 Keqing Nian
Keqing Nian Ian C. Harding
Ian C. Harding Ira M. Herman
Ira M. Herman Eno E. Ebong
Eno E. Ebong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 22 December 2020
Sec. Vascular Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.605398
This article is part of the Research Topic Involvement of Blood Brain Barrier Efficacy, Neurovascular Coupling and Angiogenesis in the Healthy and Diseased Brain View all 19 articles
Ischemic stroke, a major cause of mortality in the United States, often contributes to disruption of the blood-brain barrier (BBB). The BBB along with its supportive cells, collectively referred to as the “neurovascular unit,” is the brain’s multicellular microvasculature that bi-directionally regulates the transport of blood, ions, oxygen, and cells from the circulation into the brain. It is thus vital for the maintenance of central nervous system homeostasis. BBB disruption, which is associated with the altered expression of tight junction proteins and BBB transporters, is believed to exacerbate brain injury caused by ischemic stroke and limits the therapeutic potential of current clinical therapies, such as recombinant tissue plasminogen activator. Accumulating evidence suggests that endothelial mechanobiology, the conversion of mechanical forces into biochemical signals, helps regulate function of the peripheral vasculature and may similarly maintain BBB integrity. For example, the endothelial glycocalyx (GCX), a glycoprotein-proteoglycan layer extending into the lumen of bloods vessel, is abundantly expressed on endothelial cells of the BBB and has been shown to regulate BBB permeability. In this review, we will focus on our understanding of the mechanisms underlying BBB damage after ischemic stroke, highlighting current and potential future novel pharmacological strategies for BBB protection and recovery. Finally, we will address the current knowledge of endothelial mechanotransduction in BBB maintenance, specifically focusing on a potential role of the endothelial GCX.
Despite significant progression in treatment strategies, stroke remains one of the most common causes of death worldwide and accounts for 55% of all neurological disabilities (Posada-Duque et al., 2014). In the United States alone, approximately 800,000 people suffer from stroke yearly, accounting for $34 billion in medical expenses and lost productivity (Kassner and Merali, 2015; Shi et al., 2016). The majority of observed strokes are classified as ischemic strokes, which occur when a clot within a blood vessel interrupts the cerebral blood flow that supplies vital ions, oxygen, and nutrients to the brain (Obermeier et al., 2013). It is well established that one of the hallmarks of acute ischemic stroke is the disruption of the blood-brain barrier (BBB; Sifat et al., 2017), which leads to the subsequent disruption of ion homeostasis and transporter functions in the brain. The mechanisms of BBB damage in the setting of stroke include modification of tight junction (TJ) proteins, modulation of transporters expression, and inflammatory damage (Abdullahi et al., 2018). Structural injury of TJs combined with BBB transporter dysfunction can collectively lead to increased paracellular solute permeability, ultimately resulting in tissue edema and exacerbating brain injury associated with cognitive impairment (Abdullahi et al., 2018). Therefore, it is necessary to develop therapeutic strategies protecting against BBB dysfunction.
One area of BBB maintenance that has received limited attention is its regulation via endothelial mechanotransduction, the conversion of mechanical forces into endothelial cell (EC) biochemical signals (Lam et al., 2012). In the peripheral vasculature, endothelial mechanotransduction has been shown to support proper vessel function via the regulation of inflammation, vessel tone, and permeability (Chien, 2008; Jones, 2010; Glen et al., 2012). However, while several studies have demonstrated the presence and function of endothelial mechanotransducers in the BBB (Zaragoza et al., 2012; Conway et al., 2013), more research is necessary to determine the extent and mechanisms by which endothelial mechanotransduction precisely regulates BBB function.
In this review, we first highlight the current understanding of BBB structure. Then, we describe ischemic stroke and its relationship with BBB dysfunction, noting current and potential future therapeutic strategies. Finally, we will put emphasis on studies implicating endothelial mechanotransduction in BBB function and its possibility as a target for novel approaches for BBB protection.
The blood-brain barrier is an important dynamic and metabolic interface that precisely regulates ion homeostasis in the central nervous system (CNS) and protects delicate neural tissue from potentially toxic substances and pathogens (Jiang et al., 2018). For example, the BBB controls the entry and exit of essential nutrients and waste materials via the expression of various channels and transporters to regulate CNS concentrations of ions, neurotransmitters, neuroactive agents, and many other molecules (Jeong et al., 2006). Typically, the BBB primarily refers to the endothelium of the CNS vasculature. However, the development and maintenance of the BBB not only requires ECs and their associated TJs but also requires supporting pericytes, astrocytes, neurons, and extracellular matrix (ECM) that surround the BBB (Table 1). This intricate cellular grouping is referred to as the “neurovascular unit (NVU)” and each cell within the NVU helps promote the proper function of the BBB (Figure 1; Obermeier et al., 2013). Here, we will discuss the function of each cell type in relation to BBB function.

Table 1. Components and their functions of the NVU.
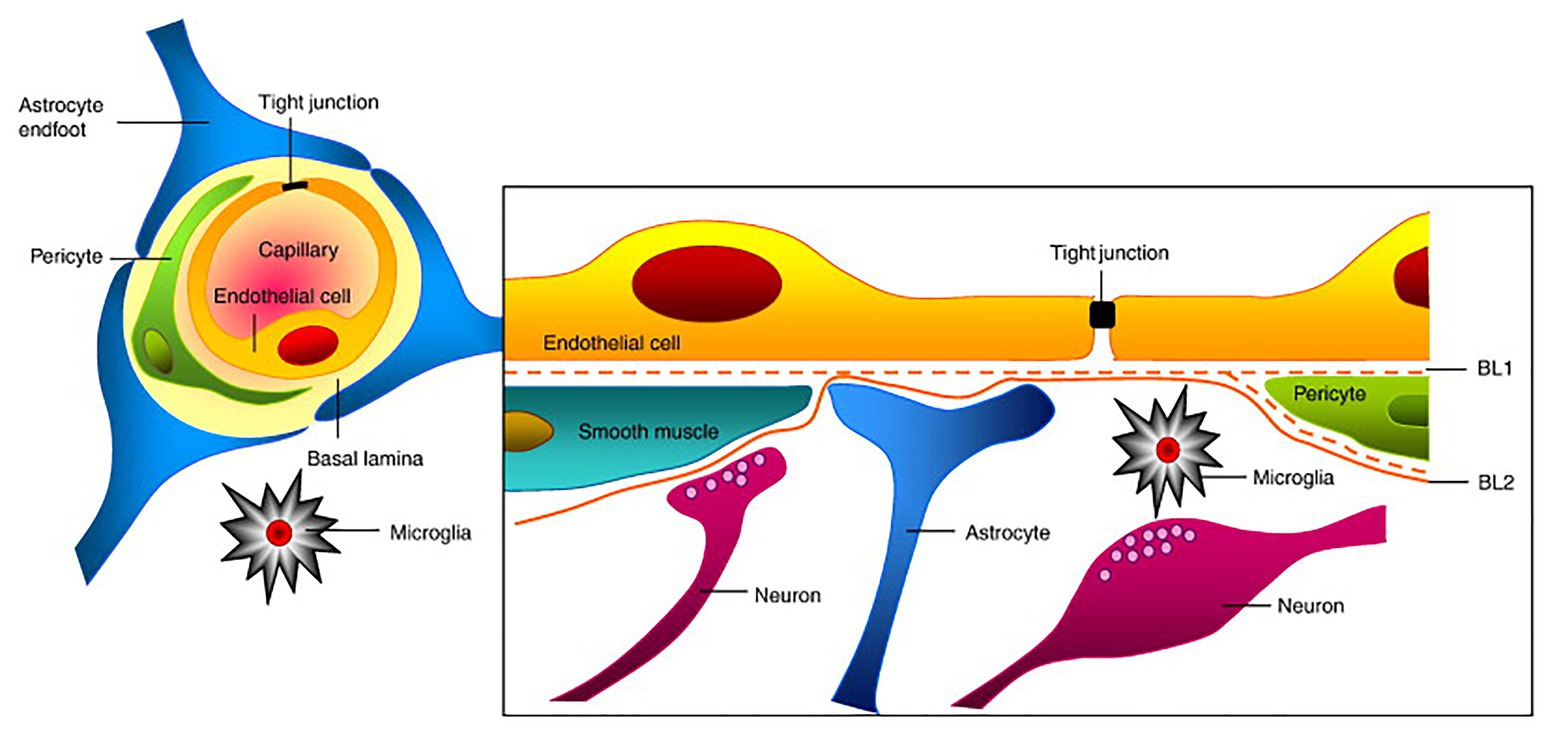
Figure 1. Structure of the neurovascular unit (NVU). Endothelial cells (ECs) form the inner most layer of the NVU and are connected to each other via tight junctions (TJs). Surrounding the ECs are pericytes in the case of capillaries or smooth muscle cells in the case of larger cerebrovasculature such as veins and arteries. Additionally, astrocytes and neurons of the NVU extend foot-processes or axons, respectively, that also help regulate NVU function. Finally, microglia, which are the brain’s resident immune cells, also contribute to NVU regulation. Collectively, ECs and these supportive cells help maintain the proper function of the blood-brain barrier (BBB). This figure is reprinted from Abbott et al. (2010)) “Structure and function of the blood-brain barrier,” Neurobiology of Disease, 37 (1): 12–25, with permission from Elsevier.
While ECs of the BBB largely mimic their counterparts in the peripheral vasculature, BBB endothelium also present several phenotypic differences that make them ideal for permeability regulation. For example, BBB ECs have increased mitochondrial content leading to an increased production of biological energy. This energy is required to strictly regulate transport processes through the expression of many receptors and ion channels. Additionally, BBB ECs are characterized by a lack of fenestrations, minimal pinocytotic activity with a small number of endocytotic vesicles, unique receptor-mediated endocytosis, and a heightened presence of TJ proteins. Collectively, these properties help restrict BBB permeability (Bicker et al., 2014; Knowland et al., 2014).
Pericytes of the NVU are located along the basement membrane of BBB ECs, encircling the vessel wall and promoting overall BBB function (Figure 1; Dalkara et al., 2011). For example, pericytes induce occludin and multidrug resistance-associated protein (MRP) expression in ECs and contribute to blood flow regulation via their contractile nature. Pericytes also exist in the peripheral vasculature, but the CNS microvasculature has the highest degree of pericyte coverage, potentially contributing to the observed vascular permeability and small vessel stability (Daneman et al., 2010). In addition to their impact on BBB EC function, pericytes exhibit phagocytic activity relevant to the clearance of toxic proteins and can also regulate the influx of immune cells into the nervous system (Rustenhoven et al., 2017). They also display an ability to self-renew and differentiate into neural and vascular lineage cells in the setting of stroke, thereby functioning as pluripotent stem cells (Nakagomi et al., 2015).
Another NVU cell type implicated in promoting BBB function are astrocytes. Astrocytes are the most common glial cell in the brain and have a variety of different morphologies and phenotypes dependent on their location within the brain and association with other cell types. Their close interaction with ECs within the BBB, particularly via astrocyte end feet, strengthens the regulation and maturation of the BBB and has been shown to contribute to cerebral blood flow control (Liu and Chopp, 2016). For example, astrocytes release many bioactive substances and regulatory factors that promote BBB function (Alvarez et al., 2013) including sonic hedgehog (Shh), which regulates TJ development and BBB permeability; nitric oxide (NO), which regulates vasodilation (Iadecola and Nedergaard, 2007); and vascular endothelial growth factor (VEGF), which is involved in angiogenesis and vasogenic edema during stroke (Davis et al., 2010). As previously mentioned, astrocytes regulate EC function mainly through their astrocytic end-feet, which extend from their cell body and connect to the basolateral surface of ECs (Figure 1). One astrocyte end-foot protein strongly implicated in BBB function is the water channel aquaporin 4 (AQP4). AQP4, which is involved in the pathogenesis of cerebral edema, facilitates water movement through the plasma membrane of several cell types in the brain, including ECs, and therefore contributes to permeability regulation (Ishida et al., 2006). Additionally, astrocytes play a substantial role in mediating neuroinflammation and thus are significant in neuroinflammatory pathologies including ischemic stroke (Colombo and Farina, 2016).
Neurons have also been reported to regulate BBB function. Although, while direct neuronal contact with the endothelium has been implicated, the incorporation of neurons into the NVU is thought to mainly occur via astrocytes. When necessary, neurons will communicate with astrocytes to influence BBB function. For instance, neurons have been suggested to tightly regulate vascular tone and cerebral blood flow via astrocytes, which is justifiable given the metabolic demands of nervous tissue; however, neuronal contributions to other BBB structures and function, such as TJ regulation, remain unknown (Koehler et al., 2006).
Although a non-cellular component, the ECM, which is composed of several major proteins including hyaluronan, lecticans, collagen IV, and fibronectin, is an integral component of the BBB and NVU. The ECM functions by connecting and functionally separating brain capillary ECs from pericytes and astrocytes, thereby allowing proper cellular orientation (Zobel et al., 2016). In addition, the ECM mediates the movement of cells within and outside of the NVU and maintains brain homeostasis due to its buffering properties. It is well established that disruption of the ECM and alterations in matrix adhesion receptors, such as integrins, contributes to increased BBB permeability during stroke (del Zoppo and Milner, 2006; Sandoval and Witt, 2008). The basement membrane, a thin layer of ECM that lines the parenchymal side of NVU, connects ECs to astrocytic end-feet and has been implicated in BBB maintenance. Specifically, damage of the basement membrane caused by increased expression of matrix metalloproteinases (MMPs) is believed to be related to alterations in BBB permeability in numerous pathologies (Wang and Shuaib, 2007; Sandoval and Witt, 2008; Lau et al., 2013).
Finally, the NVU also contains microglia, which are the primary immune cells of the CNS (Graeber and Streit, 2010). While the impact of microglia on BBB function in physiological conditions is not well known, activation of microglia via neuronal or BBB damage has been shown to contribute to and exacerbate BBB dysfunction (da Fonseca et al., 2014; Thurgur and Pinteaux, 2019). For example, microglial activation resulting in the release of pro-inflammatory cytokines, such as IL-1β and TNF-α, can lead to increased BBB permeability via disrupted TJs and increased neutrophil recruitment (Zhou et al., 2006; Chen et al., 2019). Thus, microglia are important to consider when discussing CNS pathologies associated with neuroinflammation.
Tight junctions are large, multiprotein junctional cell-cell complexes composed of three major membrane proteins, claudins, occludin, and junction adhesion molecules (JAMs), along with the accessory proteins zonula occludins (ZO; Figure 2). TJs are particularly significant in regulating permeability by inducing EC polarization and restricting the paracellular movement of ions, such as Na+ and Cl−, as well as macromolecules across the BBB. TJ efficacy in the BBB, specifically compared to the peripheral vasculature, is evidenced by a considerably high value of BBB transendothelial electrical resistance (TEER) and low paracellular permeability (Ronaldson and Davis, 2015).
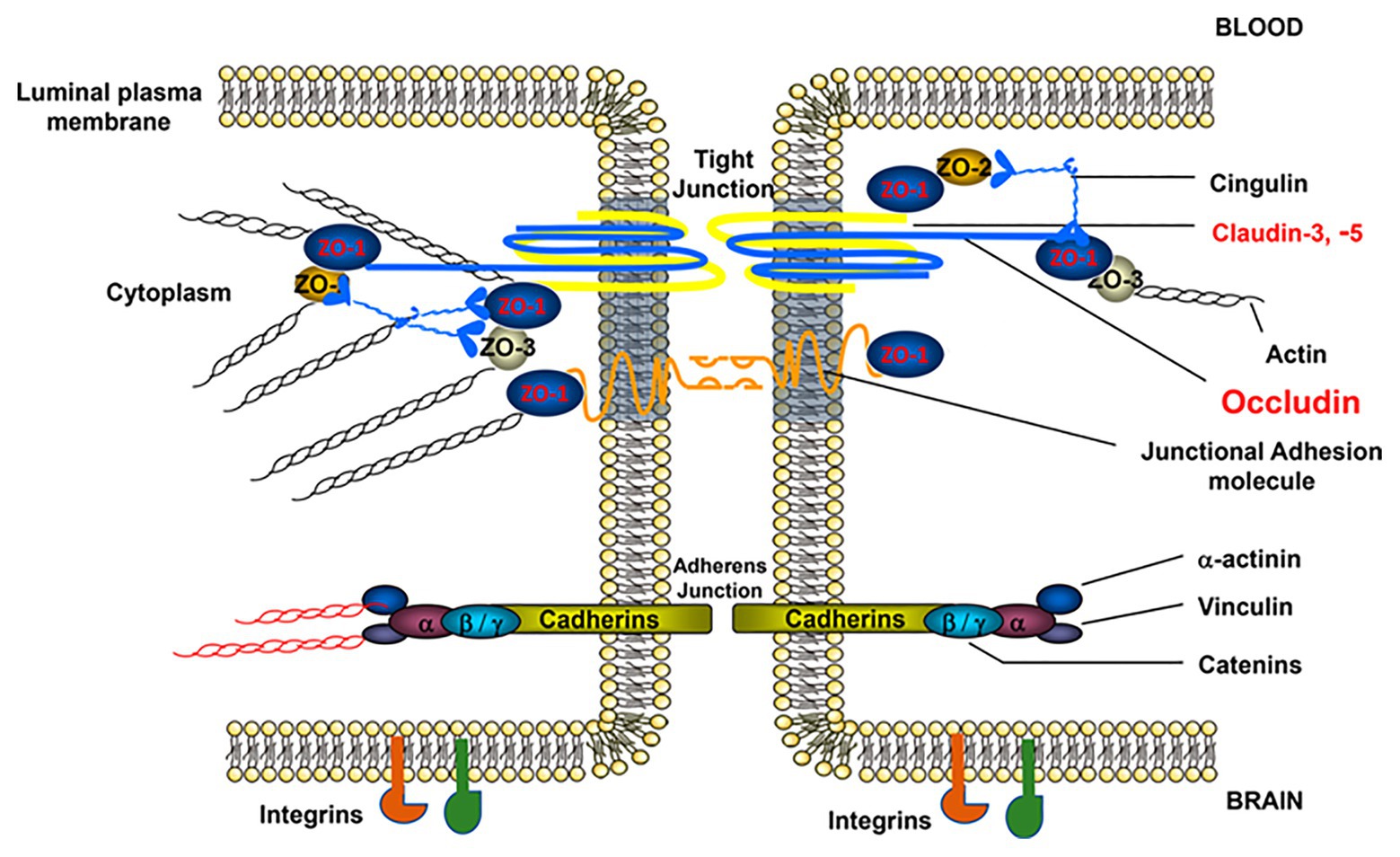
Figure 2. Structure of brain EC-cell junctions. EC-to-EC junctions consist of permeability regulating protein complexes in both tight and adherens junctions (AJs). TJs, which are more strongly associated with permeability regulation, consist of the occludin, junctional adhesion molecule, and claudin families of proteins linked to the cytoskeleton via zona occludens (ZO) proteins. AJs, which also play a role in permeability regulation, are composed of vascular endothelial (VE)-cadherin dimers similarly linked to the cytoskeleton via an array of linker proteins, such as catenin. In addition to these major junctional components, EC junctions also contain numerous supporting proteins such as cingulin, vinculin, and integrins, among others. This figure is reprinted from a previous publication (Abdullahi et al., 2018), with permission from the previous publisher, the American Physiological Society.
The tight junction protein family claudins are 20–24-kDa proteins with four transmembrane domains (Figure 2). To date, more than 24 members have been identified (Ballabh et al., 2004). However, in brain ECs, the major isoforms are claudin-1, -3, -5 and -12, all of which play important roles in barrier function (Yang and Rosenberg, 2011). Claudin-5, in particular, significantly limits paracellular diffusion of small substances in the BBB (Greene et al., 2019). Specifically, increased claudin-5 can increase TEER and decrease BBB permeability. To regulate TJ function, the carboxy terminal (intracellular tail) of claudins bind to cytoplasmic proteins including ZO-1, ZO-2, and ZO-3, forming TJ scaffolds between extracellular neighboring proteins and the cell cytoskeleton, thus providing an anchorage to maintain cell-cell contacts (Figure 2; Honda et al., 2006).
A second major transmembrane TJ protein is occludin, a 65-kDa protein involved in TJ stabilization to regulate BBB functional integrity (Figure 2; Nag et al., 2007). Similar to other TJ proteins, occludin is highly expressed in cerebral endothelium but more sparsely distributed in non-neural endothelia (Nag et al., 2007) and is associated with increased electrical resistance in TJ-containing tissues and decreased paracellular diffusion (Nag et al., 2007). As with claudins, ZO proteins also localize occludin to the cellular membrane, creating an intercellular bridge (Figure 2). Interestingly, the function of occludins is suggested to be a function of dynamic phosphorylation, which can affect the assembly/disassembly of TJs (Dorfel and Huber, 2012). Combined with claudins and junctional adhesion molecules, occludins within the BBB strictly regulate BBB permeability to promote CNS homeostasis.
Adherens junctions (AJs), located toward the basolateral side of EC TJs, are adhesive cell-cell interactions composed of cadherins along with their associated proteins, which are important for both the localization and stabilization of AJs (Figure 2). The transmembrane cadherin protein and, more specifically, cadherin-mediated signaling plays a key role in numerous EC processes, including endothelial layer integrity and the organization of microvessels during development (Lampugnani and Dejana, 2007). The regulation of these cadherin functions also relies on other AJ components, namely α-actinin and vinculin (Figure 2), which similarly have been implicated in maintaining BBB function (Abu Taha and Schnittler, 2014). Out of the numerous cadherin proteins in humans, VE-cadherin is specifically expressed by ECs and is required for endothelial survival and stabilization (Tietz and Engelhardt, 2015) and is also associated with paracellular permeability (Beard et al., 2011). Interestingly, despite the correlation between VE-cadherin expression and permeability regulation, one particular study suggested that partial loss of VE-cadherin may lead to long-term stroke protection (Gertz et al., 2016).
Endothelial cells of the BBB express many transporters that, in addition to junctional proteins, play an important role in regulating the transport of endogenous and exogenous materials, such as glucose, which cannot permeate cell-cell junctions. Major transporters include nutrient transporters, ion transporters, and the ATP-binding cassette (ABC) transporters, which will be discussed here (Table 2; Jiang et al., 2018).
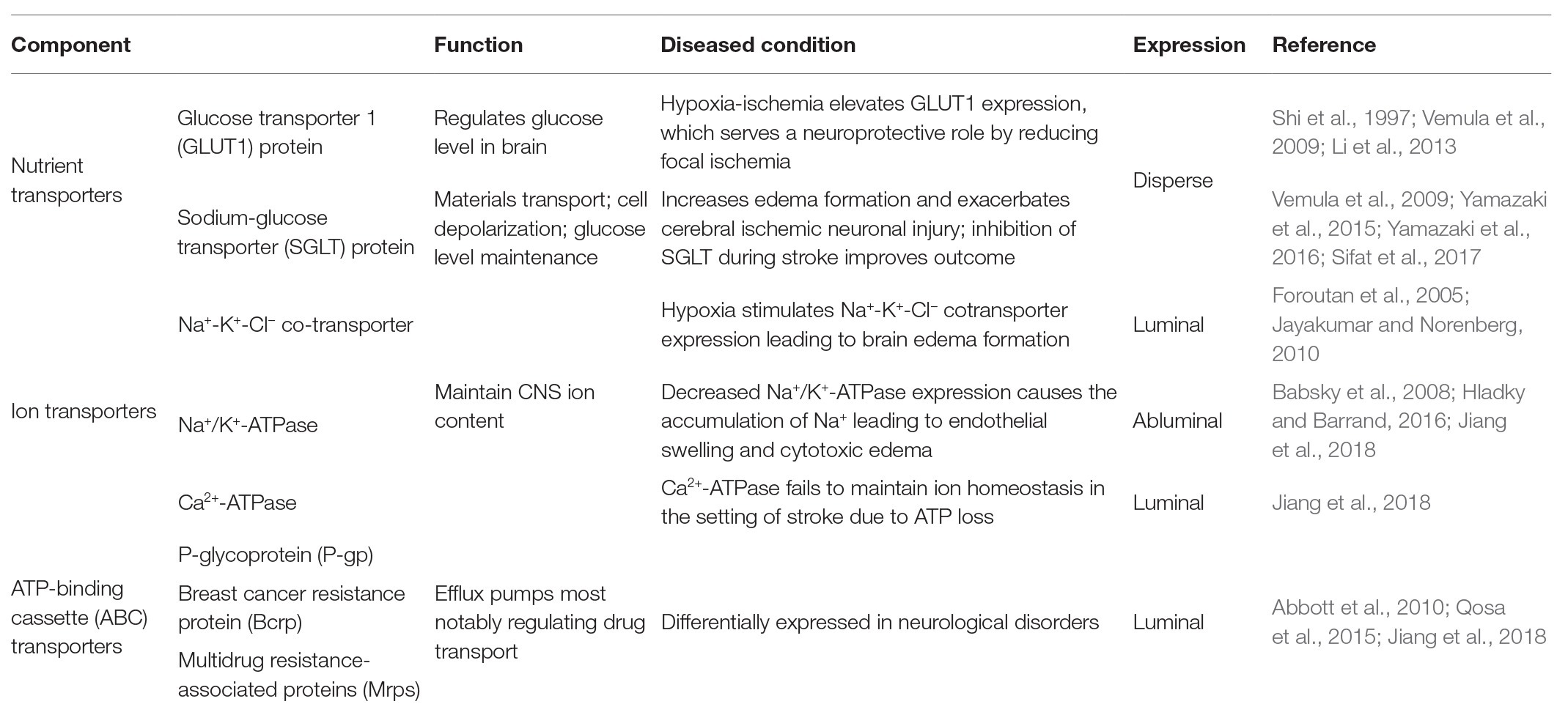
Table 2. EC transporters and their functions.
D-glucose is the main energy source for brain metabolism and a continuous supply is therefore required to maintain normal brain function. The main glucose transporter in the BBB is the glucose transporter 1 (GLUT1) protein (Table 2), which is expressed and localized to brain ECs. Many pathophysiological conditions, including chronic hypoxia and ischemia, are capable of altering glucose transporter expression, including GLUT1. However, whether these changes in expression are beneficial or detrimental to BBB and neuronal function varies (Vemula et al., 2009). For example, one study found that hypoxia-ischemia can elevate GLUT1 expression, which in previous studies has been demonstrated to serve a neuroprotective role by reducing focal ischemic lesion size (Shi et al., 1997; Li et al., 2013). Another type of glucose transporter expressed in the BBB is the sodium-glucose transporter (SGLT) protein (Table 2), which contributes to materials transport and cell depolarization and has been shown to help maintain glucose levels during stroke (Yamazaki et al., 2015). However, contrary to the observed beneficial effects of GLUT1, several studies have shown that SGLT is relevant to increased edema formation and can actually exacerbate cerebral ischemic neuronal injury due to the influx of sodium ions (Yamazaki et al., 2016; Sifat et al., 2017). Other studies have corroborated these findings, suggesting that inhibition of SGLT during stroke may improve stroke outcome (Vemula et al., 2009). Thus, regulation of glucose transporters within the BBB can have complex consequences on CNS homeostasis.
Ion transporters, which are predominantly expressed at BBB ECs’ abluminal surface, have important effects on vectorial transport across the cell membrane and, subsequently, on CNS ion homeostasis and fluid movement. The major ions in the nervous system are Na+, K+, Cl−, and Ca2+, all of which are critical to the regulation of neuronal activity (Hladky and Barrand, 2016). The transport of Na+, Cl−, and other ions combined with the associated water influx is responsible for approximately 30% of the interstitial fluid in a healthy brain. One common ion transporter is the Na+-K+-Cl− co-transporter (NKCC; Table 2), which is important in maintaining CNS homeostasis (Jayakumar and Norenberg, 2010). It has been reported that hypoxia can stimulate the expression of the Na+-K+-Cl− cotransporter, leading to ischemia-induced brain edema formation (Foroutan et al., 2005; Jayakumar and Norenberg, 2010). As such, in stroke patients, the entry of extracellular Na+ caused by the alteration of these ion transporters, such as decreased Na+/K+-ATPase expression and function, has a profound impact on endothelial swelling and cytotoxic edema (Babsky et al., 2008; Hladky and Barrand, 2016). Other ion transporters are also implicated in stroke and BBB dysfunction (Hom et al., 2007; O’Donnell, 2014; Jiang et al., 2018). For example, in an ischemic episode, the Na+/K+-ATPase and Ca2+-ATPase ion transporters (Table 2) fail to conserve ionic balance due to ATP depletion caused by the blockage of oxidative phosphorylation (Jiang et al., 2018). Collectively, these studies highlight the importance of ion transporters in regulating BBB function and in pathophysiological events such as stroke.
The ABC transporters (Table 2), which are located at the EC luminal membrane, are a protein superfamily that transports neurotoxins against concentration gradients, thereby requiring biological energy. This protein superfamily contains 48 members and is divided into seven sub-families (Jiang et al., 2018). Major ABC transporters relevant to the BBB include P-glycoprotein (P-gp; Table 2), which transports a variety of structurally diverse compounds; breast cancer resistance protein (Bcrp; Table 2), which is able to recognize variable organic anions; and MRPs (Table 2), which are associated with cellular efflux of anionic drugs as well as their metabolites (Abbott et al., 2010). While ABC transporters protect the CNS by restricting toxin permeability through the BBB, they have also been found to hinder therapeutic strategies targeting the CNS as they similarly limit the transport of drugs through the BBB. It has also been shown that ABC transporter expression varies in neurological disorders such as Alzheimer’s, Parkinson’s, and stroke (Qosa et al., 2015). Thus, while the role of ABC transporters in both proper BBB function and neurological disorders is complex, it is clear that alterations in their expression can have significant consequences on BBB and neural tissue function.
Stroke is the second leading cause of death in the world behind cardiovascular diseases and annually contributes to 5.5 million deaths globally (Alishahi et al., 2019). Stroke incidence has also increased more than 100% in developing countries, suggesting that stroke prevalence will likely increase over the coming years (Tsai et al., 2016). At present, there are two major types of stroke: ischemic strokes and hemorrhagic strokes. In this review, we focus on ischemic strokes.
Ischemic strokes amount to approximately 87% of all strokes and are classified as strokes that occur due to vessel obstruction via a blood clot (Sevick et al., 2017). These clots can either form directly at the site of insult or result from the rupture of a larger clot at an upstream region of the vasculature. When ischemic strokes occur, blood flow to the brain is interrupted leading to decreased oxygen delivery and reduced nutritional supply (e.g., glucose) to the affected part of the brain (Chauhan and Debette, 2016). The affected areas of the brain resulting from ischemic stroke are referred to as the ischemic, or infarct, core, and penumbra, which collectively form the term “brain ischemia” (Figure 3; Bandera et al., 2006). The ischemic or infarct core is defined as the area of irreversible tissue injury, typically characterized by blood flow with perfusion rates below 10 ml/100 g of tissue per minute (Latchaw et al., 2003; Bandera et al., 2006). Outside of the infarct core is the penumbra, which also contains the benign oligemia (Figure 3), and is defined as functionally impaired tissue that is still viable. Typically, tissue within the penumbra is perfused at rates below 25 ml/100 g tissue per minute (Bandera et al., 2006; Fisher and Bastan, 2012). While tissue within the benign oligemia will recover spontaneously, tissue outside of the benign oligemia but within the penumbra requires clinical intervention. Therefore, therapeutics aimed at preventing CNS damage as result of ischemic stroke target these areas of the brain, whereas the infarct core is beyond treatment due to the rapid development of necrosis (Bandera et al., 2006). The first contributors to the rate of infarct progression are the degree of collateral arterial circulation, duration of insult, and the functional and metabolic cellular state (Bandera et al., 2006). Collectively, these factors determine the likelihood of success of common ischemic stroke therapeutics.
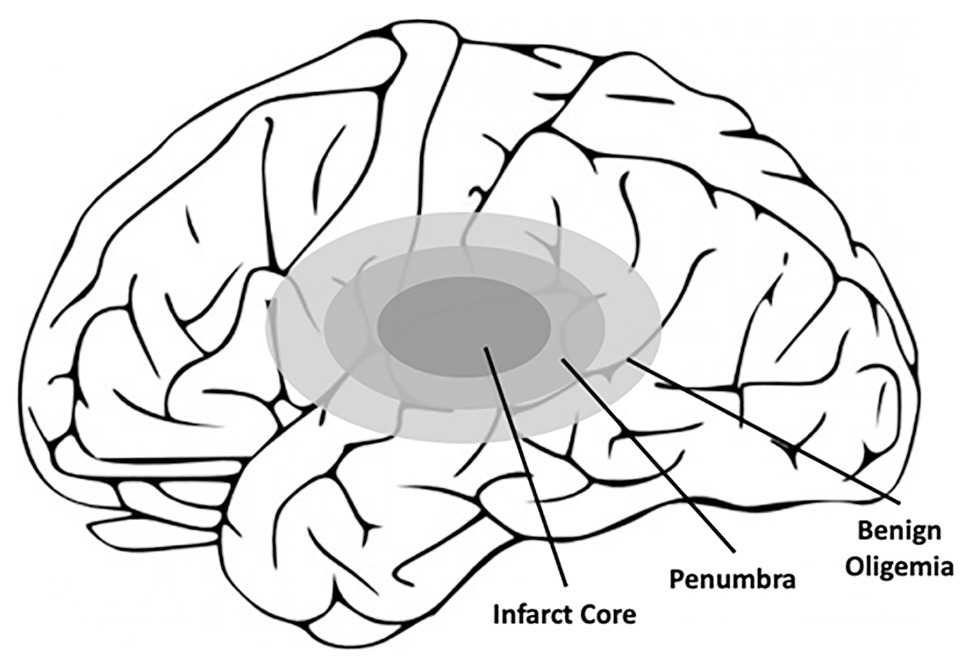
Figure 3. Schematic of the affected areas of the brain after ischemic stroke. Closest to the location of the blood clot, and thus the loss of blood flow, is the infarct core, which contains irreversibly damaged tissue. Outside of the infarct core lies the penumbra, which also contains the benign oligemia, and defines the area of reversible tissue damage. While the benign oligemia specifies tissue that will recover function on its own, tissue within the penumbra but outside of the benign oligemia requires therapeutic intervention for full recovery.
In addition to the initial insults of ischemic stroke, following ischemic stroke metabolic disturbances and energy imbalance can cause secondary injury such as inflammation and gliosis leading to further neuronal and vascular cell death. Inflammation, in particular, is believed to play a critical role in the pathogenesis of ischemic stroke. For example, the initial ischemic insult leads to the activation of microglia (Ma et al., 2017) and astrocytes (Choudhury and Ding, 2016) as well as the recruitment and infiltration of inflammatory cells such as leukocytes (Gronberg et al., 2013). Interestingly, while some studies have indicated beneficial impacts of several of these inflammatory mechanisms on brain recovery, overall the inflammatory response is believed to have a detrimental effect, particularly under prolonged inflammation (Jin et al., 2010). This is supported by the effective use of anti-inflammatory therapeutics to limit neurological deficits in animal models of ischemic stroke (Lakhan et al., 2009), although these therapeutics have not translated into the clinic. These and other therapeutic options for ischemic stroke will be further discussed later in the review.
Although not well studied, endothelial mechanotransduction within the BBB may also play a major role in ischemic stroke pathology and potentially its initial occurrence. In the peripheral vasculature, endothelial exposure to physiological shear stress promotes vessel function through the regulation of vascular permeability (Himburg et al., 2004; Warboys et al., 2010), vasoconstriction/vasodilation (Uematsu et al., 1995; Muller et al., 1997), and inflammatory phenotype (Chen et al., 2003). Recent studies have similarly demonstrated that brain-derived ECs similarly benefit from shear stress exposure via observed increases in barrier integrity (Colgan et al., 2007; Siddharthan et al., 2007; Cucullo et al., 2011; Walsh et al., 2011). Thus, stroke-induced hypoperfusion may lead to inadequate mechanotransduction that could further compromise BBB integrity. Additionally, mechanotransducers, such as the endothelial glycocalyx (GCX), have been shown to be degraded in response to ischemia-reperfusion (Rubio-Gayosso et al., 2006), which may exacerbate issues of proper mechanotransduction. Impaired mechanotransduction may even contribute to the occurrence of ischemic stroke, as the development of blood vessel plaques, which can rupture and lead to ischemic stroke, are known to occur in areas of disturbed blood flow characterized by altered mechanotransduction and degraded GCX layers (van den Berg et al., 2009; Cancel et al., 2016; Harding et al., 2018; Mitra et al., 2018). It is therefore vital to understand the impact of endothelial mechanotransduction in both stroke pathology and occurrence, which will be addressed later in the review.
A major hallmark of stroke is its associated BBB disruption, which is initiated due to ischemia but continual deteriorates with sustained hypoperfusion. This deterioration is largely attributed to a lack of nutrients (e.g., oxygen and glucose), but altered mechanotransduction may also play a role. This is evidenced by the fact that even after the restoration of blood flow, albeit below baseline levels, BBB permeability does not revert but instead persists. Furthermore, degradation of the mechanotransductive GCX layer has been shown to be degraded in ischemia/reperfusion (Chappell et al., 2009; van Golen et al., 2012), implicating its potential role in BBB maintenance. The exact time course of increased BBB permeability in ischemic stroke is widely debated. Initial studies in animal models identified a biphasic nature of BBB permeability, in which initial increases in permeability are followed by a reduction in permeability to baseline levels but eventual return to increased permeability (Kuroiwa et al., 1985; Huang et al., 1999). While more recent studies have supported this theory (Pillai et al., 2009), other studies, including in human subjects, suggest that BBB permeability remains elevated post-stroke potentially for several weeks (Strbian et al., 2008; Merali et al., 2017). Regardless, initial breakdown of the BBB is believed to occur at least partially through the overexpression of matrix metalloproteinases (MMPs), which can have numerous detrimental effects including the degradation of the endothelial GCX, a known mechanotransducer (Ramnath et al., 2014; Yang and Rosenberg, 2015; Zhang et al., 2018). GCX degradation has been shown to disrupt junctional protein expression and function (Thi et al., 2004; Mensah et al., 2017). Following this initial breakdown, a sustained increase in permeability likely occurs due to a neuroinflammatory response, which, combined with other consequences such as brain edema, contributes to longer-term permanent loss of neurological function (Abdullahi et al., 2018). To better understand the observed increase in BBB permeability following stroke, we will discuss the mechanisms of this process, focusing on the role of junctional and transporter proteins.
The overall increase in BBB permeability in stroke is largely attributed to differences in junctional protein expression and function. For example, stroke is associated with decreased expression of the TJ proteins claudin-5, occludin, and ZO-1, among others (Jiao et al., 2011). This is evidenced by the increased uptake of 14C-sucrose, a membrane impermeant marker, in the brain parenchyma after stroke (Hau et al., 2004; McCaffrey et al., 2009; Abdullahi et al., 2018). It has also been suggested that occludin redistribution, in addition to changes in expression, induced by VEGF may occur and is associated with increased paracellular permeability in ECs (Murakami et al., 2009). Caveolin-1, a coat protein of pinocytotic caveolae vesicles implicated in molecular trafficking, mediates the redistribution and localization of junctional proteins, such as ZO-1, claudin-5, and occludin, as previously mentioned (Choi et al., 2016; Abdullahi et al., 2018; Zhang et al., 2018). Additionally, while TJs are more widely known for their role in maintaining BBB permeability, the AJ protein VE-cadherin has also been shown to have decreased expression after stroke, contributing to the overall increase in permeability (Abdullahi et al., 2018). The observed changes in VE-cadherin expression following stroke may be regulated by sphingosine kinase (SphK2), as SphK2-null mice display reduced levels of VE-cadherin and other junctional proteins (Wacker et al., 2012).
Initial disruption in junctional protein expression is believed to be due to MMPs and, on a lesser basis, reactive oxygen species (ROS). This initial, short-term increase in permeability in some cases reverts to baseline levels and is thus often classified as “reversible” (Figure 4). However, as the molecular response shifts in response to stroke, longer-term BBB opening, referred to as “irreversible,” can persist for several days or longer (Figure 4). What determines the length of increased BBB permeability is not fully known, but molecules such as MMPs and cytokines are implicated in the process. For example, MMP-2 activation is believed to be responsible for initial disruption of TJ proteins (Figure 4). However, in later phases of BBB disruption resulting from stroke, MMP-9 expression is induced and results in more intense and irreversible damage (Figure 4; Yang and Rosenberg, 2015). The role of MMPs in stroke-induced BBB dysfunction was evidenced by Shuai et al., who found that stroke results in decreased ZO-1 expression and translocation of ZO-1 from the cell junctions to the cytoplasm, which occurs due to increased MMP-2/9 and caveolin-1 expression (Zhang et al., 2018). Another study suggested that reduction in brain permeability is highly associated with decreased expression of MMP-2/9 (Zeng et al., 2020). In addition to increased MMP expression and activity, increased concentrations of ROS such as superoxide, which is substantially produced by the NADPH oxidase (Nox) proteins (Revuelta et al., 2019), has similarly been shown to reduce the expression of junctional proteins such as claudin-5 and occludin. For example, Zhanying et al. found that upregulation of TJ protein expression owing to attenuated generation of ROS contributed to improvement of BBB function (Ye et al., 2019). ROS has also been shown to cause caspase-3-mediated damage of TJs and microvascular endothelial hyperpermeability in vitro (Alluri et al., 2014). In addition to the short- and long-term impacts of MMPs and ROS on BBB integrity, other inflammatory regulators, such as the cytokines TNF-α (Yang et al., 1999; Pan and Kastin, 2007) and IL-1β (Anthony et al., 1997; Ferrari et al., 2004), as well as neutrophils (Rosell et al., 2008; Joice et al., 2009; Jickling et al., 2015) have been implicated in BBB maintenance following stroke. Due to the proven detrimental effects of these factors, therapeutics targeting these molecules have drawn significant interest to maintain BBB integrity.
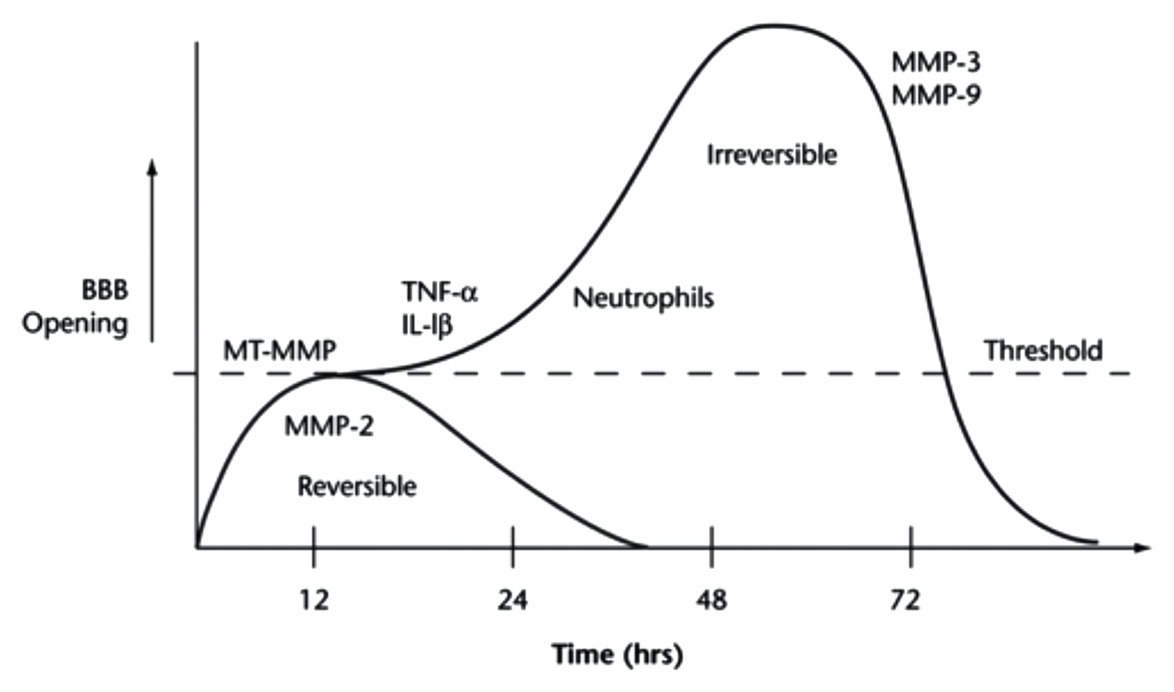
Figure 4. The time course of BBB opening and associated expression of pro-inflammatory mediators of BBB damage. Initial opening of the BBB is likely caused, at least in part, by increases in MMPs, specifically MMP-2. This initial phase occurs over ~24 h and is referred to as “reversible.” Over time, continuous production of pro-inflammatory molecules, such as TNF-alpha, IL-1beta, and MMPs 3 and 9, can lead to prolonged opening (72+ h) of the BBB, potentially contributing to long-term tissue damage. This phase is thus referred to as “irreversible.” Reprinted from Yang and Rosenberg, “Matrix metalloproteinases as therapeutic targets for stroke,” Brain Research, 2015, 1,623:30–38, with permission from Elsevier.
Changes in the expression and function of endogenous BBB transporters also impact stroke-induced BBB permeability and the resulting pathophysiological processes such as edema. Major BBB transporters associated with stroke include the glucose transporter proteins and ion transporters. For instance, the GLUT1 and GLUT3 glucose transporters are significantly elevated in the short and long term time periods following severe hypoxic-ischemic insults (Vannucci et al., 1998). Increased expression of the SGLT glucose transporter has also been observed and inhibition of this and the GLUT1 transporter can reduce edema formation despite the beneficial nutrient transport roles of these proteins (Vemula et al., 2009). Also implicated in permeability regulation in stroke and subsequent edema formation are ion transporters, particularly the Na+ transporters. For instance, increased expression and phosphorylation of the Na+-K+-Cl− transporter is associated with severe ischemic conditions (Foroutan et al., 2005). Furthermore, the inhibition of various ion transporters, including Na+-K+-Cl− (Huang et al., 2019), NHE1 (Park et al., 2010), and KCa3.1 (Chen et al., 2015), have been shown to reduce edema or infarct size, highlighting their importance in stroke. In addition to alternations in specific transporter proteins, elevation of pinocytosis within the endothelium occurs post ischemia-reperfusion (Shinozuka et al., 2013), further contributing to increased permeability (Xia et al., 2012). A study by Knowland et al. (2014) even suggested that increased transcellular transport via caveolae and other pinocytotic vesicles occurs before TJ disruption.
In addition to the common permeability pathways controlled by junctional proteins and EC transporters, other molecules, such as integrins and EC adhesion molecules, also play a role in regulating BBB permeability after stroke, albeit to a lesser extent. Integrins, transmembrane proteins that interact with the ECM to stabilize ECs to their environment, have been shown to contribute to BBB permeability regulation. For example, Osada et al. (2011) demonstrated that antibody neutralization of b1-integrin leads to a significant decrease in BBB barrier integrity, specifically through a reduction in transepithelial electrical resistance and increased permeability. Studies suggest that integrins are able to regulate BBB integrity through both the downstream regulation of TJ proteins, specifically claudin-5 (Osada et al., 2011), and by promoting EC-astrocyte interactions, which are crucial for proper BBB function (Almutairi et al., 2016).
Endothelial cell adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1), also play a role in regulating transport in the BBB in particular through their role in leukocyte transendothelial migration. After focal brain ischemia, EC adhesion molecules, mainly ICAM-1, are upregulated potentially via the local and systemic release of pro-inflammatory factors (Lindsberg et al., 1996). Combined with increased leukocyte production in response to stroke (Ross et al., 2007), this promotes increased transendothelial migration of leukocytes, such as neutrophils. While short term increases in neutrophils and other white blood cells may help resolve vascular and CNS damage, prolonged neutrophil accumulation, and activity actually worsens long term outcomes (Buck et al., 2008; Kumar et al., 2013).
Current therapeutic strategies for ischemic stroke fall into four categories: clinical care, neuroprotection, neurorestoration strategies, and rehabilitation therapy (Patel and McMullen, 2017). To date, the only major treatment strategies approved by the United States Food and Drug Administration (FDA) and used in the clinic are thrombolysis via tissue plasminogen activator (r-TPA) and endovascular treatment. However, while these treatments have been successful in improving clinical outcome, they are not able to resolve prolonged neuronal dysfunction and degeneration of still viable tissue. Therefore, many studies and experimental trials have been focused on developing neuroprotective therapies to retain function of viable tissue after an ischemic episode. Yet, while many animal studies have demonstrated promising results, they have met limited success in clinical trials. In this review, we will first address stroke management in the clinic and then elaborate on both previous and promising neuroprotective treatment options currently being assessed (Table 3). Furthermore, we will briefly discuss neurorestorative therapies.

Table 3. Current and promising therapeutics for treatment of ischemic stroke.
Two major thrombolytic therapeutic strategies, either via the use of pharmacological agents or via mechanical thrombectomy (Jovin et al., 2015; Saver et al., 2016), are currently used for recanalization and reperfusion for stroke patients (Table 3). The treatment of choice for each patient depends on the time to treatment and the etiology of the injury. At present, the most common pharmacological treatment is thrombolysis via r-tPA, known as alteplase (Table 3), which is approved by the FDA (Sifat et al., 2017). When administered intravenously, alteplase provides local fibrinolytic effects by first binding to fibrin clots and then hydrolyzing peptide bonds in surrounding plasminogen, thus converting it to plasmin and subsequently disintegrating the clot (Bansal et al., 2013; Clarke and Ayala, 2019). While studies have demonstrated that alteplase does reduce tissue infarct volume, it does not alter other associated pathophysiologies such as ischemic brain edema (Broocks et al., 2020). While r-tPA has been demonstrated as an efficient and cost-effective thrombolytic agent, its short therapeutic time window of up to 4.5 h, association with severe deleterious events, especially hemorrhagic transformation and brain injury, and candidates’ eligibility for treatment (e.g., delayed identification in the emergency department; Sung et al., 2013) greatly limit the safety and application of r-tPA (Mendez et al., 2018). Regarding deleterious events associated with tPA, it has been proposed that tPA exacerbates neuron death in the hippocampus due to inter-neuronal laminin degradation and pro-survival cell-matrix signaling disruption (Lo et al., 2004). Additionally, evidence suggests that a compromised BBB may limit the efficacy of r-tPA because it may exacerbate hemorrhagic transformation and promote brain edema and neuroinflammation. Due to these limitations, alternatives to alteplase, such as tenecteplase, have been investigated but have not shown any significant improvements in clinical trials (Logallo et al., 2017). Still, it is clear that areas for improvement exist.
Aside from r-TPA, other minor pharmacological treatment options are available, most notably aspirin. Aspirin treatment, which is only used for secondary prophylaxis, must be delivered within 24–48 h after stroke onset but has been reported to lower the potential risk of recurrent stroke and vascular events than non-users (Kong et al., 2019). However, it is suggested to be useless for the treatment of an ongoing acute ischemic stroke and, thus, alternative pharmacological therapeutic options remain the primary mode of treatment (Powers et al., 2018).
In addition to these pharmacological treatment options, endovascular mechanical thrombectomy treatment (Table 3), which utilizes micronavigation of catheters into the cerebral vasculature to mechanically disrupt clots (Akins et al., 2014), provides a more direct way to the occlusive lesion and has a longer therapeutic time window-up to 8 h (Jovin et al., 2015). Furthermore, it is reported that endovascular treatment combined with intravenous pharmacological thrombolysis may provide increased efficacy to many patients with ischemic stroke (Lowhagen Henden et al., 2017). Endovascular mechanical thrombectomy has also shown great benefits in large vessel occlusion via a combination of contact aspiration and stent retrievers (Mokin et al., 2014; Lapergue et al., 2020). However, despite the potential benefits, endovascular mechanical thrombectomy treatment is limited by high costs, its need for trained personnel, and its inability to restore tissue function within the penumbra.
Neuroprotective treatments are defined as therapeutics administered during the acute ischemic phase with the overall goal of protecting from further neuronal tissue injury. These therapeutics typically target the penumbra as this tissue is still viable but may require clinical intervention for complete functional recovery. Neuroprotective treatments as defined here are in contrast with neurorestoration techniques, which aim to restore tissue functionality through the stimulation of neurogenesis and neuroplasticity. There are two types of neuroprotection: pharmacologic neuroprotection and non-pharmacologic neuroprotection. Non-pharmacologic neuroprotective methods, such as transcranial laser therapy (Patel and McMullen, 2017), have shown some promise but will not be discussed in this review. For pharmacologic neuroprotection, our understanding of the mechanisms of tissue damage following ischemic stroke has advanced, providing scientists with viable targets for neuroprotective therapeutics. Many developed and tested therapeutics have targeted the modulation of inflammation, oxidative stress, excitotoxicity, or apoptosis, all of which have been implicated in post-stroke tissue damage (Goenka et al., 2019; Wang et al., 2019). However, despite the relatively high levels of therapeutic benefits identified by these treatments in animal models, limited success has been observed in the clinic despite the hundreds of clinical trials run. The low degree of therapeutic translation from animal models to human subjects is likely due to a combination of reasons relating to differences between animal models and human subjects (e.g., age of treatment and presence of comorbidities), the employed experimental methods, the quality of experimental or clinical trial design, the outcome measures utilized, and others. Thus, future research efforts attempting to identify neuroprotective therapeutics not only need to identify the novel therapeutics themselves but also enforce proper pre-clinical and clinical methods to properly evaluate the efficacies of these treatment options. Here, we will discuss several drugs that may prove to be promising therapeutics or may provide insight into the development of other future therapeutics.
While the only FDA-approved pharmacological therapeutic for ischemic stroke is tPA, a variety of other neuroprotective molecules have been approved for clinical use for other indications or in other countries. Still, the therapeutic efficacy of these treatments is controversial. For example, cerebrolysin (Table 3) is a mixture of brain-derived neuropeptide that exhibits anti-excitotoxicity, antioxidant, and anti-apoptotic activity that is approved for the treatment of stroke in more than 40 countries (Goenka et al., 2019). Several animal studies have demonstrated that cerebrolysin can improve outcome from stroke, for example, by reducing infarct size (Ren et al., 2007; Zhang et al., 2010). However, clinical trials for cerebrolysin have returned mixed results. Specifically, while the drug has demonstrated safety in Phase 1 and Phase 2 clinical trials, several later stage trials failed to demonstrate any improvement in neurological outcome. Although, a combination of cerebrolysin and physical rehabilitation therapy was shown to improve motor recovery in individuals with severe motor impairment.
Magnesium sulfate (Table 3), which modulates excitotoxicity, has also demonstrated success in animal studies but has been met with both success and failure in a clinical setting. An initial pilot clinical trial from 2001 found a significant correlation between magnesium sulfate and improved neurological outcome (Lampl et al., 2001). However, another larger future study identified no relationship between the drug and neurological outcomes (Muir et al., 2004). Because the administration time in this study was in some cases significantly delayed, an additional clinical trial utilizing magnesium sulfate treatment in the field within 2 h of stroke onset was therefore performed and demonstrated a significant improvement in neurological outcomes (Saver et al., 2015). Other promising drugs, such as minocycline (Table 3), an antibiotic drug approved in the United States for numerous non-stroke indications, and edaravone (Table 3), an antioxidant therapy, have similarly demonstrated promise in animal trials but mixed results in clinical trials (Fagan et al., 2010; Sharma et al., 2011; Kimura et al., 2012; Kikuchi et al., 2013; Karsy et al., 2017). These studies, among many others, demonstrate both the difficulty of identifying promising stroke therapeutics and the importance of proper pre-clinical and clinical study design. Currently, new therapeutics are being tested in clinical trials around the world that will hopefully prove to be more successful than previous waves.
One such molecule is Dl-3-n-butylphthalide (NBP; Table 3), a synthetic compound that possesses anti-inflammatory, anti-oxidative, and anti-apoptotic properties and has been approved for treatment of ischemic strokes in China (Wang et al., 2019). Animal models evaluating NBP as a stroke therapeutic have found positive results. For example, Wang et al. (2019) identified increased neurological functional recovery after NBP treatment for ischemic stroke in mice, which was associated with increased white matter integrity and the upregulation of the TJ protein occludin. Similarly, Jiamin et al. demonstrated that NBP greatly reduces BBB permeability via the upregulation of claudin-5 and ZO-1 and downregulation of caveolin-1 in ischemic stroke mice (Li et al., 2019). These and other results have supported the use of NBP as a therapeutic, which is currently being evaluated in the clinic in a Phase 2 trial. Another promising therapeutic option, liraglutide (Table 3), a glucagon-like peptide-1 receptor agonist, is supported by a significant body of work which has identified both its beneficial effects on neurological function in animal models of stroke and its mechanism. For instance, numerous studies have demonstrated improved behavioral scores/neurological deficits and reduced infarct size in animal models of stroke treated with liraglutide (Sato et al., 2013; Zhu et al., 2016). These improved outcomes are believed to be due to reductions in oxidative stress, improved mitochondrial function, and reduced cell apoptosis via neuronal sirtuin 1 and activation of the PI3K/AKT and MAPK pathways (Sato et al., 2013; Zhu et al., 2016; He et al., 2020b). A phase 3 clinical trial evaluating the efficacy of liraglutide for treatment of ischemic stroke is currently underway. Another glucagon-like peptide-1 receptor agonist, exenatide (Table 3), has also met success in animal models and has an ongoing phase 3 clinical trial. Other recent therapeutic options, such as alpha lipoic acid, verapamil, soyateltide, and JPI-289, have also recently demonstrated success in animal models and currently have ongoing clinical trials. However, whether these new therapeutics will have higher success than previous neuroprotective therapies is still yet to be seen.
While neuroprotection focuses on preventing further damage to tissue, neurorestoration aims to restore function to damaged neurons via the use of neurotrophins, a group of proteins associated with the maintenance and survival of the CNS. The most abundant neurotrophin in the adult brain is brain-derived neurotrophic factor (BDNF), which participates in proliferation and neuronal differentiation, among other functions (Liu et al., 2020). Another common and well-studied neurotrophin, neurotrophin-3 (NT-3; Table 3), is specifically involved in cell proliferation as well as the processes of memory and learning (Vilar and Mira, 2016). As a stroke therapeutic, NT-3 has demonstrated excellent translational potential. For example, studies have shown that peripheral infusion of NT-3 can increase sensorimotor function after stroke. Additionally, phase 1 and phase 2 clinical trials have already been performed for NT-3, demonstrating its safety as a therapeutic (Duricki et al., 2019). Combined with NT-3’s ability to be transported to the CNS through the BBB, these findings make it a promising future therapeutic. Metformin, another potential neurorestorative therapy, has also been shown to induce recovery of memory and learning by increasing the expression of the neutrophin BDNF (Ghadernezhad et al., 2016). Interestingly, metformin has also been shown to regenerate the GCX (Eskens et al., 2013; Targosz-Korecka et al., 2020). However, whether or not metformin-induced GCX regeneration plays a role in neurorestoration following stroke has not been studied. Other therapies, such as copolymer-1 (Cop-1), can similarly increase NT-3 and other neurotrophins to promote neurogenesis (Cruz et al., 2015). However, while some of these treatments have demonstrated efficacy in vitro or in animal models, further research is necessary to support their testing in clinical trials.
One regulatory mechanism of vascular function that has been well studied in the peripheral vasculature but not in the context of the brain vasculature (i.e., the BBB), neither in physiological nor pathological conditions, is endothelial mechanobiology. Endothelial mechanobiology is the process by which ECs sense mechanical forces and convert these forces into biochemical signals (Davies, 1995; Ando and Yamamoto, 2009). These forces include shear stress created by blood flow and tension created by vessel contraction/dilation and changes in the ECM (He et al., 2020a). In the peripheral vasculature, endothelial mechanobiology promotes proper function of the vasculature by regulating permeability, vascular tone, inflammatory state, and other important vascular functions implicated in vascular disease (Chien, 2008; Jones, 2010; Glen et al., 2012). For example, in the peripheral vasculature, both in vitro and in vivo studies have demonstrated that exposure to physiological levels of fluid shear stresses strengthens vascular barrier integrity (Himburg et al., 2004; Warboys et al., 2010). Via a novel in vivo/ex vivo permeability technique, Himburg et al. (2004) specifically found that increasing levels of physiological time-averaged shear stress leads to decreased endothelial permeability. Shear stress exposure has also been shown to increase nitric oxide production (Uematsu et al., 1995) and reduce the production of reactive oxygen species (Chen et al., 2003) when compared to samples maintained in static, no-flow conditions. In contrast, impaired mechanotransduction in the peripheral vasculature may promote the development of cardiovascular diseases such as atherosclerosis. In the case of atherosclerosis, a disease characterized by plaque formation in vessel walls, plaque development preferentially occurs in areas of the vasculature that experience abnormal flow patterns. Blood flow in these regions is typically characterized by multi-directional flow and low time-averaged shear stresses, leading to impaired mechanotransduction (Zarins et al., 1983; Ku et al., 1985).
Endothelial mechanotransduction has also been proven beneficial in the BBB. Specifically, several studies have demonstrated that exposure of brain EC monocultures or co-cultures to shear stress leads to increased TEER, reduced permeability, and increased expression of junctional proteins (Colgan et al., 2007; Siddharthan et al., 2007; Cucullo et al., 2011; Walsh et al., 2011). For example, Cucullo et al. (2011) found that shear stress application of 6.2 dynes/cm2 to a co-culture of human brain microvascular ECs and astrocytes leads to increased expression of TJ proteins, such as occludin and claudin-5, while simultaneously upregulating protective transporter proteins such as the ABC transporter family. This led to reduced permeability of numerous molecules including mannitol and d-glucose. Although, while this and other studies have correlated shear stress exposure with increased barrier integrity, many of these studies utilize sub-physiological shear stress magnitudes or do not include important supportive cells such as pericytes and astrocytes. Furthermore, while the impacts of shear stress exposure have been demonstrated, the mechanostransducers responsible for the observed impacts are largely unknown. In this section, we will address a prominent endothelial mechanotransducing structure, the GCX, and elaborate upon the potential of this and other mechanotransducers as future therapeutic targets for the treatment of stroke and other diseases associated with BBB dysfunction.
The endothelial GCX is a transmembrane, proteoglycan-glycoprotein layer extending from the luminal surface of ECs with a reported thickness ranging from 20 nm to 11 mm in vitro and in vivo, depending on the size and location of the vessel, as well as the method of GCX preservation and visualization (Figure 5; Chignalia et al., 2016; Mitra et al., 2017; Harding et al., 2019). Due to its position, the GCX serves as a barrier to vascular permeability (Butler et al., 2020), can regulate the movement and absorption of blood-borne molecules, and can even affect the resistance to blood flow (Mitra et al., 2017; Zhu et al., 2018). These and other GCX functions are understandably largely dependent on the structural composition and stability of the GCX itself.
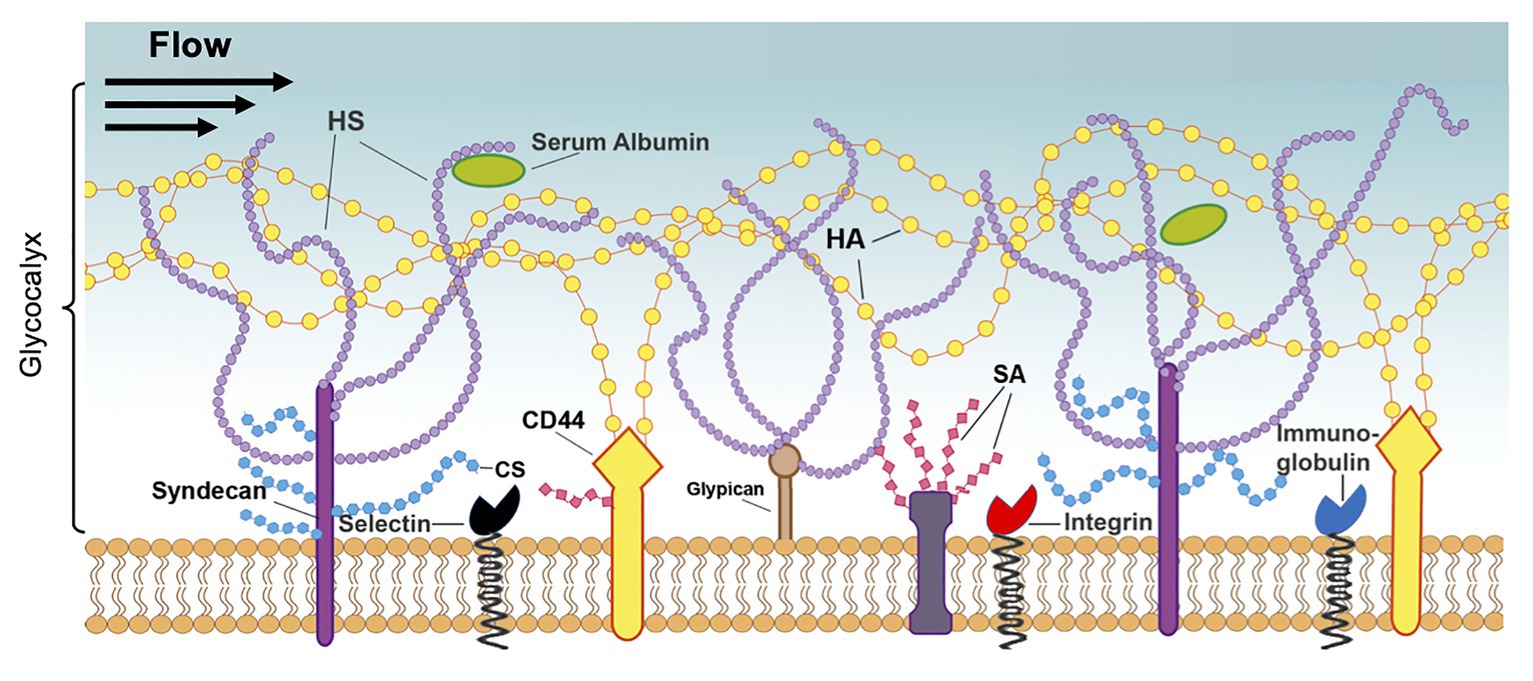
Figure 5. Structure of the endothelial glycocalyx. The glycocalyx, a mechanotransducer implicated in BBB regulation, extends from the membrane of ECs into the vessel lumen and is largely composed of proteoglycans and glycoproteins. The proteoglycans consist of the core proteins, syndecans, and glypicans, along with their attached glycosaminoglycans: heparan sulfate (HS), chondroitin sulfate (CS), and hyaluronic acid (HA). These chains are also modified by sialic acid (SA), which provides a net negative charge to the structure. Additionally, the glycocalyx contains glycoproteins, such as integrins and immunoglobulins, and soluble proteins, such as albumin. Reprinted from Harding et al., 2019 “Endothelial barrier reinforcement relies on flow-regulated glycocalyx, a potential therapeutic target,” Biorheology, 56 (2–3): 131–149, with permission from IOS Press. The publication is available at IOS Press through http://dx.doi.org/10.3233/BIR-180205.
Glycoproteins of the GCX are protein-glycan conjugates that facilitate interactions with surrounding molecules and cells and include common adhesion molecules located at the base of the GCX. Three types of adhesion molecules play an important role in GCX structure: the selectin family, including E-selectin and P-selectin, which is highly associated with the interaction of leukocytes with the endothelium; the integrin family, which regulates platelet-EC interactions; and the immunoglobulin superfamily, which serve as ligands for integrins and additional mediators of adhesion to the endothelium (Figure 5; Mitra et al., 2017). Another major GCX component, proteoglycans, are core proteins to which many glycosaminoglycan (GAG) chains are covalently attached (Figure 5; Uchimido et al., 2019). Therefore, these proteins control the incorporation of extracellular GCX components into the EC body. The syndecan family of proteoglycans, which is composed of four members, is a unique transmembrane proteoglycan family involved in many biological processes like wound healing and inflammation (Chignalia et al., 2016). The glypican family, another proteoglycan family, is composed of six members and can be bound to glycosyl-phosphatidylinositol anchors within the cell membrane, which may contribute to its functional roles (Figure 5). The GAGs attached to these proteoglycans are composed of repeating disaccharide units and include heparan sulfate, which is the most common GAG on the GCX, chondroitin sulfate, which is believed to be the second most common GAG, and hyaluronic acid, which contributes to the structural integrity of the GAG-protein matrix. Additionally, GAGs and other GCX constituents are extensively modified with sialic acid residues, imparting a net negative charge to the structure. Collectively, these GCX components are highly associated with EC protection in the peripheral vasculature and several studies have even implicated the GCX as a critical component of the BBB, as discussed below (Ando et al., 2018).
Previous investigations in the peripheral vasculature, mainly using transmission electron microscopy, identified robust expression of the GCX ex vivo (Luft, 1966; van den Berg et al., 2003; Ueda et al., 2004; Ebong et al., 2011). Subsequent studies have implicated GCX of the peripheral vasculature in regulating key EC functions. For instance, abundant GCX expression has been shown to reduce endothelial permeability in multiple vascular beds, including the mesenteric capillaries and glomerular vasculature (Adamson, 1990; Singh et al., 2007). GCX expression in the peripheral vasculature has also been shown to mediate shear stress-induced increases in nitric oxide formation, which promotes vasodilation and in general is associated with vascular health (Ebong et al., 2014; Harding et al., 2018). Specifically, shear stress exposure was shown to increase endothelial nitric oxide synthase activation and expression in a heparan sulfate-dependent nature (Harding et al., 2018). However, whether the GCX is expressed at the BBB and, if so, its thickness and composition had until recently not been studied. The first confirmation of GCX expression in the BBB came in 2017 by Yoon et al. (2017) who identified GCX expression in vivo via fluorescence microscopy. By labeling the GCX with wheat germ agglutinin and performing 2-photon microscopy, which allows for in vivo imaging at sufficient depth, Yoon identified a fluorescent dextran permeation-resistant GCX layer of approximately 1 mm in thickness. In agreement with these findings, another investigation found that GCX within the BBB resists permeation of larger molecules, such as dextran, but not smaller molecules, such as fluorescein and Alexa Fluor antibodies (Kutuzov et al., 2018). The Yoon study also suggested that GCX expression varies in the cerebrovasculature, with the highest expression in arteries and no detectable expression in veins and venules. In 2018, Ando et al. (2018) validated the presence of the GCX in the BBB using electron microscopy. Specifically, transmission electron microscopy demonstrated a robust expression of the GCX in brain capillaries that was greater than the expression in the peripheral vasculature of similar diameter. Furthermore, the study determined that GCX of the BBB is more resistant to inflammation-induced degradation, which is known to occur in numerous indications. These results suggest that GCX integrity may play an even more significant role in regulating BBB function when compared to the peripheral vasculature.
In addition to these studies confirming the presence of the GCX in vivo, other studies have implicated the GCX in regulation of BBB function in physiological conditions. For example, DeOre et al. (2020) recently found that CD44, a hyaluronic acid binding protein, promotes BBB function by regulating BBB permeability in response to fluid shear stress. Specifically, CD44 knockdown led to increased permeability and decreased TEER after 24 h of flow exposure at 0.7 dynes/cm2, demonstrating the importance of CD44 expression in physiological conditions for proper mechanotransduction. In contrast to the beneficial effects of GCX expression in physiological conditions, GCX degradation has been identified in numerous pathologies, such as stroke and traumatic brain injury (TBI), suggesting that GCX degradation may play a functional role in the development of these conditions. For example, one study identified a degraded GCX layer in individuals with cerebral small vessel disease coinciding with white matter lesions (Martens et al., 2013). Another study in rats found that TBI significantly elevates levels of syndecan-1, a major GCX component, in serum samples compared to healthy controls (Jepsen et al., 2014). This is in agreement with other studies, including one using human subjects that identified increased syndecan shedding in response to TBI (Gonzalez Rodriguez et al., 2018). It was even found that repeated, low intensity TBI is sufficient to reduce both GCX thickness and density, which correlated with downstream behavioral deficits (Hall et al., 2017). GCX degradation is also associated with ischemia/reperfusion as occurs in ischemic stroke (Chappell et al., 2009; van Golen et al., 2012). Furthermore, a recent study by Zhu et al. (2018) found that hyaluronic acid degradation in rats via hyaluronidase treatment increases BBB permeability. While these studies demonstrate a correlation between GCX degradation and several neurological conditions, few studies have directly confirmed the role of the GCX in BBB function in physiological conditions. Furthermore, although GCX degradation is associated with ischemia/reperfusion as occurs in ischemic stroke, to our knowledge, no studies have extensively investigated the relationship between GCX expression and ischemic stroke pathology. Thus, future research should focus in these areas.
In addition to the GCX, other endothelial mechanosensors are involved in the maintenance of BBB integrity and permeability. For example, it has been demonstrated that ion channels, particularly the transient receptor potential (TRP) channels, which are mechanosensitive, non-selective, calcium-permeable, cation channels, are heavily expressed on brain microvascular ECs (Chang et al., 2018) and serve as critical regulators of the intact BBB. Specifically, studies have demonstrated that transient receptor potential polycystin-2 (TRPP2; encoded by the PKD2 gene; AbouAlaiwi et al., 2009) and transient receptor potential channel 1 (TRPC1), which are responsible for calcium influx mediation, are highly involved in the response for BBB damage induced by TBI (Berrout et al., 2012). Both channels were suggested to modulate stretch-induced injury via nitric oxide (NO) production and action stress fiber formation in brain microvessel ECs. Marked decrease of the injury-induced calcium response, NO production, and stress fiber formation were observed after interfering with the function of TRPP2 and TRPC1 channel via TRPP2 and TRPC1 channel blockers or siRNA knockdown (Berrout et al., 2012). Ellaine et al. also demonstrated that stretch injury and oxygen glucose deprivation induced a significant increase in calcium ion concentration inside ECs (Salvador et al., 2015). Collectively, these studies suggest that ion channels may serve as potential therapeutic targets for BBB disruption. VE-cadherin has also been shown to regulate BBB function via endothelial mechanotransduction (Walsh et al., 2011). Specifically, Walsh et al. (2011) demonstrated for the first time that VE-cadherin transmits shear signals to occludin via Rac1, leading to BBB stabilization. This mechanosensory function of VE-cadherin likely occurs as part of the junctional mechanosensory complex, which has previously been shown to mediate EC responses to fluid shear stress (Conway et al., 2013).
To date, limited therapies targeting GCX restoration or protection exist. However, recent in vitro and in vivo animal studies have demonstrated potential promise for GCX-targeted therapeutics aiming to restore endothelial and vascular function associated with endothelial mechanotransduction. For example, spingosine-1-phosphate (S1P), which is a membrane phospholipid metabolite, can protect against shedding of the GCX and induce biosynthesis of GCX components after their shedding (Zeng et al., 2015; Araibi et al., 2020). Specifically, Zeng et al. (2015) found that S1P induces the synthesis of syndecan-1 and incorporates heparan sulfate and chondroitin sulfate chains on ECs. This function was modulated by the phosphoinositide 3-kinase (PI3K) pathway. However, other findings have suggested that S1P induction of cardioprotection against ischemia-reperfusion injury is not relevant to the integrity of syndecan-1 in adult rats but instead via alternative S1P functions (Araibi et al., 2020). Future examination of the effects of S1P will provide greater insight into the mechanisms and potential usage of S1P for GCX recovery and potentially for conditions such as ischemic stroke associated with GCX degradation.
Another treatment strategy referred to as GAG replacement includes GCX repair through the administration of exogenous GAGs, such as heparan sulfate. These include sulodexide, which contains heparin (80%) and dermatan sulfate (20%), and can lead to the reconstruction of endothelium barrier function and inhibition of GCX degrading enzymes (Mitra et al., 2017). Similarly, experiments performed by our research group have shown that heparan sulfate-degraded rat ECs can be treated with exogenous heparan sulfate and S1P to restore GCX structure. Furthermore, not only was this treatment able to restore GCX structure, but it was also able to restore EC function, particularly cell-cell gap junction communication (Mensah et al., 2017). Taken together, these studies demonstrate the potential for GCX protection as a novel approach for treatment of diseases associated with vascular dysfunction. Future work further identifying the mechanisms of GCX-induced regulation of vascular function will corroborate the GCX as a target for vascular related disease. Combining these findings with the development of accurate therapeutics to protect or restore GCX expression may thus aid in the identification of GCX-based therapeutics for BBB protection or regeneration in diseases such as ischemic stroke.
Over the past decade, our understanding of normal BBB and NVU functions and the mechanisms underlying BBB disruption during stroke has rapidly increased. It is well known that TJ proteins are complex and dynamic in nature and can be modulated in response to ischemic stroke while alterations of TJs can promote BBB permeability and oxidative stress-associated injury. In addition, endogenous BBB transporters further play a role in regulating BBB permeability and may be disrupted during ischemic stroke. Many therapeutic strategies have been investigated for their usage for ischemic stroke but with the exception of thrombolysis via recombinant tissue plasminogen activator (r-tPA) and endovascular mechanical thrombectomy treatment, these treatments have failed to pass clinical trials. New treatments targeting neuroprotection and neurorestoration, such as NBP and neurotrophins, respectively, are thus needed to help protect tissue injury from increasing in the setting of ischemic stroke. Additionally, endothelial mechanobiology, notably the endothelial GCX mechanotransducer, has been found to modulate BBB integrity and permeability, suggesting its high potential as a therapeutic target for BBB protection and recovery following ischemic stroke. However, the complete role of the GCX in BBB regulation has not been comprehensively studied, and few drugs have been developed for the protection or restoration of GCX components. Future research should further clarify the role of the GCX and other mechanotransducers in regulating BBB function and advance the development of novel therapeutics that target mechanotransducers in hopes of alleviating the impact of ischemic stroke.
KN, ICH, IMH, and EE were involved in the genesis of the review topic. KN and ICH drafted the manuscript. KN, ICH, EE, and IMH reviewed, edited, and approved the manuscript. All authors contributed to the article and approved the submitted version.
This manuscript was made possible by funding from the National Institutes of Health (K01 HL125499 awarded to EE), the American Heart Association (18PRE33960461 awarded to ICH), and Tufts University Office of the Provost (Inflammation-Based Collaborative Grant awarded to lead Principal Investigator (PI) IMH and co-PIs Luis Dorfmann and Tinatin Chabrashvili. The funders had no role in data or information collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank members of the Ebong lab, specifically Ronodeep Mitra and Chinedu Okorafor, for providing input on the manuscript.
TJ, Tight junction; EC, Endothelial cell; AJ, Adherens junction; GCX, Glycocalyx; NBP, Dl-3-n-butylphthalide; Cop-1, Copolymer 1; GAG, Glycosaminoglycan.
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Abdullahi, W., Tripathi, D., and Ronaldson, P. T. (2018). Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 315, C343–C356. doi: 10.1152/ajpcell.00095.2018
AbouAlaiwi, W. A., Takahashi, M., Mell, B. R., Jones, T. J., Ratnam, S., Kolb, R. J., et al. (2009). Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 104, 860–869. doi: 10.1161/CIRCRESAHA.108.192765
Abu Taha, A., and Schnittler, H. J. (2014). Dynamics between actin and the VE-cadherin/catenin complex: novel aspects of the ARP2/3 complex in regulation of endothelial junctions. Cell Adhes. Migr. 8, 125–135. doi: 10.4161/cam.28243
Adamson, R. H. (1990). Permeability of frog mesenteric capillaries after partial pronase digestion of the endothelial glycocalyx. J. Physiol. 428, 1–13. doi: 10.1113/jphysiol.1990.sp018197
Akins, P. T., Amar, A. P., Pakbaz, R. S., Fields, J. D., and Investigators, S. (2014). Complications of endovascular treatment for acute stroke in the SWIFT trial with solitaire and Merci devices. AJNR Am. J. Neuroradiol. 35, 524–528. doi: 10.3174/ajnr.A3707
Alishahi, M., Ghaedrahmati, F., Kolagar, T. A., Winlow, W., Nikkar, N., Farzaneh, M., et al. (2019). Long non-coding RNAs and cell death following ischemic stroke. Metab. Brain Dis. 34, 1243–1251. doi: 10.1007/s11011-019-00423-2
Alluri, H., Stagg, H. W., Wilson, R. L., Clayton, R. P., Sawant, D. A., Koneru, M., et al. (2014). Reactive oxygen species-caspase-3 relationship in mediating blood-brain barrier endothelial cell hyperpermeability following oxygen-glucose deprivation and reoxygenation. Microcirculation 21, 187–195. doi: 10.1111/micc.12110
Almutairi, M. M., Gong, C., Xu, Y. G., Chang, Y., and Shi, H. (2016). Factors controlling permeability of the blood-brain barrier. Cell. Mol. Life Sci. 73, 57–77. doi: 10.1007/s00018-015-2050-8
Alvarez, J. I., Katayama, T., and Prat, A. (2013). Glial influence on the blood brain barrier. Glia 61, 1939–1958. doi: 10.1002/glia.22575
Ando, Y., Okada, H., Takemura, G., Suzuki, K., Takada, C., Tomita, H., et al. (2018). Brain-specific ultrastructure of capillary endothelial glycocalyx and its possible contribution for blood brain barrier. Sci. Rep. 8:17523. doi: 10.1038/s41598-018-35976-2
Ando, J., and Yamamoto, K. (2009). Vascular mechanobiology: endothelial cell responses to fluid shear stress. Circ. J. 73, 1983–1992. doi: 10.1253/circj.cj-09-0583
Anthony, D. C., Bolton, S. J., Fearn, S., and Perry, V. H. (1997). Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood-brain barrier permeability in rats. Brain 120, 435–444. doi: 10.1093/brain/120.3.435
Araibi, H., van der Merwe, E., Gwanyanya, A., and Kelly-Laubscher, R. (2020). The effect of sphingosine-1-phosphate on the endothelial glycocalyx during ischemia-reperfusion injury in the isolated rat heart. Microcirculation 27:e12612. doi: 10.1111/micc.12612
Babsky, A. M., Topper, S., Zhang, H., Gao, Y., James, J. R., Hekmatyar, S. K., et al. (2008). Evaluation of extra- and intracellular apparent diffusion coefficient of sodium in rat skeletal muscle: effects of prolonged ischemia. Magn. Reson. Med. 59, 485–491. doi: 10.1002/mrm.21568
Ballabh, P., Braun, A., and Nedergaard, M. (2004). The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol. Dis. 16, 1–13. doi: 10.1016/j.nbd.2003.12.016
Bandera, E., Botteri, M., Minelli, C., Sutton, A., Abrams, K. R., and Latronico, N. (2006). Cerebral blood flow threshold of ischemic penumbra and infarct core in acute ischemic stroke: a systematic review. Stroke 37, 1334–1339. doi: 10.1161/01.STR.0000217418.29609.22
Bansal, S., Sangha, K. S., and Khatri, P. (2013). Drug treatment of acute ischemic stroke. Am. J. Cardiovasc. Drugs 13, 57–69. doi: 10.1007/s40256-013-0007-6
Beard, R. S. Jr., Reynolds, J. J., and Bearden, S. E. (2011). Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood 118, 2007–2014. doi: 10.1182/blood-2011-02-338269
Berrout, J., Jin, M., and O’Neil, R. G. (2012). Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood-brain barrier endothelial cells. Brain Res. 1436, 1–12. doi: 10.1016/j.brainres.2011.11.044
Bicker, J., Alves, G., Fortuna, A., and Falcao, A. (2014). Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: a review. Eur. J. Pharm. Biopharm. 87, 409–432. doi: 10.1016/j.ejpb.2014.03.012
Broocks, G., Kniep, H., Kemmling, A., Flottmann, F., Nawabi, J., Elsayed, S., et al. (2020). Effect of intravenous alteplase on ischaemic lesion water homeostasis. Eur. J. Neurol. 27, 376–383. doi: 10.1111/ene.14088
Buck, B. H., Liebeskind, D. S., Saver, J. L., Bang, O. Y., Yun, S. W., Starkman, S., et al. (2008). Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 39, 355–360. doi: 10.1161/STROKEAHA.107.490128
Butler, M. J., Down, C. J., Foster, R. R., and Satchell, S. C. (2020). The pathological relevance of increased endothelial glycocalyx permeability. Am. J. Pathol. 190, 742–751. doi: 10.1016/j.ajpath.2019.11.015
Cancel, L. M., Ebong, E. E., Mensah, S., Hirschberg, C., and Tarbell, J. M. (2016). Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis 252, 136–146. doi: 10.1016/j.atherosclerosis.2016.07.930
Chang, S. L., Huang, W., Mao, X., and Mack, M. L. (2018). Ethanol’s effects on transient receptor potential channel expression in brain microvascular endothelial cells. J. NeuroImmune Pharmacol. 13, 498–508. doi: 10.1007/s11481-018-9796-3
Chappell, D., Jacob, M., Hofmann-Kiefer, K., Rehm, M., Welsch, U., Conzen, P., et al. (2009). Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc. Res. 83, 388–396. doi: 10.1093/cvr/cvp097
Chauhan, G., and Debette, S. (2016). Genetic risk factors for ischemic and hemorrhagic stroke. Curr. Cardiol. Rep. 18:124. doi: 10.1007/s11886-016-0804-z
Chen, A. Q., Fang, Z., Chen, X. L., Yang, S., Zhou, Y. F., Mao, L., et al. (2019). Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 10:487. doi: 10.1038/s41419-019-1716-9
Chen, X. L., Varner, S. E., Rao, A. S., Grey, J. Y., Thomas, S., Cook, C. K., et al. (2003). Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J. Biol. Chem. 278, 703–711. doi: 10.1074/jbc.M203161200
Chen, Y. J., Wallace, B. K., Yuen, N., Jenkins, D. P., Wulff, H., and O’Donnell, M. E. (2015). Blood-brain barrier KCa3.1 channels: evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke 46, 237–244. doi: 10.1161/STROKEAHA.114.007445
Chien, S. (2008). Role of shear stress direction in endothelial mechanotransduction. Mol. Cell. Biomech. 5, 1–8. doi: 10.3970/mcb.2008.005.001
Chignalia, A. Z., Yetimakman, F., Christiaans, S. C., Unal, S., Bayrakci, B., Wagener, B. M., et al. (2016). The glycocalyx and trauma: a review. Shock 45, 338–348. doi: 10.1097/SHK.0000000000000513
Choi, K. H., Kim, H. S., Park, M. S., Kim, J. T., Kim, J. H., Cho, K. A., et al. (2016). Regulation of caveolin-1 expression determines early brain edema after experimental focal cerebral ischemia. Stroke 47, 1336–1343. doi: 10.1161/STROKEAHA.116.013205
Choudhury, G. R., and Ding, S. (2016). Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol. Dis. 85, 234–244. doi: 10.1016/j.nbd.2015.05.003
Clarke, J. D., and Ayala, J. P. (2019). Hemifacial angioedema following alteplase for acute stroke. Pract. Neurol. 19, 272–273. doi: 10.1136/practneurol-2018-002112
Colgan, O. C., Ferguson, G., Collins, N. T., Murphy, R. P., Meade, G., Cahill, P. A., et al. (2007). Regulation of bovine brain microvascular endothelial tight junction assembly and barrier function by laminar shear stress. Am. J. Physiol. Heart Circ. Physiol. 292, H3190–H3197. doi: 10.1152/ajpheart.01177.2006
Colombo, E., and Farina, C. (2016). Astrocytes: key regulators of neuroinflammation. Trends Immunol. 37, 608–620. doi: 10.1016/j.it.2016.06.006
Conway, D. E., Breckenridge, M. T., Hinde, E., Gratton, E., Chen, C. S., and Schwartz, M. A. (2013). Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr. Biol. 23, 1024–1030. doi: 10.1016/j.cub.2013.04.049
Cruz, Y., Lorea, J., Mestre, H., Kim-Lee, J. H., Herrera, J., Mellado, R., et al. (2015). Copolymer-1 promotes neurogenesis and improves functional recovery after acute ischemic stroke in rats. PLoS One 10:e0121854. doi: 10.1371/journal.pone.0121854
Cucullo, L., Hossain, M., Puvenna, V., Marchi, N., and Janigro, D. (2011). The role of shear stress in blood-brain barrier endothelial physiology. BMC Neurosci. 12:40. doi: 10.1186/1471-2202-12-40
da Fonseca, A. C., Matias, D., Garcia, C., Amaral, R., Geraldo, L. H., Freitas, C., et al. (2014). The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 8:362. doi: 10.3389/fncel.2014.00362
Dalkara, T., Gursoy-Ozdemir, Y., and Yemisci, M. (2011). Brain microvascular pericytes in health and disease. Acta Neuropathol. 122, 1–9. doi: 10.1007/s00401-011-0847-6
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Davies, P. F. (1995). Flow-mediated endothelial mechanotransduction. Physiol. Rev. 75, 519–560. doi: 10.1152/physrev.1995.75.3.519
Davis, B., Tang, J., Zhang, L., Mu, D., Jiang, X., Biran, V., et al. (2010). Role of vasodilator stimulated phosphoprotein in VEGF induced blood-brain barrier permeability in endothelial cell monolayers. Int. J. Dev. Neurosci. 28, 423–428. doi: 10.1016/j.ijdevneu.2010.06.010
del Zoppo, G. J., and Milner, R. (2006). Integrin-matrix interactions in the cerebral microvasculature. Arterioscler. Thromb. Vasc. Biol. 26, 1966–1975. doi: 10.1161/01.ATV.0000232525.65682.a2
DeOre, B. J., Partyka, P. P., Fan, F., and Galie, P. A. (2020). CD44 regulates blood-brain barrier integrity in response to fluid shear stress. bioRxiv [Preprint]. doi: 10.1101/2020.01.28.924043
Dorfel, M. J., and Huber, O. (2012). Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J. Biomed. Biotechnol. 2012:807356. doi: 10.1155/2012/807356
Duricki, D. A., Drndarski, S., Bernanos, M., Wood, T., Bosch, K., Chen, Q., et al. (2019). Stroke recovery in rats after 24-hour-delayed intramuscular neurotrophin-3 infusion. Ann. Neurol. 85, 32–46. doi: 10.1002/ana.25386
Ebong, E. E., Lopez-Quintero, S. V., Rizzo, V., Spray, D. C., and Tarbell, J. M. (2014). Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr. Biol. 6, 338–347. doi: 10.1039/c3ib40199e
Ebong, E. E., Macaluso, F. P., Spray, D. C., and Tarbell, J. M. (2011). Imaging the endothelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy. Arterioscler. Thromb. Vasc. Biol. 31, 1908–1915. doi: 10.1161/ATVBAHA.111.225268
Eskens, B. J., Zuurbier, C. J., van Haare, J., Vink, H., and van Teeffelen, J. W. (2013). Effects of two weeks of metformin treatment on whole-body glycocalyx barrier properties in db/db mice. Cardiovasc. Diabetol. 12:175. doi: 10.1186/1475-2840-12-175
Fagan, S. C., Waller, J. L., Nichols, F. T., Edwards, D. J., Pettigrew, L. C., Clark, W. M., et al. (2010). Minocycline to improve neurologic outcome in stroke (MINOS): a dose-finding study. Stroke 41, 2283–2287. doi: 10.1161/STROKEAHA.110.582601
Ferrari, C. C., Depino, A. M., Prada, F., Muraro, N., Campbell, S., Podhajcer, O., et al. (2004). Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am. J. Pathol. 165, 1827–1837. doi: 10.1016/S0002-9440(10)63438-4
Fisher, M., and Bastan, B. (2012). Identifying and utilizing the ischemic penumbra. Neurology 79(Suppl. 1), S79–S85. doi: 10.1212/WNL.0b013e3182695814
Foroutan, S., Brillault, J., Forbush, B., and O’Donnell, M. E. (2005). Moderate-to-severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na+-K+-Cl− cotransporter. Am. J. Phys. Cell Physiol. 289, C1492–C1501. doi: 10.1152/ajpcell.00257.2005
Gertz, K., Kronenberg, G., Uhlemann, R., Prinz, V., Marquina, R., Corada, M., et al. (2016). Partial loss of VE-cadherin improves long-term outcome and cerebral blood flow after transient brain ischemia in mice. BMC Neurol. 16:144. doi: 10.1186/s12883-016-0670-8
Ghadernezhad, N., Khalaj, L., Pazoki-Toroudi, H., Mirmasoumi, M., and Ashabi, G. (2016). Metformin pretreatment enhanced learning and memory in cerebral forebrain ischaemia: the role of the AMPK/BDNF/P70SK signalling pathway. Pharm. Biol. 54, 2211–2219. doi: 10.3109/13880209.2016.1150306
Glen, K., Luu, N. T., Ross, E., Buckley, C. D., Rainger, G. E., Egginton, S., et al. (2012). Modulation of functional responses of endothelial cells linked to angiogenesis and inflammation by shear stress: differential effects of the mechanotransducer CD31. J. Cell. Physiol. 227, 2710–2721. doi: 10.1002/jcp.23015
Goenka, L., Uppugunduri Satyanarayana, C. R., Suresh Kumar, S., and George, M. (2019). Neuroprotective agents in acute ischemic stroke-A reality check. Biomed. Pharmacother. 109, 2539–2547. doi: 10.1016/j.biopha.2018.11.041
Gonzalez Rodriguez, E., Cardenas, J. C., Cox, C. S., Kitagawa, R. S., Stensballe, J., Holcomb, J. B., et al. (2018). Traumatic brain injury is associated with increased syndecan-1 shedding in severely injured patients. Scand. J. Trauma Resusc. Emerg. Med. 26:102. doi: 10.1186/s13049-018-0565-3
Graeber, M. B., and Streit, W. J. (2010). Microglia: biology and pathology. Acta Neuropathol. 119, 89–105. doi: 10.1007/s00401-009-0622-0
Greene, C., Hanley, N., and Campbell, M. (2019). Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS 16:3. doi: 10.1186/s12987-019-0123-z
Gronberg, N. V., Johansen, F. F., Kristiansen, U., and Hasseldam, H. (2013). Leukocyte infiltration in experimental stroke. J. Neuroinflammation 10:115. doi: 10.1186/1742-2094-10-115
Hall, A. A., Mendoza, M. I., Zhou, H., Shaughness, M., Maudlin-Jeronimo, E., McCarron, R. M., et al. (2017). Repeated low intensity blast exposure is associated with damaged endothelial glycocalyx and downstream behavioral deficits. Front. Behav. Neurosci. 11:104. doi: 10.3389/fnbeh.2017.00104
Harding, I. C., Mitra, R., Mensah, S. A., Herman, I. M., and Ebong, E. E. (2018). Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J. Transl. Med. 16:364. doi: 10.1186/s12967-018-1721-2
Harding, I. C., Mitra, R., Mensah, S. A., Nersesyan, A., Bal, N. N., and Ebong, E. E. (2019). Endothelial barrier reinforcement relies on flow-regulated glycocalyx, a potential therapeutic target. Biorheology 56, 131–149. doi: 10.3233/BIR-180205
Hau, V. S., Huber, J. D., Campos, C. R., Davis, R. T., and Davis, T. P. (2004). Effect of lambda-carrageenan-induced inflammatory pain on brain uptake of codeine and antinociception. Brain Res. 1018, 257–264. doi: 10.1016/j.brainres.2004.05.081
He, M., Martin, M., Marin, T., Chen, Z., and Gongol, B. (2020a). Endothelial mechanobiology. APL Bioeng. 4:010904. doi: 10.1063/1.5129563
He, W., Wang, H., Zhao, C., Tian, X., Li, L., and Wang, H. (2020b). Role of liraglutide in brain repair promotion through Sirt1-mediated mitochondrial improvement in stroke. J. Cell. Physiol. 235, 2986–3001. doi: 10.1002/jcp.29204
Himburg, H. A., Grzybowski, D. M., Hazel, A. L., LaMack, J. A., Li, X. M., and Friedman, M. H. (2004). Spatial comparison between wall shear stress measures and porcine arterial endothelial permeability. Am. J. Physiol. Heart Circ. Physiol. 286, H1916–H1922. doi: 10.1152/ajpheart.00897.2003
Hladky, S. B., and Barrand, M. A. (2016). Fluid and ion transfer across the blood-brain and blood-cerebrospinal fluid barriers; a comparative account of mechanisms and roles. Fluids Barriers CNS 13:19. doi: 10.1186/s12987-016-0040-3
Hom, S., Fleegal, M. A., Egleton, R. D., Campos, C. R., Hawkins, B. T., and Davis, T. P. (2007). Comparative changes in the blood-brain barrier and cerebral infarction of SHR and WKY rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1881–R1892. doi: 10.1152/ajpregu.00761.2005
Honda, M., Nakagawa, S., Hayashi, K., Kitagawa, N., Tsutsumi, K., Nagata, I., et al. (2006). Adrenomedullin improves the blood-brain barrier function through the expression of claudin-5. Cell. Mol. Neurobiol. 26, 109–118. doi: 10.1007/s10571-006-9028-x
Huang, H., Bhuiyan, M. I. H., Jiang, T., Song, S., Shankar, S., Taheri, T., et al. (2019). A novel Na+-K+-Cl− cotransporter 1 inhibitor STS66* reduces brain damage in mice after ischemic stroke. Stroke 50, 1021–1025. doi: 10.1161/STROKEAHA.118.024287
Huang, Z. G., Xue, D., Preston, E., Karbalai, H., and Buchan, A. M. (1999). Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can. J. Neurol. Sci. 26, 298–304. doi: 10.1017/s0317167100000421
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376. doi: 10.1038/nn2003
Ishida, H., Takemori, K., Dote, K., and Ito, H. (2006). Expression of glucose transporter-1 and aquaporin-4 in the cerebral cortex of stroke-prone spontaneously hypertensive rats in relation to the blood-brain barrier function. Am. J. Hypertens. 19, 33–39. doi: 10.1016/j.amjhyper.2005.06.023
Jayakumar, A. R., and Norenberg, M. D. (2010). The Na-K-Cl Co-transporter in astrocyte swelling. Metab. Brain Dis. 25, 31–38. doi: 10.1007/s11011-010-9180-3
Jeong, S. M., Hahm, K. D., Shin, J. W., Leem, J. G., Lee, C., and Han, S. M. (2006). Changes in magnesium concentration in the serum and cerebrospinal fluid of neuropathic rats. Acta Anaesthesiol. Scand. 50, 211–216. doi: 10.1111/j.1399-6576.2006.00925.x
Jepsen, C. H., deMoya, M. A., Perner, A., Sillesen, M., Ostrowski, S. R., Alam, H. B., et al. (2014). Effect of valproic acid and injury on lesion size and endothelial glycocalyx shedding in a rodent model of isolated traumatic brain injury. J. Trauma Acute Care Surg. 77, 292–297. doi: 10.1097/TA.0000000000000333
Jiang, X., Andjelkovic, A. V., Zhu, L., Yang, T., Bennett, M. V. L., Chen, J., et al. (2018). Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 163–164, 144–171. doi: 10.1016/j.pneurobio.2017.10.001
Jiao, H., Wang, Z., Liu, Y., Wang, P., and Xue, Y. (2011). Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J. Mol. Neurosci. 44, 130–139. doi: 10.1007/s12031-011-9496-4
Jickling, G. C., Liu, D., Ander, B. P., Stamova, B., Zhan, X., and Sharp, F. R. (2015). Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J. Cereb. Blood Flow Metab. 35, 888–901. doi: 10.1038/jcbfm.2015.45
Jin, R., Yang, G., and Li, G. (2010). Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol. 87, 779–789. doi: 10.1189/jlb.1109766
Joice, S. L., Mydeen, F., Couraud, P. O., Weksler, B. B., Romero, I. A., Fraser, P. A., et al. (2009). Modulation of blood-brain barrier permeability by neutrophils: in vitro and in vivo studies. Brain Res. 1298, 13–23. doi: 10.1016/j.brainres.2009.08.076
Jones, E. A. V. (2010). Mechanotransduction and blood fluid dynamics in developing blood vessels. Can. J. Chem. Eng. 88, 136–143. doi: 10.1002/cjce.20290
Jovin, T. G., Chamorro, A., Cobo, E., de Miquel, M. A., Molina, C. A., Rovira, A., et al. (2015). Thrombectomy within 8 hours after symptom onset in ischemic stroke. N. Engl. J. Med. 372, 2296–2306. doi: 10.1056/NEJMoa1503780
Karsy, M., Brock, A., Guan, J., Taussky, P., Kalani, M. Y., and Park, M. S. (2017). Neuroprotective strategies and the underlying molecular basis of cerebrovascular stroke. Neurosurg. Focus 42:E3. doi: 10.3171/2017.1.FOCUS16522
Kassner, A., and Merali, Z. (2015). Assessment of blood-brain barrier disruption in stroke. Stroke 46, 3310–3315. doi: 10.1161/STROKEAHA.115.008861
Kikuchi, K., Miura, N., Kawahara, K. I., Murai, Y., Morioka, M., Lapchak, P. A., et al. (2013). Edaravone (Radicut), a free radical scavenger, is a potentially useful addition to thrombolytic therapy in patients with acute ischemic stroke. Biomed. Rep. 1, 7–12. doi: 10.3892/br.2012.7
Kimura, K., Aoki, J., Sakamoto, Y., Kobayashi, K., Sakai, K., Inoue, T., et al. (2012). Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients—a preliminary study. J. Neurol. Sci. 313, 132–136. doi: 10.1016/j.jns.2011.09.006
Knowland, D., Arac, A., Sekiguchi, K. J., Hsu, M., Lutz, S. E., Perrino, J., et al. (2014). Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82, 603–617. doi: 10.1016/j.neuron.2014.03.003
Koehler, R. C., Gebremedhin, D., and Harder, D. R. (2006). Role of astrocytes in cerebrovascular regulation. J. Appl. Physiol. 100, 307–317. doi: 10.1152/japplphysiol.00938.2005
Kong, T., Chen, J., Sun, K., Zhang, W., Wang, J., Song, L., et al. (2019). Aspirin reduced recurrent stroke risk in patients with lacunar stroke. Acta Neurol. Scand. 140, 78–83. doi: 10.1111/ane.13105
Ku, D. N., Giddens, D. P., Zarins, C. K., and Glagov, S. (1985). Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 5, 293–302. doi: 10.1161/01.atv.5.3.293
Kumar, A. D., Boehme, A. K., Siegler, J. E., Gillette, M., Albright, K. C., and Martin-Schild, S. (2013). Leukocytosis in patients with neurologic deterioration after acute ischemic stroke is associated with poor outcomes. J. Stroke Cerebrovasc. Dis. 22, e111–e117. doi: 10.1016/j.jstrokecerebrovasdis.2012.08.008
Kuroiwa, T., Ting, P., Martinez, H., and Klatzo, I. (1985). The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 68, 122–129. doi: 10.1007/BF00688633
Kutuzov, N., Flyvbjerg, H., and Lauritzen, M. (2018). Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood-brain barrier. Proc. Natl. Acad. Sci. U. S. A. 115, E9429–E9438. doi: 10.1073/pnas.1802155115
Lakhan, S. E., Kirchgessner, A., and Hofer, M. (2009). Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J. Transl. Med. 7:97. doi: 10.1186/1479-5876-7-97
Lam, R. H., Sun, Y., Chen, W., and Fu, J. (2012). Elastomeric microposts integrated into microfluidics for flow-mediated endothelial mechanotransduction analysis. Lab Chip 12, 1865–1873. doi: 10.1039/c2lc21146g
Lampl, Y., Gilad, R., Geva, D., Eshel, Y., and Sadeh, M. (2001). Intravenous administration of magnesium sulfate in acute stroke: a randomized double-blind study. Clin. Neuropharmacol. 24, 11–15. doi: 10.1097/00002826-200101000-00003
Lampugnani, M. G., and Dejana, E. (2007). Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 120, S1–S6. doi: 10.1016/s0049-3848(07)70124-x
Lapergue, B., Labreuche, J., Blanc, R., Marnat, G., Consoli, A., Rodesch, G., et al. (2020). Combined use of contact aspiration and the stent retriever technique versus stent retriever alone for recanalization in acute cerebral infarction: the randomized ASTER 2 study protocol. J. Neurointerv. Surg. 12, 471–476. doi: 10.1136/neurintsurg-2019-014735
Latchaw, R. E., Yonas, H., Hunter, G. J., Yuh, W. T., Ueda, T., Sorensen, A. G., et al. (2003). Guidelines and recommendations for perfusion imaging in cerebral ischemia: a scientific statement for healthcare professionals by the writing group on perfusion imaging, from the Council on Cardiovascular Radiology of the American Heart Association. Stroke 34, 1084–1104. doi: 10.1161/01.STR.0000064840.99271.9E
Lau, L. W., Cua, R., Keough, M. B., Haylock-Jacobs, S., and Yong, V. W. (2013). Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat. Rev. Neurosci. 14, 722–729. doi: 10.1038/nrn3550
Li, X., Han, H., Hou, R., Wei, L., Wang, G., Li, C., et al. (2013). Progesterone treatment before experimental hypoxia-ischemia enhances the expression of glucose transporter proteins GLUT1 and GLUT3 in neonatal rats. Neurosci. Bull. 29, 287–294. doi: 10.1007/s12264-013-1298-y
Li, J., Liu, Y., Zhang, X., Chen, R., Zhang, L., Xue, J., et al. (2019). Dl-3-N-butylphthalide alleviates the blood-brain barrier permeability of focal cerebral ischemia reperfusion in mice. Neuroscience 413, 99–107. doi: 10.1016/j.neuroscience.2019.06.020
Lindsberg, P. J., Carpen, O., Paetau, A., Karjalainen-Lindsberg, M. L., and Kaste, M. (1996). Endothelial ICAM-1 expression associated with inflammatory cell response in human ischemic stroke. Circulation 94, 939–945. doi: 10.1161/01.cir.94.5.939
Liu, Z., and Chopp, M. (2016). Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 144, 103–120. doi: 10.1016/j.pneurobio.2015.09.008
Liu, W., Wang, X., O’Connor, M., Wang, G., and Han, F. (2020). Brain-derived neurotrophic factor and its potential therapeutic role in stroke comorbidities. Neural Plast. 2020, 1–13. doi: 10.1155/2020/1969482
Lo, E. H., Broderick, J. P., and Moskowitz, M. A. (2004). tPA and proteolysis in the neurovascular unit. Stroke 35, 354–356. doi: 10.1161/01.STR.0000115164.80010.8A
Logallo, N., Novotny, V., Assmus, J., Kvistad, C. E., Alteheld, L., Rønning, O. M., et al. (2017). Tenecteplase versus alteplase for management of acute ischaemic stroke (NOR-TEST): a phase 3, randomised, open-label, blinded endpoint trial. Lancet Neurol. 16, 781–788. doi: 10.1016/s1474-4422(17)30253-3
Lowhagen Henden, P., Rentzos, A., Karlsson, J. E., Rosengren, L., Leiram, B., Sundeman, H., et al. (2017). General anesthesia versus conscious sedation for endovascular treatment of acute ischemic stroke: the AnStroke trial (anesthesia during stroke). Stroke 48, 1601–1607. doi: 10.1161/STROKEAHA.117.016554
Luft, J. H. (1966). Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 25, 1773–1783.
Ma, Y., Wang, J., Wang, Y., and Yang, G. Y. (2017). The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 157, 247–272. doi: 10.1016/j.pneurobio.2016.01.005
Martens, R. J. H., Vink, H., van Oostenbrugge, R. J., and Staals, J. (2013). Sublingual microvascular glycocalyx dimensions in lacunar stroke patients. Cerebrovasc. Dis. 35, 451–454. doi: 10.1159/000348854
McCaffrey, G., Willis, C. L., Staatz, W. D., Nametz, N., Quigley, C. A., Hom, S., et al. (2009). Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J. Neurochem. 110, 58–71. doi: 10.1111/j.1471-4159.2009.06113.x
Mendez, A. A., Samaniego, E. A., Sheth, S. A., Dandapat, S., Hasan, D. M., Limaye, K. S., et al. (2018). Update in the early management and reperfusion strategies of patients with acute ischemic stroke. Crit. Care Res. Pract. 2018:9168731. doi: 10.1155/2018/9168731
Mensah, S. A., Cheng, M. J., Homayoni, H., Plouffe, B. D., Coury, A. J., and Ebong, E. E. (2017). Regeneration of glycocalyx by heparan sulfate and sphingosine 1-phosphate restores inter-endothelial communication. PLoS One 12:e0186116. doi: 10.1371/journal.pone.0186116
Merali, Z., Huang, K., Mikulis, D., Silver, F., and Kassner, A. (2017). Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS One 12:e0171558. doi: 10.1371/journal.pone.0171558
Mitra, R., O’Neil, G. L., Harding, I. C., Cheng, M. J., Mensah, S. A., and Ebong, E. E. (2017). Glycocalyx in atherosclerosis-relevant endothelium function and as a therapeutic target. Curr. Atheroscler. Rep. 19:63. doi: 10.1007/s11883-017-0691-9
Mitra, R., Qiao, J., Madhavan, S., O’Neil, G. L., Ritchie, B., Kulkarni, P., et al. (2018). The comparative effects of high fat diet or disturbed blood flow on glycocalyx integrity and vascular inflammation. Transl. Med. Commun. 3:10. doi: 10.1186/s41231-018-0029-9
Mokin, M., Khalessi, A. A., Mocco, J., Lanzino, G., Dumont, T. M., Hanel, R. A., et al. (2014). Endovascular treatment of acute ischemic stroke: the end or just the beginning? Neurosurg. Focus 36:E5. doi: 10.3171/2013.10.FOCUS13374
Muir, K. W., Lees, K. R., Ford, I., Davis, S. and Intravenous Magnesium Efficacy in Stroke Study Investigators (2004). Magnesium for acute stroke (Intravenous Magnesium Efficacy in Stroke trial): randomised controlled trial. Lancet 363, 439–445. doi: 10.1016/S0140-6736(04)15490-1
Muller, J. M., Chilian, W. M., and Davis, M. J. (1997). Integrin signaling transduces shear stress—dependent vasodilation of coronary arterioles. Circ. Res. 80, 320–326. doi: 10.1161/01.res.80.3.320
Murakami, T., Felinski, E. A., and Antonetti, D. A. (2009). Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 284, 21036–21046. doi: 10.1074/jbc.M109.016766
Nag, S., Venugopalan, R., and Stewart, D. J. (2007). Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 114, 459–469. doi: 10.1007/s00401-007-0274-x
Nakagomi, T., Kubo, S., Nakano-Doi, A., Sakuma, R., Lu, S., Narita, A., et al. (2015). Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells 33, 1962–1974. doi: 10.1002/stem.1977
O’Donnell, M. E. (2014). Blood-brain barrier Na transporters in ischemic stroke. Adv. Pharmacol. 71, 113–146. doi: 10.1016/bs.apha.2014.06.011
Obermeier, B., Daneman, R., and Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596. doi: 10.1038/nm.3407
Osada, T., Gu, Y. H., Kanazawa, M., Tsubota, Y., Hawkins, B. T., Spatz, M., et al. (2011). Interendothelial claudin-5 expression depends on cerebral endothelial cell-matrix adhesion by beta(1)-integrins. J. Cereb. Blood Flow Metab. 31, 1972–1985. doi: 10.1038/jcbfm.2011.99
Pan, W., and Kastin, A. J. (2007). Tumor necrosis factor and stroke: role of the blood-brain barrier. Prog. Neurobiol. 83, 363–374. doi: 10.1016/j.pneurobio.2007.07.008
Park, S. L., Lee, D. H., Yoo, S. E., and Jung, Y. S. (2010). The effect of Na+/H+ exchanger-1 inhibition by sabiporide on blood-brain barrier dysfunction after ischemia/hypoxia in vivo and in vitro. Brain Res. 1366, 189–196. doi: 10.1016/j.brainres.2010.09.077
Patel, R. A. G., and McMullen, P. W. (2017). Neuroprotection in the treatment of acute ischemic stroke. Prog. Cardiovasc. Dis. 59, 542–548. doi: 10.1016/j.pcad.2017.04.005
Pillai, D. R., Dittmar, M. S., Baldaranov, D., Heidemann, R. M., Henning, E. C., Schuierer, G., et al. (2009). Cerebral ischemia-reperfusion injury in rats—a 3 T MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. J. Cereb. Blood Flow Metab. 29, 1846–1855. doi: 10.1038/jcbfm.2009.106
Posada-Duque, R. A., Barreto, G. E., and Cardona-Gomez, G. P. (2014). Protection after stroke: cellular effectors of neurovascular unit integrity. Front. Cell. Neurosci. 8:231. doi: 10.3389/fncel.2014.00231
Powers, W. J., Rabinstein, A. A., Ackerson, T., Adeoye, O. M., Bambakidis, N. C., Becker, K., et al. (2018). 2018 guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 49, e46–e110. doi: 10.1161/STR.0000000000000158
Qosa, H., Miller, D. S., Pasinelli, P., and Trotti, D. (2015). Regulation of ABC efflux transporters at blood-brain barrier in health and neurological disorders. Brain Res. 1628, 298–316. doi: 10.1016/j.brainres.2015.07.005
Ramnath, R., Foster, R. R., Qiu, Y., Cope, G., Butler, M. J., Salmon, A. H., et al. (2014). Matrix metalloproteinase 9-mediated shedding of syndecan 4 in response to tumor necrosis factor alpha: a contributor to endothelial cell glycocalyx dysfunction. FASEB J. 28, 4686–4699. doi: 10.1096/fj.14-252221
Ren, J., Sietsma, D., Qiu, S., Moessler, H., and Finklestein, S. P. (2007). Cerebrolysin enhances functional recovery following focal cerebral infarction in rats. Restor. Neurol. Neurosci. 25, 25–31.
Revuelta, M., Elicegui, A., Moreno-Cugnon, L., Buhrer, C., Matheu, A., and Schmitz, T. (2019). Ischemic stroke in neonatal and adult astrocytes. Mech. Ageing Dev. 183:111147. doi: 10.1016/j.mad.2019.111147
Ronaldson, P. T., and Davis, T. P. (2015). Targeting transporters: promoting blood-brain barrier repair in response to oxidative stress injury. Brain Res. 1623, 39–52. doi: 10.1016/j.brainres.2015.03.018
Rosell, A., Cuadrado, E., Ortega-Aznar, A., Hernandez-Guillamon, M., Lo, E. H., and Montaner, J. (2008). MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39, 1121–1126. doi: 10.1161/STROKEAHA.107.500868
Ross, A. M., Hurn, P., Perrin, N., Wood, L., Carlini, W., and Potempa, K. (2007). Evidence of the peripheral inflammatory response in patients with transient ischemic attack. J. Stroke Cerebrovasc. Dis. 16, 203–207. doi: 10.1016/j.jstrokecerebrovasdis.2007.05.002
Rubio-Gayosso, I., Platts, S. H., and Duling, B. R. (2006). Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 290, H2247–H2256. doi: 10.1152/ajpheart.00796.2005
Rustenhoven, J., Jansson, D., Smyth, L. C., and Dragunow, M. (2017). Brain pericytes as mediators of neuroinflammation. Trends Pharmacol. Sci. 38, 291–304. doi: 10.1016/j.tips.2016.12.001
Salvador, E., Burek, M., and Forster, C. Y. (2015). Stretch and/or oxygen glucose deprivation (OGD) in an in vitro traumatic brain injury (TBI) model induces calcium alteration and inflammatory cascade. Front. Cell. Neurosci. 9:323. doi: 10.3389/fncel.2015.00323
Sandoval, K. E., and Witt, K. A. (2008). Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 32, 200–219. doi: 10.1016/j.nbd.2008.08.005
Sato, K., Kameda, M., Yasuhara, T., Agari, T., Baba, T., Wang, F., et al. (2013). Neuroprotective effects of liraglutide for stroke model of rats. Int. J. Mol. Sci. 14, 21513–21524. doi: 10.3390/ijms141121513
Saver, J. L., Goyal, M., van der Lugt, A., Menon, B. K., Majoie, C. B., Dippel, D. W., et al. (2016). Time to treatment with endovascular thrombectomy and outcomes from ischemic stroke: a meta-analysis. JAMA 316, 1279–1288. doi: 10.1001/jama.2016.13647
Saver, J. L., Starkman, S., Eckstein, M., Stratton, S. J., Pratt, F. D., Hamilton, S., et al. (2015). Prehospital use of magnesium sulfate as neuroprotection in acute stroke. N. Engl. J. Med. 372, 528–536. doi: 10.1056/NEJMoa1408827
Sevick, L. K., Ghali, S., Hill, M. D., Danthurebandara, V., Lorenzetti, D. L., Noseworthy, T., et al. (2017). Systematic review of the cost and cost-effectiveness of rapid endovascular therapy for acute ischemic stroke. Stroke 48, 2519–2526. doi: 10.1161/STROKEAHA.117.017199
Sharma, P., Sinha, M., Shukla, R., Garg, R. K., Verma, R., and Singh, M. K. (2011). A randomized controlled clinical trial to compare the safety and efficacy of edaravone in acute ischemic stroke. Ann. Indian Acad. Neurol. 14, 103–106. doi: 10.4103/0972-2327.82794
Shi, Y., Leak, R. K., Keep, R. F., and Chen, J. (2016). Translational stroke research on blood-brain barrier damage: challenges, perspectives, and goals. Transl. Stroke Res. 7, 89–92. doi: 10.1007/s12975-016-0447-9
Shi, J., Zhang, Y. Q., and Simpkins, J. W. (1997). Effects of 17beta-estradiol on glucose transporter 1 expression and endothelial cell survival following focal ischemia in the rats. Exp. Brain Res. 117, 200–206. doi: 10.1007/s002210050216
Shinozuka, K., Dailey, T., Tajiri, N., Ishikawa, H., Kim, D. W., Pabon, M., et al. (2013). Stem cells for neurovascular repair in stroke. J. Stem Cell Res. Ther. 4:12912. doi: 10.4172/2157-7633.S4-004
Siddharthan, V., Kim, Y. V., Liu, S., and Kim, K. S. (2007). Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 1147, 39–50. doi: 10.1016/j.brainres.2007.02.029
Sifat, A. E., Vaidya, B., and Abbruscato, T. J. (2017). Blood-brain barrier protection as a therapeutic strategy for acute ischemic stroke. AAPS J. 19, 957–972. doi: 10.1208/s12248-017-0091-7
Singh, A., Satchell, S. C., Neal, C. R., McKenzie, E. A., Tooke, J. E., and Mathieson, P. W. (2007). Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J. Am. Soc. Nephrol. 18, 2885–2893. doi: 10.1681/ASN.2007010119
Strbian, D., Durukan, A., Pitkonen, M., Marinkovic, I., Tatlisumak, E., Pedrono, E., et al. (2008). The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience 153, 175–181. doi: 10.1016/j.neuroscience.2008.02.012
Sung, S. F., Huang, Y. C., Ong, C. T., and Chen, W. (2013). Validity of a computerised five-level emergency triage system for patients with acute ischaemic stroke. Emerg. Med. J. 30, 454–458. doi: 10.1136/emermed-2012-201423
Targosz-Korecka, M., Malek-Zietek, K. E., Kloska, D., Rajfur, Z., Stepien, E. L., Grochot-Przeczek, A., et al. (2020). Metformin attenuates adhesion between cancer and endothelial cells in chronic hyperglycemia by recovery of the endothelial glycocalyx barrier. Biochim. Biophys. Acta Gen. Subj. 1864:129533. doi: 10.1016/j.bbagen.2020.129533
Thi, M. M., Tarbell, J. M., Weinbaum, S., and Spray, D. C. (2004). The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc. Natl. Acad. Sci. U. S. A. 101, 16483–16488. doi: 10.1073/pnas.0407474101
Thurgur, H., and Pinteaux, E. (2019). Microglia in the neurovascular unit: blood-brain barrier-microglia interactions after central nervous system disorders. Neuroscience 405, 55–67. doi: 10.1016/j.neuroscience.2018.06.046
Tietz, S., and Engelhardt, B. (2015). Brain barriers: crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 209, 493–506. doi: 10.1083/jcb.201412147
Tsai, C. F., Anderson, N., Thomas, B., and Sudlow, C. L. (2016). Comparing risk factor profiles between intracerebral hemorrhage and ischemic stroke in Chinese and white populations: systematic review and meta-analysis. PLoS One 11:e0151743. doi: 10.1371/journal.pone.0151743
Uchimido, R., Schmidt, E. P., and Shapiro, N. I. (2019). The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit. Care 23:16. doi: 10.1186/s13054-018-2292-6
Ueda, A., Shimomura, M., Ikeda, M., Yamaguchi, R., and Tanishita, K. (2004). Effect of glycocalyx on shear-dependent albumin uptake in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 287, H2287–H2294. doi: 10.1152/ajpheart.00808.2003
Uematsu, M., Ohara, Y., Navas, J. P., Nishida, K., Murphy, T. J., Alexander, R. W., et al. (1995). Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am. J. Physiol. 269, C1371–C1378. doi: 10.1152/ajpcell.1995.269.6.C1371
van den Berg, B. M., Spaan, J. A., and Vink, H. (2009). Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflugers Arch. 457, 1199–1206. doi: 10.1007/s00424-008-0590-6
van den Berg, B. M., Vink, H., and Spaan, J. A. (2003). The endothelial glycocalyx protects against myocardial edema. Circ. Res. 92, 592–594. doi: 10.1161/01.RES.0000065917.53950.75
van Golen, R. F., van Gulik, T. M., and Heger, M. (2012). Mechanistic overview of reactive species-induced degradation of the endothelial glycocalyx during hepatic ischemia/reperfusion injury. Free Radic. Biol. Med. 52, 1382–1402. doi: 10.1016/j.freeradbiomed.2012.01.013
Vannucci, S. J., Reinhart, R., Maher, F., Bondy, C. A., Lee, W. H., Vannucci, R. C., et al. (1998). Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res. Dev. Brain Res. 107, 255–264. doi: 10.1016/s0165-3806(98)00021-2
Vemula, S., Roder, K. E., Yang, T., Bhat, G. J., Thekkumkara, T. J., and Abbruscato, T. J. (2009). A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J. Pharmacol. Exp. Ther. 328, 487–495. doi: 10.1124/jpet.108.146589
Vilar, M., and Mira, H. (2016). Regulation of neurogenesis by neurotrophins during adulthood: expected and unexpected roles. Front. Neurosci. 10:26. doi: 10.3389/fnins.2016.00026
Wacker, B. K., Freie, A. B., Perfater, J. L., and Gidday, J. M. (2012). Junctional protein regulation by sphingosine kinase 2 contributes to blood-brain barrier protection in hypoxic preconditioning-induced cerebral ischemic tolerance. J. Cereb. Blood Flow Metab. 32, 1014–1023. doi: 10.1038/jcbfm.2012.3
Walsh, T. G., Murphy, R. P., Fitzpatrick, P., Rochfort, K. D., Guinan, A. F., Murphy, A., et al. (2011). Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J. Cell. Physiol. 226, 3053–3063. doi: 10.1002/jcp.22655
Wang, Y., Shen, Y., Liu, Z., Gu, J., Xu, C., Qian, S., et al. (2019). Dl-NBP (Dl-3-N-butylphthalide) treatment promotes neurological functional recovery accompanied by the upregulation of white matter integrity and HIF-1alpha/VEGF/Notch/Dll4 expression. Front. Pharmacol. 10:1595. doi: 10.3389/fphar.2019.01595
Wang, C. X., and Shuaib, A. (2007). Critical role of microvasculature basal lamina in ischemic brain injury. Prog. Neurobiol. 83, 140–148. doi: 10.1016/j.pneurobio.2007.07.006
Warboys, C. M., Eric Berson, R., Mann, G. E., Pearson, J. D., and Weinberg, P. D. (2010). Acute and chronic exposure to shear stress have opposite effects on endothelial permeability to macromolecules. Am. J. Physiol. Heart Circ. Physiol. 298, H1850–H1856. doi: 10.1152/ajpheart.00114.2010
Xia, C. Y., Liu, Y. H., Wang, P., and Xue, Y. X. (2012). Low-frequency ultrasound irradiation increases blood-tumor barrier permeability by transcellular pathway in a rat glioma model. J. Mol. Neurosci. 48, 281–290. doi: 10.1007/s12031-012-9770-0
Yamazaki, Y., Harada, S., Wada, T., Yoshida, S., and Tokuyama, S. (2016). Sodium transport through the cerebral sodium-glucose transporter exacerbates neuron damage during cerebral ischaemia. J. Pharm. Pharmacol. 68, 922–931. doi: 10.1111/jphp.12571
Yamazaki, Y., Ogihara, S., Harada, S., and Tokuyama, S. (2015). Activation of cerebral sodium-glucose transporter type 1 function mediated by post-ischemic hyperglycemia exacerbates the development of cerebral ischemia. Neuroscience 310, 674–685. doi: 10.1016/j.neuroscience.2015.10.005
Yang, G. Y., Gong, C., Qin, Z., Liu, X. H., and Lorris Betz, A. (1999). Tumor necrosis factor alpha expression produces increased blood-brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res. Mol. Brain Res. 69, 135–143. doi: 10.1016/s0169-328x(99)00007-8
Yang, Y., and Rosenberg, G. A. (2011). MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol. Biol. 762, 333–345. doi: 10.1007/978-1-61779-185-7_24
Yang, Y., and Rosenberg, G. A. (2015). Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 1623, 30–38. doi: 10.1016/j.brainres.2015.04.024
Ye, Z. Y., Xing, H. Y., Wang, B., Liu, M., and Lv, P. Y. (2019). DL-3-n-butylphthalide protects the blood-brain barrier against ischemia/hypoxia injury via upregulation of tight junction proteins. Chin. Med. J. 132, 1344–1353. doi: 10.1097/CM9.0000000000000232
Yoon, J. H., Lee, E. S., and Jeong, Y. (2017). In vivo imaging of the cerebral endothelial glycocalyx in mice. J. Vasc. Res. 54, 59–67. doi: 10.1159/000457799
Zaragoza, C., Marquez, S., and Saura, M. (2012). Endothelial mechanosensors of shear stress as regulators of atherogenesis. Curr. Opin. Lipidol. 23, 446–452. doi: 10.1097/MOL.0b013e328357e837
Zarins, C. K., Giddens, D. P., Bharadvaj, B. K., Sottiurai, V. S., Mabon, R. F., and Glagov, S. (1983). Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ. Res. 53, 502–514. doi: 10.1161/01.res.53.4.502
Zeng, Y., Liu, X. H., Tarbell, J., and Fu, B. (2015). Sphingosine 1-phosphate induced synthesis of glycocalyx on endothelial cells. Exp. Cell Res. 339, 90–95. doi: 10.1016/j.yexcr.2015.08.013
Zeng, C., Wang, D., Chen, C., Chen, L., Chen, B., Li, L., et al. (2020). Zafirlukast protects blood-brain barrier integrity from ischemic brain injury. Chem. Biol. Interact. 316:108915. doi: 10.1016/j.cbi.2019.108915
Zhang, S., An, Q., Wang, T., Gao, S., and Zhou, G. (2018). Autophagy- and MMP-2/9-mediated reduction and redistribution of ZO-1 contribute to hyperglycemia-increased blood-brain barrier permeability during early reperfusion in stroke. Neuroscience 377, 126–137. doi: 10.1016/j.neuroscience.2018.02.035
Zhang, C., Chopp, M., Cui, Y., Wang, L., Zhang, R., Zhang, L., et al. (2010). Cerebrolysin enhances neurogenesis in the ischemic brain and improves functional outcome after stroke. J. Neurosci. Res. 88, 3275–3281. doi: 10.1002/jnr.22495
Zhou, H., Lapointe, B. M., Clark, S. R., Zbytnuik, L., and Kubes, P. (2006). A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J. Immunol. 177, 8103–8110. doi: 10.4049/jimmunol.177.11.8103
Zhu, J., Li, X., Yin, J., Hu, Y., Gu, Y., and Pan, S. (2018). Glycocalyx degradation leads to blood-brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J. Cereb. Blood Flow Metab. 38, 1979–1992. doi: 10.1177/0271678X17726062
Zhu, H., Zhang, Y., Shi, Z., Lu, D., Li, T., Ding, Y., et al. (2016). The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci. Rep. 6:26859. doi: 10.1038/srep26859
Keywords: blood-brain barrier, ischemic stroke, endothelial cells, mechanotransduction, neuroprotection, neurovascular unit, endothelial glycocalyx
Citation: Nian K, Harding IC, Herman IM and Ebong EE (2020) Blood-Brain Barrier Damage in Ischemic Stroke and Its Regulation by Endothelial Mechanotransduction. Front. Physiol. 11:605398. doi: 10.3389/fphys.2020.605398
Edited by:
Clotilde Lecrux, McGill University, CanadaReviewed by:
Pedro Campinho, University of Lisbon, PortugalCopyright © 2020 Nian, Harding, Herman and Ebong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eno E. Ebong, ZS5lYm9uZ0Bub3J0aGVhc3Rlcm4uZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.