
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 04 September 2020
Sec. Membrane Physiology and Membrane Biophysics
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.01040
This article is part of the Research TopicKv7 Channels: Structure, Physiology and PharmacologyView all 18 articles
 Francesco Miceli1
Francesco Miceli1 Lidia Carotenuto1Vincenzo Barrese1
Lidia Carotenuto1Vincenzo Barrese1 Maria Virginia Soldovieri2Erin L. Heinzen3,4Arthur M. Mandel5Natalie Lippa4Louise Bier4David B. Goldstein4
Maria Virginia Soldovieri2Erin L. Heinzen3,4Arthur M. Mandel5Natalie Lippa4Louise Bier4David B. Goldstein4 Edward C. Cooper6
Edward C. Cooper6 Maria Roberta Cilio7
Maria Roberta Cilio7 Maurizio Taglialatela1*
Maurizio Taglialatela1* Tristan T. Sands4,5*
Tristan T. Sands4,5*Pathogenic variants in KCNQ2 and KCNQ3, paralogous genes encoding Kv7.2 and Kv7.3 voltage-gated K+ channel subunits, are responsible for early−onset developmental/epileptic disorders characterized by heterogeneous clinical phenotypes ranging from benign familial neonatal epilepsy (BFNE) to early−onset developmental and epileptic encephalopathy (DEE). KCNQ2 variants account for the majority of pedigrees with BFNE and KCNQ3 variants are responsible for a much smaller subgroup, but the reasons for this imbalance remain unclear. Analysis of additional pedigrees is needed to further clarify the nature of this genetic heterogeneity and to improve prediction of pathogenicity for novel variants. We identified a BFNE family with two siblings and a parent affected. Exome sequencing on samples from both parents and siblings revealed a novel KCNQ3 variant (c.719T>G; p.M240R), segregating in the three affected individuals. The M240 residue is conserved among human Kv7.2-5 and lies between the two arginines (R5 and R6) closest to the intracellular side of the voltage-sensing S4 transmembrane segment. Whole cell patch-clamp recordings in Chinese hamster ovary (CHO) cells revealed that homomeric Kv7.3 M240R channels were not functional, whereas heteromeric channels incorporating Kv7.3 M240R mutant subunits with Kv7.2 and Kv7.3 displayed a depolarizing shift of about 10 mV in activation gating. Molecular modeling results suggested that the M240R substitution preferentially stabilized the resting state and possibly destabilized the activated state of the Kv7.3 subunits, a result consistent with functional data. Exposure to β-hydroxybutyrate (BHB), a ketone body generated during the ketogenic diet (KD), reversed channel dysfunction induced by the M240R variant. In conclusion, we describe the first missense loss-of-function (LoF) pathogenic variant within the S4 segment of Kv7.3 identified in patients with BFNE. Studied under conditions mimicking heterozygosity, the M240R variant mainly affects the voltage sensitivity, in contrast to previously analyzed BFNE Kv7.3 variants that reduce current density. Our pharmacological results provide a rationale for the use of KD in patients carrying LoF variants in Kv7.2 or Kv7.3 subunits.
Voltage-gated potassium (K+) channels (Kv channels) regulate the resting membrane potential and set the threshold and duration of the action potential in excitable cells. Among these, Kv7.2 and Kv7.3 voltage-gated K+ subunits, encoded by the KCNQ2 and KCNQ3 genes, are expressed in the central and peripheral nervous system (Brown and Adams, 1980; Wang et al., 1998). These subunits form homo- and heterotetrameric channels underlying the M-current (IKM), a non-inactivating K+ current with slow activation and deactivation kinetics that activates at the threshold potential of about −60/−50 mV, thus regulating the resting membrane potential and suppressing repetitive neuronal firing (Brown and Adams, 1980).
Pathogenic variants in KCNQ2 cause early-onset epilepsies with wide phenotypic heterogeneity (Allen et al., 2014; Miceli et al., 2018). Indeed, some variants have been identified in patients with benign familial neonatal epilepsy (BFNE), an autosomal-dominant epilepsy with seizures affecting otherwise healthy infants in the first days of life and spontaneously disappearing over the next several months, with mostly normal neurocognitive development (Jentsch, 2000; Singh et al., 2003; Soldovieri et al., 2006). At the severe end of the KCNQ2 spectrum is an early-onset developmental and epileptic encephalopathy (DEE) characterized by recurrent seizures starting in the neonatal period and neurodevelopmental disability (Weckhuysen et al., 2012). While more than 300 pathogenic variants have been described in KCNQ2, few variants in KCNQ3 have been detected, mostly in families with BFNE. In addition, de novo variants in KCNQ3 have been rarely described in children with DEE (Allen et al., 2013; Grozeva et al., 2015; Miceli et al., 2015; Ambrosino et al., 2018; Lauritano et al., 2019), intellectual disability (ID) apparently without epilepsy (Rauch et al., 2012; Deciphering Developmental Disorders, 2017), cortical visual impairment (Bosch et al., 2016), and in patients with ID and autism (Sands et al., 2019).
In most children affected with KCNQ2- or KCNQ3-related BFNE, seizures are controlled with conventional antiepileptic drugs, including sodium channel blockers (Sands et al., 2016). Instead, few options are available for patients with the most severe forms of KCNQ2- or KCNQ3-related disorders; in addition to sodium-channel blockers such as carbamazepine and phenytoin which appear to be highly effective (Pisano et al., 2015), ezogabine, a selective Kv7 channel activator, has been shown to improve seizure control and development in patients with Kv7.2 loss-of-function (LoF) variants (Millichap et al., 2016). Unfortunately, because of its unfavorable risk/benefit ratio, ezogabine has been withdrawn from the market. Among non-pharmacological therapies, ketogenic diet (KD) has been recently shown to be particularly effective in children with DEE caused by KCNQ2 variants (Ko et al., 2018), but the mechanisms of action are not completely understood and there are no data on the effects on KCNQ3 related disorders. KD is a low carbohydrate, high-fat, adequate-protein diet regimen that shifts the primary fuel source for neuronal activity from glucose to endogenous ketones: acetone, acetoacetate, and β-hydroxybutyrate (BHB). KD likely improves seizure control through a variety of mechanisms such as inhibition of the glycolysis, disruption of glutamatergic synaptic transmission, and activation of ATP-sensitive potassium channels (Lutas and Yellen, 2013). Recently, BHB has been shown to directly and specifically activate Kv7 channels containing Kv7.3 subunits by increasing current sensitivity to voltage (Manville et al., 2018, 2020).
In the present manuscript, we report the clinical, molecular and functional properties of a BFNE family carrying a novel KCNQ3 variant (c.719T>G; p.M240R) segregating with epilepsy in the three affected individuals. Whole cell patch-clamp recordings in Chinese hamster ovary (CHO) cells revealed that homomeric Kv7.3 M240R channels were not functional, whereas heteromeric channels incorporating Kv7.3 M240R mutant subunits with Kv7.2 and Kv7.3 displayed a depolarizing shift of about 10 mV in activation gating, consistent with a LoF in vitro effect. Consistent with these functional data, molecular modeling suggested that the M240R substitution preferentially stabilized the resting state and possibly destabilize the activated state of the Kv7.3 subunits. Finally, we demonstrate that BHB was able to reverse the functional alterations observed in heteromeric channels carrying Kv7.3 M240R subunits.
The BFNE family was referred for an epilepsy genetics research study by their treating clinician and written informed consent was obtained. Exome sequencing was performed in both parents and each sibling, and the presence of c.719T > G (p.M240R) in affected members was confirmed by Sanger sequencing in a clinical genetics laboratory. The study was approved by the human research ethics committee of Columbia University Irving Medical Center.
Mutations were engineered in KCNQ3 human cDNA cloned into pcDNA3.1 by QuikChange site-directed mutagenesis (Agilent Technologies Italia SpA, Milan, Italy), as previously described (Miceli et al., 2013). Channel subunits were expressed in CHO cells by transient transfection. CHO cells were grown in 100 mm plastic Petri dishes in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% foetal bovine serum (FBS), L-glutamine (0.1 mM), penicillin (50 U/ml), and streptomycin (50 μg/ml) in a humidified atmosphere at 37°C with 5% CO2. For electrophysiological experiments, cells were seeded on glass coverslips (Carolina Biological Supply Company, Burlington, NC, United States) and transfected on the next day with the appropriate cDNAs using Lipofectamine 2000 (Invitrogen, Milan, Italy) according to the manufacturer’s protocol. A plasmid encoding for enhanced green fluorescent protein (Clontech laboratories, Inc., Palo Alto, CA, United States) was used as transfection marker; total cDNA in the transfection mixture was kept constant at 4 μg.
Currents from CHO cells were recorded at room temperature (20–22°C) 1–2 days after transfection, using a commercially available amplifier (Axopatch 200A, Molecular Devices, Union City, CA, United States) and the whole-cell configuration of the patch-clamp technique, with glass micropipettes of 3–5 MΩ resistance. The extracellular solution contained (in millimolar): 138 NaCl, 2 CaCl2, 5.4 KCl, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. The pipette (intracellular) solution contained (in millimolar): 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, 5 Mg-ATP, pH 7.3–7.4 with KOH. The pCLAMP software (version 10.0.2) was used for data acquisition and analysis. Current densities (expressed in picoamperes per picofarad) were calculated as peak K+ currents divided by C. Data were acquired at 0.5–2 kHz and filtered at 1–5 kHz with the four-pole lowpass Bessel filter of the amplifier. No corrections were made for liquid junction potentials. To generate conductance-voltage curves, the cells were held at −80 mV, then depolarized for 1.5 s from −80 mV to +20/+80 mV in 10 mV increments, followed by an isopotential pulse at 0 mV of 300 ms duration; the current values recorded at the beginning of the 0 mV pulse were measured, normalized, and expressed as a function of the preceding voltages. Data were fit to a Boltzmann distribution of the following form y = max/[1 + exp(V1/2−V)/k], where V is the test potential, V1/2 the half-activation potential, and k the slope factor.
For current-activation kinetics analysis, the current traces recorded in response to incremental voltage steps were fitted to a double-exponential function and then a single time constant, representing the weighted average of the slow and fast components, was obtained by using the following equation: τ = (τf Af + τs As)/(Af + As), where Af and As indicate the amplitude of the fast and slow exponential components, τf and τs the time constants of each component (Miceli et al., 2013).
Three-dimensional models of Kv7.3 subunits in different gating states were generated by using as templates the coordinates of resting and activated states of the Kv1.2/2.1 chimera (Long et al., 2007; PDB accession number 2R9R; 26% of sequence identity with Kv7.3) subjected to long (>200 μs) molecular dynamic simulations (Jensen et al., 2012). Modeling of the S1-S4 VSD in each state was performed with SWISS-MODEL, as described (Sands et al., 2019). The models were optimized through all-atom energy minimization by using the GROMOS96 implementation of Swiss-PDBViewer, and analyzed using both the DeepView module of Swiss-PDBViewer (version 4.0.11) and PyMOL2.
Data are expressed as the mean ± SEM. Statistically significant differences between the data were evaluated with the Student’s t-test (p < 0.05).
A term male neonate was hospitalized for seizures starting in the first week of life. The seizures lasted less than 1 min, recurring every 2 h. Electroencephalography (EEG) recorded variable lateralization of ictal onset. Clinically, there was bilateral arm stiffening, rightward head version, and right leg stiffening, lasting 45 s. Workup for acute etiologies, including an MRI of the brain, was unrevealing. The child was treated with phenobarbital, but continued to have seizures after hospital discharge. He was cross-titrated onto levetiracetam from 3 weeks of age and seizures stopped around that time. Levetiracetam was weaned at 12 months. At 18 months he was diagnosed with isolated language delay (one standard deviation below expected) and started speech therapy. There was no concern for autism.
Nineteen months later, a female sibling was born. She began having episodes on the 5th day of life, characterized by limb stiffening and eye rolling. EEG confirmed seizures. Workup included a normal MRI of the brain. She was treated with phenobarbital and levetiracetam, which were weaned off at 4 and 6 months, respectively. She had recurrence with two back-to-back seizures at 22 months, and she was started on oxcarbazepine with seizure freedom. Follow-up EEGs have been normal. Developmental milestones have all been met on time as of 26 months.
Family history was notable for an isolated seizure in the father (case I-1) during infancy. The father’s interictal EEG was normal, the seizure did not recur, and he was not treated with anti-seizure medication. No seizure was ever described in the mother (case I-2; Figure 1A).
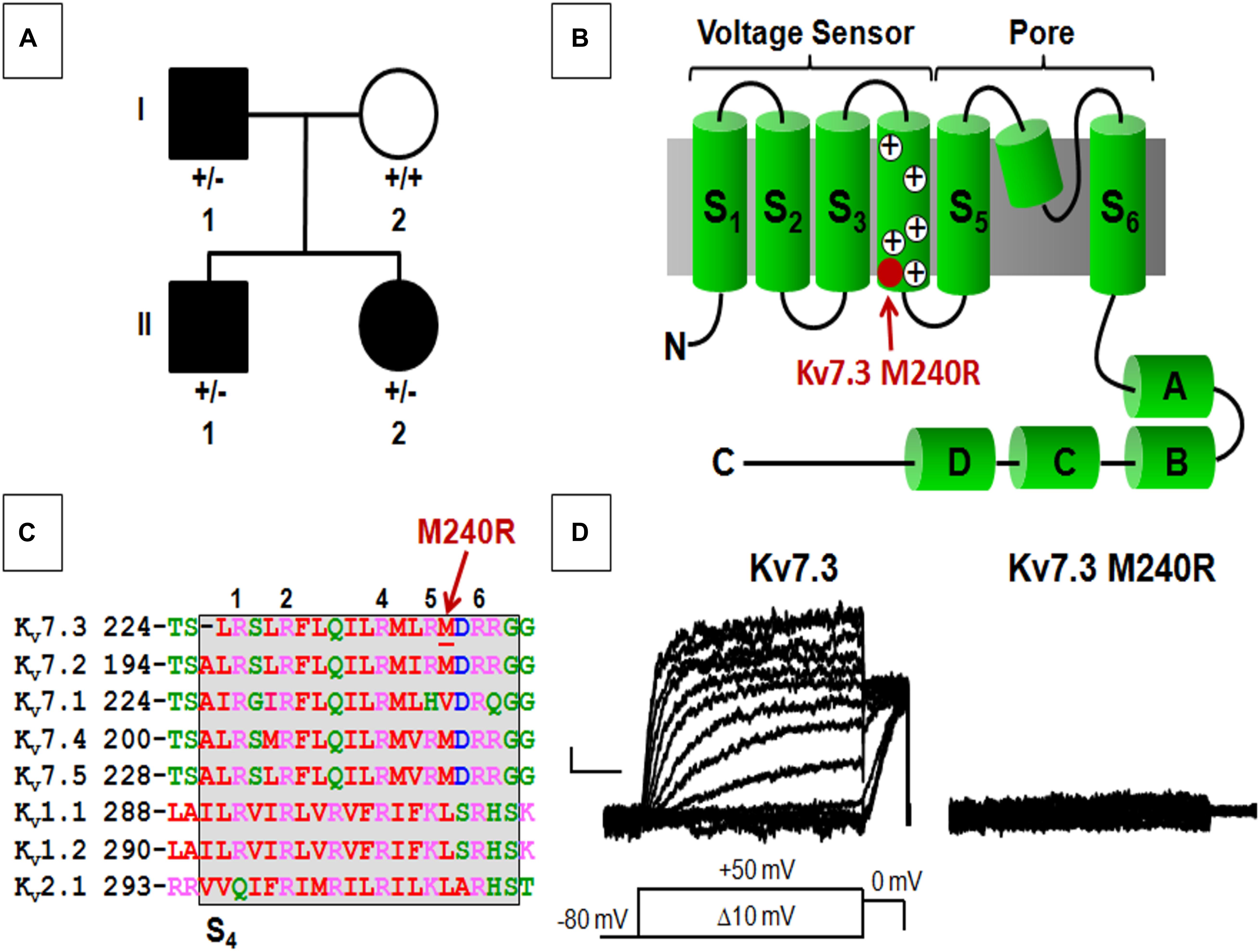
Figure 1. Pedigree, schematic representation of a single Kv7.3 subunit, and functional analysis of mutation at position 240. (A) Pedigree of the family. (B) Topological representation of a single Kv7 subunit. Arrows indicate the location of the mutation investigated (shaded in red). (A–D) Indicate the four putative alpha-helical domains identified in the Kv7 C-terminal region. (C) Sequence alignment of the S4 segments of the indicated Kv subunits (www.ebi.ac.uk/Tools/psa/). Residues are colored according to the following scheme: magenta represents basic; blue represents acid; red represents non-polar; green represents polar. 1, 2, 4, 5, and 6 refer to the positively charged arginines numbered according to their position along the S4 primary sequence. (D) Macroscopic currents from the indicated homomeric channels. Current scale, 50 pA; time scale, 0.2 s.
Exome sequencing, performed in all four family members, demonstrated a novel missense variant in KCNQ3, c.719T > G (p.M240R), in the two siblings and the affected father (Figure 1A), thus segregating with the phenotype. The variant is absent from gnomAD and predicted to be deleterious (polyphen-2, 0.989; SIFT, 0.9122; CADD score 27.3). The M240 residue lies between the R5 (R239) and R6 (R242) positions of the voltage-sensing S4 transmembrane segment within the voltage-sensing domain (VSD; Figure 1B). This non-polar residue is conserved among human Kv7.2-5 subunits, but not in Kv7.1 and other Kv channels such as Kv1.1, Kv1.2, and Kv2.1, although residues with similar physicochemical properties are present at this position (Figure 1C).
Heterologous expression of wild-type Kv7.3 subunits led to the appearance of voltage-dependent K+ −selective currents characterized by a rather slow time-course of activation and deactivation and a threshold for current activation of around −50 mV; the macroscopic K+ currents density at + 20 mV was 12.6 ± 1.1 pA/pF, similarly to previously reported values (Wang et al., 1998; Sands et al., 2019). By contrast, no measurable currents were recorded in cells expressing Kv7.3 M240R subunits, consistent with the variant causing a complete LoF effect (Figure 1D and Table 1).
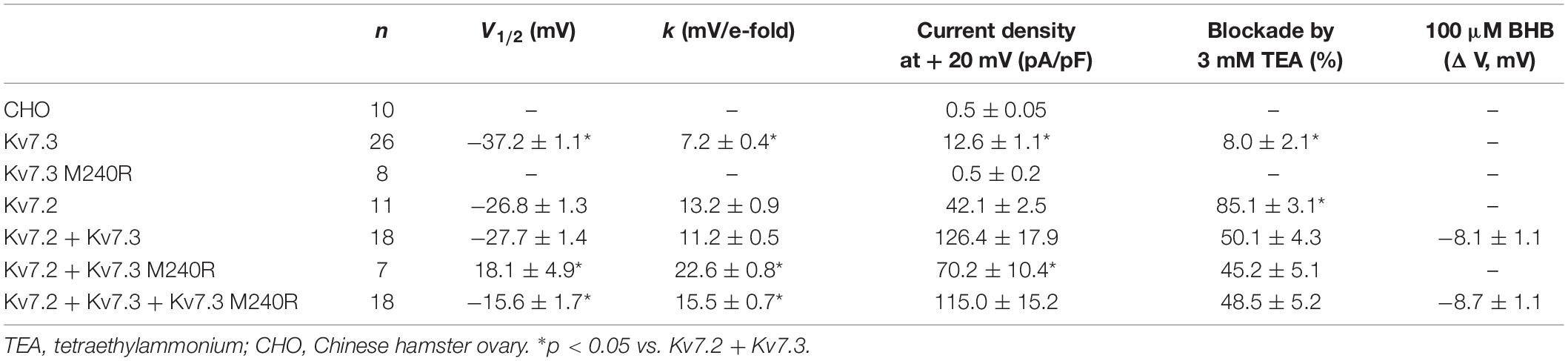
Table 1. Biophysical and pharmacological properties of mutant Kv7.3 channels.
Kv7.3 subunits assemble with Kv7.2 subunits to form IKM (Wang et al., 1998). Co-expression of Kv7.2 and Kv7.3 subunits generated currents which were considerably larger than those recorded upon expression as Kv7.2 or Kv7.3 subunits alone; in addition, currents from Kv7.2 + Kv7.3 heteromeric channels displayed an increased sensitivity to blockade by tetraethylammonium (TEA) when compared to homomeric Kv7.3 channels (Table 1). When compared to currents from wild-type Kv7.2 + Kv7.3 subunits, co-expression of Kv7.3 M240R with Kv7.2 subunits caused a marked rightward shift (by about + 40 mV) of the activation gating, a significant decrease of slope of the G/V curve (see section Materials and Methods for details), a reduced current density by about 50% at depolarized potential, with no change in sensitivity to TEA blockade (Figures 2A–C and Table 1). The marked rightward shift of the G/V curves of the heteromeric Kv7.2 + Kv7.3 M240R channels markedly reduced current density at the physiologically relevant potentials of −50/−40 mV (Figure 2C). The activation kinetics of Kv7.2 + Kv7.3-M240R currents were slower than those of Kv7.2 + Kv7.3, particularly at more depolarized potentials. In fact, τactivation at + 20 mV were 142 ± 19 ms and 274 ± 71 ms, for Kv7.2 + Kv7.3 and Kv7.2 + Kv7.3-M240R, respectively. In order to assess the influence of the depolarizing pulse length on the current steady-state properties from Kv7.2 + Kv7.3-M240R-expressing channels, additional experiments in which the pulse length was increased from 1.5 to 3 s were performed. The results obtained revealed no significant difference in the V1/2 and k values on Kv7.2 + Kv7.3-M240R current using the two protocols; indeed, the V1/2 and the k values were, respectively, + 12.2 ± 7.6 and + 10.2 ± 5.4 mV (n = 4; p > 0.05), and 23.0 ± 3.6 and 23.5 ± 1.4 mV/e-fold (n = 4; p > 0.05) for Kv7.2 + Kv7.3- and Kv7.2 + Kv7.3-M240R-expressing cells.
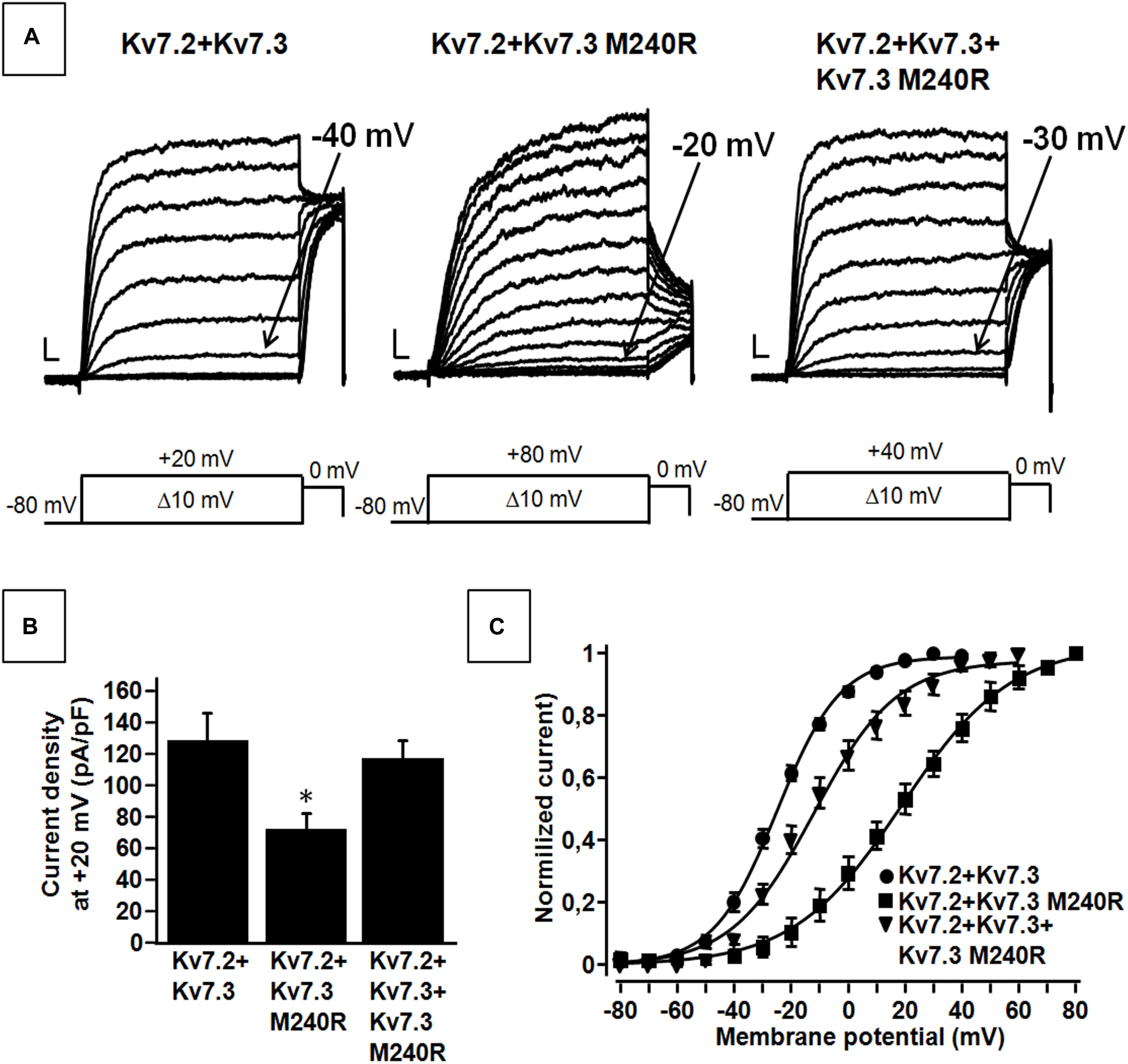
Figure 2. Functional properties of heteromeric channels incorporating subunits carrying the benign familial neonatal epilepsy (BFNE) variant. (A) Macroscopic current traces from the indicated heteromeric channels in response to the indicated voltage protocol. Current scale, 200 pA; time scale, 0.1 s. (B) Current density from the indicated heteromeric channels calculated at +20 mV. The asterisk indicates a statistically-significant value versus Kv7.2 + Kv7.3. (C) Conductance/voltage curves for Kv7.2 + Kv7.3, Kv7.2 + Kv7.3 M240R, Kv7.2 + Kv7.3 + Kv7.3M240R. Continuous lines indicate Boltzmann fits of the experimental data. Current scale, 200 pA; time scale, 0.1 s. Each data point is the mean (SEM) of 7–18 cells recorded in at least three separate experimental sessions.
Altogether these data suggest that Kv7.3 M240R subunits are able to form heteromeric channels with Kv7.2 subunits, although their currents display gating properties very different from Kv7.2 + Kv7.3 heteromeric channels. To replicate in vitro the genetic combination occurring in the affected family members who are heterozygous for the pathogenic allele (Individuals I-1, II-1, and II-2), functional studies were also carried out upon transfection of cDNAs for Kv7.2 + Kv7.3 + Kv7.3 M240R at a cDNA ratio of 1:0.5:0.5. The current density measured in CHO cells expressing heteromeric channels formed by the described subunit combinations was very similar to that recorded in cells expressing Kv7.2 + Kv7.3 subunits at a transfection ratio 1:1 (mimicking a healthy individual) (Figures 2A,B and Table 1) and no difference in the activation kinetics was observed between the Kv7.2 + Kv7.3 and Kv7.2 + Kv7.3 + Kv7.3 M240R currents; indeed the τactivation at + 20 mV were 142 ± 19 ms and 128 ± 28 ms, respectively. By contrast, the midpoint of activation of the currents recorded upon Kv7.2 + Kv7.3 + Kv7.3 M240R subunit co-expression was right-shifted by about 10 mV when compared to Kv7.2 + Kv7.3 (Figures 2A–C and Table 1), thus confirming LoF effects of the mutant subunits also when incorporated into channel tetramers with Kv7.2 + Kv7.3 subunits.
The herein identified M240R variant introduces an extra positively charged residue into the S4 segment of Kv7.3 subunits, between R5 and R6. To achieve a better understanding of the possible role of the M240 residue in the gating process and of the structural consequences of its replacement with an R, we built homology models of a Kv7.3 subunit in both resting and activated states, as previously described (Jensen et al., 2012; Sands et al., 2019). Our structural models suggested that, no electrostatic interaction between the M240 residue side chain and surrounding protein residues occurs in both the resting and the activated VSD configurations (Figure 3A); in particular, in the resting VSD state and in two of the four subunits, the M240 side chain points toward the N-terminal region of the same subunits, whereas in the other two subunits it points in the opposite direction, namely toward the S5 segment (Figure 3B). Substitution of the non-polar M residue at position 240 with an R introduces a novel electrostatic interaction between R240 side chain and a highly conserved negatively charged N-terminal glutamate (E116) in one Kv7.3 subunit. This interaction only occurred in the resting state of the VSD (Long et al., 2007) (Figure 3C). Instead, VSD movement during the activation process translated the R240 side chain in a tight pocket surrounded by non-polar residues, possibly destabilizing the tight network of hydrophobic interactions within this pocket. As a result, our model suggests that the M240R substitution may preferentially stabilize the closed state and possibly destabilize the activated state of the Kv7.3 subunits.
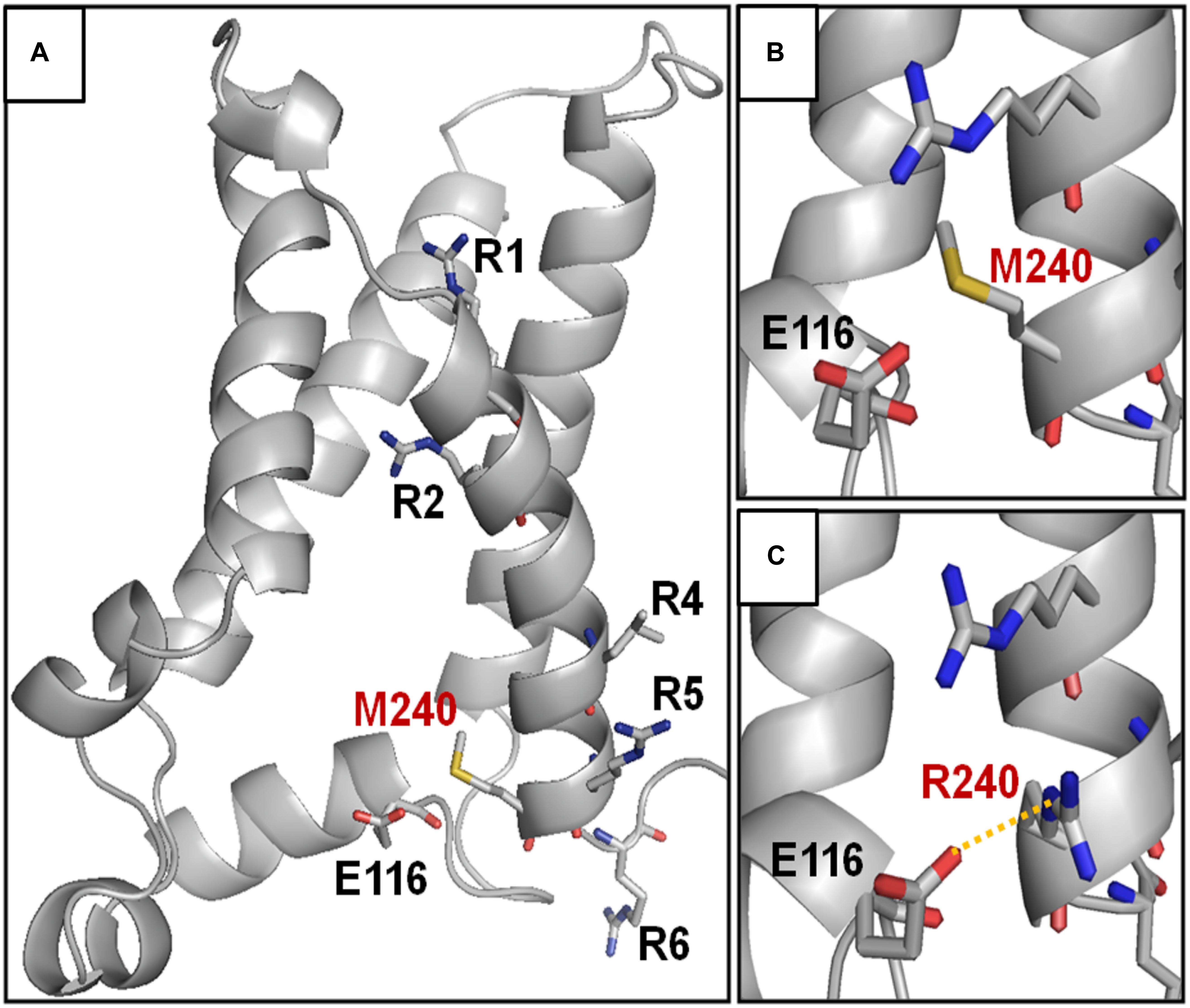
Figure 3. Structural modeling of homomeric Kv7.3 channel subunits in resting gating states. (A) Homology model of a homotetrameric Kv7.3 channel subunits, obtained as described in the section “Materials and Methods.” (B,C) Enlarged views showing the N-terminal domain encompassing the E116 residue in the wild-type (B) or M240R mutant (C) subunits, showing the occurrence the E116-R240 polar interaction (highlighted in yellow).
The results described suggest that heteromeric channels incorporating Kv7.3 M240R subunits display a reduced sensitivity to voltage, strongly suggesting a LoF pathogenic mechanism. Given that BHB has been recently shown to potentiate Kv7.3 and Kv7.2 + Kv7.3 currents by a mechanism opposite to that introduced by the M240R variant (Manville et al., 2018, 2020), we further evaluated its ability to counteract in vitro the described functional alteration. We tested the effects of the BHB using a voltage protocol in which Kv7.2 + Kv7.3 and Kv7.2 + Kv7.3 + Kv7.3 M240R currents were activated by 3 s voltage ramps from −80 to +20 mV. Perfusion with 100 μM BHB enhanced ramp-evoked Kv7.2 + Kv7.3 and Kv7.2 + Kv7.3 + Kv7.3 M240R currents; this effect was reversible since the currents returned to basal values after about 10 s upon BHB removal from the bath (Figure 4A). In addition, steady-state experiments revealed that at −40 mV, a membrane potential value close to the activation threshold, 100 μM BHB increased Kv7.2 + Kv7.3 and Kv7.2 + Kv7.3 + Kv7.3 M240R currents (Figure 4B) and also caused a 10 mV negative shift in the G/V curve (Figure 4C). Interestingly, the V1/2 value of Kv7.2 + Kv7.3 + Kv7.3 M240R currents upon BHB exposure was similar to that of Kv7.2 + Kv7.3-expressing cells (Figure 4C and Table 1), suggesting the ability of the BHB to restore Kv7.2 + Kv7.3 + Kv7.3 M240R currents to that of wild-type.
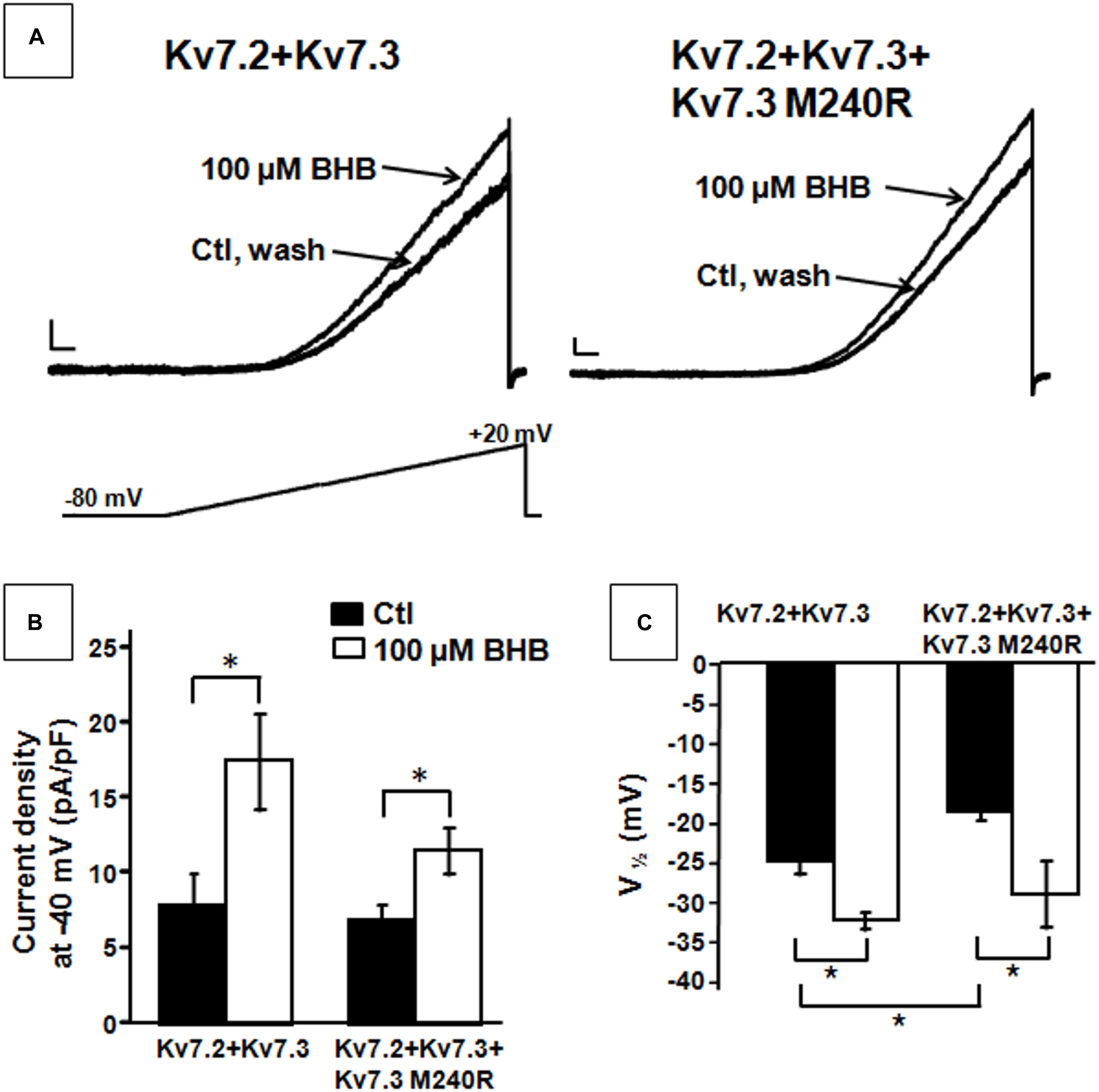
Figure 4. Effect of β-hydroxybutyrate (BHB) on heteromeric Kv7.2 + Kv7.3, Kv7.2 + Kv7.3 + Kv7.3 M240R channels. (A) Current responses from the indicated heteromeric channels to voltage ramps from −80 to +20 mV. (B,C) Quantification of the effects of 100 μM BHB on the indicated heteromeric channels. Data are expressed as current density calculated at −40 mV (B) and in the V1/2 values. Current scale, 200 pA; time scale, 200 ms. * indicates p < 0.05 vs. corresponding controls.
Inherited variants in KCNQ2 or KCNQ3 cause BFNE, a neonatal-onset familial epilepsy, characterized by recurrent focal tonic seizures in otherwise well infants (Sands et al., 2016). While seizure onset is most often in the first days of life, seizures can present later in infancy (Zara et al., 2013), as demonstrated by the father in our pedigree, or not at all, as penetrance is incomplete. Seizures tend to remit over the first year, but ∼30% of individuals have seizures later in life (Grinton et al., 2015), as illustrated by case II-2.
Most BFNE families carry pathogenic variants in KCNQ2, whereas only a small percentage carry KCNQ3 variants (Grinton et al., 2015; Sands et al., 2016; Miceli et al., 2017, 2018). KCNQ2 variants responsible for BFNE are missense, stop-gain, frameshift, splice variants, and deletions randomly distributed along the entire primary sequence of the subunit. In contrast, BFNE-causing KCNQ3 variants are all missense, affecting specific residues located in the pore region of the channel (S5–S6 and intervening loop; Table 2). Prior studies have reported 13 such variants, each affecting a different Kv7.3 residue located in and around the pore (Table 2); in addition, four Kv7.3 variants affecting residues in the long C-terminus have also been described, although the pathogenic role of these variants appears questionable due to their ample representation in the gnomAD population database without the benefit of supportive functional data (N821S, Bassi et al., 2005; R780C, Zara et al., 2013), or with functional data that fail to support a disease association (N468S, Singh et al., 2003; P574S, Miceli et al., 2009). Out of the 13 KCNQ3 variants causing BFNE, those nine with supportive functional data lie within a span of 51 residues (V279 to R330) in this region. Functional analyses of these variants to date, e.g., V279F, I317T, R330C, and R330L, have mostly demonstrated a reduction in maximal current with no effect on the voltage-dependence of activation (Soldovieri et al., 2014; Miceli et al., 2015; Maljevic et al., 2016), consistent with these variants being in the pore. Only W309R induced both a reduction in currents (by about 60%) and a rather small (3–4 mV) rightward shift in the V1/2 value, when expressed together with Kv7.2 and Kv7.3 (Uehara et al., 2008).
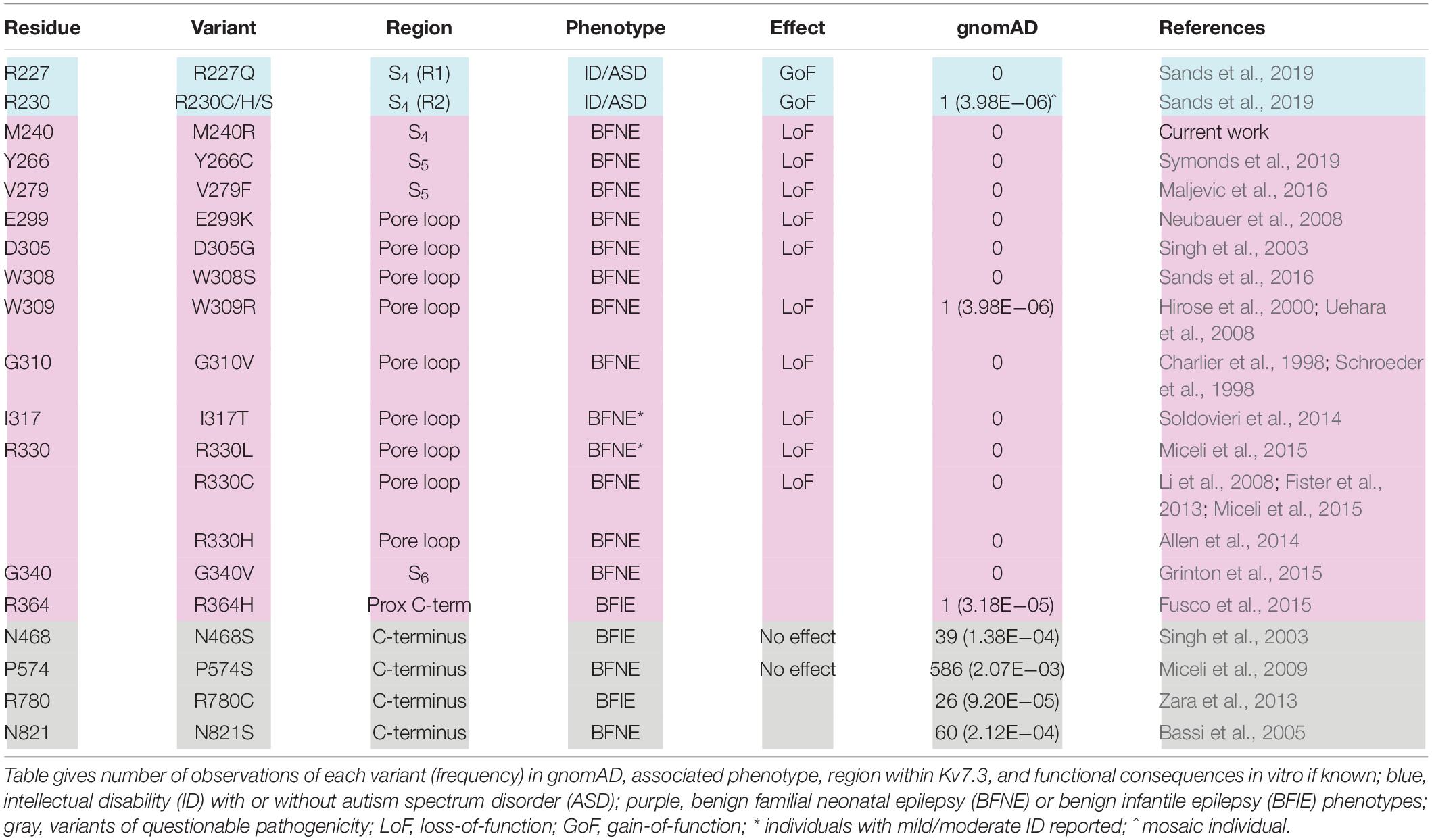
Table 2. Missense variants reported for KCNQ3.
In the present work, we report a BFNE family carrying the first variant in KCNQ3 located in the S4 helix of the VSD (M240R). Functional studies revealed that the M240R variant abolishes channel function in homomeric configuration, but that this is partially rescued in heteromeric channels with wild-type Kv7.2 and/or Kv7.2/3 subunits. Differently from all other Kv7.3 BFNE variants characterized to date, M240R subunits, when expressed with Kv7.2 and Kv7.3 subunits, show a significant decrease of channel sensitivity to voltage of about 10 mV, without major changes in pore properties and heteromerization. These functional results suggest that the introduction of an additional positively charged residue at the bottom of S4 in Kv7.3 subunits destabilizes the activated conformation of the voltage sensor, thereby impeding pore opening. Structural modeling provides a potential explanation for such a conclusion; indeed, introduction of an R at position 240 stabilizes the VSD resting state by forming a novel strong ionic interaction with an highly conserved residue in the N-terminus; in addition, the larger R side chain may destabilize the tight network of hydrophobic interaction occurring in the activated state. Further studies using residue swapping or charge reversion are needed to confirm the potential interaction between the R240 and E116 residues. The available data do not allow us to determine whether the G/V shift introduced by the mutation originates from the participation of the 240 residue in voltage-sensing or in the subsequent steps along the activation pathway leading to pore opening. However, the fact that recent structural data from human Kv7.1 (Sun and MacKinnon, 2020), reveal that the V241 residue, corresponding to the M240 residue in Kv7.3, is located within a key region for VSD-pore electro-mechanical coupling (Hou et al., 2020), raises the possibility that this residue may play a similar functional role also in Kv7.3. Therefore, a LoF mechanism appears mainly responsible for BFNE pathogenesis in our family. Similar conclusions have been reached for BFNE-causing variants in Kv7.2, where haploinsufficiency, corresponding to an IKM reduction of only ∼25%, appears responsible for disease pathogenesis (Jentsch, 2000).
Benign familial neonatal epilepsy with normal neurocognitive development is not the only phenotype associated with variants in Kv7.3. In fact, individuals with mild/moderate ID have been described in BFNE pedigrees (I317T, R330L; Soldovieri et al., 2014; Miceli et al., 2015); moreover, de novo variants in KCNQ3 have been described in children with DEE (Allen et al., 2013; Grozeva et al., 2015; Miceli et al., 2015; Ambrosino et al., 2018; Lauritano et al., 2019), ID apparently without epilepsy (Rauch et al., 2012; Deciphering Developmental Disorders, 2017), cortical visual impairment (Bosch et al., 2016), and in patients with ID and autism (Sands et al., 2019). Such phenotypic heterogeneity is at least in part correlated with variant-specific functional effects, by yet unknown mechanisms; in fact, opposite to BFNE variants causing LoF, gain-of-function (GoF) variants cause non-verbal ID, autism, and prominent sleep-activated multifocal epileptiform EEG discharges without neonatal seizures (Singh et al., 2003; Sands et al., 2019).
The observation that epilepsy-associated variants in KCNQ2 are more than 10 times more frequent than those in KCNQ3, suggests that KCNQ3 tolerates variation better than KCNQ2. Several pieces of evidence support this view; as an example, while heterozygous frameshift variants in KCNQ2 are frequent causes of BFNE (Miceli et al., 2018), no heterozygous pathogenic frameshift KCNQ3 variant has ever been associated with a human phenotype. In addition, no individual carrying KCNQ2 frameshift variants in homozygosity has ever been described, as minimal KCNQ2 residual activity is probably essential for survival (Lauritano et al., 2019); by contrast, two recent studies reported the occurrence of homozygous frameshift variants in KCNQ3 (each inherited from an asymptomatic parent) in patients with developmental delay and neonatal seizures (Kothur et al., 2018; Lauritano et al., 2019), demonstrating that, homozygous variants in KCNQ3 are compatible with life. Moreover, a distinct functional role of these two genes emerging from developmental and genetic studies in humans, appears also to be recapitulated in mice. In fact, while KCNQ2 homozygous KO mice died at birth, homozygous KCNQ3 KO mice showed no seizures and survived until adulthood (Tzingounis and Nicoll, 2008; Kim et al., 2016); similarly, conditional deletion of KCNQ2 in cortical pyramidal neurons increased neuronal excitability and decreased lifespan, whereas deletion of KCNQ3 neither increased pyramidal neurons excitability nor affected mice survival (Soh et al., 2014).
Notably, in heteromeric channels, the functional effects of the M240R variant in Kv7.3 herein described in a BFNE family are quantitatively and qualitatively very similar to those triggered by the R213Q variant in Kv7.2, the latter identified in sporadic patients with DEE (Weckhuysen et al., 2012; Millichap et al., 2016). The fact that similar, dramatic in vitro functional consequences are associated to a severe phenotype in Kv7.2 and to self-limiting epilepsy in Kv7.3, further supports the hypothesis that Kv7.3 is more tolerant than Kv7.2 to genetic changes causing LoF effects. Among many others, such as inclusion/exclusion of both genes in panels for DEE, diagnostic NGS coverage, and epistatic compensation, an attractive explanation for such distinct functional role lies in the different developmental pattern of expression, with KCNQ2 being expressed at earlier stages of development when compared to KCNQ3, in both mice and humans (Tinel et al., 1998; Kanaumi et al., 2008).
In patients with KCNQ2- and KCNQ3-related disorders, seizures are controlled with sodium channel blockers in most patients (Pisano et al., 2015; Sands et al., 2016). However, a percentage continues to have seizures, which may contribute to cognitive impairment. In addition, successful management of seizures does not address the neurodevelopmental disability that occurs in KCNQ2 and KCNQ3 DEEs. Efficacy of the KD, with greater than 90% seizure reduction, has been reported in patients with monogenic DEEs, including KCNQ2-DEE (Ko et al., 2018) and Dravet syndrome (Knupp and Wirrell, 2018). Our finding that the endogenous ketone BHB can activate heteromeric Kv7.2 + Kv7.3 channels containing Kv7.3 M240R subunits to the same extent as wild-type Kv7.2 + Kv7.3 channels (Manville et al., 2018, 2020), thereby counteracting the underlying pathophysiology, suggests that the KD could represent precision medicine for more severe phenotypes caused by KCNQ2 and KCNQ3 LoF variants. Further work is required to identify additional patients carrying KCNQ3 or KCNQ2 pathogenic LoF variants retaining responsiveness to BHB in vitro and who may benefit from KD treatment in vivo.
In conclusion, we describe the first missense LoF pathogenic variant located in the S4 segment of KCNQ3 as a cause of BFNE. In contrast to BFNE-KCNQ3 variants previously described, functional and modeling data suggest that the M240R variant primarily affects the voltage sensitivity of heteromeric channels. Our study provides a pharmacological rationale for investigating the use of KD in patients with DEE caused by KCNQ3 and KCNQ2 LoF variants.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Columbia University Irving Medical Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
FM, MT, and TS conceived the study, analyzed the data, and wrote the manuscript. EC, MC, and DG analyzed the data and wrote the manuscript. FM, LC, VB, MS, EH, AM, NL, and LB performed the research and analyzed the data. All authors contributed to the article and approved the submitted version.
The present work was supported by the Telethon Foundation (GGP15113) and by the Italian Ministry for University and Research (PRIN 2017ALCR7C) to MT, the Italian Ministry for University and Research (Project Scientific Independence of Researchers 2014 RBSI1444EM and PRIN 2017YH3SXK) to FM, the Italian Ministry of Health Ricerca Finalizzata Giovani Ricercatori 2016 (project GR-2016-02363337), the Italian Ministry for University and Research (PRIN 2017ALCR7C), and Research Funds from University of Molise to MS. This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Thomas J. Jentsch, Department of Physiology and Pathology of Ion Transport, Leibniz-Institut für Molekulare Pharmakologie, Berlin for sharing Kv7.2 and Kv7.3 cDNAs and David E. Shaw, D. E. Shaw Research, New York, for the coordinates of the Kv1.2/2.1 chimera.
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Allen, N. M., Mannion, M., Conroy, J., Lynch, S. A., Shahwan, A., Lynch, B., et al. (2014). The variable phenotypes of KCNQ-related epilepsy. Epilepsia 55, e99–e105. doi: 10.1111/epi.12715
Ambrosino, P., Freri, E., Castellotti, B., Soldovieri, M. V., Mosca, I., Manocchio, L., et al. (2018). Kv7.3 Compound Heterozygous Variants in Early Onset Encephalopathy Reveal Additive Contribution of C-Terminal Residues to PIP2-Dependent K(+) Channel Gating. Mol. Neurobiol. 55, 7009–7024. doi: 10.1007/s12035-018-0883-5
Bassi, M. T., Balottin, U., Panzeri, C., Piccinelli, P., Castaldo, P., Barrese, V., et al. (2005). Functional analysis of novel KCNQ2 and KCNQ3 gene variants found in a large pedigree with benign familial neonatal convulsions (BFNC). Neurogenetics 6, 185–193. doi: 10.1007/s10048-005-0012-2
Bosch, D. G., Boonstra, F. N., de Leeuw, N., Pfundt, R., Nillesen, W. M., de Ligt, J., et al. (2016). Novel genetic causes for cerebral visual impairment. Eur. J. Hum. Genet. 24, 660–665. doi: 10.1038/ejhg.2015.186
Brown, D. A., and Adams, P. R. (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. doi: 10.1038/283673a0
Charlier, C., Singh, N. A., Ryan, S. G., Lewis, T. B., Reus, B. E., Leach, R. J., et al. (1998). A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 18, 53–55. doi: 10.1038/ng0198-53
Deciphering Developmental Disorders (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Fister, P., Soltirovska-Salamon, A., Debeljak, M., and Paro-Panjan, D. (2013). Benign familial neonatal convulsions caused by mutation in KCNQ3, exon 6: a European case. Eur. J. Paediatr. Neurol. 17, 308–310. doi: 10.1016/j.ejpn.2012.10.007
Fusco, C., Frattini, D., and Bassi, M. T. (2015). A novel KCNQ3 gene mutation in a child with infantile convulsions and partial epilepsy with centrotemporal spikes. Eur. J. Paediatr. Neurol. 19, 102–103. doi: 10.1016/j.ejpn.2014.08.006
Grinton, B. E., Heron, S. E., Pelekanos, J. T., Zuberi, S. M., Kivity, S., Afawi, Z., et al. (2015). Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia 56, 1071–1080. doi: 10.1111/epi.13020
Grozeva, D., Carss, K., Spasic-Boskovic, O., Tejada, M. I., Gecz, J., Shaw, M., et al. (2015). Targeted Next-Generation Sequencing Analysis of 1,000 Individuals with Intellectual Disability. Hum. Mutat. 36, 1197–1204. doi: 10.1002/humu.22901
Hirose, S., Zenri, F., Akiyoshi, H., Fukuma, G., Iwata, H., Inoue, T., et al. (2000). A novel mutation of KCNQ3 (c.925T–>C) in a Japanese family with benign familial neonatal convulsions. Ann. Neurol. 47, 822–826. doi: 10.1002/1531-8249(200006)47:6<822::aid-ana19>3.0.co;2-x
Hou, P., Kang, P. W., Kongmeneck, A. D., Yang, N. D., Liu, Y., Shi, J., et al. (2020). Two-stage electro-mechanical coupling of a KV channel in voltage-dependent activation. Nat. Commun. 11:676. doi: 10.1038/s41467-020-14406-w
Jensen, M. O., Jogini, V., Borhani, D. W., Leffler, A. E., Dror, R. O., and Shaw, D. E. (2012). Mechanism of voltage gating in potassium channels. Science 336, 229–233. doi: 10.1126/science.1216533
Jentsch, T. J. (2000). Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21–30. doi: 10.1038/35036198
Kanaumi, T., Takashima, S., Iwasaki, H., Itoh, M., Mitsudome, A., and Hirose, S. (2008). Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 30, 362–369. doi: 10.1016/j.braindev.2007.11.003
Kim, K. S., Duignan, K. M., Hawryluk, J. M., Soh, H., and Tzingounis, A. V. (2016). The voltage activation of cortical KCNQ channels depends on global PIP2 levels. Biophys. J. 110, 1089–1098. doi: 10.1016/j.bpj.2016.01.006
Knupp, K. G., and Wirrell, E. C. (2018). Treatment strategies for dravet syndrome. CNS Drugs 32, 335–350. doi: 10.1007/s40263-018-0511-y
Ko, A., Jung, D. E., Kim, S. H., Kang, H. C., Lee, J. S., Lee, S. T., et al. (2018). The Efficacy of Ketogenic Diet for Specific Genetic Mutation in Developmental and Epileptic Encephalopathy. Front. Neurol. 9:530. doi: 10.3389/fneur.2018.00530
Kothur, K., Holman, K., Farnsworth, E., Ho, G., Lorentzos, M., Troedson, C., et al. (2018). Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure 59, 132–140. doi: 10.1016/j.seizure.2018.05.005
Lauritano, A., Moutton, S., Longobardi, E., Tran Mau-Them, F., Laudati, G., Nappi, P., et al. (2019). A novel homozygous KCNQ3 loss-of-function variant causes non-syndromic intellectual disability and neonatal-onset pharmacodependent epilepsy. Epilepsia Open 4, 464–475. doi: 10.1002/epi4.12353
Li, H., Li, N., Shen, L., Jiang, H., Yang, Q., Song, Y., et al. (2008). A novel mutation of KCNQ3 gene in a Chinese family with benign familial neonatal convulsions. Epilepsy Res. 79, 1–5. doi: 10.1016/j.eplepsyres.2007.12.005
Long, S. B., Tao, X., Campbell, E. B., and MacKinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. doi: 10.1038/nature06265
Lutas, A., and Yellen, G. (2013). The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40. doi: 10.1016/j.tins.2012.11.005
Maljevic, S., Vejzovic, S., Bernhard, M. K., Bertsche, A., Weise, S., Docker, M., et al. (2016). Novel KCNQ3 mutation in a large family with benign familial neonatal epilepsy: a rare cause of neonatal seizures. Mol. Syndromol. 7, 189–196. doi: 10.1159/000447461
Manville, R. W., Papanikolaou, M., and Abbott, G. W. (2018). Direct neurotransmitter activation of voltage-gated potassium channels. Nat. Commun. 9:1847. doi: 10.1038/s41467-018-04266-w
Manville, R. W., Papanikolaou, M., and Abbott, G. W. (2020). M-Channel activation contributes to the anticonvulsant action of the ketone body beta-Hydroxybutyrate. J. Pharmacol. Exp. Ther. 372, 148–156. doi: 10.1124/jpet.119.263350
Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., Migliore, M., Cilio, M. R., et al. (2013). Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc. Natl. Acad. Sci. U.S.A. 110, 4386–4391. doi: 10.1073/pnas.1216867110
Miceli, F., Soldovieri, M. V., Joshi, N., Weckhuysen, S., Cooper, E., and Taglialatela, M. (2018). “KCNQ2-Related Disorders,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Miceli, F., Soldovieri, M. V., Joshi, N., Weckhuysen, S., Cooper, E. C., and Taglialatela, M. (2017). “KCNQ3-Related Disorders,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Miceli, F., Soldovieri, M. V., Lugli, L., Bellini, G., Ambrosino, P., Migliore, M., et al. (2009). Neutralization of a unique, negatively-charged residue in the voltage sensor of K V 7.2 subunits in a sporadic case of benign familial neonatal seizures. Neurobiol. Dis. 34, 501–510. doi: 10.1016/j.nbd.2009.03.009
Miceli, F., Striano, P., Soldovieri, M. V., Fontana, A., Nardello, R., Robbiano, A., et al. (2015). A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 56, e15–e20. doi: 10.1111/epi.12887
Millichap, J. J., Park, K. L., Tsuchida, T., Ben-Zeev, B., Carmant, L., Flamini, R., et al. (2016). KCNQ2 encephalopathy: features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol Genet. 2:e96. doi: 10.1212/NXG.0000000000000096
Neubauer, B. A., Waldegger, S., Heinzinger, J., Hahn, A., Kurlemann, G., Fiedler, B., et al. (2008). KCNQ2 and KCNQ3 mutations contribute to different idiopathic epilepsy syndromes. Neurology 71, 177–183. doi: 10.1212/01.wnl.0000317090.92185.ec
Pisano, T., Numis, A. L., Heavin, S. B., Weckhuysen, S., Angriman, M., Suls, A., et al. (2015). Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 56, 685–691. doi: 10.1111/epi.12984
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682. doi: 10.1016/S0140-6736(12)61480-9
Sands, T. T., Balestri, M., Bellini, G., Mulkey, S. B., Danhaive, O., Bakken, E. H., et al. (2016). Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia 57, 2019–2030. doi: 10.1111/epi.13596
Sands, T. T., Miceli, F., Lesca, G., Beck, A. E., Sadleir, L. G., Arrington, D. K., et al. (2019). Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 86, 181–192. doi: 10.1002/ana.25522
Schroeder, B. C., Kubisch, C., Stein, V., and Jentsch, T. J. (1998). Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690. doi: 10.1038/25367
Singh, N. A., Westenskow, P., Charlier, C., Pappas, C., Leslie, J., Dillon, J., et al. (2003). KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 126(Pt 12), 2726–2737. doi: 10.1093/brain/awg286
Soh, H., Pant, R., LoTurco, J. J., and Tzingounis, A. V. (2014). Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J. Neurosci. 34, 5311–5321. doi: 10.1523/JNEUROSCI.3919-13.2014
Soldovieri, M. V., Boutry-Kryza, N., Milh, M., Doummar, D., Heron, B., Bourel, E., et al. (2014). Novel KCNQ2 and KCNQ3 mutations in a large cohort of families with benign neonatal epilepsy: first evidence for an altered channel regulation by syntaxin-1A. Hum. Mutat. 35, 356–367. doi: 10.1002/humu.22500
Soldovieri, M. V., Castaldo, P., Iodice, L., Miceli, F., Barrese, V., Bellini, G., et al. (2006). Decreased subunit stability as a novel mechanism for potassium current impairment by a KCNQ2 C terminus mutation causing benign familial neonatal convulsions. J. Biol. Chem. 281, 418–428. doi: 10.1074/jbc.m510980200
Sun, J., and MacKinnon, R. (2020). Structural basis of human KCNQ1 modulation and gating. Cell 180, 340-347.e9. doi: 10.1016/j.cell.2019.12.003
Symonds, J. D., Zuberi, S. M., Stewart, K., McLellan, A., O’Regan, M., MacLeod, S., et al. (2019). Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain 142, 2303–2318. doi: 10.1093/brain/awz195
Tinel, N., Lauritzen, I., Chouabe, C., Lazdunski, M., and Borsotto, M. (1998). The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 438, 171–176. doi: 10.1016/s0014-5793(98)01296-4
Tzingounis, A. V., and Nicoll, R. A. (2008). Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc. Natl. Acad. Sci. U.S.A. 105, 19974–19979. doi: 10.1073/pnas.0810535105
Uehara, A., Nakamura, Y., Shioya, T., Hirose, S., Yasukochi, M., and Uehara, K. (2008). Altered KCNQ3 potassium channel function caused by the W309R pore-helix mutation found in human epilepsy. J. Membr. Biol. 222, 55–63. doi: 10.1007/s00232-008-9097-5
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Weckhuysen, S., Mandelstam, S., Suls, A., Audenaert, D., Deconinck, T., Claes, L. R., et al. (2012). KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25. doi: 10.1002/ana.22644
Keywords: KCNQ, BFNE, encephalopathy, channelopathies, ketogenic diet
Citation: Miceli F, Carotenuto L, Barrese V, Soldovieri MV, Heinzen EL, Mandel AM, Lippa N, Bier L, Goldstein DB, Cooper EC, Cilio MR, Taglialatela M and Sands TT (2020) A Novel Kv7.3 Variant in the Voltage-Sensing S4 Segment in a Family With Benign Neonatal Epilepsy: Functional Characterization and in vitro Rescue by β-Hydroxybutyrate. Front. Physiol. 11:1040. doi: 10.3389/fphys.2020.01040
Received: 05 June 2020; Accepted: 29 July 2020;
Published: 04 September 2020.
Edited by:
Mark S. Shapiro, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Rían Manville, University of Brighton, United KingdomCopyright © 2020 Miceli, Carotenuto, Barrese, Soldovieri, Heinzen, Mandel, Lippa, Bier, Goldstein, Cooper, Cilio, Taglialatela and Sands. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Taglialatela, bXRhZ2xpYWxAdW5pbmEuaXQ=; Tristan T. Sands, dHRzMjdAY3VtYy5jb2x1bWJpYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.