
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 14 July 2020
Sec. Renal Physiology and Pathophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00735
This article is part of the Research TopicKidney and Distant Organ Crosstalk in Health and DiseaseView all 16 articles
 Carolina Victoria Cruz Junho1
Carolina Victoria Cruz Junho1 Wellington Caio-Silva1†
Wellington Caio-Silva1† Mayra Trentin-Sonoda2†
Mayra Trentin-Sonoda2† Marcela Sorelli Carneiro-Ramos1*
Marcela Sorelli Carneiro-Ramos1*Calcium/calmodulin-dependent protein kinases (CaMKs) are key regulators of calcium signaling in health and disease. CaMKII is the most abundant isoform in the heart; although classically described as a regulator of excitation–contraction coupling, recent studies show that it can also mediate inflammation in cardiovascular diseases (CVDs). Among CVDs, cardiorenal syndrome (CRS) represents a pressing issue to be addressed, considering the growing incidence of kidney diseases worldwide. In this review, we aimed to discuss the role of CaMK as an inflammatory mediator in heart and kidney interaction by conducting an extensive literature review using the database PubMed. Here, we summarize the role and regulating mechanisms of CaMKII present in several quality studies, providing a better understanding for future investigations of CamKII in CVDs. Surprisingly, despite the obvious importance of CaMKII in the heart, very little is known about CaMKII in CRS. In conclusion, more studies are necessary to further understand the role of CaMKII in CRS.
Calmodulin (CaM) is a low-molecular-weight protein highly conserved in the eukaryotes (Clapham, 2007). CaM was discovered in 1970 as a calcium (Ca2+) regulator in the brain, responsible for the nucleotide phosphodiesterase. It was first mentioned as a Ca2+-dependent regulator (Kakiuchi and Yamazaki, 1970). Since the origin of eukaryotes, the amino acids that compound CaM have not changed at all (Friedberg and Rhoads, 2001). It plays a fundamental role in every cell by amplification of the Ca2+ signal (Clapham, 2007). Ca2+ is a versatile messenger molecule implied in many basic processes, such as contraction, potentiation, cell proliferation and apoptosis, and others (Carafoli and Krebs, 2016). To maintain a homeostasis of Ca2+ in the intracellular environment, it has many mechanisms and signaling paths that help to establish a gradient of this ion, holding at approximately 2 mM (Clapham, 2007). One of them is the CaM that triggers conformational changes in response to Ca2+ oscillations (Carafoli and Krebs, 2016). In other words, when Ca2+ binds to CaM, it induces a structural modification, forming a Ca2+/CaM complex (Clapham, 2007).
There is a vast quantity of proteins that can bind to Ca2+/CaM via an α-helical region. This region is composed of approximately 20 amino acids, positively charged and containing hydrophobic residues (Islam, 2020). To amplify the signal generated by Ca2+, this complex activates or deactivates phosphorylation pathways, targeting a protein kinase dependent on Ca2+/CaM (CaMK) (Clapham, 2007). In other words, the binding of Ca2+/CaM and phosphorylate serine/threonine residues of target proteins are the main trigger of CaMKs activation, initiating signaling activation of the substrates (Takemoto-Kimura et al., 2017).
The Ca2+/CaM-stimulated protein kinases are divided based their substrate specificity. They can be restricted to a small number of substrates (Islam, 2020) while the multifunctional kinases have wide specificity and regulate multiple functions in the same and different cell types (Skelding et al., 2011). Restricted CaMK have three main families: phosphorylase kinase (PhK), elongation factor 2 kinase (eEF2K), and myosin light chain kinase (MLCK) (Skelding and Rostas, 2012). These families do not share common domains or structures, making them more specific to certain stimuli and pathways. On the other hand, the multifunctional kinases control many cell functions in different cell types, which makes them a powerful controller of other kinases. Regulation of Ca2+ dynamics is the most basal method to control the function of a kinase, mainly intracellular concentration of Ca2+ (Skelding and Rostas, 2012). The main multifunctional families of kinases proteins are CaMKI, CaMKIV, CaMKK, and CaMKII (Hudmon and Schulman, 2002).
The CaMKI family constitutes four elements, each of them encoding a different gene: CAMK1 (CaMKIα), PNCK (CaMKIβ/Pnck), CAMK1G (CaMKIγ/CLICK3), and CAMK1D (CaMKIδ/CKLiK), which are found in higher quantity in mice brains (Picciotto et al., 1995). CaMKI function is observed in many cellular activities, including synapsis in terminal nerves, motility, axon growth, synthesis of aldosterone, and the cell cycle (Condon et al., 2002; Skelding et al., 2011). It is known that CaMKI translocates to the nucleus mediated by CRM1 complex after an influx of intracellular Ca2+ induced by potassium depolarization or glutamate (Sakagami et al., 2005). CaMKIV only encodes one gene, CAMK4, coding the monomerics isofroms: α and β (Skelding and Rostas, 2020). CaMKIV is observed in the regulation of cyclic AMP, plasticity, fear memory, inflammatory sensibility, and control of the cell cycle (Skelding and Rostas, 2020). The CaMKIV can translocate between the cytoplasm and nucleus. For that action, it involves some catalytic activity as catalytically inactive CaMKIV remains in the cytoplasm (Lemrow et al., 2004).
Ca2+/CaM-stimulated protein kinase kinase (CaMKK), CAMKK1 and CAMKK2, produce CaMKKα and CaMKKβ, respectively. They are responsible for many functions (Skelding and Rostas, 2020). Studies show this kinase’s function, suggesting that CaMKK translocates to the nucleus under stimulation, and the inhibition of translocation directly implies the deactivation of monocytic cells (Guest et al., 2008). CaMKI, CaMKIV, and CaMKK share the same signaling pathway. It is called the Ca2+/CaM-dependent kinase cascade (Tokumitsu and Soderling, 1996). This pathway is mainly related to several cellular processes, including glucose homeostasis, hematopoietic stem cell maintenance, cell proliferation, apoptosis, and normal immune cell function (Skelding and Rostas, 2012). Last but not least, among the CaMKs, we have the CaMKII. Its function is further explained in the next section. Together with this, the present study aims to focus on CaMKII relevance and how it is implied specifically during cardiorenal syndrome (CRS).
Among the CaMKs, the most abundant in the heart is CaMKII (Luczak and Anderson, 2014). CaMKII is a serine-threonine kinase, and it was first identified in the central nervous system, where it represents only 2% of the total protein. It was later discovered that this enzyme is present in many tissues, including the pancreas, where it has an important role in the secretion of insulin, and in heart tissue, where it is responsible for Ca2+ homeostasis (Yamauchi, 2005).
The functional CaMKII enzyme structure is formed by 12 subunits (dodecameric); each monomer has an N-terminal catalytic domain and a C-terminal domain with a regulatory domain in the middle as shown in Figure 1. The catalytic domain is blocked by the regulatory domain in an autoinhibitory way, keeping the enzyme inactive (Figure 1). CaMKII becomes active when the Ca2+/CaM complex binds to the binding site of the regulatory domain of CaM. This provokes a successive autophosphorylation of Thr287 monomers, causing conformational changes that expose the catalytic domain and enable the kinase activity at all (Beckendorf et al., 2018). As proposed by Beckendorf et al. (2018), the Thr287 autophosphorylation causes what they denominate “CaM trapping,” increasing the CaM binding affinity to 1000-fold, maintaining the CaMKII activity even under low Ca2+ conditions (Beckendorf et al., 2018). Besides the activation via Ca2+/CaM that is dependent on several Ca2+ factors, such as the total Ca2+ available in a dose-dependent manner and its spark frequency, amplitude, and duration, the CaMKII can be activated via post-translational modifications as, for example, by reactive oxygen species (ROS) (Beckendorf et al., 2018).
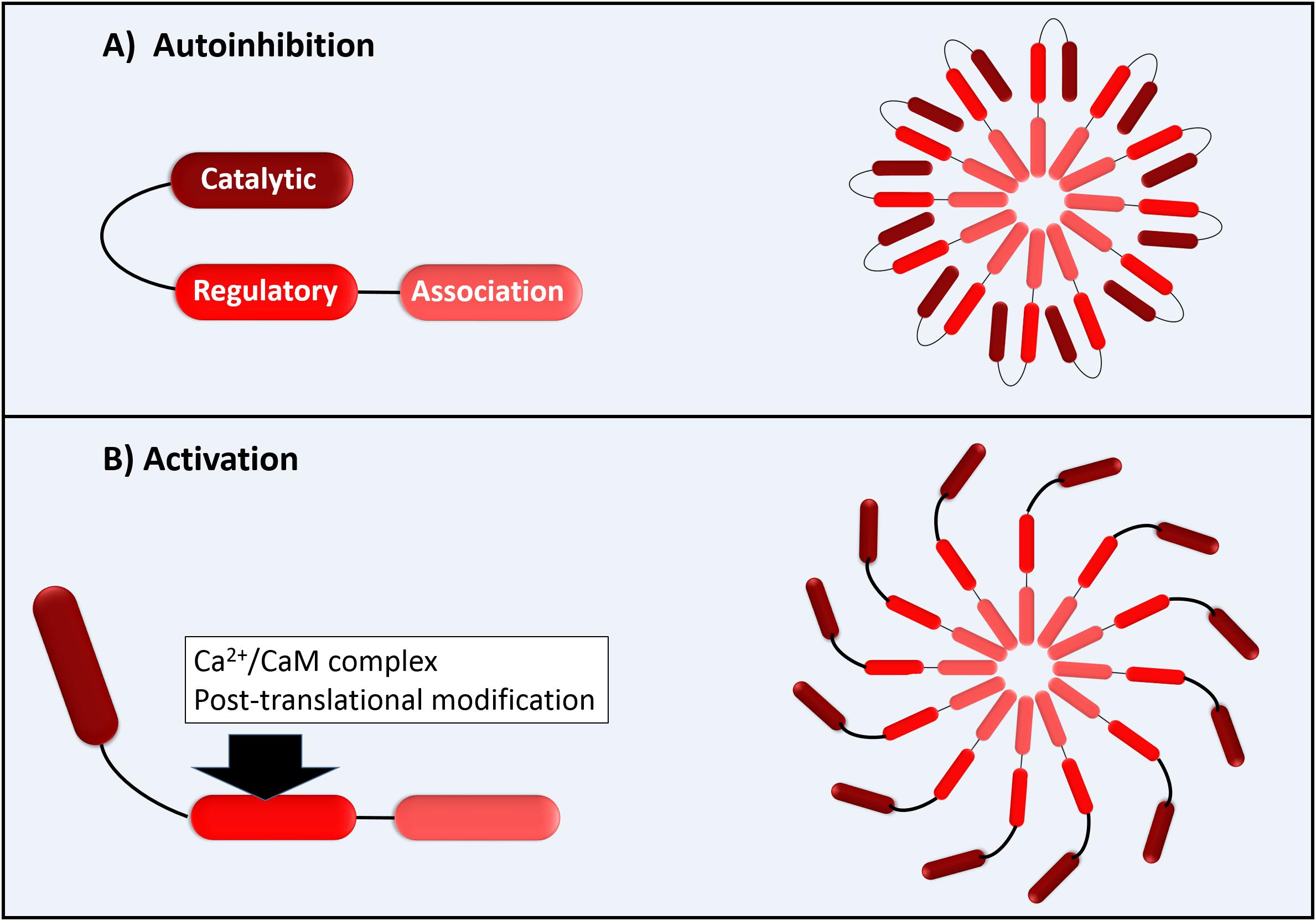
Figure 1. Schematic illustration of CaMKII with permission and adapted from Bussey and Erickson (2018). (A) Autoinhibition of each 12 CaMKII monomers; (B) Activation of CaMK monomers by Ca2+/CaM complex (direct activation) or by post-translational modifications as by ROS (autonomous activation).
CaMKII is observed in eukaryotes in four distinct isoforms (α, β, γ, δ). They encode four distinct genes (Luczak and Anderson, 2014). CaMKIIα and β are expressed mainly in the neural system while the CaMKIIδ and γ isoforms are predominantly expressed in cardiac tissue (Backs et al., 2009). This kinase modulates numerous biological processes, such as the Ca2+ homoeostasis, excitement of membrane, cell cycle, cytoskeletal organization, and gene expression (Yamauchi, 2005; Backs et al., 2009).
As the contraction is a Ca2+-dependent process, it is indisputable that CaMKII performs an important role in the heart. It regulates Ca2+-handling proteins by facilitating the L-type Ca2+ channel (LTCC) phosphorylation (Bers and Morotti, 2014); promotes phosphorylation of phospholamban (PLN) at site T17, promoting its dissociation from SERCA2a and, thus increasing SERCA2a activity; and phosphorylates ryanodine (RYR) at site S2814, consequently improving its opening probability (Mattiazzi and Kranias, 2014). Hyperphosphorylation caused by CaMKII of this site leads to an increase of spontaneous elementary Ca2+-release events from the sarcoplasmic reticulum especially during diastole (Mustroph et al., 2017). In pathological situations, increased diastolic Ca2+ in cytosol can activate the electrogenic NCX, which can cause delayed afterdepolarizations. The last ones can trigger atrial and ventricular arrhythmias (Mustroph et al., 2017). In other words, CaMKII is a pro-arrhythmogenic protein in the heart. Besides arrhythmias, CaMKII is strongly increased during myocardial injury (Ren et al., 2003), atrial fibrillation (Liu et al., 2019), cardiac hypertrophy (Kamada et al., 2019), ischemia/reperfusion injury (Rajtik et al., 2016), and heart failure (HF) (Beckendorf et al., 2018). Its inhibition has been constantly suggested as treatment for these pathologies (Rokita and Anderson, 2012; Cipolletta et al., 2015).
There are studies using transgenic (TG) overexpression of CaMKIIδ, to evaluate its effect during pathogenesis. TG mice developed severe HF and susceptibility to induced ventricular arrhythmias via programmed electrical stimulation (Maier and Bers, 2007). The cardiomyocytes isolated from TG mice presented increased diastolic Ca2+ leak together with prolonged action potential and increased incidence of early afterdepolarization (a peculiarity of CaMK-induced increased late INa) (Wagner et al., 2006).
In summary, TG mice led to a worsening of the arrhythmogenic profile of the mice in addition to increasing their risk of life. On the other hand, the silencing of CaMKII brings hope for the treatment of chronic or acute heart diseases, presenting itself as a potential clinical therapist against these pathologies.
The kidneys and the heart share an important role in the biochemical maintenance of homeostatic function of extracellular fluid. In general, while the heart is responsible for providing nutrients and oxygen-rich fluids to the body through blood flow, the kidney is responsible for providing electrolytes, acid-base homeostasis, vitamin D activation, and erythropoietin synthesis (Ronco and Di Lullo, 2014). Understanding how the kidney and heart relate has been a challenge since the Middle Ages, when Aetius of Amida initially attempted to explain fluid overlay by attributing it to kidney hardening (Diamandopoulos, 1999).
Fast-forwarding to contemporary times, several studies approach this topic with more precision. Nowadays, it is well established that an adverse imbalance in hemodynamics caused by impaired kidney function can directly impact the heart (Anavekar et al., 2004). The pathological crosstalk between both organs is called cardiorenal syndrome (CRS), in which cardiac and renal dysfunction overlap; a disorder in one organ leads to acute or chronic dysfunction of the other organ (Ronco, 2011; Ronco and Di Lullo, 2014).
CRS is divided into two major groups, the cardiorenal and nephron-cardiac, dependent on the origin of the primary pathology, both of which can be acute or chronic (Ronco and Di Lullo, 2014). Type 1 and 2 CRS are considered cardiorenal and are characterized by a loss of cardiac function leading to renal injury (Damman et al., 2007). CRS type 1, known as acute, is described by acute cardiac injury leading a renal one through hemodynamic mechanisms (Di Lullo et al., 2017). The presence of acute decompensated HF leads to decreased renal function due to low renal arterial flow and decrease of the glomerular filtration rate. Once hemodynamic parameters are restored, renal and cardiac homeostasis is also restored (Hanada et al., 2012). Type 2 CRS rises as a consequence of chronic abnormalities in cardiac function that cause renal injury or dysfunction. Examples of such abnormalities are conditions including atrial fibrillation, congenital heart disease, pericarditis constrictions, and chronic cardiac ischemia. Determining whether the CRS is a type 1 or 2 represents a challenge for clinicians since the majority of diagnostics are made when both organs are already injured.
The other two types of CRS (3 and 4) are described as nephron-cardiac syndromes, where the renal injury leads to cardiac dysfunction (Damman et al., 2007). Type 3 CRS is defined as acute nephron-cardiac syndrome, occurring when acute renal failure leads to the development of acute cardiac injury. It is intimately related to events triggering increased inflammatory processes, such as oxidative stress and secretion of neurohormones (Di Lullo et al., 2017). Type 4 CRS is studied as chronic nephron-cardiac disease, initiated by chronic kidney disease (CKD), leading to cardiovascular disease. Approximately 70–80% of patients with end-stage renal disease present type 4 CRS, presenting cardiac complications, such as infarct and long-term arrhythmias (Di Lullo et al., 2017).
Finally, CRS type 5 is a systemic disorder that reaches both organs simultaneously. Many factors have been suggested to contribute to these conditions, for instance, sepsis, infections, drugs, toxins, and diabetes. It is important to note that acute type 5 CRS can overlap a chronic injury (Ronco and Di Lullo, 2014). CRS type 5 results in cardiac and renal dysfunction coming from a larger and systematic situation. Hence, differently from other types of CRS, type 5 is relatively easier to identify the starting point of the CRS.
The study of CRS is of great relevance for clinical treatment, considering that cardiovascular diseases represent the main cause of death in the United States for at least the last 15 years, according to the Centers for Disease Control and Prevention (CDC)1. In addition, incidence of CVDs have been increasing alongside incidence of CKDs worldwide (Thomas et al., 2017), showing the importance of studies regarding CRS type 4, for example. Besides, CKDs significantly affects the regulation of cardiac Ca2+ by mechanisms not yet clarified (Ke et al., 2020).
CaMKII has already been described as a cause of many heart dysfunctions, such as arrhythmia, hypertrophy, and infarction (Yoo et al., 2018; Rusciano et al., 2019), and has been demonstrated to play a critical role in CRS types 1 and 2. The CaMKII inhibitor (CaMK2n) has been shown to protect cardiac dysfunction and ameliorate injuries observed in metabolic syndrome (Prasad et al., 2015). However, Alfazema and collaborators showed, recently, in a translational study, that deletion of CaMK2n1, diminishes CaMKII activity in the kidney and heart without affecting adipose tissue (Alfazema et al., 2019). Yet there are few studies involving both organs in a systemic profile.
It is known that elements, such as the immune system, can mediate the communication between them. In an inflammatory process as observed during CKD and chronic heart failure (CHF), cytokines are released by circulating and tissue-resident inflammatory cells (monocytes mainly) and play an important role in the progression of these diseases (Yogasundaram et al., 2019). Since Ca2+ has been associated with several events of the inflammatory response, including the activation of T cells and awakening memory (Boubali et al., 2012). The CaMKII ability to act as an intracellular sensor of Ca2+ makes it a crucial regulator in the inflammatory process (Rusciano et al., 2019). On the other hand, the Ca2+-independent form of CaMKIIγ also modulates cell death and T cell memory formation (Bui et al., 2000). Studies have already shown that CaMKII is capable of modulating NF-κB, IL-10, IL-2, and IL-4 production (Rusciano et al., 2019).
In CRS, the tissue injury is strongly followed by inflammation. Many inflammatory cytokines are enhanced in experimental models of renal ischemia (TNF-a, IL-1, and IL-6) and also markers, including the factor nuclear kappa B (NF-kB), which is very important for cell signaling during inflammatory processes (Trentin-Sonoda et al., 2015). Given that, some studies present the participation of inflammation as a cause of CKD progression in CHF patients (House et al., 2019; Marsico et al., 2019). During cardiac ischemia, myocytes release inflammatory cytokines (Colombo et al., 2012), and they can reach renal tissue, inducing local inflammation, apoptosis, or oxidative stress (Ronco and Di Lullo, 2014).
Colombo et al. (2012) suggest that inflammation during CRS is controlled by positive feedback mechanisms. Inflammation initiates vascular dysfunction, reducing the myocardial contractility and increasing myocardial cell death, linking CRS to apoptosis (Virzì et al., 2015b). Inflammation also causes progressive renal dysfunction and fibrosis, which continues to injure the organ, maintaining the cycle. Additionally, inflammation leads to the release of renin, activating the renin-angiotensin-aldosterone system (RAAS), which activates the sympathetic nervous system (SNS) by increasing serum norepinephrine concentrations and is the cause of ROS release from the inflammatory cells (Bongartz et al., 2005).
A previous study suggests that the toll-like receptor (TLR) pathway is linked with the activation of CaMKII in a model of myocardial infarction (Singh et al., 2012). Recently, Wu and collaborators demonstrated that miR-148a attenuates ischemia/reperfusion injury in liver, once CaMKIIα represses the TLR4 signaling pathway in vivo and in vitro, decreasing the production of pro-inflammatory factors (Zheng et al., 2018). Moreover, activation of CaMKII in macrophages is initiated by a significant trigger elevation of intracellular Ca2+. This progresses to the activation of myeloid differentiation factor 88 (MyD88)- dependent and Toll/interleukin-1 receptor domain, prompting proinflammatory factors. Ca2+/CaMKII is fundamental for macrophage response once it requires the complete activation of the TLR pathway (Liu X. et al., 2008).
As mentioned above and summarized by Rusciano et al. (2019) there is evidence that suggests the important role of CaMKII in many cardiac pathologies involving inflammation due its ability to enhance pro-inflammatory signaling and its responsiveness to inflammation by dysregulating the Ca2+ balance. In the kidneys, CaMKIIβ expression is associated to aldosterone-induced fibrosis (Park et al., 2018; Zhang et al., 2018) and also shows that the increase in mitochondrial fragmentation observed in hyperglycemia stress-mediated renal damage is due to the JNK-CaMKII-Fis1 pathway (Zhang et al., 2018).
As discussed above, silencing CaMKII has great therapeutic value. There are many blockers and inhibitors used nowadays in research. The most known are KN-93 and AC3-I.
The first inhibitor described was KN-62 in 1990 (Tokumitsu et al., 1990). One year later, in 1991, Sumi M and collaborators described a new and more selective inhibitor called KN-93 (Sumi et al., 1991). They act by blocking the enveloping of Ca2+/CaM around the CaM-binding segment and automatically freeing this segment from the catalytic domain (Pellicena and Schulman, 2014). In other words, it is a CaM-competitive CaMKII inhibitor. It was already discovered that KN-93 can directly block the potassium current IKr and potassium voltaged channels, preventing arrhythmic properties of CamKII (Mustroph et al., 2017). An important item to bring up is that KN-62/93 binds to the holoenzyme and directly steps in the interaction of Ca2+/CaM but does not directly bind to CaM (Sumi et al., 1991). Besides belonging to a family of CaM antagonists, KN-93 cannot avoid the activity of autophosphorylation of CaMKII (Wong et al., 2019).
Some studies have proven the efficiency of KN-93 in heart pathologies in several animal models. Under in vitro and in vivo stimulations with isoproterenol, arrhythmias have been abolished after using NK-93 (Sag et al., 2011). It also prevents arrhythmias after models of acidosis, DOX-induced, NO-donor SNAP (Mustroph et al., 2017). On some models, the KN-93 does not seem to prevent but to slow the arrhythmia (as longer cycle length) without marked alterations in baseline ECG characteristics (Hoeker et al., 2016). KN-93 also reduces diastolic cytosolic [Ca2+] after induced HF (Sag et al., 2011). It is also an attenuator of Ca2+-leak in a diabetes model of GlcNAcase inhibition (Erickson et al., 2013) and myocardial hypertrophy. Another study shows the high capacity to inhibit the binding of CaM with NaV1.5, increasing the calcium release from RYR2 in cardiomyocytes independent from CamKII (Johnson et al., 2019). This suggests that KN-93 has interactions with the CaM-Ca2+ binding; however, to inhibit CaMKII specifically, more affinized compound is necessary.
Experiments using TG RYR-mutant mice (S2814D mutant) are naturally more susceptible to atrial fibrillation, and because of this, it is used as a well-established model. Atrial fibrillation, phospholamban phosphorylation, and diastolic Ca2+-leak are reduced after prior injection of KN-93 in these TG mice (Voigt et al., 2012). Cardiomyocytes isolated from TG CaMKIIδc knockout mice completely recover the contraction ability after acidosis injury (Mustroph et al., 2017).
There is also a highly specific inhibitor of CaMKII called autocamtide-3-derived inhibitory peptide (AC3-I). It is the most used one in studies that require the blocking of CaMKII, and it is resistant even to proteolysis. As mentioned, AC3-I is derived from autocamtide-3. The last one is a substrate for CaMKII and acts mainly in the Thr-9 phosphorylation site substituted with Ala (Maier and Bers, 2007). As the research regarding CaMKII increases, its therapeutic use implicated in pharmaceuticals has been studied more. Several studies, using these inhibitors mentioned above, impart a cardio protection.
As mentioned, the AC3-I blocker, is a highly specific inhibitor of CaMKII. This is the main reason it is used more in the studies concerning the participation of CaMKII in many cell functions. It can inhibit CAMKII more selectively than CaMKIV (more and a hundredfold). Studies using this blocker also imply cardio protection: preventing hypertrophy, reducing ventricular arrhythmias, improving mechanical function, reducing RyR2 lacking, and decreasing mortality of diabetic mice (Sag et al., 2011).
Some studies have proven the efficiency of AC3-I in pathologies in some animal models. As cited with KN-93, the AC3-I also reduces atrial fibrillation in TG RYR knockout mice (Chelu et al., 2009) and seems to protect the heart from the same atrial fibrillation in Ang II models in vivo and in vitro (Purohit et al., 2013). There is a study from our group using interference RNA (RNAi) to block the expression of CaMKIIδ. It demonstrates that CaMKIIδ is fundamental for cardiomyocyte hypertrophy; once blocking the expression, the LPS-induced hypertrophy is reverted (Cruz Junho et al., 2019).
There are several models of heart problems leading to CaMKII. CRS, already cited, seems to be one of them. Both kidneys and heart share many mechanisms of homeostasis, and any injury to one can lead to one in the other. It is known that CaMKII is increased in many models of heart injury, and some models of CRS can cause arrhythmias and AP chances as well as contraction irregularities (Navarro-García et al., 2018; Alarcon et al., 2019). CaMKII could be crucial to the progression of CRS, more specifically related to the progression from acute HF to chronic (types 1 and 2). In this scenario, the inhibition of CaMKII would be cardioprotective.
The close relation between inflammation and oxidative stress in pathophysiological processes also makes the balance between oxidant and antioxidant forces and, therefore, oxidative stress, one of the most important mechanisms as has been demonstrated in heart and kidney injury studies (Li et al., 2017; Oliveira et al., 2017; Liu and Liu, 2018; Songbo et al., 2019).
Even though physiological levels of oxidative species are necessary for cellular function, the oxidative stress caused by the overproduction of these molecules in both organs leads to a series of structural abnormalities via immune system activation and fibrotic promotion (Virzì et al., 2015a). Some of these pathologies include left ventricle hypertrophy, atherosclerosis, endothelial dysfunction, and fibrosis in the heart while in the kidney ROS promotes interstitial fibrosis and increased inflammation (Kumar et al., 2019). A study involving patients with CRS type 3 shows that they have an increased level of inflammatory and oxidative stress factors, including IL-6, myeloperoxidase, nitric oxide (NO), copper/zinc superoxide dismutase (SOD), and endogenous peroxidase (Virzì et al., 2015a). Oxidative stress triggers an inflammatory response, and this response induces more oxidative stress. This stress may be maintaining the previously mentioned cycle of damage. While Ca2+ is associated with inflammation, studies have shown a connection between oxidative stress and CaMKII activation (Erickson et al., 2008). Erickson et al. (2008) also demonstrate a dynamic mechanism for CaMKII activation, which occurs via oxidation of the methionine residue site on the CaMKII regulatory domain; this oxidation-dependent CaMKII activation is important to Ang II and apoptosis since CaMKII remains active after ROS oxidation even in the absence of the Ca2+/CaM complex. Some proteins maintain a redox sensor that regulates the cell response to oxidative stress (Kim et al., 2014). CaM is one of these proteins, and this oxidation leads to a regulatory cascade response with specific targets, including CaMKII (Snijder et al., 2011), plasma membrane Ca2+ (Anbanandam et al., 2005), and nitric oxide synthase (NOS) (Montgomery et al., 2003). As mentioned above, one of the mechano-chemotransductions that ROS induces Ca2+ release by CaMKII involves NOS (Jian et al., 2014). The endothelial dysfunction caused by oxidative stress leads to uncoupling of endothelial NOS (eNOS), leading to the production of more ROS (Münzel et al., 2017). In pathological conditions, such as inflammation, the vasculature expresses the inducible form of NOS (iNOS) (Münzel et al., 2017). This isoform of NOS produces an excessive amount of NO that mediates impaired vasoconstriction, which may be further worsened by the decreased of eNOS activity (Jian et al., 2014). The continuous exposure of NO induced by pro-inflammatory mediators inhibits endothelium-dependent relaxation by impairing the via CaMKII-dependent activation of eNOS (Kassler et al., 1997). In addition, studies have shown the role of NOS in the kidney, demonstrating that, when NOS activity is compromised, there are a series of renal dysfunctions that reduce glomerular perfusion and filtration, which may lead to a progressive scenario of hypertension and kidney injuries (Carlstrom and Montenegro, 2019).
Oxidative stress in CaMKII by methionine-oxidized CaMKII was also observed in patients with atrial fibrillation (Purohit et al., 2013; Yoo et al., 2018), which also demonstrates that oxidative stress can act in part through increased constitutive activity of CaMKII, creating a highly vulnerable substrate within the HF that promotes atrial fibrillation beyond fibrosis. On the other hand, Kong et al. (2018) shows that the oxidative stress in mitochondria can be reduced after the inhibition of CaMKII, alleviating the myocardial ischemia-reperfusion injury. In addition to the redox balance, other factors indirectly contribute to cardiac and renal alterations. Many approaches have been studied in order to set a start point for the CRS. One of them is epigenetics factors.
Epigenetics is the area of biology that studies changes in the functioning of a gene that is not caused by alterations in the DNA sequence and that perpetuate in the meiotic or mitotic cell divisions (Wu and Morris, 2001). Epigenetic modifications are highly coordinated processes of change that are not restricted to a specific phase of life. These characteristics are fundamental to diseases acquired throughout life. Epigenetic changes are divided into DNA methylation, histone modification, and noncoding RNA expression (Liu L. et al., 2008).
Studies have been developed to innovate the way to prevent CRS. Slowly, epigenetics is gaining space, and traditional mechanisms (such as RAAS and inflammation) are being replaced by other patterns of findings and prevention. Imaging the scale of modifications and mutations in a syndrome such as CRS, numerous cell lines may be altered and reprogrammed, in both heart and kidney. Studies have pointed out the role of epigenetics in the development of CRS (Gaikwad et al., 2010). In types 3 and 4, for example, renal failure increases cardiac histone H3 epigenetics, evidencing the crosstalk between renal failure and the transcription of cardiomyopathy-related genes (Gaikwad et al., 2010). It is important to mention that epigenetics in CRS itself are little studied when compared to the traditional mechanisms even though it is very promising. The focus of studies is linked to inflammation and oxidative stress, which we know to be the consequences of CRS. Abnormal defects in DNA methylation, histone modifications, and microRNA (miR) participate in renal injury (Beckerman et al., 2014); however, none of them are related to post-progression of HF. During HF independent of renal injury, we can note the expression of transcription factors, angiogenic factors, and natriuretic factors, often used as biomarkers of this condition. Epigenetic modifications regulate them. Pathological hypertrophy and compromised contractility are described to increase DNA methylation levels. Inhibition of DNA methylation has already been suggested as treatment for CHF (Yang et al., 2015); however, it should be carefully studied once the DNA methylation is comprehended.
Ca2+ signaling is involved in epigenetic regulation, and the study of its signaling can contribute to the development of new therapeutic strategies (Awad et al., 2015; Puri, 2020). CaMKIIδ has been already described as fundamental for cardiac hypertrophy development (Cruz Junho et al., 2019). This mechanism occurs after CaMKIIδ selectively phosphorylates HDAC4. During cardiac hypertrophy, there is an activation of fetal cardiac genes, an important mechanism regulated by CaMKIIδ-mediated H3 chromatin regulation. With the recent development of epigenetic studies, the use of ChIP-seq to evaluate H3 phosphorylation and binding of CaMKIIδ across the genome enables a profound knowledge of the genome-wide functional effect of H3 alteration by CaMKIIδ (Awad et al., 2015). This connection can lead to CRS types 1 and 2 and can be strongly linked to the diagnosis of heart dysfunction. In addition, it is extremely important to highlight the role of micro RNA (miR). miR is a short and noncoding RNA that interacts with the 3-untranslated region (UTR) of mRNAs blocking gene expression, degrading mRNAs and regulating protein expression (Winter et al., 2009; Krol et al., 2010; Qi et al., 2019; Yang et al., 2019).
To identify signaling pathways, there is a serviceable tool called gene set analysis (GSA). It uses statistical analysis to predefine gene sets involved in a specific cellular process. Thus, GSA is especially useful to infer functions of different miRNAs. For example, miR-185 has a key role during cardiac hypertrophy. This miRNA targets pro-hypertrophic genes, such as RhoA, Cdc42, and Stim1 in the heart. Given that, miR-185 is also a potent therapeutic target for cardiac diseases (Brown et al., 2006; Carè et al., 2007; Liu et al., 2011; Zhang et al., 2015). In addition, Kim et al. (2015) show that miR-185 not only acts as the genes mentioned, but also targeting genes involved in Ca2+-associated pathological hypertrophy, including CamKII. It is worth mentioning the role of miR-1, once alterations in its expression or inhibition have been discovered in many cardiac pathologies (Yang et al., 2007; Lu et al., 2009).
Recently, Zhang et al. proposed a possible mechanism by which Lycium barbarum polysaccharides (LBP) restore cardiac contractility induced by miR-1 overexpression. The authors show that LBPs prevent the reduction of CaM and cardiac myosin light chain kinase and their corresponding downstream proteins, including CaMKII due to miR-1 overexpression (Zhang et al., 2018).
Regarding inflammatory processes, miR-625-5p has been illustrated to inhibit inflammatory response in human bronchial epithelial cells and is downregulated in heart diseases once miR-625-5p is able to inhibit STAT3 and reduce the expression of CaMKII. Moreover, miR-625-5p attenuated Ang II-induced cardiac hypertrophy through CaMKII/STAT3 (Qian et al., 2019).
In relation to kidney disease, Park et al. (2018) shows that miR-34c-5p and CaMKII are involved in aldosterone-induced fibrosis in the kidneys. In addition, recent studies have focused on MiR regulation and exosomes, specialized nanosized membranous vesicles, in different experimental models. These membrane-bound vesicles (30–100 nm) are released from different cell types and deliver bioactive molecules, including microRNAs (miRs). Recently, literature demonstrated the regulation of oxidative stress in cardiac stem cells through the miR-214/CaMKII pathway after using exosomes derived from miR-214-enriched bone marrow-derived mesenchymal cells (Wang et al., 2018).
In addition to miRs, the role of long noncoding RNAs (lncRNAs) is well known. lncRNAs are transcribed RNA molecules >200 nucleotides in length without known protein-coding function, regulating gene expression at epigenetic, transcriptional, and post-transcriptional levels (Mercer et al., 2009). Previous studies report that lncRNAs play critical roles in the modulation of heart development and cardiovascular diseases (Wang et al., 2014, 2015; Zhang et al., 2016, 2017). For example, Shao et al. (2017) demonstrated that the expression of long noncoding RNAs TINCR was downregulated in the heart after a transverse aortic constriction (TAC) model (Shao et al., 2017). More recently, the same group showed that TINCR could epigenetically inhibit the transcription of CaMKII inhibiting cardiac hypertrophy induced by angiotensin II (Shao et al., 2017).
The evidence in the literature suggests that CaMKII is a key molecule for understanding the physiology and physiopathology of cardiovascular diseases as well as a prominent target for new strategies of treatment.
In this review, we aimed to stimulate a discussion on the role of CaM as an inflammatory mediator in the systemic profile of CRS via regulation of CaMKII. The literature suggests that the Ca2+/CaM complex might be an important modulator of inflammation and oxidative stress via CaMKII in the kidney–heart interaction (summarized in Figure 2), and CaMKII regulation by pre- or post-translational mechanisms is essential for cardiac or renal homeostasis. Additionally, we have observed that CaM/CaMKII has been extensively studied in cardiovascular diseases; however, there is still the necessity of exploring how CaM could integrate Ca2+ signal in different scenarios, such as CRS.
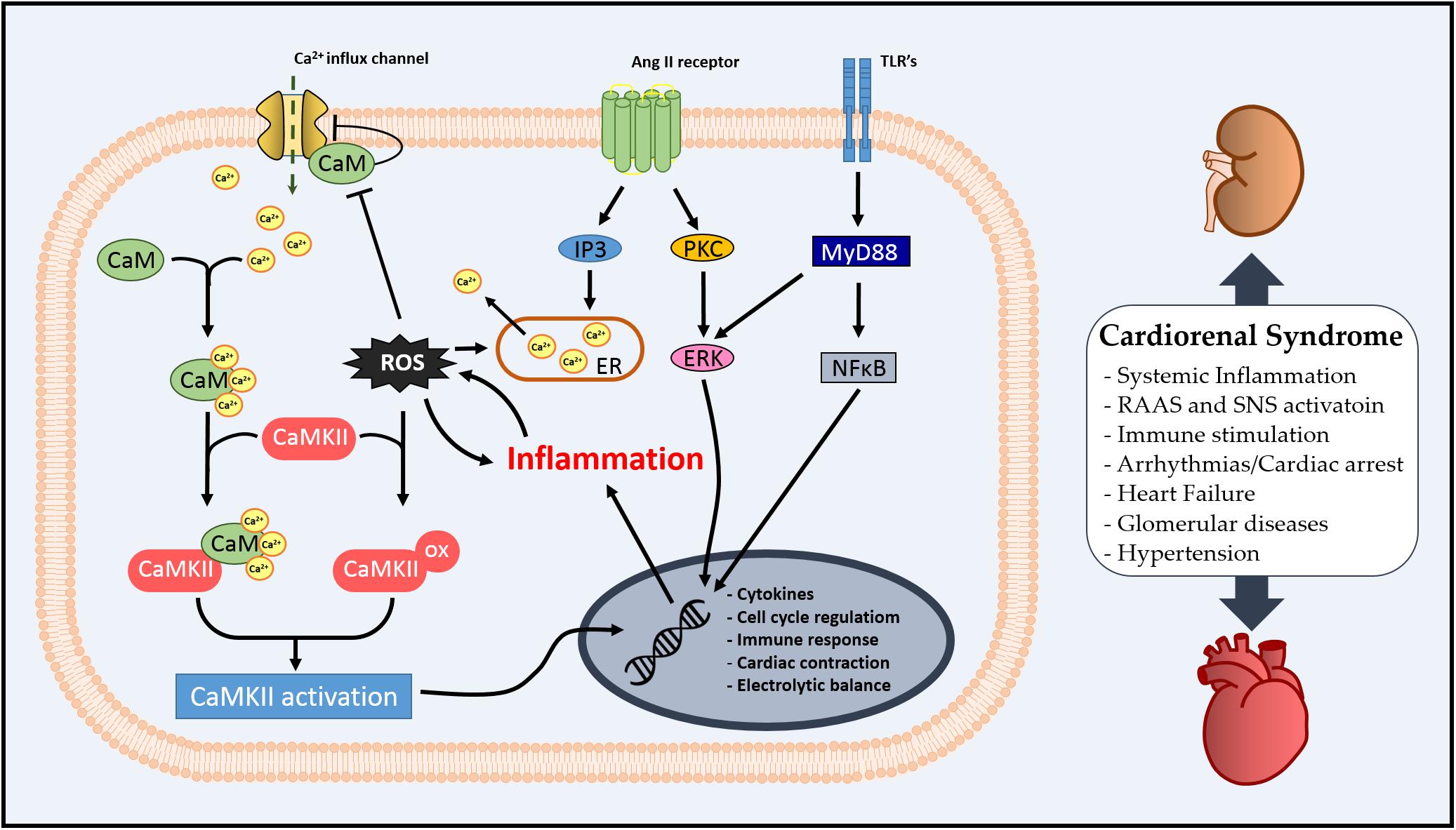
Figure 2. Schematic illustration of cellular Ca2+/calmodulin-dependent (CaMK) II involvement in cardiorenal syndrome (CRS).
CJ and MC-R proposed the idea and writing. All authors contributed to the article and approved the submitted version.
This work was supported by FAPESP: São Paulo Research Foundation, Brazil, Grants: 2008/10175-4, 2015/19105-7, 2014/16908-4, and 2018/3089-6.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alarcon, M. M. L., Trentin-Sonoda, M., Panico, K., Schleier, Y., Duque, T., Moreno-Loaiza, O., et al. (2019). Cardiac arrhythmias after renal I/R depend on IL-1β. J. Mol. Cell. Cardiol. 131, 101–111. doi: 10.1016/j.yjmcc.2019.04.025
Alfazema, N., Barrier, M., De Procé, S. M., Menzies, R. I., Carter, R., Stewart, K., et al. (2019). Camk2n1 is a negative regulator of blood pressure, left ventricular mass, insulin sensitivity, and promotes adiposity. Hypertension 74, 687–696. doi: 10.1161/HYPERTENSIONAHA.118.12409
Anavekar, N. S., McMurray, J. J. V., Velazquez, E. J., Solomon, S. D., Kober, L., Rouleau, J. L., et al. (2004). Relation between renal dysfunction and cardiovascular outcomes after myocardial infarction. N. Engl. J. Med. 351, 1285–1295. doi: 10.1056/NEJMoa041365
Anbanandam, A., Bieber Urbauer, R. J., Bartlett, R. K., Smallwood, H. S., Squier, T. C., and Urbauer, J. L. (2005). Mediating molecular recognition by methionine oxidation: conformational switching by oxidation of methionine in the carboxyl-terminal domain of calmodulin. Biochemistry 44, 9486–9496. doi: 10.1021/bi0504963
Awad, S., Al-Haffar, K. M. A., Marashly, Q., Quijada, P., Kunhi, M., Al-Yacoub, N., et al. (2015). Control of histone H3 phosphorylation by CaMKII. in response to haemodynamic cardiac stress. J. Pathol. 235, 606–618. doi: 10.1002/path.4489
Backs, J., Backs, T., Neef, S., Kreusser, M. M., Lehmann, L. H., Patrick, D. M., et al. (2009). The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. U.S.A. 106, 2342–2347. doi: 10.1073/pnas.0813013106
Beckendorf, J., van den Hoogenhof, M. M. G., and Backs, J. (2018). Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 113, 1–12. doi: 10.1007/s00395-018-0688-8
Beckerman, P., Ko, Y. A., and Susztak, K. (2014). Epigenetics: a new way to look at kidney diseases. Nephrol. Dial. Transplant. 29, 1821–1827. doi: 10.1093/ndt/gfu026
Bers, D. M., and Morotti, S. (2014). Ca2+ current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front. Pharmacol. 5:144. doi: 10.3389/fphar.2014.00144
Bongartz, L. G., Cramer, M. J., Doevendans, P. A., Bram, B. A., and Joles, J. A. (2005). The severe cardiorenal syndrome: ‘Guyton revisited. Eur. Heart J. 25, 11–17. doi: 10.1093/eurheartj/ehi020
Boubali, S., Liopeta, K., Virgilio, L., Thyphronitis, G., Mavrothalassitis, G., Dimitracopoulos, G., et al. (2012). Calcium/calmodulin-dependent protein kinase II regulates IL-10 production by human T lymphocytes: a distinct target in the calcium dependent pathway. Mol. Immunol. 52, 51–60. doi: 10.1016/j.molimm.2012.04.008
Brown, J. H., Del Re, D. P., and Sussman, M. A. (2006). The Rac and Rho Hall of Fame. Circ. Res. 98, 730–742. doi: 10.1161/01.RES.0000216039.75913.9e
Bui, J. D., Calbo, S., Hayden-Martinez, K., Kane, L. P., Gardner, P., and Hedrick, S. M. (2000). A role for CaMKII in T cell memory. Cell 100, 457–467. doi: 10.1016/S0092-8674(00)80681-9
Bussey, C. T., and Erickson, J. R. (2018). Physiology and pathology of cardiac CaMKII. Curr. Opin. Physiol. 1, 52–58. doi: 10.1016/j.cophys.2017.07.003
Carafoli, E., and Krebs, J. (2016). Why calcium? How calcium became the best communicator. J. Biol. Chem. 291, 20849–20857. doi: 10.1074/jbc.R116.735894
Carè, A., Catalucci, D., Felicetti, F., Bonci, D., Addario, A., Gallo, P., et al. (2007). MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 13, 613–618. doi: 10.1038/nm1582
Carlstrom, M., and Montenegro, M. F. (2019). Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J. Intern. Med. 285, 2–18. doi: 10.1111/joim.12818
Chelu, M. G., Sarma, S., Sood, S., Wang, S., Van Oort, R. J., Skapura, D. G., et al. (2009). Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951. doi: 10.1172/jci37059
Cipolletta, E., Rusciano, M. R., Maione, A. S., Santulli, G., Sorriento, D., Del Giudice, C., et al. (2015). Targeting the CaMKII/ERK interaction in the heart prevents cardiac hypertrophy. PLoS One 10:e0130477. doi: 10.1371/journal.pone.0130477
Colombo, P. C., Ganda, A., Lin, J., Onat, D., Harxhi, A., Iyasere, J. E., et al. (2012). Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail. Rev. 17, 177–190. doi: 10.1007/s10741-011-9261-3
Condon, J. C., Pezzi, V., Drummond, B. M., Yin, S., and Rainey, W. E. (2002). Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology 143, 3651–3657. doi: 10.1210/en.2001-211359
Cruz Junho, C. V., Trentin-Sonoda, M., Alvim, J. M., Gaisler-Silva, F., and Carneiro-Ramos, M. S. (2019). Ca2+/calmodulin-dependent kinase ii delta b is essential for cardiomyocyte hypertrophy and complement gene expression after LPS and HSP60 stimulation in vitro. Braz. J. Med. Biol. Res. 52:e8732. doi: 10.1590/1414-431x20198732
Damman, K., Navis, G., Voors, A. A., Asselbergs, F. W., Smilde, T. D. J., Cleland, J. G. F., et al. (2007). Worsening renal function and prognosis in heart failure: systematic review and meta-analysis. J. Card. Fail. 13, 599–608. doi: 10.1016/j.cardfail.2007.04.008
Di Lullo, L., Bellasi, A., Barbera, V., Russo, D., Russo, L., Di Iorio, B., et al. (2017). Pathophysiology of the cardio-renal syndromes types 1–5: An uptodate. Indian Heart J. 69, 255–265. doi: 10.1016/j.ihj.2017.01.005
Diamandopoulos, A. (1999). Plenary lecture. Twelve centuries of nephrological writings in The Graeco-Roman world of the Eastern Mediterranean (from Hippocrates to Aetius Amidanus). Nephrol. Dial. Transplant. 14, 2–9. doi: 10.1093/ndt/14.suppl_2.2
Erickson, J. R., Ling, M., Joiner, A., Guan, X., Kutschke, W., Yang, J., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Friedberg, F., and Rhoads, A. R. (2001). Evolutionary aspects of calmodulin. IUBMB Life 51, 215–221. doi: 10.1080/152165401753311753
Gaikwad, A. B., Sayyed, S. G., Lichtnekert, J., Tikoo, K., and Anders, H. J. (2010). Renal failure increases cardiac histone H3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am. J. Pathol. 176, 1079–1083. doi: 10.2353/ajpath.2010.090528
Guest, C. B., Deszo, E. L., Hartman, M. E., York, J. M., Kelley, K. W., and Freund, G. G. (2008). Ca2+/Calmodulin-dependent kinase kinase α is expressed by monocytic cells and regulates the activation profile. PLoS One 3:e1606. doi: 10.1371/journal.pone.0001606
Hanada, S., Takewa, Y., Mizuno, T., Tsukiya, T., Taenaka, Y., and Tatsumi, E. (2012). Effect of the technique for assisting renal blood circulation on ischemic kidney in acute cardiorenal syndrome. J. Artif. Organs 15, 140–145. doi: 10.1007/s10047-011-0613-5
Hoeker, G. S., Hanafy, M. A., Oster, R. A., Bers, D. M., and Pogwizd, S. M. (2016). Reduced arrhythmia inducibility with calcium/calmodulin-dependent protein kinase II inhibition in heart failure rabbits. J. Cardiovasc. Pharmacol. 67, 260–265. doi: 10.1097/FJC.0000000000000343
House, A. A., Wanner, C., Sarnak, M. J. I, Piña, L., McIntyre, C. W., Komenda, P., et al. (2019). Heart failure in chronic kidney disease: conclusions from a kidney disease: improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 95, 1304–1317. doi: 10.1016/j.kint.2019.02.022
Hudmon, A., and Schulman, H. (2002). Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 364, 593–611. doi: 10.1042/BJ20020228
Islam, M. S. (2020). Calcium signaling: from basic to bedside. Adv. Exp. Med. Biol. 1131, 1–6. doi: 10.1007/978-3-030-12457-1_1
Jian, Z., Han, H., Zhang, T., Puglisi, J., Izu, L. T., Shaw, J. A., et al. (2014). Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci. Signal 7:ra27. doi: 10.1126/scisignal.2005046
Johnson, C. N., Pattanayek, R., Potet, F., Rebbeck, R. T., Blackwell, D. J., Nikolaienko, R., et al. (2019). The CaMKII inhibitor KN93-calmodulin interaction and implications for calmodulin tuning of NaV1.5 and RyR2 function. Cell Calcium 82:102063. doi: 10.1016/j.ceca.2019.102063
Kakiuchi, S., and Yamazaki, R. (1970). Calcium dependent phosphodiesterase activity and its activating factor (PAF) from brain. Studies on cyclic 3’,5’-nucleotide phosphodiesterase (III). Biochem. Biophys. Res. Commun. 41, 1104–1110. doi: 10.1016/0006-291X(70)90199-3
Kamada, R., Yokoshiki, H., Mitsuyama, H., Watanabe, M., Mizukami, K., Tenma, T., et al. (2019). Arrhythmogenic β-adrenergic signaling in cardiac hypertrophy: the role of small-conductance calcium-activated potassium channels via activation of CaMKII. Eur. J. Pharmacol. 844, 110–117. doi: 10.1016/j.ejphar.2018.12.011
Kassler, P., Bauersachs, J., Busse, R., and Schini-Kerth, V. B. (1997). Inhibition of inducible nitric oxide synthase restores endothelium- dependent relaxations in proinflammatory mediator-induced blood vessels. Arterioscler. Thromb. Vasc. Biol. 17, 1746–1755. doi: 10.1161/01.ATV.17.9.1746
Ke, H. Y., Chin, L. H., Tsai, C. S., Lin, F. Z., Chen, Y. H., Chang, Y. L., et al. (2020). Cardiac calcium dysregulation in mice with chronic kidney disease. J. Cell. Mol. Med. 24, 3669–3677. doi: 10.1111/jcmm.15066
Kim, G., Weiss, S. J., and Levine, R. L. (2014). Methionine oxidation and reduction in proteins. Biochim. Biophys. Acta 1840, 901–905. doi: 10.1016/j.bbagen.2013.04.038
Kim, J. O., Song, D. W., Kwon, E. J., Hong, S.-E., Song, H. K., Min, C. K., et al. (2015). miR-185 Plays an Anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS One 10:e0122509. doi: 10.1371/journal.pone.0122509
Kong, L. H., Chen, Y. L., Sun, N., Wei, M., Zhu, J. X., and Su, X. L. (2018). Inhibition of CaMKII alleviates myocardial ischemia? Reperfusion injury by reducing mitochondrial oxidative stress in isolated perfused rat heart. Nan Fang Yi Ke Da Xue Xue Bao 38, 181–186. doi: 10.3969/J.ISSN.1673-4254.2018.02.10
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. doi: 10.1038/nrg2843
Kumar, U., Wettersten, N., and Garimella, P. S. (2019). Cardiorenal syndrome: pathophysiology. Cardiol. Clin. 37, 251–265. doi: 10.1016/j.ccl.2019.04.001
Lemrow, S. M., Anderson, K. A., Joseph, J. D., Ribar, T. J., Noeldner, P. K., and Means, A. R. (2004). Catalytic activity is required for calcium/calmodulin-dependent protein kinase IV to enter the nucleus. J. Biol. Chem. 279, 11664–11671. doi: 10.1074/jbc.M312613200
Li, M. S., Adesina, S. E., Ellis, C. L., Gooch, J. L., Hoover, R. S., and Williams, C. R. (2017). NADPH oxidase-2 mediates zinc deficiency-induced oxidative stress and kidney damage. Am. J. Physiol. Physiol. 312, C47–C55. doi: 10.1152/ajpcell.00208.2016
Liu, H. J., and Liu, B. (2018). Inhibition of MicroRNA-23 contributes to the isoflurane-mediated cardioprotection against oxidative stress. Cardiovasc. Toxicol. 18, 450–458. doi: 10.1007/s12012-018-9455-1
Liu, L., Li, Y., and Tollefsbol, T. O. (2008). Gene-environment interactions and epigenetic basis of human diseases. Curr. Issues Mol. Biol. 10, 25–36.
Liu, M., Lang, N., Chen, X., Tang, Q., Liu, S., Huang, J., et al. (2011). MiR-185 targets RhoA and Cdc42 expression and inhibits the proliferation potential of human colorectal cells. Cancer Lett. 301, 151–160. doi: 10.1016/j.canlet.2010.11.009
Liu, X., Yao, M., Li, N., Wang, C., Zheng, Y., and Cao, X. (2008). CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 112, 4961–4970. doi: 10.1182/blood-2008-03-144022
Liu, Z., Finet, J. E., Wolfram, J. A., Anderson, M. E., Ai, X., and Donahue, J. K. (2019). Calcium/calmodulin-dependent protein kinase II causes atrial structural remodeling associated with atrial fibrillation and heart failure. Hear. Rhythm 16, 1080–1088. doi: 10.1016/j.hrthm.2019.01.013
Lu, Y., Zhang, Y., Shan, H., Pan, Z., Li, X., and Li, B. (2009). MicroRNA-1 downregulation by propranolol in a rat model of myocardial infarction: a new mechanism for ischaemic cardioprotection. Cardiovasc. Res. 84, 434–441. doi: 10.1093/cvr/cvp232
Luczak, E. D., and Anderson, M. E. (2014). CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 73, 112–116. doi: 10.1016/j.yjmcc.2014.02.004
Maier, L. S., and Bers, D. M. (2007). Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 73, 631–640. doi: 10.1016/j.cardiores.2006.11.005
Marsico, F., Paolillo, S., Gargiulo, P., Parisi, V., Nappi, C., Assante, R., et al. (2019). Renal function and cardiac adrenergic impairment in patients affected by heart failure. J. Nucl. Cardiol. doi: 10.1007/s12350-019-01975-7 [Epub ahead of print].
Mattiazzi, A., and Kranias, E. G. (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5:5. doi: 10.3389/fphar.2014.00005
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi: 10.1038/nrg2521
Montgomery, H. J., Bartlett, R., Perdicakis, B., Jervis, E., Squier, T. C., and Guillemette, J. G. (2003). Activation of constitutive nitric oxide synthases by oxidized calmodulin mutants. Biochemistry 42, 7759–7768. doi: 10.1021/bi027097h
Münzel, T., Camici, G. G., Maack, C., Bonetti, N. R., Fuster, V., and Kovacic, J. C. (2017). Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J. Am. Coll. Cardiol. 70, 212–229. doi: 10.1016/j.jacc.2017.05.035
Mustroph, J., Neef, S., and Maier, L. S. (2017). CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 176, 22–31. doi: 10.1016/j.pharmthera.2016.10.006
Navarro-García, J. A., Fernández-Velasco, M., Delgado, C., Delgado, J. F., Kuro-o, M., Ruilope, L. M., et al. (2018). PTH, vitamin D, and the FGF-23–klotho axis and heart: going beyond the confines of nephrology. Eur. J. Clin. Invest. 48:e12902. doi: 10.1111/eci.12902
Oliveira, R. C., Brito, M. V., Ribeiro, R. F., Oliveira, L. O., Monteiro, A. M., Brandão, F. M., et al. (2017). Influence of remote ischemic conditioning and tramadol hydrochloride on oxidative stress in kidney ischemia/reperfusion injury in rats. Acta Cir. Bras. 32, 229–235. doi: 10.1590/S0102-865020170030000007
Park, E.-J., Jung, H. J., Choi, H.-J., Cho, J.-I., Park, H.-J., and Kwon, T.-H. (2018). miR-34c-5p and CaMKII are involved in aldosterone-induced fibrosis in kidney collecting duct cells. Am. J. Physiol. Physiol. 314, F329–F342. doi: 10.1152/ajprenal.00358.2017
Pellicena, P., and Schulman, H. (2014). CaMKII inhibitors: from research tools to therapeutic agents. Front. Pharmacol. 5:21. doi: 10.3389/fphar.2014.00021
Picciotto, M. R., Zoli, M., Bertuzzi, G., and Nairn, A. C. (1995). Immunochemical localization of calcium/calmodulin-dependent protein kinase I. Synapse 20, 75–84. doi: 10.1002/syn.890200111
Prasad, A. M., Morgan, D. A., Nuno, D. W., Ketsawatsomkron, P., Bair, T. B., Venema, A. N., et al. (2015). Calcium/Calmodulin−dependent kinase II inhibition in smooth muscle reduces angiotensin II–induced hypertension by controlling aortic remodeling and baroreceptor function. J. Am. Heart Assoc. 4:e001949. doi: 10.1161/JAHA.115.001949
Puri, B. K. (2020). Calcium signaling and gene expression. Adv. Exp. Med. Biol. 1131, 537–545. doi: 10.1007/978-3-030-12457-1_22
Purohit, A., Rokita, A. G., Guan, X., Chen, B., Koval, O. M., Voigt, N., et al. (2013). Oxidized Ca2+/Calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128, 1748–1757. doi: 10.1161/CIRCULATIONAHA.113.003313
Qi, H., Ren, J., Mingyao, E., Zhang, Q., Cao, Y., Ba, L., et al. (2019). MiR-103 inhibiting cardiac hypertrophy through inactivation of myocardial cell autophagy via targeting TRPV3 channel in rat hearts. J. Cell. Mol. Med. 23, 1926–1939. doi: 10.1111/jcmm.14095
Qian, F. H., Deng, X., Zhuang, Q. X., Wei, B., and Zheng, D. D. (2019). MiR-625-5p suppresses inflammatory responses by targeting AKT2 in human bronchial epithelial cells. Mol. Med. Rep. 19, 1951–1957. doi: 10.3892/mmr.2019.9817
Rajtik, T., Carnicka, S., Szobi, A., Giricz, Z., O-Uchi, J., Hassova, V., et al. (2016). Oxidative activation of CaMKIIδ in acute myocardial ischemia/reperfusion injury: a role of angiotensin AT1 receptor-NOX2 signaling axis. Eur. J. Pharmacol. 771, 114–122. doi: 10.1016/j.ejphar.2015.12.024
Ren, G., Dewald, O., and Frangogiannis, N. (2003). Inflammatory mechanisms in myocardial infarction. Curr. Drug Target Inflamm. Allergy 2, 242–256. doi: 10.2174/1568010033484098
Rokita, A. G., and Anderson, M. E. (2012). New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin- dependent kinase II (CaMKII). Circulation 126, 2125–2139. doi: 10.1161/CIRCULATIONAHA.112.124990
Ronco, C. (2011). The cardiorenal syndrome: basis and common ground for a multidisciplinary patient-oriented therapy. Cardiorenal Med. 1, 3–4. doi: 10.1159/000323352
Ronco, C., and Di Lullo, L. (2014). Cardiorenal syndrome. Heart Fail. Clin. 10, 251–280. doi: 10.1016/j.hfc.2013.12.003
Rusciano, M. R., Sommariva, E., Douin-Echinard, V., Ciccarelli, M., Poggio, P., and Maione, A. S. (2019). CaMKII activity in the inflammatory response of cardiac diseases. Int. J. Mol. Sci. 20:4374. doi: 10.3390/ijms20184374
Sag, C. M., Köhler, A. C., Anderson, M. E., Backs, J., and Maier, L. S. (2011). CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 51, 749–759. doi: 10.1016/j.yjmcc.2011.07.016
Sakagami, H., Kamata, A., Nishimura, H., Kasahara, J., Owada, Y., Takeuchi, Y., et al. (2005). Prominent expression and activity-dependent nuclear translocation of Ca 2+ /calmodulin-dependent protein kinase Iδ in hippocampal neurons. Eur. J. Neurosci. 22, 2697–2707. doi: 10.1111/j.1460-9568.2005.04463.x
Shao, M., Chen, G., Lv, F., Liu, Y., Tian, H., Tao, R., et al. (2017). LncRNA TINCR attenuates cardiac hypertrophy by epigenetically silencing CaMKII. Oncotarget 8, 47565–47573. doi: 10.18632/oncotarget.17735
Singh, M. V., Swaminathan, P. D., Luczak, E. D., Kutschke, W., Weiss, R. M., and Anderson, M. E. (2012). MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J. Mol. Cell. Cardiol. 52, 1135–1144. doi: 10.1016/j.yjmcc.2012.01.021
Skelding, K. A., and Rostas, J. A. P. (2012). The role of molecular regulation and targeting in regulating calcium/calmodulin stimulated protein kinases. Adv. Exp. Med. Biol. 740, 703–730. doi: 10.1007/978-94-007-2888-2_31
Skelding, K. A., and Rostas, J. A. P. (2020). Regulation of multifunctional calcium/calmodulin stimulated protein kinases by molecular targeting. Adv. Exp. Med. Biol. 1131, 649–679. doi: 10.1007/978-3-030-12457-1_26
Skelding, K. A., Rostas, J. A. P., and Verrills, N. M. (2011). Controlling the cell cycle: the role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 10, 631–639. doi: 10.4161/cc.10.4.14798
Snijder, J., Rose, R. J., Raijmakers, R., and Heck, A. J. R. (2011). Site-specific methionine oxidation in calmodulin affects structural integrity and interaction with Ca2+/calmodulin-dependent protein kinase II. J. Struct. Biol. 174, 187–195. doi: 10.1016/j.jsb.2010.12.002
Songbo, M., Lang, H., Xinyong, C., Bin, X., Ping, Z., and Liang, S. (2019). Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 307, 41–48. doi: 10.1016/j.toxlet.2019.02.013
Sumi, M., Kiuchi, K., Ishikawa, T., Ishii, A., Hagiwara, M., Nagatsu, T., et al. (1991). The newly synthesized selective Ca2+ calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 181, 968–975. doi: 10.1016/0006-291X(91)92031-E
Takemoto-Kimura, S., Suzuki, K., Horigane, S. I., Kamijo, S., Inoue, M., Sakamoto, M., et al. (2017). Calmodulin kinases: essential regulators in health and disease. J. Neurochem. 141, 808–818. doi: 10.1111/jnc.14020
Thomas, B., Matsushita, K., Abate, K. H., Al-Aly, Z., Ärnlöv, J., Asayama, K., et al. (2017). Global cardiovascular and renal outcomes of reduced GFR. J. Am. Soc. Nephrol. 28, 2167–2179. doi: 10.1681/ASN.2016050562
Tokumitsu, H., and Soderling, T. R. (1996). Requirements for calcium and calmodulin in the calmodulin kinase activation cascade. J. Biol. Chem. 271, 5617–5622. doi: 10.1074/jbc.271.10.5617
Tokumitsu, H. H., Chijiwa, T., Hagiwara, M., Mizutani, A., Terasawa, M., and Hidaka, H. (1990). KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 65:4315.
Trentin-Sonoda, M., da Silva, R. C., Kmit, F. V., Abrahão, M. V., Monnerat Cahli, G., Brasil, G. V., et al. (2015). Knockout of toll-like receptors 2 and 4 prevents renal ischemia-reperfusion-induced cardiac hypertrophy in mice. PLoS One 10:e0139350. doi: 10.1371/journal.pone.0139350
Virzì, G. M., Clementi, A., De Cal, M., Brocca, A., Day, S., Pastori, S., et al. (2015a). Oxidative stress: dual pathway induction in cardiorenal syndrome type 1 pathogenesis. Oxid. Med. Cell. Longev. 2015:391790. doi: 10.1155/2015/391790
Virzì, G. M., De Cal, M., Day, S., Brocca, A., Cruz, D. N., Castellani, C., et al. (2015b). Pro-apoptotic effects of plasma from patients with cardiorenal syndrome on human tubular cells. Am. J. Nephrol. 41, 474–484. doi: 10.1159/000438459
Voigt, N., Li, N., Wang, Q., Wang, W., Trafford, A. W., Abu-Taha, I., et al. (2012). Enhanced sarcoplasmic reticulum Ca 2+ Leak and increased Na +-Ca 2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/JCI26620
Wang, K., Liu, C. Y., Zhou, L. Y., Wang, J. X., Wang, M., Zhao, B., et al. (2015). APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat. Commun. 6:6779. doi: 10.1038/ncomms7779
Wang, K., Liu, F., Zhou, L. Y., Long, B., Yuan, S. M., Wang, Y., et al. (2014). The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 114, 1377–1388. doi: 10.1161/CIRCRESAHA.114.302476
Wang, Y., Zhao, R., Liu, D., Deng, W., Xu, G., Liu, W., et al. (2018). Exosomes derived from miR-214-enriched bone marrow-derived mesenchymal stem cells regulate oxidative damage in cardiac stem cells by targeting CaMKII. Oxid. Med. Cell. Longev 2018:4971261. doi: 10.1155/2018/4971261
Winter, J., Jung, S., Keller, S., Gregory, R. I., and Diederichs, S. (2009). Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 11, 228–234. doi: 10.1038/ncb0309-228
Wong, M. H., Samal, A. B., Lee, M., Vlach, J., Novikov, N., Niedziela-Majka, A., et al. (2019). The KN-93 molecule inhibits Calcium/Calmodulin-dependent protein kinase II (CaMKII) activity by binding to Ca2+/CaM. J. Mol. Biol. 431, 1440–1459. doi: 10.1016/j.jmb.2019.02.001
Wu, C. T., and Morris, J. R. (2001). Genes, genetics, and epigenetics: a correspondence. Science 293, 1103–1105. doi: 10.1126/science.293.5532.1103
Yamauchi, T. (2005). Neuronal Ca2+/Calmodulin-dependent protein kinase II—discovery, progress in a quarter of a century, and perspective: implication for learning and memory. Biol. Pharm. Bull. 28, 1342–1354. doi: 10.1248/bpb.28.1342
Yang, B., Lin, H., Xiao, J., Lu, Y., Luo, X., Li, B., et al. (2007). The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 13, 486–491. doi: 10.1038/nm1569
Yang, J., Xu, W. W., and Hu, S. J. (2015). Heart failure: advanced development in genetics and epigenetics. BioMed Res. Int. 2015:352734. doi: 10.1155/2015/352734
Yang, T., Liu, T., Cao, C., and Xu, S. (2019). miR-200a-5p augments cardiomyocyte hypertrophy induced by glucose metabolism disorder via the regulation of selenoproteins. J. Cell. Physiol. 234, 4095–4103. doi: 10.1002/jcp.27206
Yogasundaram, H., Chappell, M. C., Braam, B., and Oudit, G. Y. (2019). Cardiorenal syndrome and heart failure—challenges and opportunities. Can. J. Cardiol. 35, 1208–1219. doi: 10.1016/j.cjca.2019.04.002
Yoo, S., Aistrup, G., Shiferaw, Y., Ng, J., Mohler, P. J., Hund, T. J., et al. (2018). Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 3:e120728. doi: 10.1172/jci.insight.120728
Zhang, M., Gu, H., Chen, J., and Zhou, X. (2016). Involvement of long noncoding RNA MALAT1 in the pathogenesis of diabetic cardiomyopathy. Int. J. Cardiol. 202, 753–755. doi: 10.1016/j.ijcard.2015.10.019
Zhang, R., Xu, Y., Niu, H., Tao, T., Ban, T., Zheng, L., et al. (2018). Lycium barbarum polysaccharides restore adverse structural remodelling and cardiac contractile dysfunction induced by overexpression of microRNA-1. J. Cell. Mol. Med. 22, 4830–4839. doi: 10.1111/jcmm.13740
Zhang, Y., Feng, J., Wang, Q., Zhao, S., Yang, S., Tian, L., et al. (2018). Hyperglycaemia stress-induced renal injury is caused by extensive mitochondrial fragmentation, attenuated MKP1 signalling, and activated JNK-CaMKII-Fis1 biological axis. Cell. Physiol. Biochem 51, 1778–1798. doi: 10.1159/000495681
Zhang, Y., Zhang, M., Xu, W., Chen, J., and Zhou, X. (2017). The long non-coding RNA H19 promotes cardiomyocyte apoptosis in dilated cardiomyopathy. Oncotarget 8, 28588–28594. doi: 10.18632/oncotarget.15544
Zhang, Z., Liu, X., Feng, B., Liu, N., Wu, Q., Han, Y., et al. (2015). STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 34, 4808–4820. doi: 10.1038/onc.2014.404
Keywords: CaMKII, cardiorenal syndrome, inflammation, immune system, cardiovascular diseases
Citation: Junho CVC, Caio-Silva W, Trentin-Sonoda M and Carneiro-Ramos MS (2020) An Overview of the Role of Calcium/Calmodulin-Dependent Protein Kinase in Cardiorenal Syndrome. Front. Physiol. 11:735. doi: 10.3389/fphys.2020.00735
Received: 16 March 2020; Accepted: 08 June 2020;
Published: 14 July 2020.
Edited by:
Jonatan Barrera-Chimal, Universidad Nacional Autónoma de México, MexicoReviewed by:
Przemyslaw Radwanski, The Ohio State University, United StatesCopyright © 2020 Junho, Caio-Silva, Trentin-Sonoda and Carneiro-Ramos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela Sorelli Carneiro-Ramos, bWFyY2VsYS5yYW1vc0B1ZmFiYy5lZHUuYnI=; bXNvcmVsbGlAZ21haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.