
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 03 September 2020
Sec. Vascular Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.01067
This article is part of the Research Topic Vascular Adjustments in Cardiovascular Disorders View all 17 articles
 Gabriela M. Silva1
Gabriela M. Silva1
 Maria S. França-Falcão2
Maria S. França-Falcão2
 Natália Tabosa M. Calzerra3
Natália Tabosa M. Calzerra3
 Mickael S. Luz2
Mickael S. Luz2
 Danilo Duarte A. Gadelha4
Danilo Duarte A. Gadelha4
 Camille M. Balarini4*
Camille M. Balarini4* Thyago M. Queiroz1
Thyago M. Queiroz1
Atherosclerosis is the leading cause of vascular disease worldwide and contributes significantly to deaths from cardiovascular complications. There is a remarkably close relationship between atherosclerotic plaque formation and the activation of renin-angiotensin system (RAS). However, depending on which RAS pathway is activated, pro‐ or anti-atherogenic outcomes may be observed. This brief review focuses on the role of three of the most important pieces of RAS axis, angiotensin II (Ang-II), angiotensin converting enzyme type 2 (ACE2), and angiotensin 1–7 (Ang-1–7) and their involvement in atherosclerosis. We focused on the effects of these molecules on vascular function and inflammation, which are important determinants of atherogenesis. Furthermore, we highlighted potential pharmacological approaches to treat this disorder.
Cardiovascular diseases remain the leading cause of adult death worldwide (Herrington et al., 2016). Nowadays, it is already established that hypertension is a modifiable risk factor for cardiovascular diseases and the reduction in blood pressure is accompanied by a reduction in cardiovascular risk (Herrington et al., 2016). On the other hand, the persistent burden of cardiovascular events despite a highly effective control of conventional risk factors, suggests that other mechanisms might underlie a proportion of these events (Libby et al., 2019).
Atherosclerosis can be considered the primary origin of most cardiovascular diseases (Husain et al., 2015). As previously reviewed by us and by others, atherosclerosis consists of an inflammatory response of arterial wall to injuries. This inflammation is often initiated by endothelial dysfunction and progresses to cellular adhesion molecules (CAM) expression, adhesion of circulating leukocytes to the endothelial cells (Koleva et al., 2016), leucocyte migration and the formation of a fibrous cap around a lipidic core, which compromises vascular lumen (Freitas-Lima et al., 2015). In addition to its traditional role in hypertension, the long-term blood pressure control system (the renin-angiotensin system – RAS) is directly involved in the development of atherosclerotic lesions due to its mainly effects on endothelial function, inflammation, fibrosis, coagulation balance, plaque stability, and structural remodeling (Montezano et al., 2014; Husain et al., 2015).
Along with the classic cascade in RAS, which involves the conversion of angiotensinogen to angiotensin I (Ang-I) by renin, followed by its cleavage to angiotensin II (Ang-II) by angiotensin converting enzyme (ACE), other peptides and enzymes related to RAS are important in atherogenesis (Figure 1; Montezano et al., 2014). In this context, we highlight the role of the angiotensin converting enzyme type 2 (ACE2), which is typically responsible to form angiotensin 1–7 (Ang-1–7) from Ang-II. The heptapeptide is described to oppose Ang-II effects by mediating vasodilation, growth-inhibition, anti-inflammatory responses, and anti-thrombotic effects (Montezano et al., 2014). Considering that, this review is devoted to summarize the effects of Ang-II, ACE2, and Ang-1–7 in atherosclerosis, highlighting the promising interventions that could lead to RAS modulation and atherosclerosis treatment.
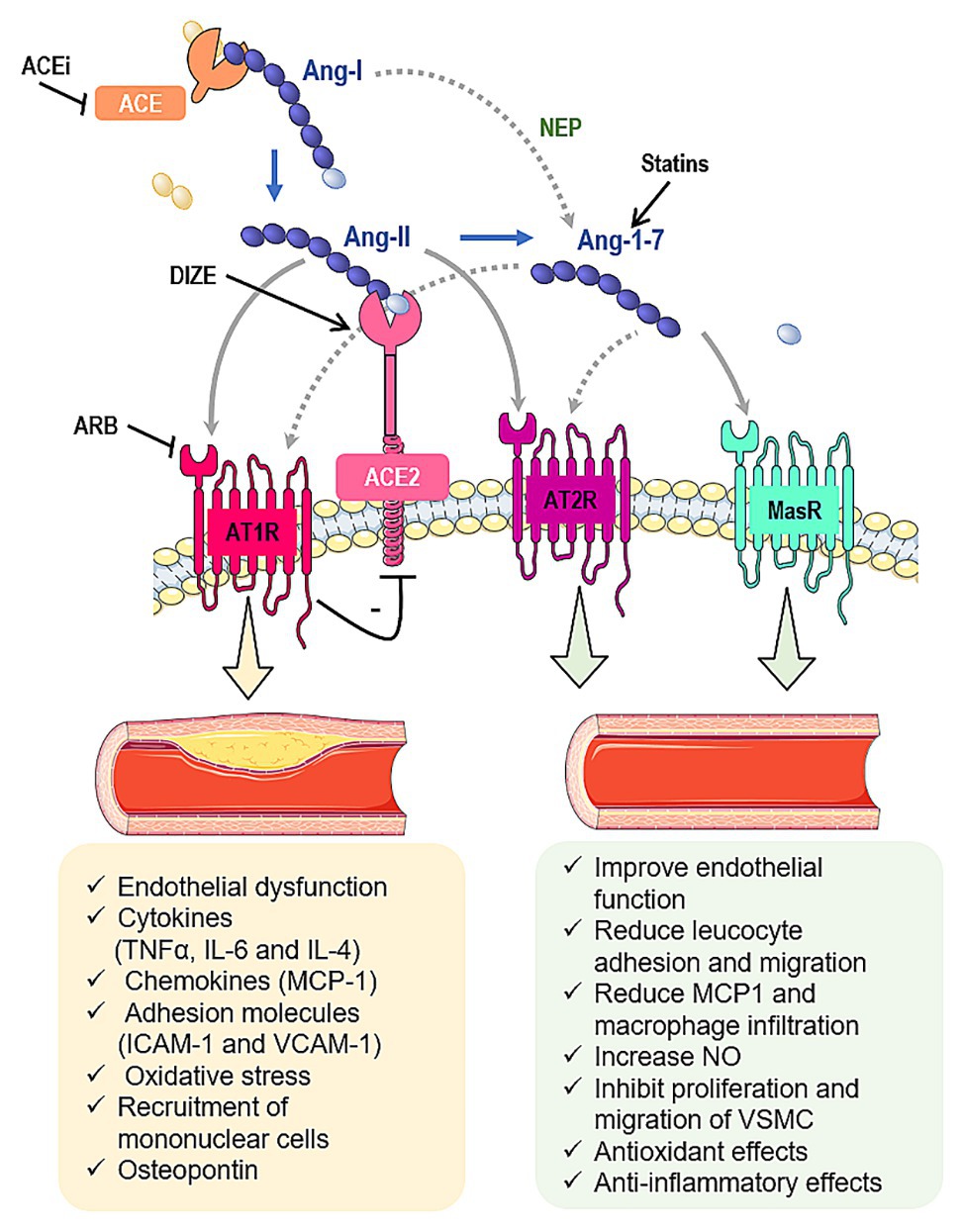
Figure 1. Interactions between the renin-angiotensin system (RAS) and atherosclerosis. Angiotensin II (Ang-II) is formed from the angiotensin I (Ang-I) cleavage by angiotensin converting enzyme (ACE). Ang-II can bind to Ang-II type 1 (AT1R) or type 2 (AT2R) receptors. Ang-II undergoes the action of angiotensin converting enzyme type 2 (ACE2) to be converted into angiotensin 1–7 (Ang-1-7), which classically interacts with Mas receptor (MasR). Furthermore, Ang-1–7 can bind to AT2R or it can induce the β-arrestin pathway through its interaction with AT1R. Ang-1–7 also can be produced by neprylisin (NEP) from Ang-I. In summary, the ACE/Ang-II/AT1R pathway induces atherosclerosis while the ACE2/Ang-1–7/MasR and Ang-II and Ang-1–7/AT2R pathways inhibit the atherosclerotic events. In addition, some pharmacological tools have been shown to interfere in some components of RAS cascade and prevent the atherosclerosis, such as statins, ACE inhibitors (ACEi), angiotensin receptor blockers (ARB) and diminazene aceturate (DIZE). Gray arrows indicate classic pathways while gray dotted arrows indicate alternative pathways. Black lines indicate potential pharmacological approaches to stimulate/increase (arrows) or block/decrease (lines) components of RAS.
Ang-II is the main effector of RAS (Colafella et al., 2019). The effects of Ang-II are mediated by its binding into the angiotensin type 1 and type 2 receptors (AT1R and AT2R, respectively). These receptors are G protein-coupled receptors that tend to present opposing activities (Kellici et al., 2015). AT1R is primarily responsible for the classic pro-hypertensive activity of Ang-II, whereas the AT2R is reported to present antagonistic effects to the AT1R (Figure 1; Ding et al., 2016).
It has been shown that Ang-II directly induces endothelial dysfunction and increases endothelial oxidative stress through the production of reactive oxygen species (ROS) such as superoxide anions (O2 −) derived from the complex enzyme nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase). This occurs predominantly through interaction with endothelial AT1R (Ziegler et al., 2020), which mediates increase in Ca2+ concentration in endothelial cells, promoting activation of calmodulin and interaction with the Nox5/Ca2+ calmodulin binding domain (Montezano et al., 2010; Piqueras and Sanz, 2020). Nox5 is a member of the NADPH oxidase family which is not found in rodents but is highly expressed in coronary arteries obtained from individuals with coronary artery disease (Guzik et al., 2008; Gray and Jandeleit-Dahm, 2015). In atherosclerosis, oxidative and inflammatory processes involve increased expression and activation of Nox5 in both vascular cells and resident macrophages (Touyz et al., 2019).
Activation of Nox5 mediated by Ang-II produces O2 −, activates RhoA and leads to the subsequent stimulation of Rho-associate kinase in human umbilical arterial endothelial cells culture (Escudero et al., 2015). The RhoA/ROCK pathway is an upstream regulator of mitogen-activated protein kinases (MAPKs – p38MAPK and ERK1/2), which promotes transactivation of several transcription factors, including NF-κB (Piqueras and Sanz, 2020). NF-κB regulates the expression of numerous genes, such as cytokines, tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), chemokines (monocyte chemoattractant protein – MCP-1), adhesion molecules (P-selectin, ICAM-1, and VCAM-1), the inflammatory enzyme cyclooxygenase type 2 (COX-2), and angiotensinogen (Durante et al., 2012; Liang et al., 2015). Moreover, activation of NF-κB seems to be an important signal transducer involved in the upregulation of oxidized low-density lipoprotein (ox-LDL)–mediated AT1R expression (Figure 2; Li et al., 2000).
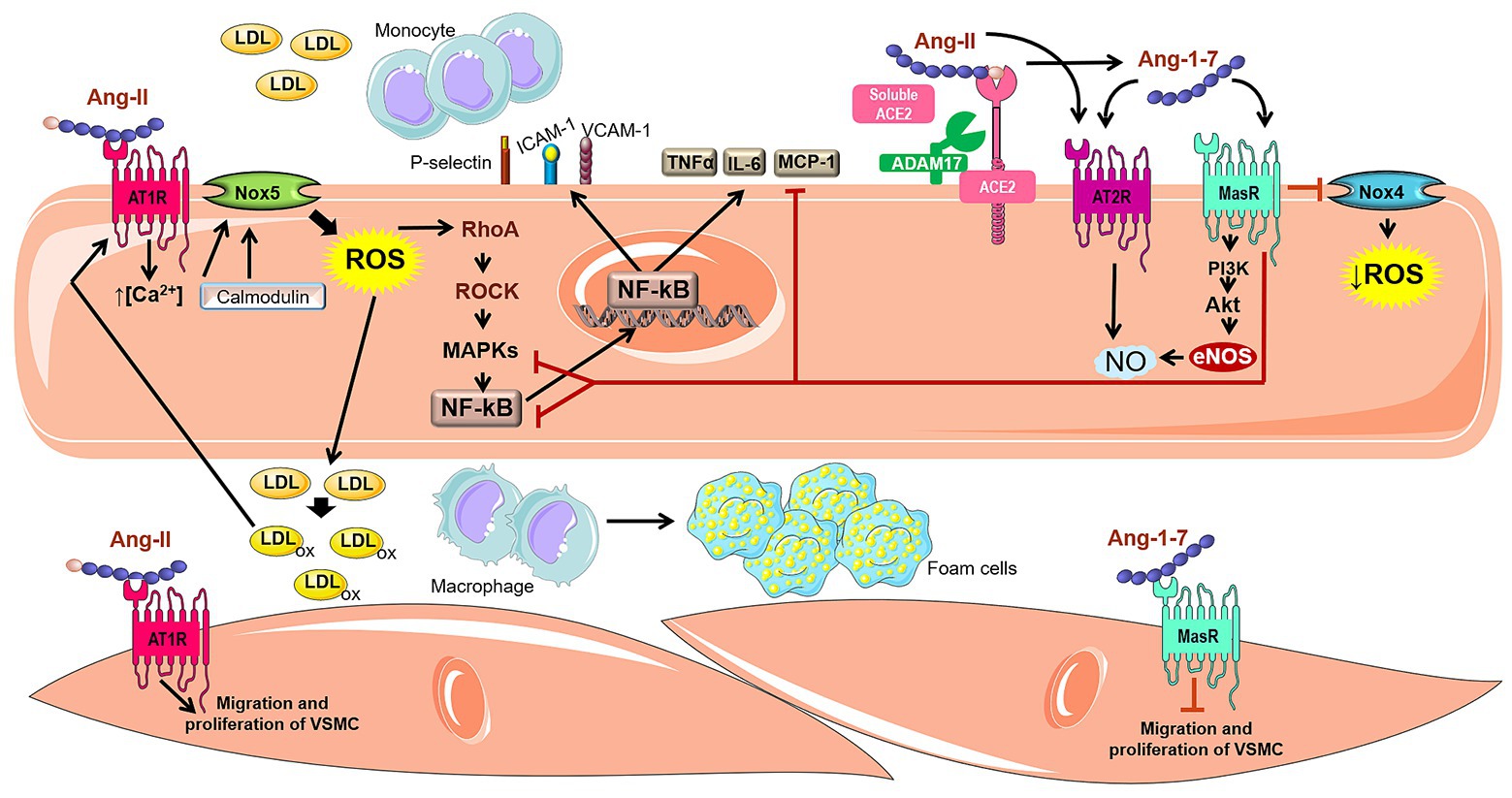
Figure 2. Involvement of Ang-II, ACE2, and Ang-1–7 in atherogenic pathways. The Ang-II binding into AT1R can activate Nox5 through a calcium/calmodulin-dependent pathway. The activated Nox5 induces the formation of ROS and stimulates the RhoA/ROCK pathway, which in turn, activates MAPKs and induces the transactivation of several transcription factors such as NF-κB. The expression of several genes is regulated by NF-κB, for instance cytokines (TNF-α and IL-6), chemokines (MCP-1), adhesion molecules (P-selectin, ICAM-1 and VCAM-1), which are involved in Ang-II-induced migration of mononuclear leukocytes. In addition, Ang-II is cleaved by ACE2 and produces Ang-1–7, an important RAS counter-regulator. Ang-1–7 shows the potential to negatively regulate atherogenic pathways, inducing anti-inflammatory effect, weakening monocyte migration and decrease of vascular lipids accumulation. These actions attributed to Ang-1–7 are related to the reduction of oxidative stress and the synthesis of inflammatory cytokines due to inhibition of the Nox4 and NF-κB-mediated pathways. Furthermore, Ang-1–7 stimulates the PI3K/Akt pathway, leading to phosphorylation of eNOS and NO formation, which improves the endothelial function. Ang-1–7 is also capable of promoting endothelial activation of AT2R, which also stimulates the NO cascade. In VSMC, Ang-1–7 inhibits muscle cell migration and proliferation, in contrast to Ang-II which possess proliferative and hypertrophic effects.
It is likely that TNF-α, released upon Ang-II stimulation of the AT1R, in combination with IL-4 acts as a paracrine molecule, inducing selective adhesion of mononuclear cells to the arterial endothelium through increased expression of CAM, and the release of varied chemokines involved in the recruitment of mononuclear cells (Piqueras and Sanz, 2020). Shu et al. (2019) demonstrated that Ang-II induces monocyte chemotactic protein-induced protein expression (MCPIP1) through an AMPK/p38 MAPK dependent pathway. The increase in MCPIP1 expression triggered apoptosis in macrophages, contributing to atherosclerotic plaque vulnerability.
In addition, Ang-II induces the expression of osteopontin, a multifunctional protein found in many cell types, including macrophages, endothelial cells, smooth muscle cells (SMCs), and epithelial cells. Osteopontin is found in atherosclerotic lesions, especially in association with macrophages and foam cells, suggesting that this protein plays an important role in the development and progression of atherosclerosis (Ding et al., 2016). The molecular mechanisms related to osteopontin involve recruitment of inflammatory cells and migration of foam cells through the binding to integrins (Giachelli and Steitz, 2000).
Ang-II also up-regulates the LOX-1 gene. LOX is a transmembrane glycoprotein that serves as a receptor for oxidized LDL (Lubrano and Balzan, 2016). In the endothelium, binding of oxLDL to LOX-1 causes increase in leukocyte adhesion molecules, activates apoptosis pathways, increases ROS and induces endothelial dysfunction. In a pro-inflammatory environment, LOX-1 is positively regulated in macrophages and is associated with more than 40% of oxLDL uptake, contributing to the formation of foam cells (Kattoor et al., 2019). In addition, oxLDL increases the generation of ACE, which in turn induces the Ang-II formation. This octapeptide increases the expression of LOX-1, which positively regulates the expression of AT1R, contributing to a self-perpetuating pro-atherogenic cycle. It has also been reported that ACE inhibitors and AT1R blockers (ARBs) decrease the expression of LOX-1 (Lubrano and Balzan, 2016).
According to experimental and clinical data, ACE inhibitors and ARBs appear to have beneficial anti-atherosclerotic effects (Tousoulis et al., 2015). Studies have shown that enalapril ameliorated oxidative vascular injury, suppressed NADPH oxidase activity, decreased inflammatory mediators and regulated the antioxidant defense system in apolipoprotein E-deficient mice (ApoE-KO; Suarez-Martinez et al., 2014; Husain et al., 2015), an animal model commonly used to study atherosclerosis.
It has been shown that the ARB olmesartan significantly reduced vascular inflammation in hypertensive patients, with a significant reduction in serum levels of many inflammatory markers, such as C-reactive protein, TNF-α, IL-6, and MCP-1 (Fliser et al., 2004; Durante et al., 2012). Moreover, long-term therapy with valsartan has been associated with atherosclerosis regression in individuals with thickening of the carotid wall. These effects were accompanied by concomitant improvements in oxidative stress markers, inflammation, and peripheral smooth muscle function (Ramadan et al., 2016).
The first evidence of a relationship between ACE2 and atherosclerosis was demonstrated by Zulli et al. (2006). They have shown the immunolocalization of ACE2 protein in macrophages and SMC actin-positive cells from rabbit atherosclerotic plaques. After this study, several experimental and clinical evidence have confirmed the involvement of ACE2 in atherosclerosis, suggesting its anti-atherogenic role (Dong et al., 2008; Lovren et al., 2008).
Dong et al. (2008) have found that ACE2 overexpression on aortic plaques attenuate the progression of early lesions in rabbits that underwent to endothelial injury and received atherogenic diet, probably by conversion of Ang-II to Ang-1–7. In this scenario, there was a reduction in local inflammation, lipid deposition, macrophage infiltration, and MCP-1 expression, in addition to an increase in collagen content, resulting in stabilized plaques. Similar results were found in rabbits fed with a high-cholesterol diet. The anti-atherosclerotic effects of ACE2 were associated with inhibition of proliferation and migration of vascular SMC and improvement of endothelial function. Additionally, ACE2 produced down-regulation of ERK1/2, p38 MAPK, JAK-STAT, and Ang-ll/ROS/NF-κB signaling pathways and upregulation of the PI3K-Akt pathway (Zhang et al., 2010).
Likewise, overexpression of ACE2 in ApoE-KO mice attenuated atherosclerotic lesion size and improved endothelial homeostasis, at least in part, through a mechanism that involves reduction of Ang-II-induced ROS generation (Lovren et al., 2008). In accordance to these data, Zhang et al. (2015a) also have shown that inhibition of inflammatory response, such as reduction of Ang-II-induced expression of adhesion molecules and cytokines prevent atherosclerotic plaque evolution in ApoE-KO animals overexpressing ACE2.
The protective role of ACE2 on atherosclerosis was also supported by the use of ACE2-deficient mice model (ACE2-KO). ACE2-deficiency in both LDL receptor-deficient mice (LDLR-KO) and ApoE-KO backgrounds resulted in larger atherosclerotic lesions when compared to their respective controls. Furthermore, the increased atherosclerotic vulnerability was associated to intraplaque inflammatory profile (Thomas et al., 2010; Thatcher et al., 2011; Sahara et al., 2014). On the other hand, the protective role of ACE2 on atherosclerosis in humans is not well-established yet.
In 2008, Sluimer and colleagues demonstrated the presence of ACE2 in humans. They detected ACE2 protein in human veins, healthy and atherosclerotic arteries, expressed in endothelial cells, SMCs, and macrophages. In addition, they found ACE2 messenger RNA (mRNA) and protein in early and advanced atherosclerotic lesion from humans. Despite total protein expression of ACE2 was similar during all stages of atherosclerosis, ACE2 activity was lower in advanced lesions, suggesting differential regulation of ACE2 in progression of atherosclerosis (Sluimer et al., 2008).
Anguiano et al. (2016) have found that baseline circulating ACE2 activity was enhanced in chronic kidney disease patients with atherosclerotic plaques when compared to patients with no plaque, suggesting that higher circulating ACE2 activity is associated with higher risk for silent atherosclerosis. Accordingly, Zhou et al. (2020) have shown an increase in circulating ACE2 protein levels in women with coronary heart disease (CHD) when compared to healthy group. This increase was associated with multi-vessel lesions, corroborating with the reports by Anguiano et al. (2016) and indicating the ACE2 as a compensatory mechanism in coronary atherosclerosis.
ACE2 is an integral cell membrane protein that can undergo cleavage or shedding and release its catalytically active ectodomain into surrounding milieu. The main promoter of ACE2 shedding is A Disintegrin and Metalloprotease 17 (ADAM17), which has been involved in atherosclerosis (Canault et al., 2006, 2007). This evidence and the results found by Zhou et al. (2020) allowed these authors to conclude that the increase in circulating ACE2 level is due to increasing tissue ACE2 synthesis from mRNA and augmented ACE2 protein shedding followed by its increase in circulation. All together these data show the increased circulating ACE2 protein levels or activity as biomarkers of atherosclerosis and encourage further studies in this direction.
Some therapeutic strategies for atherosclerosis targeting ACE2 have been thought, either with new drugs or drugs already used in the clinic. A recent study has demonstrated that overexpression of ACE by plasmid-mediated transfection in both primary monocytes and THP-1 cells leads to a marked decrease of ACE2 mRNA expression and induces a pro-atherogenic phenotype with elevated gene expression of the cellular adhesion molecules ICAM-1, VCAM-1, and macrophage colony-stimulating factor (MCSF). All these effects were partly reversed by captopril and losartan (Trojanowicz et al., 2017).
In that context, Zhang et al. (2015b) have shown that losartan inhibited the evolution of atherosclerotic plaques in high-cholesterol fed rabbits as well as increased the ACE2 protein expression in the plaques. In addition, Ang-II downregulated ACE2 protein expression and activity in SMC cell culture and losartan significantly blocked Ang-II-induced reduction of both ACE2 protein and activity. These data indicate that Ang-II generation by ACE can affect the expression and activity of ACE2 and ACE inhibitors or AT1R antagonists can upregulate ACE2 and favor its anti-atherogenic effects.
ACE2-activating drugs also seem promising, with emphasis on diminazene aceturate (DIZE) (Qaradakhi et al., 2020), which has several protective effects, such as improvement of metabolic profile and reduction of lipogenesis in mice (Macedo et al., 2015), anti-hypertensive effects in renovascular hypertensive rats (De Maria et al., 2016), and improvement of pulmonary endothelial function in Sprague Dawley rats (Shenoy et al., 2013). Thatcher et al. (2014) have found that DIZE decreases formation and severity of Ang-II-induced abdominal aortic aneurysms (AAA). Ang-II-induced AAA is characterized by progressive leukocyte accumulation, extracellular matrix degradation, luminal expansion, and thrombus (Saraff et al., 2003), being closely related to atherosclerosis. In addition, Fraga-Silva et al. (2015) have demonstrated that DIZE enhances the stability of atherosclerotic plaques in ApoE-KO mice and reduces the expression of ICAM-1 and VCAM-1. Although the mentioned studies have been performed on animal models, they suggest DIZE as a potential drug for the treatment of atherosclerosis and related cardiovascular diseases.
Ang-1–7 was investigated three decades ago as an important counter-regulator component of RAS, promoting hypotension and bradycardia after microinjection in dorsal motor nucleus of the vagus (Santos et al., 1988; Campagnole-Santos et al., 1989). The classical formation of Ang-1–7 occurs through ACE2 action on Ang-II. Alternatively, Ang-1–7 is formed by the cleavage of Ang-1–9 facilitated by ACE. Moreover, Ang-I can be directly converted into Ang-1–7 by action of neutral endopeptidase (neprylisin – NEP; Santos et al., 2003; Santos, 2014).
The formation of Ang-1–7 in vascular endothelium was first identified by Santos et al. (1992) using human aortic and human umbilical vein endothelial cells (HUVEC; Santos et al., 1992). Robust studies have shown that Ang-1–7 induces MasR activation, a G protein-coupled receptor which stimulates the PI3K/Akt pathway leading to phosphorylation of endothelial nitric oxide (NO) synthase and consequent NO production and releasing (Sampaio et al., 2007). Of note, Ang-1–7 is able to promote AT2R endothelial activation, which stimulates the bradykinin–NO cascade (Walters et al., 2005; Villela et al., 2015). NO is one of the most important factors released by endothelium. This gas is involved in vascular homeostasis and its decrease induces endothelial dysfunction (Cheng et al., 2009; Forstermann and Sessa, 2012), which is the key factor in atherogenesis (Qaradakhi et al., 2016).
Studies have demonstrated that Ang-1–7 stimulates endothelial cells function restoration by increasing NO bioavailability (Pignone et al., 2007; Sampaio et al., 2007). In addition, Ang-1–7 downregulates adhesion molecules such as VCAM-1 and ICAM-1 in endothelium by preventing both the phosphorylation of p38 MAPK and the expression of NF-κB (Anton et al., 2007; Zhang et al., 2013; Liang et al., 2015). Moreover, Ang-1–7 induces proliferation of endothelial progenitor cells in the injured vascular tissue triggered by atherogenesis (Wang et al., 2010; Zhang et al., 2015c).
During the vascular inflammation, many cytokines and inflammatory cells are required to begin and maintain atherosclerosis progression. In this context, Ang-1–7 has been described to induce anti-inflammatory phenotypes which contribute to restrain vascular lipid accumulation (Yang et al., 2013; Jiang et al., 2014). Yang et al. (2015a) found that Ang-1–7 treatment reduced the oxidative stress and macrophage infiltration due to decreasing in Nox4 (a subunit of NADPH oxidase complex) and NF-κB in aorta from ApoE-KO (Figure 2; Yang et al., 2015a). Another interesting study revealed that Ang-1–7 administration induced a remarkable decrease in the expression of pro-inflammatory cytokines such as IL-6, TNF-α, and MCP-1 in both aortic plaque and macrophages from ApoE-KO (Yang et al., 2013). Furthermore, in the same mouse model, pretreatment with AVE0991, a MasR agonist, reduced activated CD4+ T cells (Jawien et al., 2012a) and IL-12 (Jawien et al., 2012b). All these findings corroborate with an anti-inflammatory effect of Ang-1–7/MasR pathway in atherosclerosis.
In contrast to Ang-II-induced proliferative and hypertrophic effects, Ang-1–7 inhibits the migration and proliferation of vascular SMCs (Jiang et al., 2014; McKinney et al., 2014). This effect was described by Yang et al. (2013), showing that Ang-1–7 induces activation of MasR/ERK1,2/p38 and MasR/JAK/STAT pathways in vascular SMCs to mitigate the atherosclerotic plaque formation (Yang et al., 2013). Furthermore, Ang-1–7 has demonstrated a potential to negatively regulate the vascular fibrosis, as can be noticed by decreasing in matrix metalloproteases (MMP) MMP-2/MMP-9 in atherosclerotic plaques (Yang et al., 2013). Accordingly, Ang-1–7 treatment promoted a reduction in the neointimal layer growth by structural recovery of endothelium and showed atheroprotective properties attributed to its binding to both AT2R and MasR (Faria-Silva et al., 2005; Tesanovic et al., 2010). In addition, Ang-1–7 reduced atherosclerotic lesion formation by decrease in collagen accumulation through activation of AT2R (Dandapat et al., 2008). Conversely, it was described an increase in collagen content after Ang-1–7 administration, resulting in the increase of plaque stability (Yang et al., 2013). Similarly, the treatment with an Ang-1–7 antagonist, A779, induced a decline in plaque stability and reduction in collagen level (Yang et al., 2015b). Moreover, the heptapeptide can play a role as a β-arrestin-biased AT1R agonist without induce the Gq subunit activation, suggesting an additional antihypertrophic effect attributed to this peptide (Teixeira et al., 2017; Paz Ocaranza et al., 2020)
Interestingly, increase in plasmatic Ang-1–7 has been involved in regulation of lipid metabolism. It promoted a reduction in triglycerides and cholesterol levels, together with a decrease in adipose tissue mass as well as an improvement of glucose metabolism (Santos et al., 2010). The authors have suggested an involvement of adiponectin in the regulation of the glucose and lipid metabolism induced by Ang-1–7 (Santos et al., 2010). Curiously, the knocking out of MasR promoted opposing effects, once it augmented cholesterol and triglycerides levels and worsened the carbohydrate metabolism (Santos et al., 2008).
Some therapeutic strategies have been validated to positively modulate the RAS. The statins, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGcoA-reductase) inhibitors, have emerged due to its pleiotropic properties demonstrating additional effects apart from those of decreasing cholesterol levels (Zhang et al., 2015c). Treatment with statins such as atorvastatin and rosuvastatin have promoted an upregulation of ACE2/Ang-1–7 axis, reducing the proliferation of vascular SMCs and intimal thickening, respectively (Li et al., 2000; Suski et al., 2014), effects that are closely related to atherogenesis. The mechanisms by which the statins act to promote these effects are still unclear; however, studies have revealed that HMGcoA-reductase inhibitors decrease the activation of NF-κB induced by TNF-α and Ang-II, factors responsible to stimulate the migration and proliferation of vascular wall (Ortego et al., 1999; Friedrich et al., 2006; Tristano et al., 2007; Suski et al., 2014). Furthermore, authors have demonstrated that atorvastatin induced an increase in ACE2 protein expression in heart and kidney from high cholesterol-fed rabbits and augmented the occupancy of histone H3 acetylation (H3-Ac) mark on ACE2 promoter region in heart, demonstrating direct or indirect ACE2 epigenetic upregulation (Tikoo et al., 2015).
The role of statins on RAS components also have been observed in clinical trials as showed by Schindler et al. (2014) that identified, for the first time, an increase of Ang-1–7 level in hypercholesterolemic subjects after atorvastatin treatment. Altogether, those responses suggest an important role of statins on RAS components, including a decrease in Ang-II and, apparently, an upregulation in the ACE2/Ang-1–7 axis. This fact could be crucial to the atherosclerosis and cardiovascular diseases therapy.
In conclusion, here we briefly reviewed the role played by RAS components such as Ang-II, ACE2, and Ang-1–7 in atherosclerosis development. According to what is expected to components of RAS, Ang-II is considered to have pro-atherogenic effects while ACE2 and Ang-1–7 anti-atherogenic profiles. In addition to the direct pressure-related roles of these peptides, their effects on atherosclerosis involve modulation of endothelial function, oxidative stress, inflammation, cellular migration and proliferation, as well as plaque stability. Pharmacological strategies currently used to modulate the pressor effects of RAS components can offer beneficial outcomes in atherosclerosis. Moreover, we highlight the role played by statins, which have been identified to increase the RAS compensatory components (ACE2 and Ang-1–7), and induce an additional effect against the plaque formation. For this reason, the HMGcoA-reductase inhibitors should be considered when clinical decisions are made.
CB, TQ and MF-F conceived the manuscript and revised it critically. CB and NC prepared the figures. GS, NC, ML and DG drafted the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant number 424668/2016-5) to MF-F, (429767/2016-1) to CB, and (436605/2018-0) to TQ. This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES) – Finance Code 001.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Anguiano, L., Riera, M., Pascual, J., Valdivielso, J. M., Barrios, C., Betriu, A., et al. (2016). Circulating angiotensin converting enzyme 2 activity as a biomarker of silent atherosclerosis in patients with chronic kidney disease. Atherosclerosis 253, 135–143. doi: 10.1016/j.atherosclerosis.2016.08.032
Anton, L., Merrill, D. C., Neves, L. A. A., and Brosnihan, K. B. (2007). Angiotensin-(1–7) inhibits in vitro endothelial cell tube formation in human um- bilical vein endothelial cells through the AT(1–7) receptor. Endocrine 32, 212–218. doi: 10.1007/s12020-007-9022-1
Campagnole-Santos, M. J., Diz, D. I., Santos, R. A., Khosla, M. C., Brosnihan, K. B., and Ferrario, C. M. (1989). Cardiovascular effects of angiotensin-(1–7) injected into the dorsal medulla of rats. Am. J. Phys. 257, 324–329. doi: 10.1152/ajpheart.1989.257.1.H324
Canault, M., Leroyer, A. S., Peiretti, F., Lesèche, G., Tedgui, A., Bonardo, B., et al. (2007). Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am. J. Pathol. 171, 1713–1723. doi: 10.2353/ajpath.2007.070021
Canault, M., Peiretti, F., Kopp, F., Bonardo, B., Bonzi, M. F., Coudeyreet, J. C., et al. (2006). The TNF alpha convert-ing enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis 187, 182–191. doi: 10.1016/j.atherosclerosis.2005.08.031
Cheng, Z., Yang, X., and Wang, H. (2009). Hyperhomocysteinemia and endothelial dysfunction. Curr. Hypertens. Rev. 5, 158–165. doi: 10.2174/157340209788166940
Colafella, K. M. M., Bovée, D. M., and Danser, A. H. J. (2019). The renin angiotensin aldosterone system and its therapeutic targets. Exp. Eye Res. 186:107680. doi: 10.1016/j.exer.2019.05.020
Dandapat, A., Hu, C. P., Chen, J., Liu, Y., Khan, J. A., Remeo, F., et al. (2008). Over-expression of angiotensin II type 2 receptor (agtr2) decreases collagen accumulation in atherosclerotic plaque. Biochem. Biophys. Res. Commun. 366, 871–877. doi: 10.1016/j.bbrc.2007.11.061
De Maria, M. L. A., Araújo, L. D., Fraga-Silva, R. A., Pereira, L. A. S., Ribeiro, H. J., Menezes, G. B., et al. (2016). Anti-hypertensive effects of diminazene aceturate: an angiotensin-converting enzyme 2 activator in rats. Protein Pept. Lett. 23, 9–16. doi: 10.2174/0929866522666151013130550
Ding, Y., Chen, J., Cui, G., Wei, Y., Lu, C., Wang, L., et al. (2016). Pathophysiological role of osteopontin and angiotensin II in atherosclerosis. Biochem. Biophys. Res. Commun. 471, 5–9. doi: 10.1016/j.bbrc.2016.01.142
Dong, B., Zhang, C., Feng, J. B., Zhao, Y. X., Li, S. Y., Yang, Y. P., et al. (2008). Overexpression of ACE2 enhances plaque stability in a rabbit model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 1270–1276. doi: 10.1161/ATVBAHA.108.164715
Durante, A., Peretto, G., Laricchia, A., Ancona, F., Spartera, M., Mangieri, A., et al. (2012). Role of the renin-angiotensin-aldosterone system in the pathogenesis of atherosclerosis. Curr. Pharm. Des. 18, 981–1004. doi: 10.2174/138161212799436467
Escudero, P., Maranon, A. M., Collado, A., Gonzalez-Navarro, H., Hermenegildo, C., Peiro, C., et al. (2015). Combined sub-optimal doses of rosuvastatin and bexarotene impair angiotensin II-induced arterial mononuclear cell adhesion through inhibition of Nox5 signaling pathways and increased RXR/PPARα and RXR/PPARγ interactions. Antioxid. Redox Signal. 22, 901–920. doi: 10.1089/ars.2014.5969
Faria-Silva, R., Duarte, F. V., and Santos, R. A. (2005). Short-term angiotensin (1-7) receptor MAS stimulation improves endothelial function in normotensive rats. Hypertension 46, 948–952. doi: 10.1161/01.HYP.0000174594.17052.33
Fliser, D., Buchholz, K., and Haller, H. (2004). Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation 110, 1103–1107. doi: 10.1161/01.CIR.0000140265.21608.8E
Forstermann, U., and Sessa, W. C. (2012). Nitric oxide synthases: regulation and function. Eur. Heart J. 33, 829–837. doi: 10.1093/eurheartj/ehr304
Fraga-Silva, R. A., Montecucco, F., Costa-Fraga, F. P., Nencioni, A., Caffa, I., Bragina, M. E., et al. (2015). Diminazene enhances stability of atherosclerotic plaques in ApoE-deficient mice. Vasc. Pharmacol. 74, 103–113. doi: 10.1016/j.vph.2015.08.014
Freitas-Lima, L. C., Braga, V. A., de França-Silva, M., Cruz, J. C., Sousa-Santos, S. H., Monteiro, M. M. O., et al. (2015). Adipokines, diabetes and atherosclerosis: an inflammatory association. Front. Physiol. 6:304. doi: 10.3389/fphys.2015.00304
Friedrich, E. B., Clever, Y. P., Wassmann, S., Nikos, W., Michael, B., and Georg, N. (2006). Role of integrin-linked kinase in vascular smooth muscle cells: regulation by statins and angiotensin II. Biochem. Biophys. Res. Commun. 349, 883–889. doi: 10.1016/j.bbrc.2006.07.217
Giachelli, C. M., and Steitz, S. (2000). Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol. 19, 615–622. doi: 10.1016/S0945-053X(00)00108-6
Gray, S., and Jandeleit-Dahm, K. (2015). The role of NADPH oxidase in vascular disease–hypertension, atherosclerosis and stroke. Curr. Pharm. Des. 21, 5933–5944. doi: 10.2174/1381612821666151029112302
Guzik, T. J., Chen, W., Gongora, M. C., Guzik, B., Lob, H. E., Mangalat, D., et al. (2008). Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 52, 1803–1809. doi: 10.1016/j.jacc.2008.07.063
Herrington, W., Lacey, B., Sherliker, P., Armitage, J., and Lewington, S. (2016). Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 118, 535–546. doi: 10.1161/CIRCRESAHA.115.307611
Husain, K., Hernandez, W., Ansari, R. A., and Ferder, L. (2015). Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 6:209. doi: 10.4331/wjbc.v6.i3.209
Jawien, J., Toton-Zuranska, J., Gajda, M., Niepsuj, A., Gebska, A., Kus, K., et al. (2012a). Angiotensin-(1–7) receptor Mas agonist ameliorates progress of atherosclerosis in apoE-knockout mice. J. Physiol. Pharmacol. 63, 77–85.
Jawien, J., Toton-Zuranska, J., Kus, K., Pawlowska, M., Olszanecki, R., and Korbut, R. (2012b). The effect of AVE 0991, nebivolol and doxycycline on inflammatory mediators in an apoE-knockout mouse model of atherosclerosis. Med. Sci. Monit. 18, 389–393. doi: 10.12659/msm.883478
Jiang, F., Yang, J., Zhang, Y., Dong, M., Wang, S., Zhang, Q., et al. (2014). Angiotensin-converting enzyme 2 and angiotensin 1-7: novel therapeutic targets. Nat. Rev. Cardiol. 11, 413–426. doi: 10.1038/nrcardio.2014.59
Kattoor, A. J., Kanuri, S. H., and Mehta, J. L. (2019). Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 26, 1693–1700. doi: 10.2174/0929867325666180508100950
Kellici, T. F., Tzakos, A. G., and Mavromoustakos, T. (2015). Rational drug design and synthesis of molecules targeting the angiotensin II type 1 and type 2 receptors. Molecules 20, 3868–3897. doi: 10.3390/molecules20033868
Koleva, D. I., Orbetzova, M. M., Nikolova, J. G., and Deneva, T. I. (2016). Pathophysiological role of adiponectin, leptin and asymmetric dimethylarginine in the process of atherosclerosis. Folia Med. 58, 234–240. doi: 10.1515/folmed-2016-0039
Li, D., Saldeen, T., Romeo, F., and Mehta, J. L. (2000). Oxidized LDL upregulates angiotensin II type 1 receptor expression in cultured human coronary artery endothelial cells: the potential role of transcription factor NF-κB. Circulation 102, 1970–1976. doi: 10.1161/01.CIR.102.16.1970
Liang, B., Wang, X., Zhang, N., Yang, H., Bai, R., Liu, M., et al. (2015). Angiotensin-(1–7) attenuates angiotensin II-induced ICAM-1, VCAM-1, and MCP-1 expression via the MAS receptor through suppression of P38 and NF-kappaB pathways in HUVECs. Cell. Physiol. Biochem. 35, 2472–2482. doi: 10.1159/000374047
Libby, P., Pasterkamp, G., Crea, F., and Jang, I. K. (2019). Reassessing the mechanisms of acute coronary syndromes. Circ. Res. 124, 150–160. doi: 10.1161/CIRCRESAHA.118.311098
Lovren, F., Pan, Y., Quan, A., Teoh, H., Wang, G., Shukla, P. C., et al. (2008). Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 295, H1377–H1384. doi: 10.1152/ajpheart.00331.2008
Lubrano, V., and Balzan, S. (2016). Roles of LOX-1 in microvascular dysfunction. Microvasc. Res. 105, 132–140. doi: 10.1016/j.mvr.2016.02.006
Macedo, S. M., Guimarares, T. A., Andrade, J. M. O., Guimaraes, A. L. S., Paula, A. M. B. B., Ferreira, A. J., et al. (2015). Angiotensin converting enzyme 2 activator (DIZE) modulates metabolic profiles in mice, decreasing lipogenesis. Protein Pept. Lett. 22, 332–340. doi: 10.2174/0929866522666150209125401
McKinney, C. A., Fattah, C., Loughrey, C. M., Milligan, G., and Nicklin, S. A. (2014). Angiotensin-(1-7) and angiotensin-(1-9): function in cardiac and vascular remodelling. Clin. Sci. (Lond.) 126, 815–827. doi: 10.1042/CS20130436
Montezano, A. C., Burger, D., Paravicini, T. M., Chignalia, A. Z., Yusuf, H., Almasri, M., et al. (2010). Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ. Res. 106, 1363–1373. doi: 10.1161/CIRCRESAHA.109.216036
Montezano, A. C., Nguyen, D. C. A., Rios, F. J., and Touyz, R. M. (2014). Angiotensin II and vascular injury. Curr. Hypertens. Rep. 16:431. doi: 10.1007/s11906-014-0431-2
Ortego, M., Bustos, C., Hernandez-Presa, M. A., Tuñón, J., Díaz, C., Hernández, G., et al. (1999). Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis 147, 253–261. doi: 10.1016/S0021-9150(99)00193-8
Paz Ocaranza, M., Riquelme, J. A., García, L., Jalil, J. E., Chiong, M., and Santos, R. A. S. (2020). Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 17, 116–129. doi: 10.1038/s41569-019-0244-8
Pignone, A., Rosso, A. D., Brosnihan, K. B., Perfetto, F., Livi, R., and Fiori, G. (2007). Reduced circulating levels of angiotensin-(1–7) in systemic sclerosis: a new pathway in the dysregulation of endothelial-dependent vascular tone control. Ann. Rheum. Dis. 66, 1305–1310. doi: 10.1136/ard.2006.064493
Piqueras, L., and Sanz, M. J. (2020). Angiotensin II and leukocyte trafficking: new insights for an old vascular mediator. Role of redox-signaling pathways. Free Radic. Biol. Med. doi: 10.1016/j.freeradbiomed.2020.02.002 [Epub ahead of print]
Qaradakhi, T., Apostolopoulos, V., and Zulli, A. (2016). Angiotensin (1-7) and alamandine: similarities and differences. Pharmacol. Res. 111, 820–826. doi: 10.1016/j.phrs.2016.07.025
Qaradakhi, T., Gadanec, L. K., McSweeney, K. R., Tacey, A., Apostolopoulos, V., Levinger, I., et al. (2020). The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 47, 751–758. doi: 10.1111/1440-1681.13251
Ramadan, R., Dhawan, S. S., Binongo, J. N. G., Alkhoder, A., Jones, D. P., Oshinski, J. N., et al. (2016). Effect of angiotensin II type I receptor blockade with valsartan on carotid artery atherosclerosis: a double blind randomized clinical trial comparing valsartan and placebo (EFFERVESCENT). Am. Heart J. 174, 68–79. doi: 10.1016/j.ahj.2015.12.021
Sahara, M., Ikutomi, M., Morita, T., Minami, Y., Nakajima, T., Hirata, Y., et al. (2014). Deletion of angiotensin-converting enzyme 2 promotes the development of atherosclerosis and arterial neointima formation. Cardiovasc. Res. 101, 236–246. doi: 10.1093/cvr/cvt245
Sampaio, W. O., Souza dos Santos, R. A., Faria-Silva, R., da Mata Machado, L. T., Schiffrin, E. L., and Touyz, R. M. (2007). Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 49, 185–192. doi: 10.1161/01.HYP.0000251865.35728.2f
Santos, R. A. (2014). Angiotensin-(1-7). Hypertension 63, 1138–1147. doi: 10.1161/HYPERTENSIONAHA.113.01274
Santos, R. A., Brosnihan, K. B., Chappell, M. C., Pesquero, J., Chernicky, C. L., Greene, L. J., et al. (1988). Converting enzyme activity and angiotensin metabolism in the dog brain-stem. Hypertension 11, 153–157. doi: 10.1161/01.hyp.11.2_pt_2.i153
Santos, R. A., Brosnihan, K. B., Jacobsen, D. W., DiCorleto, P. E., and Ferrario, C. M. (1992). Production of angiotensin-(1-7) by human vascular endothelium. Hypertension 19, II56–II61. doi: 10.1161/01.hyp.19.2_suppl.ii56
Santos, R. A., Simões e Silva, A. C., Maric, C., Silva, D. M., Machado, R. P., de Buhr, I., et al. (2003). Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. U. S. A. 100, 8258–8263. doi: 10.1073/pnas.1432869100
Santos, S. H., Braga, J. F., Mario, E. G., Pôrto, L. C., Rodrigues-Machado, M. da. G., Murari, A., et al. (2010). Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1–7). Arterioscler. Thromb. Vasc. Biol. 30, 953–961. doi: 10.1161/ATVBAHA.109.200493
Santos, S. H., Fernandes, L. R., Mario, E. G., Ferreira, A. V., Pôrto, L. C., Alvarez-Leite, J. I., et al. (2008). Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes 57, 340–347. doi: 10.2337/db07-0953
Saraff, K., Babamusta, F., Cassis, L. A., and Daugherty, A. (2003). Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64
Schindler, C., Guenther, K., Hermann, C., Ferrario, C. M., Schroeder, C., Haufe, S., et al. (2014). Statin treatment in hypercholesterolemic men does not attenuate angiotensin II-induced venoconstriction. PLoS One 9:e103909. doi: 10.1371/journal.pone.0103909
Shenoy, V., Gjymishka, A., and Jarajapu, Y. P. (2013). Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am. J. Respir. Crit. Care Med. 187, 648–657. doi: 10.1164/rccm.201205-0880OC
Shu, S., Zhang, Y., Li, W., Wang, L., Wu, Y., Yuan, Z., et al. (2019). The role of monocyte chemotactic protein-induced protein 1 (MCPIP1) in angiotensin II-induced macrophage apoptosis and vulnerable plaque formation. Biochem. Biophys. Res. Commun. 515, 378–385. doi: 10.1016/j.bbrc.2019.05.145
Sluimer, J. C., Gasc, J. M., Hamming, I., van Goor, H., Michaud, A., van den Akker, L. H., et al. (2008). Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J. Pathol. 215, 273–279. doi: 10.1002/path.2357
Suarez-Martinez, E., Husain, K., and Ferder, L. (2014). Adiponectin expression and the cardioprotective role of the vitamin D receptor activator paricalcitol and the angiotensin converting enzyme inhibitor enalapril in ApoE-deficient mice. Ther. Adv. Cardiovasc. Dis. 8, 224–236. doi: 10.1177/1753944714542593
Suski, M., Gebska, A., Olszanecki, R., Stachowicz, A., Uracz, D., Madej, J., et al. (2014). Influence of atorvastatin on angiotensin I metabolism in resting and TNF-alpha-activated rat vascular smooth muscle cells. J. Renin-Angiotensin-Aldosterone Syst. 15, 378–383. doi: 10.1177/1470320313475907
Teixeira, L. B., Parreiras-E-Silva, L. T., Bruder-Nascimento, T., Duarte, D. A., Simões, S. C., Costa, R. M., et al. (2017). Ang-(1-7) is an endogenous beta-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 7:11903. doi: 10.1038/s41598-017-12074-3
Tesanovic, S., Vinh, A., Gaspari, T. A., Casley, D., and Widdop, R. E. (2010). Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 30, 1606–1613. doi: 10.1161/ATVBAHA.110.204453
Thatcher, S. E., Zhang, X., Howatt, D. A., Lu, H., Gurley, S. B., Daugherty, A., et al. (2011). Angiotensin converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor −/− mice. Arterioscler. Thromb. Vasc. Biol. 31, 758–765. doi: 10.1161/ATVBAHA.110.221614
Thatcher, S. E., Zhang, X., Howatt, D. A., Yiannikouris, F., Gurley, S. B., Ennis, T., et al. (2014). Angiotensin-converting enzyme 2 decreases formation and severity of angiotensin II-induced abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 34, 2617–2623. doi: 10.1161/ATVBAHA.114.304613
Thomas, M. C., Pickering, R. J., Tsorotes, D., Koitka, A., Sheehy, K., Bernardi, S., et al. (2010). Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 107, 888–897. doi: 10.1161/CIRCRESAHA.110.219279
Tikoo, K., Patel, G., Kumar, S., Karpe, P. A., Sanghavi, M., Malek, V., et al. (2015). Tissue specific up regulation of ACE2 in rabbit model of atherosclerosis by atorvastatin: role of epigenetic histone modifications. Biochem. Pharmacol. 93, 343–351. doi: 10.1016/j.bcp.2014.11.013
Tousoulis, D., Psaltopoulou, T., Androulakis, E., Papageorgiou, N., Papaioannou, S., Oikonomou, E., et al. (2015). Oxidative stress and early atherosclerosis: novel antioxidant treatment. Cardiovasc. Drugs Ther. 29, 75–88. doi: 10.1007/s10557-014-6562-5
Touyz, R. M., Anagnostopoulou, A., Rios, F., Montezano, A. C., and Camargo, L. L. (2019). NOX5: molecular biology and pathophysiology. Exp. Physiol. 104, 605–616. doi: 10.1113/EP086204
Tristano, A. G., Castejon, A. M., Castro, A., and Cubeddu, L. X. (2007). Effects of statin treatment and withdrawal on angiotensin II-induced phosphorylation of p38 MAPK and ERK1/2 in cultured vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 353, 11–17. doi: 10.1016/j.bbrc.2006.11.044
Trojanowicz, B., Ulrich, C., Kohler, F., Bode, V., Seibert, E., Fiedler, R., et al. (2017). Monocytic angiotensin-converting enzyme 2 relates to atherosclerosis in patients with chronic kidney disease. Nephrol. Dial. Transplant. 32, 287–298. doi: 10.1093/ndt/gfw206
Villela, D., Leonhardt, J., Patel, N., Joseph, J., Kirsch, S., Hallberg, A., et al. (2015). Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin. Sci. (Lond.) 128, 227–234. doi: 10.1042/CS20130515
Walters, P. E., Gaspari, T. A., and Widdop, R. E. (2005). Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 45, 960–966. doi: 10.1161/01.HYP.0000160325.59323.b8
Wang, Y., Qian, C., Roks, A. J., Westermann, D., Schumacher, S. M., Escher, F., et al. (2010). Circulating rather than cardiac angiotensin-(1–7) stimulates cardioprotection after myocardial infarction. Circ. Heart Fail. 3, 286–293. doi: 10.1161/CIRCHEARTFAILURE.109.905968
Yang, J., Sun, Y., Dong, M., Yang, X., Meng, X., Niu, R., et al. (2015a). Comparison of angiotensin-(1-7), losartan and their combination on atherosclerotic plaque formation in apolipoprotein E knockout mice. Atherosclerosis 240, 544–549. doi: 10.1016/j.atherosclerosis.2015.02.055
Yang, J., Yang, X., Meng, X., Dong, M., Guo, T., Kong, J., et al. (2015b). Endogenous activated angiotensin-(1–7) plays a protective effect against atherosclerotic plaques unstability in high fat diet fed ApoE knockout mice. Int. J. Cardiol. 184, 645–652. doi: 10.1016/j.ijcard.2015.03.059
Yang, J. M., Dong, M., Meng, X., Zhao, Y. X., Yang, X. Y., Liu, X. L., et al. (2013). Angiotensin-(1-7) dose-dependently inhibits atherosclerotic lesion formation and enhances plaque stability by targeting vascular cells. Arterioscler. Thromb. Vasc. Biol. 33, 1978–1985. doi: 10.1161/ATVBAHA.113.301320
Zhang, C., Zhao, Y. X., Zhang, Y. H., Zhu, L., Deng, B. P., Zhou, Z. L., et al. (2010). Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc. Natl. Acad. Sci. U. S. A. 107, 15886–158891. doi: 10.1073/pnas.1001253107
Zhang, F., Liu, J., Li, S. F., Song, J. X., Ren, J. Y., and Chen, H. (2015c). Angiotensin-(1-7): new perspectives in atherosclerosis treatment. J. Geriatr. Cardiol. 12, 676–682. doi: 10.11909/j.issn.1671-5411.2015.06.014
Zhang, F., Ren, J., Chan, K., and Chen, H. (2013). Angiotensin-(1–7) regulates angiotensin II-induced VCAM-1 expression on vascular endothelial cells. Biochem. Biophys. Res. Commun. 430, 642–646. doi: 10.1016/j.bbrc.2012.11.098
Zhang, Y. H., Hao, Q. Q., Wang, X. Y., Chen, X., Wang, N., Zhu, L., et al. (2015b). ACE2 activity was increased in atherosclerotic plaque by losartan: possible relation to anti-atherosclerosis. J. Renin-Angiotensin-Aldosterone Syst. 16, 292–300. doi: 10.1177/1470320314542829
Zhang, Y. H., Zhang, Y. H., Dong, X. F., Hao, Q. Q., Zhou, X. M., Yu, Q. T., et al. (2015a). ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 64, 253–260. doi: 10.1007/s00011-015-0805-1
Zhou, X., Zhang, P., Liang, T., Chen, Y., Liu, D., and Yu, H. (2020). Relationship between circulating levels of angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis and coronary heart disease. Heart Vessel. 35, 153–161. doi: 10.1007/s00380-019-01478-y
Ziegler, T., Abdel Rahman, F., Jurisch, V., and Kupatt, C. (2020). Atherosclerosis and the capillary network; pathophysiology and potential therapeutic strategies. Cell 9:50. doi: 10.3390/cells9010050
Keywords: angiotensin converting enzyme type 2, angiotensin II, angiotensin 1–7, atherosclerosis, endothelial dysfunction, inflammation
Citation: Silva GM, França-Falcão MS, Calzerra NTM, Luz MS, Gadelha DDA, Balarini CM and Queiroz TM (2020) Role of Renin-Angiotensin System Components in Atherosclerosis: Focus on Ang-II, ACE2, and Ang-1–7. Front. Physiol. 11:1067. doi: 10.3389/fphys.2020.01067
Edited by:
Luciana Venturini Rossoni, University of São Paulo, BrazilReviewed by:
Daniella Bonaventura, Federal University of Minas Gerais, BrazilCopyright © 2020 Silva, França-Falcão, Calzerra, Luz, Gadelha, Balarini and Queiroz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camille M. Balarini, Y2FtaWxsZS5iYWxhcmluaUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.