 Aaazh M. Alghamdi
Aaazh M. Alghamdi Mark R. Boyett3
Mark R. Boyett3 Jules C. Hancox
Jules C. Hancox Henggui Zhang
Henggui Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 03 December 2020
Sec. Computational Physiology and Medicine
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.546508
This article is part of the Research TopicCardiac Pacemaking in Health and Disease: From Genes to FunctionView all 17 articles
The function of the sinoatrial node (SAN), the pacemaker of the heart, declines with age, resulting in increased incidence of sinoatrial node dysfunction (SND) in older adults. The present study assesses potential ionic mechanisms underlying age associated SND. Two group studies have identified complex and various changes in some of membrane ion channels in aged rat SAN, the first group (Aging Study-1) indicates a considerable changes of gene expression with up-regulation of mRNA in ion channels of Cav1.2, Cav1.3 and KvLQT1, Kv4.2, and the Ca2+ handling proteins of SERCA2a, and down-regulation of Cav3.1, NCX, and HCN1 and the Ca2+-clock proteins of RYR2. The second group (Aging Study-2) suggests a different pattern of changes, including down regulation of Cav1.2, Cav1.3 and HCN4, and RYR2, and an increase of NCX and SERCA densities and proteins. Although both data sets shared a similar finding for some specific ion channels, such as down regulation of HCN4, NCX, and RYR2, there are contradictory changes for some other membrane ion channels, such as either up-regulation or down-regulation of Cav1.2, NCX and SERCA2a in aged rat SAN. The present study aims to test a hypothesis that age-related SND may arise from different ionic and molecular remodeling patterns. To test this hypothesis, a mathematical model of the electrical action potential of rat SAN myocytes was modified to simulate the functional impact of age-induced changes on membrane ion channels and intracellular Ca2+ handling as observed in Aging Study-1 and Aging Study-2. The role and relative importance of each individually remodeled ion channels and Ca2+-handling in the two datasets were evaluated. It was shown that the age-induced changes in ion channels and Ca2+-handling, based on either Aging Study-1 or Aging Study-2, produced similar bradycardic effects as manifested by a marked reduction in the heart rate (HR) that matched experimental observations. Further analysis showed that although the SND arose from an integrated action of all remodeling of ion channels and Ca2+-handling in both studies, it was the change to ICaL that played the most important influence.
The sinoatrial node (SAN) is the primary pacemaker of the heart. It is situated in the superior right atrium and produces a sequence of auto-rhythmic electrical activities that control the heartbeat (Baruscotti and Robinson, 2007). Dysfunction of the sinoatrial node (SND) associated with “sick sinus syndrome” (SSS) manifests as pathological bradycardia or systolic pauses (Haqqani and Kalman, 2007), producing inadequate blood supply to satisfy the demands of the body. This condition leads to symptoms such as dizziness and syncope (Haqqani and Kalman, 2007), though the initial stages of SND may be latent and asymptomatic (Choudhury et al., 2015).
Various physiological and pathological mechanisms (intrinsic, extrinsic or a combination of the two) are responsible for SND (Semelka et al., 2013). Among them is the failure of a specific part of the SAN to achieve impulse generation (Fenske et al., 2013), or the impulse conduction from the SAN to the surrounding atrial muscle, due to genetic mutations (Butters et al., 2010; Choudhury et al., 2018) or ischemic conditions (Bai et al., 2017). Increased fibrosis and degenerative changes in physiological properties of the SAN may also cause SND (Baruscotti and Robinson, 2007; Zhang et al., 2007; Choudhury et al., 2015).
Sinoatrial node dysfunction is also associated with aging (Tohno et al., 2014; Alghamdi et al., 2018). Whilst it can occur at any age, the occurrence of SND significantly increases with age (Härtel and Talvensaari, 1975). In a retrospective research study of 277 participants with compromised bradycardia, 51% of the cases were attributed to extrinsic causes (such as adverse drug reactions, imbalance of electrolytes or acute myocardial infarction, etc.), and 49% were attributable to either intrinsic or idiopathic (Sodeck et al., 2007) conditions, such as cardiac ischemia, gene mutations, excessive training and particularly aging (Semelka et al., 2013; Choudhury et al., 2015). More than 600,000 pacemaker implants are performed worldwide each year (Lamas et al., 2000; Wood and Ellenbogen, 2002; Bai et al., 2017), of which older adults account for the largest percentage.
Experimental data from animal models (Alings and Bouman, 1993; Satoh et al., 2005; Jones et al., 2007; Moghtadaei et al., 2016; Du et al., 2017) and human studies (Opthof, 1994; Dobrev, 2009) have demonstrated an association between SND and aging. It has been shown that during the aging process, changes occur to SAN function, manifested by increased pacemaking cycle length (CL) (i.e., slower heart rate) and reduced conduction of action potentials [i.e., an increased sinoatrial node-atrium conduction time (SACT)]. Both of these contribute to the reduced aerobic capacity of older adults, leading to an increased incidence of abnormal pacemaking and atrial arrhythmia (Sharpe et al., 2017). Further investigations have also revealed that such aging-associated SND is related to changes in the cellular electrical properties of the SAN (see Supplementary Table 1 in the Appendix) (Congxin Huang et al., 2006; Zhang et al., 2007; Hao et al., 2011; Larson et al., 2013; Tohno et al., 2014; Huang et al., 2015; Moghtadaei et al., 2016; Du et al., 2017) in a similar way to aging-induced cellular changes in atrial and ventricular cells (Congxin Huang et al., 2006) and intercellular electrical coupling (Jones et al., 2004). A list of aging-related changes in mRNA/proteins of ion channels, Ca2+ handling, intercellular coupling and tissue fibrosis population of the SAN for different species is given in Supplementary Table 1 in the Appendix.
Complex and variable patterns of changes to ion channels and calcium-handling proteins have been observed in aging-induced SND rat models (Tellez et al., 2011; Hatch, 2012; Huang et al., 2016). In their study, Tellez et al. (2011) (Aging Study-1) found that aging-SND was associated with a considerable changes of gene expression in the SAN, including: an increase in the relative abundance of mRNA for many ion channels [e.g., Nav1.5, Navβ1 and Cav1.2, Cav1.3 and KvLQT1, Kv4.2 (which are responsible for IKs and Ito)] in addition to an increase of the Ca2+ handling proteins, SERCA2a, that responsible for Ca2+-uptake process. The study also showed, a decrease in some other ion channels (e.g., in Cav3.1, NCX, and HCN1) and in the Ca2+ clock (RYR2) gene expressions of the SAN during aging. This implied changes of them during aging, though there is no explicit link between mRNA levels and channel activity (Brioschi et al., 2009). However, other studies (Aging Study-2) showed conflicting data, suggesting a trend of down-regulation of some ion channels (e.g., decreases of Cav1.2, HCN4, and RyR2 densities and proteins, and an increase of NCX and SERCA densities and proteins) in aged SAN cells (Hatch, 2012; Huang et al., 2016).
The ionic mechanisms that underlie the aging associated SND are as yet unclear. It is possible that SND is associated with different “patterns” of ionic and molecular remodeling mechanisms, including up-regulation or down-regulation of particular ion channels as identified in previous studies (Tellez et al., 2011; Hatch, 2012; Huang et al., 2016). The aims of the present study were to:
1. determine whether the age-induced changes in ion channels as identified in Aging Study-1 and Aging Study-2 were sufficient to account for the observed SND;
2. investigate the relative roles of particular remodeled ion channels on modulation of the characteristics of pacemaking APs, including the cycle length (CL), action potential duration (APD50), peak amplitude (PA), maximal upstroke velocity (dV/dtmax), and maximal diastolic potential (MDP); and
3. address the question of how differently remodeled ICa,L could lead to SND.
The consequence of aging-induced electrophysiological changes in ion channel currents in rat SAN cells was investigated using the model developed by Tao et al. (2011) for rat pacemaking APs. The model was based on modifications of rabbit SAN models (Zhang et al., 2000; Kurata et al., 2002) by incorporating experimental data for major ion channels obtained from rat SAN cells by Satoh (2003a, b) and Shinagawa et al. (2000). The data included aging-induced remodeling in key currents – ICa,L, IKs, IKr, and If – which were modeled by incorporation into classical Hodgkin-Huxley formulations.
In the Tao et al. model, the membrane potential Vm is dependent on a set of voltage-gated ion-channel currents, exchanger currents, and ionic pump currents, in the form of the following equations (1) and (2):
where Cm is the cell capacitance (set at 32pF); If the hyperpolarization-activated current; ICa,L and ICa,T the inward L-type and T-type Ca2+ currents; IKr and IKs the rapid and slow delayed rectifier K+ currents; Ito the Ca2+-independent transient outward K+ current; Ist the sustained inward current (carried by Na+); Isus the sustained component of 4-AP sensitive current; IK,ACh the muscarinic K+ channel current; INaK the Na+-K+ pump current; INaCa the Na+-Ca2+ exchanger current; and Ib,Na the background inward Na+ current. Details of the equations and parameters of the model are documented in Tao et al. (2011).
We obtained the source code of this mathematical model via a request to the author. The original model was developed with neural-myocyte coupling and the modulation of pacemaking by nitric oxide and cyclic GMP in response to brief sympathetic stimulations observed in hypertension. As the present study was focused on studying the effects of aging on the morphology and characteristics of the pacemaking APs, therefore the neural-myocyte coupling was removed from the model, resulting in the simulated cycle length of the pacemaking APs at 230 ms, which is slightly different to that of the original model at 257 ms, but is still within the experimental range value of the rat SAN in Shinagawa et al. (2000). Numerically, the Hodgkin-Huxley equations for the rat SAN cell model were solved with a time step of 0.01 ms, which was sufficiently small to ensure a stable numerical solution.
To simulate the functional impact of aging-induced changes of membrane ion channels and intracellular Ca2+-handling on cardiac pacemaking activity, experimental data from two independent groups of studies on aging rat SAN cells, as identified by Tellez et al. (2011) (denoted Aging Study-1); and Hatch (2012) and Huang et al. (2016) (collectively denoted Aging Study-2) for adult and older adult rat SAN were incorporated into the model Tao et al. (2011). As some of the experimental data were derived from mRNA gene expression or protein levels, their incorporation was carried out with an assumption on that there is a correlation between the gene expression or protein levels and certain ion channel current densities as we did in our previous studies (Hao et al., 2011; Choudhury et al., 2018). Both datasets were obtained from SAN cells isolated from the right atrium of Wistar Hannover rats with ages of ≥25 or ≥24 months (equivalent to about a 70-year-old human) (Tellez et al., 2011; Hatch, 2012). In both studies, cellular APs and ECGs were recorded. These data showed reductions in heart rates in the older adult rat SAN preparations by 18 (Tellez et al., 2011) and 11% (Hatch, 2012), respectively, as compared with adult preparations. Such a change in heart rate is associated with changes in cellular ion channels, as summarized in Supplementary Table 2 (see Appendix).
Changes of different ion channels in the aging condition as shown in Supplementary Table 2 were incorporated into the Tao et al. model through modifying the maximal channel conductance value of a particular ion channel equation. To simulate the effects of the aging-induced changes of the expression for RyR and SERCA, we modified the Tao et al. model which includes the Ca2+ handling equations from the Zhang et al. (2000) and Kurata et al. (2002) models of the rabbit SA node cells, by changing the flux of the Ca2+ release from the SR (Jrel) and Ca2+ uptake by the SERCA (Pup) based on scaling factors as listed in Supplementary Table 2. The equations for the SR Ca2+ release and SERCA Ca2+ uptake are as follows:
In simulations, AP characteristics including CL (and hence HR), maximal diastolic potential (MDP), peak amplitude (PA) and action potential duration (APD50) were computed in adult and older adult conditions, which were compared with the relevant experimental data (Hatch, 2012; Huang et al., 2016) for validation. In order to elucidate the primary factor(s) responsible for the slower heart rate in the older adult cells, the relative impact of different remodeled ionic channels on CL were analyzed via two distinct methods: the inclusive, and the exclusive. With the inclusive method, all changes in the remodeled ion channels and Ca2+-handling due to aging were considered for the older adult condition. With the exclusive method, only a specific remodeled ion channel or subsets of all the remodeled ion channels were considered for the older adult condition, while the rest of the aging-induced changes were not included in the models. Details of ion channels considered in each case are described in the following sections.
Figure 1 shows the simulated effects of aging on the pacemaking AP of rat SAN cells based on the experimental data set of Aging Study-1 (left panels) and Aging Study-2 (right panels). Both aging datasets produced a similar consequence of aging-related bradycardia, i.e., a slower pacemaking rate in the older adult compared with the adult condition, although there are dramatic differences in the underlying remodeling of membrane ion channels and the Ca2+-handling (see Supplementary Table 2 in the Appendix). With the use of Aging Study-1 data (Tellez et al., 2011), the CL was increased from 230 ms in the control condition to 310 ms in the older adult condition, implying a 19% decrease in pacemaking rate (HR). Similarly, using the Aging Study-2 data, an increase of the CL from 230 to 265 ms was observed, corresponding to ∼12.9% decrease in HR. Such simulated effects of aging on slowing the cardiac pacemaking APs are in agreement with those observed experimentally (Hatch, 2012; Huang et al., 2016).
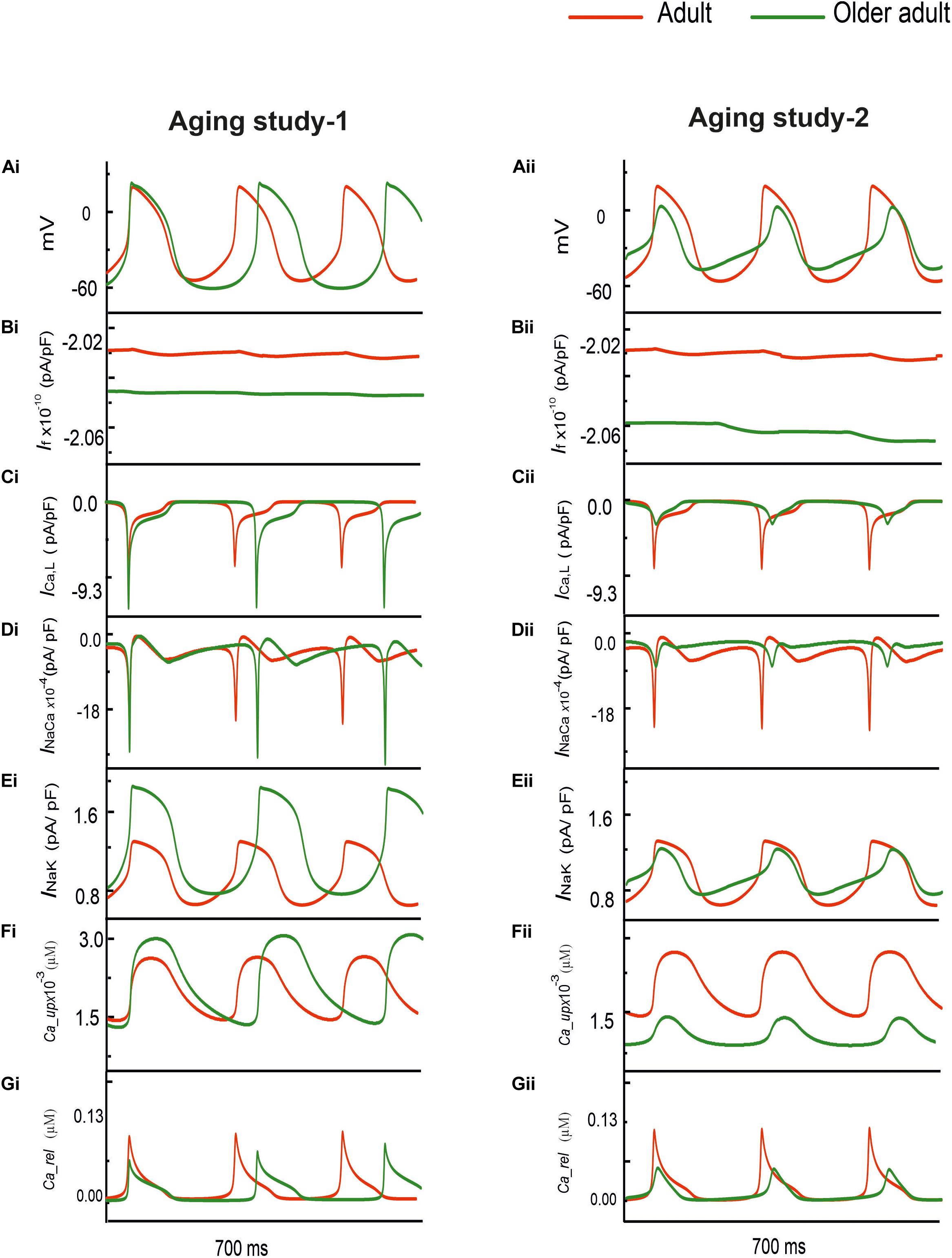
Figure 1. (Ai,Aii) Action potentials computed from rat SAN myocyte model in adult (red) and older adult (green) conditions using the Aging Study-1 (left panels) and Aging Study-2 (right panels) datasets. Simulations were conducted by using the rat SAN cell model developed by Tao et al. (2011). (Bi–Gi,Bii–Gii) Underlying ionic channel currents and Ca2+-handling during APs: If, ICaL, INaCa, INaK, Ca2+ uptake and Ca2+ release.
Although simulations using both experimental data (Tellez et al., 2011) sets showed similar effects on the reduction of the pacemaking rate, each of which produced different changes in the characteristics of the simulated APs. In the case of Aging Study-1, the simulated pacemaking APs in the older adult condition (Figure 1Ai) presented a more hyperpolarized maximum diastolic potential (MDP) (by −12.8 mV), a greater dV/dtmax (by 60%), PA (by 20%), and APD50 (by 14%). These changes in AP characteristics were attributable to changes in the underlying ion-channel currents and Ca2+-handling, which were manifested as a decreased If (Figure 1Bi), increased ICa,L (Figure 1Ci), decreased INaCa (Figure 1Di), as well as an increased INaK (Figure 1Ei) and Ca2+-uptake (Figure 1Fi), but a decreased Ca2+ release (Figure 1Gi).
However, in the case of Aging Study-2, the simulated pacemaking APs (Figure 1Aii) presented an elevated maximum diastolic potential value (by + 9.3 mV), decreased dV/dtmax (by 64%) and PA (by 70%), as well as an abbreviated APD50 (by 17%). These changes were attributable to decreased If (Figure 1Bii) and ICaL (Figure 1Cii), increased INaCa (Figure 1Dii) and Ca2+ uptake (Figure 1Fii), but decreased Ca2+ release (Figure 1Gii). The decrease in PA was attributable to the reduction of ICaL.
Simulations of the aging effect on the pacemaking APs and their characteristics were validated by quantitatively comparing the simulation data to experimental data when possible. Results are shown in Figure 2 and summarized in Supplementary Table 3 in the Appendix. In the case of Aging Study-1, simulations showed about 80 ms increase in CL, leading to a 19% reduction in HR. This is quantitatively consistent with the study of Tellez et al. (2011), who observed about 30% increase in CL that corresponded to an 18% reduction in HR. We also observed increases in APD50 and maximal dV/dtmax in simulations, that were quantitatively consistent with the results of Tellez et al. (2011).
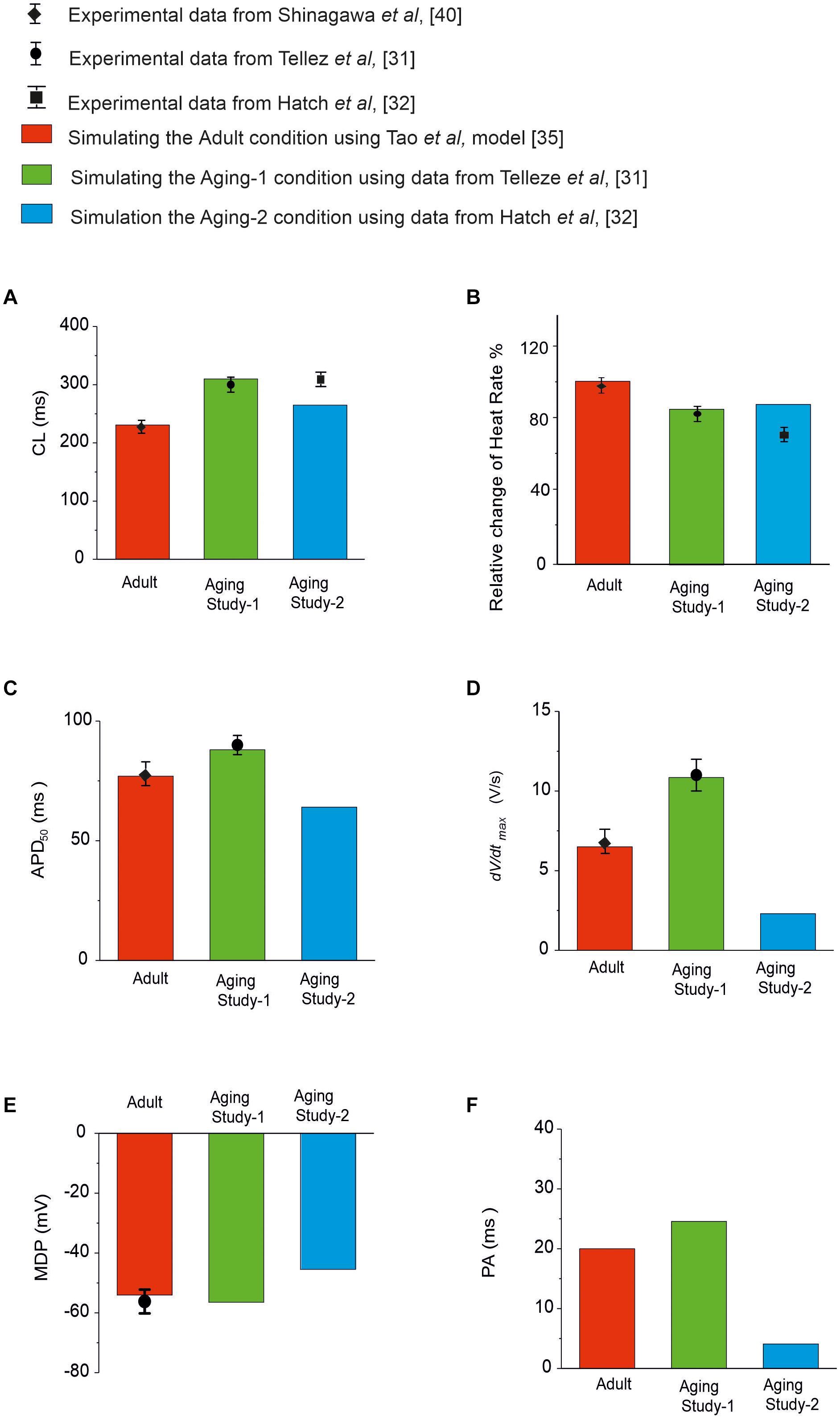
Figure 2. Quantitative comparison between experimental and simulation data of the functional effects of aging on the pacemaking APs and their characteristics. (A) CL; (B) HR; (C) APD50; (D) dV/dtmax; (E) MDP; and (F) PA. Data for adult is shown in red, and from Aging Study-1 and -2 in green and blue, respectively. Experimental data with error bars, where available, are from Tellez et al. (2011); Hatch (2012), Huang et al. (2016), and Shinagawa et al. (2000), which were all from rat SAN cells.
In Aging Study-2, our simulations showed that aging caused an increased CL by 35 ms, which corresponded to a reduction in HR of about 12.9%. This was also comparable with data from Hatch (2012), who observed a CL increase of 40% in CL; corresponding to a 30% decrease in HR. It was also comparable with the data from Huang et al. (2016), which showed an increase of the CL from 193 to 235 ms (the intrinsic HR changed from 310 to 255 bpm) with aging, corresponding to 17.7% reduction of the intrinsic HR. We also observed decreases in APD50, dV/dtmax and the amplitude of action potentials. However, there are no experimental data available for quantitative comparison with these values.
Simulation results using the data sets from Aging Study- 1 and 2 produced similar bradycardic effects that both qualitatively and quantitatively matched to their experimental observations (although limited experimental observations for Aging Study-2 were available), despite of marked differences in their underlying remodeled ion channels and Ca2+-handling were observed. In order to elucidate the primary factor(s) that contributed to aging bradycardia, effects and their relative roles of individual, or a subset of, remodeled ion channels and Ca2+-handling on pacemaking APs were simulated and analyzed using the exclusive method. Through this method, only some specific aging-induced change(s) were considered, while other remodeling factors were omitted.
After analysis of the differences and similarities in the aging-induced remodeling patterns between Aging Study- 1 and 2, three different cases were analyzed, as shown below.
Both group aging studies showed a down-regulation of HCN channels in mRNA for aging study-1 and in protein expression for aging study-2. For RyR Ca2+ release channels, also, presented a reduction of in mRNA expression in aging study-1, in protein expression and expression density of aging-2. Therefore, in Case 1, simulations were conducted to evaluate the effects of a reduced If by 16% in Aging Study-1 [this value was based on the mRNA expression data in Tellez et al. (2011) study], and 30% in Aging Study-2 [this value was based on the data of Huang et al. (2016) study], and RyR Ca2+ release from the sarcoplasmic reticulum (SR), by 80% in Aging Study-1 [this value was obtained from the mRNA expression data in Tellez et al. (2011) study], and 24% in Aging Study-2 [this value was based on the protein expression density data in Hatch (2012) study], on the pacemaking APs. Results in Figure 3 show the computed APs for the control (adult) and for the older adult conditions based on Aging Study-1 [left panels (Ai)] and Aging Study-2 [right panels (Aii)]. During the time course of APs, the time traces of If and Ca2+ release flux from the SR are also shown in panels (Bi) and (Bii), and (Ci) and (Cii), for the adult and older adult conditions, respectively.
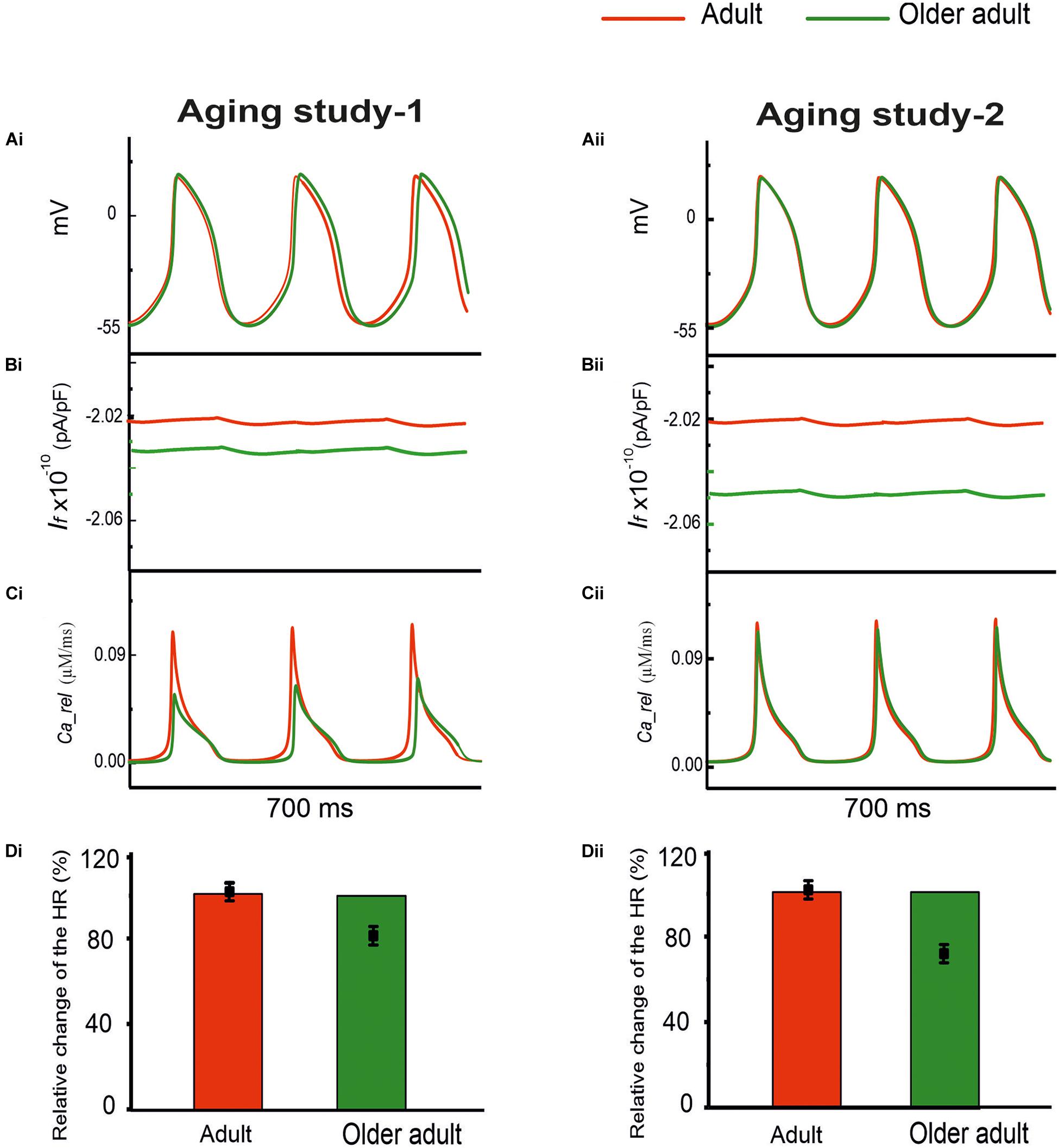
Figure 3. Simulated role of reduced If and Ca2+ release from the SR in aging bradycardia. Changes in other ion-channel currents as reported by Tellez et al. (2011) in Aging Study-1 (right panels) and by Hatch (2012) and Huang et al. (2016) in Aging Study-2 (left panels) were omitted. (Ai,Aii) APs. (Bi,Bii) Ionic current profiles for If, and (Ci,Cii) for Ca2+ release, for adult (red) and older adult (green) conditions. (Di,Dii): Bar charts showing the simulated age-dependent relative change in HR for this case; data-points with error bars show the experimental HR reduction due to aging.
With both data sets, reduced If and Ca2+ release from the SR had a limited effect on slowing down pacemaking rate. With a combined action of reduced If and Ca2+ release, the HR was reduced with the dataset from Aging Study-1, but unchanged with the dataset from Aging Study-2.
Simulation results from both datasets suggested a limited role of combined action of remodeled If and Ca2+ release in the bradycardia. Further analysis was conducted to analyze the individual role of a wide-range reduction of If or SR Ca2+ release. When If reduction alone was considered from 16% (Aging Study-1) to 30% (Aging Study-2), there was no noticeable change in spontaneous APs (data not shown). This result was in agreement with the results of Bers (2001), who observed that If played only a minor role in primary and central SAN cells, though a critical role in peripheral SAN cells.
When a reduced SR Ca2+ release alone was considered, either by 80% (Aging Study-1) or by 24% (Aging Study-2), there was a small increase in the measured CL. With an 80% reduction in the RyR2 alone, as suggested by Aging Study-1, CL was increased by 2.45%, while with a 24% reduction in the RyR2 alone as seen in Aging Study-2, no noticeable change in the CL was observed (data not shown).
To test whether or not the simulated consequences of RyR2 reduction by 80% (Aging Study-1), and 24% (Aging Study-2) are model-dependent, the Maltsev and Lakatta (2009) model for the rabbit SAN cells was also used. Results are shown in Supplementary Figure 1. It was found that a 24% reduction of the RyR2 had little effect on reducing the pacemaking rate, while 80% reduction of RyR2 produced a small, but notable change of the APs, manifested by an increased CL (by 5.2%), elevated MDP and less steep DD phase, leading to an increased time taken to producing the successive peacemaking APs. The simulation results using the Maltsev and Lakatta (2009) model for the rabbit SAN cells matched the findings using the Tao et al. model for the rat SAN cells in showing that the aging-induced change in RyR made a limited contribution to aging-associated modulation of SAN spontaneous APs.
The time traces of Ca2+ release flux during the APs are shown in panels (Ci) and (Cii) for the adult (red) and older adult (green) conditions (with a combined action of reduced If and Ca2+ release). Panels (Di) and (Dii) compare the simulation and experimental results of relative changes in HR when the age-induced remodeling of If and RyR2 alone were considered.
Both the experimental datasets showed changes to ICa,L, INaCa and Ca2+ uptake from the SR with aging, but with conflicting findings. In Aging Study-1, up-regulation of ICa,L and Ca2+ uptake, but down-regulation of INaCa, were observed; whilst in Aging Study-2, down-regulation in ICa,L and Ca2+ uptake, but an up-regulation of INaCa, were shown.
In order to investigate the role of this conflicting subset of aging-induced remodeling shown by the two sets of data, further simulations were conducted by incorporating the altered ICa,L, INaCa and Ca2+ uptake into the model to investigate their contribution to the aging bradycardia.
Results are shown in Figure 4 for the computed APs (Ai and Aii) in the adult and older adult conditions, as well as the underlying time courses of ICa,L (Bi, Bii), INaCa (Ci, Cii), and Ca2+-uptake (Di, Dii) based on data of Aging Study-1 (left panels) and -2 (right panels). Figure 4Ai shows the simulated pacemaking APs based on data from Aging Study-1, with considerations only of an increase of ICa,L by 25%, Ca2+ uptake by 15%, and a decrease in INaCa by 6% for the older adult condition [data based on the mRNA expression data in Tellez et al. (2011) study]. As compared with the adult condition, the computed CL increased by 23.4% (i.e., a decrease in HR of 11%), and this was accompanied by changes in other AP characteristics, including an increase in PA of 17.5% and in APD50 of 23.4%. In this case, the observed changes in CL and AP characteristics were comparable with those obtained when all remodeled factors were considered, as well as close to experimental observations (Tellez et al., 2011). The results seem to suggest that the aging-induced up-regulations of ICa,L and Ca2+-uptake, and down-regulation of INaCa, are sufficient to account for the aging bradycardia seen experimentally in Aging Study-1.
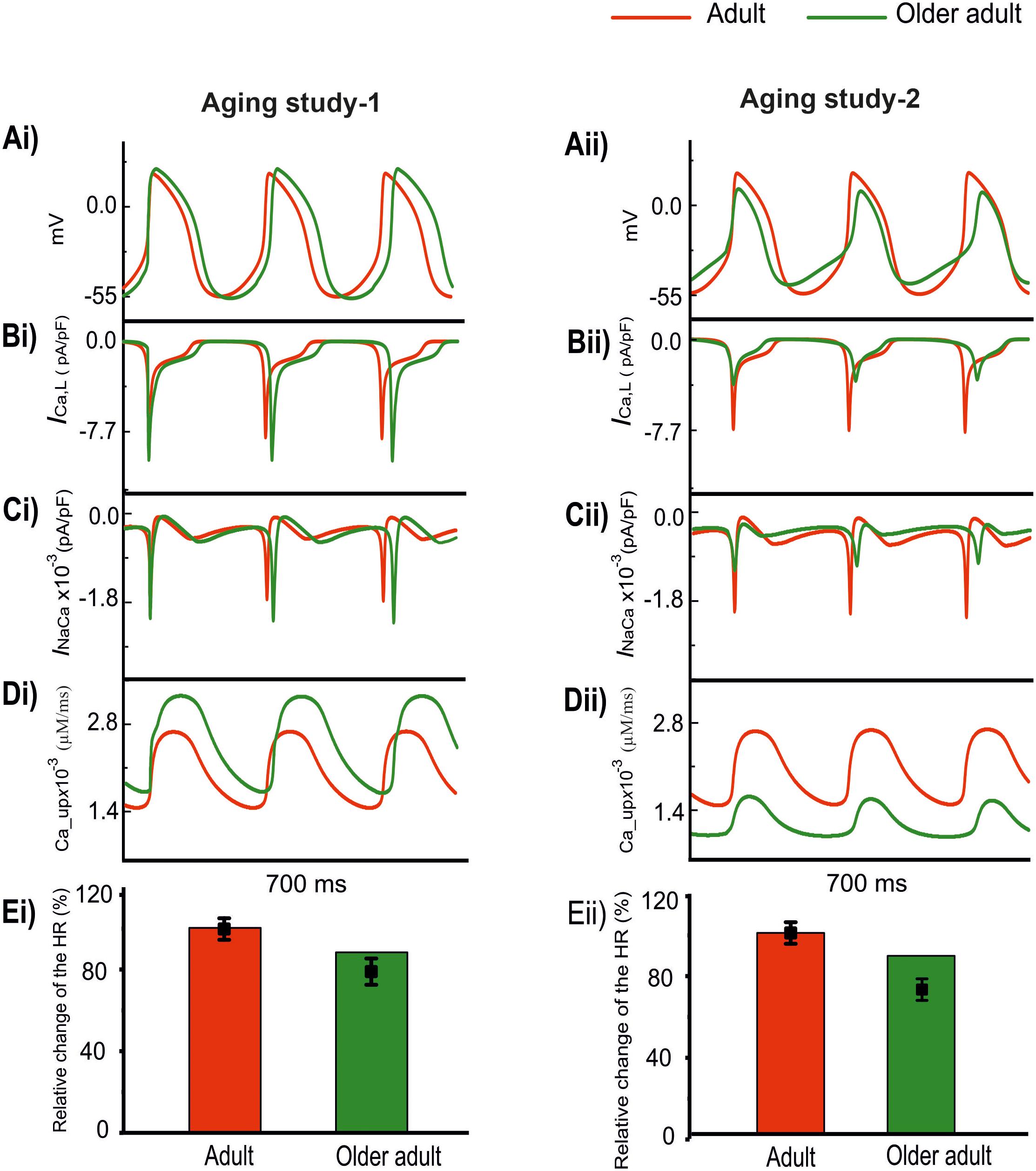
Figure 4. Effect of remodeled ICa,L, INaCa, and Ca2+ uptake on pacemaking APs (red: adult; green: older adult). Other ion channel changes, as seen by Tellez et al. (2011) in Aging Study-1 (right panels), and by Hatch (2012) in Aging Study-2 (left panels), were omitted in simulations. (Ai,Aii) APs. (Bi,Bii) ICa,L. (Ci,Cii) INaCa. (Di,Dii) Ca2+ uptake. (Ei,Eii) Relative HR and its reduction. Data-points with error bars show the experimental data.
Figure 4Aii shows simulated APs based on data from Aging Study-2 with a consideration of a decrease of ICa,L by 50%, Ca2+-uptake by 29%, and an increase of INaCa by 42% [data based on the protein expression density data in Hatch study (Hatch, 2012) as shown in Supplementary Table 2]. As compared with the APs in the adult condition, the implemented changes also reproduced bradycardia, resulting in a 9% increase in CL (i.e., a 7% HR decrease), which was accompanied by other changes in AP characteristics. These included an elevated MDP by 6 mV, a reduction of PA by 55% and an APD50 shortening by 14%. Those observed changes in APs were also close to the experimental observations of Hatch (2012), which implied that the subset of remodeled ICa,L, INaCa, and Ca2+ uptake, as seen in Aging Study-2, were also sufficient to produce the aging-related bradycardia.
The use of this subset of data from either Aging Study-1 or Aging Study-2 produced bradycardia, though there are clear differences between the two. In Aging Study-1, bradycardia was associated with an increased ICa,L (Figure 4Bi) and SR Ca2+ uptake (Figure 4Ci), and a decreased INaCa (Figure 4Di). In contrast, in Aging Study-2 it was associated with a decreased ICa,L (Figure 4Bii) and decreased SR Ca2+ uptake (Figure 4Cii), and an increased INaCa (Figure 4Dii). Quantitatively, the computed HR reductions in the older adult condition were close to the experimental data of Tellez et al. (2011) and Hatch (2012) for Aging Study-1 (Figure 4Ei) and Aging Study-2 (Figure 4Eii), respectively.
Figure 5 shows the results when aging-related remodeling of ICaL in a range based on the study of Tellez et al. (2011) and Hatch (2012) was considered. In Aging Study-1 (Figure 5Ai), the 25% up-regulated ICa,L increased the peak amplitude of the AP from 20 to 25 mV and prolonged the APD50 from 77.06 to 95.11 ms, which slowed down the time course for repolarization and consequently led to a slowing down in the pacemaking as seen in aging bradycardia when all remodeled ion channels were considered in Aging Study-1. It also produced a more hyperpolarized maximal diastolic membrane potential, which led to an increased voltage difference between the maximal diastolic membrane potential, MDP, and the take-off potential (TOP), at which a line from the MDP along the diastolic depolarization time course intersects with a vertical line drawn from the AP overshoot. This contributed to increased time taken to generate a successive action potential, though the depolarization INaCa current during the diastolic depolarization (DD) phase was increased. Overall, this led to an increased time interval between two successive APs (i.e., CL), leading to a slowed heart rate (see Table 1). Figure 5B shows how both increase and decrease of the ICa,L through changing the gCaL can affect the measured CL leading to increase the pacemaking.
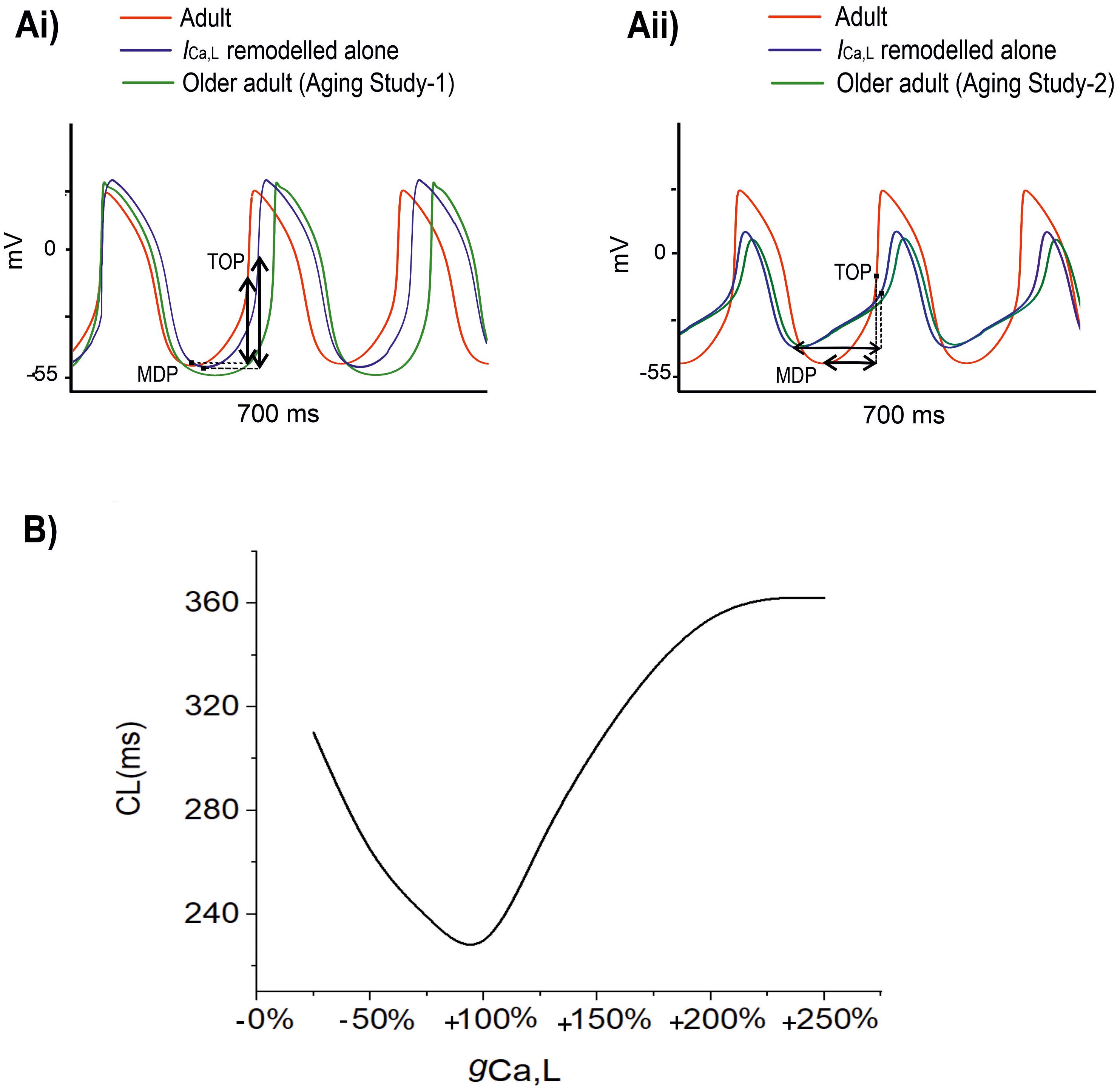
Figure 5. (A) Simulated APs when the age-related remodeling of ICa,L alone (blue) was considered [other ion channel and Ca2+ handling changes, as seen by Tellez et al. (2011) in Aging Study-1 (right panels), and by Hatch (2012) in Aging Study-2 (left panels), were ignored]. The obtained APs were compared with those of adult (red) and those with all ion channel and Ca2+ handling remodeling were considered (green). (B) Relationship between CL and increase (positive direction) or decrease (negative direction) in gCaL. It demonstrates that CL increases with either an increased or a decreased gCaL compared to the reference value of 100%. MDP: the maximal diastolic membrane potential. TOP, the take-off potential at which a line drawn from the MDP along the diastolic depolarization time course intersects with a vertical line drawn from the AP overshoot.
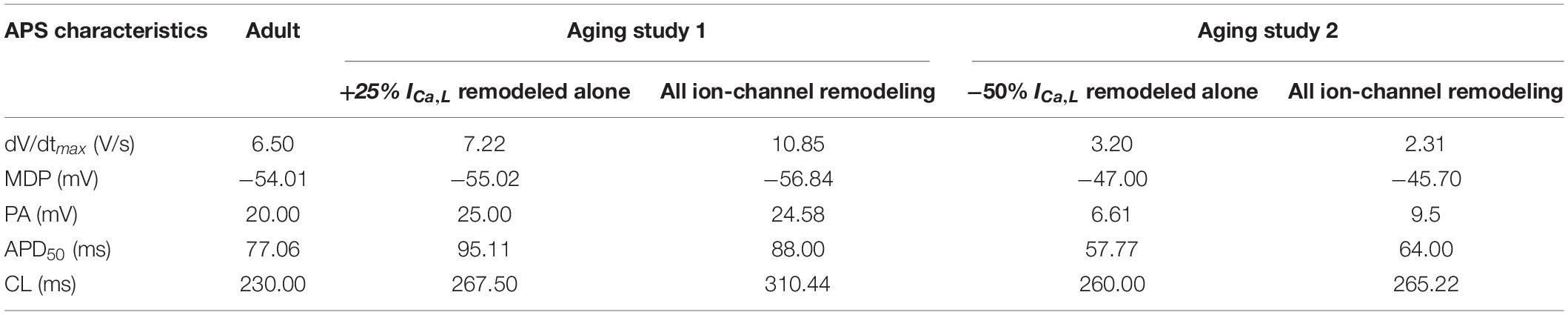
Table 1. Quantitative analysis of the role of age-related remodeling of ICa,L alone in bradycardia as compared to the case when all remodeling were considered.
In Aging Study-2 (Figure 5Aii), a decreased ICa,L also produced bradycardia. In this case, the decreased ICa,L was associated with a decreased AP peak amplitude from 20 to 6.61 mV and abbreviated APD50 from 77.06 to 57.77 ms. Both accelerated the repolarization process but produced an incomplete repolarization as a consequence of reduced AP amplitude and abbreviated APD50. Consequently, an elevated MDP was produced, leading to a reduced depolarization current in the DD phase (such as INaCa), which slowed down the time course between the MDP and the take-off potential, leading to an increased time interval between two successive pacemaking APs, resulting in bradycardia that was also comparable to the case when all remodeled ion channels were considered in Aging Study-2. Figure 5B shows how either an increase or a decreased ICa,L through changing gCaL modulates the measured CL leading to bradycardiac pacemaking.
In Aging Study-1, when the age-related increase in SERCA2a (by 15%), or the decrease in NCX (by 6%) alone was considered, negligible effects in modulating the pacemaking APs was observed. Similarly, in Aging Study-2, when a decrease in SERCA2a (by 29%) or an increase in NCX (by 42%) alone was considered, their effect on the pacemaking APs was also limited.
These simulations implied that based on experimental data of Aging Study- 1 and 2 the altered ICa,L produced notable effects on the modulation of the pacemaking AP profiles and rates, leading to bradycardia, though through different actions of either up-regulation or down-regulation of it.
Aging Study-1 also found changes in other ion channels, including IKr, IKs, Ito, INak, and ICa,T, which were absent in Aging Study-2. Further simulations were conducted to investigate the effects of such a subset of remodeled ion channels on aging bradycardia. In simulations, we considered the integral action of changes to IKr (by a 6% reduction), IKs (by a 25% increase), Ito (by a 60% increase), INaK (by a 50% increase), and ICa,T (by a 12% decrease).
Simulation results of APs for the adult and older adult conditions are shown in Figure 6A. In this case, the remodeled ion channels produced a 9.13% increase in CL (i.e., a 6.9% decrease in HR). It also produced changes in other AP characteristics, including a more hyperpolarized MDP (by −3 mV), and a prolonged APD50 (by 26 ms).
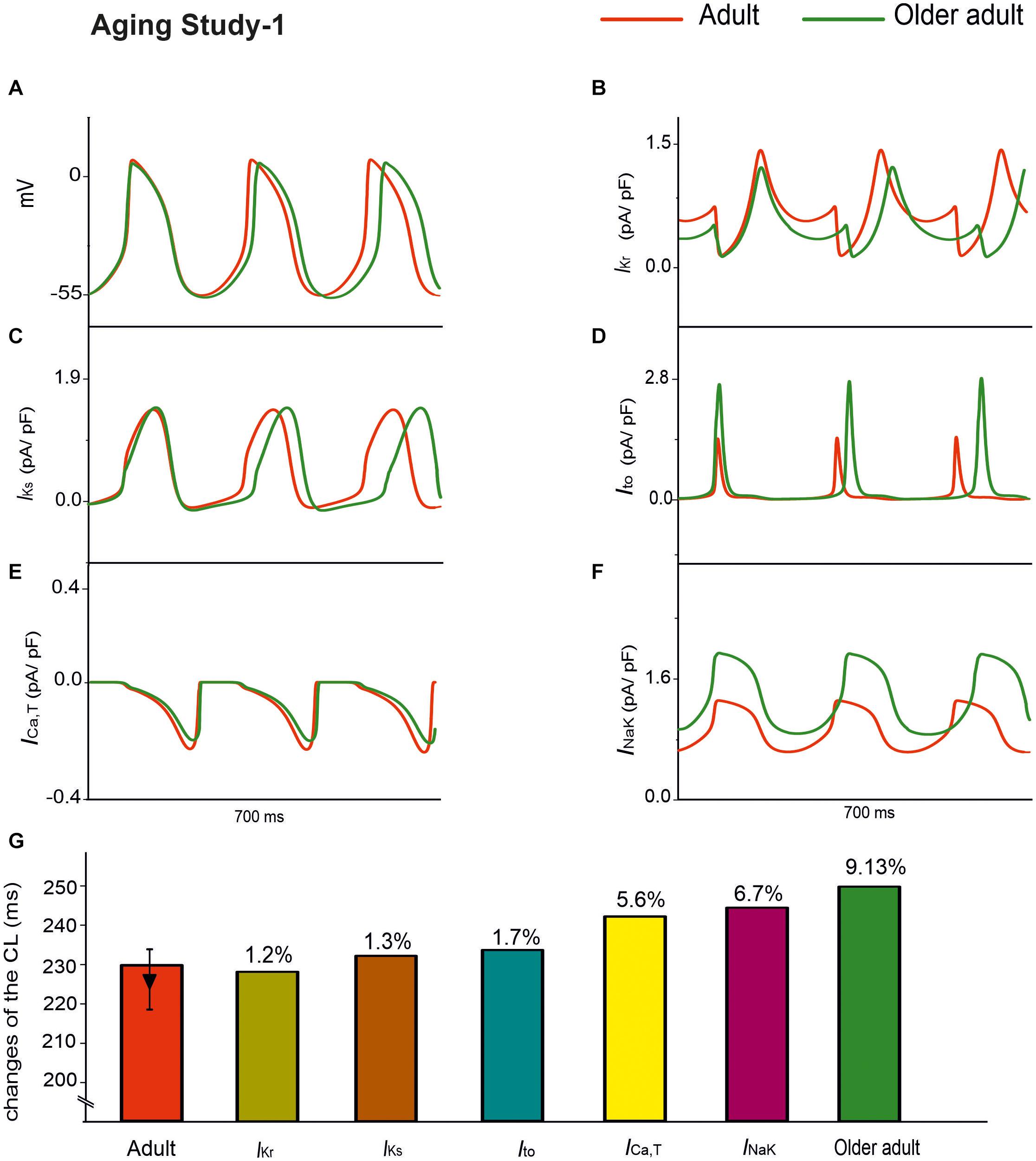
Figure 6. APs for the adult and older adult conditions with the subset of changes to the ion-channel currents involved in Aging Study-1, namely IKr, IKs, Ito, ICa,T, and INaK (A). Panels (B–F) show the time traces for the relevant ion-channel currents, namely IKr, IKs, Ito, ICa,T and INaK in the adult and older adult conditions. (G) Illustrates the role of the individual remodeled ionic channels on CL values.
Panels B–F in Figure 6 show the time traces of the relevant ion-channel currents for IKr, IKs, Ito, ICa,T, and INaK in the adult and older adult conditions. It was shown that during APs, the decreased of the peak amplitude might be attributable to the change in Ito and IKs; the more hyperpolarized MDP might be attributable to the increased INaK; the prolonged APD50 was attributable to the decreased IKr; and the decreased DD phase was partially attributable to the integral action of the decreased IKr and ICa,T. The prolonged APD50 slowed down the repolarization phase and, together with the slowed DD depolarization phase and the increased time interval required for membrane potential to reach the take-off potential from the MDP, resulted in a slowed HR.
The individual roles of each remodeled IKs, Ito, INaK, and ICa,T ion channels were also simulated, generating a 1.3, 1.7, 6.7, and 5.6% change in CL, respectively, as shown in Figure 6G. The simulation showed that the remodeled INaK and ICa,T currents alone made notable contributions to an increased CL, in which the remodeled IKr current was responsible for the increase in APD50, slowing the repolarization phase of the APs.
The main findings of this study are the following:
(1) heart-rate reduction in aged rat sinoatrial myocytes can be sufficiently accounted for by the age-induced ionic-current and Ca2+-handling remodeling of ICa,L, If, INaCa, SERCA2a, and RyR;
(2) whilst all remodeled channels and Ca2+-handling contribute to the heart-rate reduction in the older adult, the remodeled ICa,L plays the most significant role; and
(3) remodeled ICa,L, either up-regulated or down-regulated by aging, leads to a similar bradycardia effect.
Possible mechanism(s) underlying the age-related SAN dysfunction were investigated by both the inclusive and exclusive methods in simulations. With the inclusive method, when all remodeling of ion channels and Ca2+ handling identified in Aging Study-1 (Tellez et al., 2011) and Aging Study-2 (Hatch, 2012; Huang et al., 2016) were considered, a similar bradycardic effect was produced, which was manifested by a slower pacemaking rate of APs in the older adult as compared with the adult rat SAN cell. This observation suggested that the identified age-related changes in ion channels (ICa,L, If, INaCa) and Ca2+-handling (SERCA2a and RyR) in both studies were adequately responsible for the experimentally observed age-related changes in APs, which were consistent to the experimental data of the two studies. Note that, though a similar bradycardic effect was produced, changes in some other AP characteristics, such as the maximal upstroke velocity (dV/dtmax), AP amplitude and duration (APD50), were different between the two cases of simulations. While the computed dV/dtmax and APD50 in the older adult condition were increased in the case of Aging Study-1, they were decreased in the case of Aging Study-2.
The relative roles of aging-remodeled If and Ca2+-RyR release in bradycardia was investigated by using the exclusive method. When down-regulations of If and Ca2+ RyR release were considered for the older adult condition as seen in both Aging Studies 1 and 2, no apparent effect was observed on modulating the APs. This suggested a limited role of remodeled If and Ca2+ RyR release in generating the age-related bradycardic effect.
A limited role of If in aging bradycardia in slowing down the pacemaking APs as seen in the present study is consistent with the experimental observation of Shinagawa et al. (2000), who reported that If activated at a very negative threshold, −90 mV, and is little activated under the normal AP threshold range (−70 to −50 mV). As the Tao et al. model was developed for the primary pacemaking cells of the rat SAN, which has a MDP at around −55 mV, at which the activated If is small, and therefore contributes only slightly to the pacemaking APs of the rat SAN cells. Considering the regional differences in cellular electrophysiological properties of SAN between central and peripheral regions, including the MDP and ion channel current densities as seen in the rabbit heart (Zhang et al., 2000), it is expected that the aging-induced If remodeling may have a greater effect in modulating the pacemaking APs in peripheral cells as compared to the results seen in the present study using the Tao et al. model for the central SAN cells. Unfortunately, a lack of rat SAN experimental data in this regard precluded investigation of this possibility in this study. The issue certainly warrants future investigation when experimental data on the regional differences in cellular electrophysiological properties of the rat SAN become available.
In both data sets of Aging Studies 1 and 2, substantial down-regulation of RYR2 in the SAN during the aging process was observed, which was believed to be responsible for a reduced heart rate in the aged rat (Tellez et al., 2011). However, in our simulations, a reduction of SR Ca2+ release in the range from 30 to 80% as observed in the two experimental studies showed only limited effects in slowing down the heart rate. A complete block of Ca2+ release from the SR only produced a 3.5% increase in CL. This finding was in agreement with previous experimental observations, which showed that the effect of SR Ca2+ release on pacemaker activity in adult rat central SAN cells was small due to the poor development of SR in these cells (Tao et al., 2011).
There has been an ongoing debate as to the leading mechanism responsible for the pacemaking activity in the SAN (Lakatta and DiFrancesco, 2009). One theory is the role of the membrane clock, in which If plays an important role (DiFrancesco, 1993; Accili et al., 2002). The other is that the Ca2+ clock plays an important role, by which SR pumping kinetics are thought to regulate spontaneous beating in rabbit SAN (Maltsev and Lakatta, 2009). The rate of SR replenishment defines the cycle length of each natural beat. Its occurrence is based on the cytosolic Ca2+ availability and SERCA2a activity due to RYR2 release flux and Cav1.2 influx, as described by Vinogradova et al. (2000). Even though the SR’s role in initiating an AP is debatable and may be species dependent (DiFrancesco, 1993; Accili et al., 2002; Lakatta and DiFrancesco, 2009; Maltsev and Lakatta, 2009; Li et al., 2013), there are distinct changes in SR proteins that are clearly related to aging.
There are contradictory changes in ICa,L, INaCa and Ca2+ uptake from the SR between the two group datasets of the aging studies. In order to understand their relative contribution(s) to the age-related bradycardia, the exclusive method was used to investigate the collective effect of the three factors as well as their individual role. It was shown that whilst this subset of remodeling factors collectively are attributable to the bradycardic effect, it was the remodeling of ICa,L that played a primary role. In both datasets, when a down-regulation or up-regulation of ICaL alone was considered, a dramatic bradycardic effect was observed, which was comparable to the case when all ion channel and Ca2+ handling remodeling were considered. An increase in the pacemaking CL with a decrease of the channel conductance of ICaL has been shown before by Kurata et al. in the rabbit SAN cell model (Kurata et al., 2002). In the present study, we expanded their observation by showing that not only the decrease, but also the increase of ICaL can cause slowing down in the pacemaking APs as manifested by an increase in the CL.
Our results highlight the importance of the remodeled ICa,L to generate aging-related bradycardia (see Figure 7). This is consistent with a previous study, which showed that ICaL was a critical pacemaking current in adult rat SAN, as demonstrated by spontaneous beating being halted by Ca2+ antagonists (Hatch, 2012). In simulations, we also observed a complete ICa,L block abolished the pacemaker activity (data not shown), which is consistent with the experimental observation. When remodeling in SERCA2a (by 29%) or NCX alone was considered, limited effects on pacemaking APs were observed as seen in previous studies (Katz and Miledi, 1968; Szentesi et al., 2004). Collectively, our simulation results implied that the remodeled ICa,L has the most significant influence on slowing down of the pacemaking rates in the older adult rat SAN myocytes.
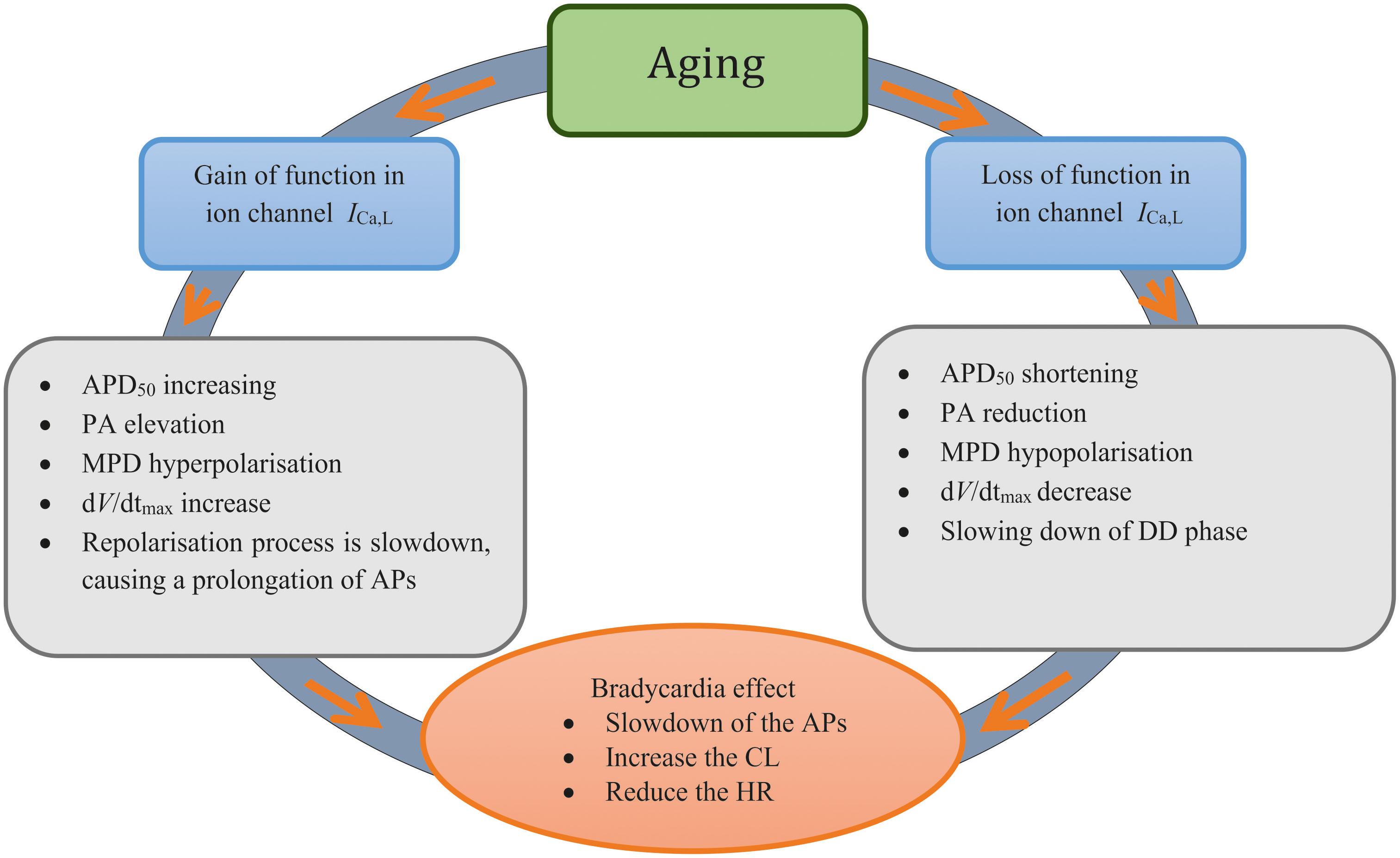
Figure 7. Diagrammatic representation to summarize the two ‘ways’ of remodeled ICa,L Investigated in this study and the mechanisms associated with each process.
The role of each individual remodeled ion channel of IKr, Ito, IKs, INaK, and ICa,T, as seen in Aging Study-1, was also investigated. The functional effect of these remodeled ion channels prolonged the time course of APs, leading to a reduced pacemaking rate, some of which was associated with a secondary action. For example, the reduction in IKr prolonged the APD50, therefore slowing down the repolarization phase of the APs. In the adult rat SAN model, IKr is considered to be another important pacemaking current. It has been observed that during diastole, IKr is slowly deactivated and the decreased outward current enables self-activated depolarization. Therefore, in combination with other currents, remodeling of this current might play a secondary role in reducing the HR of the aged SAN (Talano et al., 1978; Hagiwara et al., 1988; Zhou and Lipsius, 1994; Lei et al., 2002; Tellez et al., 2011). Decreased spontaneous activity and sinus arrest were observed in rat SAN cells when IKr was completely blocked by E-4031 (Mitsuiye et al., 2000).
The differences between Aging Study-1 and Aging Study-2 could be the result of mRNA changes not reflecting changes in protein or erroneous protein measurement. Alternatively, the differences between the two studies could reflect different remodeling patterns to age-dependent sinus node dysfunction. The literature on age-dependent sinus node dysfunction is conflicting with different studies reporting different changes (Lamas et al., 2000; Hao et al., 2011; Moghtadaei et al., 2016). Once again the differences could be the result of shortcomings in the methodologies used, but they could be genuine. Genuine differences could be the result of the use of different species in different studies. There are other possible reasons. A clue comes from the study of Moghtadaei et al. (2016) which shows that in aged mice the sinus node dysfunction depends on the frailty of the mouse. Therefore, it is possible that in the aged animal the nature of sinus node dysfunction depends on co-morbidities. For example, in a study of age-dependent sinus node dysfunction in the rat, we noted that the old male rats were obese (Yanni et al., 2010). We have independently shown that obesity in the rat causes molecular remodeling of ion channels and sinus node dysfunction (Linscheid et al., 2019; Webb et al., 2017, 2018). Therefore, the differences between studies could be the result of co-morbities which vary in different studies. In summary, it is not proven but it is feasible that there are multiple causes of sinus node dysfunction in the old animal. This computer modeling study shows that it is at least feasible that the ion channel remodeling in Aging Study-1 and Aging Study-2 causes sinus node dysfunction. There is now a need for further experimental studies of age-dependent sinus node dysfunction taking into account species, strain, gender, age, frailty and co-morbidities – any or all of these may affect sinus node function.
The rat SAN model used in our simulations had certain inherent limitations, as described previously (Tao et al., 2011). Our simulation incorporated age-induced changes in ion channels and intracellular Ca2+ handling from two independent studies into the rat SAN model (Jones et al., 2004; Tellez et al., 2011; Hatch, 2012). The data were derived from mRNA gene expression or protein levels whenever experimental data on their kinetic or current densities in the SAN cell were unavailable. Mathematical modeling of the cardiac action potential ideally should be based on electrophysiological (usually patch clamp) recording of ionic currents obtained under physiological conditions. However, when such data are incomplete or absent, measurements of ion channel protein levels can be used as they may be used as a rough measure of ion channel density. Implicit assumptions of this approach are a direct relationship between expression levels of transcript levels or membrane proteins and the measured macroscopic channel conductance, and that relative change of expression levels may reflect changes in the macroscopic channel conductance. Whilst, such assumptions have limitations because a direct correlation between the gene expression or protein levels for certain ion channel subunits and their current densities cannot be guaranteed, this is a secondary approach in cases where direct current measurement data are missing. For example, with such an assumption, data on the expression of ion channels in the human SAN obtained by the quantitative polymerase chain reaction method, especially the ratio of the expression level between the SAN and human atrial cells have been successfully used to develop a computer model for the human SAN from the human atrial cell model (Yanni et al., 2010). Moreover, this approach has also been used effectively in additional modeling studies (Hao et al., 2011; Choudhury et al., 2018), and some computer modeling of action potentials (Chandler et al., 2009; Benoist et al., 2011; Abd Allah et al., 2012; Temple et al., 2016; D’Souza et al., 2020) have been done based on the RT-qPCR measurement of ion channel mRNA levels that often reflect protein levels (Tellez et al., 2006; D’Souza et al., 2014, 2015). In the present study, we followed the same approach as implemented by these prior studies (Yanni et al., 2010; Hao et al., 2011; Choudhury et al., 2018) and successfully reproduced the bradycardia effect in the model of Aging Study -1 which is comparable to experimental data, validating again the use of this approach.
Our results have shown that the aging-induced remodeling in ICaL plays an important contribution to the aging bradycardia. It is known that L-type Ca2+ channels are multi-subunit complexes formed by different isoforms, including Cav1.2 and Cav1.3, which have different kinetics that influence their contribution to SAN pacemaking (Zuccotti et al., 2011). It would therefore be informative to further analyze their relative contributions to aging-associated bradycardia. However, due to a lack of available voltage clamp data on the activation, inactivation and channel kinetics of the two distinct ICaL components from the rat SAN cells, it was possible in the model to consider only a general form of ICaL, without differentiating effects on the two specific isoforms. The relative roles of the two ICaL components in the aging bradycardia warrants further investigation in future, when suitable experimental data become available.
Finally, the results of this study were determined at the single-cell level. Consequently, other factors associated with aging, such as remodeling the electrical coupling between cells via the gap junction (Huang et al., 2015), or connexin, and the possible incidence of fibrosis (Baruscotti and Robinson, 2007; Semelka et al., 2013), might also play a specific role in production of the bradycardia effect that is more pronounced at the tissue level. Nevertheless, without considering age-induced changes in the connexins and fibrosis, such changes in various ionic channel conductances can nevertheless increase CL and reduce HR in a manner that is quantitatively comparable with experimental data.
In this study, the mechanism underlying the age-related SAN dysfunction in a commonly used model species was elucidated. Our results suggest that while it is an integral action of all remodeled ion channels and Ca2+ handling, the remodeled ICa,L, either via a gain or loss of function, contributes primarily to age-related bradycardia. Although further work is required to establish the extent to which these findings also apply to the human SAN, aging-related bradycardia appears able to be linked to different remodeling of ICa,L, which may ultimately have utility for clinical treatment strategies.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
HZ conceived the study and contributed to data analysis and interpretation, supervision, and manuscript writing. AA conducted experiments, collected the data, and contributed to data analysis and interpretation and manuscript writing. JH contributed to data analysis and interpretation and manuscript writing. MB contributed to data analysis and interpretation and manuscript writing. All authors contributed to the article and approved the submitted version.
This work was funded by King Abdul-Aziz University KAU, Jeddah, Saudi Arabia; University of Jeddah, Jeddah, Kingdom Saudi Arabia, and supported by a scholarship from the Saudi Cultural Bureau, London, United Kingdom, and by grants from EPSRC (United Kingdom) (EP/J00958X/1 and EP/I029826/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.546508/full#supplementary-material
Abd Allah, E. S. H., Tellez, J. O., Aslanidi, O. V., Zhang, H., Dobrzynski, H., and Boyett, M. R. (2012). Postnatal development of transmural gradients in expression of ion channels and Ca2+-handling proteins in the ventricle. J. Mol. Cell. Cardiol. 53, 145–155. doi: 10.1016/j.yjmcc.2012.04.004
Accili, E. A., Proenza, C., Baruscotti, M., and DiFrancesco, D. (2002). From Funny Current to HCN channels: 20 years of excitation. Physiology 17, 32–37. doi: 10.1152/physiologyonline.2002.17.1.32
Alghamdi, A. M., Testrow, C. P., Whittaker, D. G., and Zhang, H. (2018). The mechanism underlying heart rate and pacemaking activity decline in developing sinoatrial node of the rabbit heart. Comp. Cardiol. 45, 1–4.
Alings, A. M. W., and Bouman, L. N. (1993). Electrophysiology of the aging rabbit and cat sinoatrial node a comparative study. Eur. Heart J. 14, 1278–1288. doi: 10.1093/eurheartj/14.9.1278
Bai, X., Wang, K., Yuan, Y., Li, Q., Dobrzynski, H., and Boyett, M. R. (2017). Mechanism underlying impaired cardiac pacemaking rhythm during ischemia: a simulation study. Chaos 27:093934. doi: 10.1063/1.5002664
Baruscotti, M., and Robinson, R. B. (2007). Electrophysiology and pacemaker function of the developing sinoatrial node. Am. J. Physiol. Hear. Circ. Physiol. 293, 2613–2623.
Benoist, D., Stones, R., Drinkhill, M., Bernus, O., and White, E. (2011). Arrhythmogenic substrate in hearts of rats with monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 300, H2230–H2237.
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force [Internet]. cited 4 Jul 2018]. (Developments in Cardiovascular Medicine; vol. 237). Dordrecht: Springer.
Brioschi, C., Micheloni, S., Tellez, J. O., Pisoni, G., Longhi, R., Moroni, P., et al. (2009). Distribution of the pacemaker HCN4 channel mRNA and protein in the rabbit sinoatrial node. J. Mol. Cell Cardiol. 47, 221–227. doi: 10.1016/j.yjmcc.2009.04.009
Butters, T. D., Aslanidi, O. V., Inada, S., Boyett, M. R., Hancox, J. C., Lei, M., et al. (2010). Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circ. Res. 107, 126–137. doi: 10.1161/circresaha.110.219949
Chandler, N. J., Greener, I. D., Tellez, J. O., Inada, S., Musa, H., Molenaar, P., et al. (2009). Molecular architecture of the human sinus node - insights into the function of the cardiac pacemaker. Circulation 119, 1562–1575. doi: 10.1161/circulationaha.108.804369
Choudhury, M., Black, N., Alghamdi, A., Souza, A. D., Wang, R., Yanni, J., et al. (2018). TBX18 overexpression enhances pacemaker function in a rat subsidiary atrial pacemaker model of sick sinus syndrome. J. Physiol. 596, 6141–6155. doi: 10.1113/jp276508
Choudhury, M., Boyett, M. R., and Morris, G. M. (2015). Biology of the sinus node and its disease. Arrhythmia Physiol. Rev. 4, 28–34.
Congxin Huang, M. D., Wenmao Ding, M. D., Lan Li, M. D., and Dongdong Zhao, M. C. (2006). Differences in the aging-associated trends of the monophasic action potential duration and effective refractory period of the right and left atria of the rat. Circ. J. 70, 352–357. doi: 10.1253/circj.70.352
DiFrancesco, D. (1993). Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472. doi: 10.1146/annurev.ph.55.030193.002323
Dobrev, D. (2009). Ion channel portrait of the human sinus node: useful for a better understanding of sinus node function and dysfunction in humans? Circulation 119, 1556–1558. doi: 10.1161/circulationaha.108.836866
D’Souza, A., Bucchi, A., Johnsen, A. B., Logantha, S. J., Monfredi, O., Yanni, J., et al. (2014). Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat. Commun. 5:3775.
D’Souza, A., Wang, Y., Johnsen, A. B., Olieslagers, S., Ni, H., Cox, C., et al. (2020). (Full Paper Under Revision). Circadian Control of Heart Rate Occus via an Intrinsic Sinus Node Clock and the Pacemaker Channel. Heart Rhythm, submitted.
D’Souza, A., Wegner, S., Johnsen, A. B., Gill, E., Cox, C., Dobrzynski, H., et al. (2015). Circadian control of heart rate. Circ. Res. 117, A430–A430.
Du, J., Deng, S., Pu, D., Liu, Y., Xiao, J., and She, Q. (2017). Age-dependent down-regulation of hyperpolarization-activated cyclic nucleotide-gated channel 4 causes deterioration of canine sinoatrial node function. Acta Biochim. Biophys. Sin. 49, 400–408. doi: 10.1093/abbs/gmx026
Fenske, S., Krause, S. C., Hassan, S. I. H., Becirovic, E., Auer, F., Bernard, R., et al. (2013). Sick sinus syndrome in HCN1-Deficient mice. Circulation 128, 2585–2594. doi: 10.1161/circulationaha.113.003712
Hagiwara, N., Irisawa, H., and Kameyama, M. (1988). Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 395, 233–253. doi: 10.1113/jphysiol.1988.sp016916
Hao, X., Zhang, Y., Zhang, X., Nirmalan, M., Davies, L., Konstantinou, D., et al. (2011). TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythmia Electrophysiol. 4, 397–406. doi: 10.1161/circep.110.960807
Haqqani, H. M., and Kalman, J. M. (2007). Aging and sinoatrial node dysfunction: musings on the not-so-funny side. Circulation 115, 1178–1179. doi: 10.1161/circulationaha.106.685248
Härtel, G., and Talvensaari, T. (1975). Treatment of sinoatrial syndrome with permanent cardiac pacing in 90 patients. Acta Med. Scand. 198, 341–347. doi: 10.1111/j.0954-6820.1975.tb19555.x
Hatch, F. S. (2012). Age-associated Changes to Calcium Handling Proteins Across the Whole Heart. Ph.D. thesis, University of Hull, Hull.
Huang, X., Du, Y., Yang, P., Lin, S., Xi, Y., Yang, Z., et al. (2015). Age-dependent alterations of voltage-gated Na + channel isoforms in rat sinoatrial node. Mech. Aging Dev. 152, 80–90. doi: 10.1016/j.mad.2015.10.003
Huang, X., Yang, P., Yang, Z., Zhang, H., and Ma, A. (2016). Age-associated expression of HCN channel isoforms in rat sinoatrial node. Exp. Biol. Med. 241, 331–339. doi: 10.1177/1535370215603515
Jones, S. A., Boyett, M. R., and Lancaster, M. K. (2007). Declining into failure: the age- dependent loss of the L-type calcium channel within the sinoatrial node. Circulation 115, 1183–1190. doi: 10.1161/circulationaha.106.663070
Jones, S. A., Lancaster, M. K., and Boyett, M. R. (2004). Aging-related changes of connexins and conduction within the sinoatrial node. J. Physiol. 560, 429–437. doi: 10.1113/jphysiol.2004.072108
Katz, B., and Miledi, R. (1968). The role of calcium in neuromuscular facilitation. J. Physiol. 195, 481–492. doi: 10.1113/jphysiol.1968.sp008469
Kurata, Y., Hisatome, I., Imanishi, S., and Shibamoto, T. (2002). Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Physiol. Hear. Circ. Physiol. 283, H2074–H2101.
Lakatta, E. G., and DiFrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J. Mol. Cell Cardiol. 47, 157–170. doi: 10.1016/j.yjmcc.2009.03.022
Lamas, G. A., Lee, K., Sweeney, M., Leon, A., Yee, R., Ellenbogen, K., et al. (2000). The Mode Selection Trial (MOST) in sinus node dysfunction: design, rationale, and baseline characteristics of the first 1000 patients. Am. Heart J. 140, 541–551. doi: 10.1067/mhj.2000.109652
Larson, E. D., St Clair, J. R., Sumner, W. A., Bannister, R. A., and Proenza, C. (2013). Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. Proc. Natl. Acad. Sci. U.S.A. 10, 18011–18016. doi: 10.1073/pnas.1308477110
Lei, M., Cooper, P. J., Camelliti, P., and Kohl, P. (2002). Role of the 293b-sensitive, slowly activating delayed rectifier potassium current, IKs, in pacemaker activity of rabbit isolated sino-atrial node cells. Cardiovasc. Res. 53, 68–79. doi: 10.1016/s0008-6363(01)00459-x
Li, P., Lines, G. T., Maleckar, M. M., and Tveito, A. (2013). Mathematical models of cardiac pacemaking function. Front. Phys. 1:20. doi: 10.3389/fphy.2013.00020
Linscheid, N., Logantha, S., Poulsen, P. C., Zhang, S., Schrolkamp, M., Egerod, K. L., et al. (2019). Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat. Commun. 10:2889.
Maltsev, V. A., and Lakatta, E. G. (2009). Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am. J. Physiol. Circ. Physiol. 296, H594–H615.
Mitsuiye, T., Shinagawa, Y., and Noma, A. (2000). Sustained inward current during pacemaker depolarization in mammalian sinoatrial node cells. Am. Hear. Assoc. Inc. Circ. 87, 88–91. doi: 10.1161/01.res.87.2.88
Moghtadaei, M., Jansen, H. J., Mackasey, M., Rafferty, S. A., Bogachev, O., Sapp, J. L., et al. (2016). The impacts of age and frailty on heart rate and sinoatrial node function. J. Physiol. 594, 7105–7126. doi: 10.1113/jp272979
Opthof, T. (1994). Gap junctions in the sinoatrial node: immunohistochemical localization and correlation with activation pattern. J. Cardio Electrophys. 5, 138–143. doi: 10.1111/j.1540-8167.1994.tb01153.x
Satoh, H. (2003a). Sino-atrial nodal cells of mammalian hearts: ionic currents and gene expression of pacemaker ionic channels. J Smooth Muscle Res. 39, 175–193.
Satoh, H. (2003b). Elecropharmacology of taurine on the hyperpolarization activated inward current and the sustained inward current in spontaneously beating rat sino-atrial nodal cells. J. Pharmacol. Sci. 91, 229–238. doi: 10.1254/jphs.91.229
Satoh, H., Tohno, S., Azuma, C., Minami, T., Ohishi, T., Hayashi, M., et al. (2005). Age-related attenuation in the elements in monkey sino-atrial node. Biol. Trace Elem. Res. 107, 43–52.
Semelka, M., Gera, J., and Usman, S. (2013). Sick sinus syndrome: a review. Am. Fam. Phys. 87, 691–696.
Sharpe, E. J., Larson, E. D., and Proenza, C. (2017). Cyclic AMP reverses the effects of aging on pacemaker activity and If in sinoatrial node myocytes. J. Gen. Physiol. 149, 237–247. doi: 10.1085/jgp.201611674
Shinagawa, Y., Satoh, H., and Noma, A. (2000). The sustained inward current and inward rectifier K+ current in pacemaker cells dissociated from rat sinoatrial node. J. Physiol. 523(Pt 3), 593–605. doi: 10.1111/j.1469-7793.2000.t01-2-00593.x
Sodeck, G. H., Domanovits, H., Meron, G., Rauscha, F., Losert, H., and Thalmann, M. (2007). Compromising bradycardia: management in the emergency department. Resuscitation 73, 96–102. doi: 10.1016/j.resuscitation.2006.08.006
Szentesi, P., Pignier, C., Egger, M., Kranias, E. G., and Niggli, E. (2004). Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+ -induced Ca2+ release refractoriness in heart muscle. Circ. Res. 95, 807–813.
Talano, J. V., Euler, D., Randall, W. C., Eshaghy, B., Loeb, H. S., and Gunnar, R. M. (1978). Sinus node dysfunction. Am. J. Med. 64, 773–781.
Tao, T., Paterson, D. J., and Smith, N. P. (2011). A model of cellular cardiac-neural coupling that captures the sympathetic control of sinoatrial node excitability in normotensive and hypertensive rats. Biophys. J. 101, 594–602. doi: 10.1016/j.bpj.2011.05.069
Tellez, J. O., Dobrzynski, H., Greener, I. D., Graham, G. M., Laing, E., Honjo, H., et al. (2006). Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ. Res. 99, 1384–1393. doi: 10.1161/01.res.0000251717.98379.69
Tellez, J. O., Maczewski, M., Yanni, J., Sutyagin, P., Mackiewicz, U., Atkinson, A., et al. (2011). Aging-dependent remodelling of ion channel and Ca2+ clock genes underlying sino-atrial node pacemaking. Exp. Physiol. 96, 1163–1178. doi: 10.1113/expphysiol.2011.057752
Temple, I. T., Logantha, S. J. R. J., Absi, M., Zhang, Y., Pervolaraki, E., Yanni, J., et al. (2016). Atrioventricular node dysfunction and ion channel transcriptome in pulmonary hypertension. Circ. Arrhythmia Electrophysiol. 9:e003432.
Tohno, Y., Tohno, S., Viwatpinyo, K., Minami, T., Chaisuksunt, V., Mahakkanukrauh, P., et al. (2014). Age-related changes of elements in the human sinoatrial nodes. OA Anat. 2:21.
Vinogradova, T. M., Zhou, Y. Y., Bogdanov, K. Y., Yang, D., Kuschel, M., Cheng, H., et al. (2000). Sinoatrial node pacemaker activity requires Ca(2+)/calmodulin-dependent protein kinase II activation. Circ. Res. 87, 760–767. doi: 10.1161/01.res.87.9.760
Webb, K., Absi, M., Logantha, S. J., Zaborska, K., Gurney, A., Heagerty, A., et al. (2017). Obesity increases the propensity for atrial arrhythmias. Eur. Heart J. 37:86343.
Webb, K., Logantha, S. J., Absi, M., Cartwright, E., Zhang, H., Monfredi, O., et al. (2018). Obesity causes cardiac ion channel remodelling and increases the propensity for atrial arrhythmias. Eur. Heart J. 39:5702. doi: 10.1093/eurheartj/ehy566.P5702
Wood, M. A., and Ellenbogen, K. A. (2002). Cardiology patient pages, cardiac pacemakers from the patient’s perspective. Circulation 105, 2136–2138.
Yanni, J., Tellez, J. O., Sutyagin, P. V., Boyett, M. R., and Dobrzynski, H. (2010). Structural remodelling of the sinoatrial node in obese old rats. J. Mol. Cell. Cardiol. 48, 653–662. doi: 10.1016/j.yjmcc.2009.08.023
Zhang, H., Holden, A. V., Kodama, I., Honjo, H., Lei, M., Varghese, T., et al. (2000). Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am. J. Physiol. Hear. Circ. Physiol. 279, H397–H421.
Zhang, H., Zhao, Y., Lei, M., Dobrzynski, H., Liu, J. H., and Holden, A. V. (2007). Computational evaluation of the roles of Na + current, iNa, and cell death in cardiac pacemaking and driving. Am. J. Physiol. Circ. Physiol. 292, H165–H174.
Zhou, Z., and Lipsius, S. L. (1994). T-type calcium current in latent pacemaker cells isolated from cat right atrium. J. Mol. Cell Cardiol. 26, 1211–1219. doi: 10.1006/jmcc.1994.1139
Keywords: aging, SA node, ion channel remodeling, ICaL, sick sinus syndrome (brady-arrhythmia)
Citation: Alghamdi AM, Boyett MR, Hancox JC and Zhang H (2020) Cardiac Pacemaker Dysfunction Arising From Different Studies of Ion Channel Remodeling in the Aging Rat Heart. Front. Physiol. 11:546508. doi: 10.3389/fphys.2020.546508
Received: 28 March 2020; Accepted: 13 November 2020;
Published: 03 December 2020.
Edited by:
Joseph L. Greenstein, Johns Hopkins University, United StatesReviewed by:
Yukiko Himeno, Ritsumeikan University, JapanCopyright © 2020 Alghamdi, Boyett, Hancox and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henggui Zhang, aGVuZ2d1aS56aGFuZ0BtYW5jaGVzdGVyLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.