
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol. , 06 November 2020
Sec. Clinical and Translational Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.571367
This article is part of the Research Topic The Tribute of Physiology for the Understanding of COVID-19 Disease View all 39 articles
 Matteo Briguglio1*
Matteo Briguglio1* Mauro Porta2
Mauro Porta2 Francesca Zuffada3
Francesca Zuffada3 Alberto R. Bona4Tiziano Crespi5Fabio Pino5Paolo Perazzo5Marco Mazzocchi5
Alberto R. Bona4Tiziano Crespi5Fabio Pino5Paolo Perazzo5Marco Mazzocchi5 Riccardo Giorgino6Giuseppe De Angelis7
Riccardo Giorgino6Giuseppe De Angelis7 Alfonso Ielasi8Giuseppe De Blasio9
Alfonso Ielasi8Giuseppe De Blasio9 Maurizio Turiel9
Maurizio Turiel9The rapid spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the high fatality rate of coronavirus disease 2019 (COVID-19) have been putting a strain on the world since December 2019. Infected individuals exhibit unpredictable symptoms that tend to worsen if age is advanced, a state of malnutrition persists, or if cardiovascular comorbidities are present. Once transmitted, the virus affects the lungs and in predisposed individuals can elicit a sequela of fatal cardiovascular consequences. We aim to present the pathophysiology of COVID-19, emphasizing the major cellular and clinical manifestations from a cardiological perspective. As a roaming viral particle or more likely via the Trojan horse route, SARS-CoV-2 can access different parts of the body. Cardiovascular features of COVID-19 can count myocardial injuries, vasculitis-like syndromes, and atherothrombotic manifestations. Deviations in the normal electrocardiogram pattern could hide pericardial effusion or cardiac inflammation, and dispersed microthrombi can cause ischemic damages, stroke, or even medullary reflex dysfunctions. Tailored treatment for reduced ejection fraction, arrhythmias, coronary syndromes, macrothrombosis and microthrombosis, and autonomic dysfunctions is mandatory. Confidently, evidence-based therapies for this multifaceted nevertheless purely cardiological COVID-19 will emerge after the global assessment of different approaches.
The little understanding of the natural diversity of the severe acute respiratory syndrome-related coronaviruses (SARS-CoVs) restricts the opportunities to control their zoonotic spillovers (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses, 2020). Humans are therefore increasingly affected by outbreaks that put millions of people at risk. After the plagues of severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) in 2003 and of Middle East respiratory syndrome-related coronavirus (MERS-CoV) in 2012, a familial coronavirus (SARS-CoV-2) was discovered after the first documented virus-related pneumonia in China at the end of December 2019. This new strain is primarily transmitted through respiratory droplets and is able to survive in the airway mucosa despite the presence of cleaning epithelial cells, protective lymphoid tissues, and immunocompetent nerve endings (Briguglio et al., 2020a). The optimized genomic feature to bind to the angiotensin-converting enzyme 2 (ACE2), which derives from either millions of random natural mutations during unnoticed human-to-human transmission (Xu et al., 2020a) or artificial laboratory manipulations (Andersen et al., 2020), is the major determinant for the highest viral replication (Hoffmann et al., 2020) and for the consequent respiratory (Harapan et al., 2020) and cardiovascular implications (Wu et al., 2020b). After acquiring a sufficiently high viral load in the upper cavity (Zou et al., 2020), SARS-CoV-2 infects the goblet and ciliated cells in charge of sputum expectoration (Sungnak et al., 2020). The diffusion through the mucous layer allows the ease of infection of alveolar epithelial type II cells and systemic organs that express ACE2 (Briguglio et al., 2020a). The resulting illness, named coronavirus disease 2019 (COVID-19), is multifaceted and unpredictable and can manifest with early smell disorders in over 80% of cases or result in the most severe conditions like sepsis-like shock or respiratory failure in 14% of cases (Remy et al., 2020; Wu and McGoogan, 2020). Globally, it has been observed that 1 in 16 patients has encountered fatal consequences (WHO situation report 132, May–June 2020), and several infected patients were old and malnourished (Briguglio et al., 2020d; Sattar et al., 2020). Importantly, epidemiological data have been shown that preexisting cardiovascular conditions could be another central virulence factor for disease progression. In addition, clinical findings showed that not a few numbers of COVID-19 patients encounter cardiac symptoms (Mehra et al., 2020). Since each structure and function of the cardiovascular system shows severe implications, it is crucial to discuss from a cardiological perspective the relationship between SARS-CoV-2 infection and the cardiovascular system in order to shed some light on the mechanisms that can lead to cardiac symptoms or fatal consequences in COVID-19 patients.
It is necessary to differentiate the types of SARS-CoV-2-associated damages, the various stages of the disease, and the classification of infected patients. The virus-associated damage is of two types:
•Type I damage (i.e., cytotoxicity), which is directly associated with the infiltration of the virus in those cells expressing ACE2 (pneumocytes, endothelial cells, cardiomyocytes, neuronal cells). This may lead to acute injuries in the lungs, the vasculature, the myocardium, and the brain (Kabbani and Olds, 2020; Mason, 2020).
•Type II damage, which occurs during the disease progression. It derives from hypoxemia, inflammation, and microthrombosis. In particular, pneumonia and acute respiratory distress syndrome are likely to lead to a mismatch between oxygen supply and demand (hypoxic damage). Moreover, the late increase in circulating cytokines is known to cause nonischemic multiple organ injuries (e.g., stress-cardiomyopathy, myocarditis, vasculitis-like syndromes), and the systemic inflammation or catecholamine rush are associated with plaque rupture or blood hypercoagulability (i.e., thrombi-derived ischemic damage; Basu-Ray et al., 2020; Matsushita et al., 2020; Xiong et al., 2020; Zheng et al., 2020).
Considering the disease progression, three distinct phases have been recognized, covering the early infection mechanisms, the body’s response to the viral proliferation, and the late systemic phase of the illness.
•The incubation/proliferative phase: mild-to-moderate symptoms with fever, dry cough, headache, pharyngodynia, asthenia. This phase is biochemically characterized by mild lymphopenia and variations in some coagulation parameters, such as the D-dimer, thrombocytes, and international normalized ratio (INR). Lactate dehydrogenase as well as inflammatory markers like C-reactive protein and interleukin-6 may increase (Shi et al., 2020). Therapies to boost the immune response are certainly worth considering since early B lymphocyte reduction affects antibody production (Siddiqi and Mehra, 2020). This phase usually lasts a few days (Briguglio et al., 2020a).
•The respiratory phase: moderate-to-severe respiratory symptoms like shortness of breath and measurable hypoxemia. If a dysfunctional immune system was present, SARS-CoV-2 could proliferate quickly and lead to massive impairments of infiltrated tissues. This phase is characterized by increasing circulating levels of cytokines and chemokines, such as tumor necrosis factor-α, interleukins, interferon-γ, and chemoattractant proteins (Rokni et al., 2020). As long as the disease worsens, structural consequences include multiple patchy shadows in the lungs in mildly affected individuals or pleural fluid in the most severe cases (Yang et al., 2020). This phase normally starts to aggravate around 7–14 days after onset (Briguglio et al., 2020a).
•The systemic phase: moderate-to-severe systemic implications comprising acute distress respiratory syndrome, heart failure, and multisystem organ dysfunction. Troponin I and brain natriuretic peptide may be elevated in infected patients with cardiac involvement. The coagulopathy manifests with increased D-dimer and other fibrin degradation products, low platelet counts, and increased INR and prothrombin time (Lippi et al., 2020; Thachil et al., 2020). Severe lymphopenia, kidney injury, as well as elevated liver enzymes and cytokines may be found (Shi et al., 2020). Of note, lymphocyte attachment to the activated endothelium, together with their systemic redistribution and apoptosis, is supposed to be at the basis of low lymphocyte counts (Rokni et al., 2020). This phase might be conversely replaced by a recovery phase if the virus is effectively suppressed (Lin et al., 2020).
On the clinical bases, patients can be classified according to respiratory autonomy (Briguglio et al., 2020a):
•Level 0: asymptomatic, mostly home living.
•Level 1: mild symptoms, pharyngodynia, dry cough, mild fever; these individuals should not be hospitalized.
•Level 2: moderate symptoms, high fever, persistent cough, asthenia, dyspnea; these patients might require noninvasive oxygen therapy.
•Level 3: severe symptoms; these patients require invasive oxygen therapy and intensive care support. These patients were reported to meet the diagnostic criteria for sepsis, with the impaired liver, kidney, and lung functions presenting concomitantly with cold extremities, weak peripheral pulses, shock, and severe metabolic acidosis (Li et al., 2020b).
The cardiovascular sequelae start with the viral binding to ACE2 in the lower airways, causing type I damage in pneumocytes (Leung et al., 2020). The altered diffusion of oxygen across the injured alveolar membrane is likely to ground hypoxic conditions that prevent proper tissue oxygenation. Locally, SARS-CoV-2 particles activate alveolar macrophages and T cells (Shi et al., 2020). The subsequent inflammation is known to stimulate hyaline membrane formation, wall thickening, and infiltration of circulating monocytes that differentiate into macrophages or fibroblast-like cells called fibrocytes that eventually favor fibrotic processes in the parenchyma (Pilling and Gomer, 2012). During the worsening of the respiratory phase, the overactive immunological response in the lungs alters the integrity of epithelial-endothelial barriers, with plasma components exuding in the alveolar cavity together with chemotactic monocytes and neutrophils (Li et al., 2013). In level 2 and level 3 patients, a cytokine storm might arise, being the main root for growing a worsening life-threatening systemic phase (Xu et al., 2020b). The recruitment of different leukocyte populations in the lungs could expose these cells to viral infiltration, ending up becoming Trojan horses (i.e., vectors for SARS-CoV-2, recall of the mythical subterfuge to enter the city of Troy). This mechanism was in fact shown for the familial predecessor SARS-COV-1 (Chen and Hsiao, 2004; Gu et al., 2005) and supposed for SARS-CoV-2 (Li et al., 2020b; Park, 2020), whose viral particles were found in blood samples and in the myocardium (Tavazzi et al., 2020; Wang et al., 2020b). If SARS-CoV-2 was able to infiltrate into the heart, it would be likely to elicit the secretion of cytokines from cardiac fibroblasts to subsequently increase the inflammatory milieu (van Nieuwenhoven and Turner, 2013) and to cause the recruitment of transendothelial monocytes (Lindner et al., 2014), neutrophils, and dendritic cells (Van der Borght and Lambrecht, 2018). Activated dendritic cells are known to trigger T cells (Eriksson et al., 2003), further promoting tissue damage. A plethora of immune cells, comprising macrophages and fibrocytes, may therefore populate these early myocardium lesions (Oudit et al., 2009; Pilling et al., 2009), each likely to have its own role in COVID-19-associated myocarditis and stress-cardiomyopathy (Xiong et al., 2020). Consequently, it would seem fair to assume that the myocardium of infected patients might be subjected not only to type I damage, as a consequence of direct myocardial cell injury, but also to type II damage mainly comprising the inflammation-derived grievance. Remarkably, even patients with mild respiratory symptoms can manifest early cardiovascular implications, such as acute myopericarditis (Inciardi et al., 2020), Takotsubo syndrome (Meyer et al., 2020), or acute myocardial infarction (Stefanini et al., 2020). Fulminant myocarditis was reported in level 2 patients (Hu et al., 2020; Zeng et al., 2020), and supraventricular tachycardia, decompensated heart failure, and cardiogenic shock were observed in aggravating level 3 patients (Fried et al., 2020). It is generally agreed that the lymphocytic count mirrors the nutritional status of the host (Briguglio et al., 2019), and it may be useful in predicting the patient’s reservoirs against the infection since these cells decline as long as COVID-19 worsens (Peteranderl and Herold, 2017; Chan et al., 2020). This attenuated immune potential of the host increases the susceptibility to disease complications, and the coupling of severe pneumonia with myocardial injury is likely to lead to progressive cardiorespiratory deterioration. Severe patients were in fact reported to be 13-fold more exposed to cardiovascular complications than non-severe, with an increased troponin I and low-density epicardial adipose tissue possibly reflecting the extent of the damage to the myocardium (Hui Hui et al., 2020; Li et al., 2020a), ultimately known to be associated with a worse prognosis (Clerkin et al., 2020).
Endothelial dysfunction is a feature of COVID-19 that lingers from the proliferative to the systemic phase. If the viral load is high, probably boosted by an intense viral shedding in the blood flow (Chang et al., 2020), it is very likely that some particles directly affect the endothelium (Escher et al., 2020; Sardu et al., 2020). High levels of pro-inflammatory cytokines are associated with endothelial engrossment (Finkel et al., 1992; Cheng et al., 1999) that could progress to vasculitis-like syndromes in the vessels of the brain, the kidneys, or the gastrointestinal tract (Varga et al., 2020). In severe COVID-19 patients, the Kawasaki disease has been observed (Jones et al., 2020) together with cutaneous signs, such as the “COVID-19 toes” (Mazzotta et al., 2020) or the chilblain-like lesions (Papa et al., 2020). We can therefore assume that the endothelial dysfunctions in COVID-19 arise from both type I damage and the nonischemic type II damage. The dysfunctional endothelium elicits two events that are part of the “two-activation theory of the endothelium” (Chang, 2019): the release of inflammatory cytokines triggers the activation of inflammatory pathways, whereas the activation of the platelet and exocytosis of aberrant coagulation factors trigger the activation of microthrombotic pathways. Viruses are known to directly affect hemostasis with their ability to agglutinate platelets, cause hemolysis, and lead to the formation of procoagulant complexes with antibodies (McKay and Margaretten, 1967; van Gorp et al., 1999). This latter mechanism may be advocated for SARS-CoV-2 by recent computational modeling that showed the possibility of the virus to cause hemoglobin derangements (Liu and Li, 2020). If this were the case, then the incorporation of the virus into Trojan horses would be plausible since white cells are known to commonly engulf hemoglobin in various tissues (Briguglio et al., 2020c). Aberrant coagulation is the underlying mechanism for ischemic heart disease, stroke, and venous thromboembolism, but it has been observed also in severe influenza pneumonia and SARS-CoV-1 (Chong et al., 2004; Yang and Tang, 2016). Similarly, the development of coagulopathy appears to be a noxious complication in severe level 2 and level 3 patients (Tang et al., 2020b). Clots can be found in kidney dialysis catheters, cause strokes, or leave portions of lungs bloodless. Spleen atrophy, hilar lymph node necrosis, and hepatomegaly were also observed (Li et al., 2020b). Thrombus formation was associated with increased mortality (Zhou et al., 2020), with most of level 3 patients meeting the criteria for the disseminated intravascular coagulation (i.e., consumptive of both platelets and clotting factors; Lillicrap, 2020). Once thrombi formed in capillary beds, the remodeling processes would be associated with leukocyte polarization and late recruitment of macrophages that are in charge of cell clearance and blood flow restoration through fibrinolytic processes (Pober and Sessa, 2014). This cascade of events (Virchow’s triad) is nevertheless necessary for endothelial wall restoration (Mukhopadhyay et al., 2019). However, the immune derangements in COVID-19 are likely to alter the activation of both immune cells and the fibrinolytic system. For instance, neutrophil extracellular traps (NETs) are useful to entrap viruses in weblike structures, thus facilitating cleavage by macrophages. If neutrophils are abnormally activated, aggregated NETs and their associated antimicrobial factors may be key determinants in capillary destruction (Cicco et al., 2020), vessel obstruction (Leppkes et al., 2020), and lung injury (Wang et al., 2020a). Similarly, impaired activation of the fibrinolytic system activation can recirculate the material and thus increase the risk of distant thrombi-derived ischemic damages (i.e., disseminated intravascular microthrombosis). Although it is not known if a plaque rupture is as dangerous as the plaque before rupture (Schoenhagen et al., 2002), if circulating thrombi halt in the small coronary vessels, they can certainly contribute to myocardial injury (Hendren et al., 2020). Thromboembolic events can occur in the lungs of infected patients (Ai et al., 2020; Danzi et al., 2020), further impairing gas exchange. The pulmonary damage leads to poor perfusion in the coronary vessels, misbalance of oxygen supply/demand, reduced activity of the mitochondrial electron transport chain, acidosis, and oxidative damage from reactive oxygen species (ROS; Wu et al., 2020b), whose accumulation is also known to be elicited by the cytokine storm (Bhaskar et al., 2020). Importantly, tissue hypoxia is known to induce metabolic reprogramming in cardiomyocytes, thus being critical for the progression of numerous cardiovascular diseases (Abe et al., 2017, 2019).
Alike myocardial injuries, not all COVID-19 patients who manifest alterations in the cardiac electrophysiology, such as ST-segment or ST-T wave abnormalities, show concomitant chest tomographic opacities (Bangalore et al., 2020). It is therefore possible that in predisposed individuals, the cardiovascular system is affected before the respiratory system, with electrical dysregulations being caused by circulating levels of pro-inflammatory cytokines, stress hormones, electrolytic imbalances, or drug cardiotoxicity (Chung et al., 1990; Yokoyama et al., 1993; Hasan, 2013; Driggin et al., 2020), but SARS-CoV-2 might directly damage nerve fibers. The myocardium is innervated by sympathetic and vagal parasympathetic nerve fibers that intersect in local plexuses, ganglia, and pacemaker regions. The wide expression of ACE2 in nerve tissues and the neurotrophic nature of SARS-CoV-2 might render the cardiac nerve fibers a favorite prey (Briguglio et al., 2020a). Severe arrhythmias are nevertheless life-threatening conditions that may occur in over 30% of level 2 patients (Ferrari, 2020) and in higher rates in patients of level 3 (Huang et al., 2020). The prevalence and severity of electrocardiographic changes could reflect the progression of myocardial damage (Guo et al., 2020), but it is very likely that it is associated with disease progression. Defects of electrical impulses from the sinoatrial node to the ventricles might arise as drug-induced disorders, thereby requiring careful assessments before defining the pharmacological treatment of COVID-19 (Yogasundaram et al., 2014; Borba et al., 2020). Pulmonary stretch receptors, C-fibers in the alveolar wall, baroreceptors in the carotid sinuses, extra-carotid cardiopulmonary baroreceptors together with widespread metaboreceptors are critical for integrating breathing cycle, heart rate, and vascular resistance during ventilatory and arterial pressure changes (Sant’Ambrogio, 1982; Schelegle, 2003; Timmers et al., 2003; Kougias et al., 2010; Anand et al., 2014). Type II damages are likely to disrupt these nervous components, in turn compromising the responsiveness to local stimuli, the impulse activity in afferent glossopharyngeal and vagal fibers, and the reflexive outflow (Burki and Lee, 2010; van Gestel and Steier, 2010). The central processing would therefore receive vitiated information from the periphery, which grounds the lack of adaptation of intrapulmonary vessels of COVID-19 patients (Chu et al., 2020), with the outputs being equally artificial. For instance, it has been suggested that the state of “silent hypoxemia” (i.e., depressed dyspnea response) that was observed in a large number of COVID-19 patients could be associated with defects in the carotid body, which is known to express ACE2 (Tobin et al., 2020). The consequent poor regulation of blood displacement in the microcirculation to the lungs and the brain may therefore mirror a vitiated baroreceptor reflex and hemodynamics, as was indeed observed in a COVID-19 patient (Ribas et al., 2020). Importantly, countless cardiovascular implications have been associated with the frequent renal involvement that was observed in level 2 and level 3 patients (Ronco et al., 2020). It is reasonable to believe that kidneys are subjected to both viral infiltration and several types of type II damages (Larsen et al., 2020; Su et al., 2020). Local polarization and subsequent activation of white blood cells easily disrupt the renin-angiotensin-aldosterone system (RAAS; Strutz and Zeisberg, 2006; Chen et al., 2016; Granot et al., 2017), in turn affecting the sympathetic noradrenergic and parasympathetic cholinergic neurotransmission (Miller and Arnold, 2019). Nevertheless, this intense extended autonomic system (EAS) activation was suggested to account for the multiple organ involvement of COVID-19 (Goldstein, 2020). Other than arrhythmias, level 3 patients were reported to be subjected to more frequent vasopressor support (Goyal et al., 2020). Some of these patients showed clinical involvement of the brainstem, especially of the respiratory center (Manganelli et al., 2020), which can imply a type I damage of SARS-Cov-2 via cerebrospinal fluid diffusion (Sun and Guan, 2020) or vagus nerve retrograde transport (Tassorelli et al., 2020). The autonomic center at the level of the lower medulla expresses ACE2 (Xia and Lazartigues, 2008), and it was shown to be highly infected by familial predecessors (Netland et al., 2008; Li et al., 2016). Non-epileptic seizures due to autonomic dysfunction were indeed reported in a COVID-19 patient (Logmin et al., 2020). The systemic inflammation, ischemic thrombotic/cardio-embolic injuries, or vasculitis at the level of capillary beds beneath the ependyma of the ventricle may similarly affect brainstem functions (Benghanem et al., 2020; Mirza and Das, 2020), being all hallmarks of the systemic phase of COVID-19. Endothelial damages in these critical areas are likely to affect afferent inputs from peripheral nerves, with subsequent lack of proper buffering of blood pressure fluctuations from the nucleus of the solitary tract (Cutsforth-Gregory and Benarroch, 2017). Clinically, the involvement of this medullary nucleus or of the dorsal motor nucleus of the vagus nerve might evoke nausea and vomiting frequently observed in COVID-19 patients (Goldstein, 2020). Electrical evaluation of both heart and brain activities, echocardiography, invasive hemodynamic monitoring, and serum brain natriuretic peptide can help clarify the cardiogenic component (Yufu et al., 2006; Mazeraud et al., 2016). Notably, any infection of central nervous tissues is accompanied by massive infiltration of leukocytes, such as dendritic cells from the perivascular region (Ludewig et al., 2016), that could serve as Trojan horses, further contributing to local affections.
Although it is not possible to state whether the cardiovascular implications observed in COVID-19 derive from previous conditions or depend solely on the coronavirus-associated damages, it is reasonable to assume a causal link. From a molecular point of view, the upregulation of ACE2 in some cardiovascular diseases, such as ischemic heart disease or diabetes mellitus (Zisman et al., 2003), may certainly expose the sick individuals who contract the coronavirus to poorer prognosis (Wu et al., 2020b). The subsequent binding and downregulation of ACE2 expression by SARS-CoV-2 further prevent the conversion of angiotensin II, thus worsening pulmonary and cardiovascular outcomes (Datta et al., 2020). Accordingly, a higher ACE/ACE2 ratio might be a predisposing cause of worse outcomes in COVID-19, having angiotensin II dire vasoconstriction and pro-oxidant and pro-inflammatory effects in contrast to angiotensin (1–7) that is a vasodilator, antioxidant, and anti-inflammatory (Pagliaro and Penna, 2020). Clinically, it has been proposed that the more disturbed was the hemodynamic homeostasis prior to SARS-CoV-2 infection, the more severe could be the symptoms during COVID-19 and the higher would be the risk of long-term cardiovascular consequences (Zheng et al., 2020). Concerning the Italian cohort of patients, 1 in 3 had preexisting ischemic cardiomyopathy or diabetes mellitus, 1 in 4 already suffered from atrial fibrillation, and 1 in 10 had a history of stroke (Onder et al., 2020). The preexisting myocardial metabolic imbalances or atherosclerotic lesion might have played a major role in myocardial oxygen imbalances and plaque instabilities upon the advent of the systemic phase of COVID-19 (Bonow et al., 2020). Numerous mechanical (e.g., repetitive deformations derived from the cardiac cycle) and biological forces (e.g., inflammation) are known to undermine the stability of subclinical plaques (Arroyo and Lee, 1999; Yao et al., 2019), and they all occur during infections (Madjid et al., 2007; Campbell and Rosenfeld, 2015). In addition, a preexisting poor cardiac functional reserve is more likely to lead to a sudden cardiac insufficiency in patients with COVID-19, giving also the drug-related heart damage deriving from COVID-19 treatment (Wu et al., 2020a; Zheng et al., 2020). In the past, patients with comorbid cardiovascular diseases, such as coronary artery disease or heart failure, have been already recognized to be at higher risk of contagion and exacerbation of symptoms during viral respiratory infections (Nguyen et al., 2016). Furthermore, long-term damage to the cardiovascular system has been documented in hospitalized patients recovering from pneumonia (Corrales-Medina et al., 2015), thus highlighting the cardiorespiratory deteriorations of COVID-19. It is therefore reasonable to say that any previous hypoxic/vascular condition, cardiac inflammation, or autonomic dysfunction has to be recognized as a risk factor for COVID-19 onset and cardiovascular disease progression in any individual infected with SARS-CoV-2. Notably, the highest case/fatality ratio in older adults might be due to the increasing prevalence of frailty and comorbid cardiovascular diseases in advanced age (Briguglio et al., 2020b; Moccia et al., 2020), which is known to be associated with increased ACE/ACE2 ratio (Wang et al., 2016). While it is still controversial whether RAAS inhibitors are to be administered to COVID-19 patients (Shibata et al., 2020), it is certain that the therapy with ACE inhibitors and angiotensin receptor blockers (ARBs) should definitely not be discontinued in patients with preexisting cardiovascular diseases (de Abajo et al., 2020; Vaduganathan et al., 2020).
COVID-19 is a multifaceted illness that comprises several implications of cardiological nature, including hypoxemia, sustained activation of the endothelium, nonischemic injuries, leukocyte polarization, thrombi-derived ischemic damages, dysrhythmias, and autonomic dysfunctions (Figure 1). Given these considerations, it is reasonable to conclude that the more severe autonomic dysfunctions of critically ill patients, the more complex would be the preservation of hemodynamic balances, thereby increasing the likelihood of fatal cardiovascular consequences in COVID-19 or chronic cardiovascular damages in those who survive. In these patients, long-term remote electrophysiological monitoring might be useful to provide care as necessary after discharge (Lakkireddy et al., 2020). Understanding these pathophysiological mechanisms in COVID-19 is crucial to promptly triage early risk factors, tailor treatment according to the patient’s severity and risk-benefit balance, and integrate evidence-based therapies depending on the disease phase (Carter et al., 2020; Mycroft-West et al., 2020). Drugs for COVID-19 have not been available yet (Kalil, 2020), but immunotherapies, extracorporeal membrane oxygenation, and low-molecular-weight heparin are being tested for effectiveness (Paranjpe et al., 2020; Perazzo et al., 2020; Ramanathan et al., 2020; Spyropoulos et al., 2020; Tang et al., 2020a; Thachil, 2020). In the meantime, cardiologists should stay up-to-date on recent and ongoing discoveries regarding COVID-19 and take a prominent role in the research studies or multidisciplinary teams.
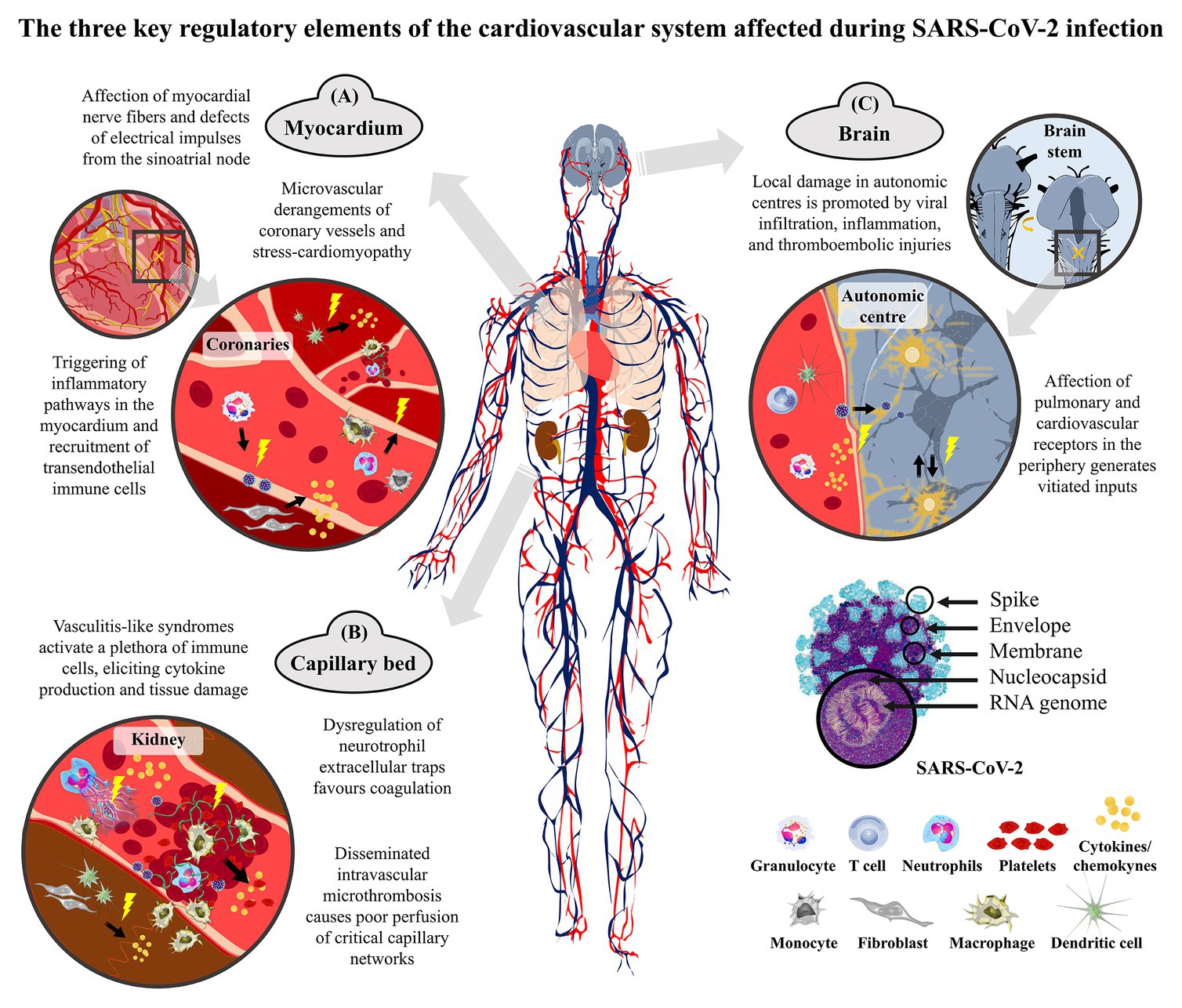
Figure 1. Representation of the cardiovascular derangements of coronavirus disease 2019 (COVID-19) that are due to either direct cellular damage of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or indirect consequences of the exaggerated host’s response. The severe acute respiratory syndrome coronavirus of 2019 (SARS-CoV-2) is a single-strand positive-sense RNA virus that spreads between humans mainly through the inhalation of respiratory droplets. Upon the collapse of the alveoli in the lungs, the virus can enter the bloodstream and distribute to systemic districts by cardiac pumping. It is likely that the transport in the blood is not as such, but carried by different types of white blood cells, such as T cells, granulocytes, and macrophages, which can therefore serve as vehicles. This Trojan route can guarantee the infiltration of the virus into normally inaccessible body districts. The systemic spread of the coronavirus elicits an exaggerated immune response in the most severe cases that strokes with hypoxic conditions. (A) In the myocardium, the excessive activation of the endothelial system upon viral damage and the enhanced inflammatory cell infiltration alter the coronary perfusion and the cardiac rhythm. Circulating monocytes and neutrophils infiltrate in the heart wall and parenchyma, with resident dendritic cells contributing to cytokine production and inflammatory/pro-fibrotic environment. (B) In the blood, the hyperactivation of both inflammatory and microthrombotic pathways leads to endothelium engrossment and coagulopathy. The activated vascular endothelium is targeted by neutrophils and monocytes, with thrombosis or bleedings being likely to occur because of the inflammation-derived imbalance between platelets, hypercoagulability, and altered fibrinolysis (fibrin in green). Thrombus-associated white blood cells produce inflammatory cytokines and proteases that contribute to local remodeling and fibroblast activation. Atherothombotic manifestations may also be promoted by dysregulation of neutrophil extracellular traps (NETs). (C) The heart is innervated by vagal postganglionic fibers from the cardio-inhibitory center and by the cardiac postganglionic fibers from the spinal cord arising from the cardio-acceleratory center of the medulla. Sympatho-inhibitory and cardio-inhibitory baroreflexes together with arterial metaboreflexes encompass inputs to neurons located in the dorsolateral nucleus of the solitary tract that integrate the vasomotor tone and the automatism of the sinus node. Damages to these reflexes disturb these central pathways and ultimately disrupt the heart beat nuclei, eventually leading to irrepressible dysautonomia.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
MB formulated the hypothesis and wrote the first draft of the manuscript. MP, FZ, AB, TC, FP, PP, MM, RG, GA, AI, GB, and MT revised the first draft and contributed to manuscript sections. All authors contributed to manuscript revision and read and approved the submitted version.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
None that interests colleagues, institutions, or agencies in supporting this hypothesis. Yet all authors thank all the health workers and volunteers in Italy at the front lines of the fight against this SARS-CoV-2.
Abe, H., Semba, H., and Takeda, N. (2017). The roles of hypoxia signaling in the pathogenesis of cardiovascular diseases. J. Atheroscler. Thromb. 24, 884–894. doi: 10.5551/jat.RV17009
Abe, H., Takeda, N., Isagawa, T., Semba, H., Nishimura, S., Morioka, M. S., et al. (2019). Macrophage hypoxia signaling regulates cardiac fibrosis via Oncostatin M. Nat. Commun. 10:2824. doi: 10.1038/s41467-019-10859-w
Ai, T., Yang, Z., Hou, H., Zhan, C., Chen, C., Lv, W., et al. (2020). Correlation of chest CT and RT-PCR testing in coronavirus disease 2019 (COVID-19) in China: a report of 1014 cases. Radiology 296, E32–E40. doi: 10.1148/radiol.2020200642
Anand, A., Srivastava, N., Barwad, P., Ramakrishnan, S., Roy, A., and Bhargava, B. (2014). Dyspnea in Eisenmenger syndrome and its amelioration by sildenafil: role of J receptors. Int. J. Cardiol. 174, 574–578. doi: 10.1016/j.ijcard.2014.04.131
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., and Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452. doi: 10.1038/s41591-020-0820-9
Arroyo, L. H., and Lee, R. T. (1999). Mechanisms of plaque rupture: mechanical and biologic interactions. Cardiovasc. Res. 41, 369–375. doi: 10.1016/s0008-6363(98)00308-3
Bangalore, S., Sharma, A., Slotwiner, A., Yatskar, L., Harari, R., Shah, B., et al. (2020). ST-segment elevation in patients with Covid-19 - a case series. N. Engl. J. Med. 382, 2478–2480. doi: 10.1056/NEJMc2009020
Basu-Ray, I. S., Almaddah, N. K., Adeboye, A., and Soos, M. P. (2020). Cardiac manifestations of coronavirus (COVID-19) [online]. Treasure Island (FL): StatPearls Publishing https://www.ncbi.nlm.nih.gov/books/NBK556152/ (Accessed June 3 2020).
Benghanem, S., Mazeraud, A., Azabou, E., Chhor, V., Shinotsuka, C. R., Claassen, J., et al. (2020). Brainstem dysfunction in critically ill patients. Crit. Care 24:5. doi: 10.1186/s13054-019-2718-9
Bhaskar, S., Sinha, A., Banach, M., Mittoo, S., Weissert, R., Kass, J. S., et al. (2020). Cytokine storm in COVID-19-Immunopathological mechanisms, clinical considerations, and therapeutic approaches: the REPROGRAM consortium position paper. Front. Immunol. 11:1648. doi: 10.3389/fimmu.2020.01648
Bonow, R. O., O’Gara, P. T., and Yancy, C. W. (2020). Cardiology and COVID-19. JAMA 324, 1131–1132. doi: 10.1001/jama.2020.15088
Borba, M. G. S., Val, F. F. A., Sampaio, V. S., Alexandre, M. A. A., Melo, G. C., Brito, M., et al. (2020). Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection: a randomized clinical trial. JAMA Netw. Open 3:e208857. doi: 10.1001/jamanetworkopen.2020.8857
Briguglio, M., Bona, A. R., Porta, M., Dell’osso, B., Pregliasco, F. E., and Banfi, G. (2020a). Disentangling the hypothesis of host dysosmia and SARS-CoV-2: the bait symptom that hides neglected neurophysiological routes. Front. Physiol. 11:671. doi: 10.3389/fphys.2020.00671
Briguglio, M., Gianola, S., Aguirre, M. F. I., Sirtori, P., Perazzo, P., Pennestri, F., et al. (2019). Nutritional support for enhanced recovery programs in orthopedics: future perspectives for implementing clinical practice. Nutr. Clin. Metab. 33, 190–198. doi: 10.1016/j.nupar.2019.04.002
Briguglio, M., Giorgino, R., Dell’Osso, B., Cesari, M., Porta, M., Lattanzio, F., et al. (2020b). Consequences for the elderly after COVID-19 isolation: FEaR (frail elderly amid restrictions). Front. Psychol. 11:565052. doi: 10.3389/fpsyg.2020.565052
Briguglio, M., Hrelia, S., Malaguti, M., De Vecchi, E., Lombardi, G., Banfi, G., et al. (2020c). Oral supplementation with sucrosomial ferric pyrophosphate plus L-ascorbic acid to ameliorate the martial status: a randomized controlled trial. Nutrients 12:386. doi: 10.3390/nu12020386
Briguglio, M., Pregliasco, F. E., Lombardi, G., Perazzo, P., and Banfi, G. (2020d). The Malnutritional status of the host as a virulence factor for new coronavirus SARS-CoV-2. Front. Med. 7:146. doi: 10.3389/fmed.2020.00146
Burki, N. K., and Lee, L. Y. (2010). Mechanisms of dyspnea. Chest 138, 1196–1201. doi: 10.1378/chest.10-0534
Campbell, L. A., and Rosenfeld, M. E. (2015). Infection and atherosclerosis development. Arch. Med. Res. 46, 339–350. doi: 10.1016/j.arcmed.2015.05.006
Carter, P., Anderson, M., and Mossialos, E. (2020). Health system, public health, and economic implications of managing COVID-19 from a cardiovascular perspective. Eur. Heart J. 41, 2516–2518. doi: 10.1093/eurheartj/ehaa342
Chan, K. W., Wong, V. T., and Tang, S. C. W. (2020). COVID-19: an update on the epidemiological, clinical, preventive and therapeutic evidence and guidelines of integrative Chinese-Western medicine for the management of 2019 novel coronavirus disease. Am. J. Chin. Med. 48, 737–762. doi: 10.1142/S0192415X20500378
Chang, J. C. (2019). Sepsis and septic shock: endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb. J. 17:10. doi: 10.1186/s12959-019-0198-4
Chang, L., Yan, Y., and Wang, L. (2020). Coronavirus disease 2019: coronaviruses and blood safety. Transfus. Med. Rev. 34, 75–80. doi: 10.1016/j.tmrv.2020.02.003
Chen, P. C., and Hsiao, C. H. (2004). Re: to KF, Tong JH, Chan PK, et al. tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in-situ hybridization study of fatal cases. J Pathol 2004; 202: 157-163. J. Pathol. 203, 729–730. doi: 10.1002/path.1575
Chen, K., Wang, J. M., Yuan, R., Yi, X., Li, L., Gong, W., et al. (2016). Tissue-resident dendritic cells and diseases involving dendritic cell malfunction. Int. Immunopharmacol. 34, 1–15. doi: 10.1016/j.intimp.2016.02.007
Cheng, X. S., Shimokawa, H., Momii, H., Oyama, J., Fukuyama, N., Egashira, K., et al. (1999). Role of superoxide anion in the pathogenesis of cytokine-induced myocardial dysfunction in dogs in vivo. Cardiovasc. Res. 42, 651–659. doi: 10.1016/S0008-6363(98)00317-4
Chong, P. Y., Chui, P., Ling, A. E., Franks, T. J., Tai, D. Y., Leo, Y. S., et al. (2004). Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: challenges in determining a SARS diagnosis. Arch. Pathol. Lab. Med. 128, 195–204. doi: 10.1043/1543-2165(2004)128<195:AODDTS>2.0.CO;2
Chu, H., Chan, J. F., Wang, Y., Yuen, T. T., Chai, Y., Hou, Y., et al. (2020). Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 71, 1400–1409. doi: 10.1093/cid/ciaa410
Chung, M. K., Gulick, T. S., Rotondo, R. E., Schreiner, G. F., and Lange, L. G. (1990). Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes. Impairment of signal transduction. Circ. Res. 67, 753–763. doi: 10.1161/01.res.67.3.753
Cicco, S., Cicco, G., Racanelli, V., and Vacca, A. (2020). Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediat. Inflamm. 2020:7527953. doi: 10.1155/2020/7527953
Clerkin, K. J., Fried, J. A., Raikhelkar, J., Sayer, G., Griffin, J. M., Masoumi, A., et al. (2020). COVID-19 and cardiovascular disease. Circulation 141, 1648–1655. doi: 10.1161/CIRCULATIONAHA.120.046941
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (2020). The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544. doi: 10.1038/s41564-020-0695-z
Corrales-Medina, V. F., Alvarez, K. N., Weissfeld, L. A., Angus, D. C., Chirinos, J. A., Chang, C. C., et al. (2015). Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA 313, 264–274. doi: 10.1001/jama.2014.18229
Cutsforth-Gregory, J. K., and Benarroch, E. E. (2017). Nucleus of the solitary tract, medullary reflexes, and clinical implications. Neurology 88, 1187–1196. doi: 10.1212/WNL.0000000000003751
Danzi, G. B., Loffi, M., Galeazzi, G., and Gherbesi, E. (2020). Acute pulmonary embolism and COVID-19 pneumonia: a random association? Eur. Heart J. 41:1858. doi: 10.1093/eurheartj/ehaa254
Datta, P. K., Liu, F., Fischer, T., Rappaport, J., and Qin, X. (2020). SARS-CoV-2 pandemic and research gaps: understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 10, 7448–7464. doi: 10.7150/thno.48076
de Abajo, F. J., Rodriguez-Martin, S., Lerma, V., Mejia-Abril, G., Aguilar, M., Garcia-Luque, A., et al. (2020). Use of renin-angiotensin-aldosterone system inhibitors and risk of COVID-19 requiring admission to hospital: a case-population study. Lancet 395, 1705–1714. doi: 10.1016/S0140-6736(20)31030-8
Driggin, E., Madhavan, M. V., Bikdeli, B., Chuich, T., Laracy, J., Biondi-Zoccai, G., et al. (2020). Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J. Am. Coll. Cardiol. 75, 2352–2371. doi: 10.1016/j.jacc.2020.03.031
Eriksson, U., Ricci, R., Hunziker, L., Kurrer, M. O., Oudit, G. Y., Watts, T. H., et al. (2003). Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 9, 1484–1490. doi: 10.1038/nm960
Escher, R., Breakey, N., and Lammle, B. (2020). Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 190:62. doi: 10.1016/j.thromres.2020.04.014
Ferrari, F. (2020). COVID-19: updated data and its relation to the cardiovascular system. Arq. Bras. Cardiol. 114, 823–826. doi: 10.36660/abc.20200215
Finkel, M. S., Oddis, C. V., Jacob, T. D., Watkins, S. C., Hattler, B. G., and Simmons, R. L. (1992). Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 257, 387–389. doi: 10.1126/science.1631560
Fried, J. A., Ramasubbu, K., Bhatt, R., Topkara, V. K., Clerkin, K. J., Horn, E., et al. (2020). The variety of cardiovascular presentations of COVID-19. Circulation 141, 1930–1936. doi: 10.1161/CIRCULATIONAHA.120.047164
Goldstein, D. S. (2020). The extended autonomic system, dyshomeostasis, and COVID-19. Clin. Auton. Res. 30, 299–315. doi: 10.1007/s10286-020-00714-0
Goyal, P., Choi, J. J., Pinheiro, L. C., Schenck, E. J., Chen, R., Jabri, A., et al. (2020). Clinical characteristics of Covid-19 in New York City. N. Engl. J. Med. 382, 2372–2374. doi: 10.1056/NEJMc2010419
Granot, T., Senda, T., Carpenter, D. J., Matsuoka, N., Weiner, J., Gordon, C. L., et al. (2017). Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity 46, 504–515. doi: 10.1016/j.immuni.2017.02.019
Gu, J., Gong, E., Zhang, B., Zheng, J., Gao, Z., Zhong, Y., et al. (2005). Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 202, 415–424. doi: 10.1084/jem.20050828
Guo, T., Fan, Y., Chen, M., Wu, X., Zhang, L., He, T., et al. (2020). Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 811–818. doi: 10.1001/jamacardio.2020.1017
Harapan, H., Itoh, N., Yufika, A., Winardi, W., Keam, S., Te, H., et al. (2020). Coronavirus disease 2019 (COVID-19): a literature review. J. Infect. Public Health 13, 667–673. doi: 10.1016/j.jiph.2020.03.019
Hasan, W. (2013). Autonomic cardiac innervation: development and adult plasticity. Organogenesis 9, 176–193. doi: 10.4161/org.24892
Hendren, N. S., Drazner, M. H., Bozkurt, B., and Cooper, L. T. Jr. (2020). Description and proposed Management of the Acute COVID-19 cardiovascular syndrome. Circulation 141, 1903–1914. doi: 10.1161/CIRCULATIONAHA.120.047349
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kruger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8. doi: 10.1016/j.cell.2020.02.052
Hu, H., Ma, F., Wei, X., and Fang, Y. (2020). Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur. Heart J. doi: 10.1093/eurheartj/ehaa190 [Epub ahead of print]
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Hui Hui, Y. Z., Yang, X., Wang, X., He, B., Li, L., Li, H., et al. (2020). Clinical and radiographic features of cardiac injury in patients with 2019 novel coronavirus pneumonia. medRxiv [Preprint]. doi: 10.1101/2020.02.24.20027052.
Inciardi, R. M., Lupi, L., Zaccone, G., Italia, L., Raffo, M., Tomasoni, D., et al. (2020). Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 819–824. doi: 10.1001/jamacardio.2020.1096
Jones, V. G., Mills, M., Suarez, D., Hogan, C. A., Yeh, D., Bradley Segal, J., et al. (2020). COVID-19 and Kawasaki disease: novel virus and novel case. Hosp. Pediatr. 10, 537–540. doi: 10.1542/hpeds.2020-0123
Kabbani, N., and Olds, J. L. (2020). Does COVID19 infect the brain? If so, smokers might be at a higher risk. Mol. Pharmacol. 97, 351–353. doi: 10.1124/molpharm.120.000014
Kalil, A. C. (2020). Treating COVID-19-off-label drug use, compassionate use, and randomized clinical trials during pandemics. JAMA 323, 1897–1898. doi: 10.1001/jama.2020.4742
Kougias, P., Weakley, S. M., Yao, Q., Lin, P. H., and Chen, C. (2010). Arterial baroreceptors in the management of systemic hypertension. Med. Sci. Monit. 16, RA1–RA8.
Lakkireddy, D. R., Chung, M. K., Gopinathannair, R., Patton, K. K., Gluckman, T. J., Turagam, M., et al. (2020). Guidance for cardiac electrophysiology during the COVID-19 pandemic from the Heart Rhythm Society COVID-19 task force; electrophysiology section of the American College of Cardiology; and the electrocardiography and arrhythmias Committee of the Council on clinical cardiology, American Heart Association. Heart Rhythm. 17, e233–e241. doi: 10.1016/j.hrthm.2020.03.028
Larsen, C. P., Bourne, T. D., Wilson, J. D., Saqqa, O., and Sharshir, M. A. (2020). Collapsing glomerulopathy in a patient with coronavirus disease 2019 (COVID-19). Kidney Int. Rep. 5, 935–939. doi: 10.1016/j.ekir.2020.04.002
Leppkes, M., Knopf, J., Naschberger, E., Lindemann, A., Singh, J., Herrmann, I., et al. (2020). Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 58:102925. doi: 10.1016/j.ebiom.2020.102925
Leung, J. M., Yang, C. X., Tam, A., Shaipanich, T., Hackett, T. L., Singhera, G. K., et al. (2020). ACE-2 expression in the small airway epithelia of smokers and COPD patients: implications for COVID-19. Eur. Respir. J. 55:2000688. doi: 10.1183/13993003.00688-2020
Li, H., Liu, L., Zhang, D., Xu, J., Dai, H., Tang, N., et al. (2020b). SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet 395, 1517–1520. doi: 10.1016/S0140-6736(20)30920-X
Li, G., Liu, Y., Zhu, Y., Liu, A., Xu, Y., Li, X., et al. (2013). ACE2 activation confers endothelial protection and attenuates neointimal lesions in prevention of severe pulmonary arterial hypertension in rats. Lung 191, 327–336. doi: 10.1007/s00408-013-9470-8
Li, K., Wohlford-Lenane, C., Perlman, S., Zhao, J., Jewell, A. K., Reznikov, L. R., et al. (2016). Middle East respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human Dipeptidyl peptidase 4. J. Infect. Dis. 213, 712–722. doi: 10.1093/infdis/jiv499
Li, B., Yang, J., Zhao, F., Zhi, L., Wang, X., Liu, L., et al. (2020a). Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 109, 531–538. doi: 10.1007/s00392-020-01626-9
Lillicrap, D. (2020). Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J. Thromb. Haemost. 18, 786–787. doi: 10.1111/jth.14781
Lin, L., Lu, L., Cao, W., and Li, T. (2020). Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerg. Microbes Infect. 9, 727–732. doi: 10.1080/22221751.2020.1746199
Lindner, D., Zietsch, C., Tank, J., Sossalla, S., Fluschnik, N., Hinrichs, S., et al. (2014). Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol. 109:428. doi: 10.1007/s00395-014-0428-7
Lippi, G., Plebani, M., and Henry, B. M. (2020). Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin. Chim. Acta 506, 145–148. doi: 10.1016/j.cca.2020.03.022
Liu, W., and Li, H. (2020). COVID-19: Attacks the 1-Beta Chain of Hemoglobin and Captures the Porphyrin to Inhibit Human Heme Metabolism. ChemRxiv [Preprint]. doi: 10.26434/chemrxiv.11938173.v9.
Logmin, K., Karam, M., Schichel, T., Harmel, J., and Wojtecki, L. (2020). Non-epileptic seizures in autonomic dysfunction as the initial symptom of COVID-19. J. Neurol. 267, 2490–2491. doi: 10.1007/s00415-020-09904-2
Ludewig, P., Gallizioli, M., Urra, X., Behr, S., Brait, V. H., Gelderblom, M., et al. (2016). Dendritic cells in brain diseases. Biochim. Biophys. Acta 1862, 352–367. doi: 10.1016/j.bbadis.2015.11.003
Madjid, M., Vela, D., Khalili-Tabrizi, H., Casscells, S. W., and Litovsky, S. (2007). Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries: clues to the triggering effect of acute infections on acute coronary syndromes. Tex. Heart Inst. J. 34, 11–18.
Manganelli, F., Vargas, M., Iovino, A., Iacovazzo, C., Santoro, L., and Servillo, G. (2020). Brainstem involvement and respiratory failure in COVID-19. Neurol. Sci. 41, 1663–1665. doi: 10.1007/s10072-020-04487-2
Mason, R. J. (2020). Pathogenesis of COVID-19 from a cell biologic perspective. Eur. Respir. J. 55:2000607. doi: 10.1183/13993003.00607-2020
Matsushita, K., Marchandot, B., Jesel, L., Ohlmann, P., and Morel, O. (2020). Impact of COVID-19 on the cardiovascular system: a review. J. Clin. Med. 9:1407. doi: 10.3390/jcm9051407
Mazeraud, A., Pascal, Q., Verdonk, F., Heming, N., Chretien, F., and Sharshar, T. (2016). Neuroanatomy and physiology of brain dysfunction in sepsis. Clin. Chest Med. 37, 333–345. doi: 10.1016/j.ccm.2016.01.013
Mazzotta, F., Troccoli, T., and Bonifazi, E. (2020). A new vasculitis at the time of COVID-19. Eur. J. Pediat. Dermatol. 30, 75–78. doi: 10.26326/2281-9649.30.2.2103
McKay, D. G., and Margaretten, W. (1967). Disseminated intravascular coagulation in virus diseases. Arch. Intern. Med. 120, 129–152. doi: 10.1001/archinte.1967.00300020001001
Mehra, M. R., Desai, S. S., Kuy, S., Henry, T. D., and Patel, A. N. (2020). Cardiovascular disease, drug therapy, and mortality in Covid-19. N. Engl. J. Med. 382:e102. doi: 10.1056/NEJMoa2007621
Meyer, P., Degrauwe, S., Van Delden, C., Ghadri, J. R., and Templin, C. (2020). Typical takotsubo syndrome triggered by SARS-CoV-2 infection. Eur. Heart J. 41:1860. doi: 10.1093/eurheartj/ehaa306
Miller, A. J., and Arnold, A. C. (2019). The renin-angiotensin system in cardiovascular autonomic control: recent developments and clinical implications. Clin. Auton. Res. 29, 231–243. doi: 10.1007/s10286-018-0572-5
Mirza, M., and Das, J. M. (2020). Neuroanatomy, Area Postrema [Online]. Treasure Island (FL): StatPearls Publishing: StatPearls [Internet]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK544249/ [Accessed 2020 April 29 2020].
Moccia, F., Gerbino, A., Lionetti, V., Miragoli, M., Munaron, L. M., Pagliaro, P., et al. (2020). COVID-19-associated cardiovascular morbidity in older adults: a position paper from the Italian society of cardiovascular researches. Geroscience 42, 1021–1049. doi: 10.1007/s11357-020-00198-w
Mukhopadhyay, S., Johnson, T. A., Duru, N., Buzza, M. S., Pawar, N. R., Sarkar, R., et al. (2019). Fibrinolysis and inflammation in venous thrombus resolution. Front. Immunol. 10:1348. doi: 10.3389/fimmu.2019.01348
Mycroft-West, C., Su, D., Elli, S., Guimond, S., Miller, G., Turnbull, J., et al. (2020). The 2019 coronavirus (SARS-CoV-2) surface protein (Spike) S1 Receptor Binding Domain undergoes conformational change upon heparin binding. bioRxiv [Preprint]. doi: 10.1101/2020.02.29.971093.
Netland, J., Meyerholz, D. K., Moore, S., Cassell, M., and Perlman, S. (2008). Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 82, 7264–7275. doi: 10.1128/JVI.00737-08
Nguyen, J. L., Yang, W., Ito, K., Matte, T. D., Shaman, J., and Kinney, P. L. (2016). Seasonal influenza infections and cardiovascular disease mortality. JAMA Cardiol. 1, 274–281. doi: 10.1001/jamacardio.2016.0433
Onder, G., Rezza, G., and Brusaferro, S. (2020). Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA 323, 1775–1776. doi: 10.1001/jama.2020.4683
Oudit, G. Y., Kassiri, Z., Jiang, C., Liu, P. P., Poutanen, S. M., Penninger, J. M., et al. (2009). SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 39, 618–625. doi: 10.1111/j.1365-2362.2009.02153.x
Pagliaro, P., and Penna, C. (2020). ACE/ACE2 ratio: a key also in 2019 coronavirus disease (Covid-19)? Front. Med. 7:335. doi: 10.3389/fmed.2020.00335
Papa, A., Salzano, A. M., Di Dato, M. T., and Varrassi, G. (2020). Images in practice: painful cutaneous Vasculitis in a SARS-Cov-2 IgG-positive child. Pain Ther. 21, 1–3. doi: 10.1007/s40122-020-00174-4
Paranjpe, I., Fuster, V., Lala, A., Russak, A., Glicksberg, B. S., Levin, M. A., et al. (2020). Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. J. Am. Coll. Cardiol. 76, 122–124. doi: 10.1016/j.jacc.2020.05.001
Park, M. D. (2020). Macrophages: a Trojan horse in COVID-19? Nat. Rev. Immunol. 20:351. doi: 10.1038/s41577-020-0317-2
Perazzo, P., Giorgino, R., Briguglio, M., Zuffada, M., Accetta, R., Mangiavini, L., et al. (2020). From standard to escalated anticoagulant prophylaxis in fractured older adults with SARS-CoV-2 undergoing accelerated orthopedic surgery. Front. Med. doi: 10.3389/fmed.2020.566770 [Epub ahead of print]
Peteranderl, C., and Herold, S. (2017). The impact of the interferon/TNF-related apoptosis-inducing ligand signaling axis on disease progression in respiratory viral infection and beyond. Front. Immunol. 8:313. doi: 10.3389/fimmu.2017.00313
Pilling, D., Fan, T., Huang, D., Kaul, B., and Gomer, R. H. (2009). Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS One 4:e7475. doi: 10.1371/journal.pone.0007475
Pilling, D., and Gomer, R. H. (2012). Differentiation of circulating monocytes into fibroblast-like cells. Methods Mol. Biol. 904, 191–206. doi: 10.1007/978-1-61779-943-3_16
Pober, J. S., and Sessa, W. C. (2014). Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 7:a016345. doi: 10.1101/cshperspect.a016345
Ramanathan, K., Antognini, D., Combes, A., Paden, M., Zakhary, B., Ogino, M., et al. (2020). Planning and provision of ECMO services for severe ARDS during the COVID-19 pandemic and other outbreaks of emerging infectious diseases. Lancet Respir. Med. 8, 518–526. doi: 10.1016/S2213-2600(20)30121-1
Remy, K. E., Brakenridge, S. C., Francois, B., Daix, T., Deutschman, C. S., Monneret, G., et al. (2020). Immunotherapies for COVID-19: lessons learned from sepsis. Lancet Respir. Med. 8, 946–949. doi: 10.1016/S2213-2600(20)30217-4
Ribas, V. R., Ribas, R. M. G., Barros, M. G. S., Ribas, K., Neto, N. A., Barros, M. G. S., et al. (2020). Hemodynamics, baroreflex index and blood biomarkers of a patient who died after being affected by COVID-19: case report. Hematol. Transfus Cell Ther. 42, 206–211. doi: 10.1016/j.htct.2020.06.004
Rokni, M., Ghasemi, V., and Tavakoli, Z. (2020). Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: comparison with SARS and MERS. Rev. Med. Virol. 30:e2107. doi: 10.1002/rmv.2107
Ronco, C., Reis, T., and Husain-Syed, F. (2020). Management of acute kidney injury in patients with COVID-19. Lancet Respir. Med. 8, P738–P742. doi: 10.1016/S2213-2600(20)30229-0
Sant’Ambrogio, G. (1982). Information arising from the tracheobronchial tree of mammals. Physiol. Rev. 62, 531–569. doi: 10.1152/physrev.1982.62.2.531
Sardu, C., Gambardella, J., Morelli, M. B., Wang, X., Marfella, R., and Santulli, G. (2020). Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J. Clin. Med. 9:1417. doi: 10.3390/jcm9051417
Sattar, N., McInnes, I. B., and McMurray, J. J. V. (2020). Obesity a risk factor for severe COVID-19 infection: multiple potential mechanisms. Circulation 142, 4–6. doi: 10.1161/CIRCULATIONAHA.120.047659
Schelegle, E. S. (2003). Functional morphology and physiology of slowly adapting pulmonary stretch receptors. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 270, 11–16. doi: 10.1002/ar.a.10004
Schoenhagen, P., Tuzcu, E. M., and Ellis, S. G. (2002). Plaque vulnerability, plaque rupture, and acute coronary syndromes: (multi)-focal manifestation of a systemic disease process. Circulation 106, 760–762. doi: 10.1161/01.cir.0000025708.36290.05
Shi, Y., Wang, Y., Shao, C., Huang, J., Gan, J., Huang, X., et al. (2020). COVID-19 infection: the perspectives on immune responses. Cell Death Differ. 27, 1451–1454. doi: 10.1038/s41418-020-0530-3
Shibata, S., Arima, H., Asayama, K., Hoshide, S., Ichihara, A., Ishimitsu, T., et al. (2020). Hypertension and related diseases in the era of COVID-19: a report from the Japanese Society of Hypertension Task Force on COVID-19. Hypertens. Res. 43, 1028–1046. doi: 10.1038/s41440-020-0515-0
Siddiqi, H. K., and Mehra, M. R. (2020). COVID-19 illness in native and immunosuppressed states: a clinical-therapeutic staging proposal. J. Heart Lung Transplant. 39, 405–407. doi: 10.1016/j.healun.2020.03.012
Spyropoulos, A. C., Ageno, W., and Barnathan, E. S. (2020). Hospital-based use of thromboprophylaxis in patients with COVID-19. Lancet 395:e75. doi: 10.1016/S0140-6736(20)30926-0
Stefanini, G. G., Montorfano, M., Trabattoni, D., Andreini, D., Ferrante, G., Ancona, M., et al. (2020). ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation 141, 2113–2116. doi: 10.1161/CIRCULATIONAHA.120.047525
Strutz, F., and Zeisberg, M. (2006). Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 17, 2992–2998. doi: 10.1681/ASN.2006050420
Su, H., Yang, M., Wan, C., Yi, L. X., Tang, F., Zhu, H. Y., et al. (2020). Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 98, 219–227. doi: 10.1016/j.kint.2020.04.003
Sun, T., and Guan, J. (2020). Novel coronavirus and central nervous system. Eur. J. Neurol. doi: 10.1111/ene.14227 [Epub ahead of print]
Sungnak, W., Huang, N., Becavin, C., Berg, M., Queen, R., Litvinukova, M., et al. (2020). SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 26, 681–687. doi: 10.1038/s41591-020-0868-6
Tang, N., Bai, H., Chen, X., Gong, J., Li, D., and Sun, Z. (2020a). Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 18, 1094–1099. doi: 10.1111/jth.14817
Tang, N., Li, D., Wang, X., and Sun, Z. (2020b). Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 18, 844–847. doi: 10.1111/jth.14768
Tassorelli, C., Mojoli, F., Baldanti, F., Bruno, R., and Benazzo, M. (2020). COVID-19: what if the brain had a role in causing the deaths? Eur. J. Neurol. 27, e41–e42. doi: 10.1111/ene.14275
Tavazzi, G., Pellegrini, C., Maurelli, M., Belliato, M., Sciutti, F., Bottazzi, A., et al. (2020). Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 22, 911–915. doi: 10.1002/ejhf.1828
Thachil, J. (2020). The versatile heparin in COVID-19. J. Thromb. Haemost. 18, 1020–1022. doi: 10.1111/jth.14821
Thachil, J., Tang, N., Gando, S., Falanga, A., Cattaneo, M., Levi, M., et al. (2020). ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 18, 1023–1026. doi: 10.1111/jth.14810
Timmers, H. J., Wieling, W., Karemaker, J. M., and Lenders, J. W. (2003). Denervation of carotid baro- and chemoreceptors in humans. J. Physiol. 553, 3–11. doi: 10.1113/jphysiol.2003.052415
Tobin, M. J., Laghi, F., and Jubran, A. (2020). Why COVID-19 silent hypoxemia is baffling to physicians. Am. J. Respir. Crit. Care Med. 202, 356–360. doi: 10.1164/rccm.202006-2157CP
Vaduganathan, M., Vardeny, O., Michel, T., McMurray, J. J. V., Pfeffer, M. A., and Solomon, S. D. (2020). Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N. Engl. J. Med. 382, 1653–1659. doi: 10.1056/NEJMsr2005760
Van der Borght, K., and Lambrecht, B. N. (2018). Heart macrophages and dendritic cells in sickness and in health: a tale of a complicated marriage. Cell. Immunol. 330, 105–113. doi: 10.1016/j.cellimm.2018.03.011
van Gestel, A. J., and Steier, J. (2010). Autonomic dysfunction in patients with chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 2, 215–222. doi: 10.3978/j.issn.2072-1439.2010.02.04.5
van Gorp, E. C., Suharti, C., ten Cate, H., Dolmans, W. M., van der Meer, J. W., ten Cate, J. W., et al. (1999). Review: infectious diseases and coagulation disorders. J. Infect. Dis. 180, 176–186. doi: 10.1086/314829
van Nieuwenhoven, F. A., and Turner, N. A. (2013). The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 58, 182–188. doi: 10.1016/j.vph.2012.07.003
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A. S., et al. (2020). Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418. doi: 10.1016/S0140-6736(20)30937-5
Wang, X. L., Iwanami, J., Min, L. J., Tsukuda, K., Nakaoka, H., Bai, H. Y., et al. (2016). Deficiency of angiotensin-converting enzyme 2 causes deterioration of cognitive function. NPJ Aging Mech. Dis. 2:16024. doi: 10.1038/npjamd.2016.24
Wang, J., Li, Q., Yin, Y., Zhang, Y., Cao, Y., Lin, X., et al. (2020a). Excessive neutrophils and neutrophil extracellular traps in COVID-19. Front. Immunol. 11:2063. doi: 10.3389/fimmu.2020.02063
Wang, W., Xu, Y., Gao, R., Lu, R., Han, K., Wu, G., et al. (2020b). Detection of SARS-CoV-2 in different types of clinical specimens. JAMA. 323, 1843–1844. doi: 10.1001/jama.2020.3786
Wu, Z., and McGoogan, J. M. (2020). Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242. doi: 10.1001/jama.2020.2648
Wu, L., O’Kane, A. M., Peng, H., Bi, Y., Motriuk-Smith, D., and Ren, J. (2020b). SARS-CoV-2 and cardiovascular complications: from molecular mechanisms to pharmaceutical management. Biochem. Pharmacol. 178:114114. doi: 10.1016/j.bcp.2020.114114
Wu, C. I., Postema, P. G., Arbelo, E., Behr, E. R., Bezzina, C. R., Napolitano, C., et al. (2020a). SARS-CoV-2, COVID-19, and inherited arrhythmia syndromes. Heart Rhythm. 17, 1456–1462. doi: 10.1016/j.hrthm.2020.03.024
Xia, H., and Lazartigues, E. (2008). Angiotensin-converting enzyme 2 in the brain: properties and future directions. J. Neurochem. 107, 1482–1494. doi: 10.1111/j.1471-4159.2008.05723.x
Xiong, T. Y., Redwood, S., Prendergast, B., and Chen, M. (2020). Coronaviruses and the cardiovascular system: acute and long-term implications. Eur. Heart J. 41, 1798–1800. doi: 10.1093/eurheartj/ehaa231
Xu, J., Li, Y., Gan, F., Du, Y., and Yao, Y. (2020a). Salivary glands: potential reservoirs for COVID-19 asymptomatic infection. J. Dent. Res. 99:989. doi: 10.1177/0022034520918518
Xu, Z., Shi, L., Wang, Y., Zhang, J., Huang, L., Zhang, C., et al. (2020b). Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422. doi: 10.1016/S2213-2600(20)30076-X
Yang, W., Sirajuddin, A., Zhang, X., Liu, G., Teng, Z., Zhao, S., et al. (2020). The role of imaging in 2019 novel coronavirus pneumonia (COVID-19). Eur. Radiol. 30, 4874–4882. doi: 10.1007/s00330-020-06827-4
Yang, Y., and Tang, H. (2016). Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 13, 432–442. doi: 10.1038/cmi.2016.1
Yao, B. C., Meng, L. B., Hao, M. L., Zhang, Y. M., Gong, T., and Guo, Z. G. (2019). Chronic stress: a critical risk factor for atherosclerosis. J. Int. Med. Res. 47, 1429–1440. doi: 10.1177/0300060519826820
Yogasundaram, H., Putko, B. N., Tien, J., Paterson, D. I., Cujec, B., Ringrose, J., et al. (2014). Hydroxychloroquine-induced cardiomyopathy: case report, pathophysiology, diagnosis, and treatment. Can. J. Cardiol. 30, 1706–1715. doi: 10.1016/j.cjca.2014.08.016
Yokoyama, T., Vaca, L., Rossen, R. D., Durante, W., Hazarika, P., and Mann, D. L. (1993). Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J. Clin. Invest. 92, 2303–2312. doi: 10.1172/JCI116834
Yufu, K., Takahashi, N., Nakagawa, M., Hara, M., Saikawa, T., and Yoshimatsu, H. (2006). Brain natriuretic peptide and cardiac autonomic function in type 2 diabetic patients. Diabetes Res. Clin. Pract. 72, 12–19. doi: 10.1016/j.diabres.2005.08.011
Zeng, J. H., Liu, Y. X., Yuan, J., Wang, F. X., Wu, W. B., Li, J. X., et al. (2020). First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection 48, 773–777. doi: 10.1007/s15010-020-01424-5
Zheng, Y. Y., Ma, Y. T., Zhang, J. Y., and Xie, X. (2020). COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 17, 259–260. doi: 10.1038/s41569-020-0360-5
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., et al. (2020). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062. doi: 10.1016/S0140-6736(20)30566-3
Zisman, L. S., Keller, R. S., Weaver, B., Lin, Q., Speth, R., Bristow, M. R., et al. (2003). Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme homologue ACE2. Circulation 108, 1707–1712. doi: 10.1161/01.CIR.0000094734.67990.99
Keywords: cardiovascular system, coronavirus, SARS-CoV-2, COVID-19, infections, virulence, host-pathogen interactions, quality of health care
Citation: Briguglio M, Porta M, Zuffada F, Bona AR, Crespi T, Pino F, Perazzo P, Mazzocchi M, Giorgino R, De Angelis G, Ielasi A, De Blasio G and Turiel M (2020) SARS-CoV-2 Aiming for the Heart: A Multicenter Italian Perspective About Cardiovascular Issues in COVID-19. Front. Physiol. 11:571367. doi: 10.3389/fphys.2020.571367
Edited by:
Emmanuelle Vidal-Petiot, Assistance Publique Hopitaux De Paris, FranceReviewed by:
Pasquale Pagliaro, University of Turin, ItalyCopyright © 2020 Briguglio, Porta, Zuffada, Bona, Crespi, Pino, Perazzo, Mazzocchi, Giorgino, De Angelis, Ielasi, De Blasio and Turiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Briguglio, bWF0dGVvLmJyaWd1Z2xpb0BncnVwcG9zYW5kb25hdG8uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.