 Xiaodong Peng
Xiaodong Peng Linling Li
Linling Li Mengxia Zhang
Mengxia Zhang Rong Bai
Rong Bai Nian Liu
Nian Liu- Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing, China
Sodium-glucose cotransporter 2 inhibitors (SGLT2i) are a novel class of glucose-lowering agents that significantly improve the prognosis of patients with type 2 diabetes (T2D) and heart failure. SGLT2i has recently been implicated in the treatment of atrial fibrillation (AF) with clinical data demonstrating that these agents decrease the incidence of AF events in patients with T2D. Fundamental findings have suggested that SGLT2i may alleviate atrial electrical and structural remodeling. The underlying mechanisms of SGLT2i are likely associated with balancing the sodium and calcium handling disorders and mitigating the mitochondrial dysfunction in atrial myocytes. This review illustrates the advances in understanding the underlying mechanisms of SGLT2i as an evolving treatment modality for AF.
Introduction
Atrial fibrillation (AF) is a prominent public health and economic issue globally. It is estimated that about 33.5 million people worldwide suffered from AF in 2010, and the number of affected patients is predicted to reach 12.1 and 14–17 million by the year 2030 in the United States and Europe, respectively, exerting tremendous pressure on the global economy (Colilla et al., 2013; Chugh et al., 2014; Zoni-Berisso et al., 2014). In clinical practice, AF is often comorbid with heart failure (HF) and type 2 diabetes (T2D), accelerating the progression of these diseases. Moreover, patients with AF have an increased (fivefold) risk of suffering from stroke, resulting in severe disability or death (Pistoia et al., 2016).
Some progress has been made in our understanding of the pathophysiology of AF. The ectopic beats from the pulmonary veins are called “triggers,” considered to be signals for the initiation of AF (Haïssaguerre et al., 1998; Santangeli and Marchlinski, 2017). Therefore, pulmonary vein isolation by insulating pulmonary veins from the atrium becomes the cornerstone of catheter ablation for the prevention of AF. The “substrate” has been well-recognized for supporting and maintaining AF, which is caused by electrical and structural remodeling in the atrium (Allessie et al., 2002). In addition to the deposition of elastic fibers and collagen in the atrium, the arrhythmogenesis of atrial myocytes has gained increasing attention in the field. In atrial myocytes, the energetic deficits and oxidative stress caused by mitochondrial dysfunction and alterations in Na+ and Ca2+ handling are critical pathophysiological hallmarks (Poulet et al., 2015; Dobrev and Wehrens, 2017; Karam et al., 2017). Amelioration of these deficits is considered a promising therapeutic strategy for the treatment of AF.
Sodium-glucose cotransporter 2 inhibitors (SGLT2i) are novel glucose-lowering agents used in the treatment of T2D. SGLT2 is a low-affinity, high-capacity glucose transporter located in the proximal tubule in the kidneys and is responsible for glucose reabsorption into the body from urine (Scheen, 2014). The (Empagliflozin) Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME), a breakthrough in the treatment of T2D, revealed that empagliflozin reduces the risk of major adverse cardiovascular events by 38% relative to placebo (Zinman et al., 2015). In the subgroup analysis of the EMPA-REG OUTCOME trial, patients with AF especially benefited from the use of empagliflozin compared with patients without AF (Böhm et al., 2020). Moreover, the Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI 58) has shown that dapagliflozin decreases the incidence of reported episodes of AF events in high-risk patients with T2D (Zelniker et al., 2019). A cohort study that enrolled more than 150,000 cases demonstrated that patients with T2D treated with SGLT2i were less likely to develop AF (Chen et al., 2020). A recent meta-analysis incorporating 10 studies published between 2014 and 2019 revealed that AF was significantly reduced after the management of SGLT2i (Cheung et al., 2020). Accumulating evidence suggests that the cardioprotective effects of SGLT2i are independent of their glucose-lowering properties (Juni et al., 2019; Li et al., 2019). Currently, SGLT2i is thought to balance Na+ and Ca2+ homeostasis and rescue mitochondrial function. In this review, we discuss the potential therapeutic mechanisms of SGLT2i as novel targets for the treatment of AF (Figure 1).
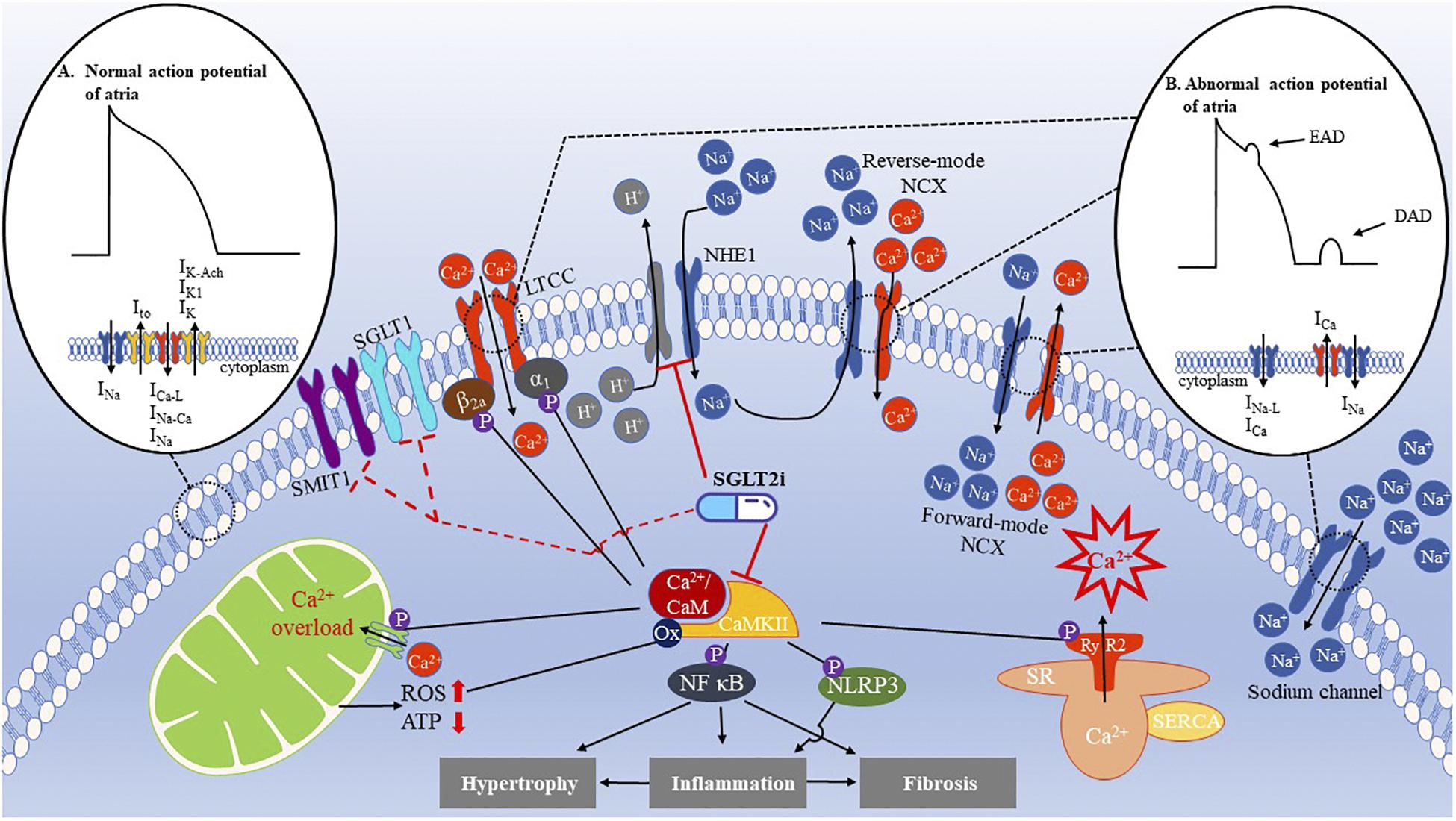
Figure 1. Ion disorder and mitochondrial dysfunction in AF and cardioprotective effects of SGLT2i. (A) The normal action potential (AP) of atria is initiated by the rapid influx of Na+ current (INa) to form an upstroke AP. Subsequently, transient outward current (Ito) achieves rapid repolarization, followed by inward L-type Ca2+ currents (ICa–L), INa, and Na+/Ca2+ exchange current (INa–Ca) as well as outward currents, such as inward rectifying potassium (IK1), delayed rectifier potassium (IK), and acetylcholine-sensitive potassium (IK–Ach) currents, which maintain the AP plateau. Afterward, IK1, IK, and IK–Ach completely achieve repolarization. This period is called the action potential duration (APD). (B) Abnormal AP is caused by irregular ion handling. For instance, increased late Na+ current (INa–L) and Ca2+ current (ICa) during the AP plateau or three phases can shorten the APD and generate early after-depolarizations (EAD). Moreover, excessive Ca2+ release from the SR activates the forward-mode NCX and increases the inward currents leading to delayed after-depolarizations (DADs). In patients with AF, the NHE1 is upregulated, enhancing the influx of Na+. As a result, intracellular Ca2+ levels increase, effectively impacting the inflammatory, fibrosis, and hypertrophic pathways that contribute to atrial remodeling. CaMKII is an important regulator of Ca2+ handling, which phosphorylates the ryanodine receptor 2 (RyR2) to exacerbate the Ca2+ leak from the SR, causing a Ca2+ overload. In addition, CaMKII also phosphorylates the L-type Ca2+ channel (LTCC) CaV1.2 β2a subunits and CaV1.3 α1 subunit to increase Ca2+ entry, activating the NF-κB and NLRP3 inflammasomes to initiate hypertrophy, inflammation, and fibrosis of atria. Mitochondrial Ca2+ uniporter (MCU) is phosphorylated by CaMKII, which causes a Ca2+ overload in the mitochondria, producing more ROS, which, in turn, oxidizes CaMKII to further activate CaMKII. These effects can be eliminated by SGLT2i. SGLT1 and sodium-myoinositol cotransporter 1 (SMIT1) are potential targets of SGLT2i.
Na+ and Ca2+ Handling in Healthy and Fibrillating Atrium: Therapeutic Target of Sodium-Glucose Cotransporter 2 Inhibitors
Normal Na+ and Ca2+ Handling in Cardiac Myocytes
The delicate balance of Na+ and Ca2+ fluxes between the intracellular and extracellular spaces in cardiac myocytes is pivotal to ensure normal contractile function and cardiac rhythm. During the plateau phase of the cardiac action potential, a small amount of Ca2+ enters the cardiac myocytes through L-type Ca2+ channels, causing a large Ca2+ release into the cytosol via the RyR2 channel. This channel is located in the membrane of the sarcoplasmic reticulum (SR), which is called Ca2+-induced Ca2+ release (CICR) (Ibrahim et al., 2012). The cytosolic Ca2+ increases to 1 μM from 150 nM in baseline and activates the contractile apparatus (Bers, 2002). This process is also named cardiac excitation-contraction (E-C) coupling. Next, the elevated cytosolic Ca2+ needs to be promptly resumed to 150 nM during the diastolic period to maintain the normal relaxation property of cardiac myocytes. The majority of Ca2+ is taken up again into the SR by sarcoplasmic/endoplasmic reticulum Ca2+ ATPase isoform 2a (SERCA2a) (Bers, 2008). The remaining Ca2+ is mainly extruded into the extracellular fluid via a forward-mode Na+/Ca2+ exchanger (NCX).
Na+ hemostasis in cardiac myocytes is regulated by the Na+ channels and transporters that couple the Na+ influx to either co- or countertransport of other ions, such as NCX, Na+/H+ exchange (NHE), and Na+/K+-ATPase (NKA). Na+ channels and NCX are the dominant Na+ influx and efflux pathways in the atrium, respectively. There is a tight coupling between Na+ and Ca2+ homeostasis via the NCX in the microdomain beneath the T-tube. The NCX may operate in both forward and reverse directions, depending on the electrochemical gradients and membrane potential. Elevated intracellular Ca2+ levels activate the forward mode of NCX, extruding the Ca2+ in exchange for Na+ with a stoichiometry of 1:3 that generates a net inward current. The transient inward current results in delayed after-depolarizations (DADs) during the diastolic period and triggers activity once it reaches the threshold of the Na+ channel (Voigt et al., 2012). Na+ elevation in the cytosol may limit Ca2+ extrusion via the forward mode of NCX and could even facilitate Ca2+ influx through the reverse mode (Baartscheer et al., 2003). Both scenarios suggest that elevated cytosolic Na+ enhances intracellular Ca2+ overload via the NCX pathway, subsequently resulting in cardiac dysfunction and arrhythmogenesis.
Abnormal Na+ and Ca2+ Handling During Atrial Fibrillation
A growing body of evidence has shown that the development and maintenance of AF are associated with dysregulation of the Ca2+ and Na+ handling in atrial myocytes. Ca2+ handling disorder may promote triggered activity, conduction abnormalities, and atrial remodeling (Jalife and Kaur, 2015). The latter two points may facilitate reentry and form the substrate for the maintenance of AF. In patients with and animal models of AF, the Ca2+/calmodulin-dependent protein kinase type-II (CaMKII) complex is activated and phosphorylates several Ca2+-handling proteins, such as RyR2 and phospholamban (PLN) in atrial myocytes. This enhances the Ca2+ leak from the SR, resulting in the triggered activity that serves as the initiation of AF (Li et al., 2012). Elevated cytosolic Ca2+ levels may promote profibrotic and inflammatory pathways that contribute to atrial remodeling, accelerating the formation of the substrate for AF. For instance, Ca2+ mobilization is crucial for the assembly and activation of an inflammatory signaling complex called the NLRP3 inflammasome (Murakami et al., 2012). NLRP3 exacerbates the Ca2+ leak from the SR, leading to ectopic firing in the atrium and enhances pyroptosis mediated by caspase-1, accelerating the formation of AF-maintaining substrates. In addition, intracellular Ca2+ leaks disturb mitochondrial function, leading to an increase in reactive oxygen species (ROS) production and impairment of energetic metabolism, resulting in the oxidation of the calcium-handling proteins, such as RyR2 and exacerbation of the Ca2+ leak, subsequently promoting the initiation and maintenance of AF.
Na+ overload underlies the development of AF. The late Na+ current (INa–L) is enhanced in AF, driving NCX in the reverse mode and increasing the cytosolic Ca2+ levels (Song et al., 2008). Normalizing Na+ overload by selectively suppressing INa–L, such as ranolazine, is a promising therapeutic strategy for AF. Notably, activation of NHE promotes cardiac hypertrophy and HF via the Na+ overload-induced Ca2+ overload pathway (Karmazyn et al., 2008). In the atrial tachycardia-induced canine model, NHE is upregulated and the NHE1 inhibitor prevents effective refractory period abbreviations, delaying the development of atrial contractile dysfunction (Jayachandran et al., 2000). The underlying mechanisms are likely associated with the alleviation of the harmful consequences of increased cytosolic Na+ levels.
The Protective Effect of Sodium-Glucose Cotransporter 2 Inhibitors on Disorganized Ca2+/Na+ Balance
SGLT2i inhibits SGLT2 in the proximal tubules to prevent the reabsorption of glucose and Na+, facilitating their excretion in urine. SGLT2i may decrease the cytosolic Na+ levels at a tissue and cellular level due to its pharmacological action. Karg et al. (2018) reported that SGLT2 inhibition with dapagliflozin resulted in a significant decrease in the Na+ content of the skin. Moreover, INa–L was remarkably reduced in empagliflozin-treated T2D rats (Lee et al., 2019). In isolated ventricular myocytes, Baartscheer et al. (2017) observed that empagliflozin significantly decreased intracellular Na+ and Ca2+ levels within 10 min. This effect was not influenced by extracellular glucose levels. In line with the lack of SGLT2 in cardiac myocytes, it is speculated that SGLT2 is not responsible for the action of empagliflozin. Intriguingly, the authors found that pretreatment with cariporide, an NHE1 inhibitor, abolished the effects of empagliflozin. Thus, it is proposed that empagliflozin is a novel NHE1 inhibitor. Subsequently, Uthman et al. (2018a) confirmed that empagliflozin, dapagliflozin, and canagliflozin directly inhibit the cardiac NHE flux and reduce cytosolic Na+ levels. Molecular docking studies demonstrated that all three SGLT2i efficiently bind to the Na+-binding pocket of NHE with high affinities (Uthman et al., 2018a). The authors postulated that SGLT2i reduces cytosolic Na+ levels by blocking NHE, decreasing intracellular Ca2+ concentration, and ultimately optimizing cardiac function. In the aging and AF atria, NHE activity is upregulated, which may produce Na+ overload and contribute to the development of AF (hui et al., 2008). It is reasonable that SGLT2i can prevent AF by ameliorating Na+ overload induced by the upregulated NHE activity in the atrium despite the lack of direct evidence. Ye et al. (2018) also showed that dapagliflozin could increase phosphorylated adenosine monophosphate kinase (AMPK) to reduce the mRNA expression of NHE1 in cardiac fibroblasts. Compelling evidence shows that NHE is a novel target for SGLT2i. Acute inhibition of NHE by SGLT2i may diminish the incidence of the triggered activity induced by Na+ overload, which has generally been recognized as the primary mechanism for AF. Long-term inhibition of NHE by SGLT2i may reverse the atrial remodeling and alleviate the substrate for AF maintenance through the alleviation of the abnormal downstream NHE signaling. However, some have pointed out that atrial remodeling could not be prevented through NHE1 inhibition (Blaauw et al., 2004). Further studies are warranted to directly explore the effect of SGLT2i on NHE in the fibrillating atria.
There are two additional members of the SGLT family expressed in the heart. One of them is SGLT1, which is upregulated in some diseases, such as T2D (Velmurugan et al., 2019). SGLT1 contributes to the Na+ influx in cardiac myocytes, and this action becomes dominant under pathological conditions. Sawa et al. (2020) report that pretreatment with KGA-2727, a selective SGLT1 inhibitor, protects against myocardial infarction–induced ventricular remodeling and HF in mice. SGLT inhibitors can be more or less selective for SGLT2 vs. SGLT1. The half-maximal inhibitory concentration IC50 of empagliflozin for SGLT1 is 8.3 μmol/L. Thus, SGLT2i may mitigate Na+ overload via SGLT1. Sodium-myoinositol cotransporter 1 (SMIT1) is another example that barely regulates glucose uptake under physiological conditions. However, it activates NADPH oxidase 2 (NOX2) and causes Na+ overload when it is overexpressed (Van Steenbergen et al., 2017). It is proposed that SGLT2i may benefit cardiac function by targeting SMIT1.
Several studies have shown that acute administration of SGLT2i promptly improves cardiac function in patients with HF. Lehrke et al. (2019) reports that treatment with empagliflozin leads to a rapid and sustained improvement of diastolic function in patients with T2D, independent of hemodynamic changes. In the ventricular strips isolated from patients with HF, Pabel et al. (2018) observed that empagliflozin significantly reduced diastolic tension, whereas systolic force was not changed. Furthermore, in anesthetized HF with preserved ejection fraction (HFpEF) rats, intravenous injection of empagliflozin significantly improved diastolic function, whereas systolic contractility was unaffected. Thus, the direct action of SGLT2i on cardiac myocytes targeting Ca2+ handling is speculated. In streptozotocin-induced diabetic rats, Lee et al. (2019) found that chronic administration of empagliflozin for 4 weeks significantly reversed the Ca2+ dysregulation in cardiac myocytes, including attenuation of the SR Ca2+ leak, an increase in Ca2+ transient amplitude, and restoration of SR Ca2+ content. Next, they revealed that decreased phosphorylated RyR2 at serine 2808 (RyR2-pS2808) and increased protein expression of SERCA2a were responsible for the action of empagliflozin. In the ventricular myocytes isolated from patients with HF, Mustroph et al. (2018) observed that exposure to empagliflozin for 24 h remarkably downregulated the activity of CaMKII. Consequently, the detrimental effects of CaMKII overactivity on Ca2+ handling in the failing ventricular myocytes were reversed. It is notable that acute (30 min) empagliflozin exposure did not alter CaMKII activity and did not improve Ca2+ handling disorder in failing ventricular myocytes, whereas the elevated cytosolic Na+ levels were attenuated. Mustroph et al. (2018) suggest that the direct inhibitory effects of empagliflozin on CaMKII or RyR2 were unlikely, and it may be initiated by the attenuation of Na+ overload.
Atrial fibrosis plays an essential role in the pathophysiology of AF, especially for the maintenance of AF (Nattel, 2017). SGLT2i ameliorates adverse cardiac fibrosis and remodeling in patients and animal models (Lee et al., 2017; Uthman et al., 2018b). Recently, Shao et al. (2019) report that empagliflozin significantly reduced the incidence of AF induced by the burst pacing in a rat model of T2D. The underlying mechanism is related to the amelioration of atrial structural and electrical remodeling by empagliflozin. Several studies have shown that the CaMKII cascade on Ca2+ regulation may play an essential role in the atrial remodeling of AF (Mustroph et al., 2017). Byrne et al. (2020) showed that empagliflozin attenuated the NLRP3 level and then mitigated the inflammatory response in mice with HF, reducing cardiac fibrosis and remodeling. Sato et al. (2018) observed that treatment with dapagliflozin might improve systemic metabolic parameters and decrease the epicardial adipose tissue volume in T2D patients, which is highly associated with the increasing incidence and severity of AF. Another obesity study in a rabbit model demonstrated that obesity caused atrial remodeling with prolonged refractoriness, upregulated calcium-handling proteins, and advanced fibrosis. Treatment with SGLT2i prevented this remodeling and led to decreased atrial arrhythmogenesis (Cheng et al., 2020).
Mitochondria and Atrial Fibrillation: Sodium-Glucose Cotransporter 2 Inhibitors Play a Role in Dissolving the Contact
Mitochondrial Function and Ca2+ Cycling
In mitochondria, acetyl-CoA derived from glucose initiates the tricarboxylic acid cycle (TAC), which produces the ATP used for cellular activities. At the same time, ROS that causes an imbalance in ion handling and deterioration of cellular function is also generated through oxidative phosphorylation. Ca2+ is considered a vital regulator of mitochondrial metabolism. Mitochondrial Ca2+ uniporter (MCU) anchored on the inner mitochondrial membrane mainly takes charge of Ca2+ uptake from the cytoplasm during diastole. The mitochondrial membrane potential generated by proton-ATPase drives Ca2+ across the inner membrane via MCU. The free energy of ATP hydrolysis (ΔGATP) supports the proton pump to maintain the electrochemical proton gradients and transport of Ca2+, which can be disordered when ΔGATP is decreased due to mitochondrial dysfunction (Rizzuto et al., 2012; Consolini et al., 2017). Hyperactivation of NHE1 is associated with mitochondrial Ca2+ overload in the rat model that were subjected to coronary artery ligation during the post-infarction remodeling, which further facilitates the opening of the mitochondrial permeability transition pore (MPTP), increasing mitochondrial fragility (Javadov et al., 2005). In the mitochondria, Ca2+ acts as a second messenger to participate in the TAC. On the one hand, Ca2+ is necessary for the activation of ATP synthase and enzymes, such as pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase (O-Uchi et al., 2014). Thus, Ca2+ disorders in mitochondria attenuate the electron transport throughout the respiratory chain reducing the ATP production. On the other hand, Ca2+-dependent NADH can further generate NADPH, which eliminates accumulating ROS and maintains the balance between oxidation and antioxidation by sustaining a reduced form of glutathione. Meanwhile, Ca2+ also activates catalase and glutathione reductase directly to decrease hydrogen peroxide and enhance superoxide dismutase (SOD) (Zhang et al., 2016). Ca2+ efflux is primarily mediated by the NCX that is located on the mitochondrial membrane (mitoNCX).
Mitochondrial Dysfunction and Atrial Fibrillation
AF is tightly connected with the ROS accumulation and increased oxidative stress caused by metabolic dysfunction of mitochondria. Mitochondrial dysfunction participates in AF via the Ca2+ signaling-dependent pathway in atrial myocytes (Opacic et al., 2016). Ca2+ overload accelerates ROS production by enhancing the electron flow into the respiratory chain and inhibiting complexes I and III on the chain (Brookes et al., 2004). Subsequently, increased ROS oxidizes Ca2+ handling proteins, such as RyR2 and CaMKII, inducing the overactivation of these proteins, disrupting Ca2+ homeostasis (Mustroph et al., 2017). The latter further facilitates ROS production in mitochondria, resulting in a vicious circle.
The dynamic mitochondrial abnormality is another trait related to AF. Fission and fusion are the two sides of mitochondrial dynamics. During mitochondrial fusion, the outer membranes are fused through the mediation of mitofusins 1/2 (Mfn1/2), which are dependent on GTPases. Second, optic atrophy 1 (OPA1) promotes the fusion of inner membranes. Thus, the exchange of material becomes easier, and mitochondria can continue to sustain their function, including metabolism and ATP production. Mitochondrial membranes are linked to the SR via Mfn2 in AF and when under oxidative stress, forming a construction called mitochondria-associated membrane (MAM), where Ca2+ microdomains are created. Ca2+ signaling is well-regulated by Mfn2 at the MAM in response to local changes in cytoplasmic Ca2+ induced by transient stress. However, mitochondrial fragmentation and cell death are initiated if the rapid stimulation and oxidative stress persist, which may be a target for AF prevention (Redpath et al., 2013).
For fission, dynamin-related protein 1 (Drp1) is recruited to the surface of mitochondria to initiate this process (Meyer et al., 2017). If mitochondrial fusion is prevented, mass transfer between two mitochondria could be ineffective, leading to a metabolic disorder that results in energetic deficits (Chen et al., 2011). Reduced mitochondrial fusion or increased fission would cause mitochondrial dysfunction related to AF. Mitochondrial dynamics are regulated by a subfamily of the Ras GTPases (Miro proteins), which are Ca2+-dependent (Reis et al., 2009). The movement of mitochondria can be prevented by the Miro proteins due to Ca2+ overload, which further exacerbates the effects of metabolic and dynamic disorders. Other studies have also shown that mitochondrial dysfunction underlies atrial remodeling in experimental and clinical AF. Atrial biopsies from patients with AF display aberrant ATP levels, mitochondrial stress–related chaperone (heat shock protein 10 and 60) upregulation and mitochondrial network fragmentation. Partial blocking or downregulation of MCU may attenuate atrial remodeling by preventing increased mitochondrial Ca2+ influx (Wiersma et al., 2019).
Sodium-Glucose Cotransporter 2 Inhibitors Restore the Metabolic and Dynamic Functions of Mitochondria During Atrial Fibrillation
Improvement of mitochondrial metabolism has been recognized as a therapeutic mechanism of SGLT2i. In a high-fat diet mouse model, canagliflozin protected against obesity-related metabolic disorders by improving mitochondrial function and fatty acid oxidation in adipose tissue (Wei et al., 2020). Sa-Nguanmoo et al. (2017) demonstrated that dapagliflozin remarkably decreased mitochondrial ROS production and mitochondrial membrane depolarization in the brain of obese rats. In ventricular myocytes isolated from overweight insulin-resistant rats, Durak et al. (2018) found that dapagliflozin suppressed oxidative stress in mitochondria reversing the prolonged ventricular-repolarization. SGLT2i also alleviates mitochondrial fusion and fission abnormalities. Increased phosphorylation of Drp1 at S616 and decreased phosphorylation at S637 are strong indicators of overactive mitochondrial fission resulting from myocardial injury (Shen et al., 2018). Empagliflozin increases the AMP/ATP ratio and subsequently activates the AMPK. The AMPK reduces Drp1 phosphorylation at S616 while augmenting the S637 phosphorylation, which suppresses aberrant mitochondrial fission. This enables the resumption of cellular metabolism, improving the diabetic myocardial microvascular injury in mice (Zhou et al., 2018). Empagliflozin also ameliorates the respiratory activities of complex I + II by increasing the uncoupling of the inner membrane, attenuating ROS production, and mitochondrial Ca2+ overload following acute ischemic reperfusion injury (Jespersen et al., 2017). Mfn2 and OPA1 protein expression are decreased in mice that are fed high-fat diets, causing tubular cell dilation and detachment. After treating cells with ipragliflozin for 16 weeks, these alterations were reversed (Takagi et al., 2018).
More recently, Shao et al. (2019) provided direct evidence that SGLT2i can restore mitochondrial function, ameliorate electrical and structural remodeling of the atria, and prevent AF in a T2D rat model. The rat model showed an enlarged left atrial diameter and promoted AF inducibility. These detrimental effects were prevented by treatment with empagliflozin for 8 weeks. In the atrial tissue, mitochondrial respiratory function, mitochondrial membrane potential, and energy generation were damaged. Empagliflozin treatment mitigated the mitochondrial biogenesis by upregulating the expression of peroxisome proliferator-activated receptor-c coactivator 1α (PGC-1α), nuclear respiratory factor-1 (NRF-1), and mitochondrial transcription factor A (TFAM). This effectively improved the mitochondrial function. These three proteins are essential parts of mitochondrial DNA (mtDNA) transcription, which regulates ATP synthesis. Reduced ATP contributes to atrial remodeling during AF (Dong et al., 2016). Shao et al. (2019) proposed that the improvement of mitochondrial function and biogenesis in T2D by SGLT2i is responsible for the prevention of T2D-related AF.
To date, the underlying mechanisms of improved mitochondrial function by SGLT2i have not been completely investigated. NHE1 is expressed in the membrane of cardiac mitochondria. Downregulation of NHE1 inhibits Ca2+-induced MPTP opening and reduces the release of ROS in ventricular myocytes in patients with HF. As mentioned above, mitochondrial function is tightly influenced by Na+ and Ca2+ levels in the mitochondria. SGLT2i are novel NHE blockers and are likely involved in the improvement of mitochondrial function in AF by modulating the mitochondrial Na+ and Ca2+ flux.
Conclusion
SGLT2i are a new class of diabetic medications used for the treatment of T2D. In the last few years, compelling clinical evidence showed that SGLT2i reduces the incidence of AF in patients with T2D and HF. The potential role of SGLT2i in the treatment of AF is emerging. Based on the current knowledge of AF pathophysiology and the pharmacological actions of SGLT2i, it is speculated that SGLT2i potentially prevents AF by ameliorating ion handling and mitochondrial dysfunction. Understanding the therapeutic mechanisms of SGLT2i in AF has gained considerable interest and has developed rapidly. It is expected that a collaborative effort between clinical and basic science researchers will continue to enable the development of a promising treatment for AF.
Author Contributions
XP wrote the manuscript. LL, MZ, QZ, KW, RB, and YR took part in preparing the manuscript. NL prepared and reviewed the manuscript before publication. All authors confirmed that they have read and approved the manuscript and they have met the criteria for authorship.
Funding
This work was supported by the National Science Foundation of China (Grant Nos. 81870244 and 81770318).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allessie, M., Ausma, J., and Schotten, U. (2002). Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 54, 230–246. doi: 10.1016/s0008-6363(02)00258-4
Baartscheer, A., Schumacher, C., Belterman, C., Coronel, R., and Fiolet, J. (2003). [Na+] i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc. Res. 57, 986–995. doi: 10.1016/s0008-6363(02)00848-9
Baartscheer, A., Schumacher, C. A., Wust, R. C., Fiolet, J. W., Stienen, G. J., Coronel, R., et al. (2017). Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 60, 568–573. doi: 10.1007/s00125-016-4134-x
Bers, D. M. (2008). Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49. doi: 10.1146/annurev.physiol.70.113006.100455
Blaauw, Y., Beier, N., Van Der Voort, P., Van Hunnik, A., Schotten, U., and Allessie, M. A. (2004). Inhibitors of the Na+/H+ exchanger cannot prevent atrial electrical remodeling in the goat. J. Cardiovasc. Electrophysiol. 15, 440–446. doi: 10.1046/j.1540-8167.2004.03498.x
Böhm, M., Slawik, J., Brueckmann, M., Mattheus, M., George, J. T., Ofstad, A. P., et al. (2020). Efficacy of empagliflozin on heart failure and renal outcomes in patients with atrial fibrillation: data from the EMPA-REG OUTCOME trial. Eur. J. Heart Fail. 22, 126–135. doi: 10.1002/ejhf.1663
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M., and Sheu, S.-S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833.
Byrne, N. J., Matsumura, N., Maayah, Z. H., Ferdaoussi, M., Takahara, S., Darwesh, A. M., et al. (2020). Empagliflozin blunts worsening cardiac dysfunction associated with reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) inflammasome activation in heart failure. Circ. Heart Fail. 13:e006277. doi: 10.1161/CIRCHEARTFAILURE.119.006277
Chen, H.-Y., Huang, J.-Y., Siao, W.-Z., and Jong, G.-P. (2020). The association between SGLT2 inhibitors and new-onset arrhythmias: a nationwide population-based longitudinal cohort study. Cardiovasc. Diabetol. 19:73.
Chen, Y., Liu, Y., and Dorn, G. W. II (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331. doi: 10.1161/CIRCRESAHA.111.258723
Cheng, W. H., Lo, L. W., Chou, Y.-H., Liu, S.-H., Tsai, T.-Y., Lin, W.-L., et al. (2020). D-PO06-009 - empagliflozin ameliorates the atrial remodeling and arrhythmogenesis in overweight rabbit model. Heart Rhythm 17, S574. doi: 10.1016/j.hrthm.2020.04.011
Cheung, C. C., George Ou, D. K., Chen, P. P., Papatheodorou, S., and Leung, J. M. (2020). D-PO06-076 - Arrhythmia Outcomes In Patients Treated With Sodium Glucose Cotransporter-2 Inhibitors: systematic Review And Meta-analysis. Heart Rhythm 17, S600–S601. doi: 10.1016/j.hrthm.2020.04.011
Chugh, S. S., Havmoeller, R., Narayanan, K., Singh, D., Rienstra, M., Benjamin, E. J., et al. (2014). Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation 129, 837–847. doi: 10.1161/CIRCULATIONAHA.113.005119
Colilla, S., Crow, A., Petkun, W., Singer, D. E., Simon, T., and Liu, X. (2013). Estimates of current and future incidence and prevalence of atrial fibrillation in the U.S. adult population. Am. J. Cardiol. 112, 1142–1147. doi: 10.1016/j.amjcard.2013.05.063
Consolini, A. E., Ragone, M. I., Bonazzola, P., and Colareda, G. A. (2017). Mitochondrial bioenergetics during ischemia and reperfusion. Adv. Exp. Med. Biol. 982, 141–167. doi: 10.1007/978-3-319-55330-6_8
Dobrev, D., and Wehrens, X. H. (2017). Calcium-mediated cellular triggered activity in atrial fibrillation. J. Physiol. 595, 4001–4008. doi: 10.1113/jp273048
Dong, J., Zhao, J., Zhang, M., Liu, G., Wang, X., Liu, Y., et al. (2016). β3-adrenoceptor impairs mitochondrial biogenesis and energy metabolism during rapid atrial pacing-induced atrial fibrillation. J. Cardiovasc. Pharmacol. Ther. 21, 114–126. doi: 10.1177/1074248415590440
Durak, A., Olgar, Y., Degirmenci, S., Akkus, E., Tuncay, E., and Turan, B. (2018). A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 17, 144–144. doi: 10.1186/s12933-018-0790-0
Haïssaguerre, M., Jaïs, P., Shah, D. C., Takahashi, A., Hocini, M., Quiniou, G., et al. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666. doi: 10.1056/NEJM199809033391003
hui, Y., junzhu, C., and jianhua, Z. (2008). Gap junction and Na+-H+ exchanger alternations in fibrillating and failing atrium. Int. J. Cardiol. 128, 147–149. doi: 10.1016/j.ijcard.2007.06.070
Ibrahim, M., Navaratnarajah, M., Siedlecka, U., Rao, C., Dias, P., Moshkov, A. V., et al. (2012). Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca(2+)-induced Ca(2+)release in a rodent model of heart failure. Eur. J. Heart Fail. 14, 571–580. doi: 10.1093/eurjhf/hfs038
Jalife, J., and Kaur, K. (2015). Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 25, 475–484. doi: 10.1016/j.tcm.2014.12.015
Javadov, S., Huang, C., Kirshenbaum, L., and Karmazyn, M. (2005). NHE-1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J. Mol. Cell. Cardiol. 38, 135–143. doi: 10.1016/j.yjmcc.2004.10.007
Jayachandran, J. V., Zipes, D. P., Weksler, J., and Olgin, J. E. (2000). Role of the Na(+)/H(+) exchanger in short-term atrial electrophysiological remodeling. Circulation 101, 1861–1866. doi: 10.1161/01.cir.101.15.1861
Jespersen, N. R., Lassen, T., Hjortbak, M. V., Stottrup, N., and Botker, H. (2017). Sodium glucose transporter 2 (SGLT2) inhibition does not protect the myocardium from acute ischemic reperfusion injury but modulates post-ischemic mitochondrial function. Cardiovasc. Pharmacol. Open Access. 6, 2–4.
Juni, R. P., Kuster, D. W., Goebel, M., Helmes, M., Musters, R. J., van der Velden, J., et al. (2019). Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl. Sci. 4, 575–591. doi: 10.1016/j.jacbts.2019.04.003
Karam, B. S., Chavez-Moreno, A., Koh, W., Akar, J. G., and Akar, F. G. (2017). Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 16:120.
Karg, M. V., Bosch, A., Kannenkeril, D., Striepe, K., Ott, C., Schneider, M. P., et al. (2018). SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: a randomised controlled trial. Cardiovasc. Diabetol. 17:5. doi: 10.1186/s12933-017-0654-z
Karmazyn, M., Kilić, A., and Javadov, S. (2008). The role of NHE-1 in myocardial hypertrophy and remodelling. J. Mol. Cell. Cardiol. 44, 647–653. doi: 10.1016/j.yjmcc.2008.01.005
Lee, T. I., Chen, Y. C., Lin, Y. K., Chung, C. C., Lu, Y. Y., Kao, Y. H., et al. (2019). Empagliflozin attenuates myocardial sodium and calcium dysregulation and reverses cardiac remodeling in streptozotocin-induced diabetic rats. Int. J. Mol. Sci. 20:1680. doi: 10.3390/ijms20071680
Lee, T. M., Chang, N. C., and Lin, S. Z. (2017). Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 104, 298–310. doi: 10.1016/j.freeradbiomed.2017.01.035
Lehrke, M., Marx, N., Bohm, M., Altiok, E., Keszei, A. P., Schuh, A., et al. (2019). Empagliflozin leads to a rapid and sustained improvement of diastolic function in patients with type 2 diabetes independent of hemodynamic changes. Paper Presented at the Circulation, Aachen.
Li, C., Zhang, J., Xue, M., Li, X., Han, F., Liu, X., et al. (2019). SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 18:15.
Li, N., Wang, T., Wang, W., Cutler, M. J., Wang, Q., Voigt, N., et al. (2012). Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ. Res. 110, 465–470. doi: 10.1161/circresaha.111.253229
Meyer, J. N., Leuthner, T. C., and Luz, A. L. (2017). Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391, 42–53. doi: 10.1016/j.tox.2017.07.019
Murakami, T., Ockinger, J., Yu, J., Byles, V., McColl, A., Hofer, A. M., et al. (2012). Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 109, 11282–11287. doi: 10.1073/pnas.1117765109
Mustroph, J., Neef, S., and Maier, L. S. (2017). CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 176, 22–31. doi: 10.1016/j.pharmthera.2016.10.006
Mustroph, J., Wagemann, O., Lücht, C. M., Trum, M., Hammer, K. P., Sag, C. M., et al. (2018). Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 5, 642–648. doi: 10.1002/ehf2.12336
Nattel, S. (2017). Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin. Electrophysiol. 3, 425–435. doi: 10.1016/j.jacep.2017.03.002
Opacic, D., van Bragt, K. A., Nasrallah, H. M., Schotten, U., and Verheule, S. (2016). Atrial metabolism and tissue perfusion as determinants of electrical and structural remodelling in atrial fibrillation. Cardiovasc. Res. 109, 527–541. doi: 10.1093/cvr/cvw007
O-Uchi, J., Ryu, S.-Y., Jhun, B. S., Hurst, S., and Sheu, S.-S. (2014). Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid. Redox Signal. 21, 987–1006. doi: 10.1089/ars.2013.5681
Pabel, S., Wagner, S., Bollenberg, H., Bengel, P., Kovács, Á, Schach, C., et al. (2018). Empagliflozin directly improves diastolic function in human heart failure. Eur. J. Heart Fail. 20, 1690–1700. doi: 10.1002/ejhf.1328
Pistoia, F., Sacco, S., Tiseo, C., Degan, D., Ornello, R., and Carolei, A. (2016). The Epidemiology of Atrial Fibrillation and Stroke. Cardiol. Clin. 34, 255–268. doi: 10.1016/j.ccl.2015.12.002
Poulet, C., Wettwer, E., Grunnet, M., Jespersen, T., Fabritz, L., Matschke, K., et al. (2015). Late sodium current in human atrial cardiomyocytes from patients in sinus rhythm and atrial fibrillation. PLoS One 10:e0131432. doi: 10.1371/journal.pone.0131432
Redpath, C. J., Bou Khalil, M., Drozdzal, G., Radisic, M., and McBride, H. M. (2013). Mitochondrial hyperfusion during oxidative stress is coupled to a dysregulation in calcium handling within a C2C12 cell model. PLoS One 8:e69165. doi: 10.1371/journal.pone.0069165
Reis, K., Fransson, Å., and Aspenström, P. (2009). The Miro GTPases: at the heart of the mitochondrial transport machinery. FEBS Lett. 583, 1391–1398. doi: 10.1016/j.febslet.2009.04.015
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Sa-Nguanmoo, P., Tanajak, P., Kerdphoo, S., Jaiwongkam, T., Pratchayasakul, W., Chattipakorn, N., et al. (2017). SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 333, 43–50. doi: 10.1016/j.taap.2017.08.005
Santangeli, P., and Marchlinski, F. E. (2017). Techniques for the provocation, localization, and ablation of non-pulmonary vein triggers for atrial fibrillation. Heart Rhythm 14, 1087–1096. doi: 10.1016/j.hrthm.2017.02.030
Sato, T., Aizawa, Y., Yuasa, S., Kishi, S., Fuse, K., Fujita, S., et al. (2018). The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 17:6.
Sawa, Y., Saito, M., Ishida, N., Ibi, M., Matsushita, N., Morino, Y., et al. (2020). Pretreatment with KGA-2727, a selective SGLT1 inhibitor, is protective against myocardial infarction-induced ventricular remodeling and heart failure in mice. J. Pharmacol. Sci. 142, 16–25. doi: 10.1016/j.jphs.2019.11.001
Scheen, A. J. (2014). Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin. Pharmacokinet. 53, 213–225. doi: 10.1007/s40262-013-0126-x
Shao, Q., Meng, L., Lee, S., Tse, G., Gong, M., Zhang, Z., et al. (2019). Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 18:165. doi: 10.1186/s12933-019-0964-4
Shen, Y.-L., Shi, Y.-Z., Chen, G.-G., Wang, L.-L., Zheng, M.-Z., Jin, H.-F., et al. (2018). TNF-α induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int. J. Mol. Med. 41, 2317–2327. doi: 10.3892/ijmm.2018.3385
Song, Y., Shryock, J. C., and Belardinelli, L. (2008). An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 294, H2031–H2039. doi: 10.1152/ajpheart.01357.2007
Takagi, S., Li, J., Takagaki, Y., Kitada, M., Nitta, K., Takasu, T., et al. (2018). Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Invest. 9, 1025–1032. doi: 10.1111/jdi.12802
Uthman, L., Baartscheer, A., Bleijlevens, B., Schumacher, C. A., Fiolet, J. W. T., Koeman, A., et al. (2018a). Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 61, 722–726. doi: 10.1007/s00125-017-4509-7
Uthman, L., Baartscheer, A., Schumacher, C. A., Fiolet, J. W., Kuschma, M. C., Hollmann, M. W., et al. (2018b). Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front. Physiol. 9:1575. doi: 10.3389/fphys.2018.01575
Van Steenbergen, A., Balteau, M., Ginion, A., Ferté, L., Battault, S., Ravenstein, C. M., et al. (2017). Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci. Rep. 7:41166. doi: 10.1038/srep41166
Velmurugan, S., Hoskins, A., Despa, F., and Despa, S. (2019). SGLT-mediated Na+ overload results in oxidative stress and abnormal SR Ca2+ release in diabetic hearts. Circ. Res. 125(Suppl._1):A766.
Voigt, N., Li, N., Wang, Q., Wang, W., Trafford, A. W., Abu-Taha, I., et al. (2012). Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. doi: 10.1161/circulationaha.111.067306
Wei, D., Liao, L., Wang, H., Zhang, W., Wang, T., and Xu, Z. (2020). Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via PPARα in vivo and in vitro. Life Sci. 247:117414. doi: 10.1016/j.lfs.2020.117414
Wiersma, M., van Marion, D., Wüst, R. C., Houtkooper, R. H., Zhang, D., de Groot, N., et al. (2019). Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 8:1202. doi: 10.3390/cells8101202
Ye, Y., Jia, X., Bajaj, M., and Birnbaum, Y. (2018). Dapagliflozin attenuates Na(+)/H(+) Exchanger-1 in cardiofibroblasts via AMPK activation. Cardiovasc. Drugs Ther. 32, 553–558. doi: 10.1007/s10557-018-6837-3
Zelniker, T. A., Bonaca, M. P., Mosenzon, O., Kuder, J. F., Murphy, S. A., Furtado, R. H., et al. (2019). Effect of dapagliflozin on atrial fibrillation/flutter in patients with type 2 diabetes mellitus: insights from the DECLARE-TIMI 58 Trial. Circulation 140(Suppl._1):A15581.
Zhang, J., Wang, X., Vikash, V., Ye, Q., Wu, D., Liu, Y., et al. (2016). ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016:4350965. doi: 10.1155/2016/4350965
Zhou, H., Wang, S., Zhu, P., Hu, S., Chen, Y., and Ren, J. (2018). Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 15, 335–346. doi: 10.1016/j.redox.2017.12.019
Zinman, B., Wanner, C., Lachin, J. M., Fitchett, D., Bluhmki, E., Hantel, S., et al. (2015). Empagliflozin, Cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 373, 2117–2128. doi: 10.1056/NEJMoa1504720
Keywords: sodium-glucose cotransporter 2 inhibitors, atrial fibrillation, calcium handling, sodium-hydrogen exchanger isoform 1, mitochondria
Citation: Peng X, Li L, Zhang M, Zhao Q, Wu K, Bai R, Ruan Y and Liu N (2020) Sodium-Glucose Cotransporter 2 Inhibitors Potentially Prevent Atrial Fibrillation by Ameliorating Ion Handling and Mitochondrial Dysfunction. Front. Physiol. 11:912. doi: 10.3389/fphys.2020.00912
Received: 03 April 2020; Accepted: 08 July 2020;
Published: 04 August 2020.
Edited by:
Ademuyiwa S. Aromolaran, Masonic Medical Research Institute (MMRI), United StatesReviewed by:
Antonius Baartscheer, University of Amsterdam, NetherlandsCoert J. Zuurbier, Amsterdam University Medical Center (UMC), Netherlands
Copyright © 2020 Peng, Li, Zhang, Zhao, Wu, Bai, Ruan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nian Liu, bGl1bmlhbjE5NzNAaG90bWFpbC5jb20=