
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 20 October 2020
Sec. Mitochondrial Research
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.566506
This article is part of the Research TopicMitochondria in Renal Health and DiseaseView all 10 articles
 Sakdithep Chaiyarit
Sakdithep Chaiyarit Visith Thongboonkerd*
Visith Thongboonkerd*Mitochondrion is a pivotal intracellular organelle that plays crucial roles in regulation of energy production, oxidative stress, calcium homeostasis, and apoptosis. Kidney stone disease (nephrolithiasis/urolithiasis), particularly calcium oxalate (CaOx; the most common type), has been shown to be associated with oxidative stress and tissue inflammation/injury. Recent evidence has demonstrated the involvement of mitochondrial dysfunction in CaOx crystal retention and aggregation as well as Randall’s plaque formation, all of which are the essential mechanisms for kidney stone formation. This review highlights the important roles of mitochondria in renal cell functions and provides the data obtained from previous investigations of mitochondria related to kidney stone disease. In addition, mechanisms for the involvement of mitochondrial dysfunction in the pathophysiology of kidney stone disease are summarized. Finally, future perspectives on the novel approach to prevent kidney stone formation by mitochondrial preservation are discussed.
Mitochondrion is a unique and dynamic intracellular organelle that varies in shape, size, and number among various cell types. It is the only subcellular organelle that has double membranes and contains its own genome. Unlike nuclear DNA, mitochondrial DNA (mtDNA) is circular double-stranded DNA that is not protected by histones (Sharma and Sampath, 2019; Yan et al., 2019). Generally, mitochondria are the foremost generator of cellular energy in human cells by converting oxygen and nutritional elements to adenosine triphosphate (ATP) via oxidative phosphorylation pathway. Tricarboxylic acid (TCA) cycle (Kreb’s cycle or citric acid cycle) and electron transport chain (ETC) are the two major metabolic processes that play pivotal roles in this energy production mechanism (Brookes et al., 2004; Pieczenik and Neustadt, 2007). Additionally, mitochondria also involve regulation of cellular oxidative stress, calcium homeostasis, apoptosis-signaling pathway, and aging processes in all cells and tissues (Bhargava and Schnellmann, 2017; Duann and Lin, 2017). Mitochondrial dysfunction, therefore, can lead to development of various metabolic diseases and other disorders, such as diabetes (Forbes and Thorburn, 2018; Ducasa et al., 2019), obesity (Song et al., 2017; Schottl et al., 2020), aging (Wojtovich et al., 2012; Giorgi et al., 2018; Jang et al., 2018; Son and Lee, 2019), cancers (Anderson et al., 2018; Neagu et al., 2019; Missiroli et al., 2020), myopathies (Walters et al., 2012; Apostolopoulou et al., 2015), and neurodegenerative disorders (Muller et al., 2018; Rango and Bresolin, 2018; Takahashi and Takahashi, 2019). Interestingly, kidney stone disease (nephrolithiasis or urolithiasis) has been shown to be associated, either directly or indirectly, with mitochondrial dysfunction (Cao et al., 2004; Williams et al., 2016; Patel et al., 2018; Dominguez-Gutierrez et al., 2020).
This review highlights the important roles of mitochondria in renal cell functions and emphasizes associations between mitochondrial dysfunction and the pathophysiology of kidney stone disease as well as the potential for the novel approach to prevent kidney stone formation by preservation of mitochondrial functions.
The kidney is the second organ after the heart that contains the largest number of mitochondria per cell (Wang et al., 2010; Forbes, 2016). This organ and its related urinary tract system require sufficient energy for body homeostasis, blood filtration, nutrient reabsorption, regulation of body fluid and electrolyte, acid-base balance, and blood pressure regulation (Bhargava and Schnellmann, 2017). The most important source of cellular energy is generated by mitochondria, which are the crucial intracellular organelles to support the renal cell functions. In particular, proximal renal tubular cells are the mitochondria-rich cells that have higher density of mitochondria than other cells lining along the nephron because they require much more energy for their functions, including but not limited to reabsorption of water, glucose, ions, and nutrients (Forbes, 2016). Also, intercalated cells of the collecting ducts are the other mitochondria-rich cells playing roles in acid-base balance and regulation of sodium, chloride, and potassium transports (Roy et al., 2015).
ATP molecules are generated mainly by electron transfer across the mitochondrial inner membrane (MIM) via ETC during oxidative phosphorylation. In addition, TCA cycle also plays pivotal roles in mitochondrial energy production (Forbes, 2016). The TCA cycle produces the following coenzymes in mitochondrial matrix, including three molecules of nicotine adenine dinucleotide (NADH) and one molecule of flavin adenine dinucleotide (FADH2), from acetate in the form or acetyl coenzyme A (acetyl CoA), which is converted from pyruvate via pyruvate dehydrogenase after glycolysis of one molecule of glucose (Pieczenik and Neustadt, 2007; Ralto et al., 2020). In addition to glycolysis, fatty acid oxidation is another mechanism that can produce even more acetyl CoA molecules for entering into the TCA cycle. As a result, fatty acid oxidation generates more NADH and FADH2 molecules compared to glycolysis (Sharpe and McKenzie, 2018). Electrons from NADH and FADH2 are transferred to NADH dehydrogenase (complex I) and succinate dehydrogenase (complex II), respectively, of ETC in the MIM. The electrons were then shuttled to ubiquinol-cytochrome c reductase (complex III) by ubiquinone (coenzyme Q10; CoQ10) and transferred to cytochrome c oxidase (COX; complex IV) by cytochrome c (Bhargava and Schnellmann, 2017). During electron transfer through the ETC in the MIM, protons are actively pumped by complexes I, III, and IV through mitochondrial intermembrane space. Finally, ATP synthase (complex V) uses excess protons in mitochondrial intermembrane space to phosphorylate adenosine diphosphate (ADP) to ATP (Bhargava and Schnellmann, 2017). The requirement for ATP in the kidney is cell type-specific as tubular cells need high energy to mediate the active transport function inside the renal cortex. In contrast, cells in the glomerular segment require lower energy for filtration and other passive processes. Overall, a large number of mitochondria and their high activities are required by renal tubular cells to maintain several renal functions.
Oxidative stress is mostly induced by excessive cytoplasmic and mitochondrial reactive oxygen species (ROS) emission (Khand et al., 2002; Duann and Lin, 2017). Electron transfer at complexes I and III has been proposed as the main source of ROS overproduction (Ray et al., 2012). Mitochondria can consume oxygen for ATP production, leading to generation of ROS such as superoxide (O2−) and hydrogen peroxide (H2O2). These ROS are important for cellular signaling pathways as well as cell cycles (Deng et al., 2003; Chang et al., 2005; An et al., 2013), apoptosis (An et al., 2013), protein kinases, and protein phosphatase (Brookes et al., 2002). Under physiologic condition, mitochondria have highly efficient antioxidant systems to keep the balance with ROS (Stowe and Camara, 2009). For example, superoxide dismutase rapidly reduces O2− to H2O2 faster than the rate of O2− production (Bhargava and Schnellmann, 2017). Subsequently, glutathione peroxidase completes the reduction processes by converting H2O2 to water (Fernandez-Checa et al., 1998; Ribas et al., 2014). During oxidative stress (pathological condition), excessive ROS emission occurs because the mitochondrial scavenging system is overwhelmed by the perpetual increase in ROS production, the so-called ROS-induced ROS release (RIRR; Zorov et al., 2014). As a result, mitochondrial functions are altered, leading to mtDNA damage and oxidative modifications of mitochondrial proteins and enzymes in TCA cycle and ETC (Khand et al., 2002; Bhargava and Schnellmann, 2017; Duann and Lin, 2017).
In oxidative stress condition, the nuclear factor erythroid 2-related factor 2 (NRF2) can trigger transcription factors of genes encoding antioxidant enzymes to cope with the ROS overproduction (Ruiz et al., 2013). This underscores the important regulatory roles of the mitochondrial antioxidant systems to maintain intracellular ATP production for all cellular events and to preserve mitochondrial functions. In particular, glutathione redox cycle is the critical antioxidant mechanism found in cytoplasm and intracellular organelles, such as nucleus, endoplasmic reticulum, and mitochondria (Ribas et al., 2014; Reiter et al., 2018). Mitochondrial glutathione (mGSH) is the reduced form of glutathione found in mitochondrial matrix and can be oxidized to glutathione disulfide (GSSG) by superoxide anions (Fernandez-Checa et al., 1998; Ribas et al., 2014). GSSG can be reversed to mGSH by glutathione reductase that requires NADPH from the pentose phosphate pathway (Baudouin-Cornu et al., 2012; Lushchak, 2012; Bajic et al., 2019). These two mechanisms are the pivotal processes for preventing oxidative stress and preserving mitochondrial functions.
Mitochondria have been found to also involve program cell death or apoptosis (Scatena, 2012). The apoptotic cell death is considered when the cell morphology/biology is changed with membrane blebbing, cell shrinkage, and nuclear DNA fragments (Indran et al., 2011). Eventually, apoptotic cells are eliminated by neighboring and/or immune cells to avoid tissue inflammation and damage. Apoptotic cell death mechanism has two main pathways, intrinsic (via mitochondria) and extrinsic (via death receptors) pathways (Grancara et al., 2016; Abate et al., 2020). However, activation of caspases is the common and key final process that both pathways share together. Caspases are classified into two groups, based on their activities, as the initiator caspases (caspase-2, -8, -9, and -10) and the effector caspases (caspase-3, -6, and -7).
In the intrinsic pathway, mitochondrial cytochrome c is released to the cytoplasm and binds with apoptotic protease activating factor 1 (APAF-1) and ATP, resulting in recruitment of procaspase-9 to form an apoptosome, in which procaspase-9 is cleaved to caspase-9. The caspase-9 then converts procaspase-3 to effector caspase-3, resulting in the completion of cellular apoptosis (Pradelli et al., 2010). For the extrinsic pathway, extracellular ligands bind to the death receptors, leading to formation of death-inducing signaling complex that then cleaves procaspase-8 to caspase-8 (Franklin, 2011). The caspase-8 can directly stimulate the effector caspase-3 that subsequently degrades a variety of proteins during apoptosis. Moreover, the effector caspase-8 has been reported to induce cytochrome c release from mitochondria by increasing permeability of mitochondrial outer membrane (MOM) (Elmore, 2007; Indran et al., 2011). Using both intrinsic and extrinsic pathways, mitochondria thus serve as the important intracellular organelles for regulation of the apoptotic cell death.
Previous studies have shown that the pathogenic processes of kidney stone disease are associated with oxidative stress condition (Hirose et al., 2010; Niimi et al., 2012). The involvement of oxidative stress in kidney stone disease has been found in various in vitro studies (Chaiyarit and Thongboonkerd, 2012; Peerapen et al., 2018) and animal models (Hirose et al., 2010; Niimi et al., 2014) as well as stone formers (patients with kidney stones) (Ma et al., 2014; Ceban et al., 2016). Such oxidative induction leads to several downstream cascades, particularly inflammatory response and tissue injury (Williams et al., 2016; Yasui et al., 2017; Dominguez-Gutierrez et al., 2020). More importantly, there is increasing evidence demonstrating that the tissue injury induced by oxidative stress enhances retention of the causative crystals [especially calcium oxalate (CaOx)] inside renal tubules and/or kidney interstitium (parenchyma) that is one of the crucial steps for kidney stone formation (Cao et al., 2004; Khan, 2013, 2014). While a number of reports have shown overproduction of ROS in renal tubular epithelial cells followed by cellular injury, many antioxidant compounds have been introduced to cope with such oxidative stress, to reduce cellular/tissue injury, and to inhibit kidney stone formation (Muthukumar and Selvam, 1998; Itoh et al., 2005; Holoch and Tracy, 2011; Fishman et al., 2013; Zhai et al., 2013; Yang et al., 2016; Zeng et al., 2019). Because cellular oxidation is mostly associated with mitochondrial activities, mitochondria are thus directly involved in such cellular oxidative stress.
Calcium is the most common cation that is precipitated in the urine as the crystalline forms with other anions, especially oxalate and phosphate. Such calcium-containing crystals are commonly found in the urine of kidney stone formers (Yasui et al., 2017). Among all chemical types of kidney stones, CaOx is the most common one found worldwide (Vinaiphat et al., 2017; Vinaiphat and Thongboonkerd, 2017). After crystallization, CaOx crystals can adhere on the surface of renal tubular cells and are then internalized into the cells via macropinocytosis (Kanlaya et al., 2013) for subsequent elimination through degradation and/or dissolution (Chaiyarit et al., 2016). The end products of such elimination processes are free calcium and oxalate ions. When the intracellular calcium level is changed, mitochondria play role as one among other mechanisms to regulate intracellular calcium homeostasis. Normally, calcium can promote mitochondrial energy production. On the other hand, calcium overload may cause mitochondrial dysfunction and ROS overproduction (Brookes et al., 2004; Muller et al., 2018). The excess ROS can cause mtDNA damage followed by alterations in mitochondrial fission/fusion process, leading to cellular injury, apoptosis, inflammatory response, and finally the disease progression (Brookes et al., 2004; Suarez-Rivero et al., 2016; Srinivasan et al., 2017; Muller et al., 2018; Yan et al., 2019).
Mitochondrial dysfunction has been recognized as one of the important keys in the etiology of kidney stone disease nearly four decades ago (Laxmanan et al., 1986; Harrison et al., 1988). Calcium dense deposits have been demonstrated inside the enlarged mitochondria in renal tubular cells of the stone formers by electron microscopy without crystalline structure observed (Harrison et al., 1988). Several other investigations have also provided evidence showing the ability of mitochondria to bind with oxalate and CaOx crystals (Laxmanan et al., 1986; Govindaraj and Selvam, 2001, 2002; Selvam and Kalaiselvi, 2003; Hirose et al., 2012; Kohri et al., 2012; Roop-Ngam et al., 2012). The binding mechanism between mitochondria and CaOx crystals has been suggested to be mediated through peroxidation of mitochondrial proteins and lipids, which further promote nucleation and aggregation of CaOx crystals (Govindaraj and Selvam, 2001). The involvement of mitochondria in early phase of kidney stone formation has gained a wide attention because mitochondrial proteins have been found in stone matrices (Govindaraj and Selvam, 2001, 2002) and the mitochondrial fragments have been found together with the crystals along distal renal tubular lumens, suggesting their roles in crystal nucleation (Hirose et al., 2012).
Although mitochondria have been found to directly interact with oxalate ion and CaOx crystals, previous investigations of mitochondria have focused only to their roles in regulation of oxidative stress and tissue injury that commonly occur in kidney stone disease. CaOx crystals have been found to induce oxidative stress in renal tubular cells leading to mitochondrial dysfunction and renal cell injury (Khan, 2013, 2014). Renal tubular cell injury and the defective mitochondria and other intracellular organelles are evidence of aggravated CaOx crystal retention inside the renal tissue, which is considered as one of the important steps for kidney stone development (Govindaraj and Selvam, 2002; Yasui et al., 2017). Following this line of investigations, several previous studies on kidney stone disease thus explored the effects of mitochondrial injury, ROS overproduction, loss of the mitochondrial membrane potential, and mitochondrial swelling (Jonassen et al., 2005; Mcmartin, 2009; Chaiyarit and Thongboonkerd, 2012; Niimi et al., 2012; Yasui et al., 2017; Peng et al., 2019; Wu et al., 2019). Moreover, CaOx crystals can induce production of mitochondrial O2− that further activates cyclophilin D (CypD), which is a component of mitochondrial permeability transition pore (mPTP) (Niimi et al., 2012). CypD plays a role in opening such pore and thus affects the permeability of mitochondria at MIM and MOM (Niimi et al., 2012). Additionally, CaOx induces cytochrome c release to cytoplasm for further activation of caspases and related signaling pathways, resulting in apoptosis of renal tubular cells (Jonassen et al., 2003; Niimi et al., 2012). On the other hand, mGSH maintains the mitochondrial integrity and functions, and can also reduce oxalate deposition in hyperoxaluria condition (Muthukumar and Selvam, 1998). Vice versa, mGSH reduction can induce mitochondrial dysfunction and may contribute to the development of CaOx kidney stones (Muthukumar and Selvam, 1998).
Our previous mitochondrial proteome study has highlighted the response of renal tubular epithelial cells of the distal nephron segment to CaOx crystals (Chaiyarit and Thongboonkerd, 2012). Using two-dimensional gel electrophoresis followed by quadrupole time-of-flight tandem mass spectrometry (Q-TOF MS/MS), a total of 15 mitochondrial proteins were identified to have differential expression levels after the distal renal tubular cells were exposed to CaOx crystals. Among these, proteins that played roles in maintaining mitochondrial functions (i.e., pyruvate dehydrogenase, ATP synthase, and NADH dehydrogenase) and cell death (ezrin) were increased (Chaiyarit and Thongboonkerd, 2012). In combination with comprehensive bioinformatics analysis of other large proteome datasets together with functional validation, the additional results indicate the association between mitochondrial dysfunction and oxidative stress-induced renal tubular cell injury (Peerapen et al., 2018).
Mitochondria have also been investigated in Randall’s plaque model of kidney stone pathogenesis. Mitochondrial dysfunction has been found to be related not only to renal tubular injury but also to the impaired immune response and inflammation by decreasing monocytes’ mitochondrial functions in the CaOx stone formers, leading to the decline of crystal elimination that further enhances tissue inflammation (Williams et al., 2016; Patel et al., 2018; Dominguez-Gutierrez et al., 2020). Progressive tissue inflammation together with supersaturation of calcium phosphate then induces Randall’s plaque formation and finally kidney stone development (Williams et al., 2016; Patel et al., 2018; Dominguez-Gutierrez et al., 2020). To cope with the impaired immune response and tissue inflammation as well as the interstitial plaque development, various antioxidants or free radical scavengers (Muthukumar and Selvam, 1998; Kohri et al., 2012; Zhai et al., 2013; Aggarwal et al., 2016; Chhiber et al., 2016) and polysaccharide compounds (Veena et al., 2007; Sun et al., 2016; Guo et al., 2018; Sun et al., 2019) have been shown to serve as the therapeutic/preventive strategies to rescue/prevent kidney stone formation.
Oxalate and calcium can alter mitochondrial activities, leading to changes in metabolic status that may induce loss or alterations of mitochondrial functions on energy production, ROS regulation, and intracellular calcium homeostasis, all of which affect mitochondrial biogenesis (Veena et al., 2008; Hirose et al., 2010; Niimi et al., 2012; Sun et al., 2017). The dynamic processes between mitochondrial fusion and fission generate various by-products, most of which are mitochondrial fragments and ROS (Aparicio-Trejo et al., 2018; Janikiewicz et al., 2018; Geto et al., 2020). As aforementioned, several lines of evidence have suggested the involvement of mitochondrial dysfunction in the initial phase of kidney stone disease. There are three main mechanisms proposed for the involvement of mitochondrial dysfunction in the pathophysiology of kidney stone disease.
Mitochondrial dysfunction can increase retention of CaOx crystals inside renal tubular lumens of the distal nephron segment. These stagnant crystals subsequently form the stone nidus, which is the central part of the stone generated from crystal aggregates that finally become the macroscopic stone. The loss or defect of mitochondrial energy production can enhance metabolic process in TCA cycle and ETC, resulting in the increase of ROS production and reduction of antioxidant enzymes (Brookes et al., 2004; Chaiyarit and Thongboonkerd, 2012; Peerapen et al., 2018). Consequently, the excess mitochondrial free radicals can damage mitochondrial membranes (MIM and MOM). As a result, mitochondrial ROS, cytochrome c, calcium, and other proinflammatory factors are further released to the cytoplasm (Cao et al., 2016; Fong-Ngern et al., 2017). High level of cytoplasmic ROS can induce lipid peroxidation, which damages cell membranes and further enhances crystal deposition on apical surfaces of renal tubular cells (Cao et al., 2016; Fong-Ngern et al., 2017). Additionally, apoptotic signaling cascades are activated by cytochrome c that also upsurges renal tubular cell injury, leading to crystal adhesion and intrarenal crystal retention (Cao et al., 2016; Fong-Ngern et al., 2017). The accumulated crystals can be further enlarged and self-aggregated, leading to the stone formation (Figure 1).
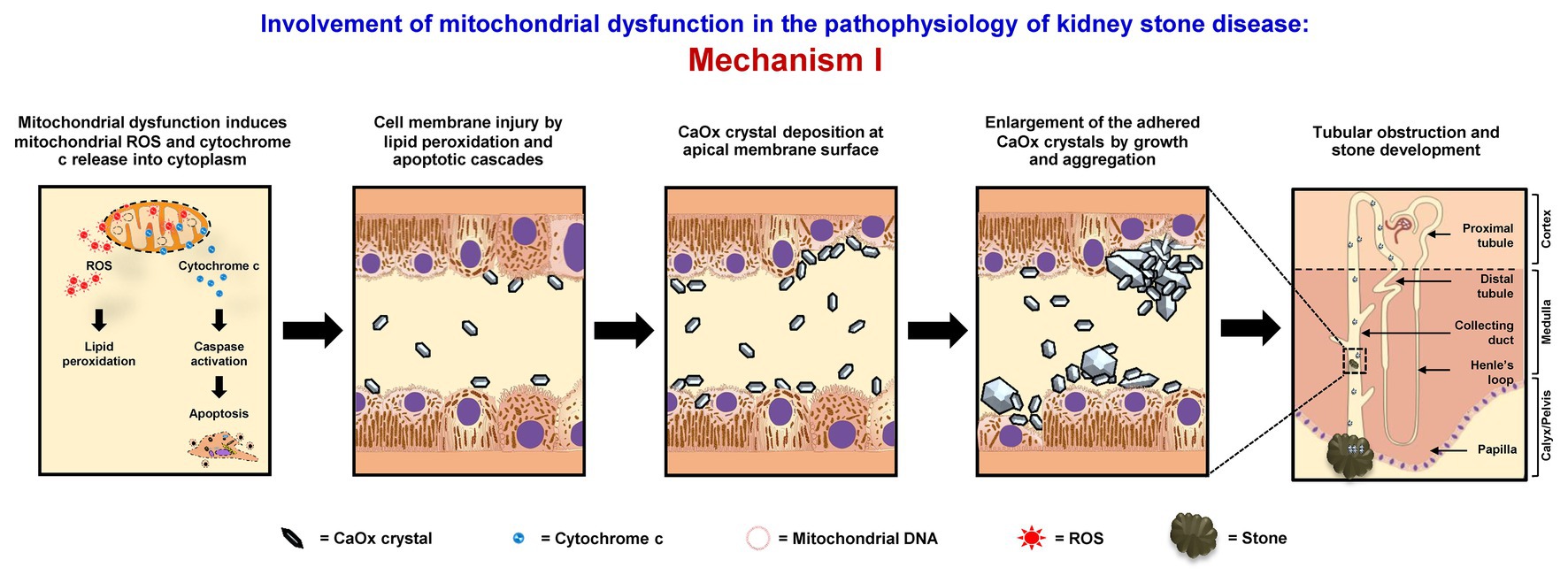
Figure 1. Involvement of mitochondrial dysfunction in the pathophysiology of kidney stone disease: Mechanism I – Mitochondrial dysfunction induces the release of mitochondrial reactive oxygen species (ROS) and cytochrome c to cytoplasm. The lipid peroxidation and apoptotic cascades can then cause cell membrane injury that enhances calcium oxalate (CaOx) crystal deposition on apical surfaces of renal tubular cells of the distal nephron segment. The accumulated crystals can be further enlarged and self-aggregated, leading to the stone formation. The right panel shows the macroscopic diagram to demonstrate the locales (related to the nephron segments) where the proposed mechanism occurs.
Mitochondrial dysfunction causes the release of cytochrome c and induces apoptotic cell death, associated with cell shrinkage, blebbing of plasma membranes, organelle condensation, and fragmentation. Thereafter, apoptotic bodies, cell debris, and fragmented subcellular organelles are released into tubular lumen and then bind with oxalate and CaOx crystals as shown by several studies (Govindaraj and Selvam, 2001, 2002; Selvam and Kalaiselvi, 2003; Hirose et al., 2012; Kohri et al., 2012). Also, fragmented mitochondria and mitochondrial proteins (i.e., 48-kDa protein) have been found in the stone core matrices and thus may get involved in the stone nidus formation (Govindaraj and Selvam, 2001, 2002). These mitochondrial components, membrane fragments, and other cellular debris can directly serve as the raw materials for the stone nidus formation in the distal nephron segment and further promote crystal nucleation, growth, and aggregation. The large aggregates may obstruct tubular lumen and/or migrate (by renal tubular fluid flow) to the calyx and pelvis to form the stone (Evan, 2010; Khan et al., 2016) (Figure 2).
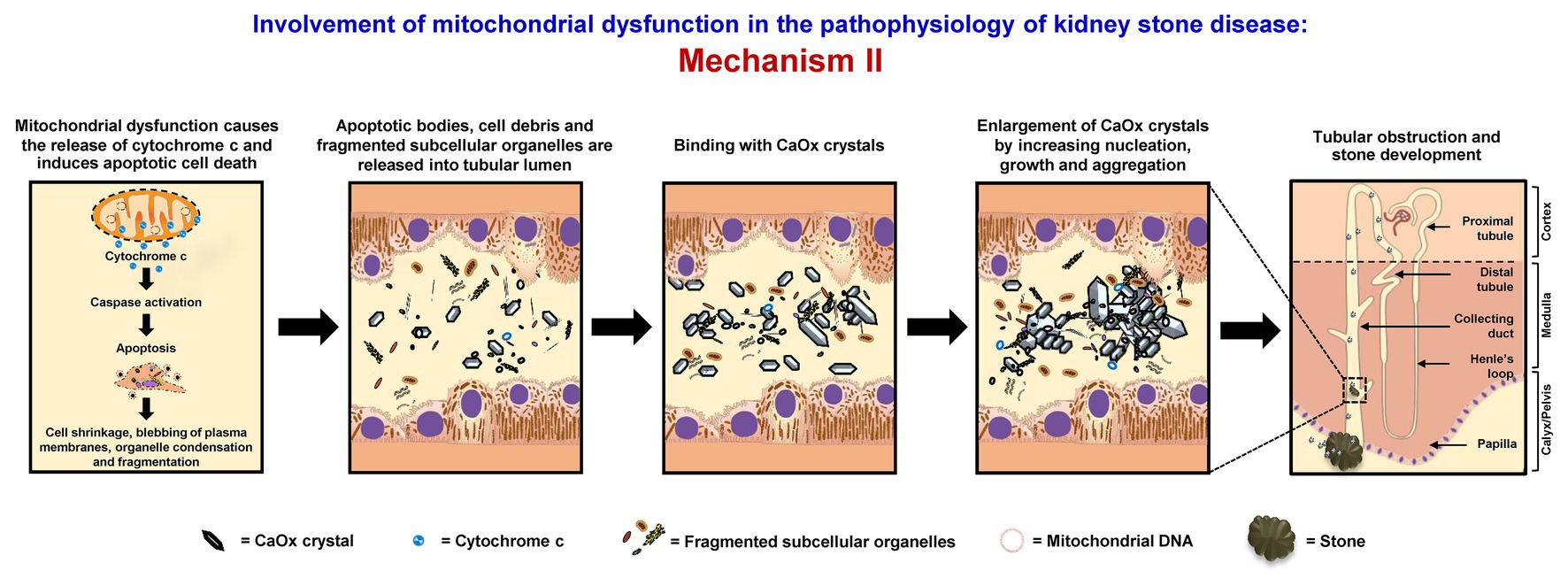
Figure 2. Involvement of mitochondrial dysfunction in the pathophysiology of kidney stone disease: Mechanism II – Mitochondrial dysfunction causes the release of cytochrome c and induces apoptotic cell death associated with cell shrinkage, blebbing of plasma membranes, organelle condensation, and fragmentation. The resulting apoptotic bodies, cell debris, and fragmented subcellular organelles are released into tubular lumens and can then bind with CaOx crystals. Moreover, fragmented mitochondria and mitochondrial proteins together with the membrane fragments and other cellular debris can serve as the raw materials for the stone nidus formation in the distal nephron and further promote crystal nucleation, growth, and aggregation. The large aggregates may obstruct tubular lumen and/or migrate (by renal tubular fluid flow) to the calyx and pelvis to form the stone. The right panel shows the macroscopic diagram to demonstrate the locales (related to the nephron segments) where the proposed mechanism occurs.
Mitochondrial dysfunction can initiate interstitial stone formation (Randall’s plaque model). In oxidative stress condition, mitochondrial damage can lead to disruption of mitochondrial membrane integrity followed by the release of ROS, mtDNA, ATP, and fragmented mitochondria to the cytoplasm. These components can then induce secretion of the proinflammatory cytokines and trigger the inflammatory cascades (West, 2017). The proinflammatory cytokines that are secreted in response to mitochondrial damage also recruit various immune cells, including tissue macrophages, to this interstitial locale. Migration of these inflammatory cells can cause tissue inflammation (Singhto et al., 2013, 2018). Moreover, the renal interstitium is commonly supersaturated with calcium phosphate, which is another common crystalline compound found in kidney stones. Together with tissue inflammation, Randall’s plaque rich with calcium phosphate starts to form (Khan, 2013, 2014). Some of these plaques can erode into the urinary space or renal pelvis, where supersaturation of CaOx is very common in the stone formers. At this locale (mostly lateral to the renal papilla), Randall’s plaque can serve as the nidus or stem for further development or growth of CaOx stone (Khan et al., 2016; Bird and Khan, 2017; Wiener et al., 2018) (Figure 3).
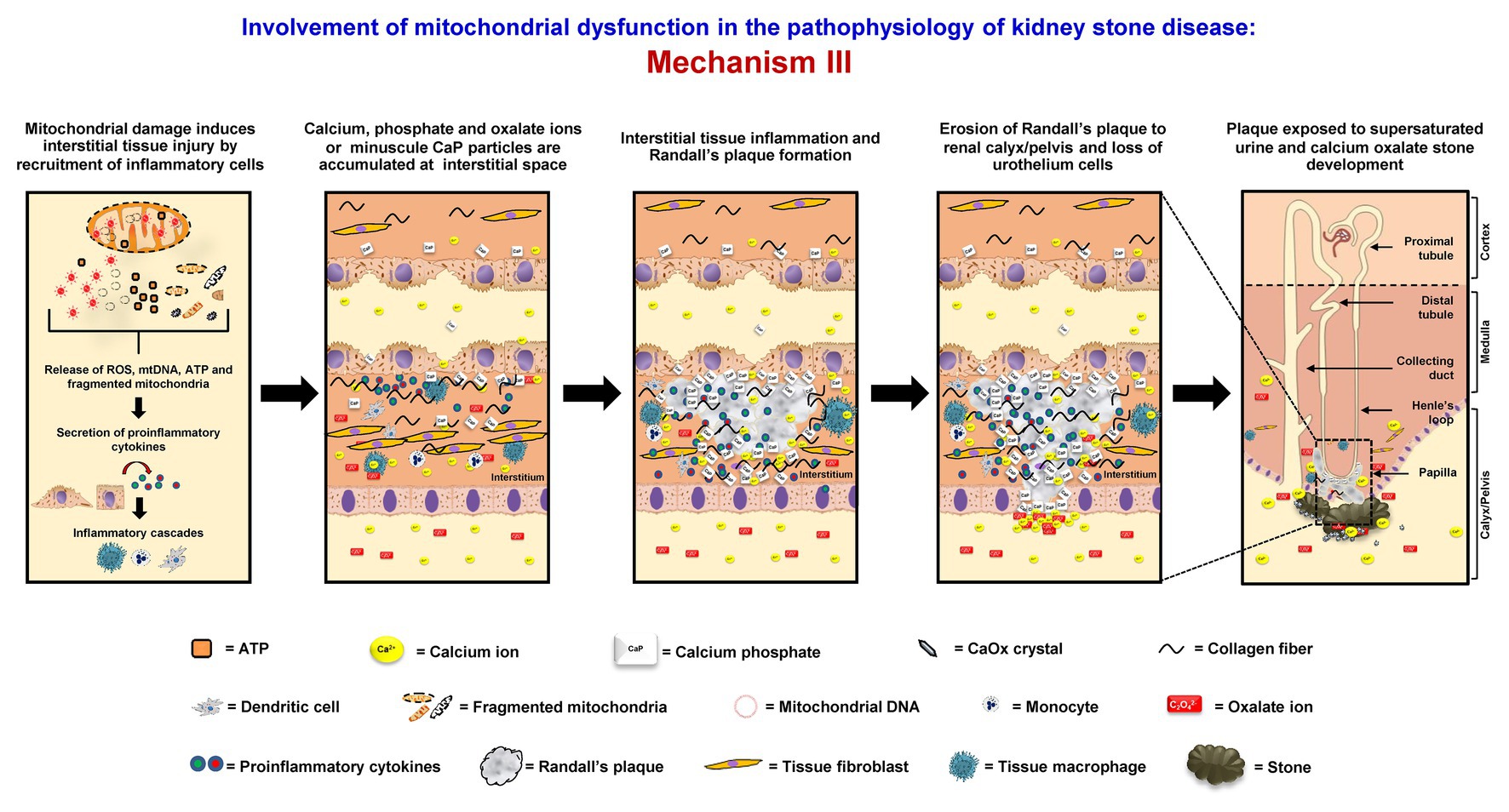
Figure 3. Involvement of mitochondrial dysfunction in the pathophysiology of kidney stone disease: Mechanism III – Mitochondrial damage induces the disruption of mitochondrial membrane integrity followed by the release of ROS, mitochondrial DNA (mtDNA), adenosine triphosphate (ATP), and fragmented mitochondria to the cytoplasm. These components can then induce secretion of the proinflammatory cytokines, trigger the inflammatory cascades, and further enhance interstitial tissue inflammation. Together with supersaturation of calcium phosphate, which is common in the renal interstitium, Randall’s plaque containing mainly calcium phosphate (hydroxyapatite) starts to form. Some of these plaques can erode into the urinary space or renal pelvis, where supersaturation of CaOx is very common in the stone formers. At this locale (mostly lateral to the renal papilla), the Randall’s plaque can serve as the nidus or stem for further development or growth of CaOx stone. The right panel shows the macroscopic diagram to demonstrate the locales (related to the nephron segments) where the proposed mechanism occurs.
In summary, mitochondria may be considered as the central intracellular organelles that play pivotal roles in kidney stone pathophysiology. Alterations in their main functions, including energy production and regulation of oxidative stress and intracellular calcium homeostasis, are associated with kidney tissue injury and inflammatory response, leading to CaOx crystal nucleation, growth, aggregation, and deposition that are the key processes for kidney stone formation. Furthermore, mitochondrial fragmented products and proteins can bind directly with CaOx crystals and thus play roles in the stone nidus formation. In addition to the intratubular crystal deposition and nidus formation, mitochondrial dysfunction is also associated with Randall’s plaque formation by enhancing tissue inflammation and interstitial deposition of calcium phosphate (hydroxyapatite).
It should be noted that mitochondrial dysfunction alone is not sufficient to induce kidney stone formation, of which mechanisms are multifactorial. For example, mtDNA mutations or defective nuclear-encoded mitochondrial proteins have been extensively studied in several mitochondrial disorders, e.g., Alzheimer’s disease, epilepsy, mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS syndrome), etc. (El-Hattab et al., 2015; Annesley and Fisher, 2019). Nevertheless, there is no evidence that link between these mitochondrial disorders and kidney stone disease. Although, MELAS syndrome also affects acid-base balanced regulated by kidney cells, the main target of this mitochondrial disorder is the neurological system, whereas kidney stone formation requires the intrarenal microenvironment involving several factors, e.g., supersaturation of CaOx, calcium phosphate, and other causative crystalline compounds, low urinary flow, imbalance of stone inhibitors and promoters, tubular cells injury, oxidative stress, inflammatory cascades, etc. (Bird and Khan, 2017).
Based on the aforementioned pathogenic mechanisms, recovering the mitochondrial functions by antioxidants may be the effective approach for prevention of new or recurrent kidney stone formation. Although antioxidant agents have been recommended to preserve mitochondrial activities and to antagonize ROS overproduction in various diseases, their efficacy and adverse events are still ambiguous and need to be further elucidated. Additionally, previous evidence has demonstrated that CypD (Duann and Lin, 2017) and Bcl-2 interacting protein 3 (BNIP3) (Chaanine et al., 2016; Peng et al., 2019) are related to mitochondrial dysfunction via alterations of mitochondrial membrane activities and death signaling release, respectively. To recover the mitochondrial functions and to reduce oxidative stress, inhibition of CypD activation using cyclosporin A (Duann and Lin, 2017) and N-methyl-4-isoleucine cyclosporine (Niimi et al., 2014) has been reported. In addition, expression and translocation of BNIP3 to mitochondria have been reported as the cell death regulatory factors for mitochondrial dysfunction (Zhang and Ney, 2009). This protein also involves mitochondrial membrane potential, mitochondrial transition pore forming, oxidative stress, calcium homeostasis, and inflammation in various cell types (Gao et al., 2020). Therefore, therapeutic application should be considered to combine antioxidants with other promising compounds (i.e., to inhibit activation of CypD, BNIP3 and other related molecules) for prevention of new and/or recurrence stone formation in the future. More importantly, their efficacies and adverse events must be evaluated in large cohorts.
Although the roles for mitochondrial dysfunction related to oxidative stress and CaOx crystal deposition have been well documented, the roles for excessive calcium that is also common in the stone formers (Canales et al., 2010; Chutipongtanate et al., 2012) may be overlooked. Recently, calcium is recognized as a mitochondrial regulator involving several steps of energy production (Gincel et al., 2001; Calderon-Cortes et al., 2008; Pivovarova and Andrews, 2010; Kaufman and Malhotra, 2014; Ummarino, 2017; Ham et al., 2019; Lambert et al., 2020). On the other hand, mitochondria also play roles in regulation of calcium homeostasis (Gincel et al., 2001; Calderon-Cortes et al., 2008; Pivovarova and Andrews, 2010; Kaufman and Malhotra, 2014; Ummarino, 2017; Ham et al., 2019; Lambert et al., 2020). A previous study on ethylene glycol induced kidney stone disease in rats has shown that only CaOx crystals, but not oxalate ion alone, could weaken mitochondrial functions (Mcmartin and Wallace, 2005). Nevertheless, the association among mitochondrial dysfunction, intracellular or mitochondrial calcium concentration, and the stone pathogenesis remains unclear and should be further elucidated. Having done so, the findings to be obtained may lead to the new strategy to cope with mitochondrial dysfunction during the stone development and ultimately to efficient prevention of kidney stone formation.
SC and VT drafted the manuscript, read and approved the final manuscript, and are responsible for all aspects of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by Mahidol University research grant and the Thailand Research Fund (IRN60W0004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acetyl CoA, acetyl coenzyme A; ADP, adenosine diphosphate; ATP, adenosine triphosphate; BNIP3, Bcl-2 interacting protein 3; CaOx, calcium oxalate; CypD, cyclophilin D; ETC, electron transport chain; FADH2, flavin adenine dinucleotide; GSSG, glutathione disulfide; H2O2, hydrogen peroxide; mGSH, mitochondrial glutathione; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; mtDNA, mitochondrial DNA; NADH, nicotine adenine dinucleotide; O2−, superoxide; ROS, reactive oxygen species; TCA, tricarboxylic acid.
Abate, M., Festa, A., Falco, M., Lombardi, A., Luce, A., Grimaldi, A., et al. (2020). Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 98, 139–153. doi: 10.1016/j.semcdb.2019.05.022
Aggarwal, D., Gautam, D., Sharma, M., and Singla, S. K. (2016). Bergenin attenuates renal injury by reversing mitochondrial dysfunction in ethylene glycol induced hyperoxaluric rat model. Eur. J. Pharmacol. 791, 611–621. doi: 10.1016/j.ejphar.2016.10.002
An, J. J., Shi, K. J., Wei, W., Hua, F. Y., Ci, Y. L., Jiang, Q., et al. (2013). The ROS/JNK/ATF2 pathway mediates selenite-induced leukemia NB4 cell cycle arrest and apoptosis in vitro and in vivo. Cell Death Dis. 4:e973. doi: 10.1038/cddis.2013.475
Anderson, R. G., Ghiraldeli, L. P., and Pardee, T. S. (2018). Mitochondria in cancer metabolism, an organelle whose time has come? Biochim. Biophys. Acta Rev. Cancer 1870, 96–102. doi: 10.1016/j.bbcan.2018.05.005
Annesley, S. J., and Fisher, P. R. (2019). Mitochondria in health and disease. Cells 8:680. doi: 10.3390/cells8070680
Aparicio-Trejo, O. E., Tapia, E., Sanchez-Lozada, L. G., and Pedraza-Chaverri, J. (2018). Mitochondrial bioenergetics, redox state, dynamics and turnover alterations in renal mass reduction models of chronic kidney diseases and their possible implications in the progression of this illness. Pharmacol. Res. 135, 1–11. doi: 10.1016/j.phrs.2018.07.015
Apostolopoulou, M., Corsini, A., and Roden, M. (2015). The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 45, 745–754. doi: 10.1111/eci.12461
Bajic, V. P., Van Neste, C., Obradovic, M., Zafirovic, S., Radak, D., Bajic, V. B., et al. (2019). Glutathione “Redox Homeostasis” and its relation to cardiovascular disease. Oxidative Med. Cell. Longev. 2019, 1–14. doi: 10.1155/2019/5028181
Baudouin-Cornu, P., Lagniel, G., Kumar, C., Huang, M. E., and Labarre, J. (2012). Glutathione degradation is a key determinant of glutathione homeostasis. J. Biol. Chem. 287, 4552–4561. doi: 10.1074/jbc.M111.315705
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646. doi: 10.1038/nrneph.2017.107
Bird, V. Y., and Khan, S. R. (2017). How do stones form? Is unification of theories on stone formation possible? Arch. Esp. Urol. 70, 12–27.
Brookes, P. S., Levonen, A. L., Shiva, S., Sarti, P., and Darley-Usmar, V. M. (2002). Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 33, 755–764. doi: 10.1016/s0891-5849(02)00901-2
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M. W., and Sheu, S. S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833. doi: 10.1152/ajpcell.00139.2004
Calderon-Cortes, E., Cortes-Rojo, C., Clemente-Guerrero, M., Manzo-Avalos, S., Villalobos-Molina, R., Boldogh, I., et al. (2008). Changes in mitochondrial functionality and calcium uptake in hypertensive rats as a function of age. Mitochondrion 8, 262–272. doi: 10.1016/j.mito.2008.04.005
Canales, B. K., Anderson, L., Higgins, L., Ensrud-Bowlin, K., Roberts, K. P., Wu, B., et al. (2010). Proteome of human calcium kidney stones. Urology 76, 1017.e1013–1017.e1020. doi: 10.1016/j.urology.2010.05.005
Cao, L. C., Honeyman, T. W., Cooney, R., Kennington, L., Scheid, C. R., and Jonassen, J. A. (2004). Mitochondrial dysfunction is a primary event in renal cell oxalate toxicity. Kidney Int. 66, 1890–1900. doi: 10.1111/j.1523-1755.2004.00963.x
Cao, Y., Liu, W., Hui, L., Zhao, J., Yang, X., Wang, Y., et al. (2016). Renal tubular injury induced by ischemia promotes the formation of calcium oxalate crystals in rats with hyperoxaluria. Urolithiasis 44, 389–397. doi: 10.1007/s00240-016-0876-7
Ceban, E., Banov, P., Galescu, A., and Botnari, V. (2016). Oxidative stress and antioxidant status in patients with complicated urolithiasis. J. Med. Life 9, 259–262.
Chaanine, A. H., Kohlbrenner, E., Gamb, S. I., Guenzel, A. J., Klaus, K., Fayyaz, A. U., et al. (2016). FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 311, H1540–H1559. doi: 10.1152/ajpheart.00549.2016
Chaiyarit, S., Singhto, N., and Thongboonkerd, V. (2016). Calcium oxalate monohydrate crystals internalized into renal tubular cells are degraded and dissolved by endolysosomes. Chem. Biol. Interact. 246, 30–35. doi: 10.1016/j.cbi.2015.12.018
Chaiyarit, S., and Thongboonkerd, V. (2012). Changes in mitochondrial proteome of renal tubular cells induced by calcium oxalate monohydrate crystal adhesion and internalization are related to mitochondrial dysfunction. J. Proteome Res. 11, 3269–3280. doi: 10.1021/pr300018c
Chang, H. H., Guo, M. K., Kasten, F. H., Chang, M. C., Huang, G. F., Wang, Y. L., et al. (2005). Stimulation of glutathione depletion, ROS production and cell cycle arrest of dental pulp cells and gingival epithelial cells by HEMA. Biomaterials 26, 745–753. doi: 10.1016/j.biomaterials.2004.03.021
Chhiber, N., Kaur, T., and Singla, S. (2016). Rottlerin, a polyphenolic compound from the fruits of Mallotus phillipensis (Lam.) Mull.Arg., impedes oxalate/calcium oxalate induced pathways of oxidative stress in male wistar rats. Phytomedicine 23, 989–997. doi: 10.1016/j.phymed.2016.06.005
Chutipongtanate, S., Fong-Ngern, K., Peerapen, P., and Thongboonkerd, V. (2012). High calcium enhances calcium oxalate crystal binding capacity of renal tubular cells via increased surface annexin A1 but impairs their proliferation and healing. J. Proteome Res. 11, 3650–3663. doi: 10.1021/pr3000738
Deng, X., Gao, F., and May, W. S. Jr. (2003). Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood 102, 3179–3185. doi: 10.1182/blood-2003-04-1027
Dominguez-Gutierrez, P. R., Kwenda, E. P., Khan, S. R., and Canales, B. K. (2020). Immunotherapy for stone disease. Curr. Opin. Urol. 30, 183–189. doi: 10.1097/MOU.0000000000000729
Duann, P., and Lin, P. H. (2017). Mitochondria damage and kidney disease. Adv. Exp. Med. Biol. 982, 529–551. doi: 10.1007/978-3-319-55330-6_27
Ducasa, G. M., Mitrofanova, A., and Fornoni, A. (2019). Crosstalk between lipids and mitochondria in diabetic kidney disease. Curr. Diab. Rep. 19:144. doi: 10.1007/s11892-019-1263-x
El-Hattab, A. W., Adesina, A. M., Jones, J., and Scaglia, F. (2015). MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 116, 4–12. doi: 10.1016/j.ymgme.2015.06.004
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Evan, A. P. (2010). Physiopathology and etiology of stone formation in the kidney and the urinary tract. Pediatr. Nephrol. 25, 831–841. doi: 10.1007/s00467-009-1116-y
Fernandez-Checa, J. C., Garcia-Ruiz, C., Colell, A., Morales, A., Mari, M., Miranda, M., et al. (1998). Oxidative stress: role of mitochondria and protection by glutathione. Biofactors 8, 7–11. doi: 10.1002/biof.5520080102
Fishman, A. I., Green, D., Lynch, A., Choudhury, M., Eshghi, M., and Konno, S. (2013). Preventive effect of specific antioxidant on oxidative renal cell injury associated with renal crystal formation. Urology 82, 489.e481–489.e487. doi: 10.1016/j.urology.2013.03.065
Fong-Ngern, K., Vinaiphat, A., and Thongboonkerd, V. (2017). Microvillar injury in renal tubular epithelial cells induced by calcium oxalate crystal and the protective role of epigallocatechin-3-gallate. FASEB J. 31, 120–131. doi: 10.1096/fj.201600543R
Forbes, J. M. (2016). Mitochondria-power players in kidney function? Trends Endocrinol. Metab. 27, 441–442. doi: 10.1016/j.tem.2016.05.002
Forbes, J. M., and Thorburn, D. R. (2018). Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 14, 291–312. doi: 10.1016/j.cca.2019.07.005
Franklin, J. L. (2011). Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid. Redox Signal. 14, 1437–1448. doi: 10.1089/ars.2010.3596
Gao, A., Jiang, J., Xie, F., and Chen, L. (2020). Bnip3 in mitophagy: novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 506, 72–83. doi: 10.1016/j.cca.2020.02.024
Geto, Z., Molla, M. D., Challa, F., Belay, Y., and Getahun, T. (2020). Mitochondrial dynamic dysfunction as a main triggering factor for inflammation associated chronic non-communicable diseases. J. Inflamm. Res. 13, 97–107. doi: 10.2147/JIR.S232009
Gincel, D., Zaid, H., and Shoshan-Barmatz, V. (2001). Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358, 147–155. doi: 10.1042/0264-6021:3580147
Giorgi, C., Marchi, S., Simoes, I. C. M., Ren, Z., Morciano, G., Perrone, M., et al. (2018). Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell Mol. Biol. 340, 209–344. doi: 10.1016/bs.ircmb.2018.05.006
Govindaraj, A., and Selvam, R. (2001). Increased calcium oxalate crystal nucleation and aggregation by peroxidized protein of human kidney stone matrix and renal cells. Urol. Res. 29, 194–198. doi: 10.1007/s002400100177
Govindaraj, A., and Selvam, R. (2002). An oxalate-binding protein with crystal growth promoter activity from human kidney stone matrix. BJU Int. 90, 336–344. doi: 10.1046/j.1464-410x.2002.02849.x
Grancara, S., Ohkubo, S., Artico, M., Ciccariello, M., Manente, S., Bragadin, M., et al. (2016). Milestones and recent discoveries on cell death mediated by mitochondria and their interactions with biologically active amines. Amino Acids 48, 2313–2326. doi: 10.1007/s00726-016-2323-z
Guo, D., Yu, K., Sun, X. Y., and Ouyang, J. M. (2018). Structural characterization and repair mechanism of Gracilaria lemaneiformis sulfated polysaccharides of different molecular weights on damaged renal epithelial cells. Oxidative Med. Cell. Longev. 2018, 1–15. doi: 10.1155/2018/7410389
Ham, J., Lim, W., Park, S., Bae, H., You, S., and Song, G. (2019). Synthetic phenolic antioxidant propyl gallate induces male infertility through disruption of calcium homeostasis and mitochondrial function. Environ. Pollut. 248, 845–856. doi: 10.1016/j.envpol.2019.02.087
Harrison, D. J., Inglis, J. A., and Tolley, D. A. (1988). Percutaneous renal biopsy specimens in stone formers. J. Clin. Pathol. 41, 971–974.
Hirose, M., Tozawa, K., Okada, A., Hamamoto, S., Higashibata, Y., Gao, B., et al. (2012). Role of osteopontin in early phase of renal crystal formation: immunohistochemical and microstructural comparisons with osteopontin knock-out mice. Urol. Res. 40, 121–129. doi: 10.1007/s00240-011-0400-z
Hirose, M., Yasui, T., Okada, A., Hamamoto, S., Shimizu, H., Itoh, Y., et al. (2010). Renal tubular epithelial cell injury and oxidative stress induce calcium oxalate crystal formation in mouse kidney. Int. J. Urol. 17, 83–92. doi: 10.1111/j.1442-2042.2009.02410.x
Holoch, P. A., and Tracy, C. R. (2011). Antioxidants and self-reported history of kidney stones: the National Health and Nutrition Examination Survey. J. Endourol. 25, 1903–1908. doi: 10.1089/end.2011.0130
Indran, I. R., Tufo, G., Pervaiz, S., and Brenner, C. (2011). Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 1807, 735–745. doi: 10.1016/j.bbabio.2011.03.010
Itoh, Y., Yasui, T., Okada, A., Tozawa, K., Hayashi, Y., and Kohri, K. (2005). Preventive effects of green tea on renal stone formation and the role of oxidative stress in nephrolithiasis. J. Urol. 173, 271–275. doi: 10.1097/01.ju.0000141311.51003.87
Jang, J. Y., Blum, A., Liu, J., and Finkel, T. (2018). The role of mitochondria in aging. J. Clin. Invest. 128, 3662–3670. doi: 10.1172/JCI120842
Janikiewicz, J., Szymanski, J., Malinska, D., Patalas-Krawczyk, P., Michalska, B., Duszynski, J., et al. (2018). Mitochondria-associated membranes in aging and senescence: structure, function, and dynamics. Cell Death Dis. 9:332. doi: 10.1038/s41419-017-0105-5
Jonassen, J. A., Cao, L. C., Honeyman, T., and Scheid, C. R. (2003). Mechanisms mediating oxalate-induced alterations in renal cell functions. Crit. Rev. Eukaryot. Gene Expr. 13, 55–72. doi: 10.1615/critreveukaryotgeneexpr.v13.i1.50
Jonassen, J. A., Kohjimoto, Y., Scheid, C. R., and Schmidt, M. (2005). Oxalate toxicity in renal cells. Urol. Res. 33, 329–339. doi: 10.1007/s00240-005-0485-3
Kanlaya, R., Sintiprungrat, K., Chaiyarit, S., and Thongboonkerd, V. (2013). Macropinocytosis is the major mechanism for endocytosis of calcium oxalate crystals into renal tubular cells. Cell Biochem. Biophys. 67, 1171–1179. doi: 10.1007/s12013-013-9630-8
Kaufman, R. J., and Malhotra, J. D. (2014). Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta 1843, 2233–2239. doi: 10.1016/j.bbamcr.2014.03.022
Khan, S. R. (2013). Reactive oxygen species as the molecular modulators of calcium oxalate kidney stone formation: evidence from clinical and experimental investigations. J. Urol. 189, 803–811. doi: 10.1016/j.juro.2012.05.078
Khan, S. R. (2014). Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis. Transl. Androl. Urol. 3, 256–276. doi: 10.3978/j.issn.2223-4683.2014.06.04
Khan, S. R., Pearle, M. S., Robertson, W. G., Gambaro, G., Canales, B. K., Doizi, S., et al. (2016). Kidney stones. Nat. Rev. Dis. Primers. 2:16008. doi: 10.1038/nrdp.2016.8
Khand, F. D., Gordge, M. P., Robertson, W. G., Noronha-Dutra, A. A., and Hothersall, J. S. (2002). Mitochondrial superoxide production during oxalate-mediated oxidative stress in renal epithelial cells. Free Radic. Biol. Med. 32, 1339–1350. doi: 10.1016/s0891-5849(02)00846-8
Kohri, K., Yasui, T., Okada, A., Hirose, M., Hamamoto, S., Fujii, Y., et al. (2012). Biomolecular mechanism of urinary stone formation involving osteopontin. Urol. Res. 40, 623–637. doi: 10.1007/s00240-012-0514-y
Lambert, J. P., Murray, E. K., and Elrod, J. W. (2020). MCUB and mitochondrial calcium uptake ‐ modeling, function, and therapeutic potential. Expert Opin. Ther. Targets 24, 163–169. doi: 10.1080/14728222.2020.1732926
Laxmanan, S., Selvam, R., Mahle, C. J., and Menon, M. (1986). Binding of oxalate to mitochondrial inner membranes of rat and human kidney. J. Urol. 135, 862–865.
Lushchak, V. I. (2012). Glutathione homeostasis and functions: potential targets for medical interventions. J. Amino Acids 2012, 1–26. doi: 10.1155/2012/736837
Ma, M. C., Chen, Y. S., and Huang, H. S. (2014). Erythrocyte oxidative stress in patients with calcium oxalate stones correlates with stone size and renal tubular damage. Urology 83, 510.e519–510.e517. doi: 10.1016/j.urology.2013.09.050
Mcmartin, K. (2009). Are calcium oxalate crystals involved in the mechanism of acute renal failure in ethylene glycol poisoning? Clin. Toxicol. 47, 859–869. doi: 10.3109/15563650903344793
Mcmartin, K. E., and Wallace, K. B. (2005). Calcium oxalate monohydrate, a metabolite of ethylene glycol, is toxic for rat renal mitochondrial function. Toxicol. Sci. 84, 195–200. doi: 10.1093/toxsci/kfi062
Missiroli, S., Genovese, I., Perrone, M., Vezzani, B., Vitto, V. A. M., and Giorgi, C. (2020). The role of mitochondria in inflammation: from cancer to neurodegenerative disorders. J. Clin. Med. 9:740. doi: 10.3390/jcm9030740
Muller, M., Ahumada-Castro, U., Sanhueza, M., Gonzalez-Billault, C., Court, F. A., and Cardenas, C. (2018). Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 12:470. doi: 10.3389/fnins.2018.00470
Muthukumar, A., and Selvam, R. (1998). Role of glutathione on renal mitochondrial status in hyperoxaluria. Mol. Cell. Biochem. 185, 77–84.
Neagu, M., Constantin, C., Popescu, I. D., Zipeto, D., Tzanakakis, G., Nikitovic, D., et al. (2019). Inflammation and metabolism in cancer cell-mitochondria key player. Front. Oncol. 9:348. doi: 10.3389/fonc.2019.00348
Niimi, K., Yasui, T., Hirose, M., Hamamoto, S., Itoh, Y., Okada, A., et al. (2012). Mitochondrial permeability transition pore opening induces the initial process of renal calcium crystallization. Free Radic. Biol. Med. 52, 1207–1217. doi: 10.1016/j.freeradbiomed.2012.01.005
Niimi, K., Yasui, T., Okada, A., Hirose, Y., Kubota, Y., Umemoto, Y., et al. (2014). Novel effect of the inhibitor of mitochondrial cyclophilin D activation, N-methyl-4-isoleucine cyclosporin, on renal calcium crystallization. Int. J. Urol. 21, 707–713. doi: 10.1111/iju.12425
Patel, M., Yarlagadda, V., Adedoyin, O., Saini, V., Assimos, D. G., Holmes, R. P., et al. (2018). Oxalate induces mitochondrial dysfunction and disrupts redox homeostasis in a human monocyte derived cell line. Redox Biol. 15, 207–215. doi: 10.1016/j.redox.2017.12.003
Peerapen, P., Chaiyarit, S., and Thongboonkerd, V. (2018). Protein network analysis and functional studies of calcium oxalate crystal-induced cytotoxicity in renal tubular epithelial cells. Proteomics 18:e1800008. doi: 10.1002/pmic.201800008
Peng, Y., Fang, Z., Liu, M., Wang, Z., Li, L., Ming, S., et al. (2019). Testosterone induces renal tubular epithelial cell death through the HIF-1alpha/BNIP3 pathway. J. Transl. Med. 17:62. doi: 10.1186/s12967-019-1821-7
Pieczenik, S. R., and Neustadt, J. (2007). Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 83, 84–92. doi: 10.1016/j.yexmp.2006.09.008
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Pradelli, L. A., Beneteau, M., and Ricci, J. E. (2010). Mitochondrial control of caspase-dependent and -independent cell death. Cell. Mol. Life Sci. 67, 1589–1597. doi: 10.1007/s00018-010-0285-y
Ralto, K. M., Rhee, E. P., and Parikh, S. M. (2020). NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 16, 99–111. doi: 10.1038/s41581-019-0216-6
Rango, M., and Bresolin, N. (2018). Brain mitochondria, aging, and Parkinson’s disease. Genes 9:250. doi: 10.3390/genes9050250
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990. doi: 10.1016/j.cellsig.2012.01.008
Reiter, R. J., Tan, D. X., Rosales-Corral, S., Galano, A., Zhou, X. J., and Xu, B. (2018). Mitochondria: central organelles for melatonin's antioxidant and anti-aging actions. Molecules 23:509. doi: 10.3390/molecules23020509
Ribas, V., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2014). Glutathione and mitochondria. Front. Pharmacol. 5:151. doi: 10.3389/fphar.2014.00151
Roop-Ngam, P., Chaiyarit, S., Pongsakul, N., and Thongboonkerd, V. (2012). Isolation and characterizations of oxalate-binding proteins in the kidney. Biochem. Biophys. Res. Commun. 424, 629–634. doi: 10.1016/j.bbrc.2012.07.015
Roy, A., Al-Bataineh, M. M., and Pastor-Soler, N. M. (2015). Collecting duct intercalated cell function and regulation. Clin. J. Am. Soc. Nephrol. 10, 305–324. doi: 10.2215/CJN.08880914
Ruiz, S., Pergola, P. E., Zager, R. A., and Vaziri, N. D. (2013). Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 83, 1029–1041. doi: 10.1038/ki.2012.439
Scatena, R. (2012). Mitochondria and cancer: a growing role in apoptosis, cancer cell metabolism and dedifferentiation. Adv. Exp. Med. Biol. 942, 287–308. doi: 10.1007/978-94-007-2869-1_13
Schottl, T., Pachl, F., Giesbertz, P., Daniel, H., Kuster, B., Fromme, T., et al. (2020). Proteomic and metabolite profiling reveals profound structural and metabolic reorganization of adipocyte mitochondria in obesity. Obesity 28, 590–600. doi: 10.1002/oby.22737
Selvam, R., and Kalaiselvi, P. (2003). Oxalate binding proteins in calcium oxalate nephrolithiasis. Urol. Res. 31, 242–256. doi: 10.1007/s00240-003-0316-3
Sharma, P., and Sampath, H. (2019). Mitochondrial DNA integrity: role in health and disease. Cells 8:100. doi: 10.3390/cells8020100
Sharpe, A. J., and Mckenzie, M. (2018). Mitochondrial fatty acid oxidation disorders associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) deficiency. Cells 7:46. doi: 10.3390/cells7060046
Singhto, N., Kanlaya, R., Nilnumkhum, A., and Thongboonkerd, V. (2018). Roles of macrophage exosomes in immune response to calcium oxalate monohydrate crystals. Front. Immunol. 9:316. doi: 10.3389/fimmu.2018.00316
Singhto, N., Sintiprungrat, K., and Thongboonkerd, V. (2013). Alterations in macrophage cellular proteome induced by calcium oxalate crystals: the association of HSP90 and F-actin is important for phagosome formation. J. Proteome Res. 12, 3561–3572. doi: 10.1021/pr4004097
Son, J. M., and Lee, C. (2019). Mitochondria: multifaceted regulators of aging. BMB Rep. 52, 13–23. doi: 10.5483/BMBRep.2019.52.1.300
Song, W., Owusu-Ansah, E., Hu, Y., Cheng, D., Ni, X., Zirin, J., et al. (2017). Activin signaling mediates muscle-to-adipose communication in a mitochondria dysfunction-associated obesity model. Proc. Natl. Acad. Sci. U. S. A. 114, 8596–8601. doi: 10.1073/pnas.1708037114
Srinivasan, S., Guha, M., Kashina, A., and Avadhani, N. G. (2017). Mitochondrial dysfunction and mitochondrial dynamics-the cancer connection. Biochim. Biophys. Acta Bioenerg. 1858, 602–614. doi: 10.1016/j.bbabio.2017.01.004
Stowe, D. F., and Camara, A. K. (2009). Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid. Redox Signal. 11, 1373–1414. doi: 10.1089/ars.2008.2331
Suarez-Rivero, J. M., Villanueva-Paz, M., De La Cruz-Ojeda, P., De La Mata, M., Cotan, D., Oropesa-Avila, M., et al. (2016). Mitochondrial dynamics in mitochondrial diseases. Diseases 5:1. doi: 10.3390/diseases5010001
Sun, X. Y., Ouyang, J. M., Bhadja, P., Gui, Q., Peng, H., and Liu, J. (2016). Protective effects of degraded soybean polysaccharides on renal epithelial cells exposed to oxidative damage. J. Agric. Food Chem. 64, 7911–7920. doi: 10.1021/acs.jafc.6b03323
Sun, X. Y., Yu, K., and Ouyang, J. M. (2017). Time-dependent subcellular structure injuries induced by nano-/micron-sized calcium oxalate monohydrate and dihydrate crystals. Mater. Sci. Eng. C Mater. Biol. Appl. 79, 445–456. doi: 10.1016/j.msec.2017.05.081
Sun, X. Y., Zhang, H., Liu, J., and Ouyang, J. M. (2019). Repair activity and crystal adhesion inhibition of polysaccharides with different molecular weights from red algae Porphyra yezoensis against oxalate-induced oxidative damage in renal epithelial cells. Food Funct. 10, 3851–3867. doi: 10.1039/c8fo02556h
Takahashi, M., and Takahashi, K. (2019). Water-soluble CoQ10 as a promising anti-aging agent for neurological dysfunction in brain mitochondria. Antioxidants 8:61. doi: 10.3390/antiox8030061
Ummarino, D. (2017). Calcium: mitochondrial calcium efflux essential for heart function. Nat. Rev. Cardiol. 14:317. doi: 10.1038/nrcardio.2017.73
Veena, C. K., Josephine, A., Preetha, S. P., Rajesh, N. G., and Varalakshmi, P. (2008). Mitochondrial dysfunction in an animal model of hyperoxaluria: a prophylactic approach with fucoidan. Eur. J. Pharmacol. 579, 330–336. doi: 10.1016/j.ejphar.2007.09.044
Veena, C. K., Josephine, A., Preetha, S. P., and Varalakshmi, P. (2007). Effect of sulphated polysaccharides on erythrocyte changes due to oxidative and nitrosative stress in experimental hyperoxaluria. Hum. Exp. Toxicol. 26, 923–932. doi: 10.1177/0960327107087792
Vinaiphat, A., Aluksanasuwan, S., Manissorn, J., Sutthimethakorn, S., and Thongboonkerd, V. (2017). Response of renal tubular cells to differential types and doses of calcium oxalate crystals: integrative proteome network analysis and functional investigations. Proteomics 17:1700192. doi: 10.1002/pmic.201700192
Vinaiphat, A., and Thongboonkerd, V. (2017). Prospects for proteomics in kidney stone disease. Expert Rev. Proteomics 14, 185–187. doi: 10.1080/14789450.2017.1283222
Walters, A. M., Porter, G. A. Jr., and Brookes, P. S. (2012). Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 111, 1222–1236. doi: 10.1161/CIRCRESAHA.112.265660
Wang, Z., Ying, Z., Bosy-Westphal, A., Zhang, J., Schautz, B., Later, W., et al. (2010). Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 92, 1369–1377. doi: 10.3945/ajcn.2010.29885
West, A. P. (2017). Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 391, 54–63. doi: 10.1016/j.tox.2017.07.016
Wiener, S. V., Ho, S. P., and Stoller, M. L. (2018). Beginnings of nephrolithiasis: insights into the past, present and future of Randall’s plaque formation research. Curr. Opin. Nephrol. Hypertens. 27, 236–242. doi: 10.1097/MNH.0000000000000414
Williams, J., Holmes, R. P., Assimos, D. G., and Mitchell, T. (2016). Monocyte mitochondrial function in calcium oxalate stone formers. Urology 93, 224–226. doi: 10.1016/j.urology.2016.03.004
Wojtovich, A. P., Nadtochiy, S. M., Brookes, P. S., and Nehrke, K. (2012). Ischemic preconditioning: the role of mitochondria and aging. Exp. Gerontol. 47, 1–7. doi: 10.1016/j.exger.2011.11.001
Wu, J., Tao, Z., Deng, Y., Liu, Q., Liu, Y., Guan, X., et al. (2019). Calcifying nanoparticles induce cytotoxicity mediated by ROS-JNK signaling pathways. Urolithiasis 47, 125–135. doi: 10.1007/s00240-018-1048-8
Yan, C., Duanmu, X., Zeng, L., Liu, B., and Song, Z. (2019). Mitochondrial DNA: distribution, mutations, and elimination. Cells 8:379. doi: 10.3390/cells8040379
Yang, X., Ding, H., Qin, Z., Zhang, C., Qi, S., Zhang, H., et al. (2016). Metformin prevents renal stone formation through an antioxidant mechanism in vitro and in vivo. Oxidative Med. Cell. Longev. 2016, 1–10. doi: 10.1155/2016/4156075
Yasui, T., Okada, A., Hamamoto, S., Ando, R., Taguchi, K., Tozawa, K., et al. (2017). Pathophysiology-based treatment of urolithiasis. Int. J. Urol. 24, 32–38. doi: 10.1111/iju.13187
Zeng, X., Xi, Y., and Jiang, W. (2019). Protective roles of flavonoids and flavonoid-rich plant extracts against urolithiasis: a review. Crit. Rev. Food Sci. Nutr. 59, 2125–2135. doi: 10.1080/10408398.2018.1439880
Zhai, W., Zheng, J., Yao, X., Peng, B., Liu, M., Huang, J., et al. (2013). Catechin prevents the calcium oxalate monohydrate induced renal calcium crystallization in NRK-52E cells and the ethylene glycol induced renal stone formation in rat. BMC Complement. Altern. Med. 13:228. doi: 10.1186/1472-6882-13-228
Zhang, J., and Ney, P. A. (2009). Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 16, 939–946. doi: 10.1038/cdd.2009.16
Keywords: antioxidant, calcium, calcium oxalate, mitochondria, nephrolithiasis, oxidative stress, reactive oxygen species, urolithiasis
Citation: Chaiyarit S and Thongboonkerd V (2020) Mitochondrial Dysfunction and Kidney Stone Disease. Front. Physiol. 11:566506. doi: 10.3389/fphys.2020.566506
Edited by:
Egor Plotnikov, Lomonosov Moscow State University, RussiaReviewed by:
Carlos Palmeira, University of Coimbra, PortugalCopyright © 2020 Chaiyarit and Thongboonkerd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Visith Thongboonkerd, dnRob25nYm9AeWFob28uY29t; dGhvbmdib29ua2VyZEBkci5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.