 Hana Starobova
Hana Starobova Evelyn Israel Nadar
Evelyn Israel Nadar Irina Vetter
Irina Vetter
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 19 August 2020
Sec. Integrative Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.01016
This article is part of the Research Topic The Perplexing Physiology of the Nociceptive System View all 13 articles
Pain is a fundamental feature of inflammation. The immune system plays a critical role in the activation of sensory neurons and there is increasing evidence of neuro-inflammatory mechanisms contributing to painful pathologies. The inflammasomes are signaling multiprotein complexes that are key components of the innate immune system. They are intimately involved in inflammatory responses and their activation leads to production of inflammatory cytokines that in turn can affect sensory neuron function. Accordingly, the contribution of inflammasome activation to pain signaling has attracted considerable attention in recent years. NLRP3 is the best characterized inflammasome and there is emerging evidence of its role in a variety of inflammatory pain conditions. In vitro and in vivo studies have reported the activation and upregulation of NLRP3 in painful conditions including gout and rheumatoid arthritis, while inhibition of NLRP3 function or expression can mediate analgesia. In this review, we discuss painful conditions in which NLRP3 inflammasome signaling has been pathophysiologically implicated, as well as NLRP3 inflammasome-mediated mechanisms and signaling pathways that may lead to the activation of sensory neurons.
Chronic pain is a dynamic process involving ongoing changes and adaption within the peripheral and central nervous systems and accompanies a vast number of inflammatory and non-inflammatory pathological states such as cancer, diabetes or rheumatoid arthritis.
Sensory neurons, including nociceptors, express a variety of ion channels and receptors involved in transformation of external stimuli into electrical signals, such the Transient Receptor Potential channels TRPV1,TRPV2, TRPV3, TRPA1, and TRPM8; purine receptor 3 (P2X3); voltage-gated sodium channel (Nav) subtypes Nav1.7, Nav1.8, and Nav1.9 and voltage-gated potassium channels (Kv). The activation of these channels and receptors by thermal, mechanical or chemical nociceptive stimuli generate an action potential that is transmitted along the axons to the central nervous system. The immune system and neuro-inflammation play a crucial role in sensitization of the peripheral and central nervous systems, including activation of nociceptors. Specifically, microglia, satellite glia cells, Schwann cells, oligodendrocytes, keratinocytes, macrophages or T-cells releasing pro-inflammatory signaling molecules, such as interleukin-1β (IL-1β) or adenosine-triphosphate (ATP), are implicated in the pathology of various painful conditions (Ji et al., 2016; Inoue and Tsuda, 2018). Moreover, the depletion of macrophages reduces hyperalgesia in different rodent models of neuropathic pain (Barclay et al., 2007; Mert et al., 2009; Kobayashi et al., 2015; Zhang et al., 2016) and macrophages were shown to directly regulate the peristaltic activity of the colon via interaction with the enteric neurons and to release a variety of pain mediators such as nerve growth factor, tumor necrosis factor or IL-1β (Muller et al., 2014; McMahon et al., 2015).
Inflammasomes are a group of pattern recognition receptors (PRR) that detect pathogen- or damage-associated molecular patterns (PAMPs and DAMPs) and activate inflammatory responses. To date, there are six known inflammasome multi-protein complexes: NACHT, LRR and PYD/CARD domains-containing proteins - NLRP1, NLRP3, NLRP6, NLRP12, and NLRC4, and AIM2 (absent in melanoma 2) (Strowig et al., 2012). While the NLR family of the inflammasomes are fundamental component of the innate immune system, recognizing a broad spectrum of viral and bacterial stimuli, such as bacterial or viral RNA, lipopolysaccharides or uric acid crystals (Franchi et al., 2009b), AIM2 recognizes double stranded DNA released during various bacterial and viral infections (Sharma et al., 2019). Out of all the NLR inflammasomes, the NLRP3 inflammasome is amongst the best characterized, being predominantly expressed by immune cells such as macrophages and neutrophils and to a lesser extent by dendritic cells, microglia and dorsal root ganglia (Guarda et al., 2011b; Gustin et al., 2015; Fann et al., 2018). The activation of NLRP3 leads to release of IL-1β, a cytokine that is known to directly sensitize nociceptors and to cause pain (Binshtok et al., 2008). Increased NLRP3 expression and activation, together with the release of IL-1β, has been linked to pathogenesis of several painful inflammatory and non-inflammatory conditions such as rheumatoid arthritis, gout, neuropathic pain and diabetic wound healing (Table 1). In this review, we discuss the mechanisms leading to NLRP3 inflammasome activation in painful conditions, the resulting signaling pathways that can lead to the sensitization or activation of sensory neurons, as well as the therapeutic potential of NLRP3 inflammasome inhibition to mediate analgesia.
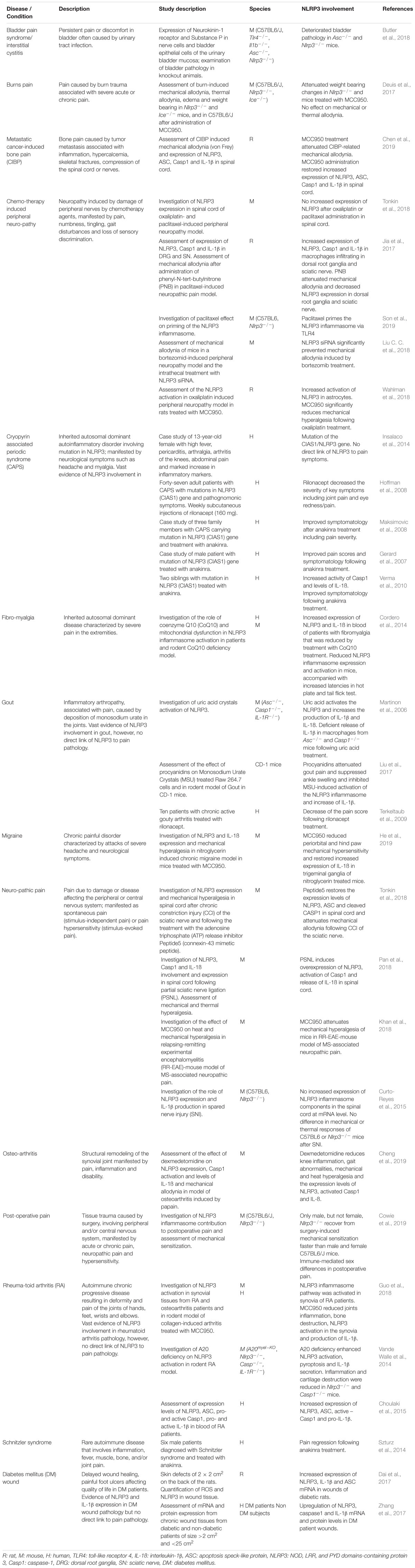
Table 1. NLRP3 in pain pathology.
The activation of the NLRP3 inflammasome requires, with some exceptions, two signals. The first signal leads to priming of the NLRP3 inflammasome by endogenous molecules (e.g., tumor necrosis factor (TNF) and IL-1β) or microbial components such as lipopolysaccharides. These compounds activate their cognate receptors, leading to nuclear factor-êB-dependent increase of NLRP3 and pro-IL-1β expression (Figure 1A) (Bauernfeind et al., 2009; Franchi et al., 2009a). The second signal causes the direct activation of NLRP3 and can involve several cellular signaling events such as potassium ion efflux, changes in calcium signaling, increase in production of reactive oxygen species by mitochondria or lysosomal leakage and release of cathepsin-B (Figure 1A; Brough et al., 2003; Bauernfeind et al., 2011; Munoz-Planillo et al., 2013; Orlowski et al., 2015). In the canonical NLRP3 signaling pathway, the activation of NLRP3 leads to assembly of a multi-protein complex that recruits caspase-1 via caspase activation and recruitment domain (CARD) interactions to promote caspase activation. Activated caspase-1 then cleaves pro-IL-1β and pro-interleukin-18 (pro-IL-18) into their active forms (Hu et al., 2013; Lu et al., 2014, 2015, 2016). Additionally, active caspase-1 cleaves the pore former pro-gasdermin D into its active form, inducing pyroptosis, a form of inflammatory cell death (Sagulenko et al., 2013).
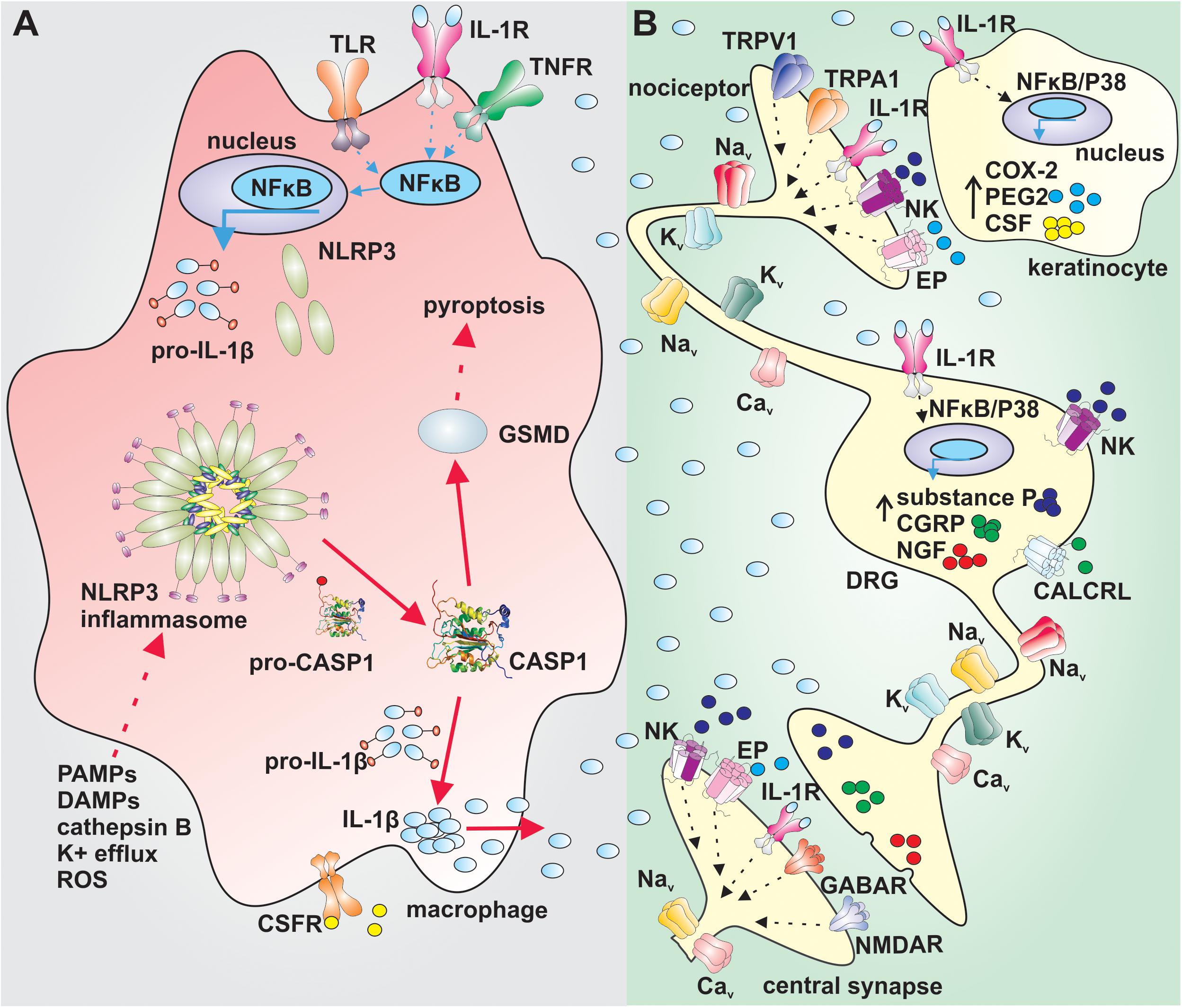
Figure 1. Putative mechanism of sensory nerve sensitization by IL-1β. (A) Priming of the NLRP3 inflammasome via activation of TLRs, IL-1R or TNFR in NFκB dependent manner in macrophages leads to increased expression of NLRP3 and pro-IL1β (blue arrows). The canonical activation of the NLRP3 (red arrows) by PAMPs, DAMPs, cathepsin B, K+ efflux or ROS leads to assembly of the NLRP3 inflammasome complex, activation of CASP1 and cleavage of pro-IL-1β and GSMD. Active IL-1β is released by macrophages and exerts its effect via IL-1R on sensory nerves and surrounding cells. Active GSMD initiates pyroptosis. (B) IL-1β increases the gene expression of COX-2, PEG2, and CSF in keratinocytes in a NFκB/P38 mitogen kinase dependent manner. PEG2 sensitize peripheral nerves and CSF regulates differentiation, proliferation and survival of macrophages. IL-1β increases excitability of nociceptors by altering the function of tetrodotoxin-resistant voltage-gated sodium channels via IL-1R and increases the gene expression in dorsal root ganglia in a NFκB/P38 mitogen kinase dependent manner. Substance P, CGRP, and NGF sensitize further peripheral nerves. IL-1β: interleukin 1β, TLR: toll-like receptor; IL-1R: interleukin-1 receptor; TNFR: tumor necrosis factor α receptor; NFκB: nuclear factor κB; NLRP3: NACHT, LRR, and PYD domains-containing protein 3; PAMPs: pathogen-associated molecular pattern; DAMPs: damage-associated molecular pattern; ROS: reactive oxygen species; CASP1: caspase 1; CSFR: colony-stimulating factor receptor; GSMD: gasdermin D; Nav: voltage gated sodium channel; EP: prostaglandin E receptor; NK: neurokinin receptor; CGRP: calcitonin gene-related peptide; CALCRL: calcitonin gene-related peptide receptor; NGF: nerve growth factor; COX-2: cyclooxygenase type 2; PEG2: prostaglandin E2; CSF: colony stimulating factor; NMDAR: N-methyl-D-aspartate receptor; Kv: voltage gated potassium channel; Cav: voltage gated calcium channel; TRPV1: transient receptor potential channel vaniloid; TRPA1: transient receptor potential channel ankyrin; GABAR: gamma-aminobutyric acid receptor.
The NLRP3 inflammasome can also be activated via non-canonical signaling pathways that were observed as a preferable response to gram-negative bacteria in mice, and are independent of toll-like receptor 4-dependent priming (TLR4) (Kayagaki et al., 2013). The non-canonical pathway involves the activation of caspase-11 by lipopolysaccharides, which leads to ATP efflux via pannexin-1 and subsequent activation of P2X-purinoceptor-7 (P2X7), efflux of intracellular potassium and activation of NLRP3. Caspase-11 also directly cleaves pro-gasdermin-D, inducing pyroptosis (Kayagaki et al., 2011; Shi et al., 2015; Yang et al., 2015).
Recently, another NLRP3 activation pathway, the so-called alternative inflammasome pathway, was discovered in human monocytes. This pathway is dependent on priming, independent of K++ efflux, involves caspase-8 activation and does not induce apoptosis-inducing speck-like protein containing CARD (ASC-speck) formation or pyroptosis (Gaidt et al., 2016).
Activation of the NLRP3 inflammasome leads to release of active IL-1β, an inflammatory cytokine that regulates the function of various cells, such as immune and neuronal cells (Strowig et al., 2012; Figure 1B). IL-1β is released at the site of inflammation and is a well-known pain-inducing molecule. Several rodent studies have shown that intraplantar or intraperitoneal injection of IL-1β causes severe hyperalgesia (Ferreira et al., 1988; Amaya et al., 2006). IL-1β likely increases the excitability of nociceptors by altering the function of several neuronal ion channels and receptors, including transient receptor potential channels (TRPA1 and TRPV1), N-methyl-D-aspartate (NMDA) and gamma-aminobutyric acid (GABA) receptors, as well as voltage- gated K+, Na+, and Ca2+ channels (Binshtok et al., 2008; Schafers and Sorkin, 2008; Ren and Torres, 2009; Malsch et al., 2014; Stemkowski et al., 2015; Zanos et al., 2018). In this context, IL-1β can be considered to be a direct neuromodulator, and although the molecular signaling pathways leading to enhanced excitability have not been studied extensively, signaling via neuronal IL-1 receptors and activation of p38-mitogen-activated protein kinase appear to contribute mechanistically. Specifically, IL-1β alters the voltage-dependence of slow inactivation of tetrodotoxin (TTX)-resistant Nav current in sensory neurons and also enhances persistent TTX-resistant current (Binshtok et al., 2008). Importantly, IL-1β-induced mechanical allodynia is also relieved in mice lacking the TTX-resistant isoform Nav1.9 (Amaya et al., 2006), supporting an important contribution of altered ion channel function to the neuromodulatory effects of IL-1β. Additionally, IL-1β also mediates production of endogenous molecules such as nerve growth factor, prostaglandin E2, cyclooxygenase-2 and calcitonin-gene related peptide that may further contribute to peripheral sensitization (Zucali et al., 1986; Safieh-Garabedian et al., 1995; Reinold et al., 2005; Figure 1B).
Increased levels of IL-1β accompany a vast number of inflammatory and non-inflammatory diseases and pathological states such as Alzheimer’s disease, cancer or rheumatoid arthritis (RA) (Mrak and Griffin, 2000; Lewis et al., 2006; Lo Gullo et al., 2014; Ruscitti et al., 2015). There is accumulating evidence for NLRP3 and cytokine expression in gout pathology as well, which is evidenced by studies conducted in animal models and human clinical studies (Martinon et al., 2006). A human monoclonal antibody targeting IL-1β that was used in randomized control trials showed significant reduction in risk of developing first gout flares as well as the prevention of recurring gout flares (Schlesinger et al., 2011). Several studies have shown increased expression of IL-1β in tissue and blood in rodent animal models of, for example, metastatic cancer-induced bone pain, chemo-therapy induced peripheral neuropathy, cryopyrin associated periodic syndrome, fibromyalgia, migraine and many other painful conditions (Verma et al., 2010; Cordero et al., 2014; Jia et al., 2017; Chen et al., 2019; He et al., 2019; Table 1). Additionally, IL-1β was found to be upregulated in the blood of patients suffering from painful conditions, and in tissues of rodents in various models of inflammatory or non-inflammatory pain (Ren and Torres, 2009; Gui et al., 2016), underpinning the importance of IL-1β in disease pathology. Only few studies used the global knockout of the IL-1β or IL1R, however, some of the studies did not assess pain behavior (Table 1). For example, a study investigating bladder pain syndrome in Il1b–/– mice showed no improvement in bladder pathology and pain (Butler et al., 2018) while studies in IL-1R–/– mice assessing gout and rheumatoid arthritis did not investigate pathology or pain improvement (Martinon et al., 2006; Vande Walle et al., 2014). Moreover, increased levels of IL-1β do not necessarily involve activation of NLRP3 as IL-1β can also be released via alternative mechanisms, including activation of the NLRP1, NLRC4, and AIM2 inflammasomes (Martinon et al., 2002; Strowig et al., 2012).
Activation of NLRP3 also leads to release of active interleukin-18 (IL-18), which has been found to mediate painful conditions such as muscle pain, cancer-induced bone pain and neuropathic pain (Liu S. et al., 2018; Vasudeva et al., 2015; Yoshida et al., 2018). However, it is currently unclear whether IL-18 can also directly contribute to nociceptor sensitization, or whether these effects are secondary to other IL-18-induced signaling events.
The NLRP3 inflammasome has been implicated in the pathology of many painful diseases and conditions such as bladder pain, neuropathic pain, rheumatoid arthritis, gout, migraine and many more (Table 1). With increasing insight into the role of the immune system in the pathology of multiple diseases, it is perhaps not surprising that NLRP3 has also been suggested as a driving factor in the development of many painful diseases. However, despite the clear links between inflammation and pain, as well as NLRP3 and inflammation, relatively few studies have directly assessed the contribution of NLRP3 signaling to pain.
A prototypical NLRP3-linked condition is cryopyrin-associated periodic syndrome (CAPS), an inherited autosomal dominant autoinflammatory disorder characterized by recurrent episodes of inflammation and fever. CAPS patients carry mutations in the NLRP3 (CIAS1) gene leading to activation of the NLRP3 inflammasome and overproduction of IL-1β. Accordingly, treatment with rilonacept, a soluble IL-1 decoy receptor, or anakinra, an IL-1 receptor antagonist, improve the symptomatology of those patients. However, there is only limited – though promising – evidence regarding efficacy of these treatments against painful symptoms often reported by CAPS patients, such as headaches and myalgia (Gerard et al., 2007; Hoffman et al., 2008; Maksimovic et al., 2008; Verma et al., 2010).
Increased expression of NLRP3 and IL-1β was also found in blood of patients suffering from fibromyalgia, and treatment with coenzyme Q10 decreased both overexpression of NLRP3 as well serum IL-1β and IL-18 levels (Cordero et al., 2014). Similar mechanisms were also observed in a rodent model of fibromyalgia, where coenzyme Q10 decreased pain behaviors in the hot plate and tail flick tests (Cordero et al., 2014).
In common inflammatory disorders characterized by pain as a predominant symptom, such as rheumatoid arthritis, osteoarthritis and gout, release of cytokines including IL-1β is well-known to contribute to pathology. Accordingly, treatments aimed at inhibiting IL-1β effects – including with biologics such as anakinra, canakinumab and rilanocept – is a common therapeutic approach that shows some promise in alleviating painful symptoms (Martinon et al., 2006; Terkeltaub et al., 2009; Vande Walle et al., 2014; Choulaki et al., 2015; Liu et al., 2017; Guo et al., 2018; Cheng et al., 2019). However, these studies provide only limited evidence about the analgesic effect of these substances. Similarly, Schnitzler syndrome, a rare autoimmune disease characterized by joint pain and arthritis, was linked to rare NLRP3 gene mutations and increased IL-1β release, and treatment with anakinra caused pain regression in six male patients (Szturz et al., 2014; Pathak et al., 2019). However, while NLRP3 has been implicated in the pathology of these conditions, IL-1β can also be released upon activation of NLRP1, NLRC4, and AIM2 inflammasomes (Martinon et al., 2002; Terkeltaub et al., 2009; Strowig et al., 2012; Liu et al., 2017; Cheng et al., 2019). Thus, the direct contribution of NLRP3 to IL-1β release, and pain, in these conditions remains to be unequivocally demonstrated. Illustrating this point is the observation that burns pain – which is accompanied by severe inflammation, including increased levels of IL-1β (Ono et al., 1995; Vindenes et al., 1998) – developed normally in animals lacking NLRP3 (Nlrp3–/–) or caspase-1/caspase11 (Ice–/–) (Deuis et al., 2017).
Varied outcomes were also reported in studies assessing the contribution of NLRP3 to neuropathic pain, which may be accompanied by significant neuro-inflammation. Increased expression and activation of NLRP3 as well as increased production of IL-1β were reported in several rodent models of nerve injury and chemotherapy-induced neuropathic pain (Jia et al., 2017; Khan et al., 2018; Liu C. C. et al., 2018; Pan et al., 2018; Tonkin et al., 2018; Wahlman et al., 2018; Son et al., 2019). However, while NLRP3 expression and activation was increased after chronic constriction injury of the sciatic nerve and partial sciatic nerve ligation, spared nerve injury did not result in elevated expression of NLRP3 inflammasome components, and pain behaviors of Nlrp3–/– mice were unaltered (Curto-Reyes et al., 2015; Pan et al., 2018; Tonkin et al., 2018). Similarly, investigations of the expression and activation of NLRP3 in models of oxaliplatin-induced neuropathy resulted in seemingly contradictory findings (Tonkin et al., 2018; Wahlman et al., 2018). These studies highlight the need for careful consideration of experimental procedures – including the type of injury, cell types included in analysis and methods used to assess expression and activity of NLRP3 – in preclinical studies addressing the contributions of the NLRP3 inflammasome to painful conditions.
While publications claiming direct or indirect NLRP3-modulating effects by analgesic compounds are plentiful, relatively few studies have assessed the contribution of NLRP3 signaling to pain using highly selective NLRP3 modulators or Nlrp3–/– animals. These approaches have revealed a key contribution of NLRP3 to migraine, with MCC950, a specific NLRP3 antagonist, reducing periorbital and hind paw mechanical hypersensitivity and reversing increased expression of IL-1β in trigeminal ganglia (He et al., 2019). Similarly, treatment with MCC950 attenuated mechanical allodynia in a rodent model of cancer-induced bone pain, and in oxaliplatin-induced peripheral neuropathy model and restored the increased expression levels of NLRP3, ASC, caspase-1 and IL-1β in spinal cord to basal levels (Wahlman et al., 2018; Chen et al., 2019). Interestingly, Cowie et al. showed that the deletion of the NLRP3 gene causes immune-mediated sex differences in pain behavior, as male Nlrp3–/– mice recovered from surgery-induced behavioral and mechanical sensitization faster than female Nlrp3–/– mice (Cowie et al., 2019).
The complex signaling cascades associated with NLRP3 activation lend themselves to pharmacological intervention at multiple points, including direct inhibition of NLRP3 activation, inhibition of inflammasome assembly, inhibition of caspase-1 or gasdermin-D, or inhibition of the biological effects of downstream effectors such as IL-1β. For the latter, a number of approved modulators – including IL-1 receptor antagonists or inhibitory antibodies – are clinically available, as outlined above. However, as IL-1β production is the result of several convergent signaling pathways, inhibition of the biological activities of IL-1β may result in unintended immunosuppressive effects. Accordingly, a number of specific NLRP3 inhibitors with various mechanisms of NLRP3 inhibition have been reported, some of which are currently in clinical development [for review see Zahid et al. (2019); Jiang et al. (2020); Olcum et al. (2020); Zhang et al. (2020))].
The small molecule MCC950 inhibits the canonical and non-canonical activation of the NLRP3 inflammasome (Coll et al., 2015) while the ketone metabolite β-hydroxybutyrate inhibits only canonical activation of the NLRP3 inflammasome via inhibition of potassium efflux (Youm et al., 2015). The type I interferons, including IFN-α and IFN-β, inhibit the NLRP3-dependent production of IL-1β and IL-18 through a yet unknown mechanism (Guarda et al., 2011a) and IFN-β therapy of multiple sclerosis in patients reduces IL-1β levels and NLRP3 protein expression (Malhotra et al., 2015). Additionally, the induction of autophagy by resveratrol, or the inhibition of NLRP3 expression by microRNA-223, inhibits NLRP3 activation and the production of IL-1β (Bauernfeind et al., 2012; Chang et al., 2015).
However, despite overwhelming evidence linking the NLRP3 inflammasome to IL-1β and IL-18 production and inflammation, and convincing evidence that these cytokines can in turn mediate pain, the therapeutic potential of compounds targeting NLRP3 inflammasome signaling pathways remain to be assessed. Certainly, for some painful conditions such as cancer-induced bone pain, chemotherapy- or multiple sclerosis-induced neuropathy, migraine and fibromyalgia, inhibition of NLRP3 signaling appears to be a promising pain treatment strategy. However, only few clinical studies using selective NLRP3 inhibitors are in progress, and additional preclinical studies are required to increase our understanding of the NLRP3-dependent mechanisms contributing to peripheral or central sensitization and pain. Additionally, as activation of the NLRP3 inflammasome regulates immune responses, it is important to consider putative side effect that could arise from NLRP3 inhibition. For example, NLRP3 activation is a first defensive response to bacterial infections, suggesting that NLRP3 inhibitors could be associated with an increased susceptibility to infections. Moreover, inhibition of NLRP3-dependent IL-1β release may affect tumor progression. For example, tumor progression and metastasis of melanoma, gastric and colon cancer were shown to be driven by IL-1β (Krelin et al., 2007; Okamoto et al., 2010; Dunn et al., 2012; Li et al., 2012), whereas the inhibition of IL-1β delayed the regression of the murine B16 melanoma hepatic metastases (Vidal-Vanaclocha et al., 1994). Moreover, the treatment of patients or mice, with metastatic breast cancer, with anakinra, a specific IL-1R inhibitor, significantly reduced the tumor growth (Holen et al., 2016; Wu et al., 2018). Therefore, use of NLRP3 or IL-1β inhibitors for cancer- or chemotherapy-associated pain requires careful investigation and consideration.
In summary, our understanding of the therapeutic potential of NLRP3 inhibition for the treatment of various painful disorders remains limited, despite emerging evidence that these targeted anti-inflammatory approaches may provide benefit. It is likely that analgesic effects will be realized for selected pain types, or subgroups of patients. Undoubtedly, with an increasing number of NLRP3 inhibitors entering clinical trials, significant insights into the therapeutic potential of these treatment approaches will be gained from additional pre-clinical and clinical studies.
NLRP3 inflammasome activation is increasingly recognized as a key molecular event underpinning numerous inflammatory conditions, including those associated with significant pain. However, our understanding of molecular mechanisms leading to activation of the NLRP3 inflammasome, and subsequent effects on sensory systems, remains limited. Accordingly, the involvement of NLRP3 in the development of specific painful conditions, and the positive as well as negative effects of NLRP3 inhibition on human health remain to be determined in future studies.
HS and EN wrote the manuscript. IV edited the manuscript. HS designed figures and tables. All authors contributed to the article and approved the submitted version.
HS was supported by the University of Queensland International Scholarship. IV was supported by a the National Health and Medical Research Council of Australia fellowship (APP1162503) and project grant (APP1186835).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Amaya, F., Wang, H., Costigan, M., Allchorne, A. J., Hatcher, J. P., Egerton, J., et al. (2006). The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 26, 12852–12860. doi: 10.1523/jneurosci.4015-06.2006
Barclay, J., Clark, A. K., Ganju, P., Gentry, C., Patel, S., Wotherspoon, G., et al. (2007). Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain 130, 225–234. doi: 10.1016/j.pain.2006.11.017
Bauernfeind, F., Bartok, E., Rieger, A., Franchi, L., Nunez, G., and Hornung, V. (2011). Cutting edge: reactive oxygen species inhibitors block priming., but not activation., of the NLRP3 inflammasome. J. Immunol. 187, 613–617. doi: 10.4049/jimmunol.1100613
Bauernfeind, F., Rieger, A., Schildberg, F. A., Knolle, P. A., Schmid-Burgk, J. L., and Hornung, V. (2012). NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 189, 4175–4181. doi: 10.4049/jimmunol.1201516
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791. doi: 10.4049/jimmunol.0901363
Binshtok, A. M., Wang, H., Zimmermann, K., Amaya, F., Vardeh, D., Shi, L., et al. (2008). Nociceptors are interleukin-1beta sensors. J. Neurosci. 28, 14062–14073.
Brough, D., Le Feuvre, R. A., Wheeler, R. D., Solovyova, N., Hilfiker, S., Rothwell, N. J., et al. (2003). Ca2+ stores and Ca2+ entry differentially contribute to the release of IL-1 beta and IL-1 alpha from murine macrophages. J. Immunol. 170, 3029–3036. doi: 10.4049/jimmunol.170.6.3029
Butler, D. S. C., Ambite, I., Nagy, K., Cafaro, C., Ahmed, A., Nadeem, A., et al. (2018). Neuroepithelial control of mucosal inflammation in acute cystitis. Sci. Rep. 8:11015.
Chang, Y. P., Ka, S. M., Hsu, W. H., Chen, A., Chao, L. K., Lin, C. C., et al. (2015). Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J. Cell Physiol. 230, 1567–1579. doi: 10.1002/jcp.24903
Chen, S. P., Zhou, Y. Q., Wang, X. M., Sun, J., Cao, F., HaiSam, S., et al. (2019). Pharmacological inhibition of the NLRP3 in fl ammasome as a potential target for cancer-induced bone pain. Pharmacol. Res. 147:104339. doi: 10.1016/j.phrs.2019.104339
Cheng, F., Yan, F. F., Liu, Y. P., Cong, Y., Sun, K. F., and He, X. M. (2019). Dexmedetomidine inhibits the NF-kappaB pathway and NLRP3 inflammasome to attenuate papain-induced osteoarthritis in rats. Pharm. Biol. 57, 649–659. doi: 10.1080/13880209.2019.1651874
Choulaki, C., Papadaki, G., Repa, A., Kampouraki, E., Kambas, K., Ritis, K., et al. (2015). Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res. Ther. 17:257.
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Munoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255.
Cordero, M. D., Alcocer-Gomez, E., Culic, O., Carrion, A. M., de Miguel, M., Diaz-Parrado, E., et al. (2014). NLRP3 inflammasome is activated in fibromyalgia: the effect of coenzyme Q10. Antioxid Redox. Signal. 20, 1169–1180. doi: 10.1089/ars.2013.5198
Cowie, A. M., Menzel, A. D., O’Hara, C., Lawlor, M. W., and Stucky, C. L. (2019). NOD-like receptor protein 3 inflammasome drives postoperative mechanical pain in a sex-dependent manner. Pain 160, 1794–1816. doi: 10.1097/j.pain.0000000000001555
Curto-Reyes, V., Kirschmann, G., Pertin, M., Drexler, S. K., Decosterd, I., and Suter, M. R. (2015). Neuropathic pain phenotype does not involve the NLRP3 inflammasome and its end product interleukin-1beta in the mice spared nerve injury model. PLoS One 10:e0133707. doi: 10.1371/journal.pone.0133707
Dai, J., Zhang, X., Wang, Y., Chen, H., and Chai, Y. (2017). ROS-activated NLRP3 inflammasome initiates inflammation in delayed wound healing in diabetic rats. Int. J. Clin. Exp. Pathol. 10:9902.
Deuis, J. R., Yin, K., Cooper, M. A., Schroder, K., and Vetter, I. (2017). Role of the NLRP3 inflammasome in a model of acute burn-induced pain. Burns 43, 304–309. doi: 10.1016/j.burns.2016.09.001
Dunn, J. H., Ellis, L. Z., and Fujita, M. (2012). Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett. 314, 24–33. doi: 10.1016/j.canlet.2011.10.001
Fann, D. Y., Lim, Y. A., Cheng, Y. L., Lok, K. Z., Chunduri, P., Baik, S. H., et al. (2018). Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 55, 1082–1096. doi: 10.1007/s12035-017-0394-9
Ferreira, S., Lorenzetti, B., Bristow, A., and Poole, S. J. N. (1988). Interleukin-1β as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature 334:698. doi: 10.1038/334698a0
Franchi, L., Eigenbrod, T., and Nunez, G. (2009a). Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 183, 792–796. doi: 10.4049/jimmunol.0900173
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009b). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128. doi: 10.1111/j.1600-065x.2008.00734.x
Gaidt, M. M., Ebert, T. S., Chauhan, D., Schmidt, T., Schmid-Burgk, J. L., Rapino, F., et al. (2016). Human monocytes engage an alternative inflammasome pathway. Immunity 44, 833–846. doi: 10.1016/j.immuni.2016.01.012
Gerard, S., le Goff, B., Maugars, Y., Berthelot, J. M., and Malard, O. (2007). Lasting remission of a Muckle-Wells syndrome with CIAS-1 mutation using half-dose anakinra. Joint Bone Spine 74:659. doi: 10.1016/j.jbspin.2007.01.032
Guarda, G., Braun, M., Staehli, F., Tardivel, A., Mattmann, C., Forster, I., et al. (2011a). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. doi: 10.1016/j.immuni.2011.02.006
Guarda, G., Zenger, M., Yazdi, A. S., Schroder, K., Ferrero, I., Menu, P., et al. (2011b). Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 186, 2529–2534. doi: 10.4049/jimmunol.1002720
Gui, W. S., Wei, X., Mai, C. L., Murugan, M., Wu, L. J., Xin, W. J., et al. (2016). Interleukin-1beta overproduction is a common cause for neuropathic pain., memory deficit., and depression following peripheral nerve injury in rodents. Mol Pain 12:1744806916646784.
Guo, C., Fu, R., Wang, S., Huang, Y., Li, X., Zhou, M., et al. (2018). NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 194, 231–243. doi: 10.1111/cei.13167
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10:e0130624. doi: 10.1371/journal.pone.0130624
He, W., Long, T., Pan, Q., Zhang, S., Zhang, Y., Zhang, D., et al. (2019). Microglial NLRP3 inflammasome activation mediates IL-1beta release and contributes to central sensitization in a recurrent nitroglycerin-induced migraine model. J. Neuroinflamm. 16:78.
Hoffman, H. M., Throne, M. L., Amar, N. J., Sebai, M., Kivitz, A. J., Kavanaugh, A., et al. (2008). Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 58, 2443–2452. doi: 10.1002/art.23687
Holen, I., Lefley, D. V., Francis, S. E., Rennicks, S., Bradbury, S., Coleman, R. E., et al. (2016). IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 7, 75571–75584. doi: 10.18632/oncotarget.12289
Hu, Z., Yan, C., Liu, P., Huang, Z., Ma, R., Zhang, C., et al. (2013). Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175. doi: 10.1126/science.1236381
Inoue, K., and Tsuda, M. (2018). Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 19, 138–152. doi: 10.1038/nrn.2018.2
Insalaco, A., Prencipe, G., Buonuomo, P. S., Ceccherini, I., Bracaglia, C., Pardeo, M., et al. (2014). A novel mutation in the CIAS1/NLRP3 gene associated with an unexpected phenotype of cryopyrin-associated periodic syndromes. Clin. Exp. Rheumatol. 32, 123–125.
Ji, R. R., Chamessian, A., and Zhang, Y. Q. (2016). Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. doi: 10.1126/science.aaf8924
Jia, M., Wu, C., Gao, F., Xiang, H., Sun, N., Peng, P., et al. (2017). Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol. Pain 13:1744806917719804.
Jiang, H., Gong, T., and Zhou, R. (2020). The strategies of targeting the NLRP3 inflammasome to treat inflammatory diseases. Adv. Immunol. 145, 55–93. doi: 10.1016/bs.ai.2019.11.003
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121.
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. doi: 10.1126/science.1240248
Khan, N., Kuo, A., Brockman, D. A., Cooper, M. A., and Smith, M. T. (2018). Pharmacological inhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology 26, 77–86. doi: 10.1007/s10787-017-0401-9
Kobayashi, Y., Kiguchi, N., Fukazawa, Y., Saika, F., Maeda, T., and Kishioka, S. (2015). Macrophage-T cell interactions mediate neuropathic pain through the glucocorticoid-induced tumor necrosis factor ligand system. J. Biol. Chem. 290, 12603–12613. doi: 10.1074/jbc.m115.636506
Krelin, Y., Voronov, E., Dotan, S., Elkabets, M., Reich, E., Fogel, M., et al. (2007). Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 67, 1062–1071. doi: 10.1158/0008-5472.can-06-2956
Lewis, A. M., Varghese, S., Xu, H., and Alexander, H. R. (2006). Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 4:48.
Li, Y., Wang, L., Pappan, L., Galliher-Beckley, A., and Shi, J. (2012). IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 11:87. doi: 10.1186/1476-4598-11-87
Liu, C. C., Huang, Z. X., Li, X., Shen, K. F., Liu, M., Ouyang, H. D., et al. (2018). Upregulation of NLRP3 via STAT3-dependent histone acetylation contributes to painful neuropathy induced by bortezomib. Exp. Neurol. 302, 104–111. doi: 10.1016/j.expneurol.2018.01.011
Liu, H. J., Pan, X. X., Liu, B. Q., Gui, X., Hu, L., Jiang, C. Y., et al. (2017). Grape seed-derived procyanidins alleviate gout pain via NLRP3 inflammasome suppression. J. Neuroinflamm. 14:74.
Liu, S., Liu, Y. P., Lv, Y., Yao, J. L., Yue, D. M., Zhang, M. Y., et al. (2018). IL-18 contributes to bone cancer pain by regulating glia cells and neuron interaction. J. Pain 19, 186–195. doi: 10.1016/j.jpain.2017.10.003
Lo Gullo, A., Mandraffino, G., Imbalzano, E., Mamone, F., Aragona, C. O., D’Ascola, A., et al. (2014). Toll-like receptor 3 and interleukin 1beta expression in CD34+ cells from patients with rheumatoid arthritis: association with inflammation and vascular involvement. Clin. Exp. Rheumatol. 32, 922–929.
Lu, A., Li, Y., Schmidt, F. I., Yin, Q., Chen, S., Fu, T. M., et al. (2016). Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 23, 416–425. doi: 10.1038/nsmb.3199
Lu, A., Li, Y., Yin, Q., Ruan, J., Yu, X., Egelman, E., et al. (2015). Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 1:15013.
Lu, A., Magupalli, V. G., Ruan, J., Yin, Q., Atianand, M. K., Vos, M. R., et al. (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. doi: 10.1016/j.cell.2014.02.008
Maksimovic, L., Stirnemann, J., Caux, F., Ravet, N., Rouaghe, S., Cuisset, L., et al. (2008). New CIAS1 mutation and anakinra efficacy in overlapping of Muckle-Wells and familial cold autoinflammatory syndromes. Rheumatology (Oxford) 47, 309–310. doi: 10.1093/rheumatology/kem318
Malhotra, S., Rio, J., Urcelay, E., Nurtdinov, R., Bustamante, M. F., Fernandez, O., et al. (2015). NLRP3 inflammasome is associated with the response to IFN-beta in patients with multiple sclerosis. Brain 138, 644–652. doi: 10.1093/brain/awu388
Malsch, P., Andratsch, M., Vogl, C., Link, A. S., Alzheimer, C., Brierley, S. M., et al. (2014). Deletion of interleukin-6 signal transducer gp130 in small sensory neurons attenuates mechanonociception and down-regulates TRPA1 expression. J. Neurosci. 34, 9845–9856. doi: 10.1523/jneurosci.5161-13.2014
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241.
McMahon, S. B., La Russa, F., and Bennett, D. L. (2015). Crosstalk between the nociceptive and immune systems in host defence and disease. Nat. Rev. Neurosci. 16, 389–402.
Mert, T., Gunay, I., Ocal, I., Guzel, A. I., Inal, T. C., Sencar, L., et al. (2009). Macrophage depletion delays progression of neuropathic pain in diabetic animals. Naunyn. Schmiedebergs Arch. Pharmacol. 379, 445–452.
Mrak, R. E., and Griffin, W. S. (2000). Interleukin-1 and the immunogenetics of Alzheimer disease. J. Neuropathol. Exp. Neurol. 59, 471–476.
Muller, P. A., Koscso, B., Rajani, G. M., Stevanovic, K., Berres, M. L., Hashimoto, D., et al. (2014). Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 158, 300–313.
Munoz-Planillo, R., Kuffa, P., Martinez-Colon, G., Smith, B. L., Rajendiran, T. M., and Nunez, G. (2013). K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153.
Okamoto, M., Liu, W., Luo, Y., Tanaka, A., Cai, X., Norris, D. A., et al. (2010). Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 285, 6477–6488.
Olcum, M., Tastan, B., Ercan, I., Eltutan, I. B., and Genc, S. (2020). Inhibitory effects of phytochemicals on NLRP3 inflammasome activation: a review. Phytomedicine 75:153238.
Ono, I., Gunji, H., Zhang, J. Z., Maruyama, K., and Kaneko, F. (1995). A study of cytokines in burn blister fluid related to wound healing. Burns 21, 352–355.
Orlowski, G. M., Colbert, J. D., Sharma, S., Bogyo, M., Robertson, S. A., and Rock, K. L. (2015). Multiple cathepsins promote Pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J. Immunol. 195, 1685–1697.
Pan, Z., Shan, Q., Gu, P., Wang, X. M., Tai, L. W., Sun, M., et al. (2018). miRNA-23a/CXCR4 regulates neuropathic pain via directly targeting TXNIP/NLRP3 inflammasome axis. J. Neuroinflamm. 15:29.
Pathak, S., Rowczenio, D. M., Owen, R. G., Doody, G. M., Newton, D. J., Taylor, C., et al. (2019). Exploratory study of MYD88 L265P., rare NLRP3 variants, and clonal hematopoiesis prevalence in patients with schnitzler syndrome. Arthritis Rheumatol. 71, 2121–2125.
Reinold, H., Ahmadi, S., Depner, U. B., Layh, B., Heindl, C., Hamza, M., et al. (2005). Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. J. Clin. Invest. 115, 673–679.
Ren, K., and Torres, R. (2009). Role of interleukin-1beta during pain and inflammation. Brain Res. Rev. 60, 57–64.
Ruscitti, P., Cipriani, P., Di Benedetto, P., Liakouli, V., Berardicurti, O., Carubbi, F., et al. (2015). Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1beta via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: a possible implication for therapeutic decision in these patients. Clin. Exp. Immunol. 182, 35–44.
Safieh-Garabedian, B., Poole, S., Allchorne, A., Winter, J., and Woolf, C. J. (1995). Contribution of interleukin-1 beta to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br. J. Pharmacol. 115, 1265–1275.
Sagulenko, V., Thygesen, S. J., Sester, D. P., Idris, A., Cridland, J. A., Vajjhala, P. R., et al. (2013). AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160.
Schafers, M., and Sorkin, L. (2008). Effect of cytokines on neuronal excitability. Neurosci. Lett. 437, 188–193.
Schlesinger, N., Mysler, E., Lin, H. Y., De Meulemeester, M., Rovensky, J., Arulmani, U., et al. (2011). Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind., randomised study. Ann. Rheum. Dis. 70, 1264–1271.
Sharma, B. R., Karki, R., and Kanneganti, T. D. (2019). Role of AIM2 inflammasome in inflammatory diseases., cancer and infection. Eur. J. Immunol. 49, 1998–2011.
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665.
Son, S., Shim, D. W., Hwang, I., Park, J. H., and Yu, J. W. (2019). Chemotherapeutic agent paclitaxel mediates priming of NLRP3 inflammasome activation. Front. Immunol. 10:1108. doi: 10.3389/fimmu.2019.01108
Stemkowski, P. L., Noh, M. C., Chen, Y., and Smith, P. A. (2015). Increased excitability of medium-sized dorsal root ganglion neurons by prolonged interleukin-1beta exposure is K(+) channel dependent and reversible. J. Physiol. 593, 3739–3755.
Strowig, T., Henao-Mejia, J., Elinav, E., and Flavell, R. (2012). Inflammasomes in health and disease. Nature 481, 278–286.
Szturz, P., Sediva, A., Zurek, M., Adam, Z., Stork, J., Cermakova, Z., et al. (2014). Anakinra treatment in Schnitzler syndrome - results of the first retrospective multicenter study in six patients from the Czech Republic. Klin Onkol. 27, 111–126.
Terkeltaub, R., Sundy, J. S., Schumacher, H. R., Murphy, F., Bookbinder, S., Biedermann, S., et al. (2009). The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled., monosequence crossover., non-randomised., single-blind pilot study. Ann. Rheum. Dis. 68, 1613–1617.
Tonkin, R. S., Bowles, C., Perera, C. J., Keating, B. A., Makker, P. G. S., Duffy, S. S., et al. (2018). Attenuation of mechanical pain hypersensitivity by treatment with Peptide5., a connexin-43 mimetic peptide., involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 300, 1–12.
Vande Walle, L., Van Opdenbosch, N., Jacques, P., Fossoul, A., Verheugen, E., Vogel, P., et al. (2014). Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73.
Vasudeva, K., Vodovotz, Y., Azhar, N., Barclay, D., Janjic, J. M., and Pollock, J. A. (2015). In vivo and systems biology studies implicate IL-18 as a central mediator in chronic pain. J. Neuroimmunol. 283, 43–49.
Verma, D., Eriksson, P., Sahdo, B., Persson, A., Ejdeback, M., Sarndahl, E., et al. (2010). Two adult siblings with atypical cryopyrin-associated periodic syndrome due to a novel M299V mutation in NLRP3. Arthritis Rheum. 62, 2138–2143.
Vidal-Vanaclocha, F., Amezaga, C., Asumendi, A., Kaplanski, G., and Dinarello, C. A. (1994). Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res. 54, 2667–2672.
Vindenes, H. A., Ulvestad, E., and Bjerknes, R. (1998). Concentrations of cytokines in plasma of patients with large burns: their relation to time after injury., burn size., inflammatory variables., infection., and outcome. Eur. J. Surg. 164, 647–656.
Wahlman, C., Doyle, T. M., Little, J. W., Luongo, L., Janes, K., Chen, Z., et al. (2018). Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 159, 1025–1034.
Wu, T. C., Xu, K., Martinek, J., Young, R. R., Banchereau, R., George, J., et al. (2018). IL1 receptor antagonist controls transcriptional signature of inflammation in patients with metastatic breast cancer. Cancer Res. 78, 5243–5258.
Yang, D., He, Y., Munoz-Planillo, R., Liu, Q., and Nunez, G. (2015). Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43, 923–932.
Yoshida, S., Hagiwara, Y., Tsuchiya, M., Shinoda, M., Koide, M., Hatakeyama, H., et al. (2018). Involvement of neutrophils and interleukin-18 in nociception in a mouse model of muscle pain. Mol. Pain 14:1744806918757286.
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269.
Zahid, A., Li, B., Kombe, A. J. K., Jin, T., and Tao, J. (2019). Pharmacological inhibitors of the NLRP3 inflammasome. Front. Immunol. 10:2538. doi: 10.3389/fimmu.2019.02538
Zanos, T. P., Silverman, H. A., Levy, T., Tsaava, T., Battinelli, E., Lorraine, P. W., et al. (2018). Identification of cytokine-specific sensory neural signals by decoding murine vagus nerve activity. Proc. Natl. Acad. Sci. U.S.A. 115, E4843–E4852.
Zhang, H., Li, Y., de Carvalho-Barbosa, M., Kavelaars, A., Heijnen, C. J., Albrecht, P. J., et al. (2016). Dorsal Root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J. Pain 17, 775–786.
Zhang, X., Dai, J., Li, L., Chen, H., and Chai, Y. (2017). NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J. Diab. Res. 2017:5281358.
Zhang, X., Xu, A., Lv, J., Zhang, Q., Ran, Y., Wei, C., et al. (2020). Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur. J. Med. Chem. 185:111822.
Keywords: NOD, LRR and PYD domains-containing protein 3, neuro-inflammation, MCC950, interleukin-1β, sensory neurons, inflammatory diseases, interleukin-1 receptor
Citation: Starobova H, Nadar EI and Vetter I (2020) The NLRP3 Inflammasome: Role and Therapeutic Potential in Pain Treatment. Front. Physiol. 11:1016. doi: 10.3389/fphys.2020.01016
Received: 17 March 2020; Accepted: 24 July 2020;
Published: 19 August 2020.
Edited by:
Istvan Nagy, Imperial College London, United KingdomReviewed by:
Arpeeta Sharma, Baker Heart and Diabetes Institute, AustraliaCopyright © 2020 Starobova, Nadar and Vetter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irina Vetter, aS52ZXR0ZXJAaW1iLnVxLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.