, a SOX10 enhancer element has been identified in chick which requires ETS1, SOX9, and/or cMYB activity to drive reporter expression (Betancur et al., 2010). While such candidate-driven functional studies have provided some insights into the functions of SoxE factors within the neural crest gene regulatory network, a more comprehensive understanding of Sox targets in the neural crest remains lacking. Moreover, as SOX factors require DNA-binding partners for efficient regulation of target genes, it is also essential to identify and study the SOXE partners that play roles in the development and evolution of the neural crest.
Evolutionarily, the emergence of the neural crest coincided with duplication and diversification of an ancestral SoxE gene (Figure 1B; Tai et al., 2016). The significance of SoxE function to neural crest evolution is interesting to contemplate given the central role these factors play in the establishment of the neural crest stem cell state. As SoxB1 factors are essential regulators of pluripotency in blastula cells, it is possible that this role was handed off to SoxE factors in the neural crest. Consistent with this, recent work has shown that SoxE function can at least partially replace SoxB1 factors in maintaining the pluripotency of blastula stem cells, although SoxB1 factors are unable to replace SoxE factor function in the neural crest (Buitrago-Delgado et al., 2018). These data suggest that SoxE factors can engage in the pluripotency gene regulatory network, maintaining expression of key targets in the absence of SoxB1 factors, even in a cellular context in which they are normally not expressed. Whether this is a unique feature of SoxE factors or if other Sox subfamilies can function in a similar context remains to be seen; however, other Sox factors can substitute for SoxB1 factors during cell reprogramming (Nakagawa et al., 2008; Jauch et al., 2011). Interestingly, SOX17 sits at the top of the specification hierarchy for human primordial germ cells (Irie et al., 2015). Other pluripotency genes, such as NANOG and OCT4, but not SOX2, are also expressed in human primordial germ cells downstream of SOX17 (Tan and Tee, 2019). Thus, the SoxB1 to SoxE hand-off in the neural crest may serve as a paradigm for transitioning molecular regulatory circuitry from one Sox subfamily to another to maintain a stem cell-like state.
Why might a transition from SoxB1 to SoxE function have been important for the evolutionary emergence of neural crest stem cells? By the end of gastrulation SoxB1 factors cease to direct pluripotency and instead are expressed in, and essential to, the formation of the neuronal progenitor pool, and continue to play prominent roles in neural lineages later in development (Rex et al., 1997; Graham et al., 2003; Ellis et al., 2004; Schlosser et al., 2008). Once SoxB1 factors have transitioned to controlling the neuronal progenitor state, SoxE factors may take over the regulation of targets essential to maintaining developmental potential in the neural crest. Furthermore, the inability of SoxB1 factors to replace SoxE factor function in the neural crest implies this switch was necessary for the emergence of the neural crest and its derivatives (Buitrago-Delgado et al., 2018). Mechanistically, it is possible that SoxB1 factors are unable to correctly regulate some SoxE target genes unique to neural crest. Furthermore, this switch in Sox factor deployment may also have facilitated the subsequent lineage diversification of neural crest cells to non-neural cell types including cartilage, melanocytes, and glia, which require SoxE function for their formation (Bi et al., 1999; Britsch et al., 2001; Aoki et al., 2003). Understanding this transition, as well as why SoxE factors play essential roles in directing the development of only a subset of neural crest lineages, will require a more complete understanding of SoxE targets and partners in both neural crest stem cells and their derivatives.
An excellent context in which to investigate the roles and regulation of Sox factors in neural crest lineage diversification is the craniofacial complex, a compilation of multiple structures that together create both the form and function of the face. While development of many of these structures occur simultaneously, the molecular mechanisms that govern their development are unique. The neural crest contributes to a significant portion of the craniofacial complex, giving rise to chondrocytes, melanocytes, a majority of the peripheral nervous system, and contributing to the mesenchymal component of structures such as the tooth and palate (Chai et al., 2000; Bronner and LeDouarin, 2012). Several Sox factors are expressed throughout the facial ectoderm and mesenchyme (Table 1), many of which play essential roles in the development of specific craniofacial structures. Other Sox factors, while expressed during craniofacial development, have yet to be functionally characterized. SoxE functions within neural crest-derived cell types of the face are of particular evolutionary significance. The basal chordate amphioxus possesses a single SoxE gene and lacks neural crest cells, yet has an oral skeleton (Jandzik et al., 2015). Interestingly, expression of amphioxus SoxE within the chick neural tube is sufficient to induce neural crest formation. These amphioxus SoxE expressing cells later expressed markers for DRG lineages, including those cells positioned dorsolaterally that would typically become melanocytes (Tai et al., 2016). These data suggest that co-option of SoxE to proto neural crest-like cells may have occurred prior to genome duplication, but required the aquisition of new cis-regulatory sequences (Jandzik et al., 2015; Tai et al., 2016). It further suggests that duplication and divergence of the SoxE family was necessary for neural crest lineage diversification and the subsequent elaboration of the vertebrate head.

Table 1. Sox gene expression in various craniofacial structures in mouse embryos.
The craniofacial skeleton serves as the framework for the face. The bones of the craniofacial complex differ from long bones, such as those of the arms, legs, or ribs, in two main ways. First, a majority of anterior craniofacial bones are derived from the neural crest while long bones are derived from the mesoderm (Noden and Trainor, 2005). Second, long bones form through a process called endochondral ossification, whereas the flat bones of the face and the bones of the skull form through intramembranous ossification (Berendsen and Olsen, 2015). The small subset of endochondral facial bones included the malleus, incus, and nasal capsule (Mori-Akiyama et al., 2003). These and the facial cartilages (such as Meckel’s) form through a chondrogenic mechanism that requires Sox transcription factors.
SOX9 serves as the central transcriptional regulator for chondrogenesis (Bell et al., 1997; Bi et al., 1999). In vertebrates, Sox9 is expressed in the neural crest-derived facial mesenchyme (Wright et al., 1995; Healy et al., 1996; Spokony et al., 2002). Heterozygous Sox9 knock-out mice display cleft secondary palate, domed skull, and a short snout (Akiyama et al., 2002). Conditional deletion of Sox9 in the neural crest (Sox9fl/fl;Wnt1-Cre) results in complete loss of facial cartilages and endochondral derived bones (Mori-Akiyama et al., 2003). Likewise, in Xenopus, Sox9 morphants display gross morphological defects in their craniofacial chondrogenic elements, including complete loss of Meckel’s cartilage (Spokony et al., 2002). In the absence of Sox9, the prechondrogenic mesenchymal condensation fails to form and Col2a1, a direct target of SOX9, fails to be expressed (Lefebvre et al., 1997; Mori-Akiyama et al., 2003). The cells that should have become chondrocytes begin to express osteoblast markers (such as Runx2), indicating that SOX9 not only functions to promote chondrogenesis, but also inhibits osteoblast formation (Mori-Akiyama et al., 2003). Additionally, SOX9 is required for the expression of Sox5 and Sox6 during chondrogenesis (Akiyama et al., 2002). Sox5–/– and Sox6–/– mutants experience early postnatal lethality, but do not have major defects in chondrogenic elements. Interestingly, while Sox5–/– mice display minor defects in the craniofacial cartilage; Sox6–/– mutants do not. In contrast, Sox5–/–;Sox6–/– double mutants die at e16.5 and fail to form cartilages. While these mutants still express Sox9 and Col2a1 in prechondrogenic regions, cells fail to differentiate into chondrocytes (Smits et al., 2001). These data indicate that SoxD factors have both unique roles and redundant roles during chondrogenesis. Analysis of DNA occupancy by SOX9 and SOX5/6 in chondrogenic cells indicates shared binding at enhancers and suggests that SOX5/6 act cooperatively with SOX9 to promote gene activation (Liu and Lefebvre, 2015). Indeed, the triple combination of SOX9-SOX5-SOX6 can promote chondrogenesis in mesenchymal stem cell without the addition of growth factors, such as TGFβ3 (Raftery et al., 2020).
The pigment of the skin, hair, and choroid layer of the eye is produced by melanocytes. These cells are derived from the neural crest and localize to the vascular uvea of the eye, the basal layer of the epidermis, or hair follicles (Hirobe, 1984; Holbrook et al., 1989; Sitiwin et al., 2019). Sox10 plays a central role in the gene regulatory network (GRN) controlling melanocyte development (Figure 3), although Sox5, Sox9, and Sox18 may also play minor roles in melanogenesis (reviewed in Harris et al., 2010). Sox10, first expressed in the neural crest cells prior to migration, persists in the neural crest cells that become melanocytes (Aoki et al., 2003). Studies with mouse Dom mutants, which possess a frameshift mutation in Sox10 (Herbarth et al., 1998), demonstrate that Sox10 is required for melanocyte development. Dom mutants lack expression of Dct/Trp2, an early melanocyte marker (Southard-Smith et al., 1998). Supporting these findings, Sox10LacZ/+ heterozygous mice have fewer melanocytes and colorless zebrafish mutants (premature stop codon in sox10) have reduced melanocytes, iridoblasts, and xanthoblasts (Kelsh and Eisen, 2000; Britsch et al., 2001; Dutton et al., 2001). Overexpression of sox10 in Xenopus embryos results in a massive expansion of melanocyte precursor cells and is sufficient to induce the expression of melanocyte marker dct/trp2 in naïve ectoderm (Aoki et al., 2003). Likewise, expression of sox9 produces supernumerary melanocytes in Xenopus embryos (Taylor and LaBonne, 2005).
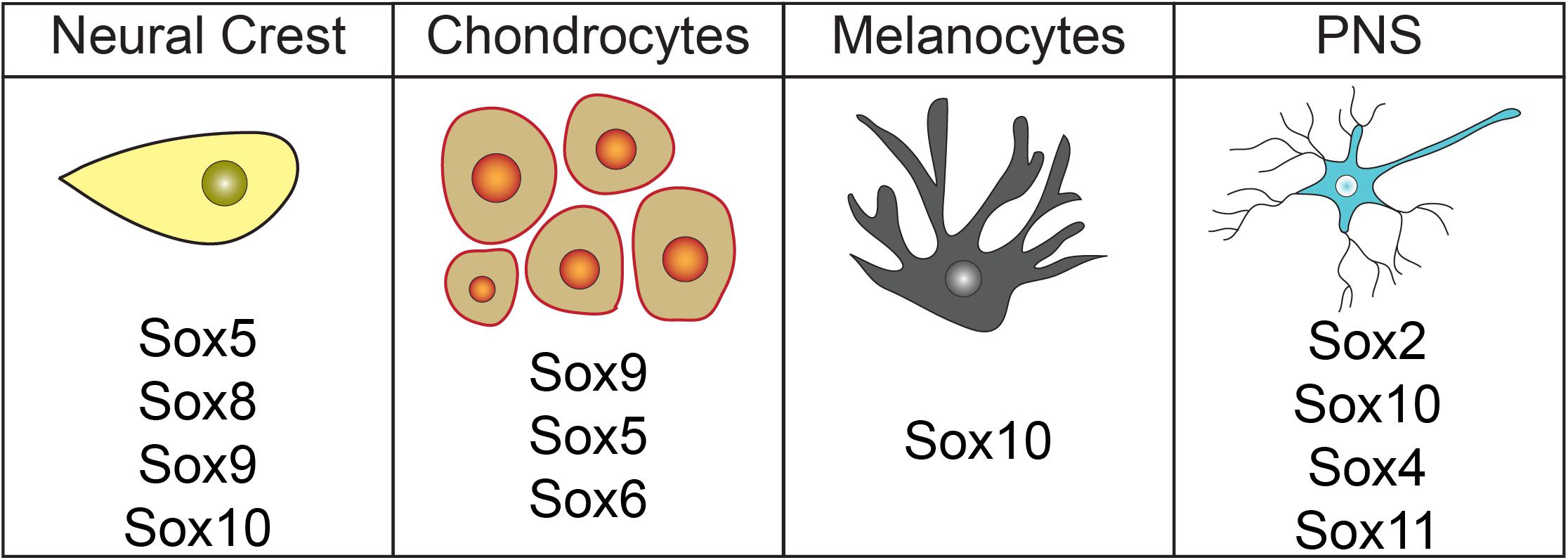
Figure 3. Sox factors involved in neural crest formation and lineage diversification. Diagram highlighting Sox factors known to have roles in neural crest cell, chondrocyte, melanocyte, and peripheral nervous system development.
In the GRN controlling melanocyte development Sox10 has been shown to directly activate Mitf, Dct/Trp2, Tyr, and Tyrp1 (Bondurand et al., 2000; Potterf et al., 2000; Jiao et al., 2004; Murisier et al., 2006, 2007). SOX10 acts synergistically with PAX3 to activate MITF expression (Bondurand et al., 2000; Potterf et al., 2000). Subsequently, MITF becomes a transcriptional partner for SOX10 and together they promote DCT/TRP2 expression (Jiao et al., 2004; Ludwig et al., 2004). In the absence of Sox10, MITF alone is unable to direct formation of pigmented cells (Hou et al., 2006). Additionally, BRG1, a key member of the chromatin remodeling SWI/SNF complex, regulates proximal and distal promotor accessibility of melanocyte-specific SOX10 target genes. SOX10 directly interacts with BRG1, possibly recruiting BRG1 to these sites (Marathe et al., 2017). Global analysis of SOX10 binding sites in an immortalized melanocyte cell line further indicates that SOX10 binds to target sites as either a monomer or homodimer and most of these sites were found at distal regulatory regions. Interestingly, target genes are both up and downregulated in Sox10LacZ/+ cells providing further evidence that SOX10 can have both activator and repressor functions (Fufa et al., 2015). Validating and integrating these targets into the existing melanocyte GRN will greatly advance our understanding of melanocyte formation and disease etiology for Waardenburg Syndrome.
SOX10 activity during melanocyte formation is regulated in several ways. First, SOX5 can bind to Sox10 target regions within promotors of melanocyte genes and recruit the transcriptional corepressors HDAC1 and CtBP2 (Stolt et al., 2008). Although it has not been demonstrate that competition for binding sites occurs in vivo, Sox5–/–, Sox10+/lacZ double mutant embryos display a less severe melanocyte phenotype than Sox10+/lacZ single mutant embryos, consistent with a role for SOX5 as a recruiter of corepressors (Stolt et al., 2008). Post-translational modification of SOX10 may also regulate its function during melanocyte formation. SOX10 can be SUMOylated at three lysine residues and this modification represses transcriptional activation of the MITF promotor in vitro (Taylor and LaBonne, 2005; Girard and Goossens, 2006).
In addition to regulating melanocyte formation during embryonic development, Sox factors are also integrally involved in postnatal melanocyte maintenance and progression of melanoma. A population of melanocyte stem cells resides at the base of hair follicle cells and contributes to the pigmentation of each hair shaft. Sox10 is expressed in these stem cells and gain and loss of Sox10 function both lead to a reduction in this cell population and the presence of white/gray hairs, consistent with disruptions in melanocyte stem cell function (Harris et al., 2013). Additionally, Sox10 is expressed in the differentiated melanocytes of hair follicles and is required for retention of melanocytes (Harris et al., 2013). SOX9 is expressed in postnatal melanocytes and can induce expression of SOX10 melanocyte target genes in B16 melanoma cells. Additionally, increased expression of SOX9 leads to enhanced melanin production (Passeron et al., 2007). Together these findings suggest a SOX9 function in adult melanocytes that parallels that of SOX10 during embryonic development. Finally, both SOX9 and SOX10 have roles in the etiology of melanoma. While a detailed description of their functions in this context is beyond the scope of this review, we highlight that these SoxE factors may have antagonistic roles in melanoma cells (Shakhova et al., 2015). Sox functions in various cancers has recently been reviewed (Grimm et al., 2019).
Neural crest cells also give rise to much of the peripheral nervous system (PNS), including the cephalic ganglia, dorsal root ganglia (DRG), Rohon-Beard cell, satellite cells, and Schwann cells (D’Amico-Martel and Noden, 1983; LeDouarin and Kalcheim, 1999). Sox2, Sox4, Sox5, Sox10, and Sox11 are each expressed in various neural crest-derived PNS cells (Britsch et al., 2001; Perez-Alcala et al., 2004; Aquino et al., 2006). While functions for Sox2, Sox4, and Sox10 have been described, only expression data for Sox5 and Sox11 has been reported (Figure 3). Schwann cell precursors, satellite glia, myelinating/non-myelinating cells, peripheral glia, and NC-derived cells within trigeminal ganglion express Sox5 (Perez-Alcala et al., 2004; Morales et al., 2007). Sox11 is expressed in several PNS cell types including DRG, cranial ganglia, and sympathetic ganglia (Hargrave et al., 1997; Kuhlbrodt et al., 1998a; Sock et al., 2004). Expression in these regions was noted to decrease over time and was very weak by e15.5 of mouse embryogenesis, suggesting that Sox11 may function in fate determination or early stages of differentiation (Hargrave et al., 1997; Kuhlbrodt et al., 1998a; Sock et al., 2004). Another SoxC subfamily member, Sox4, may possibly have an opposing role in PNS development and act as a negative regulator of Schwann cell myelination. Sox4 overexpression in Schwann cells leads to delayed myelination and hypomyelination. Interestingly, Sox4 expression is elevated in mouse models of demyelinating neuropathies (Bartesaghi et al., 2015). These Sox11 and Sox4 findings provide evidence that members of the same Sox subfamily can play divergent roles.
Sox2, which is critical for maintaining stem-cell attributes in central nervous system progenitors, also functions in a subset of PNS cells (Wakamatsu et al., 2004; Le et al., 2005; Pevny and Placzek, 2005; Aquino et al., 2006; Adameyko et al., 2012). Sox2 is expressed in neuroglial progenitors cells, but is downregulated upon differentiation (Aquino et al., 2006). In ovo electroporation of SOX2 in chick embryos results in increased proliferation of DRG cells, but blocks neuroglial progenitor differentiation to both neural and glial fates (Wakamatsu et al., 2004) while knockdown/knockout of Sox2 in the DRG neural progenitors reduces the number of DRG neurons (Cimadamore et al., 2011). Together, these data indicate that SOX2 is an essential regulator of sensory neurogenesis. In addition, SOX2, is expressed in immature Schwann cells where it suppresses expression of genes associated with Schwann cell myelination and blocks myelination, an indicator of Schwann cell differentiation and maturation (Le et al., 2005; Roberts et al., 2017; Wust et al., 2020). It is thus clear that Sox2 plays critical roles in both the developing PNS and CNS.
Importantly, the SoxE factor, Sox10, is also a critical regulator of glial cell development. Sox10 is expressed in Schwann cell precursors and sensory ganglia, and is required for the specification of all glia within the PNS (Kuhlbrodt et al., 1998b; Southard-Smith et al., 1998; Britsch et al., 2001). In the absence of Sox10 (Sox10lacZ/Sox10lacZ or colorless) cranial ganglia, enteric ganglia, and DRG numbers are reduced or display aberrant cell morphology (Kelsh and Eisen, 2000; Britsch et al., 2001; Dutton et al., 2001) and Schwann cell precursors are absent in both mutants (Kelsh and Eisen, 2000; Britsch et al., 2001; Dutton et al., 2001). These phenotypes are reminiscent of loss of neuregulin/ErbB signaling, which promotes the differentiation of neural crest into glia (Britsch et al., 2001; Britsch, 2007). ErbB3 expression is decreased in Sox10lacZ/Sox10lacZ mutants and, subsequently, migration and survival of progenitor cells is compromised (Britsch et al., 2001). In addition to regulating expression of ErbB3, in vitro studies have demonstrated that SOX10 synergies with OCT6 (POU3F1) and BRN2 (POU3F2) to activate EGR2 (KROX-20), which is essential for myelination of Schwann cells (Kuhlbrodt et al., 1998b; Le et al., 2005). SOX10 then partners with EGR2 to activate others myelin genes (LeBlanc et al., 2007). Indeed, SOX10 and EGR2 are sufficient to reprogram skin fibroblasts into Schwann cells, emphasizing the importance of both of these factors for Schwann cell development (Mazzara et al., 2017). Regulation of Sox10 in Schwann cells has been linked with eEF1A1 which, upon acetylation, removes SOX10 from the nucleus. In Schwann cells, this activity is blocked by HDAC1/2, Sox10 co-factors essential for myelination, which deacetylate eEF1A1 causing it to return to the cytoplasm and preventing nuclear export of Sox10 (Duman et al., 2020). Finally, one of the essential functions of Sox10 is to direct the neuroglial progenitor cells of the DRG toward the glial lineage. Mechanistically this is attributed to Sox10 biasing neuroglia progenitor cells toward the glial lineage (vs. sensory neurons) by promoting the ubiquitination and subsequent degradation of transcription factor Neurog2 through upregulation of Fbxo9 (Liu et al., 2020).
Cleft lip/palate is one of the most common birth defects, and numerous genes have been associated with this congenital malformation (Dixon et al., 2011). Mammalian palatogenesis begins with proliferation of the neural crest-derived cells within the palatal shelves, which leads to vertical outgrowth. The palatal shelves then elevate, sitting horizontal above the tongue, and the epithelium of the two shelves fuse. The epithelial seam formed at the midline is then removed to create a confluent mesenchyme (Bush and Jiang, 2012). A defect in any of these steps can result in a cleft secondary palate. Of the Sox transcription factors, only SOX11 has been associated with a patient presenting with cleft palate (Khan et al., 2018). Nevertheless, several Sox mutant mice (Sox11–/–, Sox9fl/fl;Wnt1-Cre, Sox2HYP, Sox5–/–) develop cleft palates (Smits et al., 2001; Mori-Akiyama et al., 2003; Sock et al., 2004; Langer et al., 2014). During e12.5-e14.5 of murine palatogenesis, expression of Sox2, Sox4, Sox5, Sox6, Sox8, Sox9, Sox11, Sox12, Sox13, and Sox21 can be detected via in situ hybridization (Table 1; Watanabe et al., 2016). Sox2 is expressed in the epithelium; Sox4/5/8/9 are expressed in the mesenchyme, while Sox6/11/12/13 are expressed in both the palatal mesenchyme and epithelium (Watanabe et al., 2016). Of note, both Sox2 and Sox11 are expressed in the palatal epithelial seam at e14.5. While it is unclear if/how Sox factors regulate palatogenesis, the spatio/temporal expression patterns make it tempting to speculate that several factors play roles in this process.
Tooth development begins as the oral epithelium thickens and the underlying neural crest-derived mesenchyme condenses around the invaginating placode. The placode continues to elongate (bud stage) and then branch (bell stage). Subsequently, cells begin to differentiate into ameleoblasts and odontoblasts (bell stage) and terminally differentiate/mineralize just prior to root formation and eruption (Thesleff, 2014). Several Sox factors are expressed throughout odontogenesis (see Kawasaki et al., 2015 for detailed expression analysis); however, functional roles for most factors have not been described (Table 1). Sox2 is expressed in the oral epithelium beginning at tooth initiation and continues to be expressed through cap stages in the lingual bud epithelium (Juuri et al., 2012; Kawasaki et al., 2015). Conditional deletion of Sox2 from the oral epithelium (Sox2fl/fl;Shh:GFP-Cre) results in only minor defects in the second and third molars; however, recombination of Shh:GFP-Cre was mosaic and some SOX2 protein still detectable (Juuri et al., 2013). In contrast, conditional deletion of Sox2 from both the oral and dental epithelium (Sox2fl/fl;Pitx2-Cre) produces abnormally shaped molars and underdeveloped incisors that regressed until undetectable at P0 (Sun et al., 2016). The regression of the incisors is consistent with a role for SOX2 in dental epithelial stem cells (DESCs). Lineage tracing experiments have revealed that Sox2 + DESCs reside in the labial cervical loop and contribute to all epithelial lineages in the mouse incisor (Juuri et al., 2012). Ablation of Sox2 prior to incisor injury dramatically decreases the ability of the incisor to regrow (Sun et al., 2016). These mutants also display reduced proliferation in labial cervical loops, suggesting that SOX2 regulates DESC proliferation (Sun et al., 2016). In addition to murine DESCs, Sox2 defines an analogous stem cell population in cartilaginous fish, which regenerate teeth successionally, suggesting that SOX2 function in DESCs is evolutionarily conserved (Martin et al., 2016).
Three pairs of salivary glands, the sublingual (SL), the submandibular (SMG), and the parotid (PG), reside inside the oral cavity and together secrete up to a quart of saliva daily (Knosp et al., 2012). Embryonically, these structures begin as placodes within the oral epithelium and then subsequently undergo elongation and branching morphogenesis (Affolter et al., 2003). Sox9, Sox10, and Sox2 are all expressed during salivary gland development (Lombaert et al., 2013; Chatzeli et al., 2017; Emmerson et al., 2017). Sox9 is expressed in the oral epithelium that gives rise to the SMG, SL, and PG. Sox9+ epithelial cells serve as the progenitor cells for the entire epithelial component of these salivary glands (Chatzeli et al., 2017). As SMG/SL/PG development progresses Sox9 becomes restricted to the distal progenitor cells of the bud. Conditional deletion of Sox9 within the oral epithelium (Sox9fl/fl;K14-Cre) results in arrested SMG, SL, and PG development at the bud stage (Chatzeli et al., 2017). In the conditional mutants, these cells fail to become specified; however, the proximal progenitors (Sox9–) are specified normally (Chatzeli et al., 2017). In the absence of distal progenitor cells, branching morphogenesis fails to occur. Embryonic expression of Sox9 ceases during lumen formation, but Sox9 becomes expressed again in the adult and contributes to regulation of the stem/progenitor cell properties of a subpopulation of salivary gland cells (Chatzeli et al., 2017; Tanaka et al., 2019). Sox10 is expressed in the distal progenitor cells of the bud and is also an acinar (mucin producing cells) progenitor marker (Lombaert et al., 2013). Sox10 lies downstream of Sox9, Sox2, Kit, and FGF signaling in these cells (Lombaert et al., 2013; Chatzeli et al., 2017; Emmerson et al., 2017). While a specific function for Sox10 within the distal/acinar progenitor cells has not been determined, it is known to be essential for branching and acinar formation of other glands (Chen et al., 2014). Sox2 is expressed in both duct and acinar progenitor cells but is only required for acinar cell formation. Sox2 promotes the expression of acinar-specific genes, including Sox10, and promotes survival of acinar progenitor cells through both maintaining proliferation and preventing apoptosis. Interestingly, parasympathetic nerves are required to maintain the Sox2 + progenitor cells, and thus are necessary for acinar cell formation (Emmerson et al., 2017).
Given the importance of Sox factors to the formation of the vertebrate craniofacial complex, it is perhaps unsurprising that a number of human syndromes presenting with craniofacial defects are linked to mutations in SOX genes (Table 2).
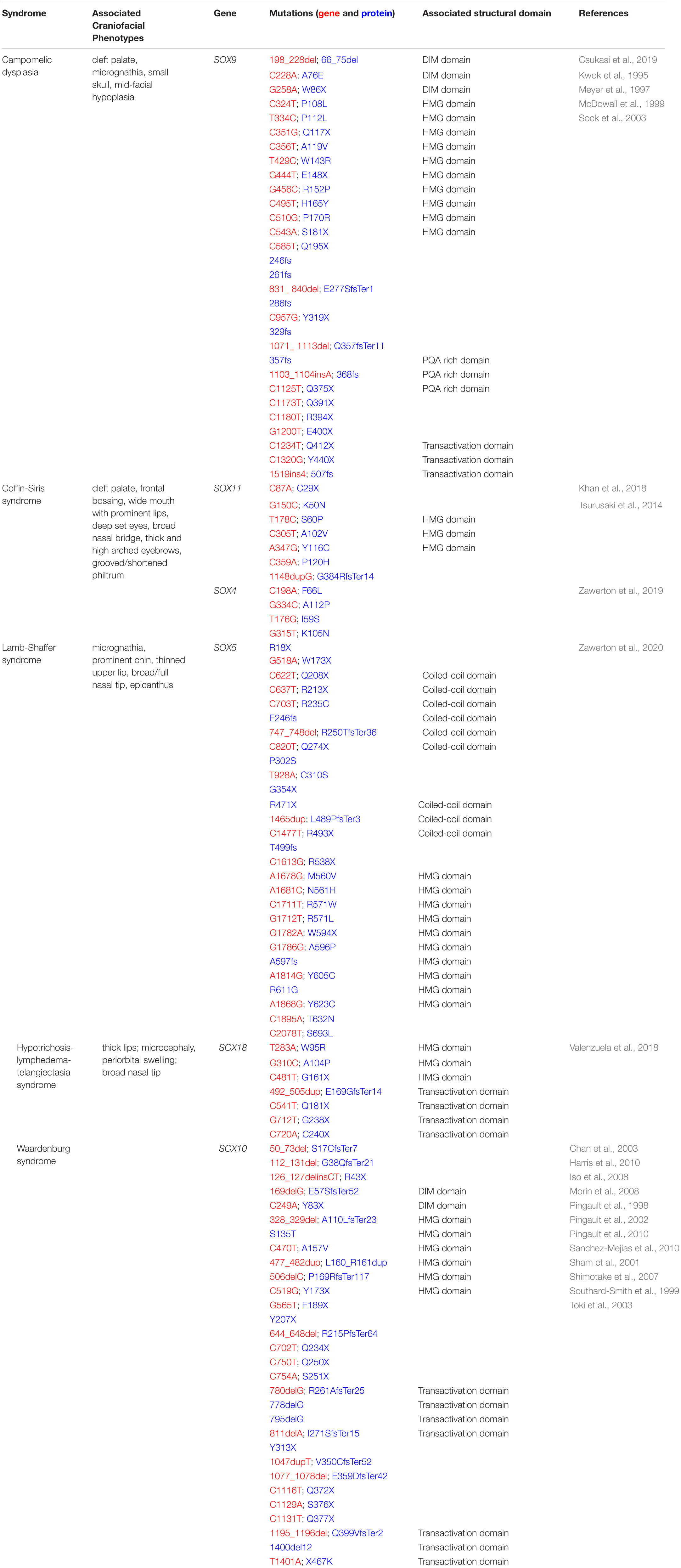
Table 2. SOXopathies with craniofacial phenotypes.
One of the most dramatic of these SOXopathies is Campomelic dysplasia (CD) which is caused by mutations in SOX9 (Foster et al., 1994; Wagner et al., 1994). Individuals with CD typically die shortly after birth and display an undermineralized skeleton and dramatic bowing of the lower limbs. Within the craniofacial complex, common CD phenotypes include cleft palate, micrognathia, a small skull, and mid-facial hypoplasia (Csukasi et al., 2019). Most CD cases are caused by a de novo mutation within a SOX9 allele (autosomal dominant). While the exact mutations within the SOX9 gene varies among CD patients, there is evidence for both haploinsufficiency and dominant negative protein function underlying the observed phenotypes (Foster et al., 1994; Csukasi et al., 2019), and in mice, loss of one allele of Sox9 phenocopies CD (Bi et al., 2001). Cell culture studies have shown that both SOX9 haploinsufficiency and dominant negative forms of SOX9 (nonsense mutations in the C-terminal transactivation domain) fail to robustly activate Col2a1 gene expression indicating that the chondrogenic program is not being fully initiated in cases of CD (Csukasi et al., 2019). Notably, although it is dysmorphic, a majority of the skeleton still forms in CD patients and animal models. Perhaps most SOX9 target genes are still activated despite loss of one functional allele. Alternately, SOX5 and SOX6 have partial functional redundant functions to SOX9 during chondrogenesis and may be able to compensate.
Waardenburg syndrome (WS) is a neurocristopathy characterized by pigment abnormalities in the hair, skin, and eyes, hearing loss, and craniofacial alterations such as hypertelorism, dystopia cantorum, nasal hypoplasia, and harelip (Banerjee, 1986; Dourmishev et al., 1999; Pingault et al., 2010; Wildhardt et al., 2013; Chen et al., 2015). There are four different subtypes of WS, two of which are associated with mutations in SOX10, type 2 and type 4 (Pingault et al., 1998; Southard-Smith et al., 1999; Bondurand et al., 2007). Individuals with type 2 present with additional neurological defects while those with type 4 also have Hirschsprung’s disease (Bondurand et al., 2007). Over 40 different SOX10 mutations have been reported across WS patients. Many mutations are truncating, causing the SOX10 transcript to undergo nonsense-mediated RNA decay resulting in a phenotype driven by haploinsufficiency (Inoue et al., 2004). A few variants have also been reported that alter the SOX10 stop codon and extend the protein (Pingault et al., 2010). For these variants, there is in vitro evidence that these elongated proteins have dominant negative effects (Inoue et al., 1999; Sham et al., 2001; Chan et al., 2003). During normal embryonic development, SOX10 is essential for neural crest stem cell formation and then subsequently for formation of specific derivatives, including melanocytes and enteric ganglia neurons (Southard-Smith et al., 1998; Britsch et al., 2001; Dutton et al., 2001). SOX10 also promotes survival and proliferation of Schwann cells (Britsch et al., 2001). In addition to SOX10, mutations in PAX3, MITF, SNAI2, EDN3, and EDNRB have all been identified as causative genetic insults for Waardenburg syndrome (Pingault et al., 2010). Together with SOX10, these genes are components of the gene regulatory networks controlling melanocyte (PAX3, MITF, SNAI2) or PNS (EDN3, EDNRB) development (Bondurand et al., 2007). Thus, WS patients with mutations in different genes can present with the same disease etiology due to disruptions in a shared gene regulatory network.
Lamb-Shaffer syndrome (LAMSHF) is classified as a neurodevelopmental disorder with common phenotypes including: developmental delays, intellectual disability, and language/motor deficits (Lamb et al., 2012). Patients also have a signature set of craniofacial features: micrognathia, prominent chin, thinned upper lip, broad/full nasal tip, and epicanthus (Zawerton et al., 2020). Mutations in SOX5, a member of the SoxD family, have been linked with LAMSHF. The observed skeletal defects such as micrognathia, broad/full nasal tip, and prominent chin are consistent with SOX5 having a role in chondrogenesis. Sox5 is strongly expressed in Meckel’s cartilage in mice (Ishikawa et al., 2018), which could explain the presentation of micrognathia, specifically. The general lack of severity of these craniofacial phenotypes is most likely due to individuals possessing other functional SOXD alleles, which is supported by Sox5–/– having only minor skeletal defects (Smits et al., 2001). In vitro studies suggest that haploinsufficiency, rather than a dominant negative effect associated with SOX5 variants, are most likely causative of LAMSHF. Furthermore, SOX5 variants with nonsense mutations or missense mutations within the HMG were localized cytoplasmically, unable to bind DNA, and failed to activate gene expression. While molecular studies demonstrated that some SOX5 variants could still activate target gene expression and other variants could not, the study could not identify any genotype-phenotype correlation among LAMSHF patients (Zawerton et al., 2020).
Another syndrome that has been associated with mutations in SOX genes, specifically Sox C family members is Coffin-Siris syndrome (CSS). Individuals with CSS have fifth fingers with clinodactyly, nail hypoplasia, microcephaly, and intellectual disabilities. Craniofacial features include cleft palate, frontal bossing, wide mouth with prominent lips, deep set eyes, broad nasal bridge, thick and high arched eyebrows, and a grooved/shortened philtrum (Tsurusaki et al., 2014; Hempel et al., 2016; Khan et al., 2018; Okamoto et al., 2018). Most patients with CSS (55–70%) have mutations in genes that encode for subunits of the BAF complex (Tsurusaki et al., 2014). Of the remaining cases, mutations in SOX11 have been identified as causal for several unrelated patients (Tsurusaki et al., 2014; Hempel et al., 2016; Khan et al., 2018; Okamoto et al., 2018). Most of the identified mutations in SOX11 lie within the HMG domain and result in decreased transcription of SOX11 target genes in vitro (Tsurusaki et al., 2014; Hempel et al., 2016). One variant has a mutation outside of this domain that is predicted to produce a truncated, non-functional protein (Khan et al., 2018). Additionally, there have been four cases of CSS where heterozygous mutations in SOX4 have been identified. Like SOX11, these mutations were within the HMG domain and variant proteins were unable to bind DNA and activate target gene expression (Zawerton et al., 2019). Little is known about the roles of SoxC factors in the context of craniofacial development. Sox4 and Sox11 are broadly expressed in the neural crest-derived facial mesenchyme, and Sox11 is, interestingly, expressed in the palatal epithelial seam (Watanabe et al., 2016). Whether Sox11 is functionally important for the removal of the epithelial seam is unknown, but this could explain occurrences of cleft palate in some CSS patients. In other cellular contexts, Sox4 and Sox11 promote proliferation (Gadi et al., 2013; Dai et al., 2017), thus it is possible that they may be regulating proliferation to some extent within the facial mesenchyme. Misregulation of proliferation could lead to phenotypes such as broad nasal bridge, shortened philtrum, and prominent lips. In the future, it would be interesting to use animal models to study the effects of single or combined loss of Sox4 and Sox11 on craniofacial development.
Lastly, mutations in Sox18 have been identified in patients with a rare condition called Hypotrichosis-lymphedema-telangiectasia syndrome (HLTS) (Irrthum et al., 2003). As the name suggests, the predominating features of these patients are sparse hair, tissue swelling due to malfunctioning lymphatic system, and presence of dilated vessels skin surface. While most case studies do not report a craniofacial phenotype with HLTS (either no phenotype or not assessed), a few patients have mild craniofacial defects that include: thick lips, microcephaly, periorbital swelling, and broad nasal tip (Bastaki et al., 2016; Valenzuela et al., 2018; Wangberg et al., 2018). Unlike the other syndromes associated with mutations in SOX genes, HLTS is associated with both autosomal dominant and autosomal recessive modes of inheritance (Irrthum et al., 2003; Wangberg et al., 2018). The autosomal dominant mode of inheritance is typically associated with nonsense mutations in the SOX18 transactivation domain while the autosomal recessive form is marked by missense mutations within the HMG domain (Valenzuela et al., 2018). Expression data in mice indicates that Sox18 primarily localizes to sites of vascularization within the developing murine craniofacial complex (Ishikawa et al., 2018). Given this expression, it is unclear how loss of Sox18 could result in phenotypes such as microcephaly or broad nasal tip. Clearly, further study into the molecular mechanisms underlying this syndrome, specifically those associated with the craniofacial defects, is necessary.
The emergence of neural crest drove the evolution of vertebrates including the elaboration of an intricate craniofacial complex. While Sox transcription factors are heavily utilized during invertebrate development, new roles have evolved for many of these factors within vertebrate cell types and structures, aided by duplication events. SoxE factors are initially required for the formation of neural crest stem cells, analogous to the role that SoxB1 factors play in blastula (inner cell mass) stem cells. Subsequently, SoxE factors are essential for the diversification of neural crest cells into a subset of non-neural lineages including cartilage, melanocytes and glia. By contrast SoxB1 factors transition to maintaining a neural progenitor state, and some SoxB1 partner pairings, including POU factors, are maintained between blastula stem cells and neural progenitor cells. Conserved SoxB1 roles within these cell populations could have necessitated the deployment of a different Sox subfamily, SoxE, in neural crest progenitors and derivatives. Evolutionarily, SoxE factor duplications at the base of the vertebrates may have helped drive neural crest lineage diversification and the development of the vertebrate craniofacial complex. Understanding the role of Sox proteins in the emergence of specialized cell types and complex forms in vertebrates will require a fuller understanding of the shared and unique functions of different Sox factors and families, and the mechanisms regulating those functions. This includes defining their transcriptional targets in different cellular contexts. Such studies should also prove to be of high clinical significance given the many congenital defects associated with Sox mutations. In particular, the plethora of craniofacial phenotypes associated with SOXopathies underscores the critical roles these factors play in the development and evolution of the vertebrate craniofacial complex.
It has been 30 years since the discovery of the SRY gene yet we are continuously learning more about the roles and regulation of this important family of transcription factors. Within the context of the neural crest and craniofacial complex, there are several outstanding questions that are ripe for study. Sox9 has been shown to have pioneer activity in hair follicle stem cells (Adam et al., 2015); however, it is unknown whether this function extends to the neural crest. One intriguing possibility is that Sox9 and other SoxE factors, through pioneer activity, set the stage in the chromatin landscape of neural crest progenitors for the subsequent adoption of specific lineage states. Recent data has shown that the chromatin of vagal neural crest is biased toward specific lineages prior to the onset of migration (Ling and Sauka-Spengler, 2019). It will be important to determine if such biases also exist in cranial neural crest and if different SoxE factors play roles in establishing these predispositions. In addition, there is evidence for direct interaction of Sox factors with epigenetic factors, such as HDACs, to regulate cell fate decisions (Duman et al., 2020). To date, studies of Sox partners/co-factors have predominately focused on other transcription factors. It is essential, however, to broaden our understanding of Sox interacting factors to include epigenetic modifiers, and to determine how these interactions shape the chromatin landscape within the neural crest and its derivatives. Finally, global histone acetylation in the neural crest differs from that of differentiated cells and actually more closely resembles that of blastula stem cells (Rao and LaBonne, 2018). To what degree are these similar epigenetic signatures mediated by Sox factors? Does switching regulation from SoxB1 in blastula stem cells to SoxE factors in neural crest stem cells lead to maintained chromatin architecture at Sox targets within the pluripotency GRN? As more large-scale sequencing experiments are conducted, and with the growing power of single cell approaches, such questions are likely to be answered in the near future.
ES wrote the manuscript. CL edited the manuscript. Both authors contributed to the article and approved the submitted version.
The authors acknowledge funding support from the NIH (1F32DE029113-ES and R01GM116538-CL), the National Science Foundation (1764421), and Simons Foundation (SFARI 597491-RWC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdelalim, E. M., Emara, M. M., and Kolatkar, P. R. (2014). The SOX transcription factors as key players in pluripotent stem cells. Stem Cells Dev. 23, 2687–2699. doi: 10.1089/scd.2014.0297
Adam, R. C., Yang, H., Rockowitz, S., Larsen, S. B., Nikolova, M., Oristian, D. S., et al. (2015). Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366–370. doi: 10.1038/nature14289
Adameyko, I., Lallemend, F., Furlan, A., Zinin, N., Aranda, S., Kitambi, S. S., et al. (2012). Sox2 and Mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 139, 397–410. doi: 10.1242/dev.065581
Affolter, M., Bellusci, S., Itoh, N., Shilo, B., Thiery, J. P., and Werb, Z. (2003). Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev. Cell 4, 11–18.
Akiyama, H., Chaboissier, M. C., Martin, J. F., Schedl, A., and de Crombrugghe, B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828. doi: 10.1101/gad.1017802
Aksoy, I., Jauch, R., Eras, V., Chng, W. B., Chen, J., Divakar, U., et al. (2013). Sox transcription factors require selective interactions with Oct4 and specific transactivation functions to mediate reprogramming. Stem Cells 31, 2632–2646. doi: 10.1002/stem.1522
Ambrosetti, D. C., Basilico, C., and Dailey, L. (1997). Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol. Cell. Biol. 17, 6321–6329. doi: 10.1128/mcb.17.11.6321
Angelozzi, M., and Lefebvre, V. (2019). SOXopathies: growing family of developmental disorders due to SOX mutations. Trends Genet. TIG 35, 658–671. doi: 10.1016/j.tig.2019.06.003
Aoki, Y., Saint-Germain, N., Gyda, M., Magner-Fink, E., Lee, Y. H., Credidio, C., et al. (2003). Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev. Biol. 259, 19–33. doi: 10.1016/s0012-1606(03)00161-1
Aquino, J. B., Hjerling-Leffler, J., Koltzenburg, M., Edlund, T., Villar, M. J., and Ernfors, P. (2006). In vitro and in vivo differentiation of boundary cap neural crest stem cells into mature Schwann cells. Exp. Neurol. 198, 438–449. doi: 10.1016/j.expneurol.2005.12.015
Banerjee, A. K. (1986). Waardenburg’s syndrome associated with ostium secundum atrial septal defect. J. R. Soc. Med. 79, 677–678.
Bartesaghi, L., Arnaud Gouttenoire, E., Prunotto, A., Medard, J. J., Bergmann, S., and Chrast, R. (2015). Sox4 participates in the modulation of Schwann cell myelination. Eur. J. Neurosci. 42, 1788–1796. doi: 10.1111/ejn.12929
Bastaki, F., Mohamed, M., Nair, P., Saif, F., Tawfiq, N., Al-Ali, M. T., et al. (2016). A novel SOX18 mutation uncovered in Jordanian patient with hypotrichosis-lymphedema-telangiectasia syndrome by whole exome sequencing. Mol. Cell. Probes 30, 18–21. doi: 10.1016/j.mcp.2015.11.005
Bell, D. M., Leung, K. K., Wheatley, S. C., Ng, L. J., Zhou, S., Ling, K. W., et al. (1997). SOX9 directly regulates the type-II collagen gene. Nat. Genet. 16, 174–178. doi: 10.1038/ng0697-174
Berendsen, A. D., and Olsen, B. R. (2015). Bone development. Bone 80, 14–18. doi: 10.1016/j.bone.2015.04.035
Betancur, P., Bronner-Fraser, M., and Sauka-Spengler, T. (2010). Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. U.S.A. 107, 3570–3575. doi: 10.1073/pnas.0906596107
Bi, W., Deng, J. M., Zhang, Z., Behringer, R. R., and de Crombrugghe, B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85–89. doi: 10.1038/8792
Bi, W., Huang, W., Whitworth, D. J., Deng, J. M., Zhang, Z., Behringer, R. R., et al. (2001). Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc. Natl. Acad. Sci. U.S.A. 98, 6698–6703. doi: 10.1073/pnas.111092198
Bondurand, N., Dastot-Le Moal, F., Stanchina, L., Collot, N., Baral, V., Marlin, S., et al. (2007). Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 81, 1169–1185. doi: 10.1086/522090
Bondurand, N., Pingault, V., Goerich, D. E., Lemort, N., Sock, E., Le Caignec, C., et al. (2000). Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum. Mol. Genet. 9, 1907–1917. doi: 10.1093/hmg/9.13.1907
Bowles, J., Schepers, G., and Koopman, P. (2000). Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 227, 239–255. doi: 10.1006/dbio.2000.9883
Britsch, S. (2007). The neuregulin-I/ErbB signaling system in development and disease. Adv. Anat. Embryol. Cell Biol. 190, 1–65. doi: 10.1007/978-3-540-37107-6_1
Britsch, S., Goerich, D. E., Riethmacher, D., Peirano, R. I., Rossner, M., Nave, K. A., et al. (2001). The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 15, 66–78. doi: 10.1101/gad.186601
Bronner, M. E., and LeDouarin, N. M. (2012). Development and evolution of the neural crest: an overview. Dev. Biol. 366, 2–9. doi: 10.1016/j.ydbio.2011.12.042
Buitrago-Delgado, E., Nordin, K., Rao, A., Geary, L., and LaBonne, C. (2015). NEURODEVELOPMENT. Shared regulatory programs suggest retention of blastula-stage potential in neural crest cells. Science 348, 1332–1335. doi: 10.1126/science.aaa3655
Buitrago-Delgado, E., Schock, E. N., Nordin, K., and LaBonne, C. (2018). A transition from SoxB1 to SoxE transcription factors is essential for progression from pluripotent blastula cells to neural crest cells. Dev. Biol. 444, 50–61. doi: 10.1016/j.ydbio.2018.08.008
Bush, J. O., and Jiang, R. (2012). Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231–243. doi: 10.1242/dev.067082
Chai, Y., Jiang, X., Ito, Y., Bringas, P. Jr., Han, J., Rowitch, D. H., et al. (2000). Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127, 1671–1679.
Chan, K. K., Wong, C. K., Lui, V. C., Tam, P. K., and Sham, M. H. (2003). Analysis of SOX10 mutations identified in Waardenburg-Hirschsprung patients: differential effects on target gene regulation. J. Cell. Biochem. 90, 573–585. doi: 10.1002/jcb.10656
Chatzeli, L., Gaete, M., and Tucker, A. S. (2017). Fgf10 and Sox9 are essential for the establishment of distal progenitor cells during mouse salivary gland development. Development 144, 2294–2305. doi: 10.1242/dev.146019
Chen, K., Zong, L., Zhan, Y., Wu, X., Liu, M., and Jiang, H. (2015). Genetic counseling for a three-generation Chinese family with Waardenburg syndrome type II associated with a rare SOX10 mutation. Int. J. Pediatr. Otorhinolaryngol. 79, 745–748. doi: 10.1016/j.ijporl.2015.03.006
Chen, Z., Huang, J., Liu, Y., Dattilo, L. K., Huh, S. H., Ornitz, D., et al. (2014). FGF signaling activates a Sox9-Sox10 pathway for the formation and branching morphogenesis of mouse ocular glands. Development 141, 2691–2701. doi: 10.1242/dev.108944
Cheung, M., and Briscoe, J. (2003). Neural crest development is regulated by the transcription factor Sox9. Development 130, 5681–5693. doi: 10.1242/dev.00808
Cheung, M., Chaboissier, M. C., Mynett, A., Hirst, E., Schedl, A., and Briscoe, J. (2005). The transcriptional control of trunk neural crest induction, survival, and delamination. Dev. Cell 8, 179–192. doi: 10.1016/j.devcel.2004.12.010
Chew, L. J., and Gallo, V. (2009). The Yin and Yang of Sox proteins: activation and repression in development and disease. J. Neurosci. Res. 87, 3277–3287. doi: 10.1002/jnr.22128
Cimadamore, F., Fishwick, K., Giusto, E., Gnedeva, K., Cattarossi, G., Miller, A., et al. (2011). Human ESC-derived neural crest model reveals a key role for SOX2 in sensory neurogenesis. Cell Stem Cell 8, 538–551. doi: 10.1016/j.stem.2011.03.011
Cizelsky, W., Hempel, A., Metzig, M., Tao, S., Hollemann, T., Kuhl, M., et al. (2013). sox4 and sox11 function during Xenopus laevis eye development. PLoS One 8:e69372. doi: 10.1371/journal.pone.0069372
Csukasi, F., Duran, I., Zhang, W., Martin, J. H., Barad, M., Bamshad, M., et al. (2019). Dominant-negative SOX9 mutations in campomelic dysplasia. Hum. Mutat. 40, 2344–2352. doi: 10.1002/humu.23888
Dai, W., Xu, X., Li, S., Ma, J., Shi, Q., Guo, S., et al. (2017). SOX4 promotes proliferative signals by regulating glycolysis through AKT activation in melanoma cells. J. Invest. Dermatol. 137, 2407–2416. doi: 10.1016/j.jid.2017.06.026
D’Amico-Martel, A., and Noden, D. M. (1983). Contributions of placodal and neural crest cells to avian cranial peripheral ganglia. Am. J. Anat. 166, 445–468. doi: 10.1002/aja.1001660406
Dixon, M. J., Marazita, M. L., Beaty, T. H., and Murray, J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167–178. doi: 10.1038/nrg2933
Dourmishev, A. L., Dourmishev, L. A., Schwartz, R. A., and Janniger, C. K. (1999). Waardenburg syndrome. Int. J. Dermatol. 38, 656–663.
Duman, M., Vaquie, A., Nocera, G., Heller, M., Stumpe, M., Siva Sankar, D., et al. (2020). EEF1A1 deacetylation enables transcriptional activation of remyelination. Nat. Commun. 11:3420.
Dutton, K. A., Pauliny, A., Lopes, S. S., Elworthy, S., Carney, T. J., Rauch, J., et al. (2001). Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 128, 4113–4125.
Ellis, P., Fagan, B. M., Magness, S. T., Hutton, S., Taranova, O., Hayashi, S., et al. (2004). SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 26, 148–165. doi: 10.1159/000082134
Emmerson, E., May, A. J., Nathan, S., Cruz-Pacheco, N., Lizama, C. O., Maliskova, L., et al. (2017). SOX2 regulates acinar cell development in the salivary gland. eLife 6:e26620.
Foster, J. W., Dominguez-Steglich, M. A., Guioli, S., Kwok, C., Weller, P. A., Stevanovic, M., et al. (1994). Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372, 525–530. doi: 10.1038/372525a0
Fufa, T. D., Harris, M. L., Watkins-Chow, D. E., Levy, D., Gorkin, D. U., Gildea, D. E., et al. (2015). Genomic analysis reveals distinct mechanisms and functional classes of SOX10-regulated genes in melanocytes. Hum. Mol. Genet. 24, 5433–5450. doi: 10.1093/hmg/ddv267
Gadi, J., Jung, S. H., Lee, M. J., Jami, A., Ruthala, K., Kim, K. M., et al. (2013). The transcription factor protein Sox11 enhances early osteoblast differentiation by facilitating proliferation and the survival of mesenchymal and osteoblast progenitors. J. Biol. Chem. 288, 25400–25413. doi: 10.1074/jbc.m112.413377
Girard, M., and Goossens, M. (2006). Sumoylation of the SOX10 transcription factor regulates its transcriptional activity. FEBS Lett. 580, 1635–1641. doi: 10.1016/j.febslet.2006.02.011
Graham, V., Khudyakov, J., Ellis, P., and Pevny, L. (2003). SOX2 functions to maintain neural progenitor identity. Neuron 39, 749–765. doi: 10.1016/s0896-6273(03)00497-5
Grimm, D., Bauer, J., Wise, P., Kruger, M., Simonsen, U., Wehland, M., et al. (2019). The role of SOX family members in solid tumours and metastasis. Semin. Cancer Biol. [Epub ahead of print].
Gubbay, J., Collignon, J., Koopman, P., Capel, B., Economou, A., Munsterberg, A., et al. (1990). A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346, 245–250. doi: 10.1038/346245a0
Haldin, C. E., and LaBonne, C. (2010). SoxE factors as multifunctional neural crest regulatory factors. Int. J. Biochem. Cell Biol. 42, 441–444. doi: 10.1016/j.biocel.2009.11.014
Hargrave, M., Wright, E., Kun, J., Emery, J., Cooper, L., and Koopman, P. (1997). Expression of the Sox11 gene in mouse embryos suggests roles in neuronal maturation and epithelio-mesenchymal induction. Dev. Dyn. 210, 79–86. doi: 10.1002/(sici)1097-0177(199710)210:2<79::aid-aja1>3.0.co;2-6
Harris, M. L., Baxter, L. L., Loftus, S. K., and Pavan, W. J. (2010). Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 23, 496–513. doi: 10.1111/j.1755-148x.2010.00711.x
Harris, M. L., Buac, K., Shakhova, O., Hakami, R. M., Wegner, M., Sommer, L., et al. (2013). A dual role for SOX10 in the maintenance of the postnatal melanocyte lineage and the differentiation of melanocyte stem cell progenitors. PLoS Genet. 9:e1003644. doi: 10.1371/journal.pgen.1003644
Haseeb, A., and Lefebvre, V. (2019). The SOXE transcription factors-SOX8, SOX9 and SOX10-share a bi-partite transactivation mechanism. Nucleic Acids Res. 47, 6917–6931. doi: 10.1093/nar/gkz523
Hattori, T., Coustry, F., Stephens, S., Eberspaecher, H., Takigawa, M., Yasuda, H., et al. (2008). Transcriptional regulation of chondrogenesis by coactivator Tip60 via chromatin association with Sox9 and Sox5. Nucleic Acids Res. 36, 3011–3024. doi: 10.1093/nar/gkn150
Healy, C., Uwanogho, D., and Sharpe, P. T. (1996). Expression of the chicken Sox9 gene marks the onset of cartilage differentiation. Ann. N. Y. Acad. Sci. 785, 261–262. doi: 10.1111/j.1749-6632.1996.tb56278.x
Heenan, P., Zondag, L., and Wilson, M. J. (2016). Evolution of the Sox gene family within the chordate phylum. Gene 575, 385–392. doi: 10.1016/j.gene.2015.09.013
Hempel, A., Pagnamenta, A. T., Blyth, M., Mansour, S., McConnell, V., Kou, I., et al. (2016). Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome. J. Med. Genet. 53, 152–162. doi: 10.1136/jmedgenet-2015-103393
Herbarth, B., Pingault, V., Bondurand, N., Kuhlbrodt, K., Hermans-Borgmeyer, I., Puliti, A., et al. (1998). Mutation of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 95, 5161–5165. doi: 10.1073/pnas.95.9.5161
Hirobe, T. (1984). Histochemical survey of the distribution of the epidermal melanoblasts and melanocytes in the mouse during fetal and postnatal periods. Anat. Rec. 208, 589–594. doi: 10.1002/ar.1092080414
Holbrook, K. A., Underwood, R. A., Vogel, A. M., Gown, A. M., and Kimball, H. (1989). The appearance, density and distribution of melanocytes in human embryonic and fetal skin revealed by the anti-melanoma monoclonal antibody, HMB-45. Anat. Embryol. 180, 443–455. doi: 10.1007/bf00305119
Hong, C. S., and Saint-Jeannet, J. P. (2005). Sox proteins and neural crest development. Semin. Cell Dev. Biol. 16, 694–703. doi: 10.1016/j.semcdb.2005.06.005
Honore, S. M., Aybar, M. J., and Mayor, R. (2003). Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Dev. Biol. 260, 79–96. doi: 10.1016/s0012-1606(03)00247-1
Hou, L., Arnheiter, H., and Pavan, W. J. (2006). Interspecies difference in the regulation of melanocyte development by SOX10 and MITF. Proc. Natl. Acad. Sci. U.S.A. 103, 9081–9085. doi: 10.1073/pnas.0603114103
Hou, L., Srivastava, Y., and Jauch, R. (2017). Molecular basis for the genome engagement by Sox proteins. Semin. Cell Dev. Biol. 63, 2–12. doi: 10.1016/j.semcdb.2016.08.005
Huang, W., Zhou, X., Lefebvre, V., and de Crombrugghe, B. (2000). Phosphorylation of SOX9 by cyclic AMP-dependent protein kinase A enhances SOX9’s ability to transactivate a Col2a1 chondrocyte-specific enhancer. Mol. Cell. Biol. 20, 4149–4158. doi: 10.1128/mcb.20.11.4149-4158.2000
Huang, Y. H., Jankowski, A., Cheah, K. S., Prabhakar, S., and Jauch, R. (2015). SOXE transcription factors form selective dimers on non-compact DNA motifs through multifaceted interactions between dimerization and high-mobility group domains. Sci. Rep. 5:10398.
Inoue, K., Khajavi, M., Ohyama, T., Hirabayashi, S., Wilson, J., Reggin, J. D., et al. (2004). Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 36, 361–369. doi: 10.1038/ng1322
Inoue, K., Tanabe, Y., and Lupski, J. R. (1999). Myelin deficiencies in both the central and the peripheral nervous systems associated with a SOX10 mutation. Ann. Neurol. 46, 313–318. doi: 10.1002/1531-8249(199909)46:3<313::aid-ana6>3.0.co;2-7
Irie, N., Weinberger, L., Tang, W. W., Kobayashi, T., Viukov, S., Manor, Y. S., et al. (2015). SOX17 is a critical specifier of human primordial germ cell fate. Cell 160, 253–268. doi: 10.1016/j.cell.2014.12.013
Irrthum, A., Devriendt, K., Chitayat, D., Matthijs, G., Glade, C., Steijlen, P. M., et al. (2003). Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am. J. Hum. Genet. 72, 1470–1478. doi: 10.1086/375614
Ishikawa, R., Kawasaki, M., Kawasaki, K., Yamada, A., Trakanant, S., Meguro, F., et al. (2018). Sox genes show spatiotemporal expression during murine tongue and eyelid development. Int. J. Dent. 2018:1601363.
Iso, M., Fukami, M., Horikawa, R., Azuma, N., Kawashiro, N., and Ogata, T. (2008). SOX10 mutation in Waardenburg syndrome type II. Am. J. Med. Genet. A 146A, 2162–2163. doi: 10.1002/ajmg.a.32403
Jandzik, D., Garnett, A. T., Square, T. A., Cattell, M. V., Yu, J. K., and Medeiros, D. M. (2015). Evolution of the new vertebrate head by co-option of an ancient chordate skeletal tissue. Nature 518, 534–537. doi: 10.1038/nature14000
Jauch, R., Aksoy, I., Hutchins, A. P., Ng, C. K., Tian, X. F., Chen, J., et al. (2011). Conversion of Sox17 into a pluripotency reprogramming factor by reengineering its association with Oct4 on DNA. Stem Cells 29, 940–951. doi: 10.1002/stem.639
Jiao, Z., Mollaaghababa, R., Pavan, W. J., Antonellis, A., Green, E. D., and Hornyak, T. J. (2004). Direct interaction of Sox10 with the promoter of murine Dopachrome Tautomerase (Dct) and synergistic activation of Dct expression with Mitf. Pigment. Cell Res. 17, 352–362. doi: 10.1111/j.1600-0749.2004.00154.x
Julian, L. M., McDonald, A. C., and Stanford, W. L. (2017). Direct reprogramming with SOX factors: masters of cell fate. Curr. Opin. Genet. Dev. 46, 24–36. doi: 10.1016/j.gde.2017.06.005
Juuri, E., Jussila, M., Seidel, K., Holmes, S., Wu, P., Richman, J., et al. (2013). Sox2 marks epithelial competence to generate teeth in mammals and reptiles. Development 140, 1424–1432. doi: 10.1242/dev.089599
Juuri, E., Saito, K., Ahtiainen, L., Seidel, K., Tummers, M., Hochedlinger, K., et al. (2012). Sox2+ stem cells contribute to all epithelial lineages of the tooth via Sfrp5+ progenitors. Dev. Cell 23, 317–328. doi: 10.1016/j.devcel.2012.05.012
Kamachi, Y., and Kondoh, H. (2013). Sox proteins: regulators of cell fate specification and differentiation. Development 140, 4129–4144. doi: 10.1242/dev.091793
Kawasaki, K., Kawasaki, M., Watanabe, M., Idrus, E., Nagai, T., Oommen, S., et al. (2015). Expression of Sox genes in tooth development. Int. J. Dev. Biol. 59, 471–478. doi: 10.1387/ijdb.150192ao
Kellerer, S., Schreiner, S., Stolt, C. C., Scholz, S., Bosl, M. R., and Wegner, M. (2006). Replacement of the Sox10 transcription factor by Sox8 reveals incomplete functional equivalence. Development 133, 2875–2886. doi: 10.1242/dev.02477
Kelsh, R. N., and Eisen, J. S. (2000). The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development 127, 515–525.
Khan, U., Study, D., Baker, E., and Clayton-Smith, J. (2018). Observation of cleft palate in an individual with SOX11 mutation: indication of a role for SOX11 in human palatogenesis. Cleft. Palate. Craniofac. J. 55, 456–461. doi: 10.1177/1055665617739312
Knosp, W. M., Knox, S. M., and Hoffman, M. P. (2012). Salivary gland organogenesis. Wiley interdisciplinary reviews. Dev. Biol. 1, 69–82. doi: 10.1002/wdev.4
Kondoh, H., and Kamachi, Y. (2010). SOX-partner code for cell specification: regulatory target selection and underlying molecular mechanisms. Int. J. Biochem. Cell Biol. 42, 391–399. doi: 10.1016/j.biocel.2009.09.003
Kuhlbrodt, K., Herbarth, B., Sock, E., Enderich, J., Hermans-Borgmeyer, I., and Wegner, M. (1998a). Cooperative function of POU proteins and SOX proteins in glial cells. J. Biol. Chem. 273, 16050–16057. doi: 10.1074/jbc.273.26.16050
Kuhlbrodt, K., Herbarth, B., Sock, E., Hermans-Borgmeyer, I., and Wegner, M. (1998b). Sox10, a novel transcriptional modulator in glial cells. J. Neurosci. 18, 237–250. doi: 10.1523/jneurosci.18-01-00237.1998
Kwok, C., Weller, P. A., Guioli, S., Foster, J. W., Mansour, S., Zuffardi, O., et al. (1995). Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sexreversal. Am. J. Hum. Genet. 57, 1028–1036.
Lakiza, O., Miller, S., Bunce, A., Lee, E. M., and McCauley, D. W. (2011). SoxE gene duplication and development of the lamprey branchial skeleton: insights into development and evolution of the neural crest. Dev. Biol. 359, 149–161. doi: 10.1016/j.ydbio.2011.08.012
Lamb, A. N., Rosenfeld, J. A., Neill, N. J., Talkowski, M. E., Blumenthal, I., Girirajan, S., et al. (2012). Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum. Mutation 33, 728–740. doi: 10.1002/humu.22037
Langer, L., Sulik, K., and Pevny, L. (2014). Cleft Palate in a Mouse Model of SOX2 Haploinsufficiency. Cleft. Palate. Craniofac. J. 51, 110–114. doi: 10.1597/12-260
Le, N., Nagarajan, R., Wang, J. Y., Araki, T., Schmidt, R. E., and Milbrandt, J. (2005). Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc. Natl. Acad. Sci. U.S.A. 102, 2596–2601. doi: 10.1073/pnas.0407836102
LeBlanc, S. E., Ward, R. M., and Svaren, J. (2007). Neuropathy-associated Egr2 mutants disrupt cooperative activation of myelin protein zero by Egr2 and Sox10. Mol. Cell. Biol. 27, 3521–3529. doi: 10.1128/mcb.01689-06
LeDouarin, N. M., and Kalcheim, C. (1999). The Neural Crest, 2nd Edn. Cambridge, MA: Cambridge University Press.
Lee, P. C., Taylor-Jaffe, K. M., Nordin, K. M., Prasad, M. S., Lander, R. M., and LaBonne, C. (2012). SUMOylated SoxE factors recruit Grg4 and function as transcriptional repressors in the neural crest. J. Cell Biol. 198, 799–813. doi: 10.1083/jcb.201204161
Lefebvre, V., Huang, W., Harley, V. R., Goodfellow, P. N., and de Crombrugghe, B. (1997). SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol. Cell. Biol. 17, 2336–2346. doi: 10.1128/mcb.17.4.2336
Lefebvre, V., Li, P., and de Crombrugghe, B. (1998). A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 17, 5718–5733. doi: 10.1093/emboj/17.19.5718
Leung, V. Y., Gao, B., Leung, K. K., Melhado, I. G., Wynn, S. L., Au, T. Y., et al. (2011). SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 7:e1002356. doi: 10.1371/journal.pgen.1002356
Li, M., Zhao, C., Wang, Y., Zhao, Z., and Meng, A. (2002). Zebrafish sox9b is an early neural crest marker. Dev. Genes Evol. 212, 203–206. doi: 10.1007/s00427-002-0235-2
Ling, I. T. C., and Sauka-Spengler, T. (2019). Early chromatin shaping predetermines multipotent vagal neural crest into neural, neuronal and mesenchymal lineages. Nat. Cell Biol. 21, 1504-1517. doi: 10.1038/s41556-019-0428-9
Liu, C. F., and Lefebvre, V. (2015). The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 43, 8183–8203. doi: 10.1093/nar/gkv688
Liu, J. A., Tai, A., Hong, J., Cheung, M. P. L., Sham, M. H., Cheah, K. S. E., et al. (2020). Fbxo9 functions downstream of Sox10 to determine neuron-glial fate choice in the dorsal root ganglia through Neurog2 destabilization. Proc. Natl. Acad. Sci. U.S.A. 117, 4199–4210. doi: 10.1073/pnas.1916164117
Liu, J. A., Wu, M. H., Yan, C. H., Chau, B. K., So, H., Ng, A., et al. (2013). Phosphorylation of Sox9 is required for neural crest delamination and is regulated downstream of BMP and canonical Wnt signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 2882–2887. doi: 10.1073/pnas.1211747110
Liu, Y. R., Laghari, Z. A., Novoa, C. A., Hughes, J., Webster, J. R., Goodwin, P. E., et al. (2014). Sox2 acts as a transcriptional repressor in neural stem cells. BMC Neurosci. 15:95. doi: 10.1186/1471-2202-15-95
Liu, Z., and Kraus, W. L. (2017). Catalytic-independent functions of PARP-1 determine Sox2 pioneer activity at intractable genomic loci. Mol. Cell. 65, e589.
Lombaert, I. M., Abrams, S. R., Li, L., Eswarakumar, V. P., Sethi, A. J., Witt, R. L., et al. (2013). Combined KIT and FGFR2b signaling regulates epithelial progenitor expansion during organogenesis. Stem Cell Rep. 1, 604–619. doi: 10.1016/j.stemcr.2013.10.013
Long, J., Wang, G., He, D., and Liu, F. (2004). Repression of Smad4 transcriptional activity by SUMO modification. Biochem. J. 379, 23–29. doi: 10.1042/bj20031867
Lovell-Badge, R. (2010). The early history of the Sox genes. Int. J. Biochem. Cell Biol. 42, 378–380. doi: 10.1016/j.biocel.2009.12.003
Ludwig, A., Rehberg, S., and Wegner, M. (2004). Melanocyte-specific expression of dopachrome tautomerase is dependent on synergistic gene activation by the Sox10 and Mitf transcription factors. FEBS Lett. 556, 236–244. doi: 10.1016/s0014-5793(03)01446-7
Marathe, H. G., Watkins-Chow, D. E., Weider, M., Hoffmann, A., Mehta, G., Trivedi, A., et al. (2017). BRG1 interacts with SOX10 to establish the melanocyte lineage and to promote differentiation. Nucleic Acids Res. 45, 6442–6458. doi: 10.1093/nar/gkx259
Martin, K. J., Rasch, L. J., Cooper, R. L., Metscher, B. D., Johanson, Z., and Fraser, G. J. (2016). Sox2+ progenitors in sharks link taste development with the evolution of regenerative teeth from denticles. Proc. Natl. Acad. Sci. U.S.A. 113, 14769–14774. doi: 10.1073/pnas.1612354113
Maruyama, M., Ichisaka, T., Nakagawa, M., and Yamanaka, S. (2005). Differential roles for Sox15 and Sox2 in transcriptional control in mouse embryonic stem cells. J. Biol. Chem. 280, 24371–24379. doi: 10.1074/jbc.m501423200
Mazzara, P. G., Massimino, L., Pellegatta, M., Ronchi, G., Ricca, A., Iannielli, A., et al. (2017). Two factor-based reprogramming of rodent and human fibroblasts into Schwann cells. Nat. Commun. 8:14088.
McCauley, D. W., and Bronner-Fraser, M. (2006). Importance of SoxE in neural crest development and the evolution of the pharynx. Nature 441, 750–752. doi: 10.1038/nature04691
McDowall, S., Argentaro, A., Ranganathan, S., Weller, P., Mertin, S., Mansour, S., et al. (1999). Functional and structural studies of wild type SOX9 and mutations causing campomelic dysplasia. J. Biol. Chem. 274, 24023–24030. doi: 10.1074/jbc.274.34.24023
Meyer, J., Sudbeck, P., Held, M., Wagner, T., Schmitz, M. L., Bricarelli, F. D., et al. (1997). Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum. Mol. Genet. 6, 91–98. doi: 10.1093/hmg/6.1.91
Morales, A. V., Perez-Alcala, S., and Barbas, J. A. (2007). Dynamic Sox5 protein expression during cranial ganglia development. Dev. Dyn. 236, 2702–2707. doi: 10.1002/dvdy.21282
Mori-Akiyama, Y., Akiyama, H., Rowitch, D. H., and de Crombrugghe, B. (2003). Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. U.S.A. 100, 9360–9365. doi: 10.1073/pnas.1631288100
Morin, M., Vinuela, A., Rivera, T., Villamar, M., Moreno-Pelayo, M. A., Moreno, F., et al. (2008). A de novo missense mutation in the gene encoding the SOX10 transcription factor in a Spanish sporadic case of Waardenburg syndrome type IV. Am. J. Med. Genet. A 146A, 1032–1037. doi: 10.1002/ajmg.a.32181
Murisier, F., Guichard, S., and Beermann, F. (2006). A conserved transcriptional enhancer that specifies Tyrp1 expression to melanocytes. Dev. Biol. 298, 644–655. doi: 10.1016/j.ydbio.2006.05.011
Murisier, F., Guichard, S., and Beermann, F. (2007). The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigment. Cell Res. 20, 173–184. doi: 10.1111/j.1600-0749.2007.00368.x
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106. doi: 10.1038/nbt1374
Noden, D. M., and Trainor, P. A. (2005). Relations and interactions between cranial mesoderm and neural crest populations. J. Anat. 207, 575–601. doi: 10.1111/j.1469-7580.2005.00473.x
Nordin, K., and LaBonne, C. (2014). Sox5 Is a DNA-binding cofactor for BMP R-Smads that directs target specificity during patterning of the early ectoderm. Dev. Cell. 31, 374–382. doi: 10.1016/j.devcel.2014.10.003
O’Donnell, M., Hong, C. S., Huang, X., Delnicki, R. J., and Saint-Jeannet, J. P. (2006). Functional analysis of Sox8 during neural crest development in Xenopus. Development 133, 3817–3826. doi: 10.1242/dev.02558
Okamoto, N., Ehara, E., Tsurusaki, Y., Miyake, N., and Matsumoto, N. (2018). Coffin-Siris syndrome and cardiac anomaly with a novel SOX11 mutation. Congenit. Anom. 58, 105–107. doi: 10.1111/cga.12242
Passeron, T., Valencia, J. C., Bertolotto, C., Hoashi, T., Le Pape, E., Takahashi, K., et al. (2007). SOX9 is a key player in ultraviolet B-induced melanocyte differentiation and pigmentation. Proc. Natl. Acad. Sci. U.S.A. 104, 13984–13989. doi: 10.1073/pnas.0705117104
Peirano, R. I., Goerich, D. E., Riethmacher, D., and Wegner, M. (2000). Protein zero gene expression is regulated by the glial transcription factor Sox10. Mol. Cell. Biol. 20, 3198–3209. doi: 10.1128/MCB.20.9.3198-3209.2000
Peirano, R. I., and Wegner, M. (2000). The glial transcription factor Sox10 binds to DNA both as monomer and dimer with different functional consequences. Nucleic Acids Res. 28, 3047–3055. doi: 10.1093/nar/28.16.3047
Perez-Alcala, S., Nieto, M. A., and Barbas, J. A. (2004). LSox5 regulates RhoB expression in the neural tube and promotes generation of the neural crest. Development 131, 4455–4465. doi: 10.1242/dev.01329
Pevny, L., and Placzek, M. (2005). SOX genes and neural progenitor identity. Curr. Opin. Neurobiol. 15, 7–13. doi: 10.1016/j.conb.2005.01.016
Pingault, V., Bondurand, N., Kuhlbrodt, K., Goerich, D. E., Prehu, M. O., Puliti, A., et al. (1998). SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18, 171–173. doi: 10.1038/ng0298-171
Pingault, V., Girard, M., Bondurand, N., Dorkins, H., Van Maldergem, L., Mowat, D., et al. (2002). SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum. Genet. 111, 198–206. doi: 10.1007/s00439-002-0765-8
Pingault, V., Ente, D., Dastot-Le Moal, F., Goossens, M., Marlin, S., and Bondurand, N. (2010). Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 31, 391–406. doi: 10.1002/humu.21211
Potterf, S. B., Furumura, M., Dunn, K. J., Arnheiter, H., and Pavan, W. J. (2000). Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum. Genet. 107, 1–6. doi: 10.1007/s004390000328
Prasad, M. S., Sauka-Spengler, T., and LaBonne, C. (2012). Induction of the neural crest state: control of stem cell attributes by gene regulatory, post-transcriptional and epigenetic interactions. Dev. Biol. 366, 10–21. doi: 10.1016/j.ydbio.2012.03.014
Raftery, R. M., Gonzalez Vazquez, A. G., Chen, G., and O’Brien, F. J. (2020). Activation of the SOX-5, SOX-6, and SOX-9 trio of transcription factors using a gene-activated scaffold stimulates mesenchymal stromal cell chondrogenesis and inhibits endochondral ossification. Adv. Healthc. Mater. 9:e1901827. doi: 10.1002/adhm.201901827
Rao, A., and LaBonne, C. (2018). Histone deacetylase activity has an essential role in establishing and maintaining the vertebrate neural crest. Development 145:dev163386. doi: 10.1242/dev.163386
Rex, M., Orme, A., Uwanogho, D., Tointon, K., Wigmore, P. M., Sharpe, P. T., et al. (1997). Dynamic expression of chicken Sox2 and Sox3 genes in ectoderm induced to form neural tissue. Dev. Dyn. 209, 323–332. doi: 10.1002/(SICI)1097-0177(199707)209:3<323::AID-AJA7>3.0.CO;2-K
Roberts, S. L., Dun, X. P., Doddrell, R. D. S., Mindos, T., Drake, L. K., Onaitis, M. W., et al. (2017). Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development 144, 3114–3125. doi: 10.1242/dev.150656
Roellig, D., Tan-Cabugao, J., Esaian, S., and Bronner, M. E. (2017). Dynamic transcriptional signature and cell fate analysis reveals plasticity of individual neural plate border cells. eLife 6:e21620. doi: 10.7554/eLife.21620.032
Rosonina, E., Akhter, A., Dou, Y., Babu, J., and Sri Theivakadadcham, V. S. (2017). Regulation of transcription factors by sumoylation. Transcription 8, 220–231. doi: 10.1080/21541264.2017.1311829
Sakai, D., Suzuki, T., Osumi, N., and Wakamatsu, Y. (2006). Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development 133, 1323–1333. doi: 10.1242/dev.02297
Sanchez-Mejias, A., Watanabe, Y., R, M. F., López-Alonso, M., Antinolo, G., Bondurand, N., Borrego, S., et al. (2010). Involvement of SOX10 in the pathogenesis of Hirschsprung disease: report of a truncating mutation in an isolated patient. J. Mol. Med. 88, 507–514. doi: 10.1007/s00109-010-0592-7
Schlierf, B., Ludwig, A., Klenovsek, K., and Wegner, M. (2002). Cooperative binding of Sox10 to DNA: requirements and consequences. Nucleic Acids Res. 30, 5509–5516. doi: 10.1093/nar/gkf690
Schlosser, G., Awtry, T., Brugmann, S. A., Jensen, E. D., Neilson, K., Ruan, G., et al. (2008). Eya1 and Six1 promote neurogenesis in the cranial placodes in a SoxB1-dependent fashion. Dev. Biol. 320, 199–214. doi: 10.1016/j.ydbio.2008.05.523
Schreiner, S., Cossais, F., Fischer, K., Scholz, S., Bosl, M. R., Holtmann, B., et al. (2007). Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development 134, 3271–3281. doi: 10.1242/dev.003350
Shakhova, O., Cheng, P., Mishra, P. J., Zingg, D., Schaefer, S. M., Debbache, J., et al. (2015). Antagonistic cross-regulation between Sox9 and Sox10 controls an anti-tumorigenic program in melanoma. PLoS Genet. 11:e1004877. doi: 10.1371/journal.pgen.1004877
Sham, M. H., Lui, V. C., Chen, B. L., Fu, M., and Tam, P. K. (2001). Novel mutations of SOX10 suggest a dominant negative role in Waardenburg-Shah syndrome. J. Med. Genet. 38:E30. doi: 10.1136/jmg.38.9.e30
Shimotake, T., Tanaka, S.-C, Fukui, R., Makino, S., and Maruyama, R. (2007). Neuroglial disorders of central and peripheral nervous systems in a patient with Hirschsprung’s disease carrying allelic SOX10 truncating mutation. J. Pediatr. Surg. 42, 725–731. doi: 10.1016/j.jpedsurg.2006.12.003
Simoes-Costa, M., and Bronner, M. E. (2015). Establishing neural crest identity: a gene regulatory recipe. Development 142, 242–257. doi: 10.1242/dev.105445
Sitiwin, E., Madigan, M. C., Gratton, E., Cherepanoff, S., Conway, R. M., Whan, R., et al. (2019). Shedding light on melanins within in situ human eye melanocytes using 2-photon microscopy profiling techniques. Sci. Rep. 9:18585. doi: 10.1038/s41598-019-54871-y
Smits, P., Li, P., Mandel, J., Zhang, Z., Deng, J. M., Behringer, R. R., et al. (2001). The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev. Cell 1, 277–290. doi: 10.1016/S1534-5807(01)00003-X
Sock, E., Pagon, R. A., Keymolen, K., Lissens, W., Wegner, M., and Scherer, G. (2003). Loss of DNA-dependent dimerization of the transcription factor SOX9 as a cause for campomelic dysplasia. Hum. Mol. Genet. 12, 1439–1447. doi: 10.1093/hmg/ddg158
Sock, E., Rettig, S. D., Enderich, J., Bosl, M. R., Tamm, E. R., and Wegner, M. (2004). Gene targeting reveals a widespread role for the high-mobility-group transcription factor Sox11 in tissue remodeling. Mol. Cell. Biol. 24, 6635–6644. doi: 10.1128/MCB.24.15.6635-6644.2004
Sock, E., Schmidt, K., Hermanns-Borgmeyer, I., Bosl, M. R., and Wegner, M. (2001). Idiopathic weight reduction in mice deficient in the high-mobility-group transcription factor Sox8. Mol. Cell. Biol. 21, 6951–6959. doi: 10.1128/MCB.21.20.6951-6959.2001
Soufi, A., Garcia, M. F., Jaroszewicz, A., Osman, N., Pellegrini, M., and Zaret, K. S. (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. doi: 10.1016/j.cell.2015.03.017
Southard-Smith, E. M., Angrist, M., Ellison, J. S., Agarwala, R., Baxevanis, A. D., Chakravarti, A., et al. (1999). The Sox10(Dom) mouse: modeling the genetic variation of Waardenburg-Shah (WS4) syndrome. Genome Res. 9, 215–225.
Southard-Smith, E. M., Kos, L., and Pavan, W. J. (1998). Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 18, 60–64. doi: 10.1038/ng0198-60
Spokony, R. F., Aoki, Y., Saint-Germain, N., Magner-Fink, E., and Saint-Jeannet, J. P. (2002). The transcription factor Sox9 is required for cranial neural crest development in Xenopus. Development 129, 421–432.
Stolt, C. C., Lommes, P., Hillgartner, S., and Wegner, M. (2008). The transcription factor Sox5 modulates Sox10 function during melanocyte development. Nucleic Acids Res. 36, 5427–5440. doi: 10.1093/nar/gkn527
Sun, Z., Yu, W., Sanz Navarro, M., Sweat, M., Eliason, S., Sharp, T., et al. (2016). Sox2 and Lef-1 interact with Pitx2 to regulate incisor development and stem cell renewal. Development 143, 4115–4126. doi: 10.1242/dev.138883
Tai, A., Cheung, M., Huang, Y. H., Jauch, R., Bronner, M. E., and Cheah, K. S. (2016). SOXE neofunctionalization and elaboration of the neural crest during chordate evolution. Sci. Rep. 6:34964. doi: 10.1038/srep34964
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Tan, H., and Tee, W. W. (2019). Committing the primordial germ cell: an updated molecular perspective. Wiley Interdiscip. Rev. Syst. Biol. Med. 11”e1436. doi: 10.1002/wsbm.1436
Tanaka, J., Mabuchi, Y., Hata, K., Yasuhara, R., Takamatsu, K., Kujiraoka, S., et al. (2019). Sox9 regulates the luminal stem/progenitor cell properties of salivary glands. Exp. Cell Res. 382:111449. doi: 10.1016/j.yexcr.2019.05.030
Tanaka, S., Kamachi, Y., Tanouchi, A., Hamada, H., Jing, N., and Kondoh, H. (2004). Interplay of SOX and POU factors in regulation of the Nestin gene in neural primordial cells. Mol. Cell. Biol. 24, 8834–8846. doi: 10.1128/MCB.24.20.8834-8846.2004
Taylor, K. M., and LaBonne, C. (2005). SoxE factors function equivalently during neural crest and inner ear development and their activity is regulated by SUMOylation. Dev. Cell 9, 593–603. doi: 10.1016/j.devcel.2005.09.016
Taylor, K. M., and LaBonne, C. (2007). Modulating the activity of neural crest regulatory factors. Curr. Opin. Genet. Dev. 17, 326–331. doi: 10.1016/j.gde.2007.05.012
Thesleff, I. (2014). Current understanding of the process of tooth formation: transfer from the laboratory to the clinic. Aust. Dent. J. 59(Suppl. 1), 48–54. doi: 10.1111/adj.12102
Toki, F., Suzuki, N., Inoue, K., Suzuki, M., Hirakata, K., Nagai, K., et al. (2003). Intestinal aganglionosis associated with the Waardenburg syndrome: report of two cases and review of the literature. Pediatr. Surg. Int. 19, 725–728. doi: 10.1007/s00383-003-1057-7
Tsurusaki, Y., Koshimizu, E., Ohashi, H., Phadke, S., Kou, I., Shiina, M., et al. (2014). De novo SOX11 mutations cause Coffin-Siris syndrome. Nat. Commun. 5:4011. doi: 10.1038/ncomms5011
Uy, B. R., Simoes-Costa, M., Koo, D. E., Sauka-Spengler, T., and Bronner, M. E. (2015). Evolutionarily conserved role for SoxC genes in neural crest specification and neuronal differentiation. Dev. Biol. 397, 282–292. doi: 10.1016/j.ydbio.2014.09.022
Valenzuela, I., Fernandez-Alvarez, P., Plaja, A., Ariceta, G., Sabate-Rotes, A., Garcia-Arumi, E., et al. (2018). Further delineation of the SOX18-related hypotrichosis, lymphedema, telangiectasia syndrome (HTLS). Eur. J. Med. Genet. 61, 269–272. doi: 10.1016/j.ejmg.2018.01.001
Wagner, T., Wirth, J., Meyer, J., Zabel, B., Held, M., Zimmer, J., et al. (1994). Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79, 1111–1120. doi: 10.1016/0092-8674(94)90041-8
Wakamatsu, Y., Endo, Y., Osumi, N., and Weston, J. A. (2004). Multiple roles of Sox2, an HMG-box transcription factor in avian neural crest development. Dev. Dyn. 229, 74–86. doi: 10.1002/dvdy.10498
Wangberg, H., Wigby, K., and Jones, M. C. (2018). A novel autosomal dominant mutation in SOX18 resulting in a fatal case of hypotrichosis-lymphedema-telangiectasia syndrome. Am. J. Med. Genet. A 176, 2824–2828. doi: 10.1002/ajmg.a.40532
Watanabe, M., Kawasaki, K., Kawasaki, M., Portaveetus, T., Oommen, S., Blackburn, J., et al. (2016). Spatio-temporal expression of Sox genes in murine palatogenesis. Gene Exp. Patterns GEP 21, 111–118. doi: 10.1016/j.gep.2016.05.002
Wildhardt, G., Zirn, B., Graul-Neumann, L. M., Wechtenbruch, J., Suckfull, M., Buske, A., et al. (2013). Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: implications for molecular genetic diagnostics. BMJ Open 3:e001917. doi: 10.1136/bmjopen-2012-001917
Williams, C. A. C., Soufi, A., and Pollard, S. M. (2019). Post-translational modification of SOX family proteins: key biochemical targets in cancer? Semin. Cancer Biol. [Epub ahead of print]. doi: 10.1016/j.semcancer.2019.09.009
Wright, E., Hargrave, M. R., Christiansen, J., Cooper, L., Kun, J., Evans, T., et al. (1995). The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat. Genet. 9, 15–20. doi: 10.1038/ng0195-15
Wust, H. M., Wegener, A., Frob, F., Hartwig, A. C., Wegwitz, F., Kari, V., et al. (2020). Egr2-guided histone H2B monoubiquitination is required for peripheral nervous system myelination. Nucleic Acids Res. 48, 8959–8976. doi: 10.1093/nar/gkaa606
Yan, Y. L., Willoughby, J., Liu, D., Crump, J. G., Wilson, C., Miller, C. T., et al. (2005). A pair of Sox: distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development 132, 1069–1083. doi: 10.1242/dev.01674
Zawerton, A., Mignot, C., Sigafoos, A., Blackburn, P. R., Haseeb, A., McWalter, K., et al. (2020). Widening of the genetic and clinical spectrum of Lamb-Shaffer syndrome, a neurodevelopmental disorder due to SOX5 haploinsufficiency. Genet. Med. 22, 524–537. doi: 10.1038/s41436-019-0657-0
Keywords: neural crest, craniofacial development, SoxB1, SoxE, stem cells
Citation: Schock EN and LaBonne C (2020) Sorting Sox: Diverse Roles for Sox Transcription Factors During Neural Crest and Craniofacial Development. Front. Physiol. 11:606889. doi: 10.3389/fphys.2020.606889
Received: 15 September 2020; Accepted: 09 November 2020;
Published: 08 December 2020.
Edited by:
Jean-Pierre Saint-Jeannet, New York University, United StatesReviewed by:
Martin Chi Hang Cheung, The University of Hong Kong, Hong KongCopyright © 2020 Schock and LaBonne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carole LaBonne, Y2xhYm9ubmVAbm9ydGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.