 Haili Tian
Haili Tian Sujuan Liu2†
Sujuan Liu2† Jun Ren
Jun Ren Jason Kai Wei Lee
Jason Kai Wei Lee Peijie Chen
Peijie Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 11 August 2020
Sec. Integrative Physiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00949
This article is part of the Research TopicNew Drug Targets for Proteotoxicity in Cardiometabolic DiseasesView all 24 articles
Skeletal muscle is the largest metabolic organ in the human body and is able to rapidly adapt to drastic changes during exercise. Histone acetyltransferases (HATs) and histone deacetylases (HDACs), which target histone and non-histone proteins, are two major enzyme families that control the biological process of histone acetylation and deacetylation. Balance between these two enzymes serves as an essential element for gene expression and metabolic and physiological function. Genetic KO/TG murine models reveal that HDACs possess pivotal roles in maintaining skeletal muscles’ metabolic homeostasis, regulating skeletal muscles motor adaptation and exercise capacity. HDACs may be involved in mitochondrial remodeling, insulin sensitivity regulation, turn on/off of metabolic fuel switching and orchestrating physiological homeostasis of skeletal muscles from the process of myogenesis. Moreover, many myogenic factors and metabolic factors are modulated by HDACs. HDACs are considered as therapeutic targets in clinical research for treatment of cancer, inflammation, and neurological and metabolic-related diseases. This review will focus on physiological function of HDACs in skeletal muscles and provide new ideas for the treatment of metabolic diseases.
Skeletal muscle is the largest metabolic organ in the human body, consuming about 18% of the entire body daily expenditure of energy (Durnin, 1981). It produces various secrete factors and participates in the interplay among multiple tissues and organs (Pedersen and Febbraio, 2012; Mizgier et al., 2019). Muscular contraction is one of the main physiological functions of skeletal muscles and plays a role in maintaining organ and systemic metabolic homeostasis (Guo et al., 2020). Proper exercises help build up body defense to combat various diseases including obesity, type 2 diabetes, Alzheimer disease, osteoarthritis, and so on (Luan et al., 2019). These important functions require delicate regulations in muscle from the level of genome to signal transduction, establishing muscle plasticity, and responses to environmental stress. Acetyl-CoA as a central metabolite of amino acid/fatty acid/glucose not only serves as fuel for energy expenditure but also bridges the gap between environmental stress and organismal response (Lempradl et al., 2015; Liu et al., 2020). This control occurs through post-translational modification on nucleosomes histone lysine residues and other signal transducing proteins, which in turn controls accessibility of DNA to regulatory factors and protein activity. Acetylation, one of the most prevalent modifications of protein, is believed to function as a key modulator of chromatin structure and signal transduction, and provides an avenue to couple extracellular stimuli with genome during muscle metabolism process by regulating acetylation and deacetylation (Haberland et al., 2009).
histone deacetylases (HDACs) as a family of protein deacetylases have been demonstrated to moderate physiological homeostasis and development by deacetylation (Haberland et al., 2009) (Table 1). In humans, there are 18 types of HDACs, which are classified into four categories based on homologous proteins in yeast, namely: Class I Rpd3-like protein (HDAC1, -2, -3, -8); Class II Hda1-like proteins are classified as Class IIa (HDAC4, -5, -7, -9) and Class IIb (HDAC6, -10); Class III Sir2p-like, nicotinamide adenine dinucleotide (NAD+)-dependent (SIRT1, -2, -3, -4, -5, -6, -7); and Class IV HDAC11, containing homologous domain with both Rpd3 and Hda1 (Seto and Yoshida, 2014). HDACs modulate gene expression and protein activity through deacetylating proteins. When histone lysine ε-amino acid in the nucleosome is acetylated, it can neutralize positive charge, before loosening chromatin structure, to promote binding of transcription factors to DNA and expression of downstream target genes. By contrast, histone deacetylation compresses chromatin structure, thereby inhibiting transcriptional gene expression (Shahbazian and Grunstein, 2007).
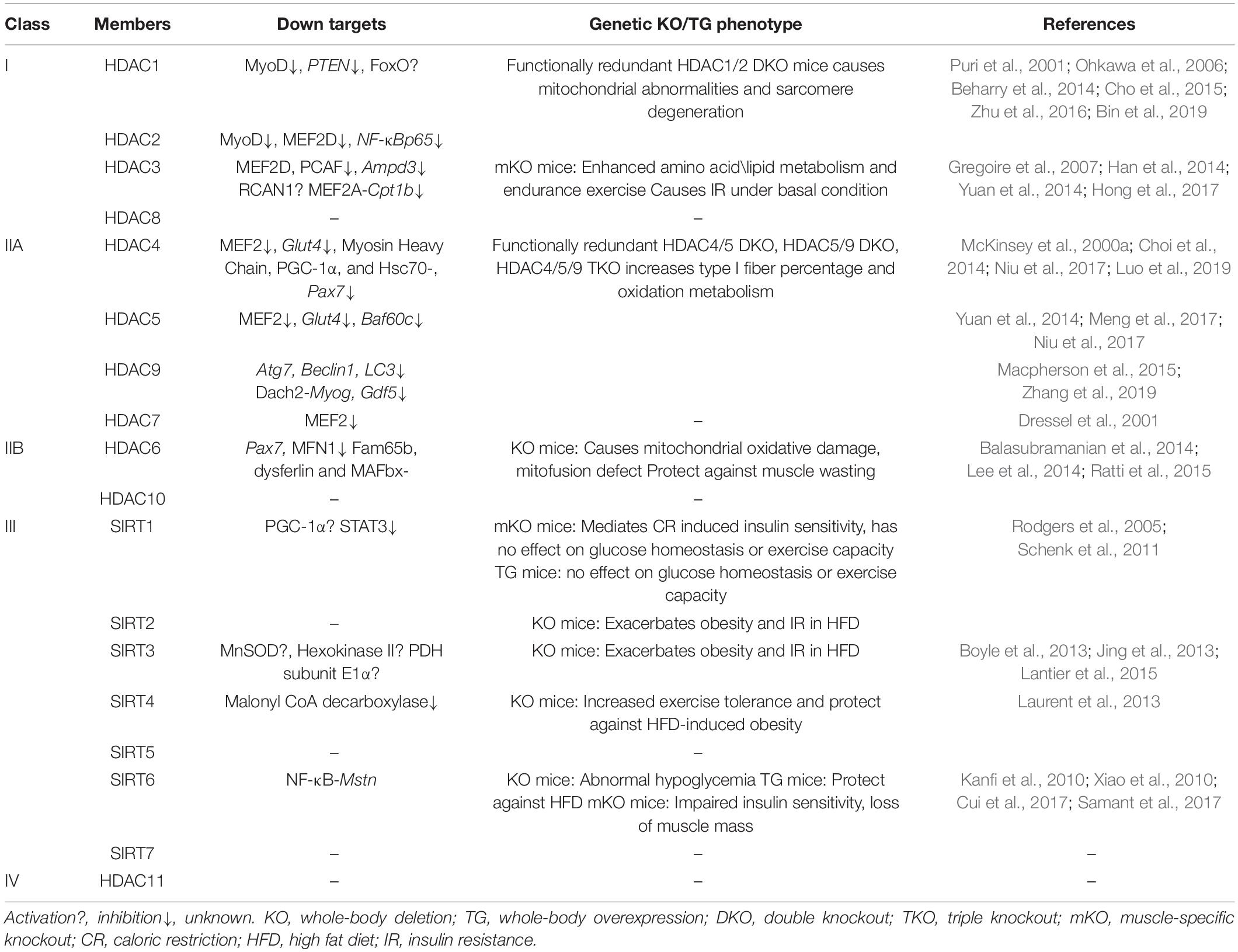
Table 1. The targets of HADCs in skeletal muscles and their physiological functions.
Biochemical properties of HDACs have been comprehensively reviewed (Shahbazian and Grunstein, 2007). However, the physiological functions of HDACs have yet to be well examined in skeletal muscles. A series of researches shed light on the potential of drugs targeting HDACs to improve muscle fitness and cardiac muscle disease, which needs further clarification of HDACs physiological function to understand the mechanisms (Bai et al., 2011; Galmozzi et al., 2013; Xie et al., 2014; Zhang and Ren, 2014; Gaur et al., 2016). Ample work has revealed the role of HDAC in muscle physiology such as skeletal muscles me olism and thus exercise capacity (McGee and Hargreaves, 2010) (Table 1). Class I HDACs can interact with myocyte enhancer factor 2 (MEF2), MyoD, regulating myogenesis, and exercise capacity (Mal et al., 2001; Puri et al., 2001; Ohkawa et al., 2006; Gregoire et al., 2007). HDAC3 participates in myocytes differentiation and is linked to skeletal muscles metabolic fuel switching (Gregoire et al., 2007; Hong et al., 2017). Class II HDACs can be phosphorylated by HDAC kinases and are transduced from nucleus to cytoplasm in response to cellular stress, mediating muscle fiber switch and affecting the pathway of insulin sensitivity such as Glut4 and AKT (Haberland et al., 2009; McGee and Hargreaves, 2010). Class III HDACs target a crucial mitochondrial biogenesis factor peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) in skeletal muscles, liver and fat tissue (White and Schenk, 2012). Therefore, we will further discuss how these HDACs influence respective downstream targets, exercise capacity, and therapeutic effects in human diseases.
In previous studies, HDAC1/2/3 was shown to be associated with the process of myogenesis or myocyte differentiation (Mal et al., 2001; Puri et al., 2001; Ohkawa et al., 2006; Gregoire et al., 2007). HDAC1 tightly binds to MyoD and deacetylates specific sites to inhibit the expression of muscle-specific genes such as MHC and MCK in myoblasts. Mimicking muscle differentiation condition by serum deprivation, HDAC1 protein gradually decreases during myocyte differentiation and transfers to bind to the tumor suppressor pRb, accompanied by isolation of HDAC1-MyoD and transcriptional activation of muscle-specific genes (Puri et al., 2001). A HDAC1 (H141A) mutant incapable of binding with MyoD loses its inhibitory property on muscle-specific genes in myoblast state (Mal et al., 2001). In the late stage of myocyte differentiation, activating factor gradually switches from MyoD to myogenin, occurring with a decrease in the MEF2D inhibitory regulator HDAC2. Simultaneous overexpression of myogenin and MEF2D can enhance the expression of the muscle-specific gene MHC in the absence of MyoD (Ohkawa et al., 2006). MEF2D, a key factor that controls myocyte differentiation, can only be effectively deacetylated by HDAC3, rather than HDAC1/2/8 (Gregoire et al., 2007). In addition, HDAC3 can inhibit autoacetylation of acetyltransferases p300 and p300/CBP-associated factor (PCAF). Thus, HDAC3 impedes MyoD-MEF2-PCAF to form a multicomplex, disturbing MEF2-dependent myogenic transcription (Gregoire et al., 2007; Figure 1).
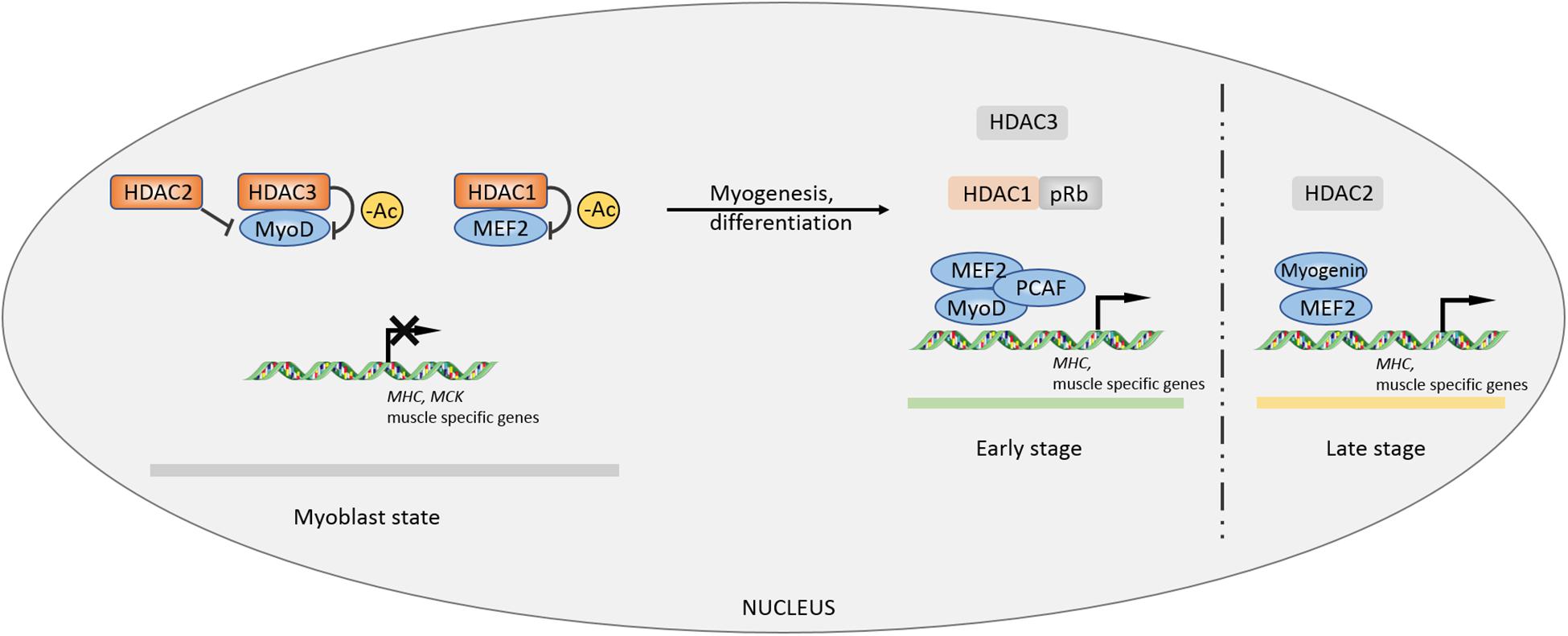
Figure 1. Controls of myogenesis by Class I HDACs. The inhibitory effects of HDACs on myogenic factors MEF2 and MyoD maintain a primary myoblast state. During myocyte differentiation and myogenic process, the inhibition is lifted by other factors, facilitating MEF2-MyoD complex formation to promote myogenesis. –Ac, deacetylation.
Loss of function of HDAC1/2 inhibits autophagy flux in skeletal muscles tissue, accompanied by decreased LC3 II/I ratio and p62 accumulation under fasting condition (Moresi et al., 2012). Toxic autophagy intermediates accumulate in muscle fibers in which class I HDACs deficiency led to impaired exercise capacity (Moresi et al., 2012). This offers convincing evidence on the role of HDAC1/2 in stabilizing basic structure and facilitating development in muscles. Genetic models suggest that Class I HDACs control the physiological homeostasis of skeletal muscles. A germline deletion shows that whole-body knockout of either HDAC1 or HDAC2 leads to mortality in mice prior to the perinatal period (Montgomery et al., 2007). However, a tissue-specific single deletion of HDAC1 or HDAC2 in myocardium does not cause any phenotype (Montgomery et al., 2007). Double knockout HDAC1/2 in the heart leads to severe cardiomyopathy in mice, indicating the obligatory role for HDAC1/2 in specific tissues (Montgomery et al., 2007). In skeletal muscle, double knockout of HDAC1/2 by myogenin-Cre causes mitochondrial abnormalities and sarcomere degeneration, disrupting fundamental structural units of myofibers (Moresi et al., 2012). Therefore, these findings demonstrate that HDAC1/2 redundantly maintains sarcomere homeostasis in skeletal muscle.
Histone deacetylases 3 is the enzymatic core of nuclear receptor corepressor (N-CoR) and silencing mediator of retinoic acid and thyroid hormone receptors (SMRT) co-repressor; correspondingly, N-CoR and SMRT co-repressor activate HDAC3 through its SANT domain enzyme activity (Karagianni and Wong, 2007). Mice suffer from insulin resistance after specifically knocking out HDAC3 in skeletal muscles-mKO, while their endurance exercise abilities are enhanced (Hong et al., 2017). It seems a self-contradictory phenomenon because former studies pointed out that increased endurance exercise capacity enhances insulin sensitivity. The study shows that insulin signaling cascades including pAKT/AKT, pIRS1-S1101, and pGSK/GSK have no change in HDAC3 mKO muscle, while glucose uptake and insulin sensitivity are impaired in glucose tolerance test (GTT) and insulin tolerance test (ITT) (Hong et al., 2017), which is called the “dissociation effect” by researchers. Lipid tends to be used as energy fuel in HDAC mKO mice, which inhibits glucose absorption but does not influence insulin signaling sensitivity (Hong et al., 2017). Further work found that the HDAC3 knockout upregulates the expression of AMP deaminase 3 (AMPD3), the first rate-limiting enzyme in purine metabolism, in skeletal muscles (Fortuin et al., 1996; Hong et al., 2017). AMPD3 can deaminate AMP to form IMP, facilitating aspartic acid to transmit into fumarate and malate, the intermediate metabolites of tricarboxylic acid cycle (TCA cycle) (Hong et al., 2017). Research studies found that exercise induced glucose labeled 13C6 expressed a lower glycolysis flux rate in muscles of HDAC3 mKO but a higher expression in the TCA cycle intermediates (Hong et al., 2017; Gong et al., 2018). Thus, a general increase in TCA cycle metabolites activates the oxidation in mitochondria. In vitro radioactive aspartic acid isotope tracer test showed that inhibiting HDAC3 or overexpressing AMPD3 can increase amino acid metabolism rate and enhance fatty acid metabolism, thereby downregulating glucose metabolism (Hong et al., 2017Figure 2). In liver, knockout HDAC3 causes severe liver steatosis and can be rescued by wild-type or catalytically inactive mutants of HDAC3, indicating an enzymatic activity independent function (Sun et al., 2013). However, the muscle fuel switching in HDAC3 mKO mice cannot be rescued by an enzymatic inactivity mutant HDAC3 (Song et al., 2019). This finding demonstrates that HDAC3 controls the fuel utilization in skeletal muscles and is dependent on its enzymatic activity. The relationship between exercise and glucose uptake is far more complex than one could expect.
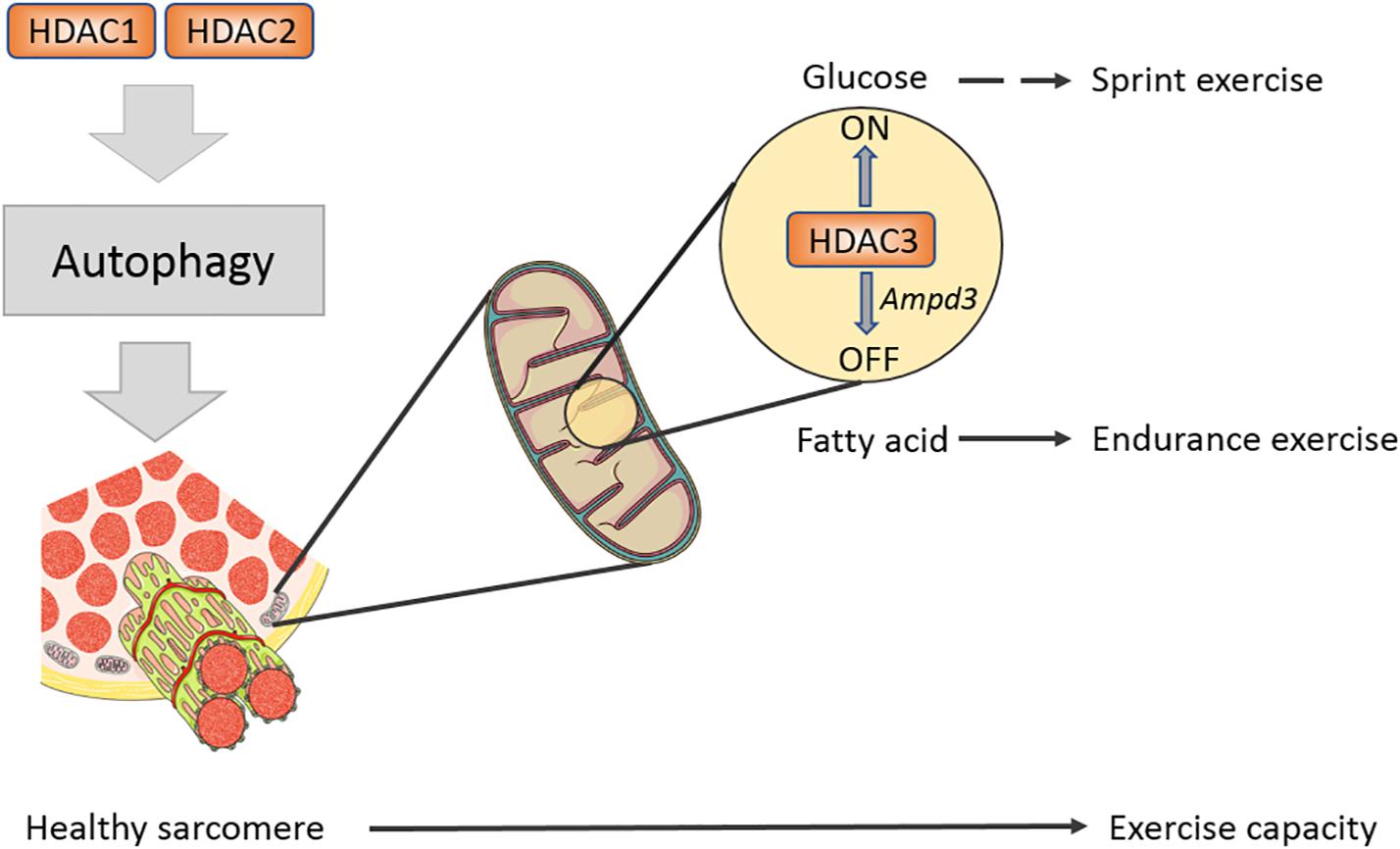
Figure 2. Class I HDACs regulates sarcomere structure and exercise capacity in mature muscle. A schematic diagram shows Class I HDACs regulate exercise capacity on different levels. HDAC1/2 have redundant roles in modulating autophagy flux in muscle, eliminating toxic factors in muscle. HDAC3 serves as a fuel switch and promotes muscle to use fatty acid to adapt endurance exercise.
The MEF2 family is a class of transcription factors that are closely associated with myogenic basic helix-loop-helix domains, including MyoD (Taylor and Hughes, 2017). MEF2 may interact with Class IIa histone deacetylase HDAC4/5 to inhibit the transcription of MEF2-dependent genes, thereby impeding the differentiation from myoblast to myotube (McKinsey et al., 2000a). During exercise, Ca2+ flows out from sarcoplasmic reticulum and activate calcium/calmodulin-dependent protein kinase (CaMK), which phosphorylates HDAC4/5 and mediates HDAC4/5 shuttle from the nucleus to the cytoplasm, thus releasing the repressive effect of HDAC4/5 on MEF2-dependent genes (McKinsey et al., 2000b; Yuan et al., 2014). Further research demonstrates that the nucleoplasmic shuttle of HDAC4 relies on 14-3-3 binding, while the shuttle of HDAC5 depends on CaMK phosphorylation at serine -259 and -498 sites first, and then exporting from the nucleus by binding with 14-3-3 (McKinsey et al., 2000b). As expected, HDAC7 in Class IIa also possesses the ability to be exported from nucleus to cytoplasm (Gao et al., 2010), suggesting that there may be redundant functions of Class IIa HDACs. Except for Class IIa, knocking out Class IIb HDAC6 in embryonic stem cells (ESCs) can promote ESCs to differentiate into myoblast cells, and transplantation of ESCs with HDAC6 knockdown can also help the regeneration of wound skeletal muscles (Lee et al., 2015).
Evidence shows that the proportion of type I fiber in skeletal muscles has no change in animal knocked out alone any member of Class IIa (Potthoff et al., 2007). However, the proportion of type I fiber is significantly increased in HDAC4/5 DKO, HDAC5/9 DKO, HDAC4/5/9 TKO mice as well as exercise capacity of skeletal muscles (Potthoff et al., 2007). Altogether, the finding indicated a functional redundant mechanism in Class II HDACs family. Previous study found that Class IIa HDACs are closely associated with the MEF2 family and can repress their downstream target genes (McKinsey et al., 2000a). Therefore, similar to Class IIa HDACs DKO, the proportion of type I fiber is decreased in the MEF2C and MEF2D knockout mouse models, overexpressing MEF2C results in increasing type I fiber and exercise capacity. HDAC4/5 not only regulates exercise capacity but also governs motor adaptation. Several studies had shown that exercise promotes phosphorylation of HDAC4/5 mediated by CaMK and adenosine monophosphate-activated protein kinase (AMPK), then increasing a MEF2-dependent transcription of Glut4 and muscle-specific genes, which participates in the adaptation and plasticity of skeletal muscles (McGee et al., 2008, 2009; McGee and Hargreaves, 2011). Besides exercise, glucose can also activate KATP channel-dependent calcium signaling and CaMK, which induce phosphorylation-dependent nuclear-cytoplasm transportation of HDAC5 (Meng et al., 2017). The leaving HDAC5 releases its repressive effect on Baf60c and enhancing insulin-independent AKT activation (Meng et al., 2017). Using specific inhibitor of class IIa HDAC activity, myosin heavy chain, PGC-1α, and heat-shock cognate (HSC70) are confirmed to be one of the substrates of HDAC4 in denervation-induced atrophy muscle in comparison with acetylated protein enrichment with untreated group (Luo et al., 2019). Taken together, Class IIa HDACs play critical roles in exercise capacity, remodeling, and nutrient sensing of skeletal muscles (Figure 3).
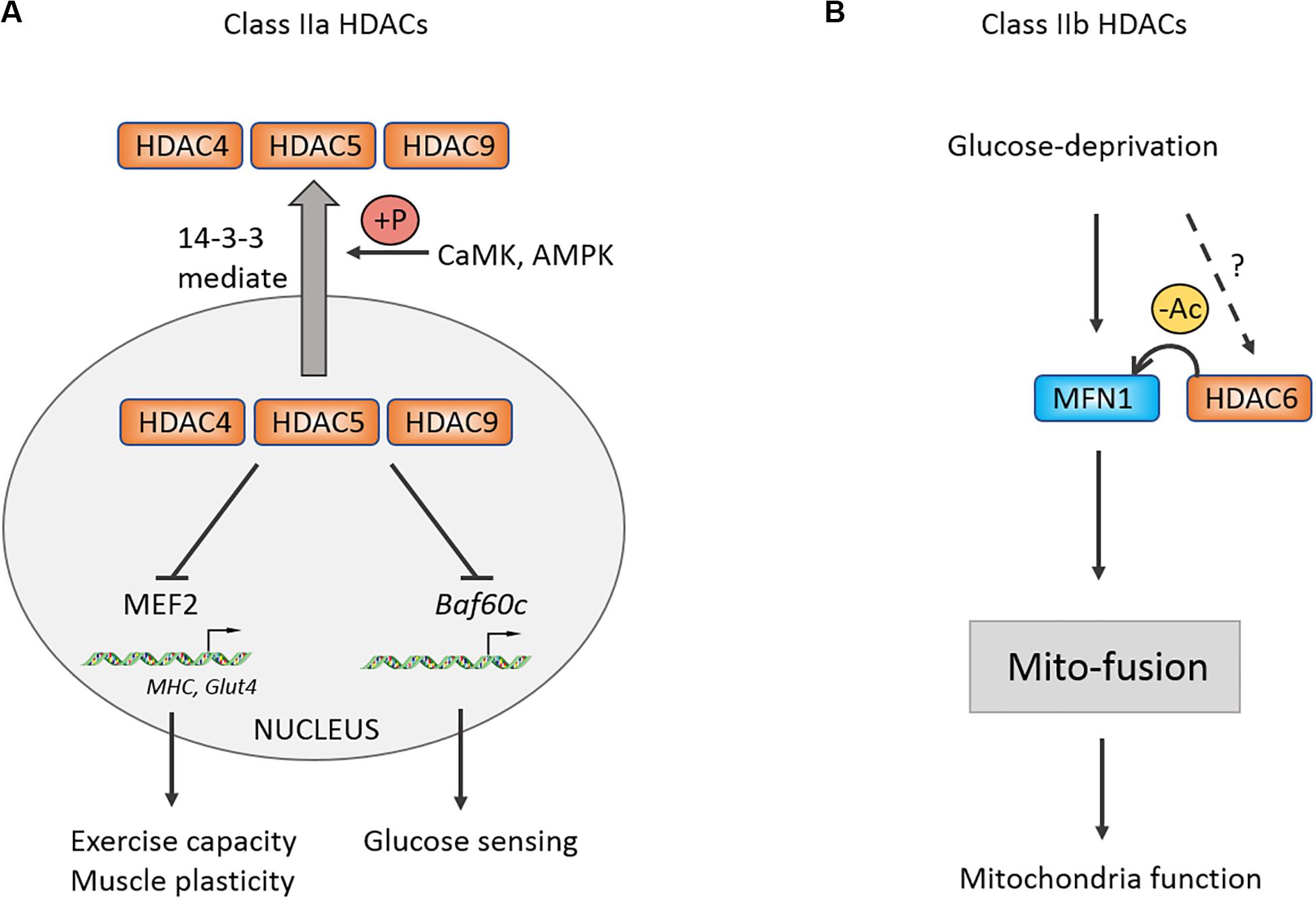
Figure 3. Control of muscle exercise adaption and mitochondrial function by Class II HDACs. (A) Schematic of the Class IIa HDACs nucleoplasm shuttle. Under stress condition, CaMK and AMPK could be activated and phosphorylate Class IIa HDACs. Phosphorylated HDACs then bind with 14-3-3 and shuttle to cytoplasm, relieving inhibitory effects on MEF2 and Baf60c. (B) Class IIb HDAC6 can deacetylate MFN1 and facilitates its mito-fusion function. +P, phosphorylation.
For Class IIb, it was revealed that glucose deprivation-induced mitochondrial fusion mediated by mitofusion1 (MFN1) is significantly disrupted in HDAC6 KO mice, causing mitochondria degeneration, which is reversed following application of MFN1 acetylation-resistant mutant (Lee et al., 2014). This study suggested that MFN1 deacetylation by HDAC6 plays an important role in mitochondrial adaptive energy production and remodeling of skeletal muscles.
Numerous studies have found that caloric restriction (CR) extends lifespan of mammals, Caenorhabditis elegans, and fruit flies, while such effect is lost in SIR2 or NPT1 mutant C. elegans strains (Imai et al., 2000; Lin et al., 2000). SIR2 encodes the silencing protein, Sir2p, and NPT1 participates in the synthesis of NAD+. NAD+ is an essential cofactor for Sir2p-Class III HDACs to exert enzyme activity rather than Class I, II, and IV relaying on the zinc (Imai et al., 2000; Lin et al., 2000). In mammals, Sir2 has 7 homologous proteins and these proteins are essential to mitochondrial energy homeostasis, antioxidant defense, cell proliferation, and DNA repair (Vargas-Ortiz et al., 2019).
Rodgers et al. (2005) reported that SIRT1 promotes the transcriptional activity of PGC-1α by deacetylating the PGC-1α at K13 site and mediates CR-induced gluconeogenesis-related genes G6P, PEPCK expression in liver. Further research showed that the NAD+/NADH ratio and the enzymatic activity of Sir2, a sensor of redox state, were decreased in differentiating myocytes, which relieved the inhibitory effect on MyoD and then promoted cell differentiation. Resveratrol (RSV), a compound that may target SIRT1-PGC-1α, can improve mitochondrial function and metabolic homeostasis, owning a potential to prolong lifespan. RSV loses its activating effect on PGC-1α in SIRT1 (–/–) MEFs (Lagouge et al., 2006). Moreover, RSV-treated mice at a dose of 4 g/kg increased their exercise capacity, oxygen consumption, and improved insulin sensitivity against high-fat induced obesity (Lagouge et al., 2006). In addition, RSV could enhance the deacetylation of PGC-1α in brown fat tissue (BAT) and skeletal muscles, which promotes mitochondrial production, oxygen consumption, and thus improves metabolic syndrome (Lagouge et al., 2006). The function of SIRT1 in regulating skeletal muscles metabolic homeostasis and exercise capacity has attracted great attention from researchers. Researchers further generated skeletal muscle-specific SIRT1-mKO mice and found that mKO mice lost the effect of CR-induced increasing insulin sensitivity and were unable to deacetylate and inactivate STAT3, resulting in upregulating the phosphatidylinositol-3-kinase (PI3K) inhibitory regulator p55α/p50α expression (Schenk et al., 2011). Although knockout or overexpression of SIRT1 in skeletal muscles has no effect on endurance capacity or glucose homeostasis in mice (Gurd et al., 2009; Philp et al., 2011; White et al., 2013; Svensson et al., 2020), some studies have shown that AMPK regulates energy metabolism of skeletal muscles partially mediated by SIRT1, and SIRT1 is significantly increased after endurance exercise (Suwa et al., 2008; Canto et al., 2009). Overexpression of Sirt1 in skeletal muscles by adeno-associated virus 1 (AAV1) promotes the expression of oxidation-related genes including Ppargc1a, Tfam, Cpt1b, and Pdk4, while it has no effect on insulin sensitivity of body (Vila et al., 2016). But overexpressing Sirt1 in liver by AAV8 can protect from fatty liver induced by high-carbohydrate food (HCD) (Vila et al., 2014). Taken together, the aforementioned studies suggest that SIRT1 possesses limited regulation capacity of skeletal muscles motility, and SIRT1 may modulate mitochondrial homeostasis and mediate skeletal muscles adaptation under certain physiological conditions, such as CR, aging, and regeneration (Gomes et al., 2013; Ryall et al., 2015; Figure 4). In addition, there may be more beneficial effects of SIRT1 on metabolism in the liver or fat tissue (Lagouge et al., 2006; Vila et al., 2014, 2016; Stefanowicz et al., 2018).
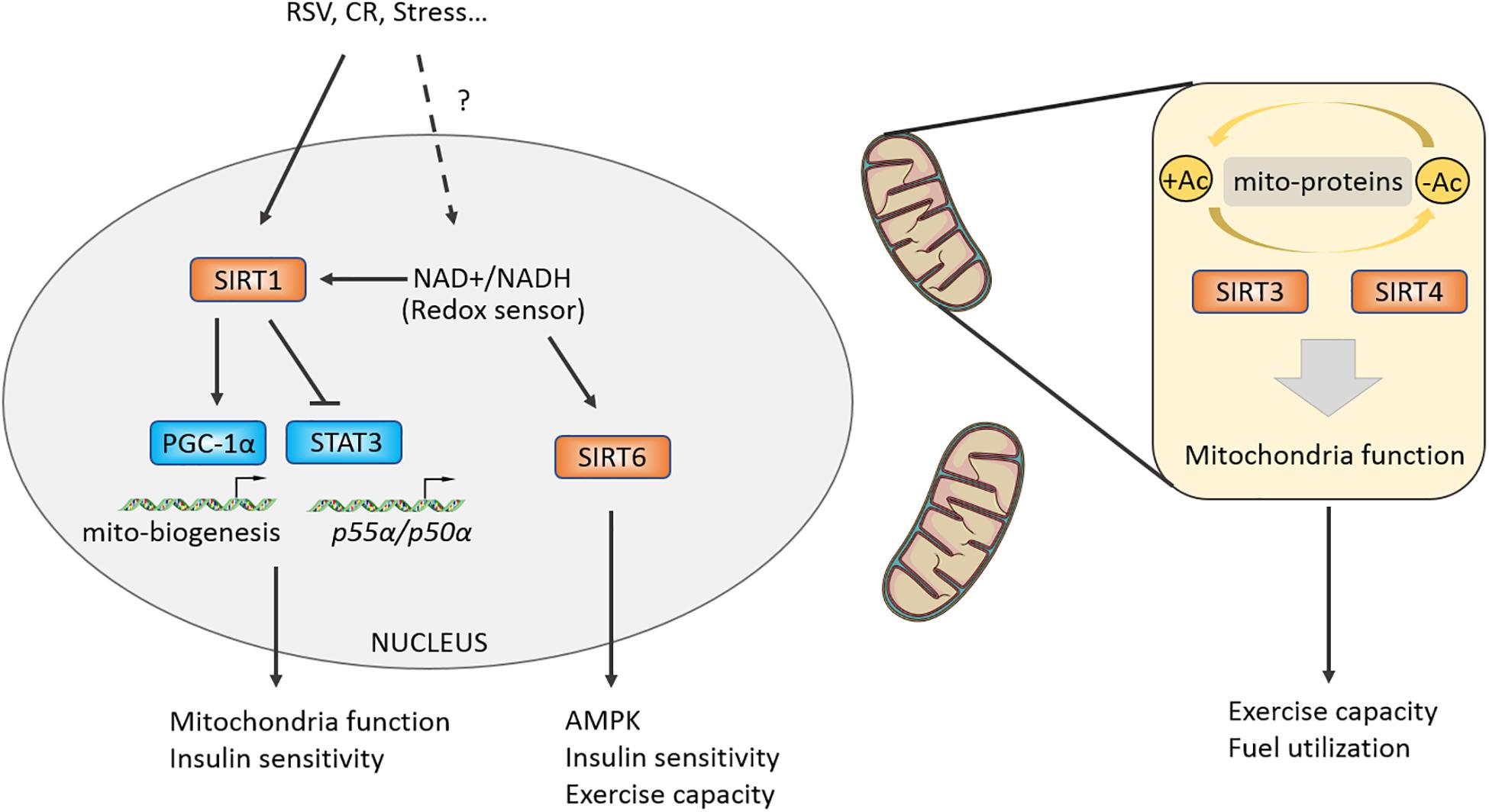
Figure 4. Class III HDACs promote a stress-induced adaptation in muscle and regulate mitochondrial function. Class III HDACs are NAD+-dependent deacetylases and may function as a redox sensor in muscle cell. SIRT1 could activate PGC-1alpha by directly deacetylasing K13 site and regulate insulin sensitivity by targeting STAT3. Mitochondria-located Class III HDACs control mitochondrial function by deacetylating certain mitochondrial proteins, modulating muscle fuel utilization and exercise capacity.
SIRT6 is another Sir2-like deacetylase located in the nucleus. Whole body deletion of SIRT6 causes 60% mice death around 4 weeks owning to hypoglycemia (Xiao et al., 2010). Nevertheless, the skeletal muscle-specific knockout of SIRT6 causes insulin resistance and impairs glucose homeostasis of mKO mice (Cui et al., 2017). Because the activity of AMPK is reduced in SIRT6-mKO mice, the glycolipids absorption and utilization of skeletal muscles are impaired, and the exercise capacity of the mice was also attenuated (Cui et al., 2017). Furthermore, researchers constructed the overexpressing SIRT6 (Sirt6BAC) mice and found that their body weight and fat content were normal, but the Sirt6BAC mice could protect from HCD-induced hyperglycemia via increasing p-AKT/AKT induced by insulin stimulation and glucose absorption (Anderson et al., 2015). Vitro experiments further demonstrated that the ability of glucose uptake was enhanced mainly in skeletal muscles, not in brain, iWAT, eWAT tissues of Sirt6BAC mice (Anderson et al., 2015). These results shows the potential effects of SIRT6 targeted in skeletal muscles on improving exercise performance and treating metabolic diseases (Figure 4).
SIRT3/4 are mitochondria-localized deacetylases and responsible for regulating the deacetylation of proteins in mitochondria and maintaining mitochondrial homeostasis. After feeding high-fat-diet (HFD), accelerated obesity, attenuated insulin sensitivity, worsened fatty liver and increased acetylation of proteins in mitochondria were observed in SIRT3 KO mice (Hirschey et al., 2011). Multi-tissue proteomics and physiological examination reveals that SIRT3 is responsible for mitochondrial acetylated proteome regulation and metabolic fuel switching in brain, heart, kidney, liver, and skeletal muscles (Dittenhafer-Reed et al., 2015). Protein enrichment analysis discovers that its regulatory proteins were mainly concentrated in lipid metabolism (Dittenhafer-Reed et al., 2015). Knocking out SIRT3 leads to reduction in the levels of deacetylation of manganese superoxide dismutase (MnSOD) mitochondrial complex II and pyruvate dehydrogenase (PDH) subunit E1α, a downregulation of hexokinase II (HK II) binding with mitochondria, resulting in impaired glucose and lipid metabolism of skeletal muscles (Lantier et al., 2015). Interestingly, SIRT3 liver-specific knockout (hep–/–) and skeletal muscle-specific knockout (skm–/–) mice did not affect glucose homeostasis under chow or HFD conditions (Fernandez-Marcos et al., 2012). The above results indicate that SIRT3 has an important role in metabolic regulation, but in specific physiological processes such as redox state, exercise, and aging, and its regulatory mechanism is yet defined in skeletal muscles (Kong et al., 2010; Robin et al., 2020; Williams et al., 2020). A recent study found that SIRT4 knockout can resist HFD-induced obesity and increase endurance exercise in mice by repressing malonyl CoA decarboxylase, which was a key enzyme controlling fatty acid beta-oxidation and was reported to regulate muscle fuel switching between carbohydrates and fatty acids (Koves et al., 2008; Laurent et al., 2013). Moreover, knockdown of SIRT4 by adenoviral shRNA can increase the mRNA and protein content of SIRT1, thereby enhancing the expression of fatty acid oxidation genes and mitochondrial oxidation capacity in hepatocytes and myotubes (Nasrin et al., 2010) (Table 1).
A comparative analysis used HDAC pan-inhibitor SAHA, a class I HDAC selective inhibitor (MS275) and a class II HDAC selective inhibitor (MC1568) to treat C2C12 separately. It was found that only MS275 could significantly stimulate mitochondria biogenesis and oxygen consumption (Galmozzi et al., 2013). In obese diabetic mice, it was found that specifically inhibiting Class I rather than Class II HDACs improved GTT and insulin sensitivity, increased oxidative metabolism of skeletal muscles and adipose tissue, and reduced body weight (Galmozzi et al., 2013). Similar to the effect of MS275, knockdown HDAC3 in C2C12 could also increase Pgc-1α, Glut4, Tfam, Idh3α transcription, suggesting that HDAC3 and its target genes played an important role in the above events (Galmozzi et al., 2013).
For Class II HDACs, studies have found that scriptaid, a Class IIa HDAC inhibitor, has similar effects to exercise. Six weeks of scriptaid administration significantly improved endurance performance, and significantly increased the whole-body energy expenditure and the expression of lipid oxidation related genes (Pdk4, Cpt1b, Pgc-1α, Pparδ, etc.) of C57BL/6 mice (Gaur et al., 2016). One possible explanation of above observation is the potential target of scriptaid inhibition of titin (a structure protein of sarcomere) deacetylation and downstream genes of Class IIa HDACs (Gaur et al., 2016; Huang et al., 2019).
In mice treated with SPT1720, another activator of SIRT1, overtly enhanced endurance exercise ability was noted. Moreover, these mice were protected from HFD-induced obesity and insulin resistance, owing to an upregulation of the oxidative metabolism in skeletal muscles, liver, and BAT tissues (Pacholec et al., 2010). In order to further explore the principle of SIRT1 activation, the researchers purified SIRT1 in vitro and added SRT1720 and its analogs SRT2183, SRT1460, and RSV, and they found that the enzymatic activity of SIRT1 was not enhanced, suggesting that these drugs may indirectly activate SIRT1 (Pacholec et al., 2010). The enzymatic activity of SIRT1 largely depends on the content of cofactor NAD+. This implied that the enhancement of SIRT1 activity may be achieved through indirect upregulation of NAD+. Study has discovered that knocking out poly (ADP-ribose) polymerase-1 (PARP-1), which is a NAD+ consuming enzyme, could increase NAD+ content and enhance the activity of SIRT1 in BAT and skeletal muscles (Bai et al., 2011). PARP-1 inhibitors can upregulate the proteins of mitochondrial respiratory chain complexes in mice, enhance the aerobic oxidation capacity of mitochondria, and improve mitochondrial defects in the primary myotubes of obese humans (Bai et al., 2011; Pirinen et al., 2014).
Resveratrol was initially reported as an SIRT1 activator that improves mitochondrial function and exercise capacity in mice, and resists HFD-induced obesity (Lagouge et al., 2006). However, subsequent studies have discovered that administering the same dose of RSV to rats or mice does not increase mitochondrial protein content (Higashida et al., 2013). By contrast, overexpression of SIRT1 in the triceps muscle of rats decreases the mitochondrial protein content (Higashida et al., 2013). In a double-blind human trial, 11 healthy and obese men were supplied for 30-day RSV, in which the data showed that RSV can improve systolic blood pressure and homeostasis model assessment (HOMA) index-indicating glucose metabolism ability, simulating the effect of CR (Timmers et al., 2011). However, some researchers found that the overexpression of SIRT1 alone cannot mimic the CR effect in transgenic mice, and the transcriptomic changes in various tissues were quite different or even opposite (Boutant et al., 2016). Such an opposite situation may explain that the genetic model and compound stimulation are not completely consistent. RSV as a potential metabolic syndrome treatment drug still needs large-scale population sample verification.
The very first mammalian histone deacetylase HDAC1 was cloned and isolated by Taunton et al. (1996), and 15,000 articles about HDACs have been published in the last 20 years. Currently, we know at least 18 HDAC proteins. They are responsible for eradicating epigenetic modifications, establishing an epigenetic off chromatin state, and regulating heritable gene expression (Yang and Seto, 2008; Haberland et al., 2009). In these processes, each HDACs may play a different role (Table 1). What kind of gene/protein is the specific downstream target of these HDACs? Is its enzyme activity related to intracellular localization and the formation of multi-protein complex? It is still one of the key and difficult issues in this field. Based on previous research experience, some techniques may be used to further experiments as follows: (1) HDACs interacting proteins/complex co-IP; (2) Protein-acetylation western; (3) Histone acetylation target gene ChIP; (4) High through put proteomics/acetylome with specific HDACs inhibitor, etc.
Histone deacetylases are potential therapeutic targets, and clinical drugs such as SAHA (Vorinostat) and FK228 (romidepsin) have been used for antitumor treatment (Bolden et al., 2006). However, they have two disadvantages: (1) great toxic and side effects and (2) difficulty in specific inhibition of HDACs activity. With the development of computer simulation technology and structural biology, it is believed that more specific HDACs inhibitors/activators can be constructed. And researches should attach importance to HDACs regulatory factors like PARP-1 when direct targeting on HDACs fails to show its effects. Further development of their inhibitors with more specificity, and trials for the treatment of metabolic diseases, may have great potential as well. Additionally, with the further development of biotechnology, some RNA therapies presented by AAV can specifically inhibit/overexpression of certain protein in skeletal muscles tissue, which may be able to target HDACs and treat related diseases (Wang et al., 2005; Long et al., 2016). However, its safety and effectiveness still need further clinical trials. Future research can discover known or unknown HDACs targeted drugs for the treatment of metabolic diseases.
HT, SL, and JR designed the literature search and wrote the review. JL, RW, and PC critically analyzed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the China Postdoctoral Science Foundation (2019M661042 to SL and 2019M651553 to HT) and National Natural Science Foundation of China (81501071 to SL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Anderson, J. G., Ramadori, G., Ioris, R. M., Galie, M., Berglund, E. D., Coate, K. C., et al. (2015). Enhanced insulin sensitivity in skeletal muscle and liver by physiological overexpression of SIRT6. Mol. Metab. 4, 846–856. doi: 10.1016/j.molmet.2015.09.003
Bai, P., Canto, C., Oudart, H., Brunyanszki, A., Cen, Y., Thomas, C., et al. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. doi: 10.1016/j.cmet.2011.03.004
Balasubramanian, A., Kawahara, G., Gupta, V. A., Rozkalne, A., Beauvais, A., Kunkel, L. M., et al. (2014). Fam65b is important for formation of the HDAC6-dysferlin protein complex during myogenic cell differentiation. Faseb J. 28, 2955–2969. doi: 10.1096/fj.13-246470
Beharry, A. W., Sandesara, P. B., Roberts, B. M., Ferreira, L. F., Senf, S. M., and Judge, A. R. (2014). HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J. Cell Sci. 127(Pt 7), 1441–1453. doi: 10.1242/jcs.136390
Bin, Y., Xiao, Y., Huang, D., Ma, Z., Liang, Y., Bai, J., et al. (2019). Theophylline inhibits cigarette smoke-induced inflammation in skeletal muscle by upregulating HDAC2 expression and decreasing NF-kappaB activation. Am. J. Physiol. Lung. Cell Mol. Physiol. 316, L197–L205. doi: 10.1152/ajplung.00005.2018
Bolden, J. E., Peart, M. J., and Johnstone, R. W. (2006). Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug. Discov. 5, 769–784. doi: 10.1038/nrd2133
Boutant, M., Kulkarni, S. S., Joffraud, M., Raymond, F., Metairon, S., Descombes, P., et al. (2016). SIRT1 gain of function does not mimic or enhance the adaptations to intermittent fasting. Cell Rep. 14, 2068–2075. doi: 10.1016/j.celrep.2016.02.007
Boyle, K. E., Newsom, S. A., Janssen, R. C., Lappas, M., and Friedman, J. E. (2013). Skeletal muscle MnSOD, mitochondrial complex II, and SIRT3 enzyme activities are decreased in maternal obesity during human pregnancy and gestational diabetes mellitus. J. Clin. Endocrino.l Metab. 98, E1601–E1609. doi: 10.1210/jc.2013-1943
Canto, C., Gerhart-Hines, Z., Feige, J. N., Lagouge, M., Noriega, L., Milne, J. C., et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060. doi: 10.1038/nature07813
Cho, O. H., Mallappa, C., Hernandez-Hernandez, J. M., Rivera-Perez, J. A., and Imbalzano, A. N. (2015). Contrasting roles for MyoD in organizing myogenic promoter structures during embryonic skeletal muscle development. Dev. Dyn. 244, 43–55. doi: 10.1002/dvdy.24217
Choi, M. C., Ryu, S., Hao, R., Wang, B., Kapur, M., Fan, C. M., et al. (2014). HDAC4 promotes Pax7-dependent satellite cell activation and muscle regeneration. EMBO Rep. 15, 1175–1183. doi: 10.15252/embr.201439195
Cui, X., Yao, L., Yang, X., Gao, Y., Fang, F., Zhang, J., et al. (2017). SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. Am. J. Physiol. Endocrinol. Metab. 313, E493–E505. doi: 10.1152/ajpendo.00122.2017
Dittenhafer-Reed, K. E., Richards, A. L., Fan, J., Smallegan, M. J., Fotuhi Siahpirani, A., Kemmerer, Z. A., et al. (2015). SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 21, 637–646. doi: 10.1016/j.cmet.2015.03.007
Dressel, U., Bailey, P. J., Wang, S. C., Downes, M., Evans, R. M., and Muscat, G. E. (2001). A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J. Biol. Chem. 276, 17007–17013. doi: 10.1074/jbc.M101508200
Durnin, J. (1981). Basal Metabolic Rate in Man. Report to FAO/WHO/UNU. Vol. 15, Rome: FAO. 507–524. doi: 10.1038/s41574-019-0230-6
Fernandez-Marcos, P. J., Jeninga, E. H., Canto, C., Harach, T., de Boer, V. C., Andreux, P., et al. (2012). Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci. Rep. 2:425. doi: 10.1038/srep00425
Fortuin, F. D., Morisaki, T., and Holmes, E. W. (1996). Subunit composition of AMPD varies in response to changes in AMPD1 and AMPD3 gene expression in skeletal muscle. Proc. Assoc. Am. Physicians 108, 329–333.
Galmozzi, A., Mitro, N., Ferrari, A., Gers, E., Gilardi, F., Godio, C., et al. (2013). Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 62, 732–742. doi: 10.2337/db12-0548
Gao, C., Liu, Y., Lam, M., and Kao, H. Y. (2010). Histone deacetylase 7 (HDAC7) regulates myocyte migration and differentiation. Biochim. Biophys. Acta 1803, 1186–1197. doi: 10.1016/j.bbamcr.2010.06.008
Gaur, V., Connor, T., Sanigorski, A., Martin, S. D., Bruce, C. R., Henstridge, D. C., et al. (2016). Disruption of the Class IIa HDAC corepressor complex increases energy expenditure and lipid oxidation. Cell Rep. 16, 2802–2810. doi: 10.1016/j.celrep.2016.08.005
Gomes, A. P., Price, N. L., Ling, A. J., Moslehi, J. J., Montgomery, M. K., Rajman, L., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638. doi: 10.1016/j.cell.2013.11.037
Gong, Y., Cao, R., Ding, G., Hong, S., Zhou, W., Lu, W., et al. (2018). Integrated omics approaches to characterize a nuclear receptor corepressor-associated histone deacetylase in mouse skeletal muscle. Mol. Cell. Endocrinol. 471, 22–32. doi: 10.1016/j.mce.2017.05.024
Gregoire, S., Xiao, L., Nie, J., Zhang, X., Xu, M., Li, J., et al. (2007). Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol. Cell Biol. 27, 1280–1295. doi: 10.1128/mcb.00882-06
Guo, S., Huang, Y., Zhang, Y., Huang, H., Hong, S., and Liu, T. (2020). Impacts of exercise interventions on different diseases and organ functions in mice. J. Sport Health Sci. 9, 53–73. doi: 10.1016/j.jshs.2019.07.004
Gurd, B. J., Yoshida, Y., Lally, J., Holloway, G. P., and Bonen, A. (2009). The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis. J. Physiol. 587(Pt 8), 1817–1828. doi: 10.1113/jphysiol.2008.168096
Haberland, M., Montgomery, R. L., and Olson, E. N. (2009). The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42. doi: 10.1038/nrg2485
Han, K. A., Kang, H. S., Lee, J. W., Yoo, L., Im, E., Hong, A., et al. (2014). Histone deacetylase 3 promotes RCAN1 stability and nuclear translocation. PLoS One 9:e105416. doi: 10.1371/journal.pone.0105416
Higashida, K., Kim, S. H., Jung, S. R., Asaka, M., Holloszy, J. O., and Han, D. H. (2013). Effects of resveratrol and SIRT1 on PGC-1alpha activity and mitochondrial biogenesis: a reevaluation. PLoS Biol. 11:e1001603. doi: 10.1371/journal.pbio.1001603
Hirschey, M. D., Shimazu, T., Jing, E., Grueter, C. A., Collins, A. M., Aouizerat, B., et al. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190. doi: 10.1016/j.molcel.2011.07.019
Hong, S., Zhou, W., Fang, B., Lu, W., Loro, E., Damle, M., et al. (2017). Dissociation of muscle insulin sensitivity from exercise endurance in mice by HDAC3 depletion. Nat. Med. 23, 223–234. doi: 10.1038/nm.4245
Huang, S., Liu, S., Niu, Y., and Fu, L. (2019). Scriptaid/exercise-induced lysine acetylation is another type of posttranslational modification occurring in titin. J. Appl. Physiol. 128, 276–285. doi: 10.1152/japplphysiol.00617.2019
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. doi: 10.1038/35001622
Jing, E., O’Neill, B. T., Rardin, M. J., Kleinridders, A., Ilkeyeva, O. R., Ussar, S., et al. (2013). Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62, 3404–3417. doi: 10.2337/db12-1650
Kanfi, Y., Peshti, V., Gil, R., Naiman, S., Nahum, L., Levin, E., et al. (2010). SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9, 162–173. doi: 10.1111/j.1474-9726.2009.00544.x
Karagianni, P., and Wong, J. (2007). HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene 26, 5439–5449. doi: 10.1038/sj.onc.1210612
Kong, X., Wang, R., Xue, Y., Liu, X., Zhang, H., Chen, Y., et al. (2010). Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 5:e11707. doi: 10.1371/journal.pone.0011707
Koves, T. R., Ussher, J. R., Noland, R. C., Slentz, D., Mosedale, M., Ilkayeva, O., et al. (2008). Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56. doi: 10.1016/j.cmet.2007.10.013
Lagouge, M., Argmann, C., Gerhart-Hines, Z., Meziane, H., Lerin, C., Daussin, F., et al. (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122. doi: 10.1016/j.cell.2006.11.013
Lantier, L., Williams, A. S., Williams, I. M., Yang, K. K., Bracy, D. P., Goelzer, M., et al. (2015). SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes 64, 3081–3092. doi: 10.2337/db14-1810
Laurent, G., German, N. J., Saha, A. K., de Boer, V. C., Davies, M., Koves, T. R., et al. (2013). SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 50, 686–698. doi: 10.1016/j.molcel.2013.05.012
Lee, J. Y., Kapur, M., Li, M., Choi, M. C., Choi, S., Kim, H. J., et al. (2014). MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J. Cell Sci. 127(Pt 22), 4954–4963. doi: 10.1242/jcs.157321
Lee, S. W., Yang, J., Kim, S. Y., Jeong, H. K., Lee, J., Kim, W. J., et al. (2015). MicroRNA-26a induced by hypoxia targets HDAC6 in myogenic differentiation of embryonic stem cells. Nucleic Acids Res. 43, 2057–2073. doi: 10.1093/nar/gkv088
Lempradl, A., Pospisilik, J. A., and Penninger, J. M. (2015). Exploring the emerging complexity in transcriptional regulation of energy homeostasis. Nat. Rev. Genet. 16, 665–681. doi: 10.1038/nrg3941
Lin, S. J., Defossez, P. A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi: 10.1126/science.289.5487.2126
Liu, S., Niu, Y., and Fu, L. (2020). Metabolic adaptations to exercise training. J. Sci. Sport Exerc. 2, 1–6. doi: 10.1007/s42978-019-0018-3
Long, C., Amoasii, L., Mireault, A. A., McAnally, J. R., Li, H., Sanchez-Ortiz, E., et al. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403. doi: 10.1126/science.aad5725
Luan, X., Tian, X., Zhang, H., Huang, R., Li, N., Chen, P., et al. (2019). Exercise as a prescription for patients with various diseases. J. Sport Health Sci. 8, 422–441. doi: 10.1016/j.jshs.2019.04.002
Luo, L., Martin, S. C., Parkington, J., Cadena, S. M., Zhu, J., Ibebunjo, C., et al. (2019). HDAC4 controls muscle homeostasis through deacetylation of myosin heavy Chain, PGC-1alpha, and Hsc70. Cell Rep. 29, 749.e12–763.e12. doi: 10.1016/j.celrep.2019.09.023
Macpherson, P. C., Farshi, P., and Goldman, D. (2015). Dach2-Hdac9 signaling regulates reinnervation of muscle endplates. Development 142, 4038–4048. doi: 10.1242/dev.125674
Mal, A., Sturniolo, M., Schiltz, R. L., Ghosh, M. K., and Harter, M. L. (2001). A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. Embo J. 20, 1739–1753. doi: 10.1093/emboj/20.7.1739
McGee, S. L., Fairlie, E., Garnham, A. P., and Hargreaves, M. (2009). Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 587(Pt 24), 5951–5958. doi: 10.1113/jphysiol.2009.181065
McGee, S. L., and Hargreaves, M. (2010). Histone modifications and skeletal muscle metabolic gene expression. Clin. Exp. Pharmacol. Physiol. 37, 392–396. doi: 10.1111/j.1440-1681.2009.05311.x
McGee, S. L., and Hargreaves, M. (2011). Histone modifications and exercise adaptations. J. Appl. Physiol. 110, 258–263. doi: 10.1152/japplphysiol.00979.2010
McGee, S. L., van Denderen, B. J., Howlett, K. F., Mollica, J., Schertzer, J. D., Kemp, B. E., et al. (2008). AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 57, 860–867. doi: 10.2337/db07-0843
McKinsey, T. A., Zhang, C. L., Lu, J., and Olson, E. N. (2000a). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111. doi: 10.1038/35040593
McKinsey, T. A., Zhang, C. L., and Olson, E. N. (2000b). Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. U.S.A. 97, 14400–14405. doi: 10.1073/pnas.260501497
Meng, Z. X., Gong, J., Chen, Z., Sun, J., Xiao, Y., Wang, L., et al. (2017). Glucose sensing by skeletal myocytes couples nutrient signaling to systemic homeostasis. Mol. Cell 66, 332.e4–344.e4. doi: 10.1016/j.molcel.2017.04.007
Mizgier, M. L., Fernandez-Verdejo, R., Cherfan, J., Pinget, M., Bouzakri, K., and Galgani, J. E. (2019). Insights on the role of putative muscle-derived factors on pancreatic beta cell function. Front. Physiol. 10:1024. doi: 10.3389/fphys.2019.01024
Montgomery, R. L., Davis, C. A., Potthoff, M. J., Haberland, M., Fielitz, J., Qi, X., et al. (2007). Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802. doi: 10.1101/gad.1563807
Moresi, V., Carrer, M., Grueter, C. E., Rifki, O. F., Shelton, J. M., Richardson, J. A., et al. (2012). Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 1649–1654. doi: 10.1073/pnas.1121159109
Nasrin, N., Wu, X., Fortier, E., Feng, Y., Bare, O. C., Chen, S., et al. (2010). SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 285, 31995–32002. doi: 10.1074/jbc.M110.124164
Niu, Y., Wang, T., Liu, S., Yuan, H., Li, H., and Fu, L. (2017). Exercise-induced GLUT4 transcription via inactivation of HDAC4/5 in mouse skeletal muscle in an AMPKalpha2-dependent manner. Biochim. Biophys. Acta 1863, 2372–2381. doi: 10.1016/j.bbadis.2017.07.001
Ohkawa, Y., Marfella, C. G., and Imbalzano, A. N. (2006). Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. Embo J. 25, 490–501. doi: 10.1038/sj.emboj.7600943
Pacholec, M., Bleasdale, J. E., Chrunyk, B., Cunningham, D., Flynn, D., Garofalo, R. S., et al. (2010). SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285, 8340–8351. doi: 10.1074/jbc.M109.088682
Pedersen, B. K., and Febbraio, M. A. (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 8, 457–465. doi: 10.1038/nrendo.2012.49
Philp, A., Chen, A., Lan, D., Meyer, G. A., Murphy, A. N., Knapp, A. E., et al. (2011). Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J. Biol. Chem. 286, 30561–30570. doi: 10.1074/jbc.M111.261685
Pirinen, E., Canto, C., Jo, Y. S., Morato, L., Zhang, H., Menzies, K. J., et al. (2014). Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041. doi: 10.1016/j.cmet.2014.04.002
Potthoff, M. J., Wu, H., Arnold, M. A., Shelton, J. M., Backs, J., McAnally, J., et al. (2007). Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Invest. 117, 2459–2467. doi: 10.1172/jci31960
Puri, P. L., Iezzi, S., Stiegler, P., Chen, T. T., Schiltz, R. L., Muscat, G. E., et al. (2001). Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 8, 885–897. doi: 10.1016/s1097-2765(01)00373-2
Ratti, F., Ramond, F., Moncollin, V., Simonet, T., Milan, G., Mejat, A., et al. (2015). Histone deacetylase 6 is a FoxO transcription factor-dependent effector in skeletal muscle atrophy. J. Biol. Chem. 290, 4215–4224. doi: 10.1074/jbc.M114.600916
Robin, J. D., Jacome Burbano, M. S., Peng, H., Croce, O., Thomas, J. L., Laberthonniere, C., et al. (2020). Mitochondrial function in skeletal myofibers is controlled by a TRF2-SIRT3 axis over lifetime. Aging Cell 19:e13097. doi: 10.1111/acel.13097
Rodgers, J. T., Lerin, C., Haas, W., Gygi, S. P., Spiegelman, B. M., and Puigserver, P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118. doi: 10.1038/nature03354
Ryall, J. G., Dell’Orso, S., Derfoul, A., Juan, A., Zare, H., Feng, X., et al. (2015). The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16, 171–183. doi: 10.1016/j.stem.2014.12.004
Samant, S. A., Kanwal, A., Pillai, V. B., and Bao, R. (2017). The histone deacetylase SIRT6 blocks myostatin expression and development of muscle atrophy. Sci. Rep. 7:11877. doi: 10.1038/s41598-017-10838-5
Schenk, S., McCurdy, C. E., Philp, A., Chen, M. Z., Holliday, M. J., Bandyopadhyay, G. K., et al. (2011). Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J. Clin. Invest. 121, 4281–4288. doi: 10.1172/jci58554
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. doi: 10.1146/annurev.biochem.76.052705.162114
Song, S., Wen, Y., Tong, H., Loro, E., Gong, Y., Liu, J., et al. (2019). The HDAC3 enzymatic activity regulates skeletal muscle fuel metabolism. J. Mol. Cell Biol. 11, 133–143. doi: 10.1093/jmcb/mjy066
Stefanowicz, M., Nikolajuk, A., Matulewicz, N., and Karczewska-Kupczewska, M. (2018). Adipose tissue, but not skeletal muscle, sirtuin 1 expression is decreased in obesity and related to insulin sensitivity. Endocrine 60, 263–271. doi: 10.1007/s12020-018-1544-1
Sun, Z., Feng, D., Fang, B., Mullican, S. E., You, S. H., Lim, H. W., et al. (2013). Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell. 52, 769–782. doi: 10.1016/j.molcel.2013.10.022
Suwa, M., Nakano, H., Radak, Z., and Kumagai, S. (2008). Endurance exercise increases the SIRT1 and peroxisome proliferator-activated receptor gamma coactivator-1alpha protein expressions in rat skeletal muscle. Metabolism 57, 986–998. doi: 10.1016/j.metabol.2008.02.017
Svensson, K., Tahvilian, S., Martins, V. F., Dent, J. R., Lemanek, A., Barooni, N., et al. (2020). Combined overexpression of SIRT1 and knockout of GCN5 in adult skeletal muscle does not affect glucose homeostasis or exercise performance in mice. Am. J. Physiol. Endocrinol. Metab. 318, E145–E151. doi: 10.1152/ajpendo.00370.2019
Taunton, J., Hassig, C. A., and Schreiber, S. L. (1996). A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272, 408–411. doi: 10.1126/science.272.5260.408
Taylor, M. V., and Hughes, S. M. (2017). Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 72, 33–44. doi: 10.1016/j.semcdb.2017.11.020
Timmers, S., Konings, E., Bilet, L., Houtkooper, R. H., van de Weijer, T., Goossens, G. H., et al. (2011). Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 14, 612–622. doi: 10.1016/j.cmet.2011.10.002
Vargas-Ortiz, K., Perez-Vazquez, V., and Macias-Cervantes, M. H. (2019). Exercise and sirtuins: a way to mitochondrial health in skeletal muscle. Int. J. Mol. Sci. 20:2717. doi: 10.3390/ijms20112717
Vila, L., Elias, I., Roca, C., Ribera, A., Ferre, T., Casellas, A., et al. (2014). AAV8-mediated Sirt1 gene transfer to the liver prevents high carbohydrate diet-induced nonalcoholic fatty liver disease. Mol. Ther. Methods Clin. Dev. 1:14039. doi: 10.1038/mtm.2014.39
Vila, L., Roca, C., Elias, I., Casellas, A., Lage, R., Franckhauser, S., et al. (2016). AAV-mediated Sirt1 overexpression in skeletal muscle activates oxidative capacity but does not prevent insulin resistance. Mol. Ther. Methods Clin. Dev. 5:16072. doi: 10.1038/mtm.2016.72
Wang, Z., Zhu, T., Qiao, C., Zhou, L., Wang, B., Zhang, J., et al. (2005). Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 23, 321–328. doi: 10.1038/nbt1073
White, A. T., McCurdy, C. E., Philp, A., Hamilton, D. L., Johnson, C. D., and Schenk, S. (2013). Skeletal muscle-specific overexpression of SIRT1 does not enhance whole-body energy expenditure or insulin sensitivity in young mice. Diabetologia 56, 1629–1637. doi: 10.1007/s00125-013-2912-2
White, A. T., and Schenk, S. (2012). NAD(+)/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 303, E308–E321. doi: 10.1152/ajpendo.00054.2012
Williams, A. S., Koves, T. R., Davidson, M. T., Crown, S. B., Fisher-Wellman, K. H., Torres, M. J., et al. (2020). Disruption of acetyl-lysine turnover in muscle mitochondria promotes insulin resistance and redox stress without overt respiratory dysfunction. Cell Metab. 31, 131.e11–147.e11. doi: 10.1016/j.cmet.2019.11.003
Xiao, C., Kim, H. S., Lahusen, T., Wang, R. H., Xu, X., Gavrilova, O., et al. (2010). SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 285, 36776–36784. doi: 10.1074/jbc.M110.168039
Xie, M., Kong, Y., Tan, W., May, H., Battiprolu, P. K., Pedrozo, Z., et al. (2014). Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129, 1139–1151. doi: 10.1161/circulationaha.113.002416
Yang, X. J., and Seto, E. (2008). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218. doi: 10.1038/nrm2346
Yuan, H., Niu, Y., Liu, X., and Fu, L. (2014). Exercise increases the binding of MEF2A to the Cpt1b promoter in mouse skeletal muscle. Acta Physiol. 212, 283–292. doi: 10.1111/apha.12395
Zhang, Y., and Ren, J. (2014). Targeting autophagy for the therapeutic application of histone deacetylase inhibitors in ischemia/reperfusion heart injury. Circulation 129, 1088–1091. doi: 10.1161/circulationaha.113.008115
Zhang, Z., Zhang, L., Zhou, Y., Li, L., Zhao, J., Qin, W., et al. (2019). Increase in HDAC9 suppresses myoblast differentiation via epigenetic regulation of autophagy in hypoxia. Cell Death Dis. 10:552. doi: 10.1038/s41419-019-1763-2
Keywords: histone deacetylases, exercise capacity, skeletal muscle, metabolism, muscle physiology
Citation: Tian H, Liu S, Ren J, Lee JKW, Wang R and Chen P (2020) Role of Histone Deacetylases in Skeletal Muscle Physiology and Systemic Energy Homeostasis: Implications for Metabolic Diseases and Therapy. Front. Physiol. 11:949. doi: 10.3389/fphys.2020.00949
Received: 27 May 2020; Accepted: 14 July 2020;
Published: 11 August 2020.
Edited by:
Brian James Morris, The University of Sydney, AustraliaReviewed by:
Sean L. McGee, Deakin University, AustraliaCopyright © 2020 Tian, Liu, Ren, Lee, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ru Wang, d2FuZ3J1QHR5LnN1cy5lZHUuY24=; Peijie Chen, Y2hlbnBlaWppZUBzdXMuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.