 Lydia Hering
Lydia Hering Masudur Rahman
Masudur Rahman Sebastian A. Potthoff
Sebastian A. Potthoff Lars C. Rump
Lars C. Rump Johannes Stegbauer
Johannes Stegbauer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 09 November 2020
Sec. Renal Physiology and Pathophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.566871
This article is part of the Research TopicHypertension and Chronic Kidney Injury or FailureView all 10 articles
The kidney is extensively innervated by sympathetic nerves playing an important role in the regulation of blood pressure homeostasis. Sympathetic nerve activity is ultimately controlled by the central nervous system (CNS). Norepinephrine, the main sympathetic neurotransmitter, is released at prejunctional neuroeffector junctions in the kidney and modulates renin release, renal vascular resistance, sodium and water handling, and immune cell response. Under physiological conditions, renal sympathetic nerve activity (RSNA) is modulated by peripheral mechanisms such as the renorenal reflex, a complex interaction between efferent sympathetic nerves, central mechanism, and afferent sensory nerves. RSNA is increased in hypertension and, therefore, critical for the perpetuation of hypertension and the development of hypertensive kidney disease. Renal sympathetic neurotransmission is not only regulated by RSNA but also by prejunctional α2-adrenoceptors. Prejunctional α2-adrenoceptors serve as autoreceptors which, when activated by norepinephrine, inhibit the subsequent release of norepinephrine induced by a sympathetic nerve impulse. Deletion of α2-adrenoceptors aggravates hypertension ultimately by modulating renal pressor response and sodium handling. α2-adrenoceptors are also expressed in the vasculature, renal tubules, and immune cells and exert thereby effects related to vascular tone, sodium excretion, and inflammation. In the present review, we highlight the role of α2-adrenoceptors on renal sympathetic neurotransmission and its impact on hypertension. Moreover, we focus on physiological and pathophysiological functions mediated by non-adrenergic α2-adrenoceptors. In detail, we discuss the effects of sympathetic norepinephrine release and α2-adrenoceptor activation on renal sodium transporters, on renal vascular tone, and on immune cells in the context of hypertension and kidney disease.
Hypertension is the second common cause for end stage renal disease (ESRD) and one of the major risk factors for morbidity and mortality worldwide (GBD 2016 Risk Factors Collaborators, 2017). The kidney is a master regulator of blood pressure homeostasis by regulating vascular tone, as well as sodium and water handling. Renal dysfunction, such as an increase in sodium and water retention, renin release, or renal vascular resistance, causes hypertension and, subsequently in the long-term run chronic kidney damage. The kidney is extensively innervated by sympathetic nerves, which are playing an important role in the regulation of blood pressure homeostasis (Dibona and Kopp, 1997; Dibona, 2000; Grassi et al., 2015). Renal nerves follow the renal arteries and innervate not only the vasculature but also the juxtaglomerular apparatus and the basement membrane of epithelial cells within the nephron. Therefore, it is not surprising that the main neurotransmitter neuropeptide Y (NPY), ATP, and norepinephrine, released at neuroeffector junctions in the kidney, mediate several physiological effects within the kidney. Sympathetic norepinephrine release induces renal vasoconstriction and stimulates renin release as well as tubular sodium and water reabsorption in the kidney. In hypertensive patients, renal sympathetic nerve activity (RSNA) is increased (Schlaich et al., 2009; Grassi et al., 2015). Thus, increased RSNA results in a reduction of renal blood flow and glomerular filtration rate (GFR), an increase in renal vascular resistance and tubular sodium and water reabsorption, and an increased release of renin, contributing to the development and maintenance of hypertension. Studies performed in patients with therapy resistant hypertension show a robust reduction in blood pressure after renal denervation, highlighting a critical crosstalk between the sympathetic nervous system and the kidney in hypertension (Schlaich et al., 2009; Kandzari et al., 2018; Vonend et al., 2018; Steinberg et al., 2020). In addition, evidence emerges that hypertension is at least in part an immune-mediated inflammatory disease. In this regard, several studies have shown a close interaction between the sympathetic nervous system and immune cell response in hypertension. Thus, reduction in RSNA by renal denervation reduces pro-inflammatory markers and immune cell migration in humans and mice (Xiao et al., 2015; Zaldivia et al., 2017).
To understand the role of RSNA in the development of hypertensive kidney disease, it is essential to know how RSNA affects mechanisms in the kidney controlling blood pressure homeostasis. The amount of neurotransmitter released from renal prejunctional nerve endings is not only controlled by the RSNA but also by prejunctional alpha2-adrenergic receptors (α2-adrenoceptors). Prejunctional α2-adrenoceptors serve as autoreceptors which, when activated by norepinephrine released from sympathetic nerve endings, inhibit the subsequent release of norepinephrine induced by a sympathetic nerve impulse (Figures 1, 2). Recent studies have highlighted the critical role of α2-adrenoceptors in the development of hypertension and kidney disease (Kim and Padanilam, 2013; Hering et al., 2020). However, α2-adrenergic receptors are not only expressed prejunctional on sympathetic nerves but also on non-adrenergic cells like immune cells, vascular smooth muscle cells (VSMCs), and renal epithelial cells. Activation of α2-adrenoceptors on these cells mediates a variety of effects, including inflammatory and fibrotic responses (Kim and Padanilam, 2013, 2015) and changes in renal vasoconstriction and VSMC turnover (Bohmann et al., 1995; Jackson et al., 2001, 2005), as well as altering sodium balance (Nord et al., 1987; Mansley et al., 2015) which may also influence blood pressure and kidney damage.
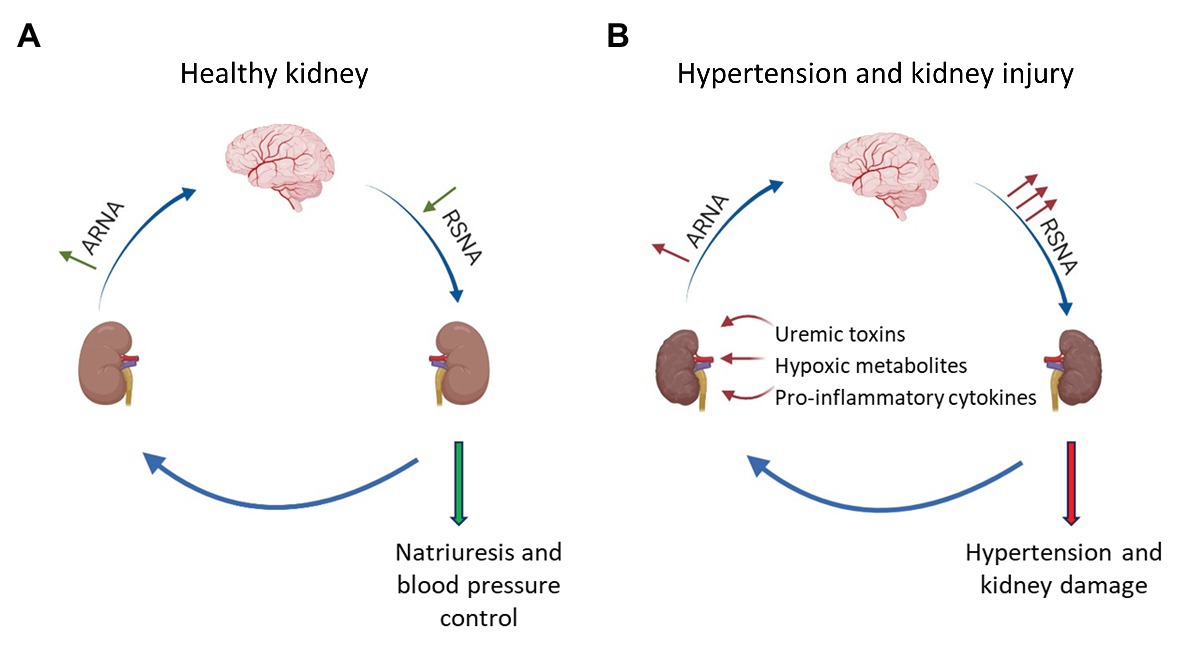
Figure 1. Interaction between afferent renal nerves and renal sympathetic nerve activity in healthy and injured kidneys. (A) In healthy kidneys, increased renal sympathetic nerve activity (RSNA) leads to an activation of α1-adrenoceptors expressed in the renal pelvis, which increases afferent renal nerve activity (ARNA). Subsequently, inhibitory neurons within the brainstem and hypothalamus decrease RSNA via a negative feedback mechanism leading to natriuresis. (B) In hypertension or kidney injury, accumulation of pro-inflammatory cytokines, uremic toxins, or ischemic metabolites activate afferent renal nerves. Under these conditions, the negative feedback mechanism is dysregulated and increased ARNA results in a further activation of renal sympathetic nerves causing the progression of hypertension and hypertensive kidney disease.
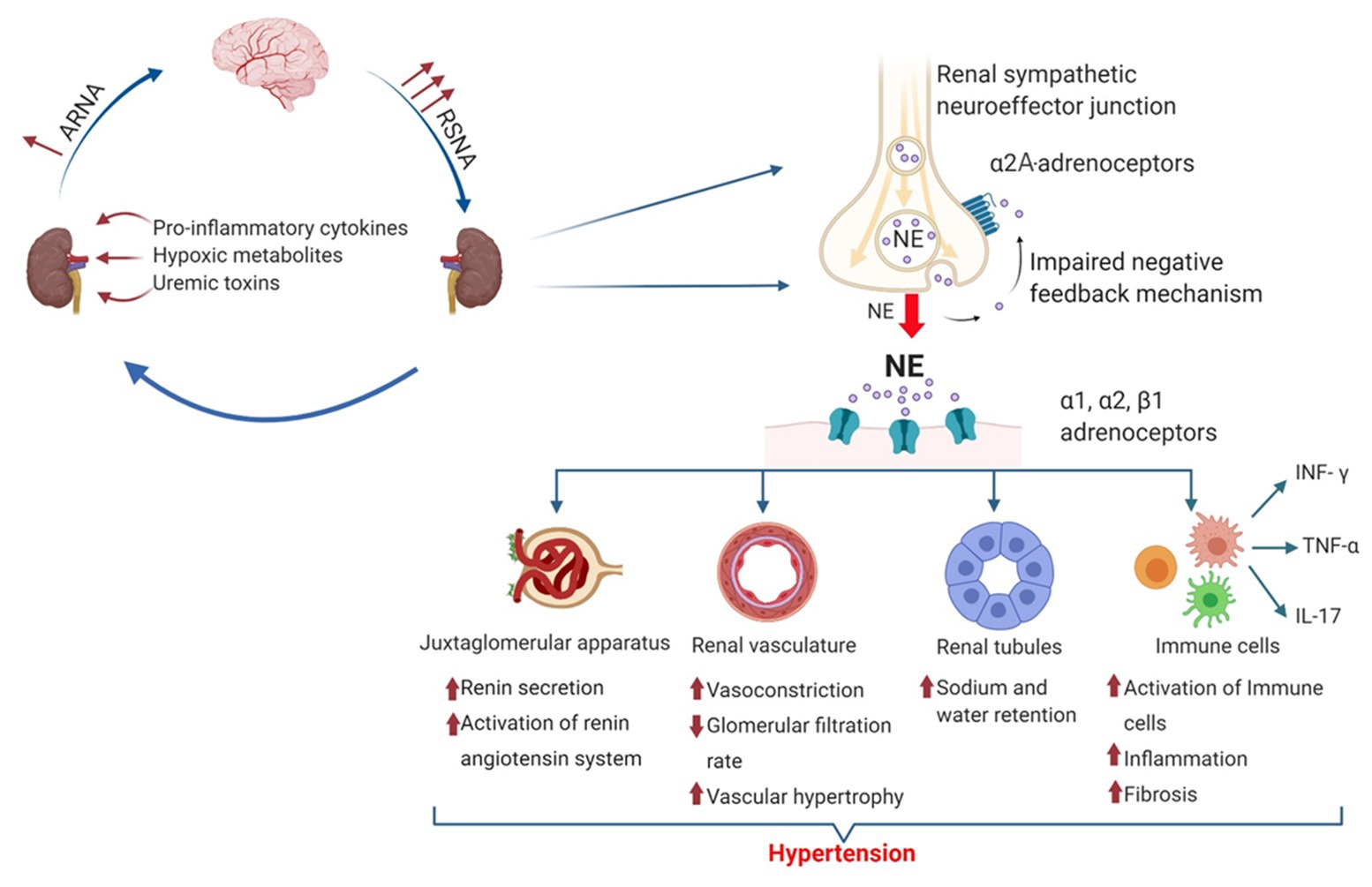
Figure 2. Mechanisms causing hypertension by increased renal sympathetic norepinephrine release. The amount of norepinephrine released by prejunctional renal sympathetic nerves is controlled by RSNA and prejunctional α2A-adrenoceptors. Norepinephrine activates different adrenoceptors and exerts various physiological and pathophysiological effects leading to hypertension and chronic kidney disease (CKD). Thereby, norepinephrine activates the renin-angiotensin system (RAS) by stimulating renin release from the juxtaglomerular apparatus via β2-adrenergic receptor activation. Activation of α1- and α2-adrenoceptors causes vasoconstriction and increases renal vascular resistance, leading to vascular hypertrophy and a reduced glomerular filtration rate (GFR). In renal tubules, NE induced α1-, α2-, and β1-adrenergic receptor activation modulates the activity of different sodium transporters such as sodium hydrogen exchanger 3 (NHE3), Na+-Cl− Co-transporter (NCC), and epithelial sodium channel (ENaC) leading to decreased sodium excretion. In addition, norepinephrine modulates immune cell function and phenotype, leading to an increased infiltration into the kidney and an increased release of various pro-inflammatory cytokines such as interferon gamma (INF-γ), tumor necrosis factor alpha (TNF-α), and interleukin-17 (IL-17) aggravating the development of hypertension and renal fibrosis.
In the present review, we will highlight the role of α2-adrenoceptors on RSNA and its impact on hypertension. Moreover, we will focus on physiological and pathophysiological effects which were mediated by non-adrenergic α2-adrenoceptors with special respect to renal epithelial cells and immune cells. Most of the mechanisms described in the present review are based on animal studies.
While this review focuses on physiological and pathophysiological effects mediated by the prejunctional and non-adrenergic α2-adrenoceptors expressed in the kidney and on immune cells, it should be noted that all three α2-adrenoceptor subtypes are widely distributed throughout the central nervous system (CNS) that is ultimately regulating sympathetic nerve activity. Central acting α2-adrenoceptor agonists such as clonidine, guanabenz, and moxonidine are effective in the treatment of hypertension (Kanagy, 2005). These sympatholytic agents cross the blood-brain barrier and interact with central α2-adrenoceptors, leading to a reduction in sympathetic nerve activity and an increase in vagal activity. This change in sympathetic tone causes lower cardiac output and heart rate, reduced renin release, and subsequently a reduction in vascular resistance leading to blood pressure reduction (Dibona and Kopp, 1997; Hein, 2006; Schlaich et al., 2012; Sata et al., 2018). In addition, sympatholytic treatment induced by moxonidine has been shown to attenuate the progression of chronic kidney disease (CKD) in hypertensive patients and rats with advanced renal failure (Amann et al., 2000; Vonend et al., 2003). Whether these effects are in part mediated by non-central effects of α2-adrencoeptor activation is still not fully understood.
Several studies have investigated the association of hypertension and polymorphism of human ADRA2 gene. Genes of α2-adrenocepotor subtypes ADRA2A (α2A-adrenoceptors), ADRA2B (α2B-adrenoceptors), and ADRA2C (α2C-adrenoceptors) are located on chromosomes 10, 2, and 4, respectively. The ADRA2A 1780 C > T (rs553668) genotype is associated with an exercise-dependent aggravation of systolic as well as diastolic blood pressures in women (Nunes et al., 2014). In addition, the described polymorphism is associated with increased platelet aggregation and a marked decrease in sodium excretion. Both findings are common in essential hypertension (Freeman et al., 1995). Moreover, the −1291 C > G (rs1800544) substitution in the ADRA2A promoter region is responsible for reduced presynaptic autoinhibition of α2A-adrenoceptors, resulting in excessive norepinephrine concentration and, therefore, in an increased vascular resistance (Kelsey et al., 2012). Deletion polymorphisms or different variants of ADRA2B and ADRA2C are known to be related to endothelial dysfunction, heart failure, and hypertension (Heinonen et al., 2002; Small et al., 2002; von Wowern et al., 2004; Matsunaga et al., 2007).
In conclusion, there is substantial evidence that genetic variability in ADRA2A and ADRA2B genes influences α2-adrenoceptor function, leading to hypertension due to modulating vascular resistance, endothelial function, and sodium homeostasis in different cohorts.
The regulation of RSNA is complex and involves central and peripheral mechanism. In general, the nerve activity of sympathetic premotor nuclei in the brainstem and hypothalamus [the rostral ventrolateral medulla (RVLM) and rostral ventromedial medulla (RVMM) as well as the paraventricular nucleus (PVN)] regulates RSNA. While the exact regulatory system of these central mechanisms is not the focus of the current review (Zheng and Patel, 2017), it is noteworthy that central nerve activity in the RVLM, RVMM, and PVN is modulated by neurotransmitters, local factors such as reactive oxygen species, cytokines, and angiotensin II (Ang II), as well as mechano- and chemo-sensitive renal afferent nerves which project to the RVLM via the nucleus tractus solitarius (NTS) and PVN. According to physiological or pathophysiological conditions, afferent renal nerve activity (ARNA) can either activate or inhibit sympathetic premotor activity and thereby RSNA via a positive or negative feedback mechanism (Dibona, 2000; Pyner, 2014; Zheng and Patel, 2017).
Under physiological conditions, RSNA is controlled by the renorenal reflex, which is considered as a negative feedback loop to maintain efferent RSNA (ERSNA) at low-levels, and thereby controlling natriuresis and blood pressure. This interaction between efferent sympathetic nerves and afferent sensory nerves is complex. Increased RSNA increases ARNA by activating mechanoceptors and chemoceptors, which in turn lowers efferent RSNA via inhibitory neurons which project to the RVLM (Dibona and Kopp, 1997; Kopp et al., 2007, 2011a). Norepinephrine acting on adrenoceptors located in the renal pelvis mediates the ERSNA-ARNA interaction. Activation of α1-adrenoceptors leads to an increase in ARNA whereas activation of α2-adrenoceptors decreases ARNA (Kopp et al., 2007). In this regard, Kopp et al. (2011b) showed that low sodium diet reduces ARNA via α2-adrenoceptor activation leading to an increase in RSNA and consequently to sodium reabsorption. In contrast, in spontaneous hypertensive rats (SHRs), this mechanism seems to be dysregulated. Thus, ARNA is reduced in SHRs due to an overactivation of α2-adrenoceptors in renal pelvic tissue. These studies suggest a direct role of renal pelvic α2-adrenoceptors in decreasing the responsiveness of ARNA to increased RSNA and thereby in the development of hypertension (Kopp et al., 2007, 2011a). In contrast, a recent study performed in α2A-adrenoceptor deficient mice showed that deletion of α2A-adrenoceptors accelerates Ang II-dependent hypertension rather than decreases blood pressure (Hering et al., 2020). This study suggests that the renorenal reflex mediated by α2-adrenoceptors is dysregulated or does not seem to play an important role in this experimental model of hypertension.
In hypertension as well as acute or chronic kidney damage, stimulation of renal nociceptive afferent nerves mediate an increase in sympathetic nerve activity leading to a further activation of RSNA and subsequently to a progression of hypertension and hypertensive kidney disease. Thus, several other factors such as pro-inflammatory cytokines (Banek et al., 2019), uremic toxins (Campese and Kogosov, 1995), and hypoxia (Dibona and Kopp, 1997; Soukhova-O'hare et al., 2006; Saha et al., 2019) can activate the chemo- and mechano-sensitive afferent nerves leading to an increased RSNA. Activation of afferent nerves under pathophysiological conditions such as acute kidney injury induced by a phenol injection into the kidney causes hypertension by increasing RSNA via afferent renal nerve stimulation (Ye et al., 2002a,b; Leong et al., 2006). The importance of an increased ARNA in the development and maintenance of hypertension in acute kidney disease or CKD is supported by several studies. For instance, in hypertensive rats treated with deoxycorticosterone acetate (DOCA) salt, increased ARNA seems to perpetuate hypertension as selective ablation of the afferent renal nerves reduces blood pressure (Banek et al., 2016). Additionally, in patients with kidney failure treated with dialysis, increased sympathetic nerve activity and thereby hypertension could only be reduced by a bilateral removal of the kidneys (Converse et al., 1992).
In summary, renal pelvic α1- and α2-adrenoceptors affect the renorenal reflex that regulates RSNA activity via ARNA under physiological conditions. In hypertension or kidney injury, ARNA is activated by other factors leading to an increase in RSNA via a positive feedback mechanism. Therefore, it seems plausible that in patients with hypertension increased RSNA is in part the consequence of increased ARNA and an important pathophysiological mechanism for the development of treatment resistant hypertension.
There are three different subtypes of α2-adrenoceptors (α2A-, α2B-, and α2C-adrenoceptors; Trendelenburg et al., 2001). The cellular distribution of these subtypes varies, but several in vivo and in vitro studies confirmed that the α2A-adrenoceptor is the predominant subtype involved in the regulation of renal and cardiac sympathetic neurotransmitter release (Hein et al., 1999; Vonend et al., 2007; Hoch et al., 2011). Based on early results from radioligand binding studies which were confirmed and expanded by deep sequencing analysis of microdissected rat renal tubules (Muntz et al., 1986; Nord et al., 1987; Lee et al., 2015), the cellular distribution of α2-adrenoceptor subtypes along the nephron is now well-described and summarized in Figure 3 and Table 1. α2B-adrenoceptors are expressed in the proximal tubule whereas the α2A-adrenoceptors are located on the connecting tubule, collecting duct and the renal pelvis (Dibona and Kopp, 1997; Kopp et al., 2007; Lee et al., 2015; Table 1). In the glomerulus, only α2B-adrenoceptors seem to be expressed. However, the exact cellular localization is not known (Lee et al., 2015). In VSMCs, all three subtypes are expressed and involved in maintaining vascular tone. However, the distribution of the α2-adrenoceptor subtypes varies based on vascular bed and size of the vessels and species. High amount of α2A-adrenoceptor is expressed in large arteries like the aorta, whereas α2B-adrenoceptor is mostly distributed in small arteries and veins contributing to vasoconstriction (Faber et al., 2001; Kanagy, 2005). In addition, most immune cells express α2-adrenoceptors, with α2A- and α2B-adrenoceptors being the predominant subtypes. Thus, α2A- and α2B-adrenoceptors were detected on macrophages, T-cells, and natural killer cells from rodents and humans (Elenkov et al., 2000; Flierl et al., 2007; Scanzano and Cosentino, 2015; Harwani, 2018).
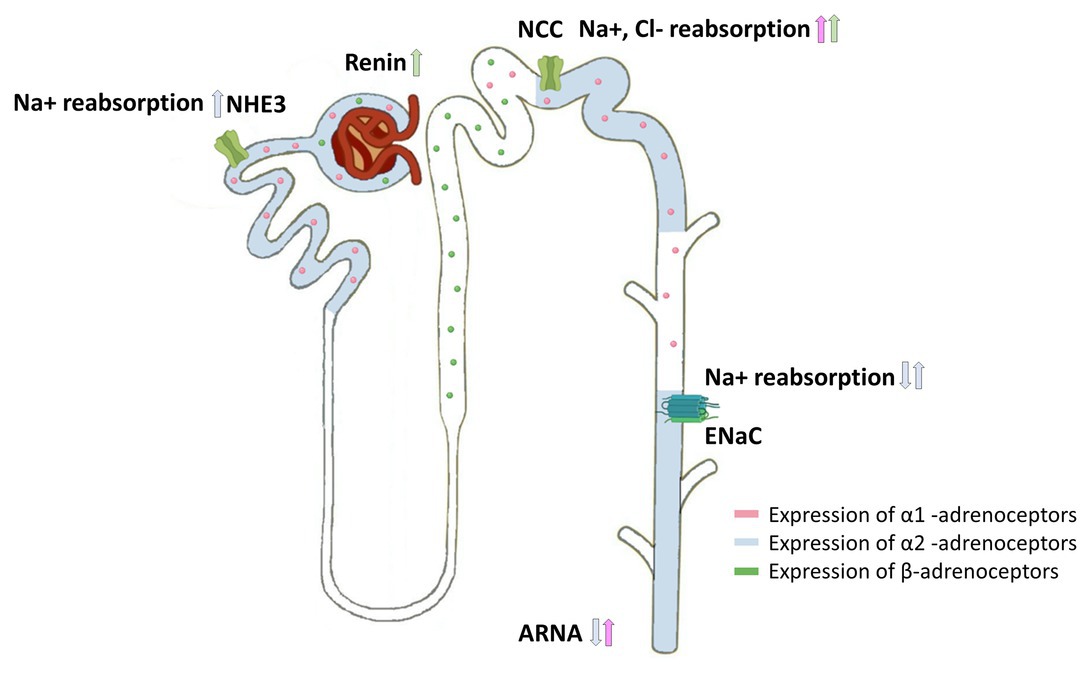
Figure 3. Cellular distribution of adrenoceptors in the nephron and their effects on sodium transporters. Expression of α1-adrenoceptors (pink), α2-adrenoceptors (blue), and β-adrenoceptors (green) along the nephron showing the influence of renal sympathetic neurotransmission on sodium handling. Norepinephrine modulates sodium handling in the proximal (Na+/H+ 1 and 3 Exchange) and distal tubule (NCC and ENaC) by activating α1-adrenoceptors, α2-adrenoceptors, or β-adrenoceptors. Furthermore, α2-adrenoceptor activation within the renal pelvis decreases ARNA, whereas α1-adrenoceptor activation increases ARNA. β-adrenergic receptor activation induces renin release.

Table 1. Gene expression levels of α1-, α2-, and β-adrenoceptor subtypes along a microdissected rat nephron are summarized.
Taken together, α2-adrenoceptor subtypes are expressed along the nephron, on VSMC and on immune cells. When examining the physiological relevance of α2-adrenoceptors, one has to consider, that physiological activity of α2-adrenoceptors is at least in part dependent on their density (Duzic et al., 1992).
The effects of α2-adrenoceptor function in renal physiology can be divided into two categories. First, prejunctional α2A- and α2C-adrenoceptors regulate renal sympathetic neurotransmitter release via an autoinhibitory feedback mechanism (Philipp et al., 2002). Activation of prejunctional α-adrenoceptors regulates not only the release of norepinephrine but also the release of ATP, neuropeptide Y (NPY; Burnstock, 1996; Lundberg, 1996; Oberhauser et al., 1999), and thereby modifies renin release, vascular tone, water and sodium handling, as well as the development of renal inflammation and fibrosis by activating different receptors (Dibona and Kopp, 1997; Amann et al., 2000; Bradley et al., 2003; Mcdonough, 2010; Sumi et al., 2010; Kim and Padanilam, 2013).
Second, norepinephrine (in part prejunctional released) activates α2-adrenoceptors expressed on non-adrenergic cells such as renal epithelial cells, VSMCs, or immune cells. This α2-adrenoceptor activation modifies vascular tone, sodium handling, tubulo-interstitial fibrosis, and inflammation within the kidney (Starke et al., 1975; Gilsbach et al., 2009, 2011; Hoch et al., 2011; Kim and Padanilam, 2013; Jang et al., 2019; Hering et al., 2020; Figure 2).
Although the effects of prejunctional released ATP and NPY are not the focus of this review, their effects on renal physiological and pathophysiological mechanisms are important. NPY and ATP, released from sympathetic neurons upon α1- and α2-adrenoceptor signaling (Bradley et al., 2003; Vonend et al., 2007; Sumi et al., 2010), are described to play a role in hypertension (Thulin and Erlinge, 1995) and renal failure (Bald et al., 1997). Additionally to the pleiotropic effects of ATP on its P2 (purinergic type 2) receptors in the kidney (Solini et al., 2015), ATP can also be sequentially hydrolyzed by CD93 to ADP and AMP with AMP being further converted to adenosine by CD73. Alterations in the balance of nucleotides to nucleosides have major impacts on renal function, the development of hypertension, renal fibrosis, and inflammation (for a better overview, please refer to Kishore et al., 2018; Perry et al., 2019).
Norepinephrine induced renal pressor response is predominantly mediated by α1-adrenoceptors. However, subpressor concentrations of Ang II revealed a role of α2-adrenoceptors in the renal vasoconstrictor response to norepinephrine (Bohmann et al., 1995). Moreover, α2-adrenoceptor activation potentiates Ang II-induced renal pressor response in vivo and in vitro of SHRs mainly through an α2-adrenoceptor-mediated RhoA-dependent mechanism (Jackson et al., 2001, 2005).
Beside a direct effect on renal vascular resistance, activation of both α1- and α2B-adrenoceptors by chronic renal sympathetic overactivity induces a phenotypic switch of VSMC into proliferative VSMC, leading to hypertrophy, renal vascular stiffness, and reduced renal blood flow (Wang et al., 2004; Huhtinen and Scheinin, 2008). This phenotypic switch is mediated by norepinephrine-induced reactive oxygen species via p38 MAPK activation (Kalyankrishna and Malik, 2003; Bleeke et al., 2004). However, the exact mechanism of how increased RSNA induces the development of preglomerular arteriolopathy is still not known. As proof that RSNA is involved in regulating renal pressor response, renal denervation reduced renal sympathetic neurotransmission results in a significant decrease of renal vascular resistance and a significant increase of renal plasma flow as well as GFR in SHR compared to non-denervated SHRs (Tomoda et al., 1997).
Summing up, α2- and α1-adrenoceptors are directly involved in renal vasoconstriction and indirectly by causing a phenotypic switch toward proliferative VSMC.
Changes in renal vascular resistance and renal blood flow have been shown to influence sodium excretion (Sparks et al., 2015). RSNA affects natriuresis in several animal models and patients with resistant hypertension (Katayama et al., 2013; Poss et al., 2015; Hering et al., 2020). Renal sympathetic nerves also innervate renal tubules. As shown in Figure 3, α1-adrenoceptors, α2-adrenoceptors, and β1-adrenergic receptors are expressed along the nephron (Dibona and Kopp, 1997; Lee et al., 2015; Sata et al., 2018; Kiuchi et al., 2019). During chronic Ang II infusion, sodium and volume excretion was significantly reduced in α2A-adrenoceptor deficient mice compared to wildtype mice (Hering et al., 2020). This impaired natriuretic response was in part caused by an increased abundance of the cleaved epithelial sodium channel (ENaC)-alpha and -gamma subtypes, both markers for ENaC activation (Nguyen et al., 2013; Veiras et al., 2020). The role of α2-adrenoceptors in controlling natriuresis is still not fully understood, as it is hard to distinguish between α2-adrenoceptor-mediated effects and effects mediated by an increased renal sympathetic norepinephrine release. In general, it is widely accepted that increased sympathetic norepinephrine release increases ENaC expression and activation (Mansley et al., 2015; Hering et al., 2020). In contrast, renal denervation has been shown to reduce ENaC and aquaporin2 expression in a mouse model of heart failure, suggesting that the amount of renal norepinephrine release is relevant for the regulation of ENaC expression (Zheng et al., 2019). Besides that, several reports demonstrate an interaction between α2-adrenoceptor activation and regulation of ENaC abundance and activation. Thus, α2-adrenoceptor activation inhibits vasopressin-induced cAMP generation (Chabardes et al., 1984; Krothapalli and Suki, 1984), which in turn decreases vasopressin induced ENaC activation (Roos et al., 2013). In contrast, activation of basolateral α2-adrenoceptors on principal cells increases ENaC activity in vitro (Mansley et al., 2015). Thus, only selective deletion of α2A-adrenoceptors from the collecting duct will show evidence about the impact of α2-adrenoceptors on ENaC function.
In addition, sympathetic norepinephrine influences the expression and activation of the Na+Cl− co-transporter (NCC) and, thereby, sodium excretion in the distal nephron and the development of hypertension. Norepinephrine stimulates NCC expression through an activation of basolateral Kir4.1/Kir5.1 potassium channel via beta-adrenergic receptor activation (Duan et al., 2019). Activation of α1-adrenoceptors inhibits the suppression of NCC during high salt intake via a WNK/SPAK/OxSR1-dependent signaling pathway in rat kidneys (Frame et al., 2019). Dephosphorylation of NCC by the protein phosphatase 1 can be inhibited through a protein kinase A-dependent activation of the protein phosphatase 1 inhibitor via β1-adrenergic receptor activation (Penton et al., 2019).
In proximal tubules, acute increase in sympathetic norepinephrine has been shown to stimulate sodium hydrogen exchanger 1 and 3 and, thereby, sodium reabsorption most likely by activating α2-adrenoceptors (Nord et al., 1987; Leong et al., 2006; Healy et al., 2014; Lee et al., 2015). In this context, short term stimulation of renal nerves has been shown to activate sodium hydrogen exchanger 3 (NHE3)-mediated sodium reabsorption and the intrarenal renin-angiotensin system (RAS). As this effect was blocked by losartan, a selective Ang II type 1 receptor blocker, the authors suggested that this mechanism is in part mediated via an intrarenal RAS activation induced by ERSA (Pontes et al., 2015). NHE3 function is negatively correlated to its phosphorylation status at the PKA site (serine 552) that effects subcellular trafficking and, therefore, its activity (Kocinsky et al., 2005). Ang II treatment decreased cAMP/PKA signaling and, therefore, the phosphorylation at serin 552 leading to increased NHE3 activity (Crajoinas et al., 2016). In contrast, increased sympathetic norepinephrine release in long-term Ang II-dependent hypertension suppresses NHE3 abundance (Nguyen et al., 2013; Hering et al., 2020; Veiras et al., 2020). This suppression of NHE3 is a compensatory natriuretic mechanism of the kidney to regulate blood pressure in chronic hypertension and override the stimulatory effect of Ang II on NHE3 (Mcdonough, 2010; Mcdonough and Nguyen, 2015).
This section highlights the effect of renal sympathetic norepinephrine release on renal sodium transport. The amount of prejunctional released norepinephrine regulates sodium homeostasis in the kidney. There is strong evidence that α2-adrenoceptors play a role in regulating ENaC function, whereas α1- and β-adrenoceptors are involved in the regulation of NCC.
Although renal sympathetic overactivity plays an important role in the progression of hypertensive kidney disease, its role in the development of fibrosis and inflammation leading to CKD is not fully understood (Veelken et al., 2008; Jang et al., 2019). The therapeutic strategy of renal denervation preventing renal failure may also be at least in part due to its protective anti-inflammatory effect attenuating renal inflammation and fibrosis (Veelken et al., 2008; Kim and Padanilam, 2013). Animal studies show robust evidence that the sympathetic nervous system interacts with the immune system and, thereby, modulates the inflammatory response in the target organ, leading to fibrosis und progression of the underlying disease (Andersson and Tracey, 2012; Carnevale et al., 2016).
In lymphoid organs, sympathetic neurons release norepinephrine which has a direct effect on immune cells by modulating T-cell polarization, lymphocyte trafficking, and proliferation as well as cytokine production via adrenoceptor activation. Moreover, immune cell trafficking depends also on regional blood flow which is under tight control of the local sympathetic nerve activity (Elenkov et al., 2000). Although all three adrenoceptor subtypes are expressed within the immune system, β-adrenergic receptor-mediated effects are studied most extensively with special interest for the β2-adrenoceptor subtype due to its anti-inflammatory effects (Elenkov et al., 2000). The role of α2-adrenoceptors on immune cells is still not well-examined but comes into the focus of research (Elenkov et al., 2000; Flierl et al., 2007; Kim and Padanilam, 2013).
A very recent study showed that increased sympathetic nerve activity caused by experimental hypertension increases T-cell homing of effector memory T-cells in the bone marrow via β2-adrenergic receptor activation (Xiao et al., 2020). When hypertensive stimuli persist, these effector memory T-cells infiltrate into the vasculature and the kidney and release cytokines like interferon gamma (INF-γ), interleukin-17 (IL-17), and tumor necrosis factor alpha (TNF-α), which aggravate hypertension (Madhur et al., 2010). INF-γ, TNF-α, and IL-17 accelerate vascular damage and water reabsorption by affecting different sodium transporters along the distal nephron (Sriramula et al., 2008; Kamat et al., 2015; Wu et al., 2016). In addition, increased RSNA in low-dose Ang II-dependent hypertension activates antigen presenting cells and, subsequently, T-cells infiltrating hypertensive kidneys. Reduction in RSNA by renal denervation significantly reduced T-cell and macrophage infiltration, attenuated renal fibrosis, and improved renal function (Xiao et al., 2015). These results clearly demonstrate that renal sympathetic nerves mediate renal inflammation and T-cell activation in hypertension. However, there is conflicting evidence concerning the distinctive contribution of renal afferent and efferent nerves on the described anti-inflammatory effects of RDN. Xiao et al. (2015) could not find a contribution of afferent nerves on Ang II-induced hypertension and renal inflammation, whereas Banek showed evidence that renal afferent nerves modulate at least in part renal inflammation in DOCA-salt hypertension (Banek et al., 2016, 2019). In another study, performed in global α2A-deficent mice chronically infused with a high dose of Ang II, increased renal sympathetic norepinephrine release impairs renal function and aggravates hypertension as well as renal fibrosis without affecting the amount of infiltrating immune cells (Hering et al., 2020). At first glance, these results seem to be conflicting as several studies have shown that increased RSNA activates a pro-inflammatory immune cell response leading to immune cell infiltration and an aggravation of renal fibrosis and hypertension (Kim and Padanilam, 2013, 2015; Xiao et al., 2015; Banek et al., 2019). However, α2-adrenoceptors also seem to regulate immune cell function, and activation of α2-adrenoceptors seems to induce a pro-inflammatory immune response (Scanzano and Cosentino, 2015). Thus, inhibition of α2-adrenoceptor on alveolar macrophages reduces the release of several cytokines like TNF-α, IL-6, or IL-1β (Flierl et al., 2007), whereas activation of α2-adrenoceptors on macrophages has been shown to increase TNF-α production (Spengler et al., 1994). In addition, inhibition of renal a2-adrenoceptors reduces renal inflammation and infiltration of neutrophils and macrophages in obstructed murine kidneys, whereas direct infusion of norepinephrine in denervated kidneys induced a fibrotic response similar to innervated non-infused kidneys. These results suggest an important role of norepinephrine signaling through renal α2-adrenoceptors in fibrogenesis and mediating inflammation (Kim and Padanilam, 2013). In line with these observations, a recent study could show that α2A-adrenoceptor deficiency reduced lung injury in mice and decreased lung inflammation by reducing immune cell infiltration as well as decreasing pro-inflammatory cytokines (Cong et al., 2020).
Activation of α1- and α2-adrenoceptors seems to induce polarization toward the inflammatory M1 phenotype (Grisanti et al., 2011; Harwani, 2018), and activated macrophages have been shown to accelerate hypertension (Wenzel et al., 2011). In contrast to the innate immune system, adrenoceptor functions on T-cells are less well described and conflicting. On one side, activation of β2-adrenergic receptors activates the homing of CD8+ effector memory T-cells and an upregulation of CCL19 and CCL21 in hypertension. On the other side, activation of β2-adrenergic receptors in experimental autoimmune disease or in vitro reduces the T-cell response to sympathetic norepinephrine and decreases the release of INF-γ and TNF-α from CD8+ T-cells (Estrada et al., 2016; Araujo et al., 2019). Although α2-adrenoceptors are expressed on T-cells, their role in T-cell function in general and particularly in hypertension is not well understood. Early studies have shown that reduced peripheral blood T-cell mitogenesis is caused by activation of peripheral α2-adrenoceptors (Felsner et al., 1995). Activation of α2-adrenoceptors expressed on dendritic cells reduces induction of T-cell proliferation (Araujo et al., 2019). In patients undergoing surgery, activation of α2-adrenoceptors shifted the Th1/Th2 and the Treg/Th17 cytokine balance toward a Th1 and Th17 response, respectively, suggesting a pro-inflammatory rather than an anti-inflammatory effect on human T-cells (Lee et al., 2018).
In conclusion, increased sympathetic norepinephrine release seems to activate T-cell response through a complex interaction with the innate immune system leading to an aggravation of hypertension and CKD. The role of adrenergic receptors in modulating the immune response in hypertensive kidney damage needs further investigation but seems to be an interesting therapeutic approach, as selective agonists and antagonists of α- and β-adrenergic receptors are already in clinical practice.
Renal sympathetic nerve activity plays a major role in blood pressure homeostasis. Regulation of RSNA describes a complex interaction between afferent nerve activity and central mechanism. Under physiological conditions, RSNA is controlled by afferent renal mechano- and chemo-sensitive nerves by the renorenal reflex via a negative feedback mechanism. Thereby, ARNA is regulated in part by α1- or α2-adrenoceptors located in the renal pelvis. During hypertension or kidney damage, this negative feedback mechanism is disturbed. Activation of afferent renal nerves induced by several factors including uremic toxin, pro-inflammatory cytokines, and hypoxia injury increases RSNA and is therefore an important factor for the development of resistant hypertension and kidney disease.
Increased sympathetic nerve activity results in an elevated release of sympathetic neurotransmitter. Prejunctional α2-adrenoceptors control renal sympathetic neurotransmission via a negative feedback mechanism. Deletion or pharmacological inhibition of α2-adrenoceptors accelerates hypertension and kidney injury through multiple mechanisms. First, increased sympathetic neurotransmission particularly norepinephrine release increases renin release, renal vascular tone, sodium reabsorption, and inflammation through an activation of α- and β-adrenoceptors in the kidney and on immune cells. Second, non-adrenergic α2-adrenoceptor activation on renal epithelial cells, VSMCs, or immune cells directly modulates vascular tone, sodium balance, and immune cell response in the kidney. Based on this complex interaction between the well-studied function of prejunctional α2-adrenoceptors and the multiple effects of adrenoceptors activation on non-adrenergic cells in the kidney and on immune cells, the exact physiological and pathophysiological role of α2-adrenoceptor is still not fully understood and needs further studies in where α2-adrenoceptor function can be examined cell specific.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was supported through a research grant of the German Research Foundation (DFG) to JS (STE 2042/1-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Amann, K., Rump, L. C., Simonaviciene, A., Oberhauser, V., Wessels, S., Orth, S. R., et al. (2000). Effects of low dose sympathetic inhibition on glomerulosclerosis and albuminuria in subtotally nephrectomized rats. J. Am. Soc. Nephrol. 11, 1469–1478.
Andersson, U., and Tracey, K. J. (2012). Neural reflexes in inflammation and immunity. J. Exp. Med. 209, 1057–1068. doi: 10.1084/jem.20120571
Araujo, L. P., Maricato, J. T., Guereschi, M. G., Takenaka, M. C., Nascimento, V. M., De Melo, F. M., et al. (2019). The sympathetic nervous system mitigates CNS autoimmunity via beta2-adrenergic receptor signaling in immune cells. Cell Rep. 28, 3120.e5–3130.e5. doi: 10.1016/j.celrep.2019.08.042
Bald, M., Gerigk, M., and Rascher, W. (1997). Elevated plasma concentrations of neuropeptide Y in children and adults with chronic and terminal renal failure. Am. J. Kidney Dis. 30, 23–27. doi: 10.1016/s0272-6386(97)90560-6
Banek, C. T., Gauthier, M. M., Van Helden, D. A., Fink, G. D., and Osborn, J. W. (2019). Renal inflammation in DOCA-salt hypertension. Hypertension 73, 1079–1086. doi: 10.1161/HYPERTENSIONAHA.119.12762
Banek, C. T., Knuepfer, M. M., Foss, J. D., Fiege, J. K., Asirvatham-Jeyaraj, N., Van Helden, D., et al. (2016). Resting afferent renal nerve discharge and renal inflammation: elucidating the role of afferent and efferent renal nerves in deoxycorticosterone acetate salt hypertension. Hypertension 68, 1415–1423. doi: 10.1161/HYPERTENSIONAHA.116.07850
Bleeke, T., Zhang, H., Madamanchi, N., Patterson, C., and Faber, J. E. (2004). Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ. Res. 94, 37–45. doi: 10.1161/01.RES.0000109412.80157.7D
Bohmann, C., Rist, W., Schollmeyer, P., and Rump, L. C. (1995). Low concentrations of angiotensin II unmask vasoconstrictory alpha 2-adrenoceptors in isolated perfused kidneys of spontaneously hypertensive rats. Cardiovasc. Res. 30, 857–865.
Bradley, E., Law, A., Bell, D., and Johnson, C. D. (2003). Effects of varying impulse number on cotransmitter contributions to sympathetic vasoconstriction in rat tail artery. Am. J. Physiol. Heart Circ. Physiol. 284, H2007–H2014. doi: 10.1152/ajpheart.01061.2002
Burnstock, G. (1996). Development and perspectives of the purinoceptor concept. J. Auton. Pharmacol. 16, 295–302. doi: 10.1111/j.1474-8673.1996.tb00039.x
Campese, V. M., and Kogosov, E. (1995). Renal afferent denervation prevents hypertension in rats with chronic renal failure. Hypertension 25, 878–882. doi: 10.1161/01.hyp.25.4.878
Carnevale, D., Perrotta, M., Pallante, F., Fardella, V., Iacobucci, R., Fardella, S., et al. (2016). A cholinergic-sympathetic pathway primes immunity in hypertension and mediates brain-to-spleen communication. Nat. Commun. 7:13035. doi: 10.1038/ncomms13035
Chabardes, D., Montegut, M., Imbert-Teboul, M., and Morel, F. (1984). Inhibition of alpha 2-adrenergic agonists on AVP-induced cAMP accumulation in isolated collecting tubule of the rat kidney. Mol. Cell. Endocrinol. 37, 263–275. doi: 10.1016/0303-7207(84)90096-0
Cong, Z., Li, D., Lv, X., Yang, C., Zhang, Q., Wu, C., et al. (2020). alpha2A-adrenoceptor deficiency attenuates lipopolysaccharide-induced lung injury by increasing norepinephrine levels and inhibiting alveolar macrophage activation in acute respiratory distress syndrome. Clin. Sci. 134, 1957–1971. doi: 10.1042/CS20200586
Converse, R. L. Jr., Jacobsen, T. N., Toto, R. D., Jost, C. M., Cosentino, F., Fouad-Tarazi, F., et al. (1992). Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 327, 1912–1918. doi: 10.1056/NEJM199212313272704
Crajoinas, R. O., Polidoro, J. Z., Carneiro De Morais, C. P., Castelo-Branco, R. C., and Girardi, A. C. (2016). Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. Am. J. Physiol. Cell Physiol. 311, C768–C776. doi: 10.1152/ajpcell.00191.2016
Dibona, G. F. (2000). Neural control of the kidney: functionally specific renal sympathetic nerve fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1517–R1524. doi: 10.1152/ajpregu.2000.279.5.R1517
Dibona, G. F., and Kopp, U. C. (1997). Neural control of renal function. Physiol. Rev. 77, 75–197. doi: 10.1152/physrev.1997.77.1.75
Duan, X. P., Gu, L., Xiao, Y., Gao, Z. X., Wu, P., Zhang, Y. H., et al. (2019). Norepinephrine-induced stimulation of Kir4.1/Kir5.1 is required for the activation of NaCl transporter in distal convoluted tubule. Hypertension 73, 112–120. doi: 10.1161/HYPERTENSIONAHA.118.11621
Duzic, E., Coupry, I., Downing, S., and Lanier, S. M. (1992). Factors determining the specificity of signal transduction by guanine nucleotide-binding protein-coupled receptors. I. Coupling of alpha 2-adrenergic receptor subtypes to distinct G-proteins. J. Biol. Chem. 267, 9844–9851.
Elenkov, I. J., Wilder, R. L., Chrousos, G. P., and Vizi, E. S. (2000). The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol. Rev. 52, 595–638.
Estrada, L. D., Agac, D., and Farrar, J. D. (2016). Sympathetic neural signaling via the beta2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8(+) T-cell effector function. Eur. J. Immunol. 46, 1948–1958. doi: 10.1002/eji.201646395
Faber, J. E., Yang, N., and Xin, X. (2001). Expression of alpha-adrenoceptor subtypes by smooth muscle cells and adventitial fibroblasts in rat aorta and in cell culture. J. Pharmacol. Exp. Ther. 298, 441–452.
Felsner, P., Hofer, D., Rinner, I., Porta, S., Korsatko, W., and Schauenstein, K. (1995). Adrenergic suppression of peripheral blood T cell reactivity in the rat is due to activation of peripheral alpha 2-receptors. J. Neuroimmunol. 57, 27–34. doi: 10.1016/0165-5728(94)00158-k
Flierl, M. A., Rittirsch, D., Nadeau, B. A., Chen, A. J., Sarma, J. V., Zetoune, F. S., et al. (2007). Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 449, 721–725. doi: 10.1038/nature06185
Frame, A. A., Puleo, F., Kim, K., Walsh, K. R., Faudoa, E., Hoover, R. S., et al. (2019). Sympathetic regulation of NCC in norepinephrine-evoked salt-sensitive hypertension in Sprague-Dawley rats. Am. J. Physiol. Renal Physiol. 317, F1623–F1636. doi: 10.1152/ajprenal.00264.2019
Freeman, K., Farrow, S., Schmaier, A., Freedman, R., Schork, T., and Lockette, W. (1995). Genetic polymorphism of the alpha 2-adrenergic receptor is associated with increased platelet aggregation, baroreceptor sensitivity, and salt excretion in normotensive humans. Am. J. Hypertens. 8, 863–869. doi: 10.1016/0895-7061(95)00155-I
GBD 2016 Risk Factors Collaborators (2017). Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet 390, 1345–1422. doi: 10.1016/S0140-6736(17)32366-8
Gilsbach, R., Albarran-Juarez, J., and Hein, L. (2011). Pre- versus post-synaptic signaling by alpha(2)-adrenoceptors. Curr. Top. Membr. 67, 139–160. doi: 10.1016/B978-0-12-384921-2.00007-0
Gilsbach, R., Roser, C., Beetz, N., Brede, M., Hadamek, K., Haubold, M., et al. (2009). Genetic dissection of alpha2-adrenoceptor functions in adrenergic versus nonadrenergic cells. Mol. Pharmacol. 75, 1160–1170. doi: 10.1124/mol.109.054544
Grassi, G., Mark, A., and Esler, M. (2015). The sympathetic nervous system alterations in human hypertension. Circ. Res. 116, 976–990. doi: 10.1161/CIRCRESAHA.116.303604
Grisanti, L. A., Perez, D. M., and Porter, J. E. (2011). Modulation of immune cell function by alpha(1)-adrenergic receptor activation. Curr. Top. Membr. 67, 113–138. doi: 10.1016/B978-0-12-384921-2.00006-9
Harwani, S. C. (2018). Macrophages under pressure: the role of macrophage polarization in hypertension. Transl. Res. 191, 45–63. doi: 10.1016/j.trsl.2017.10.011
Healy, V., Thompson, C., and Johns, E. J. (2014). The adrenergic regulation of proximal tubular Na+/H+ exchanger 3 in the rat. Acta Physiol. 210, 678–689. doi: 10.1111/apha.12181
Hein, L. (2006). Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 326, 541–551. doi: 10.1007/s00441-006-0285-2
Hein, L., Altman, J. D., and Kobilka, B. K. (1999). Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature 402, 181–184. doi: 10.1038/46040
Heinonen, P., Jartti, L., Jarvisalo, M. J., Pesonen, U., Kaprio, J. A., Ronnemaa, T., et al. (2002). Deletion polymorphism in the alpha2B-adrenergic receptor gene is associated with flow-mediated dilatation of the brachial artery. Clin. Sci. 103, 517–524. doi: 10.1042/cs1030517
Hering, L., Rahman, M., Hoch, H., Marko, L., Yang, G., Reil, A., et al. (2020). Alpha2A-adrenoceptors modulate renal sympathetic neurotransmission and protect against hypertensive kidney disease. J. Am. Soc. Nephrol. 31, 783–798. doi: 10.1681/ASN.2019060599
Hoch, H., Stegbauer, J., Potthoff, S. A., Hein, L., Quack, I., Rump, L. C., et al. (2011). Regulation of renal sympathetic neurotransmission by renal alpha(2A)-adrenoceptors is impaired in chronic renal failure. Br. J. Pharmacol. 163, 438–446. doi: 10.1111/j.1476-5381.2011.01223.x
Huhtinen, A., and Scheinin, M. (2008). Expression and characterization of the human alpha 2B-adrenoceptor in a vascular smooth muscle cell line. Eur. J. Pharmacol. 587, 48–56. doi: 10.1016/j.ejphar.2008.03.049
Jackson, E. K., Andresen, B. T., Seasholtz, T. M., Zhu, C., and Romero, G. G. (2005). Enhanced activation of RhoA by angiotensin II in SHR preglomerular microvascular smooth muscle cells. J. Cardiovasc. Pharmacol. 45, 283–285. doi: 10.1097/01.fjc.0000155383.83927.9f
Jackson, E. K., Herzer, W. A., Kost, C. K. Jr., and Vyas, S. J. (2001). Enhanced interaction between renovascular alpha(2)-adrenoceptors and angiotensin II receptors in genetic hypertension. Hypertension 38, 353–360. doi: 10.1161/01.hyp.38.3.353
Jang, H. S., Kim, J., and Padanilam, B. J. (2019). Renal sympathetic nerve activation via alpha2-adrenergic receptors in chronic kidney disease progression. Kidney Res. Clin. Pract. 38, 6–14. doi: 10.23876/j.krcp.18.0143
Kalyankrishna, S., and Malik, K. U. (2003). Norepinephrine-induced stimulation of p38 mitogen-activated protein kinase is mediated by arachidonic acid metabolites generated by activation of cytosolic phospholipase a(2) in vascular smooth muscle cells. J. Pharmacol. Exp. Ther. 304, 761–772. doi: 10.1124/jpet.102.040949
Kamat, N. V., Thabet, S. R., Xiao, L., Saleh, M. A., Kirabo, A., Madhur, M. S., et al. (2015). Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension 65, 569–576. doi: 10.1161/HYPERTENSIONAHA.114.04975
Kanagy, N. L. (2005). Alpha(2)-adrenergic receptor signalling in hypertension. Clin. Sci. 109, 431–437. doi: 10.1042/CS20050101
Kandzari, D. E., Bohm, M., Mahfoud, F., Townsend, R. R., Weber, M. A., Pocock, S., et al. (2018). Effect of renal denervation on blood pressure in the presence of antihypertensive drugs: 6-month efficacy and safety results from the SPYRAL HTN-ON MED proof-of-concept randomised trial. Lancet 391, 2346–2355. doi: 10.1016/S0140-6736(18)30951-6
Katayama, T., Sueta, D., Kataoka, K., Hasegawa, Y., Koibuchi, N., Toyama, K., et al. (2013). Long-term renal denervation normalizes disrupted blood pressure circadian rhythm and ameliorates cardiovascular injury in a rat model of metabolic syndrome. J. Am. Heart Assoc. 2:e000197. doi: 10.1161/JAHA.113.000197
Kelsey, R. M., Alpert, B. S., Dahmer, M. K., Krushkal, J., and Quasney, M. W. (2012). Alpha-adrenergic receptor gene polymorphisms and cardiovascular reactivity to stress in black adolescents and young adults. Psychophysiology 49, 401–412. doi: 10.1111/j.1469-8986.2011.01319.x
Kim, J., and Padanilam, B. J. (2013). Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J. Am. Soc. Nephrol. 24, 229–242. doi: 10.1681/ASN.2012070678
Kim, J., and Padanilam, B. J. (2015). Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 87, 350–358. doi: 10.1038/ki.2014.300
Kishore, B. K., Robson, S. C., and Dwyer, K. M. (2018). CD39-adenosinergic axis in renal pathophysiology and therapeutics. Purinergic Signal 14, 109–120. doi: 10.1007/s11302-017-9596-x
Kiuchi, M. G., Ho, J. K., Nolde, J. M., Gavidia, L. M. L., Carnagarin, R., Matthews, V. B., et al. (2019). Sympathetic activation in hypertensive chronic kidney disease—a stimulus for cardiac arrhythmias and sudden cardiac death? Front. Physiol. 10:1546. doi: 10.3389/fphys.2019.01546
Kocinsky, H. S., Girardi, A. C., Biemesderfer, D., Nguyen, T., Mentone, S., Orlowski, J., et al. (2005). Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am. J. Physiol. Renal Physiol. 289, F249–F258. doi: 10.1152/ajprenal.00082.2004
Kopp, U. C., Cicha, M. Z., and Smith, L. A. (2011a). Impaired interaction between efferent and afferent renal nerve activity in SHR involves increased activation of alpha2-adrenoceptors. Hypertension 57, 640–647. doi: 10.1161/HYPERTENSIONAHA.110.166595
Kopp, U. C., Cicha, M. Z., Smith, L. A., Mulder, J., and Hokfelt, T. (2007). Renal sympathetic nerve activity modulates afferent renal nerve activity by PGE2-dependent activation of alpha1- and alpha2-adrenoceptors on renal sensory nerve fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1561–R1572. doi: 10.1152/ajpregu.00485.2007
Kopp, U. C., Cicha, M. Z., Smith, L. A., Ruohonen, S., Scheinin, M., Fritz, N., et al. (2011b). Dietary sodium modulates the interaction between efferent and afferent renal nerve activity by altering activation of alpha2-adrenoceptors on renal sensory nerves. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R298–R310. doi: 10.1152/ajpregu.00469.2010
Krothapalli, R. K., and Suki, W. N. (1984). Functional characterization of the alpha adrenergic receptor modulating the hydroosmotic effect of vasopressin on the rabbit cortical collecting tubule. J. Clin. Invest. 73, 740–749. doi: 10.1172/JCI111267
Lee, J. W., Chou, C. L., and Knepper, M. A. (2015). Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J. Am. Soc. Nephrol. 26, 2669–2677. doi: 10.1681/ASN.2014111067
Lee, J. M., Han, H. J., Choi, W. K., Yoo, S., Baek, S., and Lee, J. (2018). Immunomodulatory effects of intraoperative dexmedetomidine on T helper 1, T helper 2, T helper 17 and regulatory T cells cytokine levels and their balance: a prospective, randomised, double-blind, dose-response clinical study. BMC Anesthesiol. 18:164. doi: 10.1186/s12871-018-0625-2
Leong, P. K., Yang, L. E., Landon, C. S., Mcdonough, A. A., and Yip, K. P. (2006). Phenol injury-induced hypertension stimulates proximal tubule Na+/H+ exchanger activity. Am. J. Physiol. Renal Physiol. 290, F1543–F1550. doi: 10.1152/ajprenal.00392.2005
Lundberg, J. M. (1996). Pharmacology of cotransmission in the autonomic nervous system: integrative aspects on amines, neuropeptides, adenosine triphosphate, amino acids and nitric oxide. Pharmacol. Rev. 48, 113–178.
Madhur, M. S., Lob, H. E., Mccann, L. A., Iwakura, Y., Blinder, Y., Guzik, T. J., et al. (2010). Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55, 500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094
Mansley, M. K., Neuhuber, W., Korbmacher, C., and Bertog, M. (2015). Norepinephrine stimulates the epithelial Na+ channel in cortical collecting duct cells via alpha2-adrenoceptors. Am. J. Physiol. Renal Physiol. 308, F450–F458. doi: 10.1152/ajprenal.00548.2014
Matsunaga, T., Yasuda, K., Adachi, T., Gu, N., Yamamura, T., Moritani, T., et al. (2007). Alpha-adrenoceptor gene variants and autonomic nervous system function in a young healthy Japanese population. J. Hum. Genet. 52:28. doi: 10.1007/s10038-006-0076-3
Mcdonough, A. A. (2010). Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R851–R861. doi: 10.1152/ajpregu.00002.2010
Mcdonough, A. A., and Nguyen, M. T. (2015). Maintaining balance under pressure: integrated regulation of renal transporters during hypertension. Hypertension 66, 450–455. doi: 10.1161/HYPERTENSIONAHA.115.04593
Muntz, K. H., Meyer, L., Gadol, S., and Calianos, T. A. (1986). Alpha-2 adrenergic receptor localization in the rat heart and kidney using autoradiography and tritiated rauwolscine. J. Pharmacol. Exp. Ther. 236, 542–547.
Nguyen, M. T., Lee, D. H., Delpire, E., and Mcdonough, A. A. (2013). Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am. J. Physiol. Renal Physiol. 305, F510–F519. doi: 10.1152/ajprenal.00183.2013
Nord, E. P., Howard, M. J., Hafezi, A., Moradeshagi, P., Vaystub, S., and Insel, P. A. (1987). Alpha 2 adrenergic agonists stimulate Na+-H+ antiport activity in the rabbit renal proximal tubule. J. Clin. Invest. 80, 1755–1762. doi: 10.1172/JCI113268
Nunes, R. A., Barroso, L. P., Pereira Ada, C., Krieger, J. E., and Mansur, A. J. (2014). Gender-related associations of genetic polymorphisms of alpha-adrenergic receptors, endothelial nitric oxide synthase and bradykinin B2 receptor with treadmill exercise test responses. Open Heart 1:e000132. doi: 10.1136/openhrt-2014-000132
Oberhauser, V., Vonend, O., and Rump, L. C. (1999). Neuropeptide Y and ATP interact to control renovascular resistance in the rat. J. Am. Soc. Nephrol. 10, 1179–1185.
Penton, D., Moser, S., Wengi, A., Czogalla, J., Rosenbaek, L. L., Rigendinger, F., et al. (2019). Protein phosphatase 1 inhibitor-1 mediates the cAMP-dependent stimulation of the renal NaCl cotransporter. J. Am. Soc. Nephrol. 30, 737–750. doi: 10.1681/ASN.2018050540
Perry, H. M., Gorldt, N., Sung, S. J., Huang, L., Rudnicka, K. P., Encarnacion, I. M., et al. (2019). Perivascular CD73+ cells attenuate inflammation and interstitial fibrosis in the kidney microenvironment. Am. J. Physiol. Renal Physiol. 317, F658–F669. doi: 10.1152/ajprenal.00243.2019
Philipp, M., Brede, M., and Hein, L. (2002). Physiological significance of alpha(2)-adrenergic receptor subtype diversity: one receptor is not enough. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R287–R295. doi: 10.1152/ajpregu.00123.2002
Pontes, R. B., Crajoinas, R. O., Nishi, E. E., Oliveira-Sales, E. B., Girardi, A. C., Campos, R. R., et al. (2015). Renal nerve stimulation leads to the activation of the Na+/H+ exchanger isoform 3 via angiotensin II type I receptor. Am. J. Physiol. Renal Physiol. 308, F848–F856. doi: 10.1152/ajprenal.00515.2014
Poss, J., Ewen, S., Schmieder, R. E., Muhler, S., Vonend, O., Ott, C., et al. (2015). Effects of renal sympathetic denervation on urinary sodium excretion in patients with resistant hypertension. Clin. Res. Cardiol. 104, 672–678. doi: 10.1007/s00392-015-0832-5
Pyner, S. (2014). The paraventricular nucleus and heart failure. Exp. Physiol. 99, 332–339. doi: 10.1113/expphysiol.2013.072678
Roos, K. P., Bugaj, V., Mironova, E., Stockand, J. D., Ramkumar, N., Rees, S., et al. (2013). Adenylyl cyclase VI mediates vasopressin-stimulated ENaC activity. J. Am. Soc. Nephrol. 24, 218–227. doi: 10.1681/ASN.2012050449
Saha, M., Menuet, C., Sun, Q. J., Burke, P. G. R., Hildreth, C. M., Allen, A. M., et al. (2019). Respiratory sympathetic modulation is augmented in chronic kidney disease. Respir. Physiol. Neurobiol. 262, 57–66. doi: 10.1016/j.resp.2019.02.001
Sata, Y., Head, G. A., Denton, K., May, C. N., and Schlaich, M. P. (2018). Role of the sympathetic nervous system and its modulation in renal hypertension. Front. Med. 5:82. doi: 10.3389/fmed.2018.00082
Scanzano, A., and Cosentino, M. (2015). Adrenergic regulation of innate immunity: a review. Front. Pharmacol. 6:171. doi: 10.3389/fphar.2015.00171
Schlaich, M. P., Hering, D., Sobotka, P., Krum, H., Lambert, G. W., Lambert, E., et al. (2012). Effects of renal denervation on sympathetic activation, blood pressure, and glucose metabolism in patients with resistant hypertension. Front. Physiol. 3:10. doi: 10.3389/fphys.2012.00010
Schlaich, M. P., Sobotka, P. A., Krum, H., Lambert, E., and Esler, M. D. (2009). Renal sympathetic-nerve ablation for uncontrolled hypertension. N. Engl. J. Med. 361, 932–934. doi: 10.1056/NEJMc0904179
Small, K. M., Wagoner, L. E., Levin, A. M., Kardia, S. L., and Liggett, S. B. (2002). Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N. Engl. J. Med. 347, 1135–1142. doi: 10.1056/NEJMoa020803
Solini, A., Usuelli, V., and Fiorina, P. (2015). The dark side of extracellular ATP in kidney diseases. J. Am. Soc. Nephrol. 26, 1007–1016. doi: 10.1681/ASN.2014070721
Soukhova-O'hare, G. K., Roberts, A. M., and Gozal, D. (2006). Impaired control of renal sympathetic nerve activity following neonatal intermittent hypoxia in rats. Neurosci. Lett. 399, 181–185. doi: 10.1016/j.neulet.2006.01.054
Sparks, M. A., Stegbauer, J., Chen, D., Gomez, J. A., Griffiths, R. C., Azad, H. A., et al. (2015). Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J. Am. Soc. Nephrol. 26, 2953–2962. doi: 10.1681/ASN.2014080816
Spengler, R. N., Chensue, S. W., Giacherio, D. A., Blenk, N., and Kunkel, S. L. (1994). Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro. J. Immunol. 152, 3024–3031.
Sriramula, S., Haque, M., Majid, D. S., and Francis, J. (2008). Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51, 1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152
Starke, K., Endo, T., and Taube, H. D. (1975). Pre- and postsynaptic components in effect of drugs with alpha adrenoceptor affinity. Nature 254, 440–441. doi: 10.1038/254440a0
Steinberg, J. S., Shabanov, V., Ponomarev, D., Losik, D., Ivanickiy, E., Kropotkin, E., et al. (2020). Effect of renal denervation and catheter ablation vs catheter ablation alone on atrial fibrillation recurrence among patients with paroxysmal atrial fibrillation and hypertension: the ERADICATE-AF randomized clinical trial. JAMA 323, 248–255. doi: 10.1001/jama.2019.21187
Sumi, Y., Woehrle, T., Chen, Y., Yao, Y., Li, A., and Junger, W. G. (2010). Adrenergic receptor activation involves ATP release and feedback through purinergic receptors. Am. J. Physiol. Cell Physiol. 299, C1118–C1126. doi: 10.1152/ajpcell.00122.2010
Tomoda, F., Bergstrom, G., Evans, R. G., and Anderson, W. P. (1997). Evidence for decreased structurally determined preglomerular resistance in the young spontaneously hypertensive rat after 4 weeks of renal denervation. J. Hypertens. 15, 1187–1195. doi: 10.1097/00004872-199715100-00018
Trendelenburg, A. U., Klebroff, W., Hein, L., and Starke, K. (2001). A study of presynaptic alpha2-autoreceptors in alpha2A/D-, alpha2B- and alpha2C-adrenoceptor-deficient mice. Naunyn Schmiedeberg's Arch. Pharmacol. 364, 117–130. doi: 10.1007/s002100100423
Veelken, R., Vogel, E. M., Hilgers, K., Amann, K., Hartner, A., Sass, G., et al. (2008). Autonomic renal denervation ameliorates experimental glomerulonephritis. J. Am. Soc. Nephrol. 19, 1371–1378. doi: 10.1681/ASN.2007050552
Veiras, L. C., Mcfarlin, B. E., Ralph, D. L., Buncha, V., Prescott, J., Shirvani, B. S., et al. (2020). Electrolyte and transporter responses to angiotensin II induced hypertension in female and male rats and mice. Acta Physiol. 229:e13448. doi: 10.1111/apha.13448
von Wowern, F., Bengtsson, K., Lindblad, U., Rastam, L., and Melander, O. (2004). Functional variant in the (alpha)2B adrenoceptor gene, a positional candidate on chromosome 2, associates with hypertension. Hypertension 43, 592–597. doi: 10.1161/01.HYP.0000116224.51189.80
Vonend, O., Habbel, S., Stegbauer, J., Roth, J., Hein, L., and Rump, L. C. (2007). Alpha(2A)-adrenoceptors regulate sympathetic transmitter release in mice kidneys. Br. J. Pharmacol. 150, 121–127. doi: 10.1038/sj.bjp.0706961
Vonend, O., Marsalek, P., Russ, H., Wulkow, R., Oberhauser, V., and Rump, L. C. (2003). Moxonidine treatment of hypertensive patients with advanced renal failure. J. Hypertens. 21, 1709–1717. doi: 10.1097/00004872-200309000-00021
Vonend, O., Martin, O., Rump, L. C., Kroepil, P., and Stegbauer, J. (2018). Erythrocyte salt sedimentation assay does not predict response to renal denervation. Front. Med. 5:51. doi: 10.3389/fmed.2018.00051
Wang, Y., Hou, R., Li, P., Li, J., Yan, J., Yin, F., et al. (2004). Gene expression profiles in response to the activation of adrenoceptors in A7r5 aortic smooth muscle cells. Clin. Exp. Pharmacol. Physiol. 31, 602–607. doi: 10.1111/j.1440-1681.2004.04058.x
Wenzel, P., Knorr, M., Kossmann, S., Stratmann, J., Hausding, M., Schuhmacher, S., et al. (2011). Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 124, 1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470
Wu, J., Saleh, M. A., Kirabo, A., Itani, H. A., Montaniel, K. R., Xiao, L., et al. (2016). Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J. Clin. Invest. 126, 50–67. doi: 10.1172/JCI80761
Xiao, L., Do Carmo, L., Foss, J. D., Chen, W., and Harrison, D. G. (2020). Sympathetic enhancement of memory T cell homing and hypertension sensitization. Circ. Res. 126, 708–721. doi: 10.1161/CIRCRESAHA.119.314758
Xiao, L., Kirabo, A., Wu, J., Saleh, M. A., Zhu, L., Wang, F., et al. (2015). Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-induced hypertension. Circ. Res. 117, 547–557. doi: 10.1161/CIRCRESAHA.115.306010
Ye, S., Zhong, H., Duong, V. N., and Campese, V. M. (2002a). Losartan reduces central and peripheral sympathetic nerve activity in a rat model of neurogenic hypertension. Hypertension 39, 1101–1106. doi: 10.1161/01.hyp.0000018590.26853.c7
Ye, S., Zhong, H., Yanamadala, V., and Campese, V. M. (2002b). Renal injury caused by intrarenal injection of phenol increases afferent and efferent renal sympathetic nerve activity. Am. J. Hypertens. 15, 717–724. doi: 10.1016/s0895-7061(02)02959-x
Zaldivia, M. T., Rivera, J., Hering, D., Marusic, P., Sata, Y., Lim, B., et al. (2017). Renal denervation reduces monocyte activation and monocyte-platelet aggregate formation: an anti-inflammatory effect relevant for cardiovascular risk. Hypertension 69, 323–331. doi: 10.1161/HYPERTENSIONAHA.116.08373
Zheng, H., Liu, X., Katsurada, K., and Patel, K. P. (2019). Renal denervation improves sodium excretion in rats with chronic heart failure: effects on expression of renal ENaC and AQP2. Am. J. Physiol. Heart Circ. Physiol. 317, H958–H968. doi: 10.1152/ajpheart.00299.2019
Keywords: renal sympathetic nervous system, hypertension, α2-adrenoceptors, sodium transporters, renal vasculature resistance, renal sympathetic neurotransmission, immune cells, macrophages
Citation: Hering L, Rahman M, Potthoff SA, Rump LC and Stegbauer J (2020) Role of α2-Adrenoceptors in Hypertension: Focus on Renal Sympathetic Neurotransmitter Release, Inflammation, and Sodium Homeostasis. Front. Physiol. 11:566871. doi: 10.3389/fphys.2020.566871
Edited by:
Jennifer Sullivan, Augusta University, United StatesReviewed by:
Adriana Castello Costa Girardi, University of São Paulo, BrazilCopyright © 2020 Hering, Rahman, Potthoff, Rump and Stegbauer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes Stegbauer, bHlkaWEuaGVyaW5nQG1lZC51bmktZHVlc3NlbGRvcmYuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.