 Mengyuan Ge
Mengyuan Ge Flavia Fontanesi
Flavia Fontanesi Sandra Merscher
Sandra Merscher Alessia Fornoni
Alessia Fornoni
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 07 July 2020
Sec. Renal Physiology and Pathophysiology
Volume 11 - 2020 | https://doi.org/10.3389/fphys.2020.00732
This article is part of the Research Topic Mitochondria in Renal Health and Disease View all 10 articles
The kidney is one of the most energy-demanding organs that require abundant and healthy mitochondria to maintain proper function. Increasing evidence suggests a strong association between mitochondrial dysfunction and chronic kidney diseases (CKDs). Lipids are not only important sources of energy but also essential components of mitochondrial membrane structures. Dysregulation of mitochondrial oxidative metabolism and increased reactive oxygen species (ROS) production lead to compromised mitochondrial lipid utilization, resulting in lipid accumulation and renal lipotoxicity. However, lipotoxicity can be either the cause or the consequence of mitochondrial dysfunction. Imbalanced lipid metabolism, in turn, can hamper mitochondrial dynamics, contributing to the alteration of mitochondrial lipids and reduction in mitochondrial function. In this review, we summarize the interplay between renal lipotoxicity and mitochondrial dysfunction, with a focus on glomerular diseases.
Measurement of resting metabolic rates shows that the kidney and heart have the highest resting energy expenditure among major organs and tissues in adults (Wang et al., 2010). Mitochondria provide energy to support kidney functions such as removing waste and balancing fluid. The energy currency ATP is mostly generated by oxidative phosphorylation (OXPHOS) coupled to electron transport through the mitochondrial respiratory chain (MRC). Increasing evidence indicates that mitochondrial damage and dysfunction are major contributors to the pathogenesis of chronic kidney disease (CKD) (Che et al., 2014). Lipids serve as a source and reservoir of energy supply. However, lipid surplus can hamper mitochondrial dynamics, contributing to increased reactive oxygen species (ROS) production and mitochondrial dysfunction. Conversely, impaired mitochondrial function can disturb the balance between lipid availability and lipid use, further increasing lipid accumulation (Schrauwen-Hinderling et al., 2016). Excess renal lipids have been reported in CKD of different origin, suggesting the existence of renal lipotoxicity (Gai et al., 2019). Although it has been suggested that both lipid dysmetabolism and mitochondrial dysfunction contribute to CKD, it remains to be established whether renal lipotoxicity is the cause or the consequence of mitochondrial dysfunction in the pathogenesis of CKD.
While tubular cells are mainly responsible for fluid and waste balance and account for most of the energy metabolism in the kidney, mitochondrial dysfunction and lipotoxicity were recently also described in diseases of glomerular origin. This is of particular importance as, different from tubular cells, key glomerular cells forming the kidney filtration barrier such as podocytes are terminally differentiated and, once injured, are unlikely to be replaced, thus causing proteinuria and disease progression (Kriz et al., 1998). Unlike tubular epithelial cells which preferentially use fatty acid (FA) β-oxidation as the main energy source (Kang et al., 2015), podocytes, endothelial cells, and mesangial cells in glomeruli highly rely on glucose as the substrate for energy production, while FA is used as an alternative substrate (Abe et al., 2010; Fink et al., 2012; Czajka and Malik, 2016). In the diabetic kidney, glucose oxidation in glomerular cells is disrupted and could account for the switch to FA oxidation (Forbes and Thorburn, 2018). In support, Imasawa et al. reported an oxidative shift during podocyte differentiation, where the expression of most glycolytic enzymes is decreased while the expression of proteins that are involved in FA β-oxidation is up-regulated (Imasawa et al., 2017). Peroxisome proliferator-activated receptor (PPAR)-γ coactivator (PGC)-1α expression can also induce a shift from their baseline glucose preference toward FA usage in podocytes (Li et al., 2017). Glomerular diseases, either primary or secondary to systemic diseases, represent an important cause of CKD (D’Agati et al., 2011). While we previously discussed how lipid accumulation contributes to mitochondrial dysfunction (Ducasa et al., 2019a), the current review focusses on and highlights the “two-way” interaction between lipids and mitochondria. More precisely, we summarize the findings observed in the setting of glomerular diseases with the goal to link lipotoxicity and mitochondrial dysfunction to irreversible podocyte injury and glomerular diseases.
Mitochondria fulfill several pivotal roles in cellular metabolism. In aerobic organisms, mitochondria are mostly responsible for the conversion of nutrient-derived energy in the form of ATP molecules through the mitochondrion-housed pathways of the Krebs cycle, FA β-oxidation, and OXPHOS (Lenaz and Genova, 2010). The complete oxidation of sugars and fats to carbon dioxide is achieved in the Krebs cycle and is paired with the concomitant conservation of free energy in the form of the reducing equivalents, nicotinamide-adenine dinucleotide (NADH), and flavin-adenine dinucleotide (FADH2). The metabolic intermediate oxidized in the Krebs cycle, acetyl-coenzyme A (acetyl-CoA), derives from pyruvate generated by glycolysis in the cytosol and from the breakdown of FA. FA β-oxidation occurs primarily in mitochondria through a series of four enzymatic reactions that constitute the predominant energy-producing pathway in the kidney (Bhargava and Schnellmann, 2017). In mitochondria, both the Krebs cycle and FA β-oxidation generate NADH and FADH2, which in turn transfer electrons to the MRC. Here, energy is further converted into an electrochemical gradient across the mitochondrial inner membrane. This conversion is achieved by the coupling of electron transfer through the MRC components to the final electron acceptor, molecular oxygen, with proton translocation from the mitochondrial matrix to the intermembrane space. Lastly, the protein gradient is used as the source of energy to drive ATP synthesis. The MRC is formed by four multimeric complexes (CI–CIV) embedded in the mitochondrial inner membrane and two mobile electron carriers, coenzyme Q (CoQ) and cytochrome c. Together, the MRC and ATP synthase (or CV) constitute the mitochondrial OXPHOS system (Lenaz and Genova, 2010).
ROS are byproducts of mitochondrial respiration, generated by electron “leakage” to oxygen, mainly at the level of CI and CIII of the MRC, to form superoxide anion (Stowe and Camara, 2009). In vivo, superoxide radicals have a short half-life and are rapidly converted into hydrogen peroxide by the action of superoxide dismutases, SOD1 and SOD2, located in the mitochondrial intermembrane space and matrix, respectively. While at low levels, ROS fulfill physiological functions as signaling molecules, mitochondrial dysfunction is commonly associated with a cellular energy deficit and increased ROS production, leading to the oxidative damage of cellular structures (Vafai and Mootha, 2012). Free FA, when in excess, can act as mitochondrial respiration uncouplers and MRC CI inhibitors and can also cause oxidative stress. To avoid lipotoxicity, FA levels in mitochondria are tightly controlled. FAs are stored in cytosolic lipid droplets (LDs), which exhibit close physical and metabolic interactions with mitochondria (Aon et al., 2014).
Furthermore, mitochondrial dysfunction is also frequently characterized by structural and morphological alterations. Mitochondria form a complex and highly dynamic network inside cells, which is continuously shaped by the processes of mitochondrial fusion and fission (Lackner, 2014). Mitochondrial hyperbranching, fragmentation, and loss of the mitochondrial inner membrane invaginations, known as cristae, have been observed in pathological conditions associated with mitochondrial respiratory defects and stress (Vincent et al., 2016). In addition to their role in cellular energy production, mitochondria are a hub for anabolic reactions, since the Krebs cycle provides intermediates for a wide range of biosynthetic pathways. These include precursors for amino acids, nucleobases, the antioxidant molecule glutathione, and metal prosthetic group biosynthesis. Notably, the Krebs cycle intermediate citrate is exported into the cytosol, where it is used as a substrate for FA and cholesterol synthesis (Owen et al., 2002). Among lipids, the synthesis of cardiolipin (CL) takes place in mitochondria. CL is the signature lipid of the mitochondrial inner membrane, which has an essential structural role in cristae formation and is required for optimal MRC assembly and function (Claypool and Koehler, 2012).
Mitochondria are organelles of endosymbiotic origin and still retain their genome, the mitochondrial DNA (mtDNA), and gene expression machinery, the mitochondrial ribosomes. In humans, the mtDNA encodes for a complete set of tRNAs, the ribosomal RNAs, and a handful of proteins, all catalytic subunits of the OXPHOS system. It has been estimated that the mitochondrial proteome could comprise about 1,500 proteins (Vafai and Mootha, 2012). With only 13 polypeptides encoded by the mtDNA, most of these proteins, including the remaining subunits of the OXPHOS system, are encoded by nuclear genes, synthesized in the cytosol, and imported into mitochondria. Mutations in both nuclear and mitochondrial genes are causes of major energetic deficits associated with severe hereditary diseases known as mitochondrial disorders (Turnbull and Rustin, 2016). While defects in nuclear genes follow Mendelian inheritance, mutations in the mtDNA are maternally transmitted. Since each cell contains hundreds to thousands of mtDNA molecules, wildtype and mutant genomes can co-exist, a condition known as heteroplasmy. Clinical manifestation appears only when the fraction of mutated molecules reaches a certain threshold, which can be tissue-specific (Turnbull and Rustin, 2016). Primary mitochondrial disorders are multisystemic diseases, although high energy demanding organs, such as heart and brain, are frequently the most affected. Kidneys also require large amounts of energy, mainly for tubular reabsorption (Hansell et al., 2013). In several instances, renal manifestations have been reported in patients affected by mitochondrial disorders (Finsterer and Scorza, 2017). In addition to primary mitochondrial diseases, mitochondrial dysfunction has been shown to be involved in the etiology and progression of numerous human pathologies, including kidney diseases and, in particular glomerular diseases, which are the focus of this review.
Genetic defects in mtDNA or nuclear DNA (nDNA) that encode mitochondrial proteins can result in mitochondrial cytopathies. Though most mitochondrial disorders with a renal phenotype are characterized by tubular dysfunction, mtDNA 3243A > G mutation and CoQ10 biosynthesis defects (due to nDNA mutation) are associated with two mitochondrial cytopathies primarily causing glomerular diseases, most commonly focal segmental glomerulosclerosis (FSGS).
Thirteen proteins are encoded by the mitochondrial genome, all of which are subunits of the MRC (Anderson et al., 1981; Di Donato, 2009). Mutations in mtDNA cause defects in the mitochondrial OXPHOS, resulting in energy deficiency and increased ROS. The most common mtDNA point mutation 3243A > G in the tRNALEU gene is associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. Representing one of the most frequent mitochondrial disorders, MELAS syndrome could be considered as an underlying cause of primary FSGS, as several studies reported glomerular manifestation, often presents as FSGS (Kurogouchi et al., 1998; Cheong et al., 1999; Hotta et al., 2001; Gucer et al., 2005; Rudnicki et al., 2016; Narumi et al., 2018). In patients with MELAS syndrome and renal symptoms, the renal disease initially manifests with proteinuria and typical changes of the glomerular basement membrane (GBM) (Kurogouchi et al., 1998; Cheong et al., 1999; Narumi et al., 2018). Biopsy samples from these patients show abnormal mitochondria in podocytes, as well as foot process effacement (Gucer et al., 2005; Narumi et al., 2018). Patients with other point mutations (m.A4269G, m.A5728G, and m.A5843G) that affect mitochondrial tRNAs present with various severe clinical phenotypes, from fatal cardiomyopathy to multi-organ failure. These mutations are not associated with MELAS but were also reported to be associated with an FSGS phenotype (Taniike et al., 1992; Scaglia et al., 2003; Meulemans et al., 2006). Interestingly, deafness was also described in individuals with MELAS syndrome, which resembles the symptom of another rare genetic disease characterized by mutations of another important constituent of the GBM, i.e., type IV collagen (Alport syndrome). Besides, FSGS has been reported in a case of Kearns-Sayre syndrome due to mtDNA deletion (Becher et al., 1999). It is worth noting that mtDNA mutations associated with glomerular diseases affect overall mitochondrial gene expression and, in consequence, cause combined MRC enzymatic deficiencies, commonly associated with very severe outcomes.
Besides the 13 proteins encoded by the mitochondrial genome, there are over 1,158 other nuclear-encoded mitochondrial proteins (Calvo et al., 2016). The enzymes of the CoQ10 biosynthetic pathway are encoded by nuclear CoQ genes. Located mainly in the mitochondrial inner membrane, CoQ10 is a carrier that transfers electrons from complex I and II to complex III of the MRC (Ernster and Dallner, 1995). At least 15 genes are required in CoQ10 biosynthesis (Desbats et al., 2015). Mutations in these genes may cause CoQ10 deficiency, some of which were reported to be associated with glomerular involvement. In humans, prenyl (decaprenyl) diphosphate synthase subunit 1 (PDSS1) and PDSS2 are responsible for the synthesis of the prenyl side chain of CoQ10 (Saiki et al., 2005). Deficiency in CoQ10 content was reported in a patient carrying compound heterozygous mutations in PDSS2. The patient was diagnosed with Leigh’s syndrome with nephropathy (Lopez et al., 2006). Dietary supplementation with CoQ10 was shown to rescue proteinuria and interstitial nephritis in Pdss2kd/kd mice (Saiki et al., 2008) and diabetic nephropathy in db/db mice (Sourris et al., 2012). Gasser et al. analyzed patients with FSGS and collapsing glomerulopathy for PDSS2 polymorphisms. Surprisingly, they found a reduction of CoQ10 content, irrespectively of the PDSS2 haplotype (Gasser et al., 2013). Vasta et al. also reported that one patient carrying a PDSS1 mutation presented with nephrotic syndrome (Vasta et al., 2012). Pathogenic CoQ2 variants are another cause of primary CoQ10 deficiency. Patients with CoQ2 mutations have been reported to exhibit nephropathy that can be treated by CoQ10 supplementation (Starr et al., 2018). Moreover, loss-of-function mutations in CoQ6 have been described as a cause of glomerular damage, and patients manifested with FSGS in renal biopsy (Heeringa et al., 2011). It is important to note that genetic testing in patients with steroid-resistant nephrotic syndrome identified mutations in several genes involved in mitochondrial function and CoQ10 biosynthesis, including AarF domain-containing kinase 4 (ADCK4), CoQ6, CoQ2, and PDSS2 (Lovric et al., 2016). Patients who carry mutations in the ADCK4 gene may present with CoQ10 deficiency and mitochondrial nephropathy manifesting as FSGS (Ashraf et al., 2013; Yang et al., 2018; Widmeier et al., 2020). ADCK4 interacts with members of the CoQ10 biosynthesis pathway and is involved in CoQ biosynthesis, although its function remains to be characterized.
Mitochondrial dysfunction can lead to lipid metabolism disorders in several cell types. Vankoningsloo et al. studied preadipocytes incubated with inhibitors of mitochondrial respiration (antimycin A) and found that mitochondrial dysfunction induces triglyceride accumulation. This process is mediated by reduced PPARγ activity leading to decreased expression of muscle isoform of carnitine palmitoyltransferase I (CPT1) and FA β-oxidation. Direct conversion of glucose into triglycerides and activation of carbohydrate-responsive element-binding protein (ChREBP), a lipogenic transcription factor, were also observed (Vankoningsloo et al., 2005). Boren and Brindle observed that the induction of apoptosis leads to mitochondrial ROS generation in murine lymphoma cells and mouse embryo fibroblasts, which inhibits mitochondrial FA β-oxidation but increases acyl-CoA synthetase activity for FA synthesis. Thereby, the switch from FA β-oxidation into lipid synthesis results in LD accumulation in the cytoplasm (Boren and Brindle, 2012).
Mitochondrial FA β-oxidation is the major energy pathway of the kidney. Mitochondrial dysfunction leads to compromised lipid utilization, eventually resulting in lipid accumulation, which is toxic to cells in the kidney. Herman-Edelstein et al. measured genes and enzymes involved in the FA β-oxidation pathway in kidneys of patients with diabetic nephropathy compared to healthy kidneys. The genetic analysis revealed the downregulation of some key transcriptional regulators of FA β-oxidation, such as PPARα, PPARγ, CPT1, and Aconitase 2 (ACO2) (Herman-Edelstein et al., 2014). Interestingly, upregulation of low-density lipoprotein receptor (LDLR) and cluster of differentiation 36 (CD36), downregulation of liver X receptor α (LXRα), ATP-binding cassette transporter A1 (ABCA1), ATP-binding cassette transporter G1 (ABCG1), and apolipoprotein E (APOE) were also detected, indicating alterations in cholesterol metabolism (Figure 1; Herman-Edelstein et al., 2014). In addition to diabetic nephropathy phenotypes including podocyte process effacement, thickening of the GBM, and mesangial expansion, electron microscopy analysis also showed extensive accumulation of LDs in podocytes, tubular epithelial cells, mesangial cells, and fenestrated endothelial cells (Herman-Edelstein et al., 2014). Consistently, CD36, PPARα, PPARγ, and LDLR were found to be genes with the highest connectivity to diabetic nephropathy progression in a study comparing human-mouse cross-species glomerular transcriptional networks (Hodgin et al., 2013).
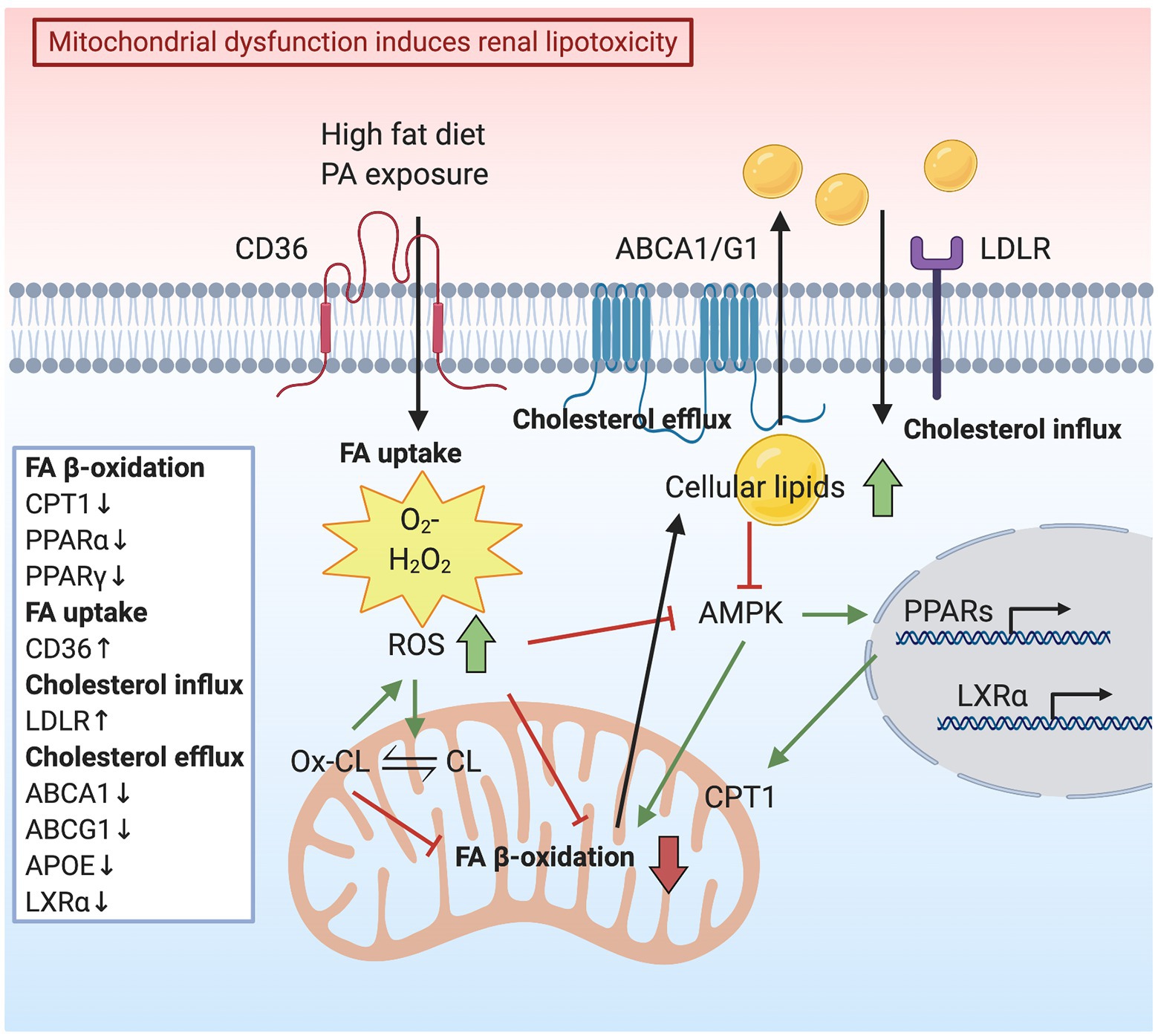
Figure 1. Schematic representation indicating how mitochondrial dysfunction induces renal lipotoxicity. High-fat diet or palmitic acid (PA) exposure increases mitochondrial reactive oxygen species (ROS) production. Consequently, elevated oxidative stress reduces mitochondrial fatty acid (FA) oxidation, resulting in lipid accumulation in the cytoplasm. Lipid accumulation is also caused by deceased cholesterol efflux mediated by ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1) and elevated cholesterol influx via low-density lipoprotein receptor (LDLR). AMP-activated protein kinase (AMPK) suppression due to a lipid surplus inhibits mitochondrial FA oxidation and lipid utilization, accompanied by downregulation of transcription factors, liver X receptor (LXR) α, peroxisome proliferator-activated receptor (PPAR) α, and PPARγ. ROS generation contributes to cardiolipin (CL) peroxidation and loss of mitochondria cristae membranes, thus impairing the capacity for energy production.
A renal-protective effect of PPARs was also reported. The study by Calkin et al. suggests that the administration of PPARα, PPARγ, and PPARα/γ agonists in experimental mouse models of diabetes has renoprotective effects (Calkin et al., 2006). Doxorubicin is an antitumor drug that induces renal injury, leading to proteinuria. Doxorubicin-induced PPARα knockout mice showed more severe podocyte foot process effacement compared with Doxorubicin-induced wildtype mice but the treatment with PPARα agonist fenofibrate reduced proteinuria and ameliorated podocyte foot process effacement (Zhou et al., 2011). Fenofibrate has also been shown to prevent high-fat diet (HFD)-induced glomerular injury in an experimental model of diabetic nephropathy (Tanaka et al., 2011). Fenofibrate treatment reduces glomerular lipid (mainly triglycerides) accumulation, attenuates oxidative stress in kidneys of HFD-fed mice, and enhances the expression of lipolytic enzymes, including CPT1. Fenofibrate also attenuates tubulointerstitial injury by enhancing renal lipolysis (Tanaka et al., 2011). In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial, a trend to renoprotection was observed in fenofibrate treated patients with diabetic nephropathy (Keech et al., 2005). PPARγ is also involved in renal lipid metabolism. In fact, PPARγ expression is reduced in glomeruli of diabetic mice, while activation of PPARγ inhibits the development of diabetic glomerular lesion (Zheng et al., 2002). Moreover, another study demonstrated that both PPARα and PPARγ agonists increase LXRα and ABCA1 gene expression and enhance apolipoprotein A1 (APOA1)-mediated cholesterol efflux from human mesangial cells (Ruan et al., 2003). However, PPARγ2 KO mice generated in an ob/ob background exhibit lipid accumulation at both the glomerular and tubular levels, accompanied by glomerular damage, in addition to the original metabolic syndrome phenotype (Martinez-Garcia et al., 2012). Located in the mitochondrial inner membrane, uncoupling proteins (UCPs) are mitochondrial transporters that control the level of coupled respiration (Rousset et al., 2004). Ke et al. reported that UCP2 expression is increased in human biopsy samples and mice kidney tissues with tubulointerstitial fibrosis but UCP2 deficient mice display reduced lipid accumulation and attenuated hypoxia in kidney tissue. Compared with wildtype mice, the expression of PPARα and CPT1α, which are the major regulators of lipid metabolism, is restored in UCP2 deficient mice (Ke et al., 2020). Both CPT1 and ACO2 are the rate-limiting enzymes of FA β-oxidation. Idrovo et al. demonstrated that stimulating CPT1 activity with a synthetic compound (C75) not only restores ATP generation but also improves renal function using a rat model of ischemia/reperfusion injury (Idrovo et al., 2012). In addition, mRNA levels of ACO1 and ACO2 are decreased in the tubulointerstitial and the glomerular compartment of non-diabetic CKD patients when compared to healthy controls (Hallan et al., 2017).
Free FAs are sources of energy to synthesize ATP by mitochondrial FA β-oxidation. Palmitic acid (PA) is the predominant circulating saturated FA, which plays an important role in podocyte injury through lipotoxicity initiated by mitochondrial superoxide overproduction (Lee et al., 2017). Podocytes are vulnerable to PA-induced oxidative damage due to impaired peroxidase activity. Podocytes incubated with PA show increased FA uptake, mitochondrial superoxide, and hydrogen peroxide (H2O2) generation, as well as elevated AMP-activated protein kinase (AMPK) α phosphorylation (Figure 1; Lee et al., 2017). Furthermore, Liu et al. demonstrated that PA induces intracellular lipid accumulation and LD formation. It also induces podocyte apoptosis through activation of the mitochondrial pathway and triggers the release of cytochrome c from the mitochondria to the cytosol (Liu et al., 2018). In mouse podocytes, PA induces apoptosis by increasing cytosolic Ca2+ concentration, resulting from Ca2+ accumulation in the mitochondria via the mitochondrial Ca2+ uniporter (Yuan et al., 2017). The depletion of endoplasmic reticulum (ER) Ca2+ is also due to oxidative stress caused by PA (Xu et al., 2015). Under diabetic conditions, podocytes are susceptible to PA-induced oxidative damage due to an inadequate activity of peroxidase, the enzyme that catalyzes the conversion of the intracellular H2O2 to water in response to excess ROS generation (Lee and Lee, 2018). On the contrary, the nontoxic mono-unsaturated FA oleic acid (OA) inhibits the PA-induced ROS formation in podocytes (Lee et al., 2017; Lee and Lee, 2018). OA attenuates ROS generation and protects the mitochondria from PA-induced oxidative stress by the restoration of AMPK activity (Palomer et al., 2018).
Oxidative stress in the form of lipid peroxidation can be caused by the generation of lipid peroxyl radicals in podocytes. Excessive accumulation of lipid peroxyl radicals contributes to podocyte injury in CKD (Kruger et al., 2018). It was demonstrated that ROS generation contributes to glomerular and tubular injury through lipid peroxidation of cell and organelle membranes. ROS-induced lipid peroxidation disrupts the structural integrity of the lipid bilayer and impairs the capacity for energy production (Baud and Ardaillou, 1993). SS-31 is a tetrapeptide that protects mitochondria cristae structure and matrix density in all kidney cells. SS-31 targets CL, the phospholipid almost exclusively present in the inner mitochondrial membrane (Szeto and Birk, 2014). CL peroxidation alters mitochondrial cristae formation (Szeto, 2014), thus disrupting the functional and structural integrity of respiratory supercomplexes (Pfeiffer et al., 2003). Moreover, during apoptosis, CL binds to cytochrome c to form peroxidase complexes capable of catalyzing CL peroxidation and promoting cytochrome c release from mitochondria (Kagan et al., 2005). SS-31 selectively binds to CL, preventing CL from converting cytochrome c into a peroxidase while maintaining its function as an electron carrier (Szeto, 2014). The study of Szeto et al. described changes in mitochondrial structure in glomerular endothelial cells, podocytes, and proximal tubular epithelial cells after HFD induction. HFD increases mitochondrial ROS generation, causing CL peroxidation and loss of mitochondria cristae membranes. Increased ROS limits mitochondrial FA β-oxidation, which in turn causes cellular lipid accumulation and inhibits AMPK activity (Figure 1; Szeto et al., 2016). By preserving normal mitochondrial cristae structure, SS-31 treatment was able to restore AMPK activity and prevent intracellular lipid accumulation, ER stress, and glomerular inflammation (Szeto et al., 2016). AMPK is a cellular energy sensor that regulates glucose, lipid, and protein metabolism. During energy shortage, AMPK is activated and promotes ATP production (Hardie et al., 2012). On the contrary, AMPK is down regulated under conditions of energetic oversupply (Picard et al., 2012). Mice fed on a HFD show increased glomerular area and matrix accumulation. A significant rise of cholesteryl esters and phospholipids in the kidneys was also observed, in association with reduced AMPK activity. Reduced AMPK activity was also described in kidneys from both diabetic mice and in patients with diabetes (Dugan et al., 2013). Conversely, the activation of AMPK reverses the accumulation of cholesteryl esters and phospholipids in kidneys of mice fed a HFD (Decleves et al., 2011, 2014). Improving mitochondrial biogenesis and function by AMPK activation was found to be a therapeutic approach in a rat model of diabetic nephropathy (Zhou et al., 2019). The administration of AMPK activators increased the expression of PGC-1α, which induces a ROS scavenging mechanism, including the expression of SOD2 in the presence of ROS, protecting cells against ROS generation and damage (Spiegelman, 2007; Zhou et al., 2019). Similarly, AMPK activation in diabetic mice restores mitochondrial content and function while improving DKD progression (Figure 1; Dugan et al., 2013).
Defects in genes relevant to lipid metabolism result in inherited diseases associated with renal lipotoxicity. Tangier disease is a rare disorder characterized by reduced levels of high-density lipoprotein (HDL) in the blood. Patients with Tangier disease have mutations in the ABCA1 gene and present with mild proteinuria and foamy podocytes in kidney biopsy (Ferrans and Fredrickson, 1975; Schaefer et al., 2010). Similarly, foamy podocytes, vacuolated tubular epithelial cells, and interstitial foam cells were described in a case report of a patient with Niemann-Pick disease, an inherited condition with an accumulation of sphingomyelin in various organs, including kidneys (Grafft et al., 2009). Caused by the accumulation of cholesterol in blood and tissue, familial lecithin-cholesterol acyltransferase (LCAT) deficiency is another genetic disorder, where some patients present with kidney injury. Renal biopsies of patients with LCAT deficiency identified LDs in mesangial and endothelial cells (Ossoli et al., 2015). Borysiewicz et al. described that patients with LCAT deficiency show disturbance of lipid metabolism and accumulation of lipids in the kidneys, eventually leading to end-stage kidney disease (Borysiewicz et al., 1982). In addition, transgenic mice with LCAT deficiency develop proteinuria and glomerulosclerosis characterized by the accumulation of free cholesterol and polar lipids in glomeruli (Lambert et al., 2001). Disturbed lipid metabolism was also reported in rare cases of lipoprotein glomerulopathy. Patients with an APOE gene mutation are characterized by proteinuria and hyperlipidemia. It has been suggested that the mutation of APOE increases the affinity of lipoproteins for glomerular capillary walls and promotes the formation of lipoprotein aggregates (Sam et al., 2006; Tsimihodimos and Elisaf, 2011). The evidence that genetic impairment of genes regulating cholesterol efflux leads to glomerular disease strongly suggests a causative link between altered renal lipid metabolism and kidney disease. This is very different from genetic disorders of hyperlipidemia, such as familial hypercholesterolemia (FH), where the high level of circulating lipids does not necessarily translate into an increased accumulation of renal lipids and does not always cause a renal phenotype. While this remains controversial, a recent association between FH and CKD was reported (Emanuelsson et al., 2018), and a patient with homozygous FH was found to develop FSGS (Elmaci et al., 2007). Similarly, the accumulation of lipids was found in the renal parenchyma of patients affected by FH (Buja et al., 1979). Although some of these studies suggest a link between systemic hyperlipidemia and CKD, most of the studies have demonstrated that statins may partially reduce proteinuria but this does not result in the prevention of CKD progression (Agarwal, 2006).
Podocytes rely on a constant supply of lipids and proteins to form foot processes (Simons et al., 1999; Fornoni et al., 2014). However, lipid overload causes lipotoxicity. Lipid accumulation and increased inflammation accelerate lipotoxicity induced glomerular disease (Martinez-Garcia et al., 2015). Among glomerular cells, podocytes are terminally differentiated cells that are most susceptible to damage caused by lipid overload. The podocyte slit diaphragm is assembled as a lipid-raft like structure, enriched with lipids including sphingolipids and cholesterol. Plasma membrane and intracellular lipids can affect podocyte function (Fornoni et al., 2014). Localized to lipid raft domain, sphingomyelinase-like phosphodiesterase 3b (SMPDL3b) is a sphingolipid-related enzyme that modifies the plasma lipid composition and modulates intracellular inflammatory pathways in podocytes (Heinz et al., 2015; Yoo et al., 2015). Moreover, cholesterol is required for the proper function of the slit diaphragm (Simons et al., 2001), while excessive accumulation of cholesterol causes podocyte injury and proteinuria (Merscher-Gomez et al., 2013; Pedigo et al., 2016).
Foam cells are macrophages that ingest low-density lipoprotein (LDL) during hyperlipidemia. Their formation represents the hallmark of atherosclerosis (Yu et al., 2013). Interestingly, foam cells also appear in various glomerular diseases, including diabetic nephropathy, FSGS, nephrotic syndrome, and Alport syndrome (Nolasco et al., 1985; Stokes et al., 2006; Wu et al., 2009; Eom et al., 2015), indicating lipid-mediated toxicity. However, it is not established whether foam cells generated during the pathogenesis of kidney diseases are renal resident cells or infiltrating macrophages from the circulation (Eom et al., 2015). It is certain that glomerular cells can uptake LDL. In fact, the activity of lipoprotein receptors in cultured epithelial cells of the human glomerulus was characterized in the early 1990s. Grone et al. demonstrated that cultured glomerular epithelial cells show lipoprotein receptor activity, which mediates the uptake of apolipoprotein B (APOB)‐ and APOE-rich lipoproteins, contributing to lipid accumulation and oxidative stress (Grone et al., 1990). In addition, the accumulation of APOB and APOE in immune deposits could lead to GBM damage (Figure 2). Heymann nephritis is a rat model of membranous nephropathy characterized by the formation of subepithelial immune deposits in the GBM (Heymann et al., 1959). In Heymann nephritis, Kerjaschki et al. demonstrated that antibodies targeting megalin (an LDL-related protein) inhibit megalin-mediated binding and clearance of APOE and APOB, leading to the accumulation of apolipoproteins in immune deposits. The lipid environment is associated with lipid peroxidation, injury of the GBM, and proteinuria (Kerjaschki et al., 1997). Oxidized LDL has also been found to directly damage podocytes via the chemokine ligand 16 (CXCL16), as well as induce ROS production in podocytes (Bussolati et al., 2005; Gutwein et al., 2009a). CXCL16 is a major scavenger receptor for oxidized LDL in human podocytes. Both glomerular levels of CXCL16 and oxidized LDL are increased in patients with diabetic nephropathy and membranous nephropathy (Gutwein et al., 2009a,b).
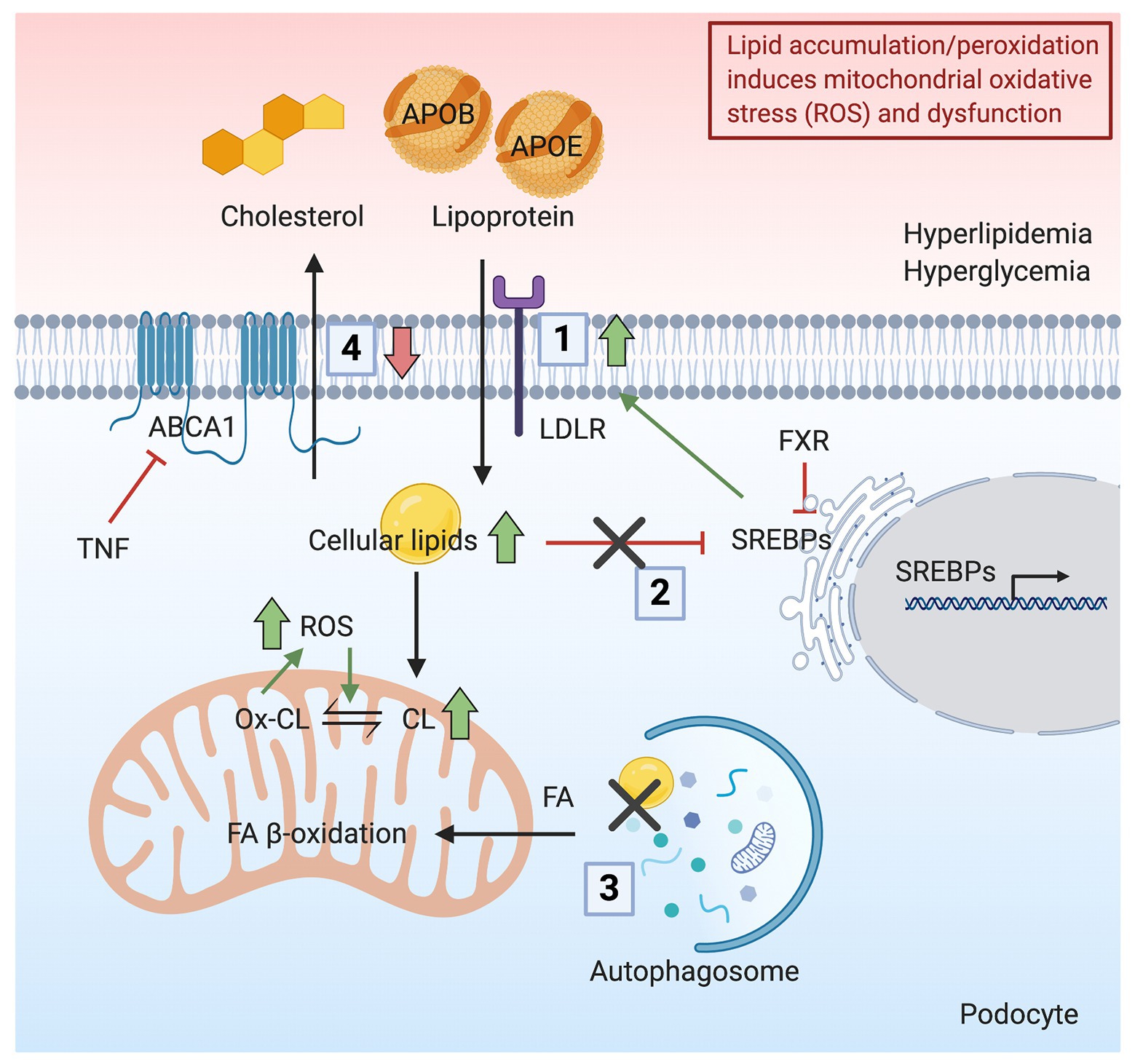
Figure 2. Schematic representation indicating how lipid accumulation/peroxidation induces mitochondrial oxidative stress and dysfunction. Podocyte lipid accumulation and increased peroxidation induces ROS production and mitochondrial dysfunction. (1) In the presence of hyperlipidemia, increased uptake of apolipoprotein B (APOB)‐ and apolipoprotein E (APOE)-rich lipoproteins occurs in podocytes through the LDLR, resulting in a lipid-rich environment. (2) Hyperglycemia disrupts the feedback regulation of sterol regulatory element-binding proteins (SREBPs), causing further lipid accumulation. (3) Cellular lipids, mainly in the form of triglycerides, stored in lipid droplets (LDs) are hydrolyzed and delivered as FAs to mitochondria by autophagic clearance. The inhibition of autophagic clearance of lipids is associated with increased lipid endocytosis. (4) Downregulation of ABCA1 impairs reverse cholesterol transport, resulting in the accumulation of cholesterol and CL and peroxidation of CL. Together, the accumulation of lipids in podocytes induces ROS production and mitochondrial dysfunction.
Experimental evidence also supports a role of lipid-lowering agents in the protection of kidney diseases. For example, treatment of patients with lipid-lowering agents such as the hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitor atorvastatin was able to lower serum lipids and glomerular lesions but to a lesser extent than the blockage of the renal angiotensin system with quinapril. In fact, the renal protective effect of atorvastatin is associated with lipid-related and non-lipid-related effects (Blanco et al., 2005). Once more, however, the renal protective effects of statin treatment remain to be confirmed in patients affected by CKD. Interestingly, hypercholesterolemia associated with nephrotic syndrome can be ameliorated by inhibiting proprotein convertase subtilisin/kexin type 9 (PCSK9), a posttranscriptional regulator of the LDL receptor. PCSK9 targets LDLR for degradation and thereby reduces the clearance of LDL from the circulation (Shrestha et al., 2019). Inhibition of PCSK9 decreases the proportion of APOB-associated cholesterol (VLDL and LDL) and increases the proportion of HDL-associated cholesterol, producing a more protective lipoprotein profile (Haas et al., 2016). Currently, the monoclonal antibodies evolocumab and alirocumab are approved PCSK9 inhibitors for therapeutic use and are effective and safe in patients with mild to moderate CKD (Schmit et al., 2019). However, it would be important to study if PCSK9 inhibitors can also alleviate proteinuria and CKD progression in patients with advanced CKD, such as nephrotic syndrome. Studies in cultured vascular smooth muscle and endothelial cells suggest that inhibition of PCSK9 attenuates ROS production, while PCSK9 overexpression increases ROS generation in a concentration-dependent manner (Ding et al., 2015). The crosstalk between PCSK9 and mtDNA damage in vascular smooth muscle cells is thought to be partially mediated by mitochondria-derived ROS (Ding et al., 2016). Lastly, more studies are needed to assess the contribution of LDL apheresis to the treatment of proteinuric kidney disease, in order to gain a better understanding of whether the antiproteinuric effects are linked to LDL apheresis or related proteins.
Lipid distribution among plasma, tissues, and cellular organelles can be altered by inflammatory stress (Ruan et al., 2009). In addition to the uptake of plasma lipids into cells via lipoprotein receptors, other factors that mediate lipid accumulation in glomerular cells, thus contributing to disease development and progression, have been described in several kidney diseases. Among them, we have demonstrated that glomerular TNF is a major driver of lipid dysmetabolism in FSGS (Pedigo et al., 2016). Our group reported the accumulation of cholesterol in kidney cortices in mouse models of diabetes (Ducasa et al., 2019b), Alport syndrome, and FSGS (Pedigo et al., 2016; Mitrofanova et al., 2018). Impaired ABCA1 mediated reverse cholesterol transport in a mouse model of FSGS was rescued by genetic ABCA1 overexpression. Similarly, treatment with hydroxypropyl-β-cyclodextrin (HPβCD) was found to reduce cholesterol accumulation in kidney cortices and was found to protect from renal failure in mouse models of FSGS, Alport syndrome, and DKD (Pedigo et al., 2016; Mitrofanova et al., 2018). Additionally, lipid accumulation is associated with the downregulation of ABCA1 in podocytes treated with sera from patients diagnosed with type 1 diabetes or type 2 diabetes (Pedigo et al., 2016; Ducasa et al., 2019b). Moreover, podocyte-specific deletion of ABCA1 renders mice susceptible to DKD progression, while the induction of ABCA1 ameliorates podocyte injury (Ducasa et al., 2019b).
Apart from failing to remove lipids from cells, lipid oversupply is another major cause of renal dysfunction (Figure 2). Sterol regulatory element-binding proteins (SREBPs) activate genes involved in the synthesis and uptake of cholesterol, FAs, triglycerides and phospholipids, endocytosis of LDL, and glucose metabolism (Edwards et al., 2000; Horton et al., 2002). In DKD, increased expression of SREBPs is associated with the accumulation of triglycerides and cholesterol in the kidney, resulting in glomerulosclerosis and proteinuria (Sun et al., 2002). Similarly, C57BL/6J mice fed on a HFD also show renal accumulation of triglycerides and cholesterol, accompanied by glomerulosclerosis due to SREBPs pathway activation (Jiang et al., 2005). Moreover, LDLR plays a role in the feedback system that modulates plasma and intracellular cholesterol homeostasis. The expression of LDLR is tightly regulated by SREBPs and SREBP cleavage-activating protein (SCAP). In the presence of high glucose or inflammatory stress, the LDLR feedback regulation is disrupted, causing intracellular cholesterol accumulation and podocyte injury (Zhang et al., 2015a,b). Interestingly, the up-regulation of hepatic PCSK9 was found to be modulated by SREBP-2 and SREBP-1c, which could furthermore dysregulate LDL cholesterol (Jeong et al., 2008; Rong et al., 2017).
The turnover of lipids in podocytes regulates podocyte health. Serezani reported that the expression of phosphatase and tensin homolog (PTEN) inhibits phagocytosis, cell growth, and cytoskeletal remodeling (Serezani et al., 2012). The downregulation of PTEN is associated with increased lipid endocytosis in podocytes from patients with obesity-related glomerulopathy, in cultured mouse podocytes with PTEN knock-down and in mice with podocyte-specific knockout of PTEN, thus enhancing the uptake of lipids and causing podocyte injury and proteinuria through oxidative stress response (Shi et al., 2019). On the contrary, elevated autophagic sequestration of LDs was reported as an adaption to alcoholic liver disease (Eid et al., 2013). In the study of Sato et al., podocytes with a higher level of autophagy are more competent to remove proteins and lipids from cells, suggesting a tendency to better prognosis (Sato et al., 2006). Autophagy related 5 (ATG5) is a critical autophagy gene during nephrogenesis. Mutation of ATG5 in the kidney epithelium leads to FSGS in mice, accompanied by enhanced ROS generation, ER stress, and mitochondrial dysfunction (Kawakami et al., 2015). Interestingly, autophagy regulates lipid metabolism by autophagic clearance of cellular lipids stored as triglycerides in LDs, which are then hydrolyzed and delivered as FAs for energy production in mitochondria (Figure 2; Singh et al., 2009).
Lipid surplus induces a vicious cycle where the accumulation of lipids induces ROS production and mitochondrial dysfunction. Conversely, dysfunctional mitochondria may decrease the lipid oxidative capacity, which additionally increases the lipid surplus (Schrauwen-Hinderling et al., 2016). In a rat model of hypertension and proteinuria, increased metabolic stress was indicated by increased oxidized lipids derived from lipid breakdown in glomeruli at the early stage, causing decreased ATP and NADH levels. The change of metabolic aspects in glomeruli from these mice was associated with the activation of phosphoprotein-dependent signaling such as mTOR and AMPK (Rinschen et al., 2019). Steinberg et al. demonstrated that AMPK activity is suppressed by TNFα signaling, resulting in decreased FA β-oxidation and excess lipids in skeletal muscle (Steinberg et al., 2006). Notably, we previously showed that serum TNFR1 and TNFR2 levels are increased in patients with DKD and FSGS, whereas TNF levels are only increased in patients with DKD. More importantly, we demonstrated that glomerular TNF expression rather than systemic TNF causes free cholesterol accumulation and podocyte injury (Pedigo et al., 2016). High lipid availability induced ROS production can furthermore cause lipid oxidation (Schrauwen-Hinderling et al., 2016). Fibroblasts from patients with Tangier disease, who have an inherited ABCA1 mutation, show an increased content of the mitochondrial-specific phospholipid CL, while patients have reduced HDL levels in the blood (Fobker et al., 2001). Our group demonstrated that ABCA1 deficiency in human podocytes leads to CL accumulation and mitochondrial dysfunction. SS-31 has been used to scavenge ROS and stabilize CL (Zhao et al., 2004). The administration of an Abca1 inducer or elamipretide (SS-31) reduces CL peroxidation and ameliorates podocyte injury, indicating that ABCA1 deficiency causes CL accumulation and peroxidation, eventually leading to mitochondrial dysfunction (Figure 2; Ducasa et al., 2019b).
Reduction of renal parenchymal lipids ameliorates mitochondrial dysfunction, thus mitigating kidney injury. FSGS with podocyte injury and depletion can be mediated by mitochondrial oxidative damage in adjacent endothelial cells via endothelin-1 (EDN1) signaling. Podocyte-derived EDN1 can act on endothelial cells, which mediates oxidative stress and endothelial dysfunction via EDN1 receptor type A (EDNRA) activation, thus promoting podocyte apoptosis. In support of this observation, mitochondrial-targeted ROS scavengers and endothelin antagonists inhibit the release of EDN1, thus reducing mitochondrial oxidative stress and endothelial cell dysfunction, thereby preventing glomerulosclerosis (Daehn et al., 2014). In DKD, glomerular endothelial mitochondrial dysfunction is also associated with increased glomerular EDNRA expression and increased circulating EDN1. EDNRA antagonist treatment prevents mitochondrial oxidative damage in endothelial cells and ameliorates diabetes-induced glomerular injury, suggesting a crosstalk between podocytes and glomerular endothelial cells (Qi et al., 2017). Interestingly, EDN1 promotes lipid accumulation during the transformation of macrophage foam cells through the downregulation of ABCG1 and impaired HDL-mediated cholesterol efflux (Lin et al., 2011). It is possible that the inhibition of EDN1 signaling could promote reduced renal lipotoxicity in addition to suppressing oxidative stress. The farnesoid X receptor (FXR) is a bile acid-activated nuclear receptor that regulates lipid metabolism by modulating renal SREBP-1 activity. Since the SREBPs play a critical role in renal lipid accumulation, the administration of FXR agonists ameliorates renal triglyceride accumulation and regulates renal lipid metabolism in HFD-fed mice by modulating FA synthesis and oxidation, improving proteinuria and kidney injury (Wang et al., 2009). In addition, administration of the dual agonist of FXR and G protein-coupled membrane receptor Takeda G protein-coupled receptor 5 (TGR5) to mice decreases proteinuria and prevents mitochondrial impairments. This dual agonist modulates renal lipid metabolism and prevents renal lipid accumulation in mouse models of nephropathy associated with diabetes and obesity. Dual agonist treatment prevents inflammation, oxidative stress, and ER stress, as well as induces mitochondrial biogenesis and metabolism pathways (Wang et al., 2017, 2018). Similarly, Gai et al. also reported that FXR activation reduces glomerulosclerosis and tubulointerstitial injury in mice fed on a HFD. FXR agonist treated mice showed improved renal function in association with reduced lipid accumulation and reduced ROS. Additionally, FXR activation attenuated the decrease in autophagosomes in obese mice, maintaining effective energy production (Gai et al., 2016).
Lipids are essential sources for mitochondrial energy production. However, abnormal lipid metabolism in many tissues or cell types has proven to be detrimental and to contribute to the pathogenesis and progression of several disorders. We first summarized glomerular diseases caused by inherited mitochondrial disparities, as well as by inherited lipid disorders, thus providing the genetic evidence in humans that both altered mitochondrial function and lipid metabolism may contribute to the pathogenesis of kidney diseases. We then elaborated on the independent impacts of lipotoxicity and mitochondrial dysfunction on kidney diseases, especially diseases characterized by glomerular injury. Finally, we reviewed the evidence that lipotoxicity can be the cause or the consequence of mitochondrial dysfunction. Lipids form the bilayer membrane structure of mitochondria. However, the accumulation of lipids induces excess ROS generation and increased mitochondrial oxidative stress. Meanwhile, accumulation and peroxidation of CL interrupt the normal structure of mitochondria and impair mitochondrial respiration and energy production (Figure 1). On the other hand, mitochondria utilize lipids as a substrate for FA β-oxidation in order to meet high energy demand. Increased ROS production, secondary to stress conditions, inhibits mitochondrial FA β-oxidation and lipid utilization, which in turn generates more ROS (Figure 2). It is clear that ROS play a critical role in the interplay of mitochondrial dysfunction and glomerular lipotoxicity. Interweaved by ROS production, a vicious cycle is formed by down-regulated FA β-oxidation, accumulation of intracellular lipids and peroxidation of lipids (especially CL). Therefore, identifying therapeutic targets that break this vicious cycle may offer novel promising therapeutic interventions for the cure of glomerular diseases.
MG and FF conceived and wrote the manuscript. SM and AF critically reviewed and improved the entire manuscript. All authors contributed to the article and approved the submitted version.
AF and SM are supported by NIH grants R01DK117599, R01DK104753, and R01CA227493. AF is also supported by U54DK083912, UM1DK100846, U01DK116101, and UL1TR000460 (Miami Clinical Translational Science Institute). FF is supported by Department of Defense (DoD) Discovery Award (PR180598). MG is supported by a predoctoral fellowship of the American Heart Association (18PRE34030042).
AF and SM are inventors on pending or issued patents (PCT/US2019/041730 and PCT/US2019/032215) aimed at diagnosing or treating proteinuric kidney diseases. They stand to gain royalties from the future commercialization of these patents. AF is vice-president of L&F Health LLC and is a consultant for ZyVersa Therapeutics, Inc. ZyVersa Therapeutics, Inc. has licensed worldwide rights to develop and commercialize hydroxypropyl-beta-cyclodextrin from L&F Research for the treatment of kidney disease. AF is founder of LipoNexT LLC. SM is a consultant for Kintai Therapeutics, Inc. and holds equity interest in L&F Research. AF and SM are supported by Hoffman-La Roche and by Boehringer Ingelheim.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the Katz family for supporting this study. Figures were created with BioRender.
Abe, Y., Sakairi, T., Kajiyama, H., Shrivastav, S., Beeson, C., and Kopp, J. B. (2010). Bioenergetic characterization of mouse podocytes. Am. J. Physiol. Cell Physiol. 299, C464–C476. doi: 10.1152/ajpcell.00563.2009
Agarwal, R. (2006). Effects of statins on renal function. Am. J. Cardiol. 97, 748–755. doi: 10.1016/j.amjcard.2005.09.110
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi: 10.1038/290457a0
Aon, M. A., Bhatt, N., and Cortassa, S. C. (2014). Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 5:282. doi: 10.3389/fphys.2014.00282
Ashraf, S., Gee, H. Y., Woerner, S., Xie, L. X., Vega-Warner, V., Lovric, S., et al. (2013). ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Invest. 123, 5179–5189. doi: 10.1172/JCI69000
Baud, L., and Ardaillou, R. (1993). Involvement of reactive oxygen species in kidney damage. Br. Med. Bull. 49, 621–629. doi: 10.1093/oxfordjournals.bmb.a072635
Becher, M. W., Wills, M. L., Noll, W. W., Hurko, O., and Price, D. L. (1999). Kearns-Sayre syndrome with features of Pearson’s marrow-pancreas syndrome and a novel 2905-base pair mitochondrial DNA deletion. Hum. Pathol. 30, 577–581. doi: 10.1016/S0046-8177(99)90204-6
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646. doi: 10.1038/nrneph.2017.107
Blanco, S., Vaquero, M., Gomez-Guerrero, C., Lopez, D., Egido, J., and Romero, R. (2005). Potential role of angiotensin-converting enzyme inhibitors and statins on early podocyte damage in a model of type 2 diabetes mellitus, obesity, and mild hypertension. Am. J. Hypertens. 18, 557–565. doi: 10.1016/j.amjhyper.2004.10.034
Boren, J., and Brindle, K. M. (2012). Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 19, 1561–1570. doi: 10.1038/cdd.2012.34
Borysiewicz, L. K., Soutar, A. K., Evans, D. J., Thompson, G. R., and Rees, A. J. (1982). Renal failure in familial lecithin: cholesterol acyltransferase deficiency. Q. J. Med. 51, 411–426.
Buja, L. M., Kovanen, P. T., and Bilheimer, D. W. (1979). Cellular pathology of homozygous familial hypercholesterolemia. Am. J. Pathol. 97, 327–357.
Bussolati, B., Deregibus, M. C., Fonsato, V., Doublier, S., Spatola, T., Procida, S., et al. (2005). Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidylinositol 3-kinase/AKT-signaling pathway. J. Am. Soc. Nephrol. 16, 1936–1947. doi: 10.1681/ASN.2004080629
Calkin, A. C., Giunti, S., Jandeleit-Dahm, K. A., Allen, T. J., Cooper, M. E., and Thomas, M. C. (2006). PPAR-alpha and -gamma agonists attenuate diabetic kidney disease in the apolipoprotein E knockout mouse. Nephrol. Dial. Transplant. 21, 2399–2405. doi: 10.1093/ndt/gfl212
Calvo, S. E., Clauser, K. R., and Mootha, V. K. (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257. doi: 10.1093/nar/gkv1003
Che, R., Yuan, Y., Huang, S., and Zhang, A. (2014). Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal Physiol. 306, F367–F378. doi: 10.1152/ajprenal.00571.2013
Cheong, H. I., Chae, J. H., Kim, J. S., Park, H. W., Ha, I. S., Hwang, Y. S., et al. (1999). Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatr. Nephrol. 13, 477–480. doi: 10.1007/s004670050641
Claypool, S. M., and Koehler, C. L. (2012). The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41. doi: 10.1016/j.tibs.2011.09.003
Czajka, A., and Malik, A. N. (2016). Hyperglycemia induced damage to mitochondrial respiration in renal mesangial and tubular cells: implications for diabetic nephropathy. Redox Biol. 10, 100–107. doi: 10.1016/j.redox.2016.09.007
Daehn, I., Casalena, G., Zhang, T., Shi, S., Fenninger, F., Barasch, N., et al. (2014). Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Invest. 124, 1608–1621. doi: 10.1172/JCI71195
D’Agati, V. D., Kaskel, F. J., and Falk, R. J. (2011). Focal segmental glomerulosclerosis. N. Engl. J. Med. 365, 2398–2411. doi: 10.1056/NEJMra1106556
Decleves, A. E., Mathew, A. V., Cunard, R., and Sharma, K. (2011). AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 22, 1846–1855. doi: 10.1681/ASN.2011010026
Decleves, A. E., Zolkipli, Z., Satriano, J., Wang, L., Nakayama, T., Rogac, M., et al. (2014). Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 85, 611–623. doi: 10.1038/ki.2013.462
Desbats, M. A., Lunardi, G., Doimo, M., Trevisson, E., and Salviati, L. (2015). Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 38, 145–156. doi: 10.1007/s10545-014-9749-9
Di Donato, S. (2009). Multisystem manifestations of mitochondrial disorders. J. Neurol. 256, 693–710. doi: 10.1007/s00415-009-5028-3
Ding, Z., Liu, S., Wang, X., Deng, X., Fan, Y., Sun, C., et al. (2015). Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid. Redox Signal. 22, 760–771. doi: 10.1089/ars.2014.6054
Ding, Z., Liu, S., Wang, X., Mathur, P., Dai, Y., Theus, S., et al. (2016). Cross-talk between PCSK9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxid. Redox Signal. 25, 997–1008. doi: 10.1089/ars.2016.6631
Ducasa, G. M., Mitrofanova, A., and Fornoni, A. (2019a). Crosstalk between lipids and mitochondria in diabetic kidney disease. Curr. Diab. Rep. 19:144. doi: 10.1007/s11892-019-1263-x
Ducasa, G. M., Mitrofanova, A., Mallela, S. K., Liu, X., Molina, J., Sloan, A., et al. (2019b). ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Invest. 129, 3387–3400. doi: 10.1172/JCI125316
Dugan, L. L., You, Y. H., Ali, S. S., Diamond-Stanic, M., Miyamoto, S., DeCleves, A. E., et al. (2013). AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Invest. 123, 4888–4899. doi: 10.1172/JCI66218
Edwards, P. A., Tabor, D., Kast, H. R., and Venkateswaran, A. (2000). Regulation of gene expression by SREBP and SCAP. Biochim. Biophys. Acta 1529, 103–113. doi: 10.1016/s1388-1981(00)00140-2
Eid, N., Ito, Y., Maemura, K., and Otsuki, Y. (2013). Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J. Mol. Histol. 44, 311–326. doi: 10.1007/s10735-013-9483-x
Elmaci, A. M., Peru, H., Akin, F., Akcoren, Z., Caglar, M., and Ozel, A. (2007). A case of homozygous familial hypercholesterolemia with focal segmental glomerulosclerosis. Pediatr. Nephrol. 22, 1803–1805. doi: 10.1007/s00467-007-0534-y
Emanuelsson, F., Nordestgaard, B. G., and Benn, M. (2018). Familial hypercholesterolemia and risk of peripheral arterial disease and chronic kidney disease. J. Clin. Endocrinol. Metab. 103, 4491–4500. doi: 10.1210/jc.2018-01058
Eom, M., Hudkins, K. L., and Alpers, C. E. (2015). Foam cells and the pathogenesis of kidney disease. Curr. Opin. Nephrol. Hypertens. 24, 245–251. doi: 10.1097/MNH.0000000000000112
Ernster, L., and Dallner, G. (1995). Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1271, 195–204. doi: 10.1016/0925-4439(95)00028-3
Ferrans, V. J., and Fredrickson, D. S. (1975). The pathology of Tangier disease. A light and electron microscopic study. Am. J. Pathol. 78, 101–158.
Fink, B. D., Herlein, J. A., O’Malley, Y., and Sivitz, W. I. (2012). Endothelial cell and platelet bioenergetics: effect of glucose and nutrient composition. PLoS One 7:e39430. doi: 10.1371/journal.pone.0039430
Finsterer, J., and Scorza, F. A. (2017). Renal manifestations of primary mitochondrial disorders. Biomed. Rep. 6, 487–494. doi: 10.3892/br.2017.892
Fobker, M., Voss, R., Reinecke, H., Crone, C., Assmann, G., and Walter, M. (2001). Accumulation of cardiolipin and lysocardiolipin in fibroblasts from Tangier disease subjects. FEBS Lett. 500, 157–162. doi: 10.1016/S0014-5793(01)02578-9
Forbes, J. M., and Thorburn, D. R. (2018). Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 14, 291–312. doi: 10.1038/nrneph.2018.9
Fornoni, A., Merscher, S., and Kopp, J. B. (2014). Lipid biology of the podocyte--new perspectives offer new opportunities. Nat. Rev. Nephrol. 10, 379–388. doi: 10.1038/nrneph.2014.87
Gai, Z., Gui, T., Hiller, C., and Kullak-Ublick, G. A. (2016). Farnesoid X receptor protects against kidney injury in uninephrectomized obese mice. J. Biol. Chem. 291, 2397–2411. doi: 10.1074/jbc.M115.694323
Gai, Z., Wang, T., Visentin, M., Kullak-Ublick, G. A., Fu, X., and Wang, Z. (2019). Lipid accumulation and chronic kidney disease. Nutrients 11:722. doi: 10.3390/nu11040722
Gasser, D. L., Winkler, C. A., Peng, M., An, P., McKenzie, L. M., Kirk, G. D., et al. (2013). Focal segmental glomerulosclerosis is associated with a PDSS2 haplotype and, independently, with a decreased content of coenzyme Q10. Am. J. Physiol. Renal Physiol. 305, F1228–F1238. doi: 10.1152/ajprenal.00143.2013
Grafft, C. A., Fervenza, F. C., Semret, M. H., Orloff, S., and Sethi, S. (2009). Renal involvement in Neimann-Pick disease. NDT Plus 2, 448–451. doi: 10.1093/ndtplus/sfp101
Grone, H. J., Walli, A. K., Grone, E., Kramer, A., Clemens, M. R., and Seidel, D. (1990). Receptor mediated uptake of apo B and apo E rich lipoproteins by human glomerular epithelial cells. Kidney Int. 37, 1449–1459. doi: 10.1038/ki.1990.135
Gucer, S., Talim, B., Asan, E., Korkusuz, P., Ozen, S., Unal, S., et al. (2005). Focal segmental glomerulosclerosis associated with mitochondrial cytopathy: report of two cases with special emphasis on podocytes. Pediatr. Dev. Pathol. 8, 710–717. doi: 10.1007/s10024-005-0058-z
Gutwein, P., Abdel-Bakky, M. S., Doberstein, K., Schramme, A., Beckmann, J., Schaefer, L., et al. (2009a). CXCL16 and oxLDL are induced in the onset of diabetic nephropathy. J. Cell. Mol. Med. 13, 3809–3825. doi: 10.1111/j.1582-4934.2009.00761.x
Gutwein, P., Abdel-Bakky, M. S., Schramme, A., Doberstein, K., Kampfer-Kolb, N., Amann, K., et al. (2009b). CXCL16 is expressed in podocytes and acts as a scavenger receptor for oxidized low-density lipoprotein. Am. J. Pathol. 174, 2061–2072. doi: 10.2353/ajpath.2009.080960
Haas, M. E., Levenson, A. E., Sun, X., Liao, W. H., Rutkowski, J. M., de Ferranti, S. D., et al. (2016). The role of proprotein convertase subtilisin/kexin type 9 in nephrotic syndrome-associated hypercholesterolemia. Circulation 134, 61–72. doi: 10.1161/CIRCULATIONAHA.115.020912
Hallan, S., Afkarian, M., Zelnick, L. R., Kestenbaum, B., Sharma, S., Saito, R., et al. (2017). Metabolomics and gene expression analysis reveal down-regulation of the citric acid (TCA) cycle in non-diabetic CKD patients. EBioMedicine 26, 68–77. doi: 10.1016/j.ebiom.2017.10.027
Hansell, P., Welch, W. J., Blantz, R. C., and Palm, F. (2013). Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 40, 123–137. doi: 10.1111/1440-1681.12034
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
Heeringa, S. F., Chernin, G., Chaki, M., Zhou, W., Sloan, A. J., Ji, Z., et al. (2011). COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Invest. 121, 2013–2024. doi: 10.1172/JCI45693
Heinz, L. X., Baumann, C. L., Koberlin, M. S., Snijder, B., Gawish, R., Shui, G., et al. (2015). The lipid-modifying enzyme SMPDL3B negatively regulates innate immunity. Cell Rep. 11, 1919–1928. doi: 10.1016/j.celrep.2015.05.006
Herman-Edelstein, M., Scherzer, P., Tobar, A., Levi, M., and Gafter, U. (2014). Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 55, 561–572. doi: 10.1194/jlr.P040501
Heymann, W., Hackel, D. B., Harwood, S., Wilson, S. G., and Hunter, J. L. (1959). Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc. Soc. Exp. Biol. Med. 100, 660–664. doi: 10.3181/00379727-100-24736
Hodgin, J. B., Nair, V., Zhang, H., Randolph, A., Harris, R. C., Nelson, R. G., et al. (2013). Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 62, 299–308. doi: 10.2337/db11-1667
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131. doi: 10.1172/JCI0215593
Hotta, O., Inoue, C. N., Miyabayashi, S., Furuta, T., Takeuchi, A., and Taguma, Y. (2001). Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 59, 1236–1243. doi: 10.1046/j.1523-1755.2001.0590041236.x
Idrovo, J. P., Yang, W. L., Nicastro, J., Coppa, G. F., and Wang, P. (2012). Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J. Surg. Res. 177, 157–164. doi: 10.1016/j.jss.2012.05.053
Imasawa, T., Obre, E., Bellance, N., Lavie, J., Imasawa, T., Rigothier, C., et al. (2017). High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J. 31, 294–307. doi: 10.1096/fj.201600293R
Jeong, H. J., Lee, H. S., Kim, K. S., Kim, Y. K., Yoon, D., and Park, S. W. (2008). Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J. Lipid Res. 49, 399–409. doi: 10.1194/jlr.M700443-JLR200
Jiang, T., Wang, Z., Proctor, G., Moskowitz, S., Liebman, S. E., Rogers, T., et al. (2005). Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 280, 32317–32325. doi: 10.1074/jbc.M500801200
Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232. doi: 10.1038/nchembio727
Kang, H. M., Ahn, S. H., Choi, P., Ko, Y. A., Han, S. H., Chinga, F., et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46. doi: 10.1038/nm.3762
Kawakami, T., Gomez, I. G., Ren, S., Hudkins, K., Roach, A., Alpers, C. E., et al. (2015). Deficient autophagy results in mitochondrial dysfunction and FSGS. J. Am. Soc. Nephrol. 26, 1040–1052. doi: 10.1681/ASN.2013111202
Ke, Q., Yuan, Q., Qin, N., Shi, C., Luo, J., Fang, Y., et al. (2020). UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis. 11:26. doi: 10.1038/s41419-019-2219-4
Keech, A., Simes, R. J., Barter, P., Best, J., Scott, R., Taskinen, M. R., et al. (2005). Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 366, 1849–1861. doi: 10.1016/S0140-6736(05)67667-2
Kerjaschki, D., Exner, M., Ullrich, R., Susani, M., Curtiss, L. K., Witztum, J. L., et al. (1997). Pathogenic antibodies inhibit the binding of apolipoproteins to megalin/gp330 in passive Heymann nephritis. J. Clin. Invest. 100, 2303–2309. doi: 10.1172/JCI119768
Kriz, W., Gretz, N., and Lemley, K. V. (1998). Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 54, 687–697. doi: 10.1046/j.1523-1755.1998.00044.x
Kruger, C., Burke, S. J., Collier, J. J., Nguyen, T. T., Salbaum, J. M., and Stadler, K. (2018). Lipid peroxidation regulates podocyte migration and cytoskeletal structure through redox sensitive RhoA signaling. Redox Biol. 16, 248–254. doi: 10.1016/j.redox.2018.02.024
Kurogouchi, F., Oguchi, T., Mawatari, E., Yamaura, S., Hora, K., Takei, M., et al. (1998). A case of mitochondrial cytopathy with a typical point mutation for MELAS, presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation. Am. J. Nephrol. 18, 551–556. doi: 10.1159/000013406
Lackner, L. (2014). Shaping the dynamic mitochondrial network. BMC Biol. 12:35. doi: 10.1186/1741-7007-1112-1135
Lambert, G., Sakai, N., Vaisman, B. L., Neufeld, E. B., Marteyn, B., Chan, C. C., et al. (2001). Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J. Biol. Chem. 276, 15090–15098. doi: 10.1074/jbc.M008466200
Lee, E., Choi, J., and Lee, H. S. (2017). Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J. Cell. Physiol. 232, 3209–3217. doi: 10.1002/jcp.25867
Lee, E., and Lee, H. S. (2018). Peroxidase expression is decreased by palmitate in cultured podocytes but increased in podocytes of advanced diabetic nephropathy. J. Cell. Physiol. 233, 9060–9069. doi: 10.1002/jcp.26875
Lenaz, G., and Genova, M. L. (2010). Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid. Redox Signal. 12, 961–1008. doi: 10.1089/ars.2009.2704
Li, S. Y., Park, J., Qiu, C., Han, S. H., Palmer, M. B., Arany, Z., et al. (2017). Increasing the level of peroxisome proliferator-activated receptor gamma coactivator-1alpha in podocytes results in collapsing glomerulopathy. JCI Insight 2:e92930. doi: 10.1172/jci.insight.92930
Lin, C. Y., Lee, T. S., Chen, C. C., Chang, C. A., Lin, Y. J., Hsu, Y. P., et al. (2011). Endothelin-1 exacerbates lipid accumulation by increasing the protein degradation of the ATP-binding cassette transporter G1 in macrophages. J. Cell. Physiol. 226, 2198–2205. doi: 10.1002/jcp.22556
Liu, T., Chen, X. M., Sun, J. Y., Jiang, X. S., Wu, Y., Yang, S., et al. (2018). Palmitic acid-induced podocyte apoptosis via the reactive oxygen species-dependent mitochondrial pathway. Kidney Blood Press. Res. 43, 206–219. doi: 10.1159/000487673
Lopez, L. C., Schuelke, M., Quinzii, C. M., Kanki, T., Rodenburg, R. J., Naini, A., et al. (2006). Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 79, 1125–1129. doi: 10.1086/510023
Lovric, S., Ashraf, S., Tan, W., and Hildebrandt, F. (2016). Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol. Dial. Transplant. 31, 1802–1813. doi: 10.1093/ndt/gfv355
Martinez-Garcia, C., Izquierdo, A., Velagapudi, V., Vivas, Y., Velasco, I., Campbell, M., et al. (2012). Accelerated renal disease is associated with the development of metabolic syndrome in a glucolipotoxic mouse model. Dis. Model. Mech. 5, 636–648. doi: 10.1242/dmm.009266
Martinez-Garcia, C., Izquierdo-Lahuerta, A., Vivas, Y., Velasco, I., Yeo, T. K., Chen, S., et al. (2015). Renal lipotoxicity-associated inflammation and insulin resistance affects actin cytoskeleton organization in podocytes. PLoS One 10:e0142291. doi: 10.1371/journal.pone.0142291
Merscher-Gomez, S., Guzman, J., Pedigo, C. E., Lehto, M., Aguillon-Prada, R., Mendez, A., et al. (2013). Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 62, 3817–3827. doi: 10.2337/db13-0399
Meulemans, A., Seneca, S., Lagae, L., Lissens, W., De Paepe, B., Smet, J., et al. (2006). A novel mitochondrial transfer RNA(Asn) mutation causing multiorgan failure. Arch. Neurol. 63, 1194–1198. doi: 10.1001/archneur.63.8.1194
Mitrofanova, A., Molina, J., Varona Santos, J., Guzman, J., Morales, X. A., Ducasa, G. M., et al. (2018). Hydroxypropyl-beta-cyclodextrin protects from kidney disease in experimental alport syndrome and focal segmental glomerulosclerosis. Kidney Int. 94, 1151–1159. doi: 10.1016/j.kint.2018.06.031
Narumi, K., Mishima, E., Akiyama, Y., Matsuhashi, T., Nakamichi, T., Kisu, K., et al. (2018). Focal segmental glomerulosclerosis associated with chronic progressive external ophthalmoplegia and mitochondrial DNA A3243G mutation. Nephron 138, 243–248. doi: 10.1159/000485109
Nolasco, F., Cameron, J. S., Reuben, R., Hartley, B., Carvalho, F., and Coelho, R. A. (1985). Interstitial foam cells in the nephrotic syndrome belong to the monocyte/macrophage lineage. Proc. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 21, 666–671.
Ossoli, A., Lucca, F., Boscutti, G., Remaley, A. T., and Calabresi, L. (2015). Familial LCAT deficiency: from pathology to enzyme replacement therapy. J. Clin. Lipidol. 10, 405–413. doi: 10.2217/clp.15.34
Owen, O., Kalhan, S., and Hanson, R. (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412. doi: 10.1074/jbc.R200006200
Palomer, X., Pizarro-Delgado, J., Barroso, E., and Vazquez-Carrera, M. (2018). Palmitic and oleic acid: the yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol. Metab. 29, 178–190. doi: 10.1016/j.tem.2017.11.009
Pedigo, C. E., Ducasa, G. M., Leclercq, F., Sloan, A., Mitrofanova, A., Hashmi, T., et al. (2016). Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J. Clin. Invest. 126, 3336–3350. doi: 10.1172/JCI85939
Pfeiffer, K., Gohil, V., Stuart, R. A., Hunte, C., Brandt, U., Greenberg, M. L., et al. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880. doi: 10.1074/jbc.M308366200
Picard, M., Jung, B., Liang, F., Azuelos, I., Hussain, S., Goldberg, P., et al. (2012). Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am. J. Respir. Crit. Care Med. 186, 1140–1149. doi: 10.1164/rccm.201206-0982OC
Qi, H., Casalena, G., Shi, S., Yu, L., Ebefors, K., Sun, Y., et al. (2017). Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes 66, 763–778. doi: 10.2337/db16-0695
Rinschen, M. M., Palygin, O., Guijas, C., Palermo, A., Palacio-Escat, N., Domingo-Almenara, X., et al. (2019). Metabolic rewiring of the hypertensive kidney. Sci. Signal. 12:eaax9760. doi: 10.1126/scisignal.aax9760
Rong, S., Cortes, V. A., Rashid, S., Anderson, N. N., McDonald, J. G., Liang, G., et al. (2017). Expression of SREBP-1c requires SREBP-2-mediated generation of a sterol ligand for LXR in livers of mice. elife 6:e25015. doi: 10.7554/eLife.25015
Rousset, S., Alves-Guerra, M. C., Mozo, J., Miroux, B., Cassard-Doulcier, A. M., Bouillaud, F., et al. (2004). The biology of mitochondrial uncoupling proteins. Diabetes 53(Suppl. 1), S130–S135. doi: 10.2337/diabetes.53.2007.S130
Ruan, X. Z., Moorhead, J. F., Fernando, R., Wheeler, D. C., Powis, S. H., and Varghese, Z. (2003). PPAR agonists protect mesangial cells from interleukin 1beta-induced intracellular lipid accumulation by activating the ABCA1 cholesterol efflux pathway. J. Am. Soc. Nephrol. 14, 593–600. doi: 10.1097/01.asn.0000050414.52908.da
Ruan, X. Z., Varghese, Z., and Moorhead, J. F. (2009). An update on the lipid nephrotoxicity hypothesis. Nat. Rev. Nephrol. 5, 713–721. doi: 10.1038/nrneph.2009.184
Rudnicki, M., Mayr, J. A., Zschocke, J., Antretter, H., Regele, H., Feichtinger, R. G., et al. (2016). MELAS syndrome and kidney disease without fanconi syndrome or proteinuria: a case report. Am. J. Kidney Dis. 68, 949–953. doi: 10.1053/j.ajkd.2016.06.027
Saiki, R., Lunceford, A. L., Shi, Y., Marbois, B., King, R., Pachuski, J., et al. (2008). Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Renal Physiol. 295, F1535–F1544. doi: 10.1152/ajprenal.90445.2008
Saiki, R., Nagata, A., Kainou, T., Matsuda, H., and Kawamukai, M. (2005). Characterization of solanesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J. 272, 5606–5622. doi: 10.1111/j.1742-4658.2005.04956.x
Sam, R., Wu, H., Yue, L., Mazzone, T., Schwartz, M. M., Arruda, J. A., et al. (2006). Lipoprotein glomerulopathy: a new apolipoprotein E mutation with enhanced glomerular binding. Am. J. Kidney Dis. 47, 539–548. doi: 10.1053/j.ajkd.2005.12.031
Sato, S., Kitamura, H., Adachi, A., Sasaki, Y., and Ghazizadeh, M. (2006). Two types of autophagy in the podocytes in renal biopsy specimens: ultrastructural study. J. Submicrosc. Cytol. Pathol. 38, 167–174.
Scaglia, F., Vogel, H., Hawkins, E. P., Vladutiu, G. D., Liu, L. L., and Wong, L. J. (2003). Novel homoplasmic mutation in the mitochondrial tRNATyr gene associated with atypical mitochondrial cytopathy presenting with focal segmental glomerulosclerosis. Am. J. Med. Genet. A 123A, 172–178. doi: 10.1002/ajmg.a.20315
Schaefer, E. J., Santos, R. D., and Asztalos, B. F. (2010). Marked HDL deficiency and premature coronary heart disease. Curr. Opin. Lipidol. 21, 289–297. doi: 10.1097/MOL.0b013e32833c1ef6
Schmit, D., Fliser, D., and Speer, T. (2019). Proprotein convertase subtilisin/kexin type 9 in kidney disease. Nephrol. Dial. Transplant. 34, 1266–1271. doi: 10.1093/ndt/gfz122
Schrauwen-Hinderling, V. B., Kooi, M. E., and Schrauwen, P. (2016). Mitochondrial function and diabetes: consequences for skeletal and cardiac muscle metabolism. Antioxid. Redox Signal. 24, 39–51. doi: 10.1089/ars.2015.6291
Serezani, C. H., Kane, S., Medeiros, A. I., Cornett, A. M., Kim, S. H., Marques, M. M., et al. (2012). PTEN directly activates the actin depolymerization factor cofilin-1 during PGE2-mediated inhibition of phagocytosis of fungi. Sci. Signal. 5:ra12. doi: 10.1126/scisignal.2002448
Shi, Y., Wang, C., Zhou, X., Li, Y., Ma, Y., Zhang, R., et al. (2019). Downregulation of PTEN promotes podocyte endocytosis of lipids aggravating obesity-related glomerulopathy. Am. J. Physiol. Renal Physiol. 318, F589–F599. doi: 10.1152/ajprenal.00392.2019
Shrestha, P., van de Sluis, B., Dullaart, R. P. F., and van den Born, J. (2019). Novel aspects of PCSK9 and lipoprotein receptors in renal disease-related dyslipidemia. Cell. Signal. 55, 53–64. doi: 10.1016/j.cellsig.2018.12.001
Simons, M., Saffrich, R., Reiser, J., and Mundel, P. (1999). Directed membrane transport is involved in process formation in cultured podocytes. J. Am. Soc. Nephrol. 10, 1633–1639.
Simons, M., Schwarz, K., Kriz, W., Miettinen, A., Reiser, J., Mundel, P., et al. (2001). Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am. J. Pathol. 159, 1069–1077. doi: 10.1016/S0002-9440(10)61782-8
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Sourris, K. C., Harcourt, B. E., Tang, P. H., Morley, A. L., Huynh, K., Penfold, S. A., et al. (2012). Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 52, 716–723. doi: 10.1016/j.freeradbiomed.2011.11.017
Spiegelman, B. M. (2007). Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found. Symp. 287, 60–63.
Starr, M. C., Chang, I. J., Finn, L. S., Sun, A., Larson, A. A., Goebel, J., et al. (2018). COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr. Nephrol. 33, 1257–1261. doi: 10.1007/s00467-018-3937-z
Steinberg, G. R., Michell, B. J., van Denderen, B. J., Watt, M. J., Carey, A. L., Fam, B. C., et al. (2006). Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 4, 465–474. doi: 10.1016/j.cmet.2006.11.005
Stokes, M. B., Valeri, A. M., Markowitz, G. S., and D’Agati, V. D. (2006). Cellular focal segmental glomerulosclerosis: clinical and pathologic features. Kidney Int. 70, 1783–1792. doi: 10.1038/sj.ki.5001903
Stowe, D. F., and Camara, A. K. (2009). Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid. Redox Signal. 11, 1373–1414. doi: 10.1089/ars.2008.2331
Sun, L., Halaihel, N., Zhang, W., Rogers, T., and Levi, M. (2002). Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J. Biol. Chem. 277, 18919–18927. doi: 10.1074/jbc.M110650200
Szeto, H. H. (2014). First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 171, 2029–2050. doi: 10.1111/bph.12461
Szeto, H. H., and Birk, A. V. (2014). Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 96, 672–683. doi: 10.1038/clpt.2014.174
Szeto, H. H., Liu, S., Soong, Y., Alam, N., Prusky, G. T., and Seshan, S. V. (2016). Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 90, 997–1011. doi: 10.1016/j.kint.2016.06.013
Tanaka, Y., Kume, S., Araki, S., Isshiki, K., Chin-Kanasaki, M., Sakaguchi, M., et al. (2011). Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 79, 871–882. doi: 10.1038/ki.2010.530
Taniike, M., Fukushima, H., Yanagihara, I., Tsukamoto, H., Tanaka, J., Fujimura, H., et al. (1992). Mitochondrial tRNA(Ile) mutation in fatal cardiomyopathy. Biochem. Biophys. Res. Commun. 186, 47–53. doi: 10.1016/S0006-291X(05)80773-9
Tsimihodimos, V., and Elisaf, M. (2011). Lipoprotein glomerulopathy. Curr. Opin. Lipidol. 22, 262–269. doi: 10.1097/MOL.0b013e328345ebb0
Turnbull, D. M., and Rustin, P. (2016). Genetic and biochemical intricacy shapes mitochondrial cytopathies. Neurobiol. Dis. 92, 55–63. doi: 10.1016/j.nbd.2015.02.003
Vafai, S. B., and Mootha, V. K. (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. doi: 10.1038/nature11707
Vankoningsloo, S., Piens, M., Lecocq, C., Gilson, A., De Pauw, A., Renard, P., et al. (2005). Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. J. Lipid Res. 46, 1133–1149. doi: 10.1194/jlr.M400464-JLR200
Vasta, V., Merritt, J. L. 2nd, Saneto, R. P., and Hahn, S. H. (2012). Next-generation sequencing for mitochondrial diseases: a wide diagnostic spectrum. Pediatr. Int. 54, 585–601. doi: 10.1111/j.1442-200X.2012.03644.x
Vincent, A. E., Ng, Y. S., White, K., Davey, T., Mannella, C., Falkous, G., et al. (2016). The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci. Rep. 6:30610. doi: 10.1038/srep30610
Wang, X. X., Jiang, T., Shen, Y., Adorini, L., Pruzanski, M., Gonzalez, F. J., et al. (2009). The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am. J. Physiol. Renal Physiol. 297, F1587–F1596. doi: 10.1152/ajprenal.00404.2009
Wang, X. X., Luo, Y., Wang, D., Adorini, L., Pruzanski, M., Dobrinskikh, E., et al. (2017). A dual agonist of farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5, INT-767, reverses age-related kidney disease in mice. J. Biol. Chem. 292, 12018–12024. doi: 10.1074/jbc.C117.794982
Wang, X. X., Wang, D., Luo, Y., Myakala, K., Dobrinskikh, E., Rosenberg, A. Z., et al. (2018). FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J. Am. Soc. Nephrol. 29, 118–137. doi: 10.1681/ASN.2017020222
Wang, Z., Ying, Z., Bosy-Westphal, A., Zhang, J., Schautz, B., Later, W., et al. (2010). Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 92, 1369–1377. doi: 10.3945/ajcn.2010.29885
Widmeier, E., Yu, S., Nag, A., Chung, Y. W., Nakayama, M., Fernandez-Del-Rio, L., et al. (2020). ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. 31, 1191–1211. doi: 10.1681/ASN.2019070756
Wu, Y., Chen, Y., Chen, D., Zeng, C., Li, L., and Liu, Z. (2009). Presence of foam cells in kidney interstitium is associated with progression of renal injury in patients with glomerular diseases. Nephron Clin. Pract. 113, c155–c161. doi: 10.1159/000232596
Xu, S., Nam, S. M., Kim, J. H., Das, R., Choi, S. K., Nguyen, T. T., et al. (2015). Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 6:e1976. doi: 10.1038/cddis.2015.331
Yang, J., Yang, Y., and Hu, Z. (2018). A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy. Biochem. Biophys. Res. Commun. 506, 444–449. doi: 10.1016/j.bbrc.2018.10.102
Yoo, T. H., Pedigo, C. E., Guzman, J., Correa-Medina, M., Wei, C., Villarreal, R., et al. (2015). Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J. Am. Soc. Nephrol. 26, 133–147. doi: 10.1681/ASN.2013111213
Yu, X. H., Fu, Y. C., Zhang, D. W., Yin, K., and Tang, C. K. (2013). Foam cells in atherosclerosis. Clin. Chim. Acta 424, 245–252. doi: 10.1016/j.cca.2013.06.006
Yuan, Z., Cao, A., Liu, H., Guo, H., Zang, Y., Wang, Y., et al. (2017). Calcium uptake via mitochondrial uniporter contributes to palmitic acid-induced apoptosis in mouse podocytes. J. Cell. Biochem. 118, 2809–2818. doi: 10.1002/jcb.25930
Zhang, Y., Ma, K. L., Liu, J., Wu, Y., Hu, Z. B., Liu, L., et al. (2015a). Dysregulation of low-density lipoprotein receptor contributes to podocyte injuries in diabetic nephropathy. Am. J. Physiol. Endocrinol. Metab. 308, E1140–E1148. doi: 10.1152/ajpendo.00591.2014
Zhang, Y., Ma, K. L., Liu, J., Wu, Y., Hu, Z. B., Liu, L., et al. (2015b). Inflammatory stress exacerbates lipid accumulation and podocyte injuries in diabetic nephropathy. Acta Diabetol. 52, 1045–1056. doi: 10.1007/s00592-015-0753-9
Zhao, K., Zhao, G. M., Wu, D., Soong, Y., Birk, A. V., Schiller, P. W., et al. (2004). Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 279, 34682–34690. doi: 10.1074/jbc.M402999200
Zheng, F., Fornoni, A., Elliot, S. J., Guan, Y., Breyer, M. D., Striker, L. J., et al. (2002). Upregulation of type I collagen by TGF-beta in mesangial cells is blocked by PPARgamma activation. Am. J. Physiol. Renal Physiol. 282, F639–F648. doi: 10.1152/ajprenal.00189.2001
Zhou, Y., Kong, X., Zhao, P., Yang, H., Chen, L., Miao, J., et al. (2011). Peroxisome proliferator-activated receptor-alpha is renoprotective in doxorubicin-induced glomerular injury. Kidney Int. 79, 1302–1311. doi: 10.1038/ki.2011.17
Keywords: kidney, glomerular diseases, lipotoxicity, mitochondrial dysfunction, oxidative stress
Citation: Ge M, Fontanesi F, Merscher S and Fornoni A (2020) The Vicious Cycle of Renal Lipotoxicity and Mitochondrial Dysfunction. Front. Physiol. 11:732. doi: 10.3389/fphys.2020.00732
Edited by:
Egor Plotnikov, Lomonosov Moscow State University, RussiaReviewed by:
Mark Cooper, Monash University, Australia*Correspondence: Alessia Fornoni, YWZvcm5vbmlAbWVkLm1pYW1pLmVkdQ==
Copyright © 2020 Ge, Fontanesi, Merscher and Fornoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.